ABSTRACT
There are currently several different classes of drugs available for lowering cholesterol levels. There are currently seven HMG-CoA reductase inhibitors (statins) approved for lowering cholesterol levels and they are the first line drugs for treating cholesterol disorders and can lower LDL-C levels by as much as 60%. Statins also are effective in reducing triglyceride levels in patients with hypertriglyceridemia. Statins lower LDL levels by inhibiting HMG-CoA reductase activity leading to decreases in hepatic cholesterol content resulting in an up-regulation of hepatic LDL receptors, which increases the clearance of LDL. The major side effects are muscle complications and an increased risk of diabetes. The different statins have varying drug interactions. Ezetimibe lowers LDL-C levels by approximately 20% by inhibiting cholesterol absorption by the intestines leading to the decreased delivery of cholesterol to the liver, a decrease in hepatic cholesterol content, and an up-regulation of hepatic LDL receptors. Ezetimibe is very useful as add on therapy when statin therapy is not sufficient or in statin intolerant patients. Ezetimibe has few side effects. Bile acid sequestrants lower LDL-C by10-30% by decreasing the absorption of bile acids in the intestine which decreases the bile acid pool consequently stimulating the synthesis of bile acids from cholesterol leading to a decrease in hepatic cholesterol content and an up-regulation of hepatic LDL receptors. Bile acid sequestrants can be difficult to use as they decrease the absorption of multiple drugs, may increase triglyceride levels, and cause constipation and other GI side effects. They do improve glycemic control in patients with diabetes, which is an additional benefit. PCSK9 inhibitors, either monoclonal antibodies or small interfering RNA, lower LDL-C by 50-60% by decreasing PCSK9, which decreases the degradation of LDL receptors. PCSK9 inhibitors also decrease Lp(a) levels. PCSK9 inhibitors are very useful when maximally tolerated statin therapy do not reduce LDL sufficiently and in statin intolerant patients. PCSK9 inhibitors have very few side effects. Bempedoic acid lowers LDL-C by 15-25% by inhibiting hepatic ATP citrate lyase activity resulting in a decrease in cholesterol synthesis in the liver, a decrease in hepatic cholesterol content, and an up-regulation of LDL receptors. Bempedoic acid is employed in patients who do not reach their LDL-C goals on maximally tolerated statin therapy or in patients who do not tolerate statins. Bempedoic acid is associated with elevations in uric acid levels and gouty attacks. Lomitapide and evinacumab are approved for lowering LDL levels in patients with homozygous familiar hypercholesterolemia, as they are not dependent on LDL receptors for decreasing LDL levels. Lomitapide inhibits microsomal triglyceride transfer protein decreasing the formation of chylomicrons in the intestine and VLDL in the liver. Lomitapide has the potential to cause liver toxicity and therefore they were approved with a risk evaluation and mitigation strategy (REMS) to reduce risk. Evinacumab is a monoclonal antibody that inhibits the activity of angiopoietin-like protein 3 resulting in the increased activity of lipoprotein lipase and endothelial cell lipase resulting in a decrease in LDL-C, HDL-C, and triglyceride levels. Mipomersen, which is no longer available, is a second-generation apolipoprotein anti-sense oligonucleotide that decreases apolipoprotein B synthesis resulting in a reduction in the formation and synthesis of VLDL and was approved for the treatment of homozygous familial hypercholesterolemia.
INTRODUCTION
This chapter will discuss the currently available drugs for lowering total cholesterol levels, especially LDL-C: statins, ezetimibe, bile acid sequestrants, PCSK9 inhibitors, bempedoic acid, lomitapide, mipomersen, and evinacumab. We will not discuss the effect of lifestyle changes or food additives, such as phytosterols, on LDL-C as this is addressed in the chapter entitled “The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels” (1). Additionally, we will not discuss guidelines for determining who to treat, how aggressively to treat, or targets of treatment as these issues are discussed in detail in the chapters entitled “Guidelines for the Management of High Blood Cholesterol” and “Approach to the Patient with Dyslipidemia” (2,3).
STATINS
Introduction
In the 1970s Dr. Akira Endo, working at Sankyo, discovered that compounds isolated from fungi inhibited the activity of HMG-CoA reductase, a key enzyme in the synthesis of cholesterol (4). Further studies at Merck led to the development of the first HMG-CoA reductase inhibitor, lovastatin, approved in 1987 for the treatment of hypercholesterolemia (5). There are currently seven HMG-CoA reductase inhibitors (statins) approved in the United States for lowering cholesterol levels. Three statins are derived from fungi (lovastatin, simvastatin, and pravastatin) and four statins are synthesized (atorvastatin, rosuvastatin, fluvastatin, and pitavastatin). Most of these statins are now generic drugs and therefore they are relatively inexpensive. Which particular statin one elects to use may depend on the degree of cholesterol lowering needed and the potential of drug-drug interactions. Statins are the first line drugs for treating elevated cholesterol levels and therefore one of the most widely utilized class of drugs. Statins have revolutionized the field of preventive cardiology and made an important contribution to the reduction in atherosclerotic cardiovascular events.
Effect on Statins on Lipid and Lipoprotein Levels
The major effect of statins is lowering LDL-C levels. The effect of the various statins at different doses on LDL-C levels is shown in Table 1. As can be seen in Table 1 different statins have varying abilities to lower LDL-C with maximal reductions of approximately 60% seen with rosuvastatin 40mg. Doubling the dose of a statin results in an approximate 6% further decrease in LDL-C levels. The percent reduction in LDL-C levels is similar in patients with high and low starting LDL-C levels but the absolute decrease is greater if the starting LDL-C is high. Because of this profound ability of statins to lower LDL-C levels, treatment with these drugs as monotherapy is often sufficient to lower LDL-C below target levels.
|
Table 1. Approximate Effect of Different Doses of Statins on LDL-C Levels
|
|
% LDL Reduction
|
Simvastatin (Zocor)
|
Atorvastatin (Lipitor)
|
Lovastatin (Mevacor)
|
Pravastatin (Pravachol)
|
Fluvastatin (Lescol)
|
Rosuvastatin (Crestor)
|
Pitavastatin (Livalo)
|
|
27
|
10mg
|
-
|
20mg
|
20mg
|
40mg
|
-
|
-
|
|
34
|
20mg
|
10mg
|
40mg
|
40mg
|
80mg
|
-
|
1mg
|
|
41
|
40mg
|
20mg
|
80mg
|
80mg
|
-
|
-
|
2mg
|
|
48
|
80mg
|
40mg
|
-
|
-
|
-
|
10mg
|
4mg
|
|
54
|
-
|
80mg
|
-
|
-
|
-
|
20mg
|
-
|
|
60
|
-
|
-
|
-
|
-
|
-
|
40mg
|
-
|
Data modified from package inserts
As would be predicted from the effect of statins on LDL-C levels, statins are also very effective in lowering non-HDL-C levels (LDL-C is the major contributor to non-HDL-C levels) (6,7). In addition, statins also lower plasma triglyceride levels (8,9). The ability of statins to lower triglyceride levels correlates with the reduction in LDL-C (9). Statins that are most efficacious in lowering LDL-C are also most efficacious in lowering plasma triglyceride and VLDL-C levels. Notably the percent reduction in plasma triglyceride levels is dependent on the baseline triglyceride levels (9). For example, in patients with normal triglyceride levels (<150mg/dL), simvastatin 80mg per day lowered plasma triglyceride levels by 11%. In contrast, if plasma triglyceride levels were elevated (> 250mg/dL), simvastatin 80mg per day lowered triglyceride levels by 40% (9). In patients with both elevated LDL-C and triglyceride levels statin therapy can be very effective in improving the lipid profile and are therefore the first line class of drugs to treat patients with mixed hyperlipidemia unless the triglyceride levels are markedly elevated (>500-1000mg/dL). As expected, given the ability of statins to lower LDL-C and triglyceride/VLDL levels, statin therapy is very effective in lowering apolipoprotein B levels (6,7).
Of note despite the ability of statins to lower LDL-C, non-HDL-C, and apolipoprotein B levels, statins do not lower Lp(a) levels and may even increase levels (10,11). Finally, statins modestly increase HDL-C levels (8,12,13). In most studies HDL-C levels increase between 5-10% with statin therapy. Interestingly, while low dose atorvastatin increases HDL levels similar to other statins at high doses the effect of atorvastatin is blunted with either very modest increases or no change observed (12).
|
Table 2. Effect of Statins on Lipid/Lipoprotein Levels
|
|
LDL-C
|
Decrease
|
|
Non-HDL-C
|
Decrease
|
|
Apolipoprotein B
|
Decrease
|
|
Triglycerides
|
Variable. If TG levels increased will decrease
|
|
HDL-C
|
Small Increase
|
|
Lp(a)
|
No change or small increase
|
Non-Lipid Effects of Statins
In addition to effects on lipid metabolism statins also have pleiotropic effects that may not be directly related to alterations in lipid metabolism (14). For example, statins are anti-inflammatory and consistently decrease CRP levels (15). Other pleiotropic effects of statins include anti-proliferative effects, antioxidant properties, anti-thrombosis, improving endothelial dysfunction, and attenuating vascular remodeling (14). Whether these pleiotropic effects contribute to the beneficial effects of statins in preventing cardiovascular disease is uncertain and much of the beneficial effect of statins on cardiovascular disease can be attributed to reductions in lipid levels.
Mechanism Accounting for the Statin Induced Lipid Effects
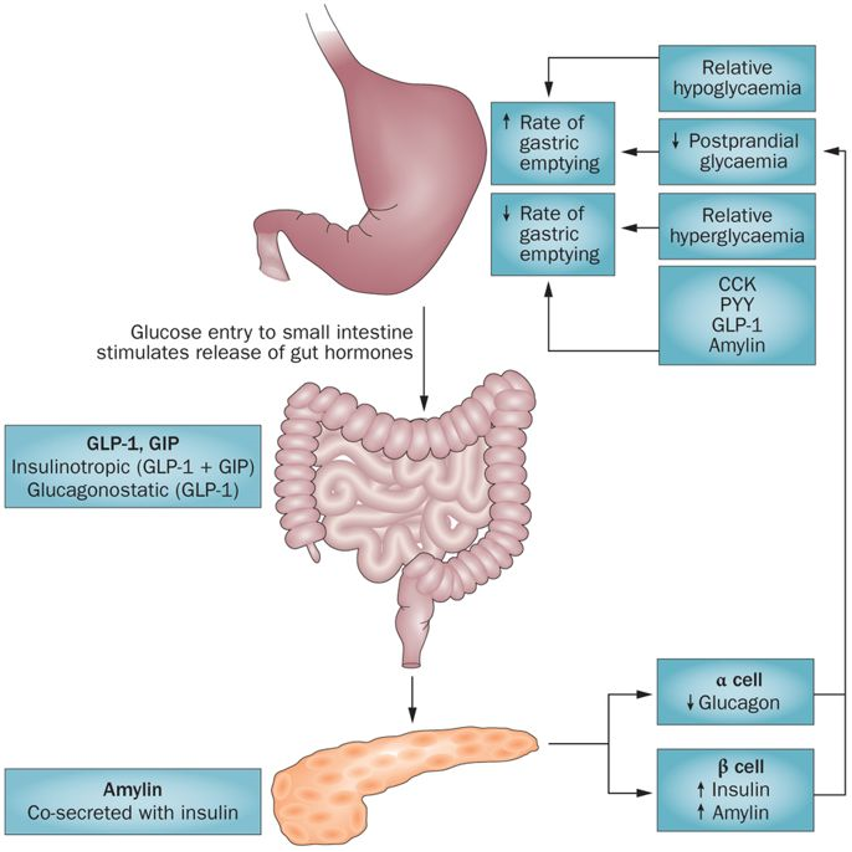
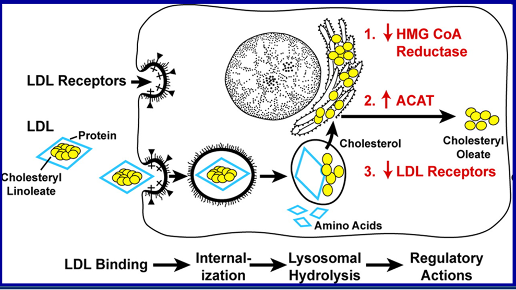
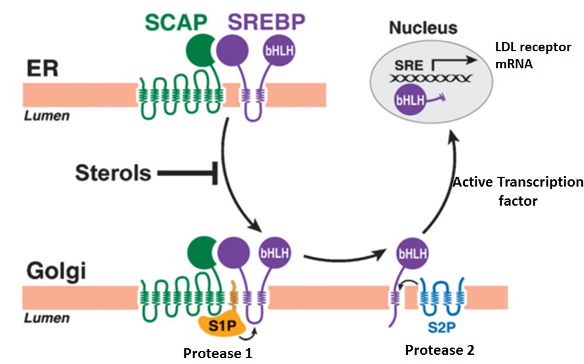
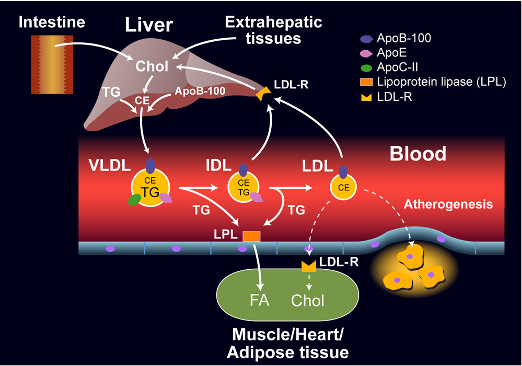
Statins are competitive inhibitors of HMG-CoA reductase, which leads to a decrease in cholesterol synthesis in the liver. This inhibition of hepatic cholesterol synthesis results in a decrease in cholesterol in the endoplasmic reticulum resulting in the movement of sterol regulatory element binding proteins (SREBPs) from the endoplasmic reticulum to the golgi where they are cleaved by proteases into active transcription factors (16). The SREBPs then translocate to the nucleus where they increase the expression of a number of genes including HMG-CoA reductase and, most importantly, the LDL receptor (16). The increased expression of HMG-CoA reductase restores hepatic cholesterol synthesis towards normal while the increased expression of the LDL receptor results in an increase in the number of LDL receptors on the plasma membrane of hepatocytes leading to the accelerated clearance of LDL (Figure 1) (16). The increased clearance of LDL accounts for the reduction in plasma LDL-C levels. In patients with a total absence of LDL receptors (Homozygous Familiar Hypercholesterolemia) statin therapy is not very effective in lowering LDL-C levels.
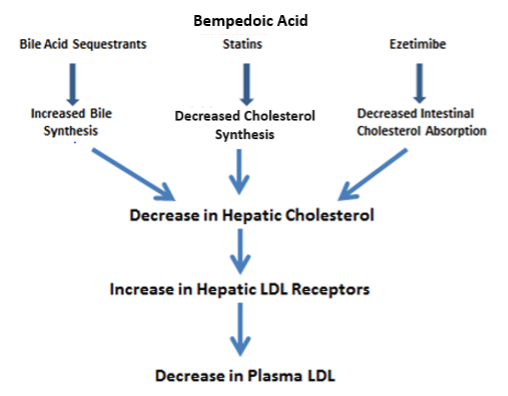
Figure 1. Mechanism for the Decrease in LDL Levels
In addition to lowering LDL and VLDL levels by accelerating the clearance of lipoproteins some studies have also shown that statins reduce the production and secretion of VLDL particles by the liver (17). This could contribute to the decrease in triglyceride levels. The mechanism by which statins increase HDL-C levels is not clear. The small increase in Lp(a) may be due to increased production as studies have shown that incubating HepG2 hepatocytes with a statin increased the expression of LPA mRNA and apolipoprotein(a) protein (18).
Pharmacokinetics and Drug Interactions
Statins have different pharmacokinetic properties which can explain clinically important differences in safety and drug interactions (19-22). Most statins are lipophilic except for pravastatin and rosuvastatin, which are hydrophilic. Lipophilic statins can enter cells more easily but the clinical significance of this difference is not clear. Most of the clearance of statins is via the liver and GI tract (19-21). Renal clearance of statins in general is low with atorvastatin having a very low renal clearance making this particular drug the statin of choice in patients with significant renal disease. The half-life of statins varies greatly with lovastatin, pravastatin, simvastatin, and fluvastatin having a short half-life (1-3 hours) while atorvastatin, rosuvastatin, and pitavastatin having a long half-life (19-22). In patient’s intolerant of statins, the use of a long-acting statin every other day or 2 times per week has been employed. Short acting statins are most effective when administered in the evening when HMG-CoA reductase activity is maximal while the efficacy of long-acting statins is equivalent whether given in the AM or PM (23). In patients who prefer to take their statin in the morning one should use a long-acting statin.
A key difference between statins is their pathway of metabolism. Simvastatin, lovastatin, and atorvastatin are metabolized by the CYP3A4 enzymes and drugs that affect the CYP3A4 pathway can alter the metabolism of these statins (19-22,24). Fluvastatin is metabolized mainly by CYP2C9 with a small contribution by CYP2C8 (19-21,24). Pitavastatin and rosuvastatin are minimally metabolized by the CYP2C9 pathway (19-21,24). Pravastatin is not metabolized at all via the CYP enzyme system (19-21).
Drugs that inhibit CYP3A4 can impede the metabolism of simvastatin, lovastatin, and to a smaller extent atorvastatin resulting in high serum levels of these drugs (19-22,24). These higher levels are associated with adverse effects particularly muscle toxicity. Drugs that inhibit CYP3A4 include intraconazole, ketoconazole, erythromycin, clarithromycin, HIV protease inhibitors (amprenavir, darunavir, fosamprenavir, indinavir, nelfinavir, ritonavir, and saquinavir), amiodarone, diltiazem, verapamil, and cyclosporine (19-22,24). It should be noted that grapefruit juice contains compounds that inhibit CYP3A4 and the consumption of grapefruit juice can significantly increase statin blood levels (25). The inhibition of CYP3A4 by grapefruit juice is dose dependent and increases with the concentration and volume of grapefruit juice ingested. One glass of grapefruit juice everyday can influence the metabolism of statins that are metabolized by the CYP3A4 pathway (25). If a patient requires treatment with a drug that inhibits CYP3A4 the clinician has a number of options to avoid potential drug interactions. One could use a statin that is not metabolized via the CYP3A4 system such as pravastatin or rosuvastatin, one could use an alternative drug to the CYP3A4 inhibitor (for example instead of using erythromycin use azithromycin), or one could temporarily suspend for a short period of time the use of the statin that is metabolized by the CYP3A4 pathway (this is particularly useful when a short course of treatment with an antifungal, antiviral, or antibiotic is required). Drugs that inhibit CYP2C9 do not seem to increase the toxicity of fluvastatin, pitavastatin, or rosuvastatin probably because metabolism via the CYP2C9 pathway is not a dominant pathway.
Most statins are transported into the liver and other tissues by organic anion transporting polypeptides, particularly OATP1B1 (19-21,24). Drugs, such as clarithromycin, ritonavir, indinavir, saquinavir, and cyclosporine that inhibit OATP1B1 can increase serum statin levels thereby increasing the risk of statin muscle toxicity (19-21,24). Fluvastatin is the statin that is least affected by OATP1B1 inhibitors. In fact, fluvastatin 40mg per day has been studied in patients receiving renal transplants concomitantly treated with cyclosporine and over a five year study period the risk of myopathy or rhabdomyolysis was not increased in the fluvastatin treated patients compared to those treated with placebo (26).
Gemfibrozil inhibits the glucuronidation of statins, which accounts for a significant portion of the metabolism of most statins (24). This can lead to the reduced clearance of statins and elevated blood levels increasing the risk of muscle toxicity (24). The only statin whose metabolism is not altered by gemfibrozil is fluvastatin (24). Notably, fenofibrate, another fibrate that has very similar effects on lipid and lipoprotein levels as gemfibrozil, does not inhibit statin glucuronidation (24). Therefore, in patients on statin therapy who also need treatment with a fibrate one should use fenofibrate and not gemfibrozil in combination with statin therapy. Studies have shown that fenofibrate combined with statins does not significantly increase toxicity (27).
There are other drug interactions with statins whose mechanisms are unknown. For example, the lopinavir/ritonavir combination used to treat HIV increases rosuvastatin levels by 2-5-fold and atazanavir/ritonavir increases rosuvastatin levels by 2-6-fold (28-32). Similarly, the tipranavir/ritonavir combination increases rosuvastatin levels 2-fold and atorvastatin levels 8-fold (31). When HIV patients are on these drugs other statins should be used to lower LDL-C levels. The use of statins in patients with HIV is discussed in detail in the Endotext chapter entitled “Lipid Disorders in People with HIV” (33).
Thus, despite the excellent safety record of statins, careful attention must be paid to the potential drug-drug interactions. For additional information see Kellick et al (22,24).
Effect of Statin Therapy on Clinical Outcomes
A large number of studies using a variety of statins in diverse patient populations have shown that statin therapy reduces atherosclerotic cardiovascular disease. The Cholesterol Treatment Trialists have published meta-analyses derived from individual subject data. Their first publication included data from 14 trials with over 90,000 subjects (34). There was a 12% reduction in all-cause mortality in the statin treated subjects, which was mainly due to a 19% reduction in coronary heart disease deaths. Non-vascular causes of death were similar in the statin and placebo groups indicating that statin therapy and lowering LDL-C did not increase the risk of death from other causes such as cancer, respiratory disease, etc. Of particular note there was a 23% decrease in major coronary events per 1 mmol/L (39mg/dL) reduction in LDL-C. Decreases in other vascular outcomes including non-fatal MI, coronary heart disease death, vascular surgery, and stroke were also reduced by 20-25% per 1 mmol/L (39mg/dL) reduction in LDL-C. Additionally, analysis of these studies demonstrated that the greater the reduction in absolute LDL-C levels the greater the decrease in cardiovascular events. For example, while a 40mg/dL decrease in LDL-C will reduce coronary events by approximately 20%, an 80mg/dL decrease in LDL-C will reduce events by approximately 40%. These results support aggressive lipid lowering with statin therapy.
Of note the decrease in the number of events begins to be seen in the first year of therapy indicating that the ability of statins to reduce events occurs relatively quickly and increases over time. The ability of statins to reduce cardiovascular events was seen in a wide diversity of patients including those with and without a history of prior cardiovascular disease, patients over age 65 and younger than age 65, males and females, and patients with and without a history of diabetes or hypertension. Additionally, the beneficial effects of statins were seen regardless of the baseline lipid levels. Subjects with elevated or low LDL-C, HDL-C, or triglyceride levels all had similar decreases in the relative risk of cardiovascular events.
A subsequent publication by the Cholesterol Treatment Trialists has focused on five studies with over 39,000 subjects that have compared usual vs. intensive statin therapy (35). It was noted that there was a 0.51mmol/L (20mg/dL) further reduction in LDL-C in the intensively treated subjects. This further decrease in LDL-C resulted in a15% reduction in cardiovascular events. The strong numerical relationship between decreases in LDL-C levels and the reduction in cardiovascular events provides evidence indicating that much of the beneficial effect of statins is accounted for by lipid lowering.
In addition, the authors added 7 additional trials to their original comparison of statin treatment vs. placebo for a total of 21 trials with over 129,000 subjects. In this larger cohort a 1mmol/L (39mg/dL) decrease in LDL was associated with a 21% reduction in major cardiovascular events. As seen previously the benefits of statin therapy were seen in a wide variety of subjects including patients older than age 75, obese patients, cigarette smokers, patients with decreased renal function, and patients with low and high HDL-C levels. Additionally, a reduction of cardiovascular events with statin therapy was seen regardless of baseline LDL-C levels.
A more recent meta-analysis by the Cholesterol Treatment Trialists examined the effect of statins in 27 trials that included 46,675 women and 127,474 men (36). They found that statin therapy was similarly effective in reducing cardiovascular events in both men and women. Thus, there is an overwhelming database of randomized clinical outcome trials showing the benefits of statin therapy in reducing cardiovascular disease, which, coupled with their excellent safety profile, has resulted in statins becoming a very widely used class of drugs and first line therapy for the prevention of cardiovascular disease.
Effect of Statins Therapy on Clinical Outcomes in Specific Patient Groups
PRIMARY PREVENTION
While there is no doubt that individuals with pre-existing cardiovascular disease require statin therapy, the use of statins for primary prevention was initially debated. There are now a large number of statin primary prevention studies. The Cholesterol Treatment Trialists reported that statin therapy was very effective in reducing cardiovascular events in subjects without a history of vascular disease and the relative risk reduction was similar to subjects with a history of cardiovascular events (35). Additionally, vascular deaths were reduced by statin treatment even in subjects without a history of vascular disease. As expected, non-vascular deaths were not altered in these subjects without a history of pre-existing vascular disease. Additionally, the Cholesterol Treatment Trialists compared the benefits of statin therapy based on baseline risk of developing cardiovascular disease (<5%, ≥5% to <10%, ≥10% to <20%, ≥20% to <30%, ≥30%) (37). The proportional reduction in major vascular events was at least as big in the two lowest risk categories as in the higher risk categories indicating that subjects at low-risk benefit from statin therapy. Similar to the Cholesterol Treatment Trialists analysis, a Cochrane review published in 2013 on the effect of statins in primary prevention patients reached the following conclusion: “Reductions in all-cause mortality, major vascular events, and revascularizations were found with no excess adverse events among people without evidence of CVD treated with statins” (38). An additional study (HOPE-3 trial), not included in the above analyses, has been completed that focused on intermediate risk patients without cardiovascular disease. In this trial 12,705 men and women who had at least one risk factor for cardiovascular disease were randomized to 10mg rosuvastatin vs. placebo (39). Rosuvastatin treatment resulted in a 27% reduction in LDL-C levels and a 24% decrease in cardiovascular events providing additional evidence that statin treatment can reduce events in primary prevention patients. It is therefore clear that statins are effective in safely reducing events in primary prevention patients.
The key issue is “which primary prevention patients should be treated” and this is still controversial. It should be noted that the higher the baseline risk the greater the absolute reduction in events with statin therapy. For example, in a high-risk patient with a 20% risk of developing a vascular event, a 25% risk reduction will result in a 15% risk of an event (absolute decrease of 5%). In contrast in a low-risk patient with a 4% risk of developing a vascular event, a 25% risk reduction will result in a 3% risk (absolute decrease of only 1%). Thus, the absolute benefit of statin therapy over the short term will depend on the risk of developing cardiovascular disease.
Additionally, based on the Cholesterol Treatment Trialists results the reduction in cardiovascular events is dependent on the absolute decrease in LDL-C levels. Thus, the effect of statin treatment will be influenced by baseline LDL-C levels. For example, a 50% decrease in LDL-C is 80mg/dL if the starting LDL is 160mg/dL and only 40mg/dL if the starting LDL-C is 80mg/dL. Based on studies showing that a decrease in LDL-C of 1 mmol/L (40mg/dL) reduces cardiovascular events by ~20% the relative benefit of statin therapy will be greater in the patient with the starting LDL-C of 160mg/dL (40% decrease in events) than in the patient with the starting LDL-C of 80mg/dL (20% decrease in events). Thus, decisions on treatment need to factor in both relative risk and baseline LDL levels.
Finally, it should be recognized that clinical trials represent short term reductions in LDL-C levels (typically 2-5 years) in a disorder that begins early in life and progresses over decades. Life-long decreases in LDL-C levels due to genetic polymorphisms are associated with a much greater reduction in cardiovascular events than would be expected based on the clinical trial results (40). These results suggest that earlier and longer lasting therapy that decreases LDL-C levels will result in a greater reduction in cardiovascular events (41). An in depth discussion of the benefits of early therapy is discussed in the following reference (42).
ELDERLY
Few studies have focused on lowering LDL-C in elderly patients, which we define as individuals greater than 75 years of age (this is based on the ACC/AHA guidelines using age 75 in their decision algorithms) (3). The Prosper Trial determined the effect of pravastatin 40mg/day (n= 2891) vs. placebo (n= 2913) on cardiovascular events in older subjects (70-82) with pre-existing vascular disease or who were at high risk for vascular disease (43). The average age in this trial was 75 years of age and approximately 45% had cardiovascular disease. As expected, pravastatin treatment lowered LDL-C by 34% compared to the placebo group. The primary end point was coronary death, non-fatal myocardial infarction, and fatal or non-fatal stroke which was reduced by 15% (HR 0.85, 95% CI 0.74-0.97, p=0.014). However, in the individuals without pre-existing cardiovascular disease pravastatin did not significantly reduce cardiovascular events (HR- 0.94; CI- 0.77–1.15). In contrast, in individuals with cardiovascular disease pravastatin therapy reduced cardiovascular events (HR- 0.78, CI- 0.66–0.93). Thus, this study demonstrated benefits of statin therapy in the elderly with cardiovascular disease but failed to demonstrate benefit in the elderly without cardiovascular disease.
A meta-analysis by the Cholesterol Treatment Trialists of 28 trials with 14,483 of 186,854 participants older than 75 years of age found a decrease in cardiovascular events in all age groups including participants older than 75 years of age (Figure 2) (44). Similar to the Prosper Trial a decrease in cardiovascular events was clearly demonstrated in individuals with pre-existing cardiovascular disease (secondary prevention) but in individuals without pre-existing cardiovascular disease (primary prevention) the decrease in cardiovascular events was not statistically significant (Figure 3). Thus, in older patients with cardiovascular disease lowering LDL-C levels with statins clearly reduces cardiovascular events but in older patients without cardiovascular disease the data demonstrating that statins reduce cardiovascular events is less robust but suggests a reduction in cardiovascular events.
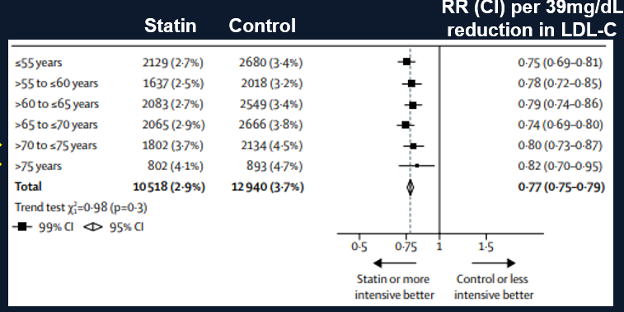
Figure 2. Effect of Statin Treatment on Major Vascular Events. Modified from (44).
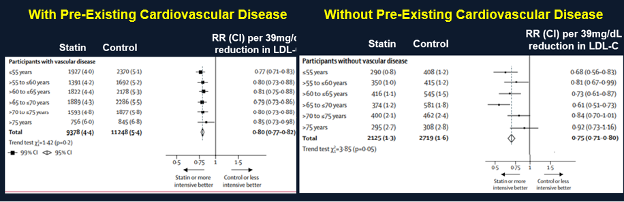
Figure 3. Effect of Statin Treatment on Major Vascular Events in Individuals With and Without Pre-Existing Cardiovascular Disease. Modified from (44).
Studies are currently underway to provide definitive information on whether statin therapy is beneficial as primary prevention in the elderly. STAREE (NCT02099123) is a multicenter randomized trial in Australia of atorvastatin 40mg vs. placebo in adults ≥ 70 years of age without cardiovascular disease and PREVENTABLE (NCT04262206) is a multicenter randomized trial in the USA of atorvastatin vs. placebo in adults ≥ 75 years of age without cardiovascular disease (45,46).
WOMEN
As noted above a meta-analysis by the Cholesterol Treatment Trialists examined the effect of statins in 27 trials that included 46,675 women and 127,474 men (36). They found that statin therapy was similarly effective in reducing cardiovascular events in both men and women.
ASIANS
Pharmacokinetic data have shown that the serum levels of statins are higher in Asians than in Caucasians (47). Moreover, Asians achieve similar LDL lowering at lower statin doses than Caucasians (47). Therefore, the statin dose used should be lower in Asians. For example, the starting dose of rosuvastatin is 5mg in Asians as compared to 10mg in Caucasians. Additionally, the maximum recommended dose of statin is lower in Japan vs. the United States (Table 3). In contrast, studies suggest that South Asian patients may be treated with atorvastatin and simvastatin at doses typically applied to white patients (48). Studies have demonstrated that statins reduce cardiovascular events in Asians (49,50)
|
Table 3. Maximum Statin Dose in Japan and United States
|
|
Statin
|
Japan
|
United States
|
|
Atorvastatin
|
40
|
80
|
|
Fluvastatin
|
60
|
80
|
|
Pravastatin
|
20
|
80
|
|
Rosuvastatin
|
20
|
40
|
|
Simvastatin
|
20
|
40
|
DIABETES
Statin trials, including both primary and secondary prevention trials, have consistently shown the beneficial effect of statins on cardiovascular disease in patients with diabetes (51). The Cholesterol Treatment Trialists analyzed data from 18,686 subjects with diabetes (mostly type 2 diabetes) from 14 randomized trials (52). In the statin treated group there was a 9% decrease in all-cause mortality, a 13% decrease in vascular mortality, and a 21% decrease in major vascular events per 1mmol/L (39mg/dL) reduction in LDL-C. The beneficial effect of statin therapy was seen in both primary and secondary prevention patients. The effect of statin treatment on cardiovascular events in patients with diabetes was similar to that seen in non-diabetic subjects. It should be noted that while the data for patients with type 2 diabetes is robust, the number of patients with type 1 diabetes in these trials is relatively small and the results less definitive. Also, of note is that information on young patients with diabetes (< age 40) is very limited. Thus, these studies indicate that statins are beneficial in reducing cardiovascular disease in patients with diabetes. For addition details on the treatment of dyslipidemia in patients with diabetes see the chapter entitled “Dyslipidemia in Patients with Diabetes” (51).
RENAL DISEASE
The Cholesterol Treatment Trialists examined the effect of renal function on statin effectiveness. They reported that the relative risk reduction for cardiovascular events was similar if the eGFR was < 60ml/min as compared to > 90 or 60-90 (35). In a follow-up analysis it was reported that the relative risk reduction per 1mMol/l (~39mg/dL) decrease in LDL-C levels with statin therapy was 0·78 for an eGFR ≥60 mL/min, 0·76 for an eGFR 45 to <60 mL/min, 0·85 for an eGFR 30 to <45 mL/min, and 0·85 for an eGFR <30 mL/min in patients not on dialysis (53). In patients on dialysis the relative risk reduction was 0·94 (99% CI 0·79-1·11). Similarly, a meta-analysis of 57 studies with >143,000 participants with renal disease not on dialysis reported a 31% reduction in major cardiovascular events in statin treated subjects compared to placebo groups (54). Thus, in patients with renal disease not on dialysis, treatment with statins is beneficial and should be utilized in this population at high risk for vascular disease.
In contrast to the above results, studies examining the role of statins in dialysis patients have not found a benefit from statin therapy. The Deutsche Diabetes Dialyse Studie (4D) randomized 1,255 type 2 diabetic subjects on hemodialysis to either 20 mg atorvastatin or placebo (55). The LDL-cholesterol reduction was similar to that seen in non-dialysis patients but there was no significant reduction in cardiovascular death, nonfatal myocardial infarction, or stroke in the atorvastatin treated compared to the placebo group. Similarly, A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis (AURORA) randomized 2,776 subjects on hemodialysis to rosuvastatin 10 mg or placebo (56). Again, the LDL-cholesterol lowering in dialysis patients was similar to that seen in other studies but there was no significant effect on the primary endpoint of cardiovascular death, nonfatal myocardial infarction, or stroke. A meta-analysis of 25 studies involving 8,289 dialysis patients found no benefit of statin therapy on major cardiovascular events, cardiovascular mortality, all-cause mortality, or myocardial infarction, despite efficacious lipid lowering. The reason for the failure of statins in patients on maintenance dialysis is unclear but could be due to a number of factors including the possibility that the marked severity of atherosclerosis in end stage renal disease may limit reversal, that different mechanisms of atherosclerosis progression occur in dialysis patients (for example an increased role for inflammation, oxidation, or thrombosis), or that cardiovascular events in this patient population may not be due to atherosclerosis. We would recommend continuing statin therapy in patients on dialysis who have been previously treated with statins but not initiating therapy in the rare statin naïve patient beginning dialysis.
Statins are primarily metabolized in the liver and therefore the need to adjust the statin dose is not usually needed in patients with renal disease until the eGFR is < 30ml/min. The effect of renal dysfunction on statin clearance varies from statin to statin (57). For some statins such as atorvastatin, there is no need to adjust the dose in renal disease because there is limited renal clearance (57). However, for other statins it is recommended to adjust the dose in patients when the eGFR is < 30ml/min. In patients with an eGFR < 30ml/min the maximum dose of rosuvastatin is 10mg, simvastatin 40mg, pitavastatin 2mg, pravastatin 20mg, lovastatin 20mg, and fluvastatin 40mg per day (57).
For additional information on the treatment of dyslipidemia in patients with renal disease see the chapter entitled “Dyslipidemia in Chronic Kidney Disease” (57).
CONGESTIVE HEART FAILURE
In the Corona study 5,011 patients with New York Heart Association class II, III, or IV ischemic, systolic heart failure (most were class III) were randomly assigned to receive 10 mg of rosuvastatin or placebo per day (58). While rosuvastatin treatment reduced LDL-C levels by 45% compared to placebo, rosuvastatin did not decrease death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke. Similarly, the GISSI-HF trial randomized 4,574 patients with class II, III, of IV congestive heart failure (most were class II) to 10mg of rosuvastatin or placebo (59). The primary endpoints were time to death, and time to death or admission to hospital for cardiovascular reasons and these were similar in the statin and placebo groups. Why statin treatment was not beneficial in patients with congestive heart failure is unknown.
LIVER DISEASE
Many patients with liver disease, particularly those with nonalcoholic fatty liver disease (NAFLD), are at high risk for cardiovascular disease and therefore require statin therapy (60). There have been concerns that these patients would not tolerate statin therapy and that statin therapy would worsen their underlying liver disease. Fortunately, there are now studies of statin therapy in patients with abnormal liver function tests and underlying liver disease at baseline (60-62). With a variety of statins, studies have demonstrated no significant worsening of liver disease and in fact several studies have suggested improvement in liver function tests with statin therapy (62). This is true for patients with hepatitis C, NAFLD/NASH, and primary biliary cirrhosis. Additionally, in the GREACE trial, statin treatment reduced cardiovascular events in patients with moderately abnormal liver function tests (transaminases < 3x the upper limit of normal) (63). Thus, in patients with mild liver disease without elevations in bilirubin or abnormalities in synthetic function, statins are safe and reduce the risk of cardiovascular disease.
For additional information on the treatment of dyslipidemia in patients with liver disease see the chapter entitled “Lipid and Lipoprotein Metabolism in Liver Disease” (64).
HIV
Patients living with HIV have an increased risk of cardiovascular disease (33). A trial randomized 7,769 participants with HIV infection with a low-to-moderate risk of cardiovascular disease to either pitavastatin 4 mg or placebo (65). The primary outcome was the occurrence of cardiovascular death, myocardial infarction, hospitalization for unstable angina, stroke, transient ischemic attack, peripheral arterial ischemia, revascularization, or death from an undetermined cause. In the pitavastatin group cardiovascular events were decreased by 35% (HR, 0.65; 95% CI, 0.48 to 0.90; P=0.002). For additional information on the use of statins in HIV patients see the Endotext chapter “Lipid Disorders in People with HIV” (33).
Statin Side Effects
An umbrella review of meta-analyses of observational studies and randomized controlled trials examined 278 unique non-CVD outcomes from 112 meta-analyses of observational studies and 144 meta-analyses of RCTs and found that the only adverse effects associated with statin therapy were the development of diabetes and muscle disorders (66). For a detailed discussion of the side effects of statin therapy a scientific statement from the American Heart Association provides a comprehensive review (67).
DIABETES
After many years of statin use it was recognized that statins increase the risk of developing diabetes. In a meta-analysis of 13 trials with over 90,000 subjects, there was a 9% increase in the incidence of diabetes during follow-up among subjects receiving statin therapy (68). All statins appear to increase the risk of developing diabetes. In comparisons of intensive vs. moderate statin therapy, Preiss et al observed that patients treated with intensive statin therapy had a 12% greater risk of developing diabetes compared to subjects treated with moderate dose statin therapy (69). Older subjects, obese subjects, and subjects with high glucose levels were at a higher risk of developing diabetes while on statin therapy (70). Thus, statins may be unmasking and accelerating the development of diabetes that would have occurred naturally in these subjects at some point in time. In patients without risk factors for developing diabetes, treatment with statins does not appear to increase the risk of developing diabetes.
In patients with diabetes, an analysis of 9 studies with over 9,000 patients with diabetes reported that the patients randomized to statin therapy had a 0.12% higher A1c than the placebo group indicating that statin therapy is associated with only a very small increase in A1c levels in patients with diabetes that is unlikely to be clinically significant (71). Individual studies, such as CARDS and the Heart Protection Study, have also shown only a very modest effect of statins on A1c levels in patients with diabetes (72,73).
The mechanism by which statins increase the risk of developing diabetes is unknown (74). A study has demonstrated that a polymorphism in the gene for HMG-CoA reductase that results in a decrease in HMG-CoA reductase activity and a small decrease in LDL levels is also associated with an increase in body weight and plasma glucose and insulin levels (75). Additionally, a cross sectional study that compared the change in BMI in individuals on statins to individuals not on statins observed an increased BMI in the subjects taking statins (+1.3 in stain users vs. + 0.4 in non-users over a 10 year period; p=0.02) (76). These observations suggest that the inhibition of HMG-CoA reductase per se may be contributing to the statin induced increased risk of diabetes via weight gain. However, studies have now shown that polymorphisms in different genes (NPC1L1 and PCSK9) that lead to a decrease in LDL-C levels are also associated with an increase in diabetes suggesting that decreases in LDL-C levels per se alter glucose metabolism and increase the risk of diabetes (74,77). How a decrease in LDL-C levels might affect glucose metabolism is unknown. Clearly further studies are required to understand the mechanisms by which statins increase the risk of developing diabetes.
In balancing the benefits and risks of statin therapy it is important to recognize that an increase in plasma glucose levels is a surrogate marker for an increased risk of developing micro and macrovascular disease (i.e., an increase in plasma glucose per se is not an event but rather increases the risk of future events). In contrast, statin therapy is preventing actual clinical events that cause morbidity and mortality. Furthermore, it may take many years for an elevated blood glucose to induce diabetic complications while the reduction in cardiovascular events with statin therapy occurs relatively quickly. Finally, the number of patients needed to treat with statins to avoid one cardiovascular event is much lower (10-20 depending on the type of patient) than the number of patients needed to treat to cause one patient to develop diabetes (100–200 for one extra case of diabetes) (74). Patients on statin therapy, particularly those with risk factors for the development of diabetes, should be periodically screened for the development of diabetes with measurement of fasting glucose or A1c levels.
CANCER
Analysis of 14 trials with over 90,000 subjects by the Cholesterol Treatment Trialists did not demonstrate an increased risk of cancer or any specific cancer with statin therapy (34). An update with an analysis of 27 trials with over 174,000 participants also did not observe an increase in cancer incidence or death (36). Additionally, no differences in cancer rates were observed with any particular statin.
COGNITIVE DYSFUNCTION
Several randomized clinical trials have examined the effect of statin therapy on cognitive function and have not indicated any increased risk (78-80). The Prosper Trial was designed to determine whether statin therapy will reduce cardiovascular disease in older subjects (age 70-82) (43). In this trial cognitive function was assessed repeatedly and no difference in cognitive decline was found in subjects treated with pravastatin compared to placebo (43,81). In the Heart Protection Study over 20,000 patients were randomized to simvastatin 40mg or placebo and again no significant differences in cognitive function was observed between the statin vs. placebo group (82). Additionally, a Cochrane review examined the effect of statin therapy in patients with established dementia and identified 4 studies with 1154 participants (83). In this analysis no benefit or harm of statin therapy on cognitive function could be demonstrated in this high-risk group of patients. Thus, randomized clinical trials do not indicate a significant association.
HEMORRAGIC STROKE
In a scientific statement from the American Heart Association on statin safety reached the following conclusions; “The available data in aggregate show no increased risk of brain hemorrhage with statin use in primary stroke prevention populations. An increased risk in secondary stroke prevention populations is possible, but the absolute risk is very small, and the benefit in reducing overall stroke and other vascular events generally outweighs that risk” (67).
LIVER DISEASE
It was in initially thought that statins induced liver dysfunction and it was recommended that liver function tests be routinely obtained while patients were taking statins. However, studies have now shown that the risk of liver function test abnormalities in patients taking statins is very small (61). For example, in a survey of 35 randomized studies involving > 74,000 subjects, elevations in transaminases were seen in 1.4% of statin treated subjects and 1.1% of controls (84). Similarly, in a meta-analysis of > 49,000 patients from 13 placebo controlled studies, the incidence of transaminase elevations greater than three times the upper limit of normal was 1.14% in the statin group and 1.05% in the placebo group (85). Moreover, even when the transaminase levels are elevated, repeat testing often demonstrates a return towards normal levels (86). The increases in transaminase levels with statin therapy are dose related with high doses of statins leading to more frequent elevations (87). At this time, routine monitoring of liver function tests in patients taking statins is no longer recommended. However, obtaining baseline liver function tests prior to starting statin therapy is indicated (61). If liver function tests are obtained during statin treatment, one should not be overly concerned with modestly elevated transaminase levels (less than 3x the upper limit of normal) (61). If the transaminase is greater than 3x the upper limit of normal the test should be repeated and if it remains > 3x the upper limit of normal, statin therapy should be stopped and the patient evaluated (61).
A more clinically important issue is whether statins lead to an increased risk of liver failure. Studies have suggested that the incidence of liver failure in patients taking statins is very similar to the rate observed in the general population (approx. 1 case per 1 million patient years) (88,89). Thus, statin therapy causing serious liver injury is a very rare event.
Non-alcoholic fatty liver disease (NAFLD) is very common and is associates with obesity, metabolic syndrome, diabetes, and cardiovascular disease. In patients with NAFLD studies have shown that statins decrease liver enzymes and reduce steatosis (90).
MUSCLE
The most common side effect of statin therapy is muscle symptoms. These can range from life threatening rhabdomyolysis to myalgias (Table 4) (91).
|
Table 4. Spectrum of Statin Induced Muscle Disorders (Adapted from J. Clinical Lipidology 8: S58-71, 2014)
|
|
Myalgia- aches, soreness, stiffness, tenderness, cramps with normal CK levels
|
|
Myopathy- muscle weakness with or without increased CK
|
|
Myositis- muscle inflammation
|
|
Myonecrosis- mild (CK >3x ULN); moderate (CK> 10x ULN); severe (CK> 50x ULN)
|
|
Rhabdomyolysis- myonecrosis with myoglobinuria or acute renal failure
|
Many patients will discontinue the use of statins due to muscle symptoms. Risk factors associated with an increased incidence of statin associated muscle symptoms are listed in Table 5 (92,93).
|
Table 5. Risk Factors for Statin Myopathy
|
|
Medications that alter statin metabolism
|
|
Older age
|
|
Female
|
|
Hypothyroidism
|
|
Excess alcohol
|
|
Vitamin D deficiency
|
|
History of muscle disorders
|
|
Renal disease
|
|
Liver disease
|
|
Personal or family history of statin intolerance
|
|
Low BMI
|
|
Polymorphism in SLCO1B1 gene
|
|
High dose statin
|
|
Drug-drug interactions
|
The Cholesterol Treatment Trialists analyzed individual participant data on the development of muscle symptoms from 19 double-blind trials of statin versus placebo with 123,940 participants and four double-blind trials of a more intensive vs. a less intensive statin regimen with 30,724 participants (94). After a median follow-up of 4.3 years 27.1% of the individuals taking a statin vs. 26.6% on placebo reported muscle pain or weakness representing a 3% increase greater than placebo (risk ratio- 1.03; 95% CI 1.01-1.06) (Table 6). The specific muscle symptoms caused by statin therapy, myalgia, muscle cramps or spasm, limb pain, other musculoskeletal pain, or muscle fatigue or weakness were similar to those caused by placebo. The increase in muscle symptoms in the statin treated individuals was manifest in the first year of therapy but in the later years muscle symptoms were similar in the statin treated and placebo groups. The relative risk of statin induced muscle symptoms was greater in women than men. Intensive statin treatment with 40-80 mg atorvastatin or 20-40 mg rosuvastatin resulted in a higher risk of muscle symptoms than less intensive or moderate-intensity regimens but different statins at equivalent LDL-C lowering doses had similar effects on muscle symptoms. This study demonstrates that there is a small increase in muscle symptoms that primarily manifests in the first year of therapy. Statin therapy caused approximately 11 additional complaints of muscle pain or weakness per 1000 patients during the first year, but little excess in later years. Of particularly note is that 26.6% of patients taking a placebo had muscle symptoms demonstrating a very high frequency of this clinical complaint. Given the high prevalence of muscle complaints and the small increase attributed to statins it is very difficult to determine if a muscle complaint is actually due to the statin, which presents great clinical difficulties in patient management.
|
Table 6. Effect of Statin vs. Placebo on Muscle Symptoms
|
|
Symptom
|
Statin Events
|
Placebo Events
|
RR (95% CI)
|
|
Myalgia
|
12.0%
|
11.7%
|
1·03 (0·99–1·08)
|
|
Other musculoskeletal pain
|
13.3
|
13.0
|
1·03 (0·99–1·08)
|
|
Any muscle pain
|
26.9%
|
26.3%
|
1·03 (1·01–1·06)
|
|
Any muscle pain or weakness
|
27.1%
|
26.6%
|
1·03 (1·01–1·06)
|
Modified from (94).
While the results of the randomized trials suggest that muscle symptoms are not frequently induced by statin therapy, in typical clinical settings a significant percentage of patients are unable to tolerate statins due to muscle symptoms (in many studies as high as 5-25% of patients) (95-97). Recently there was a randomized trial that explored the issue of myopathy with statin therapy in great detail (98). In this trial the effect of atorvastatin 80mg a day vs. placebo for 6 months on creatine kinase (CK), exercise capacity, and muscle strength was studied in 420 healthy, statin-naive subjects. Atorvastatin treatment led to a modest increase in CK levels (20.8U/L) with no change observed in the placebo group. None of the subjects had an elevation of CK > 10x the upper limits of normal. There were no changes in muscle strength or exercise capacity with atorvastatin treatment. However, myalgia was reported in 19 subjects (9.4%) in the atorvastatin group compared to 10 subjects (4.6%) in the placebo group (p=0.05). In this study “myalgia” was considered to be present if all of the following occurred: (1) subjects reported new or increased muscle pain, cramps, or aching not associated with exercise; (2) symptoms persisted for at least 2 weeks; (3) symptoms resolved within 2 weeks of stopping the study drug; and (4) symptoms reoccurred within 4 weeks of restarting the study medication. Notably these myalgias were not associated with elevated CK levels. In the atorvastatin group the myalgias tended to occur soon after therapy (average 35 days) whereas in the placebo group myalgias occur later (average 61 days). In the atorvastatin group the symptoms were predominantly localized to the legs and included aches, cramps, and fatigue, whereas in the placebo group they were more diverse including whole body fatigue, foot cramps, worsening of pain in previous injuries, and groin pain. A number of conclusions can be reached from this study. First, statin treatment does in fact increase the incidence of myalgias. Second, a substantial number of patients treated with placebo will also develop myalgias. Third, clinically differentiating statin induced myalgias from placebo induced myalgias is difficult, as there are no specific symptoms, signs, or biomarkers that clearly distinguish between the two. It should be recognized that the patient population typically treated with statins (patients 50-80 years of age) often have muscle symptoms in the absence of statin therapy and it is therefore difficult to be certain that the muscle symptoms described by the patient are actually due to statin therapy.
Additionally, when patients know that they are taking a statin they are more likely to have muscle symptoms (i.e. the nocebo effect). This was nicely demonstrated in the ASCOT-LLA extension trial (99). In the initial phase of the study the patients were randomly assigned to atorvastatin 10 mg (n= 5101) or matching placebo (n= 5079) in a double-blind fashion. During the 3.3 years of the double blinded phase adverse muscle symptoms were very similar in the atorvastatin and placebo groups (HR 1.03; p=0.72). This double-blind phase was followed by a non-blinded non-randomized extension where 6409 patients were treated with atorvastatin 10mg and 3490 were untreated. During the 2.3 years of this extension study muscle symptoms were significantly increased in the atorvastatin group (HR 1·41; p=0.006).
In a very small study in the Annals of Internal Medicine eight patients with “statin related myalgia” were re-challenged with statin or placebo and there were no statistically significant differences in the recurrence of myalgias on the statin or placebo (100). This approach has been expanded upon in other studies. In 120 patients with “statin induced myalgia” patients were randomized in a double blinded crossover trial to either simvastatin 20mg per day or placebo and the occurrence of muscle symptoms was determined (101). Only 36% of these patients were confirmed to actually have statin induced myalgia (presence of symptoms on simvastatin without symptoms on placebo). In a similar study, Nissen and colleagues studied 491 patients with “statin induced myalgia” treating with either atorvastatin 20mg per day or placebo in a double-blind crossover trial (102). In this trial 42.6% of patients were confirmed to have statin induced muscle symptoms. In a trial of 156 patients with prior statin induced muscle symptoms patients were treated with alternating periods of atorvastatin 20mg or placebo (103). In this trial no difference in muscle symptoms was found between the statin and placebo treatment periods. A smaller crossover trial in 49 patients who had stopped statin therapy also found no difference in muscle symptoms when patients were taking atorvastatin 20mg or placebo (104)
Thus, while statin induced myalgias are a real entity careful studies have shown that in the majority of patients with “statin induced muscle symptoms” the symptoms are not actually due to statin therapy. In the clinic it is difficult to be certain whether the muscle symptoms are actually due to true statin intolerance or to other factors. The approach to treating these patients will be discussed later in this chapter (Treatment of Stain Intolerant Patients). While some patients will not tolerate statin therapy due to myalgias, this side effect does not appear to result in serious morbidity or long-term consequences. In contrast, studies have found that discontinuing statins increases the risk of myocardial infarctions and death from cardiovascular disease (105,106).
Fortunately, the more serious muscle related side effects of statin therapy are rare. In a meta-analysis of 21 statin vs. placebo trials there was an excess risk of rhabdomyolysis of 1.6 patients per 100,000 patient years or a standardized rate of 0.016/patient years (86). Other studies report a rate of rhabdomyolysis between 0.03- 0.16 per 1,000 patient years (107). Similarly, the risk of statin induced myositis (muscle symptoms with an increase in CK 10 times the upper limits of normal) is also very low. In an analysis of 21 randomized trials myositis occurred in only 5 patients per 100,000 person years or 0.05/1000 patient years (86). The higher the dose of statin used the greater the risk of myositis and rhabdomyolysis. In a comparison of five trials that compared high dose statin vs. low dose statin there was an excess risk of rhabdomyolysis of 4 per 10,000 people treated (35). The likely basis for an increased risk of myositis or rhabdomyolysis is elevated statin blood levels, which are more likely to occur with high doses of statins. In the development of statins, manufacturers have studied higher doses that are not approved for clinical use. For example, simvastatin and pravastatin at 160mg per day were studied but discontinued due to an increased incidence of muscle side effects (108,109). The use of simvastatin 80mg per day, a previously approved dose, was discontinued due to an increased risk of muscle side effects. Similarly, pitavastatin at doses greater than 4mg per day was investigated, but development was abandoned when an increased risk of rhabdomyolysis was observed. Along similar lines, in many of the patients that develop rhabdomyolysis, the etiology can be linked to the use of other drugs that alter statin metabolism thereby increasing statin blood levels (93). For example, prior to drug interactions being recognized the use of cyclosporine, gemfibrozil, HIV protease inhibitors, and erythromycin in conjunction with certain statins was linked with the development of rhabdomyolysis (93). Finally, common variants in SLCO1B1, which encodes the organic anion-transporting polypeptide OATP1B1, are strongly associated with an increased risk of statin-induced myopathy (110). OATP1B1 facilitates the transport of statins into the liver and certain polymorphisms are associated with an increased risk of developing statin induced muscle disorders, due to the decreased transport of statins into the liver resulting in increased blood levels (111). The exact mechanism by which elevated blood levels induce muscle toxicity remains to be elucidated.
Recently it has been recognized that a very small number of patients taking statins develop a progressive autoimmune necrotizing myopathy, which is characterized by progressive symmetric proximal muscle weakness, elevated CK levels (typically >10x the ULN), and antibodies against HMG-CoA reductase (112). It is estimated that this occurs in 2 or 3 per 100,000 patients treated with a statin (112). This myopathy may begin soon after initiating statin therapy or develop after a patient has been on statins for many years (112). Muscle biopsy reveals necrotizing myopathy without severe inflammation (112). In contrast to the typical muscle disorders induced by statin therapy, the autoimmune myopathy progresses despite discontinuing therapy. Spontaneous improvement is not typical and most patients will need to be treated with immunosuppressive therapy (glucocorticoids plus methotrexate, azathioprine, or mycophenolate mofetil) (112). It should be recognized that this disorder can occur in individuals that have not been exposed to statin therapy (113). Statins likely potentiate the development of this disorder in susceptible individuals, perhaps by increasing HMG-CoA reductase levels.
From the above certain conclusions can be reached. First, the risk of serious muscle disorders due to statin therapy is very small, particularly if one is aware of the potential drug interactions that increase the risk. Second, the muscle toxicity is usually linked to elevated statin blood levels and the higher the dose of the statin the more likely the chance of developing toxicity. Third, myalgias in patients taking statins are very common and can be due to statin treatment. However, in the individual patient, it is very difficult to know if the myalgia is actually secondary to statin therapy and in many, if not most patients, the myalgias are not due to statin therapy. Fourth, the muscle symptoms that occur in association with statin treatment are a major reason why patients discontinue statin use and therefore better diagnostic algorithms and treatments are required to allow patients to better comply with these highly effective treatments to reduce cardiovascular disease.
Contraindications
Previously statins were contraindicated in pregnant women or lactating women. However, in July 2021 the FDA requested the removal of the strongest recommendation against using statins during pregnancy. They continue to advise against the use of statins in pregnancy given the limited data and quality of information available. The decision of whether to continue a statin during pregnancy requires shared decision-making between the patient and clinician, and healthcare professionals need to discuss the risks versus the benefits in high-risk women, such as those with homozygous FH or prior ASCVD events, that may benefit from statin therapy. For a detailed discussion of the use of statins during pregnancy see the Endotext chapter entitled “Effect of Pregnancy on Lipid Metabolism and Lipoprotein Levels” (114).
In addition, liver function tests should be obtained prior to initiating statin treatment and moderate to severe liver disease is a contraindication to statin therapy (61).
Summary
An enormous data base has accumulated which demonstrates that statins are very effective at reducing the risk of cardiovascular disease and that statins have an excellent safety profile. The risk benefit ratio of treating patients with statins is very favorable and has resulted in this class of drugs being widely utilized to lower serum lipid levels and to reduce the risk of cardiovascular disease and death.
EZETIMBE (ZETIA)
Introduction
Ezetimibe (Zetia) inhibits the absorption of cholesterol by the intestine thereby resulting in modest decreases in LDL-C levels (115). Ezetimibe is primarily used in combination with statin therapy when statin treatment alone does not lower LDL-C levels sufficiently or when patients only tolerate a low statin dose. It may also be used as monotherapy or in combination with other lipid lowering drugs to lower LDL-C levels in patients with statin intolerance. Finally, it is the drug of choice in patients with the rare genetic disorder sitosterolemia, which is discussed in detail in the chapter “Sitosterolemia” (116). Ezetimibe is relatively inexpensive as it is now a generic drug.
Effect of Ezetimibe on Lipid and Lipoprotein Levels
Pandor and colleagues have published a meta-analysis of ezetimibe monotherapy that included 8 studies with 2,722 patients (117). They reported that ezetimibe decreased LDL-C levels by 18.6%, decreased triglyceride levels by 8.1%, and increased HDL-C levels by 3% compared to placebo. In a pooled analysis by Morrone and colleagues of 27 studies with 11, 714 subjects treated with ezetimibe in combination with statin therapy similar results were observed (118). Specifically, LDL-C levels were decreased by 15.1%, non-HDL-C levels by 13.5%, triglycerides by 4.7%, apolipoprotein B levels by 10.8%, and HDL-C levels were increased by 1.6%. The combination of a high dose potent statin plus ezetimibe can lower LDL-C levels by 70% (119). A meta-analysis of the effect of ezetimibe on Lp(a) revealed that with either monotherapy or combination with statin there was no change in Lp(a) levels (120). The effect of ezetimibe on lipid parameters occurs quickly and can be seen after 2 weeks of treatment. In patients with Heterozygous Familial Hypercholesterolemia who have marked elevations in LDL-C levels, the addition of ezetimibe to statin therapy resulted in a further 16.5% decrease in LDL-C levels (121). Thus, in comparison with statins, ezetimibe treatment produces modest decreases in LDL-C levels (15-20%). In addition to these changes in lipid parameters, ezetimibe in combination with a statin decreased hs-CRP by 10-19% compared to statin monotherapy (122,123). However, ezetimibe alone does not decrease hs-CRP levels (123).
|
Table 7. Effect of Ezetimibe on Lipid/Lipoprotein Levels
|
|
LDL-C
|
Decrease
|
|
Non-HDL-C
|
Decrease
|
|
Apolipoprotein B
|
Decrease
|
|
Triglycerides
|
Small decrease
|
|
HDL-C
|
Small increase
|
|
Lp(a)
|
No change
|
Mechanisms Accounting for the Ezetimibe Induced Lipid Effects
NPC1L1 (Niemann-Pick C1-like 1 protein) is highly expressed in the intestine with the greatest expression in the proximal jejunum, which is the major site of intestinal cholesterol absorption (124,125). Knock out animals deficient in NPC1L1 have been shown to have a decrease in intestinal cholesterol absorption (124). Ezetimibe binds to NPC1L1 and inhibits cholesterol absorption (115,124,125). In animals lacking NPC1L1, ezetimibe has no effect on intestinal cholesterol absorption, demonstrating that ezetimibe’s effect on cholesterol absorption is mediated via NPC1L1 (115,125). Thus, a major site of action of ezetimibe is to block the absorption of cholesterol by the intestine (115,125). Cholesterol in the intestinal lumen is derived from both dietary cholesterol (approximately 25%) and biliary cholesterol (approximately 75%); thus the majority is derived from the bile (125). As a consequence, even in patients that have very little cholesterol in their diet, ezetimibe will decrease cholesterol absorption. While ezetimibe is very effective in blocking intestinal cholesterol absorption it does not interfere with the absorption of triglycerides, fatty acids, bile acids, or fat-soluble vitamins including vitamin D and K.
When intestinal cholesterol absorption is decreased the chylomicrons formed by the intestine contain less cholesterol and thus the delivery of cholesterol from the intestine to the liver is diminished (126). This results in a decrease in the cholesterol content of the liver, leading to the activation of SREBPs, which enhance the expression of LDL receptors resulting in an increase in LDL receptors on the plasma membrane of hepatocytes (Figure 1) (126). Thus, similar to statins the major mechanism of action of ezetimibe is to decrease the levels of cholesterol in the liver resulting in an increase in the number of LDL receptors leading to the increased clearance of circulating LDL (126). In addition, the decreased cholesterol delivery to the liver may also decrease the formation and secretion of VLDL (126).
In addition to NPC1L1 expression in the intestine this protein is also expressed in the liver where it mediates the transport of cholesterol from the bile back into the liver (127). The inhibition of NPC1L1 in the liver will result in the increased secretion of cholesterol in bile and thereby could also contribute to a decrease in the cholesterol content of the liver and an increase in LDL receptor expression and a decrease in VLDL production.
Pharmacokinetics and Drug Interactions
Following absorption by intestinal cells ezetimibe is rapidly glucuronidated. The glucuronidated ezetimibe is then secreted into the portal circulation and rapidly taken up by the liver where it is secreted into the bile and transported back to the intestine (115). This enterohepatic circulation repeatedly returns ezetimibe to its site of action (note glucuronidated ezetimibe is a very effective inhibitor of NPC1L1) (115). Additionally, this enterohepatic circulation accounts for the long duration of action of ezetimibe and limits peripheral tissue exposure (115). Ezetimibe is not significantly excreted by the kidneys and thus the dose does not need to be adjusted in patients with renal disease.
Ezetimibe is not metabolized by the P450 system and does not have many drug interactions (115). It should be noted that cyclosporine does increase ezetimibe levels.
Effect of Ezetimibe Therapy on Clinical Outcomes
There have been a limited number of ezetimibe clinical outcome trials. Two have studied the effect of ezetimibe in combination with a statin vs. placebo making it virtually impossible to determine if ezetimibe per se has beneficial effects. However, one study has compared ezetimibe plus a statin vs. a statin alone and one study compared ezetimibe vs. placebo. Finally, a study compared moderate-intensity statin with ezetimibe vs. high-intensity statin monotherapy.
SEAS TRIAL
The SEAS Trial was a randomized trial of 1,873 patients with mild-to-moderate, asymptomatic aortic stenosis (128). The patients received either simvastatin 40mg per day in combination with ezetimibe 10mg per day vs. placebo daily. The primary outcome was a composite of major cardiovascular events, including death from cardiovascular causes, aortic-valve replacement, non-fatal myocardial infarction, hospitalization for unstable angina pectoris, heart failure, coronary-artery bypass grafting, percutaneous coronary intervention, and non-hemorrhagic stroke. Secondary outcomes were events related to aortic-valve stenosis and ischemic cardiovascular events. Simvastatin plus ezetimibe lowered LDL-C levels by 61% compared to placebo. There were no significant differences in the primary outcome between the treated vs. placebo groups. Similarly, the need for aortic valve replacement was also not different between the treated and placebo groups. However, fewer patients had ischemic cardiovascular events in the simvastatin plus ezetimibe treated group than in the placebo group (hazard ratio, 0.78; 95% CI, 0.63 to 0.97; P=0.02), which was primarily accounted for by a decrease in the number of patients who underwent coronary-artery bypass grafting. The design of this study does not allow for one to determine if the beneficial effect on ischemic cardiovascular events typically produced by statin therapy was enhanced by the addition of ezetimibe.
SHARP TRIAL
The SHARP Trial was a randomized trial of 9,270 patients with chronic kidney disease (3,023 on dialysis and 6,247 not on dialysis) with no known history of myocardial infarction or coronary revascularization (129). Patients were randomly assigned to simvastatin 20 mg plus ezetimibe 10 mg daily vs. placebo. The primary outcome was first major atherosclerotic event (non-fatal myocardial infarction or coronary death, non-hemorrhagic stroke, or any arterial revascularization procedure). Treatment with simvastatin plus ezetimibe resulted in a decrease in LDL-C of 0.85 mmol/L (~34mg/dL). This decrease in LDL-C was associated with a 17% reduction in major atherosclerotic events. In patients on hemodialysis there was a 5% decrease in cardiovascular events that was not statistically significant. Unfortunately, similar to the SEAS Trial, it is impossible to determine whether the addition of ezetimibe improved outcomes above and beyond what would have occurred with statin treatment alone.
IMPROVE-IT TRIAL
The IMPROVE-IT Trial tested whether the addition of ezetimibe to statin therapy would provide an additional beneficial effect in patients with the acute coronary syndrome (130). The IMPROVE-IT Trial was a large trial with over 18,000 patients randomized to simvastatin 40mg vs. simvastatin 40mg + ezetimibe 10mg per day. On treatment LDL-C levels were 70mg/dL in the statin alone group vs. 54mg/dL in the statin + ezetimibe group. There was a small but significant 6.4% decrease in major cardiovascular events (cardiovascular death, MI, documented unstable angina requiring rehospitalization, coronary revascularization, or stroke) in the statin + ezetimibe group (HR 0.936 CI (0.887, 0.988) p=0.016). Cardiovascular death, non-fatal MI, or non-fatal stroke were reduced by 10% (HR 0.90 CI (0.84, 0.97) p=0.003). There was a significant 21% reduction in ischemic stroke (HR, 0.79; 95% CI, 0.67-0.94; P=0.008) and a nonsignificant increase in hemorrhagic stroke (HR, 1.38; 95% CI, 0.93-2.04; P=0.11) (131). Patients with a prior stroke were at a higher risk of stroke recurrence and the risk of a subsequent stroke was reduced by 40% (HR, 0.60; 95% CI, 0.38-0.95; P=0.030) with ezetimibe added to simvastatin therapy (131). In patients with diabetes or other high risk factors the benefits of adding ezetimibe to statin therapy was enhanced (132). In fact, patients without DM and at low or moderate risk demonstrated no benefit with the addition of ezetimibe to simvastatin (132). Similarly, patients who also had peripheral arterial disease or a history of cerebral vascular disease also had the greatest absolute benefits from the addition of ezetimibe (133). Thus, the addition of ezetimibe to statin therapy is of greatest benefit in patients at high risk (for example patients with diabetes, peripheral vascular disease, cerebrovascular disease, etc.).
The results of this study have a number of important implications. First, it demonstrates that combination therapy has benefits above and beyond statin therapy alone. Second, it provides further support for the hypothesis that lowering LDL per se will reduce cardiovascular events. The reduction in cardiovascular events was similar to what one would predict based on the Cholesterol Treatment Trialists results. Third, it suggests that lowering LDL levels into the 50s will have benefits above and beyond lowering LDL levels to the 70mg/dL range in patients with diabetes or other factors that result in a high risk for cardiovascular events. These results have implications for determining goals of therapy and provide support for combination therapy.
EWTOPIA 75
This was a multicenter, randomized trial in Japan that examined the preventive efficacy of ezetimibe for patients aged ≥75 years (mean age 80.6 years), with elevated LDL-C (≥140 mg/dL) without a history of coronary artery disease who were not taking lipid lowering drugs (134). Patients were randomized to ezetimibe 10mg (n=1,716) or usual care (n=1,695) and followed for 4.1 years. The primary outcome was a composite of sudden cardiac death, myocardial infarction, coronary revascularization, or stroke. In the ezetimibe group LDL-C was decreased by 25.9% and non-HDL-C by 23.1% while in the usual care group LDL-C was decreased by 18.5% and non-HDL-C by 16.5% (p<0.001 for both lipid parameters). By the end of the trial 9.6% of the patients in the usual care group and 2.1% of the ezetimibe group were taking statins. Ezetimibe reduced the incidence of the primary outcome by 34% (HR 0.66; P=0.002). Additionally, composite cardiac events were reduced by 60% (HR 0.60; P=0.039) and coronary revascularization by 62% (HR 0.38; P=0.007) in the ezetimibe group vs. the control group. There was no difference in the incidence of stroke or all-cause mortality between the groups. It should be noted that the reduction in cardiovascular events was much greater than one would expect based on the absolute difference in LDL-C levels (121mg/dL in ezetimibe group vs. 132mg/dL). As stated by the authors “Given the open-label nature of the trial, its premature termination, and issues with follow-up, the magnitude of benefit observed should be interpreted with caution.” Nevertheless, this study provides additional support that ezetimibe can reduce cardiovascular events.
RACING TRIAL
The RACING trial was a randomized, open-label trial in patients with atherosclerotic cardiovascular disease carried out in South Korea (135). Patients were randomly assigned to either rosuvastatin 10 mg with ezetimibe 10 mg (n= 1894) or rosuvastatin 20 mg (n= 1886). The primary endpoint was cardiovascular death, major cardiovascular events, or non-fatal stroke. The median LDL-C level during the study was 58mg/dL in the combination therapy group and 66mg/dL in the statin monotherapy group (p<0·0001). The primary endpoint occurred in 9.1% of the patients in the combination therapy group and 9·9% of the patients in the high-intensity statin monotherapy group (non-inferior). Non-inferiority was observed in patients with LDL-C levels < 100mg/dL and >100mg/dL and in patients greater than 75 years of age (136,137).
This study demonstrates that moderate intensity statin with ezetimibe was non-inferior to high-intensity statin therapy with regards to cardiovascular death, major cardiovascular events, or non-fatal stroke. Interestingly a lower prevalence of discontinuation or dose reduction caused by intolerance to the study drug was seen with combination therapy. This indicates that using a moderate intensity dose of a statin with ezetimibe is a useful strategy in patients that do not tolerate high intensity statin therapy.
Side Effects
Ezetimibe has not demonstrated significant side effects. In monotherapy trials, the effect on liver function tests was similar to placebo. In a meta-analysis by Toth et al. of 27 randomized trials in > 20,000 participants evaluating statin plus ezetimibe vs. statin alone the incidence of liver function test abnormalities was slightly greater in the combination therapy group (statin alone- 0.35% vs. statin plus ezetimibe 0.56%) (138). In contrast, Luo and colleagues in a meta-analysis of 20 randomized with > 14,000 subjects did not observe a difference in liver function tests in the ezetimibe plus statin vs. statin alone group (139). With regards to muscle side effects, a meta-analysis of seven randomized trials by Kashani and colleagues found that monotherapy with ezetimibe or ezetimibe in combination with a statin did not increase the risk of myositis compared to placebo or monotherapy with a statin (140). Similarly, Luo et al also did not observe that combination therapy with ezetimibe and a statin increased the risk of myositis (139). In a meta-analysis by Savarese et al. of 7 randomized long-term studies including SEAS, SHARP, and IMPROVE-IT, the incidence of cancer was similar in patients treated with ezetimibe vs. patients not treated with ezetimibe (141). This confirms a previous study that also did not demonstrate an increased cancer risk in the three largest ezetimibe trials (142). Ezetimibe does not appear to have adverse effects on fasting glucose levels or A1c levels (143).
Thus, over many years of use ezetimibe has been shown to be a very safe drug without major side effects.
Contraindications
Ezetimibe is contraindicated in patients with active liver disease. The use of ezetimibe during pregnancy and lactation has not been studied.
Summary
Ezetimibe has a modest ability to lower LDL-C levels and can be a very useful adjunct to statin therapy. When added to statin therapy it will lower the LDL-C by an additional 15-20% which is equivalent to three titrations of the statin dose (for example adding ezetimibe is equivalent to increasing atorvastatin from 10mg to 80mg per day). Additionally, the combination of a high dose of a potent statin (rosuvastatin 40mg per day) with ezetimibe was able to lower the LDL by approximately 70%, which will allow many patients to reach their LDL goal (123). In patient’s intolerant of statins who either cannot take a statin or can only take low doses of a statin, ezetimibe is extremely useful in further lowering LDL-C. The ease of taking ezetimibe, the lack of serious side effects, and that it is inexpensive as it is now a generic drug make it an obvious second choice drug after statins to lower LDL-C levels.
BILE ACID SEQUESTRANTS
Introduction
There are three bile acid sequestrants approved for use in the United States. The first bile acid sequestrant, cholestyramine (Questran), was developed in the 1950s and was the second drug available to lower cholesterol levels (niacin was the first drug). Colestipol (Colestid) was developed in the 1970s and is very similar to cholestyramine. In 2000, Colesevelam (Welchol) was approved. Colesevelam has enhanced binding and affinity for bile acids compared to cholestyramine and colestipol and therefore can be given in much lower doses reducing some side effects (144).
Cholestyramine is available as a powder and the dose ranges from 8-24 grams per day given with meals. Colestipol is available as a tablet and the dose ranges from 2-16 grams per day given with meals or granules and the dose ranges from 5-30 grams per day given with meals. The dose of colesevelam is 3.75 grams per day and can be given as tablets (take 6 tablets once daily or 3 tablets twice daily), oral suspension (take one packet once daily), or chewable bars (take one bar once daily). Because bile acid sequestrants mechanism of action starts with the binding of bile acids in the intestine (see below) these drugs are most effective when administered with meals.
Effect of Bile Acid Sequestrants on Lipid and Lipoprotein Levels
The major effect of bile acid sequestrants is to lower LDL-C levels in a dose dependent fashion. Depending upon the specific drug and dose the decrease in LDL-C ranges from approximately 5 to 30% (144-146). The effect of monotherapy with bile acid sequestrants on LDL-C levels observed in various studies is shown in table 8.
|
Table 8. Effect of Bile Acid Sequestrants on LDL-C
|
|
Drug
|
LDL lowering
|
|
Cholestyramine 4g/day
|
7% decrease
|
|
Cholestyramine 24g/day
|
28% decrease
|
|
Colestipol 4g/day
|
12% decrease
|
|
Colestipol 16g/day
|
24% decrease
|
|
Colesevelam 3.8g/day
|
15% decrease
|
|
Colesevelam 4.3g/day
|
18% decrease
|
Bile acid sequestrants are typically used in combination with statins and the addition of bile acid sequestrants to statin therapy will result in a further 10% to 25% decrease in LDL-C levels (144-146). Combination therapy can result in a 60% reduction in LDL-C levels when high doses of potent statins are combined with high doses of bile acid sequestrants. Bile acid sequestrants will also further lower LDL-C levels by as much as 18% when added to statins and ezetimibe (147). This is particularly useful in patients with Heterozygous Familial Hypercholesterolemia who can have very high LDL-C levels at baseline. Additionally, in patients who are statin intolerant, the combination of a bile acid sequestrant and ezetimibe resulted in an additional 10-20% decrease in LDL-C compared to either drug alone (148,149). Thus, both in monotherapy and in combination with other drugs that lower LDL-C levels, bile acid sequestrants are effective in lowering LDL-C levels
Bile acid sequestrants have a very modest effect on HDL-C levels, typically resulting in a 3-9% increase (144-146). The effect of bile acid sequestrants on triglyceride levels varies (144-146). In patients with normal triglyceride levels, bile acid sequestrants increase triglyceride levels by a small amount. However, as baseline triglyceride levels increase, the effect of bile acid sequestrants on plasma triglyceride levels becomes greater, and can result in substantial increases in triglyceride levels. In patients with triglycerides > 400mg/dL the use of bile acid sequestrants is contraindicated.
|
Table 9. Effect of Bile Acid Sequestrants on Lipid/Lipoprotein Levels
|
|
LDL-C
|
Decrease
|
|
Non-HDL-C
|
Decrease
|
|
Apolipoprotein B
|
Decrease
|
|
Triglycerides
|
Variable. If TG levels elevated will increase significantly
|
|
HDL-C
|
Small Increase
|
|
Lp(a)
|
No change
|
Non-Lipid Effects of Bile Acid Sequestrants
Bile acid sequestrants have been shown to reduce fasting glucose and hemoglobin A1c levels (150). Colesevelam has been most intensively studied and in a number of different studies colesevelam has decreased A1c levels by approximately 0.5-1.0% in patients also treated with a variety of glucose lowering drugs including metformin, sulfonylureas, and insulin. The Food and Drug Administration (FDA) has approved colesevelam for improving glycemic control in patients with type 2 diabetes.
Bile acid sequestrants decrease CRP. For example, Devaraj et al have shown that colesevelam decreases hs-CRP by 18% compared to placebo (151). In combination with a statin, colesevelam reduced hs-CRP levels by 23% compared to statin alone (152).
Mechanisms Accounting for Bile Acid Sequestrants Induced Lipid Effects
Bile acid sequestrants bind bile acids in the intestine, preventing their reabsorption in the terminal ileum leading to the increased fecal excretion of bile acids (153). This decrease in bile acid reabsorption reduces the size of the bile acid pool, which stimulates the conversion of cholesterol into bile acids in the liver (153). This increase in bile acid synthesis decreases hepatic cholesterol levels leading to the activation of SREBPs that up-regulate the expression of the enzymes required for the synthesis of cholesterol and the expression of LDL receptors (153). The increase in hepatic LDL receptors results in the increased clearance of LDL from the circulation leading to a decrease in serum LDL-C levels (Figure 1). Thus, similar to statins and ezetimibe, bile acids lower plasma LDL-C levels by decreasing hepatic cholesterol levels, which stimulates LDL receptor production and thereby accelerates the clearance of LDL from the blood.
The key regulator of bile acid synthesis is FXR (farnesoid X receptor), a nuclear hormone receptor that forms a heterodimer with RXR to regulate gene transcription (154,155). Bile acids down-regulate cholesterol 7α hydroxylase, the first enzyme in the bile acid synthetic pathway by several FXR mediated mechanisms. In the ileum, bile acids via FXR stimulate the production of FGF19, which is secreted into the portal vein and inhibits cholesterol 7α hydroxylase expression in the liver (154). Additionally, in the liver, bile acids activate FXR leading to the increased expression of SHP (small heterodimer partner), which inhibits the transcription of cholesterol 7α hydroxylase (155). Thus, a decrease in bile acids will lead to the decreased activation of FXR in the liver and intestines and thereby result in an increase in cholesterol 7α hydroxylase expression and the increased conversion of cholesterol to bile acids resulting in a decrease in hepatic cholesterol content.
Decreased activation of FXR can also explain the adverse effects of bile acid sequestrants on triglyceride levels (156,157). Activation of FXR increases the expression of apolipoprotein C-II, apolipoprotein A-V, and the VLDL receptor, proteins that decrease plasma triglyceride levels while decreasing the expression of apolipoprotein C-III, a protein that is associated with increases in plasma triglycerides (156,157). Thus, activation of FXR would be expected to decrease triglyceride levels as increases in apolipoprotein C-II, apolipoprotein A-V, and the VLDL receptor and decreases in apolipoprotein C-III would reduce plasma triglyceride levels. With bile acid sequestrants the activation of FXR would be reduced and decreases in the expression of apolipoprotein C-II, apolipoprotein A-V, and the VLDL receptor and increased expression of apolipoprotein C-III would increase plasma triglyceride levels.
The mechanism by which treatment with bile acid sequestrants improves glycemic control is unclear (158).
Pharmacokinetics and Drug Interactions
Bile acid sequestrants are not absorbed and not altered by digestive enzymes and thus their primary effects are localized to the intestine (144-146). It should be noted that bile acid sequestrants can indirectly have systemic effects by decreasing the reabsorption of bile acids and thereby reducing the exposure of cells to bile acids, which are biologically active compounds.
Unfortunately, in the intestine bile acid sequestrants can impede the absorption of many other drugs (144-146). This is particularly true for cholestyramine and colestipol which are used in large quantities (maximum doses- cholestyramine 24 grams per day; colestipol 30 grams per day). In contrast, colesevelam, which requires a much lower quantity of drug because of its high affinity and binding capacity for bile salts, has less of an effect on the absorption of other drugs (recommended dose of colesevelam 3.75 grams/day). Of particular note colesevelam does not interfere with absorption of statins, fenofibrate, or ezetimibe. A list of some of the drugs whose absorption is affected by cholestyramine or colestipol is shown in table 10 and a list of drugs whose absorption is affected by colesevelam is shown in table 11.
|
Table 10. Some of the Drugs Affected by Cholestyramine/Colestipol
|
|
Statins
|
Ezetimibe
|
Gemfibrozil
|
Fenofibrate
|
|
Thiazides
|
Furosemide
|
Spironolactone
|
Digoxin
|
|
Warfarin
|
L-thyroxine
|
Corticosteroids
|
Vitamin K
|
|
Cyclosporine
|
Raloxifine
|
NSAIDs
|
Sulfonylureas
|
|
Aspirin
|
Beta blockers
|
Tricyclic
|
|
|
Table 11. Some of the Drugs Affected by Colesevelam
|
|
L-thyroxine
|
Cyclosporine
|
Glimepiride
|
Glipizide
|
|
Glyburide
|
Phenytoin
|
Olmesartan
|
Warfarin
|
|
Oral contraceptives
|
|
|
|
It is currently recommended that medications should be taken either 4 hours before or 4 hours after taking bile acid sequestrants. This is particularly important with drugs that have a narrow toxic/therapeutic window, such as thyroid hormone, digoxin, or warfarin. It can be very difficult for many patients, particularly those on multiple medications, to take bile acid sequestrants given the need to separate pill ingestion.
Cholestyramine and colestipol may also interfere with the absorption of fat-soluble vitamins. Taking a multivitamin 4 hours before or after these drugs can reduce the likelihood of a vitamin deficiency.
Effect of Bile Acid Sequestrants on Clinical Outcomes
The Lipid Research Clinics Coronary Primary Prevention Trial (LRC-CPPT) of cholestyramine vs. placebo was the first large drug study to explore the effect of specifically lowering LDL-C on cardiovascular outcomes (159). LRC-CPPT was a multicenter, randomized, double-blind study in 3,806 asymptomatic middle-aged men with primary hypercholesterolemia. The treatment group received cholestyramine 24 grams per day and the control group received a placebo for an average of 7.4 years. In the cholestyramine group total and LDL-C was decreased by 8.5% and 12.6% as compared to the placebo group. In the cholestyramine group there was a 19% reduction in risk (p < 0.05) of the primary end point accounted for by a 24% reduction in definite CHD death and a 19% reduction in nonfatal myocardial infarction. In addition, the incidence rates for new positive exercise tests, angina, and coronary bypass surgery were reduced by 25%, 20%, and 21%, respectively, in the cholestyramine group. The reduction in events correlated with the decrease in LDL-C levels (160). Of note, compliance with cholestyramine 24 grams per day was limited with many patients taking much less than the prescribed doses. These results indicate that lowering LDL-C with bile acid sequestrant monotherapy reduces cardiovascular disease.
In addition to the LRC-CPPT clinical outcome study, two studies have examined the effect of cholestyramine monotherapy on angiographic changes in the coronary arteries. The National Heart, Lung, and Blood Institute Type II Coronary Intervention Study and the St Thomas Atherosclerosis Regression Study reported that cholestyramine decreased the progression of atherosclerosis (161,162). There are a number of studies that have employed bile acid sequestrants in combination with other drugs and have shown a reduction in the progression of atherosclerosis or an increase in the regression of atherosclerosis but given the use of multiple drugs it is difficult to attribute the beneficial effects to the bile acid sequestrants (163-165). Unfortunately, there are no clinical outcome studies comparing statins alone vs. statins plus bile acid sequestrants.
Side Effects
Bile acid sequestrants do not have major systemic side effects as they are not absorbed and remain in the intestinal tract. However, they do cause gastrointestinal (GI) side effects (144-146). Constipation is a very common side effect and can be severe. In addition, patients will often complain of bloating, abdominal discomfort, and aggravation of hemorrhoids. Because of GI distress, a significant number of patients will discontinue therapy with bile acid sequestrants. These GI side effects are much more common with cholestyramine and colestipol compared to colesevelam, which is much better tolerated. One can reduce or ameliorate these GI side effects by increasing hydration, adding fiber to the diet (psyllium), and using stool softeners. Notably, bile acid sequestrants do not cause liver or muscle problems.
One should also be aware that bile acid sequestrants can be difficult for many patients to take. Colestipol and colesevelam pills are large and can be difficult for some patients to swallow. Additionally, patients need to take a large number of these pills (colesevelam- 6 pills per day; colestipol- as many as 16 pills per day). The granular forms of cholestyramine and colestipol do not dissolve and are ingested as a suspension in liquid. Many patients find mixing with water leads to an unpalatable mixture that is difficult to take. Sometimes mixing with fruit juice, apple sauce, mash potatoes, etc. make the mixture more palatable. The suspension form of colesevelam with either 1.875 or 3.75 grams is preferred by many patients.
As noted, earlier bile acid sequestrants can increase triglyceride levels, particularly in patients with elevated baseline triglyceride levels.
Contraindications
Bile acid sequestrants usually should be avoided in patients with pre-existing GI disorders. Bile acid sequestrants are contraindicated in patients with recent or repeated intestinal obstruction and patients with plasma triglyceride levels > 400mg/dL. In contradistinction from other lipid lowering drugs, bile acid sequestrants are not contraindicated during pregnancy or lactation (category B) (166). In women of child bearing age who are planning to become pregnant bile acid sequestrants can be a good choice to lower LDL levels.
Summary
Bile acid sequestrants are useful secondary drugs for the treatment of elevated LDL-C levels. They are typically used in combination with statin therapy as a second line drug or as an addition to statin plus ezetimibe therapy as a third line drug. In statin intolerant patients the combination of ezetimibe and a bile acid sequestrant is frequently employed. Bile acid sequestrants can be difficult drugs for patients to take due to GI side effects, difficulty taking the medication, and the need to avoid taking these drugs with other medications. To improve compliance with these drugs the clinician needs to spend time educating the patient on how to take these drugs and how to avoid side effects. Because of these difficulties other cholesterol lowering drugs are used more commonly than bile acid sequestrants. In patients with type 2 diabetes who need an improvement in glycemic control and LDL-C lowering colesevelam can be used to target both abnormalities.
PCSK9 MONOCLONAL ANTIBODIES
Introduction
In 2015 two monoclonal antibodies that inhibit PCSK9 (proprotein convertase subtilisin kexin type 9) were approved for the lowering of LDL-C levels. Alirocumab (Praluent) is produced by Regeneron/Sanofi and evolocumab (Repatha) is produced by Amgen (167,168). Alirocumab is administered as either 75mg or 150mg subcutaneously every 2 weeks or 300mg once a month while evolocumab is administered as either 70mg subcutaneously every 2 weeks or 420mg subcutaneously once a month.
Effect of PCSK inhibitors on Lipid and Lipoprotein Levels
There are a large number of studies that have examined the effect of PCSK9 inhibitors on lipid and lipoprotein levels. A meta-analysis of 24 studies comprising 10,159 patients reported a reduction in LDL-C levels of approximately 50% and in an increase in HDL of 5-8% (169). Notably, in 12 RCTs with 6,566 patients, Lp(a) levels were reduced by 25-30% (169). The higher the baseline Lp(a) the greater the reduction with treatment (170). It should be recognized that most LDL-C lowering drugs (statins, ezetimibe, bempedoic acid, and bile acid sequestrants) do not lower Lp(a) levels. PCSK9 inhibitors have not been shown to decrease hs-CRP levels (171).
MONOTHERAPY
Both alirocumab and evolocumab have been studied as monotherapy vs. ezetimibe. In the Mendel-2 study patients were randomly assigned to evolocumab, placebo, or ezetimibe (172). In the evolocumab group, LDL-C levels decreased by 57% while in the ezetimibe group LDL-C levels decreased by 18% compared to placebo. Additionally, non-HDL-C was decreased by 49%, apolipoprotein B by 47%, triglycerides by 5.3% (NS), and Lp(a) by 18.5% while HDL levels increased by 5.5% in the evolocumab treated subjects. In a study of alirocumab vs. ezetimibe, LDL-C levels were reduced by 47% in the alirocumab group and 16% in the ezetimibe group (173). In addition, alirocumab decreased non-HDL-C by 41%, apolipoprotein B by 37%, triglycerides by 12%, and Lp(a) by 17% and increased HDL by 6%. Thus, PCSK9 monoclonal antibodies are very effective in lowering pro-atherogenic lipoproteins when used in monotherapy and have a more robust effect than ezetimibe.
IN COMBINATION WITH STATINS
In the Odyssey Combo I study, patients on maximally tolerated statin therapy were randomized to alirocumab or placebo (174). Similar to monotherapy results, when alirocumab was added to statin therapy there was a further decrease in LDL-C levels by 46%, non-HDL-C by 38%, apolipoprotein B by 36%, and Lp(a) by 15% with an increase in HDL of 7% and no change in triglyceride levels. In the Odyssey Combo II study, patients on maximally tolerated statin therapy were randomized to alirocumab vs. ezetimibe (175). Alirocumab reduced LDL levels by 51% while ezetimibe reduced LDL by 21%, demonstrating that even when added to statin therapy, alirocumab has a significantly greater ability to reduce LDL-C levels than ezetimibe. In Odyssey Combo II, non-HDL-C levels were decreased by 42%, apolipoprotein B by 41%, triglycerides by 13%, and Lp(a) by 28% while HDL increased by 9% in the alirocumab treated group. In the Laplace-2 study, evolocumab was added to various statins used at different doses (176). It didn’t make any difference which statin was being used (atorvastatin, rosuvastatin, or simvastatin) or what dose (atorvastatin 10mg or 80mg; rosuvastatin 5mg or 40mg); the addition of evolocumab resulted in an approximately 60% further decrease in LDL-C levels beyond statin alone. Additionally, the Laplace-2 trial also showed that evolocumab was much more potent than ezetimibe when added to statin therapy (evolocumab resulted in an approximately 60% decrease in LDL vs. while ezetimibe resulted in an approximately 20-25% reduction).
IN COMBINATION WITH STATINS AND EZETIMIBE
When evolocumab was added to patients receiving atorvastatin 80mg and ezetimibe 10mg there was 48% further reduction in LDL-C levels indicating that even in patients on very aggressive lipid lowering therapy the addition of a PCSK9 inhibitor can still result in a marked reduction in LDL-C (177). In addition to decreasing LDL-C there was also a 41% decrease in non-HDL-C, a 38% decrease in apolipoprotein B, and a 19% decrease in Lp(a) when evolocumab was added to statin plus ezetimibe therapy.
PATIENTS WITH HETEROZYGOUS FAMILIAL HYPERCHOLESTEROLEMIA
Both alirocumab and evolocumab have been tested in patients with Heterozygous Familial Hypercholesterolemia (178,179). In the Rutherford-2 trial, evolocumab lowered LDL-C by 60%, non-HDL-C by 56%, apolipoprotein B by 49%, Lp(a) by 31%, and triglycerides by 22% while increasing HDL by 8% (178). In the Odyssey FH I and FH II studies, alirocumab lowered LDL-C by approximately 55%, non-HDL-C by ~50%, apolipoprotein B by ~43%, Lp(a) by ~19% and triglycerides by ~14% while increasing HDL by ~7% (179). Thus, in these difficult to treat patients PCSK9 monoclonal antibodies were still very effective at lowering pro-atherogenic lipoproteins.
PATIENTS WITH HOMOZYGOUS FAMILIAL HYPERCHOLESTEROLEMIA
Evolocumab resulted in a 21-31% decrease in LDL-C levels compared to placebo in patients with Homozygous Familial Hypercholesterolemia (180,181). The response to therapy appears to be dependent on the underlying genetic cause. Patients with mutations in the LDL receptor leading to the expression of defective receptors respond to therapy whereas patients with mutations leading to negative receptors (null variants) have a poor response (180-182). Given the mechanism by which PCSK9 inhibitors lower LDL-C levels it is not surprising that patients that do not have any functional LDL receptors will not respond to therapy (see section on Mechanism of Lipid Lowering). Alirocumab decreased LDL-C by 35.6%, non-HDL-C by 32.9%, apolipoprotein B by 29.8%, and lipoprotein (a) by 28.4% (183). Given that PCSK9 monoclonal antibodies decrease LDL-C levels in some patients with Familial Hypercholesterolemia these drugs can be useful in this very difficult to treat patient population.
STATIN INTOLERANT PATIENTS
A number of studies have examined the effect of PCSK9 monoclonal antibodies in statin intolerant patients (myalgias) and compared the response to ezetimibe treatment (102,184,185). As expected, treatment with a PCSK9 inhibitor was more effective in lowering LDL-C levels than ezetimibe. Importantly, muscle symptoms were less frequent in the PCSK9 treated patients than those treated with ezetimibe, indicating that PCSK9 monoclonal antibodies will be an effective treatment choice in statin intolerant patients with myalgias.
PATIENTS WITH DIABETES
A meta-analysis of three trials with 413 patients with type 2 diabetes found that in patients with type 2 diabetes evolocumab caused a 60% decrease in LDL-C compared to placebo and a 39% decrease in LDL-C compared to ezetimibe treatment (186). In addition, in patients with type 2 diabetes, evolocumab decreased non-HDL-C 55% vs. placebo and 34% vs. ezetimibe) and Lp(a) (31% vs. placebo and 26% vs. ezetimibe). These beneficial effects were not affected by glycemic control, insulin use, renal function, and cardiovascular disease status. Thus, PCSK9 inhibitors are effective therapy in patients with type 2 diabetes and the beneficial effects on pro-atherogenic lipoproteins is similar to what is observed in non-diabetic patients.
PATIENTS WITH HYPERTRIGLYCERIDEMIA
There are no studies that have examined the effect of PCSK9 monoclonal antibodies in patients with marked elevations in triglyceride levels (>400mg/dL).
|
Table 12. Effect of PCSK9 Inhibitors on Lipid/Lipoprotein Levels
|
|
LDL-C
|
Decrease
|
|
Non-HDL-C
|
Decrease
|
|
Apolipoprotein B
|
Decrease
|
|
Triglycerides
|
No change or small decrease
|
|
HDL-C
|
Small Increase
|
|
Lp(a)
|
Decrease
|
Mechanism Accounting for the PCSK9 Inhibitor Induced Lipid Effects
The linkage of PCSK9 with lipoprotein metabolism was first identified by Abifadel and colleagues in 2003, when they demonstrated that certain mutations in PCSK9 could result in the phenotypic appearance of Familiar Hypercholesterolemia (187). Subsequent studies demonstrated that gain of function mutations in PCSK9 are an uncommon cause of Familiar Hypercholesterolemia (167,168,188). In 2005 it was shown that loss of function mutations in PCSK9 resulted in lower LDL-C levels and this decrease in LDL-C levels was associated with a reduction in the risk of cardiovascular events (189,190).
The main route of clearance of clearance of plasma LDL is via LDL receptors in the liver (191). When the LDL particle binds to the LDL receptor the LDL particle- LDL receptor complex is taken into the liver by endocytosis (191). The LDL particle and the LDL receptor then disassociate and the LDL lipoprotein particle is delivered to lysosomes where it is degraded and the LDL receptor returns to the plasma membrane (Figure 2) (191). After endocytosis LDL receptors recirculate back to the plasma membrane over 100 times.
PCSK9 is predominantly expressed in the liver and secreted into the circulation. Once extracellular, PCSK9 can bind to the LDL receptor and alter the metabolism of the LDL receptor (192,193). Instead of the LDL receptor recycling to the plasma membrane the LDL receptor bound to PCSK9 remains associated with the LDL particle and is delivered to the lysosomes where it is also degraded (Figure 4) (192,193). This results in a decrease in the number of plasma membrane LDL receptors resulting in the decreased clearance of circulating LDL leading to elevations in plasma LDL-C levels.
The PCSK9 monoclonal antibodies bind PCSK9 preventing the PCSK9 from interacting with LDL receptors and thereby preventing PCSK9 from inducing LDL receptor degradation (192,193). The decreased LDL receptor degradation results in an increase in hepatic LDL receptors on the plasma membrane leading to the increased clearance of LDL and decreases in plasma LDL-C levels (194,195). Thus, similar to statins, ezetimibe, bempedoic acid, and bile acid sequestrants, PCSK9 inhibitors are reducing plasma LDL-C levels by up-regulating hepatic LDL receptors. The difference is that PCSK9 inhibitors are decreasing the degradation of LDL receptors while statins, ezetimibe, bempedoic acid, and bile acid sequestrants stimulate the production of LDL receptors.
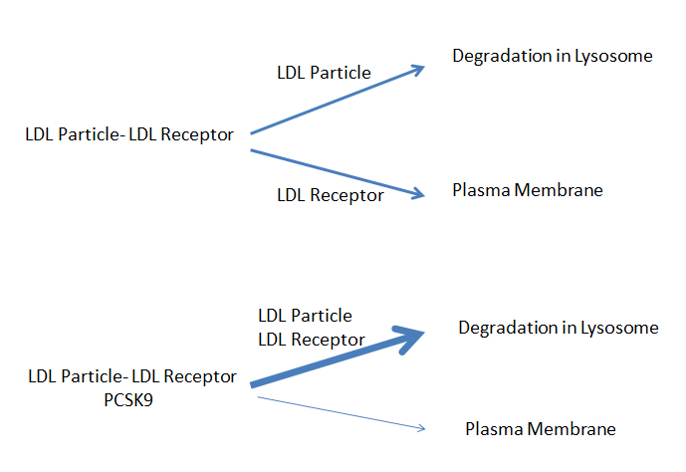
Figure 4. PCSK9 Directs LDL Receptor to Degradation in Lysosome.
The expression of PCSK9 is stimulated by SREBP-2 (192,193). Statins and other drugs that lower hepatic cholesterol levels lead to the activation of SREBP-2 and thereby increase plasma PCSK9 levels (192,193). Inhibition of PCSK9 with monoclonal antibodies is more effective in lowering plasma LDL-C levels in patients on statin therapy due to the higher levels of plasma PCSK9 in these individuals.
The mechanism by which PCSK9 inhibitors reduce Lp(a) levels is unclear. Studies have shown that PCSK9 inhibitors increase the catabolism of lipoprotein(a) particles (196,197). In some circumstances PCSK9 inhibitors may also decrease the production rate (197). It has been postulated that increasing hepatic LDL receptor levels in the setting of marked reductions in circulating LDL levels will result in the clearance of Lp(a) by liver LDL receptors (198).
Pharmacokinetics and Drug Interactions
PCSK9 monoclonal antibodies are eliminated primarily by cellular endocytosis, phagocytosis, and target-mediated clearance. They are not metabolized or cleared by the liver or kidneys and therefore there is no need to adjust the dose in patients with either liver or kidney disease. There are no interactions with the cytochrome P450 system or transport proteins and thus the risk of drug-drug interactions is minimal. Currently there are no reported drug-drug interactions with PCSK9 monoclonal antibodies.
Effect of PCSK9 Inhibitors on Clinical Outcomes
FOURIER TRIAL
The FOURIER trial was a randomized, double-blind, placebo-controlled trial of evolocumab vs. placebo in 27,564 patients with atherosclerotic cardiovascular disease and an LDL-C level of 70 mg/dL or higher who were on statin therapy (199). The primary end point was cardiovascular death, myocardial infarction, stroke, hospitalization for unstable angina, or coronary revascularization and the key secondary end point was cardiovascular death, myocardial infarction, or stroke. The median duration of follow-up was 2.2 years. Baseline LDL-C levels were 92mg/dL and evolocumab resulted in a 59% decrease in LDL levels (LDL-C level on treatment approximately 30mg/dL). Evolocumab treatment significantly reduced the risk of the primary end point (hazard ratio, 0.85; 95% confidence interval (CI), 0.79 to 0.92; P<0.001) and the key secondary end point (hazard ratio, 0.80; 95% CI, 0.73 to 0.88; P<0.001). The results were consistent across key subgroups, including the subgroup of patients in the lowest quartile for baseline LDL-C levels (median, 74 mg/dL). Of note, a similar decrease in cardiovascular events occurred in patients with diabetes treated with evolocumab and glycemic control was not altered (200). Additionally, in patients with peripheral arterial disease evolocumab also reduced cardiovascular events (201). Further analysis has shown that in the small number of patients with a baseline LDL-C level less than 70mg/dL, evolocumab reduced cardiovascular events to a similar degree as in the patients with an LDL-C greater than 70mg/dL (202). The lower the on-treatment LDL-C levels (down to levels below 20mg/dL), the lower the cardiovascular event rate, suggesting that greater reductions in LDL-C levels will result in greater reductions in cardiovascular disease (203). Finally, the relative risk reductions with evolocumab for the cardiovascular events tended to be greater in high-risk subgroups (20% for those with a more recent MI, 18% with multiple prior MI, and 21% with residual multivessel coronary artery disease), whereas the relative risk reduction was 5% to 8% in patients without these risk factors (204). This observation suggests that certain groups of patients will derive greater benefit from the addition of a PCSK9 inhibitor.
It should be noted that that the duration of the FOURIER trial was very short and it is well recognized from previous statin trials that the beneficial effects of lowering LDL-C levels take time with only modest effects observed during the first year of treatment. In the FOURIER trial the reduction of cardiovascular death, myocardial infarction, or stroke was 16% during the first year but was 25% beyond 12 months.
ODYSSEY TRIAL
The ODYSSEY trial was a multicenter, randomized, double-blind, placebo-controlled trial involving 18,924 patients who had an acute coronary syndrome 1 to 12 months earlier, an LDL-C level of at least 70 mg/dL, a non-HDL-C level of at least 100 mg/dL, or an apolipoprotein B level of at least 80 mg/dL while on high intensity statin therapy or the maximum tolerated statin dose (205). Patients were randomly assigned to receive alirocumab 75 mg every 2 weeks or matching placebo. The dose of alirocumab was adjusted to target an LDL-C level of 25 to 50 mg/dL. The primary end point was a composite of death from coronary heart disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization. During the trial LDL-C levels in the placebo group was 93-103mg/dL while in the alirocumab group LDL-C levels were 40mg/dL at 4 months, 48mg/dL at 12 months, and 66mg/dL at 48 months (the increase with time was due to discontinuation of alirocumab or a decrease in dose). The primary endpoint was reduced by 15% in the alirocumab group (HR 0.85; 95% CI 0.78 to 0.93; P<0.001). In addition, total mortality was reduced by 15% in the alirocumab group (HR 0.85; 95% CI 0.73 to 0.98). The absolute benefit of alirocumab was greatest in patients with a baseline LDL-C level greater than 100mg/dL. In patients with an LDL-C level > than 100mg/dL the number needed to treat with alirocumab to prevent an event was only 16. It should be noted that the duration of this trial was very short (median follow-up 2.8 years) which may have minimized the beneficial effects. Additionally, because alirocumab 75mg every 2 weeks was stopped if the LDL-C level was < 15mg/dL on two consecutive measurements the beneficial effects may have been blunted (7.7% of patients randomized to alirocumab were switched to placebo).
SUMMARY OF OUTCOME TRIALS
It should be noted that that the duration of the PCSK9 outcome trials were relatively short and it is well recognized from previous statin trials that the beneficial effects of lowering LDL-C levels take time with only modest effects observed during the first year of treatment. In the FOURIER trial the reduction of cardiovascular death, myocardial infarction, or stroke was 16% during the first year but was 25% beyond 12 months. In the ODYSSEY trial the occurrence of cardiovascular events was similar in the alirocumab and placebo group during the first year of the study with benefits of alirocumab appearing after year one. Thus, the long-term benefits of treatment with a PCSK9 inhibitor may be greater than that observed during these relatively short-term studies.
GLAGOV TRIAL
While not an outcome trial the GLAGOV trial provides further support for the benefits of further lowering of LDL-C levels with a PCSK9 inhibitor added to statin therapy (206). This trial was a double-blind, placebo-controlled, randomized trial of evolocumab vs. placebo in 968 patients presenting for coronary angiography. The primary efficacy measure was the change in percent atheroma volume (PAV) from baseline to week 78, measured by serial intravascular ultrasonography (IVUS) imaging. Secondary efficacy measures included change in normalized total atheroma volume (TAV) and percentage of patients demonstrating plaque regression. As expected, there was a marked decrease in LDL-C levels in the evolocumab group (Placebo 93mg/dL vs. evolocumab 37mg/dL; p<0.001). PAV increased 0.05% with placebo and decreased 0.95% with evolocumab (P < .001) while TAV decreased 0.9 mm3 with placebo and 5.8 mm3 with evolocumab (P < .001). There was a linear relationship between achieved LDL-C and change in PAV (i.e., the lower the LDL-C the greater the regression in atheroma volume down to an LDL-C of 20mg/dL). Additionally, evolocumab induced plaque regression in a greater percentage of patients than placebo (64.3% vs 47.3%; P < .001 for PAV and 61.5% vs 48.9%; P < .001 for TAV). These results demonstrate the anti-atherogenic effects of PCSK9 inhibitors. Other trials in different patient populations have also shown that treatment with PCSK9 inhibitors are anti-atherogenic (207,208).
VENOUS THROMBOEMBOLISM
In the FOURIER trial treatment with evolocumab resulted in a reduction in venous thromboembolism (VTE) (HR 0.71; 95% CI, 0.50-1.00; P=0.05) (209). Interestingly no effect was observed in the 1st year (HR, 0.96; 95% CI, 0.57-1.62) but a 46% reduction in VTE (HR, 0.54; 95% CI, 0.33-0.88; P=0.014) beyond 1 year occurred. In patients with low baseline Lp(a) levels, evolocumab reduced Lp(a) by only 7 nmol/L and had no effect on VTE risk but in patients with high baseline Lp(a) levels, evolocumab reduced Lp(a) by 33 nmol/L and risk of VTE by 48% (HR, 0.52; 95% CI, 0.30-0.89; P=0.017). In the ODYSSEY OUTCOMES trial, the risk of VTE was reduced but just missed being statistically significant (HR, 0.67; 95% CI, 0.44-1.01; P=0.06) (210). A meta-analysis of FOURIER and ODYSSEY OUTCOMES demonstrated a 31% relative risk reduction in VTE with PCSK9 inhibition (HR, 0.69; 95% CI, 0.53-0.90; P=0.007) (209).
Side Effects
The major side effect of PCSK9 monoclonal antibodies has been injection site reactions including erythema, itching, swelling, pain, and tenderness. Allergic reactions have been reported and as with any protein there is potential immunogenicity. In general side effects have been minimal, which is not surprising, as monoclonal antibodies do not typically have off target side effects. Since PCSK9 does not appear to have important functions other than regulating LDL receptor degradation, it is not surprising that inhibiting PCSK9 function has not resulted in major side effects.
A meta-analysis of 20 randomized controlled trials with 68,123 subjects found a very modest effect on fasting glucose (mean difference 1.88 mg/dL) and A1c levels (mean difference 0.032%) and did not observe an increased risk of developing diabetes (211). It should be recognized that the duration of these trials was relatively short (median follow-up 78 weeks) and therefore further long-term studies are required.
In the large outcome trials (ODYSSEY and FOURIER) there was no significant difference between the PCSK9 treated group vs. the placebo group with regard to adverse events (including new-onset diabetes and neurocognitive events). The only exception was the expected increase in injection-site reactions in the patients treated with a PCSK9 inhibitor. Additionally, in a subgroup of patients from the FOURIER trial a prospective study of cognitive function (EBBINGHAUS Study) was carried out and no significant differences in cognitive function was observed over a median of 19 months in the PCSK9 treated vs. placebo group (212). It should be recognized that while short-term treatment with PCSK9 inhibitors have not demonstrated any significant side effects it is possible that long-term use could lead to unexpected side-effects.
An issue of concern is whether lowering LDL-C to very low levels has the potential to cause toxicity. In a number of the PCSK9 studies a significant number of patients had LDL-C levels < 25mg/dL. For example, in the Odyssey long term study 37% of patients on alirocumab had two consecutive LDL-C levels below 25mg/dL and in the Osler long term study in patients treated with evolocumab 13% had values below 25mg/dL (213,214). In these short term PCSK9 studies, toxicity from very low LDL-C levels has not been observed. Additionally, in patients with Familial Hypobetalipoproteinemia LDL levels can be very low and these patients do not have any major disorders other than hepatic steatosis, which is not mechanistically due to low LDL-C levels (215). Similarly, there are rare individuals who are homozygous for loss of function mutations in the PCSK9 gene and they also do not appear to have major medical issues (168). Finally, in a number of statin trials there have been patients with very low LDL-C levels and an increased risk of side effects has not been consistently observed in those patients (216-218). Thus, with the limited data available there does not appear to be a major risk of markedly lowering LDL-C levels.
Contraindications
Other than a history of a hypersensitivity to these drugs there are currently no contraindications. There are no studies during pregnancy or lactation.
Summary
PCSK9 monoclonal antibodies robustly reduce LDL-C levels when used as monotherapy, in combination with statins, or when added to the combination of statins + ezetimibe. In distinction to most other cholesterol lowering drugs the PCSK9 inhibitors also decrease Lp(a) levels. Outcome studies have clearly demonstrated that decreasing LDL-C levels with PCSK9 inhibitors reduces cardiovascular events. The side effect profile appears to be very favorable and there are no drug-drug interactions. The major limitation is the high expense of these drugs, which has limited their widespread use.
INCLISIRAN (LEQVIO)
Introduction
Inclisiran (Leqvio) is a double-stranded, siRNA (small interfering RNA) conjugated on the sense strand with triantennary N-acetylgalactosamine (GalNAc) to facilitate uptake into hepatocytes (219). In hepatocytes, inclisiran stimulates the catalytic breakdown of PCSK9 mRNA thereby reducing the hepatic synthesis of PCSK9 and markedly decreasing plasma PCSK9 levels (219,220). The recommended dose of inclisiran is 284 mg by subcutaneous injection, followed with a repeat injection at 3 months, and then every 6 months (package insert). If a dose is missed by more than 3 months it is recommended to repeat the dosage schedule described above (package insert). It is recommended that inclisiran be administered by a healthcare professional.
Effect on Inclisiran on Lipid and Lipoprotein Levels
There have been several large trials examining the efficacy of inclisiran. The ORION-10 trial was conducted in the United States and included adults with atherosclerotic cardiovascular disease on a maximally tolerated statin with an LDL-C > 70mg/dL (220). Patients were randomized to inclisiran 284mg (n=781) at initial visit, 3 months, 9 months, and 15 months or placebo (n=780) and followed for 540 days. After 3 months the LDL-C was reduced by approximately 50% and this reduction was sustained throughout the duration of the trial (at 540 days the LDL-c was reduced by 52.3% (P<0.001)). As expected, total cholesterol (-33%), non-HDL-C (-47%), and apolipoprotein B (-43%) were also decreased. Additionally, triglyceride (-13%) and Lp(a) (-26%) levels were decreased while HDL-C levels (+5.1%) and hsCRP (+8.8%) were slightly increased. ORION-11 was a very similar trial with an identical protocol conducted in Europe and South Africa and included adults with ASCVD or an ASCVD risk equivalent on maximally tolerated statin therapy (inclisiran n=810 and placebo n=807) (220). At 540 days LDL-C was reduced by 49.9% (P<0.001). Changes in other lipid parameters were similar to those observed in ORION 10. Subgroup analysis revealed that in both the ORION 10 and 11 trials that all subgroups had a similar reduction in LDL-C levels with inclisiran therapy including subjects with diabetes, moderate renal impairment, and greater than 75 years of age (220). Statin therapy and whether statin therapy was moderate intensity or high intensity also did not affect the reduction in LDL-C (220). Additionally, in patients with renal disease, including individuals with an estimated creatinine clearance between 15-29 mL/min, the reduction in LDL-C levels with inclisiran administration were similar to individuals with normal renal function (221). The decrease in LDL-C with inclisiran treatment has been shown to persist for 4 years (222).
HETEROZYGOUS FAMILIAL HYPERCHOLESTEROLEMIA
The effect of inclisiran on LDL-C levels was determined in patients with heterozygous familial hypercholesterolemia who were randomized to receive subcutaneous injections of inclisiran 284mg (n= 242) or placebo (n=240) on days 1, 90, 270, and 450 (223). The mean baseline LDL-C level was 153±54mg/dL and 90% of the patients were receiving statins with most on high intensity statins (75%). At day 510 LDL-C levels were reduced by 47.9% compared to placebo (P<0.001). The reduction in LDL-C was similar in all genotypes of familial hypercholesterolemia. Total cholesterol was reduced by 33%, non-HDL-C by 44%, Lp(a) by 17.2%, and triglycerides by 12%. HDL-C and hsCRP were not markedly altered.
HOMOZYGOUS FAMILIAL HYPERCHOLESTEROLEMIA
A small study reported that inclisiran treatment lowered LDL-C levels in 3 of 4 patients with homozygous familiar hypercholesterolemia (17.5% to 37% decrease) but less than that seen in individuals with fully functioning LDL receptors (224). A larger more recent trial failed to demonstrate a decrease in LDL-C levels with inclisiran treatment (225). Of note there was considerable variation in the LDL-C response, which could be due to differences in genetic variants. Individuals with null-null LDL receptor variants (i.e. no functioning LDL receptors) are unlikely to respond to inclisiran due to the absence of LDL receptors and the group treated with inclisiran in this study was enriched in patients with this genotype, which could explain the absence of a significant reduction in LDL-C.
Mechanisms Accounting for Inclisiran Induced Lipid Effects
The mechanism of action of inclisiran is the same as for PCSK9 monoclonal antibodies (219). Briefly, decreasing the production of PCSK9 in the liver, the primary source of circulating PCSK9, leads to a decrease in plasma PCSK9 levels resulting in a decrease in LDL receptor degradation (219). An increase in the number of hepatic LDL receptors increases the clearance of LDL leading to a decrease in LDL-C levels (219).
Pharmacokinetics and Drug Interactions
There are no drug interactions. The reduction in LDL-C occurs within 14 days after drug administration and persists for an extended period of time allowing for administration every 6 months.
Effect of Inclisiran on Clinical Outcomes
No outcome studies are currently available. A cardiovascular outcome study (ORION-4) is ongoing and includes 15,000 patients with established ASCVD. The trial duration is five years and completion is expected in 2024 (NCT03705234) (ClinicalTrials.gov, 2020a).
Side Effects
The only adverse reactions associated with inclisiran were injection site reactions including rash, pain, and erythema (220). In an analysis of 7 studies with 3,576 patients treated with inclisiran for up to 6 years and 1,968 patients treated with placebo for up to 1.5 years, hepatic, muscle, and kidney events; incident diabetes; and elevations of creatine kinase or creatinine were not increased in patients treated with inclisiran (226).
Contraindications
In patients with severe hepatic or renal impairment inclisiran should be used with caution as there is limited data and experience in these patients. There are no studies during pregnancy or lactation.
Summary
Inclisiran very effectively lowers LDL-C levels. The major advantage of this drug compared to PCSK9 monoclonal antibodies is the ability to administer inclisiran every 6 months, which may improve compliance.
BEMPEDOIC ACID (NEXLETOL)
Introduction
Bempedoic acid was approved in the US in February 2020 and is an adenosine triphosphate-citrate lyase (ACL) inhibitor. It is administered orally once daily with or without food at a dose of 180mg (Nexletol). It is also available as a combination tablet containing 180 mg of bempedoic acid and 10 mg of ezetimibe (Nexlizet).
Effect on Bempedoic on Lipid and Lipoprotein Levels
EFFECT WITHOUT STATINS
In a study that randomized 345 patients with hypercholesterolemia (LDL-C 158mg/dL) and a history of intolerance to statin to either bempedoic acid or placebo (2:1), bempedoic acid decreased LDL-C by 21.4%, non-HDL-C by 17.9%, and apolipoprotein B by 15% (227). One third of patients were on background non-statin therapy most commonly ezetimibe and fish oil. Triglyceride levels were not altered but there was a small decrease in HDL-C levels that was statistically significant (-4.5%).
IN COMBINATION WITH STATINS
There have been two large trials that determined the effect of adding bempedoic acid to statin therapy. In a study that randomized 779 patients on maximally tolerated statin therapy +/- ezetimibe (only a small number on ezetimibe) with an LDL-C level greater than 70mg/dL (baseline LDL-C 120mg/dL) to either bempedoic acid or placebo it was observed that bempedoic acid decreased LDL-C levels by 17.4% compared to placebo (p<0.001) (228). In addition, non-HDL-C and apolipoprotein B levels were decreased by 13% compared to placebo while there was no significant change in triglyceride levels. Bempedoic acid decreased HDL-C levels by approximately 6%. In a similar study, patients with atherosclerotic cardiovascular disease, heterozygous familial hypercholesterolemia, or both with an LDL-C level greater than 70 mg/dL (baseline LDL-C 103mg/dL) while on maximally tolerated statin therapy with or without additional lipid-lowering therapy (only a small number on ezetimibe) were randomized to bempedoic acid (n= 1,488) or placebo (n= 742) (229). Compared to placebo, treatment with bempedoic acid decreased LDL-C by 18.1%, non-HDL-C by 13.5%, and apolipoprotein B by 11.9%. Triglyceride levels were unchanged but HDL-C decreased by 5.92%. Of note in both of the above studies the decrease in LDL-C was maintained over 52 weeks.
Notably, the addition of bempedoic acid to atorvastatin 80mg per day was still capable of significantly decreasing LDL-C (22%), non-HDL-C (13%), and apolipoprotein B (-15%) compared to placebo (230). The addition of bempedoic acid to high dose atorvastatin therapy did not cause meaningful changes in atorvastatin pharmacokinetics.
IN COMBINATION WITH EZETIMIBE
Patients on maximally tolerated statin therapy with LDL-C levels greater 100 mg/dL if they had cardiovascular disease and/or Familiar Hypercholesterolemia or greater than 130 mg/dL if they had multiple CVD risk factors were randomized to bempedoic acid + ezetimibe, bempedoic acid alone, ezetimibe alone, or placebo (231). The key results of this study are shown in Table 14. Changes from baseline in HDL-C and triglyceride level were modest (<10%) in all treatment groups. In another study patients with a history of statin intolerance on ezetimibe therapy were randomized to bempedoic acid (n=181) or placebo (n= 88) (232). Compared to placebo, bempedoic acid decreased LDL-C by 28.5%, non-HDL-C by -23.6%, and apolipoprotein B by -19.3%. As seen in other studies bempedoic acid did not alter triglyceride levels but slightly decreased HDL-C levels (approximately 6% decrease compared to placebo).
|
Table 14. Effect of Bempedoic Acid and Ezetimibe on Lipid Parameters (231)
|
|
|
LDL-C
|
Non-HDL-C
|
Apo B
|
hsCRP
|
|
Bempedoic acid + ezetimibe
|
-38%
|
-33.7%
|
-30.1
|
-35.1
|
|
Bempedoic acid
|
-19%
|
-15.9%
|
-17.3
|
-31.9
|
|
Ezetimibe
|
-25%
|
-21.7
|
-20.8
|
-8.2
|
Results are percent decrease compared to the placebo group.
Summary
Bempedoic acid typically lowers LDL-C by 15-25%, non-HDL-C by 10-20%, and apolipoprotein B levels by 10-20% with no significant effects on triglyceride levels. HDL-C levels decrease by 5-8% and Lp(a) are unchanged (233).
|
Table 15. Effect of Bempedoic Acid on Lipid/Lipoprotein Levels
|
|
LDL-C
|
Decrease
|
|
Non-HDL-C
|
Decrease
|
|
Apolipoprotein B
|
Decrease
|
|
Triglycerides
|
No change
|
|
HDL-C
|
Small decrease
|
|
Lp(a)
|
No change
|
Non-Lipid Effects of Bempedoic Acid
Bempedoic acid decreases hsCRP levels (see table 14, 16).
|
Table 16. Effect of Bempedoic Acid on hsCRP Levels
|
|
Reference
|
Percent decrease in hsCRP
|
|
(227)
|
-24.3
|
|
(228)
|
-8.7
|
|
(229)
|
-21.5
|
|
(230)
|
-44
|
|
(232)
|
-31
|
In the CLEAR Outcome study with a median follow-up of 3.4 years there was no difference in the development of new onset diabetes in the bempedoic acid and placebo groups (429 of 3848, -11·1% with bempedoic acid vs 433 of 3749, 11·5% with placebo; HR 0.95; 95% CI 0.83-1.09) (234). Additionally, during the study HbA1c concentrations and fasting glucose levels were similar between the bempedoic acid and placebo groups in patients who had either prediabetes or normoglycemia. In the CLEAR Outcome study in patients with diabetes the prevalence of worsening diabetes was similar in the bempedoic acid and placebo group (235,236)
Mechanisms Accounting for Bempedoic Acid Induced Lipid Effects
Bempedoic acid is a potent inhibitor of ATP-citrate lyase, which catalyzes the formation of acetyl-CoA in the cytoplasm (237). Acetyl-CoA is a precursor for the synthesis of cholesterol (figure 5). The inhibition of ATP-citrate lyase by bempedoic acid decreases cholesterol synthesis in liver reducing hepatic intracellular cholesterol levels (237). Of note, bempedoic acid is a pro-drug and conversion to its CoA-derivative by very-long-chain acyl-CoA synthetase-1 is required for inhibition of cholesterol synthesis (237). Very-long-chain acyl-CoA synthetase-1 is highly expressed in the liver but is not expressed in adipose tissue, kidney, intestine or skeletal muscle (237). The inability of bempedoic acid to be activated in muscle and inhibit cholesterol synthesis suggests that bempedoic acid is unlikely result in muscle toxicity.
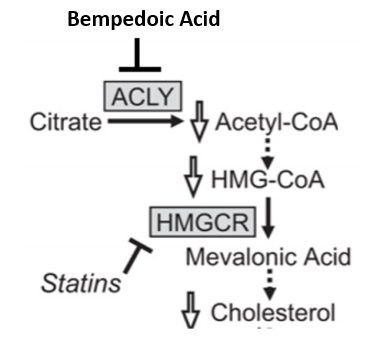
Figure 5. Inhibition of Cholesterol Synthesis by Bempedoic Acid.
The decrease in plasma LDL-C levels in patients treated with bempedoic acid is primarily due to an increase in hepatic LDL receptors secondary to the inhibition of cholesterol synthesis resulting in a reduction in hepatic cholesterol levels (237). It should be noted that bempedoic acid also decreases circulating LDL-C levels in LDL receptor deficient mice and LDL receptor deficient miniature pigs indicating that mechanisms in addition to up-regulation of hepatic LDL receptors may contribute to the decrease in LDL-C levels (237). The inhibition of hepatic cholesterol synthesis may decrease the production and secretion of VLDL, which could contribute to a decrease in LDL-C.
Pharmacokinetics and Drug Interactions
No dose adjustments are required in patients with mild or moderate renal or hepatic impairment or in the elderly (package insert). Concomitant use of bempedoic acid with simvastatin or pravastatin causes an increase in the concentrations of these drugs and therefore may increase the risk of myopathy (package insert). This drug interaction may be secondary to bempedoic acid inhibiting organic anion-transporting polypeptide OATP1B1. It is recommended to avoid concomitant use of bempedoic acid with simvastatin greater than 20 mg/day or pravastatin 40mg/day. While concomitant administration of bempedoic acid with atorvastatin or rosuvastatin elevated the area under the curve by 1.7-fold these elevations were generally within the individual statin exposures and do not impact dosing recommendations (package insert).
Effect of Bempedoic Acid on Clinical Outcomes
In animal models of atherosclerosis, treatment with bempedoic acid had favorable effects on atherosclerosis (237). Moreover, genetic variants of ATP citrate lyase that lower LDL-C levels are associated with a decrease in cardiovascular disease suggesting that bempedoic acid will have favorable effects on reducing the risk of cardiovascular disease (238).
The CLEAR Outcome trial was a double-blind, randomized, placebo-controlled trial involving patients with cardiovascular disease or at high risk of cardiovascular disease who were unable or unwilling to take statins ("statin-intolerant" patients) (239). The patients were randomized to bempedoic acid 180 mg (n= 6992) or placebo (n= 6978) and the median duration of follow-up was 40.6 months. As expected, LDL-C levels were decreased by 21% in the bempedoic group compared to placebo (29mg/dL difference). The primary endpoint, death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke, or coronary revascularization, was reduced by 13% in the bempedoic acid group (HR 0.87; 95% CI 0.79 to 0.96; P = 0.004). Bempedoic acid also decreased fatal and non-fatal myocardial infarctions and coronary revascularization but had no significant effects on fatal or nonfatal stroke, death from cardiovascular causes, and death from any cause. In the patients who were at high risk for cardiovascular disease (primary prevention), 66% had diabetes, and the primary endpoint was reduced by 30% in the bempedoic acid group (HR 0.70; 95% CI, 0.55-0.89; P = .002) (235). In patients with diabetes with or without cardiovascular disease the primary endpoint was reduced by 17% in the bempedoic acid group (HR 0.83; 95% CI 0.72-0.95) (234). This study clearly demonstrates that treatment with bempedoic acid reduces the risk cardiovascular events.
Side Effects
HYPERURICEMIA
In clinical trials, 26% of bempedoic acid-treated patients with normal baseline uric acid values experienced hyperuricemia one or more times versus 9.5% in the placebo group (package insert). In the CLEAR Outcomes trial elevated uric acid levels occurred in 10.9% of the patients on bempedoic acid compared to 5.6% taking the placebo (239). The increase in uric acid is due to bempedoic acid inhibiting renal tubular OAT2. The Increase in uric acid levels typically occurred within the first 4 weeks of treatment and persisted throughout treatment. After 12 weeks of treatment, the mean placebo-adjusted increase in uric acid compared to baseline was 0.8 mg/dL for patients treated with bempedoic acid (package insert). Elevations in blood uric acid levels may lead to the development of gout. Gout was reported in 1.5% of patients treated with bempedoic acid vs. 0.4% of patients treated with placebo. The risk for gout attacks were higher in patients with a prior history of gout (11.2% for bempedoic acid treatment vs. 1.7% in the placebo group) (package insert). In patients with no prior history of gout only 1% of patients treated with bempedoic acid and 0.3% of the placebo group had a gouty attack (package insert). In the CLEAR Outcomes trial gout was increased in the bempedoic acid group (3.1% vs. 2.1%) (239).
TENDON RUPTURE
In clinical trials tendon rupture occurred in 0.5% of patients treated with bempedoic acid vs. 0% of placebo treated patients and involved the rotator cuff (the shoulder), biceps tendon, or Achilles tendon (package insert). Tendon rupture occurred within weeks to months of starting bempedoic acid and occurred more frequently in patients over 60 years of age, in those taking corticosteroid or fluoroquinolone drugs, in patients with renal failure, and in patients with previous tendon disorders. In the CLEAR Outcomes trial tendon rupture was similar in the bempedoic acid and placebo group (bempedoic acid 1.2% and placebo 0.9%) (239).
RENAL FUNCTION
Bempedoic acid treatment resulted in a mean increase in serum creatinine of 0.05 mg/dL compared to baseline. Approximately 3.8% of patients treated with bempedoic acid had BUN levels that doubled vs. 1.5% in the placebo group and about 2.2% of patients treated with bempedoic acid had creatinine values that increased by 0.5 mg/dL vs. 1.1% in the placebo group (package insert). Renal function returned to baseline when bempedoic acid was discontinued. In the CLEAR Outcomes trial renal impairment was increased in the bempedoic acid group (11.5% vs.8.6%) as was the change from baseline creatinine (0.05±0.2 mg/dL vs. 0.01±0.2 mg/dL) (239).
CHOLELITHIASIS
In the CLEAR Outcomes trial cholelithiasis was increased in the bempedoic acid group (2.2 vs 1.2) (239).
BENIGN PROSTATIC HYPERPLASIA
Bempedoic acid was associated with an increased risk of benign prostatic hyperplasia (BPH) in men with no reported history of BPH, occurring in 1.3% of NEXLETOL-treated patients versus 0.1% of placebo-treated patients (package insert).
MISCELLANEOUS LABORATORY ABNORMALITIES
Approximately 5.1% of patients on bempedoic acid vs. 2.3% on placebo had decreases in hemoglobin levels of 2 or more g/dL and below the lower limit of normal on one or more occasion. Anemia was reported in 2.8% of patients treated with bempedoic acid and 1.9% of patients treated with placebo. Hemoglobin decrease was generally asymptomatic and did not require medical intervention (package insert).
Approximately 9.0% of bempedoic acid treated patients with a normal baseline leukocyte count decreased leukocyte count to less than the lower limit of normal on one or more occasions vs. 6.7% in the placebo group. The leukocyte decrease was generally asymptomatic and did not require medical intervention (package insert).
Approximately 10.1% of bempedoic acid treated patients vs. 4.7% in the placebo group had
increases in platelet counts of 100× 109/L or more on one or more occasion. The platelet count increase was asymptomatic, did not result in an increased risk for thromboembolic events, and did not require medical intervention (package insert).
Increases to more than 3× the upper limit of normal (ULN) in AST occurred in 1.4% of patients treated with bempedoic acid vs. 0.4% of placebo patients, and increases to more than 5× ULN occurred in 0.4% of bempedoic acid treated patients vs. 0.2% of placebo-treated patients. Increases in ALT were similar in bempedoic acid treated patients and placebo-treated patients. Elevations in transaminases were generally asymptomatic and not associated with elevations ≥2× ULN in bilirubin or with cholestasis. In most cases, the elevations were transient and resolved or improved with continued therapy or after discontinuation of therapy (package insert).
Contraindications
The use of bempedoic acid during pregnancy and lactation has not been studied (package insert).
Summary
In patients on statins and ezetimibe with an LDL-C that is not at goal the addition of bempedoic acid is a reasonable third drug. In addition, in patients that cannot tolerate statin therapy the combination of ezetimibe and bempedoic acid may allow for the lowering of LDL-C to goal. One can expect a reduction in LDL-C of approximately 15-25% with bempedoic acid monotherapy therapy or when used in combination with other LDL-C lowering drugs.
LOMITAPID (JUXTAPID)
Introduction
Lomitapide (Juxtapid), a selective microsomal triglyceride transfer protein inhibitor, was approved in December 2012 for lowering LDL-C levels in adults with Homozygous Familial Hypercholesterolemia (240-242). As will be discussed below it lowers LDL-C levels by an LDL receptor independent mechanism.
Effect on Lomitapide on Lipid and Lipoprotein Levels
The effect of lomitapide on lipid and lipoprotein levels has been studied in patients with Homozygous Familial Hypercholesterolemia. The pivotal study was a 78-week single arm open label study in 29 patients receiving treatment for Homozygous Familial Hypercholesterolemia (243). Lomitapide was initiated at 5mg per day and was up-titrated to 60mg per day based on tolerability and liver function tests. On an intention to treat basis, LDL-C was decreased by 40% and apolipoprotein B by 39%. In patients who were actually taking lomitapide, LDL-C levels were reduced by 50%. In addition to decreasing LDL-C levels, non-HDL-C levels were decreased by 50%, Lp(a) by 15%, and triglycerides by 45%. Interestingly HDL and apolipoprotein A-I levels were decreased by 12% and 14% respectively in this study. Follow-up revealed that the decrease in LDL-C could be sustained for a prolonged period of time (294 weeks) (244).
The effect of lomitapide has also been studied in patients without Homozygous Familial Hypercholesterolemia. A study by Samaha and colleagues compared the effect of ezetimibe and lomitapide in patients with elevated cholesterol levels(245). Patients were treated with ezetimibe alone, lomitapide alone, or the combination of ezetimibe and lomitapide. Ezetimibe monotherapy led to a 20–22% decrease in LDL-C levels, lomitapide monotherapy led to a dose dependent decrease in LDL-cholesterol levels (19% at 5.0 mg, 26% at 7.5 mg and 30% at 10 mg). Combined therapy produced a larger dose-dependent decrease in LDL-C levels (35%, 38% and 46%, respectively). Additionally, lomitapide decreased triglycerides by 10%, non-HDL-C by 27%, apolipoprotein B by 24%, and Lp(a) by 17%.
The above studies demonstrate that lomitapide decreases LDL-C, non-HDL-C, triglycerides, and Lp(a) levels.
Mechanism Accounting for the Lomitapide Induced Lipid Effects
Lomitapide is a selective inhibitor of microsomal triglyceride transfer protein (MTTP) (240-242). MTTP is located in the endoplasmic reticulum of hepatocytes and enterocytes where it plays a key role in transferring triglycerides onto newly synthesized apolipoprotein B leading to the formation of VLDL and chylomicrons (246). Loss of function mutations in both alleles of MTTP results in abetalipoproteinemia, which is characterized by the virtual absence of apolipoprotein B, VLDL, chylomicrons, and LDL in the plasma due to the failure of the liver and intestine to produce VLDL and chylomicrons (215). Lomitapide by inhibiting MTTP activity reduces the secretion of chylomicrons by the intestine and VLDL by the liver leading to a decrease in LDL, apolipoprotein B, triglycerides, non-HDL-C, and Lp(a) (240-242).
Pharmacokinetics and Drug Interactions
Lomitapide is extensively metabolized in the liver by the CYP3A4 pathway (240,241). Therefore, lomitapide is contraindicated in patients on strong CYP3A4 inhibitors and lower doses should be used in patients on weak inhibitors. Of particular note, in patients on atorvastatin the maximal dose of lomitapide is 30mg per day and lomitapide should not be used in patients taking more than 20mg of simvastatin (240,241). Lomitapide can increase warfarin levels and therefore close monitoring is required. Finally, given the risk of liver abnormalities (see side effect section) the avoidance of alcohol or a reduction in alcohol intake is prudent.
Effect of Lomitapide on Clinical Outcomes
There are no clinical outcome trials but it is presumed that lowering LDL-C levels in patients with Homozygous Familial Hypercholesterolemia will reduce cardiovascular events. After initiating lomitapide therapy 1.7 cardiovascular events per 1000 patient months on treatment was observed vs. 26.1 cardiovascular events per 1000 patient months in a comparison cohort (247).
Side Effects
As expected from its mechanism of action lomitapide causes side effects in the GI tract and liver. In the GI tract diarrhea, nausea, vomiting, and dyspepsia occur very commonly (240-242). In the pivotal study in patients with Homozygous Familial Hypercholesterolemia, 90% of the patients developed GI symptoms during drug titration (243). GI side effects are potentiated by high fat meals and it is therefore recommended that dietary fat be limited. Approximately 10% of patients will discontinue lomitapide, mostly from diarrhea. Lomitapide also reduces the absorption of fat soluble vitamins and therefore patients need to take vitamin supplements (240,241). Additionally, it may also block the absorption of essential fatty acids and it is therefore recommended that supplements of essential fatty acids also be provided (at least 200 mg linoleic acid, 210 mg alpha-linolenic acid (ALA), 110 mg eicosapentaenoic acid (EPA), and 80 mg docosahexaenoic acid (DHA) (240,241).
Blocking the formation of VLDL in the liver can lead to fatty liver with elevated liver enzymes (240-242). Approximately 30% of patients will develop increased transaminase levels but in the small number of patients studied this has not resulted in liver failure. After stopping the drug, the transaminases have returned to normal. Whether long term treatment with lomitapide will lead to an increase in liver disease is unknown. There is a single case of a patient with lipoprotein lipase deficiency who was treated for 13 years with lomitapide who developed steatohepatitis and fibrosis (248). In an observational study of a small number of patients on lomitapide for > 5 years liver failure or cirrhosis was not noted (249). In another study in Italy, 34 patients were treated with lomitapide for more than 9 years and elevations in hepatic fat were mild-to-moderate, hepatic stiffness remained normal, and the mean FIB-4 score remained below the fibrosis threshold (250). The studies suggest that in most patients’ severe liver disease will not develop. To reduce the risk of liver dysfunction it is important that patients avoid or limit alcohol intake and avoid drugs that inhibit Cyp3A4 activity.
Because of the high potential risk of serious complications the FDA has mandated several measures to ensure that patients are closely followed and monitored for liver toxicity ((Risk Evaluation and Mitigation Strategy (REMS) Program) (240,241). ALT, AST, alkaline phosphatase, and total bilirubin should be measured before initiating treatment. During the first year, liver function tests should be measured prior to each increase in dose or monthly, whichever occurs first. After the first year, liver function tests should be measured at least every 3 months and before any increase in dose.
Contraindications
Lomitapide should not be used during pregnancy and in patients with moderate or severe liver disease. In addition, it should not be used in patients on strong CYP3A4 inhibitors.
Summary
Lomitapide is approved only for the treatment of lipid disorders in patients with Homozygous Familiar Hypercholesterolemia. The frequent GI side effects and the potential risk of serious liver disease greatly limit the use of this drug and it should be reserved for the patients in which more benign therapies are not sufficient in lowering LDL-C into a reasonable range. It is used as an adjunct to other lipid lowering therapies and lipoprotein apheresis in patients with Homozygous Familiar Hypercholesterolemia.
MIPOMERSEN (KYNAMRO)
Introduction
Mipomersen (Kynamro) is a second generation apolipoprotein B antisense oligonucleotide that was approved in January 2013 for the treatment of patients with Homozygous Familiar Hypercholesterolemia (241,242,251). It is administered as a 200mg subcutaneous injection once a week (241,242,251). As will be discussed below, it lowers LDL-C levels by an LDL receptor independent mechanism. In May 2018 sales were discontinued due to safety concerns related to increased liver transaminases and fatty liver.
Effect on Mipomersen on Lipid and Lipoprotein Levels
In the pivotal trial, 51 patients with Homozygote Familial Hypercholesterolemia on treatment were randomized to additional treatment with mipomersen (n= 34) or placebo (n=17) and followed for 26 weeks (252). Mipomersen lowered LDL-C levels by 21% and apolipoprotein B levels by 24% compared to placebo. In addition, non-HDL-C was decreased by 21.6%, triglycerides by 17%, and Lp(a) by 23% while HDL and apolipoprotein A-I were increased by 11.2% and 3.9% respectively.
Mipomersen has also been studied in patients with Heterozygous Familial Hypercholesterolemia. In a double-blind, placebo-controlled, randomized trial, patients on maximally tolerated statin therapy were treated weekly with subcutaneous mipomersen 200 mg or placebo for 26 weeks (253). LDL-C levels decreased by 33% in the mipomersen group compared to placebo. Additionally, mipomersen significantly reduced apolipoprotein B by 26%, triglycerides by 14%, and Lp(a) by 21% compared to placebo with no significant changes in HDL-C levels. In an extension follow-up study the beneficial effects of mipomersen were maintained for at least 2 years (254).
In a meta-analysis of 8 randomized studies with 462 subjects with either non-specified hypercholesterolemia or Heterozygous Familial Hypercholesterolemia, Panta and colleagues reported that mipomersen decreased LDL-C levels by 32% compared to placebo (255). Additionally, non-HDL-C was decreased by 31%, apolipoprotein B by 33%, triglycerides by 36%, and Lp(a) by 26% with no effect on HDL-C levels.
Mechanism Accounting for the Mipomersen Induced Lipid Effects
Apolipoprotein B 100 is the main structural protein of VLDL and LDL and is required for the formation of VLDL and LDL (191). Familiar Hypobetalipoproteinemia is a genetic disorder due to a mutation of one apolipoprotein B allele that is characterized by very low concentrations of LDL and apolipoprotein B due to the decreased production of lipoproteins by the liver (215). Mipomersen, an apolipoprotein B antisense oligonucleotide, mimics Familiar Hypobetalipoproteinemia by inhibiting apolipoprotein B 100 production in the liver by pairing with apolipoprotein B mRNA preventing its translation (241,242,251). This decrease in apolipoprotein B synthesis results in a decrease in hepatic VLDL production leading to a decrease in LDL levels.
Pharmacokinetics and Drug Interactions
No significant drug interactions have been reported. Given the risk of liver abnormalities (see side effect section) the avoidance of alcohol or a reduction in alcohol intake would be prudent.
Effect of Mipomersen on Clinical Outcomes
There are no clinical outcome trials but it is presumed that lowering LDL-C levels in patients with Homozygous Familial Hypercholesterolemia will reduce cardiovascular events. In a study comparing cardiovascular events in patients with Homozygous Familial Hypercholesterolemia in the 24 months prior to initiating mipomersen therapy and after initiating mipomersen revealed a decrease in events (prior to treatment 61.5% of patients had an event vs. 9.6% after initiating mipomersen; P < .0001) (256). In this trial mipomersen resulted in a mean absolute reduction in LDL-C of 70 mg/dL (-28%), non-HDL cholesterol of 74 mg/dL (-26%), and Lp(a) of 11 mg/dL (-17%).
Side Effects
The most common side effect is injection site reactions, which occur in 75-98% of patients and typically consist of one or more of the following: erythema, pain, tenderness, pruritus, and local swelling (241,242,251). Additional, influenza like symptoms, which typically occur within 2 days after an injection, occur in 30-50% of patients and include one or more of the following: influenza-like illness, pyrexia, chills, myalgia, arthralgia, malaise or fatigue which result in a substantial percentage of patients discontinuing therapy (241,242,251).
A major safety concern is liver toxicity (241,242,251). By inhibiting VLDL formation and secretion the risk of fatty liver is increased. Fatty liver has been observed in 5-20% of patients treated with mipomersen (241,242,251). In 10-15% of patients treated with mipomersen increases in transaminases occur (241,242,251). Additionally, liver biopsies from 7 patients after a minimum of 6 months of mipomersen therapy have demonstrated the presence of fatty liver although there was no inflammation despite elevations in liver enzymes (257). Liver function should be measured prior to initiating therapy and monthly during the first year and every 3 months after the first year. Fortunately, when treatment is discontinued liver function tests and fatty liver return to normal.
Because of the potential for liver toxicity this drug is no longer available.
Contraindications
Mipomersen is contraindicated in patients in patients with liver disease or severe renal disease. Mipomersen is not recommended for use during pregnancy or lactation. In animal studies mipomersen has not resulted in fetal abnormalities.
Summary
Mipomersen was approved only for the treatment of lipid disorders in patients with Homozygous Familiar Hypercholesterolemia. The potential risk of serious liver disease greatly limits the use of this drug and therefore it was reserved for patients in which more benign therapies were not sufficient in lowering LDL-C into a reasonable range. It was used as an adjunct to other lipid lowering therapies in patients with Homozygous Familiar Hypercholesterolemia but because of safety concerns is no longer available.
EVINACUMAB (EVKEEZA)
Introduction
Evinacumab is a human monoclonal antibody against angiopoietin-like protein 3 (ANGPTL3). It is approved for the treatment of Homozygous Familial Hypercholesterolemia. Evinacumab decreases LDL-C levels by mechanisms independent of LDL receptor activity. The recommended dose of evinacumab is 15 mg/kg administered by intravenous infusion over 60 minutes every 4 weeks.
Effect on Evinacumab on Lipid and Lipoprotein Levels
HOMOZYGOUS FAMILIAL HYPERCHOLESTEROLEMIA
A double-blind, placebo-controlled trial randomly treated patients with Homozygous Familial Hypercholesterolemia with an intravenous infusion of evinacumab 15 mg/Kg every 4 weeks (n= 43) or placebo (n= 22) (258). The individuals in this trial were on lipid lowering therapy (94% were on a statin with 77% on a high-intensity statin, 77% on a PCSK9 inhibitor, 75% on ezetimibe, 25% on lomitapide, and 34% undergoing apheresis) and the mean baseline LDL-C level was approximately 250-260mg/dL. After 24 weeks of treatment patients in the evinacumab group had a 47% reduction in LDL-C levels vs. a 1.9% increase in the placebo group (table 17). This decrease in LDL-C levels was observed after 2 weeks of therapy and was observed regardless of concomitant use of other lipid lowering drugs or apheresis. Notably, in individuals with null-null LDL receptor variants evinacumab resulted in a 43% decrease in LDL-C levels indicating that evinacumab therapy was effective in the absence of functional LDL receptors. As expected, total cholesterol, non-HDL-C cholesterol, and apo B levels were also decreased. Moreover, triglyceride levels decreased 55% and HDL-C levels decreased 30% with evinacumab administration while Lp(a) levels were unchanged.
|
Table 17. Effect of Evinacumab on Lipid Levels in Homozygous Familial Hypercholesterolemia
|
|
|
LDL-C
|
Apo B
|
Non-HDL-C
|
TG
|
HDL-C
|
|
Baseline mg/dL
|
255
|
171
|
278
|
124
|
44
|
|
Evinacumab % Change
|
−47%
|
−41%
|
−50%
|
−55%
|
−30%
|
|
Placebo % Change
|
+2%
|
−5%
|
+2%
|
−5%
|
+1%
|
REFRACTORY HYPERCHOLESTEROLEMIA
In a double-blind, placebo-controlled trial, patients with refractory hypercholesterolemia with a screening LDL-C level > 70 mg/dL with atherosclerosis or LDL-C > 100 mg/dL without atherosclerosis were randomized to receive subcutaneous or intravenous evinacumab or placebo (259). The hypercholesterolemia was refractory to treatment with a PCSK9 inhibitor and a statin at a maximum tolerated dose, with or without ezetimibe. In this trial a number of different treatment regimens of evinacumab were employed (intravenous or subcutaneous; different doses) and in this summary only the results of intravenous evinacumab 15 mg/kg every 4 weeks (39 patients) vs. placebo (34 patients) will be presented. Baseline LDL-C levels were approximately 145mg/dL. After 16 weeks of treatment the LDL-C level was decreased by 50% with evinacumab administration vs. a 0.6% decrease with placebo. An extension of this trial for 72 weeks found that the reduction in LDL-C were sustained (260). The decrease in LDL-C was observed after 2 weeks of treatment. As expected, total cholesterol, non-HDL-C, and apo B levels also decreased in the evinacumab group. Evinacumab administration decreased triglyceride levels by 53% and HDL-C levels by 31%. In contrast to the results in the homozygous familiar hypercholesterolemia study described above in this study evinacumab decreased Lp(a) levels by 16%. The effect of the subcutaneous administration of evinacumab on lipid levels was similar to that observed with intravenous administration.
The effect of evinacumab on triglyceride levels in patients with marked hypertriglyceridemia is described in the Endotext chapter “Triglyceride Lowering Drugs” (261).
Mechanism Accounting for the Evinacumab Induced Lipid Effects
ANGPTL3 inhibits lipoprotein lipase (LPL) activity thereby slowing the clearance of VLDL and chylomicrons resulting in an increase in plasma triglyceride levels (262,263). Mice deficient in ANGPTL3 have lower plasma triglyceride levels while mice overexpressing ANGPTL3 have elevated plasma triglyceride levels (263). Evinacumab by inhibiting the ability of ANGPTL3 to inhibit LPL activity will accelerate the clearance of TG rich lipoproteins decreasing plasma triglyceride levels (263). Furthermore, ANGPTL3 has also been shown to reduce endothelial lipase activity (263). Endothelial lipase is a phospholipase that catabolizes phospholipids on HDL and accelerates HDL clearance (264,265). Evinacumab by inhibiting the ability of ANGPTL3 to inhibit endothelial lipase activity will lead to a decrease in HDL levels (266).
The mechanism(s) that explain the decrease in LDL-C levels with evinacumab administration is not completely understood. A study has demonstrated that the increase in endothelial lipase activity induced by evinacumab leads to VLDL remodeling and lipid depletion that increases VLDL clearance when the LDL receptor is absent (267). This decrease in VLDL, the precursor of LDL, limits LDL particle production resulting in a reduction in plasma LDL-C levels (267). Kinetic studies in four patients with homozygous familial hypercholesterolemia observed that evinacumab markedly increased the fractional catabolic rate of IDL (intermediate-density lipoprotein) and LDL apoB (268). Whether decreases in VLDL production also plays a role in the decrease in LDL-C levels with evinacumab treatment requires additional studies. It should be noted that inhibition of ANGPTL3 decreases LDL-C levels independent of LDL receptor activity (269).
Pharmacokinetics and Drug Interactions
There are no significant drug interactions.
Effect of Evinacumab on Clinical Outcomes
There are no cardiovascular outcome studies. In two patients with homozygous Familial Hypercholesterolemia evinacumab therapy markedly reduced LDL-C levels with a concomitant decrease in plaque volume determined by coronary computed tomography angiography (268).
Homozygosity for loss-of-function mutations in ANGPTL3 is associated with significantly lower plasma levels of LDL-C, HDL-C, and triglycerides (familial combined hypolipidemia) (215,263,270). Heterozygous carriers of loss-of-function mutations in ANGPTL3, which occur at a frequency of about 1:300, have significantly lower total cholesterol, LDL-C, and triglyceride levels than noncarriers (263). Moreover, patients carrying loss-of-function variants in ANGPTL3 have a significantly lower risk of coronary artery disease (271,272). Additionally, in an animal model of atherosclerosis treatment with evinacumab decreased atherosclerotic lesion area and necrotic content (271). Taken together these observations suggest that inhibiting ANGPTL3 with evinacumab will reduce cardiovascular disease.
Side Effects
Serious hypersensitivity reactions have occurred with evinacumab. In clinical trials, 1 (1%) of evinacumab treated patients experienced anaphylaxis vs. 0% of patients who received placebo (package insert).
Contraindications
Based on animal studies, evinacumab may cause fetal harm when administered to pregnant patients (package insert). Patients should be advised of the potential risks to the fetus of pregnancy. Patients who may become pregnant should be advised to use effective contraception during treatment with evinacumab and for at least 5 months following the last dose.
Summary
In patients with Homozygous Familiar Hypercholesterolemia the ability of evinacumab to lower LDL-C levels independent of LDL receptor activity makes this agent very useful in these patients. Most patients with Homozygous Familial Hypercholesterolemia do not achieve goal LDL-C levels with triple drug therapy with maximally tolerated statin therapy, ezetimibe, and a PCSK9 inhibitor and therefore the addition of evinacumab will be needed in many of these patients. Evinacumab is also effective in patients with refractory hypercholesterolemia but the drug is not yet FDA approved in this situation. Nevertheless, one can foresee in patients with refractory hypercholesterolemia at high risk for cardiovascular events the use of evinacumab. In addition to lowering LDL-C levels evinacumab also lowers triglyceride levels and could be useful in selected patients with very severe hypertriglyceridemia (261,273).
APPROACH TO TREATING PATIENTS WITH HYPERCHOLESTEROLEMIA
Introduction
The issues of deciding who to treat, how aggressive to treat, and the goals of therapy are discussed in detail in the chapter “Guidelines for the Management of High Blood Cholesterol” and therefore will not be addressed in this chapter (3). Additionally, the role of life style changes to lower LDL-C is discussed in great depth in chapter “The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels” and therefore will also not be addressed here (1). Rather we will focus on how to use the drugs discussed in this chapter to treat various categories of patients. The factors to consider when deciding which drugs are appropriate to use for lowering plasma LDL-C levels are; the efficacy in lowering LDL-C levels, the effect on other lipid and lipoprotein levels, the ability to reduce cardiovascular events, the side effects of drug therapy, the ease of complying with the drug regimen, and the cost of the drugs. Many statins and ezetimibe are generic drugs and therefore they are relatively inexpensive.
Isolated Hypercholesterolemia with Cardiovascular Disease
In patients with isolated hypercholesterolemia and cardiovascular disease, initial drug therapy should be high intensity statin therapy (atorvastatin 40-80mg or rosuvastatin 20-40mg). In patients with cardiovascular disease, one should aim to lower the LDL-C to below 70mg/dL. Many experts, based on studies comparing statin alone vs. statin + ezetimibe or statin + a PCSK9 inhibitor, would recommend a more aggressive LDL-C goal in high-risk patients (LDL-C <55mg/dL). If statin therapy alone is not sufficient adding ezetimibe, is a reasonable next step. Because a considerable amount of data indicates that the lower the LDL-C the greater the reduction in cardiovascular events many experts would use a combination of high intensity statin therapy plus ezetimibe in all high-risk patients to maximize LDL-C reduction. Ezetimibe is inexpensive, easy to take, has few side effects, will modestly lower LDL-C, and has been shown in combination with statins to further reduce cardiovascular events. High dose statin and ezetimibe will lower LDL-C by as much as 70%, which will lower LDL-C to goal in a large number of patients who do not have a genetic basis for their elevated LDL-C levels. If the combination of statin plus ezetimibe does not lower the LDL to goal one can add a third drug. If the LDL is close to goal, one could add a bile acid sequestrant such as colesevelam or bempedoic acid. If the LDL is not very close to goal one could instead use a statin +/- ezetimibe plus a PCSK9 inhibitor, which will result in marked reductions in LDL-C levels. If the patient has diabetes with a moderately elevated A1c level using a bile acid sequestrant such as colesevelam instead of ezetimibe or in combination with ezetimibe could improve both glycemic control and further lower LDL levels. If the cost of PCSK9 inhibitors decrease the earlier use of these drugs will become feasible.
Isolated Hypercholesterolemia in Primary Prevention
In patients with isolated hypercholesterolemia (LDL-C < 190mg/dL) without cardiovascular disease initial drug therapy is with a statin. The statin dose should be chosen based on the percent reduction in LDL-C required to lower the LDL-C level to below the target goal (typically < 100mg/dL but if multiple risk factors with a high risk for cardiovascular events is present many experts would aim for <70mg/dL). As discussed earlier, the side effects of statin therapy increase with higher doses so one should not automatically start with high doses, but instead should choose a dose balancing the benefits and risks. Generic statins are inexpensive drugs and are very effective in both lowering LDL-C levels and reducing cardiovascular events. Additionally, they have an excellent safety profile. If the initial statin dose does not lower LCL-C sufficiently, one can then increase the dose or add ezetimibe. If the maximal statin dose does not lower LDL-C sufficiently adding ezetimibe is a reasonable next step if the LDL-C level is in a reasonable range and an additional 20-25% reduction in LDL will be sufficient. High dose statin and ezetimibe will lower LDL-C by as much as 70%, which will lower LDL-C to goal in the majority of patients who do not have a genetic basis for their elevated LDL-C levels. If the combination of statin plus ezetimibe does not lower the LDL-C to goal one can add a third drug, such as bempedoic acid or colesevelam. If the patient has diabetes with a moderately elevated A1c level using colesevelam instead of ezetimibe or in combination with ezetimibe could improve both glycemic control and further lower LDL-C levels.
Mixed Hyperlipidemia
In patients with mixed hyperlipidemia (elevated LDL-C and triglyceride levels) Initial drug therapy should also be a generic statin unless triglyceride levels are greater than 500-1000mg/dL. If triglycerides are > 500-1000mg/dL initial therapy is directed at lowering triglyceride levels (261). In addition to lowering LDL-C levels, statins are also effective in lowering triglyceride levels particularly when the triglycerides are elevated. If LDL-C is not lowered sufficiently ezetimibe is a reasonable next step. Bile acid sequestrants are not appropriate drugs in patients with hypertriglyceridemia. The approach to the patient whose LDL-C levels are at goal but the triglycerides and non-HDL-C are still elevated is discussed in the chapter on triglyceride lowering drugs (261).
Heterozygous Familial Hypercholesterolemia
In patients with Heterozygous Familial Hypercholesterolemia or other disorders with very elevated LDL-C levels (>190mg/dL), high doses of a potent statin such as atorvastatin 40-80mg or rosuvastatin 20-40mg are the first step to lower LDL-C levels. In many patients this will not be sufficient. If the LDL-C levels are above goal then adding ezetimibe is a reasonable next step. If after ezetimibe the LDL-C is still slightly above goal triple drug therapy with bempedoic acid or a bile acid sequestrant can be employed. If on statin alone or with the combination of statin and ezetimibe the LDL-C still needs to be markedly reduced a PCSK9 inhibitor may be a better choice as these drugs can markedly lower LDL-C levels.
Homozygous Familiar Hypercholesterolemia
In patients with Homozygous Familiar Hypercholesterolemia initial therapy with a maximally tolerated statin and ezetimibe can be instituted. This will likely not result in an acceptable LDL-C level and then one can add a PCSK9 inhibitor. Because these therapies depend on LDL receptor activity to lower LDL-C a high percentage of patients will not reach goal and then one can add lomitapide and/or evinacumab, drugs that lower LDL-C levels independent of LDL receptor activity. Because side effects are fewer with evinacumab this is the preferred initial drug in most patients. Studies have shown that with the addition of evinacumab many patients will reach acceptable LDL-C levels. If LDL-C levels are still not acceptable one could then initiate lipoprotein apheresis (274).
Statin Intolerance
Statin intolerance is frequently due to myalgias but on occasion can be due other issues, such as increased liver or muscle enzymes, cognitive dysfunction, or other neurological disorders. The percentage of patients who are “statin intolerant” varies greatly but in clinical practice a significant number of patients have difficulty taking statins.
As discussed earlier it can be difficult to determine if the muscle symptoms that occur when a patient is taking a statin are actually due to the statin or are unrelated to statin use. The first step in a “statin intolerant patient” is to take a careful history of the nature and location of the muscle symptoms and the timing of onset in relation to statin use to determine whether the presentation fits the typical picture for statin induced myalgias. The characteristic findings with a statin induced myalgia are shown in table 18 and findings that are not typical for statin induced myalgia are shown in table 19. The disappearance of symptoms within a few weeks of stopping statins and the reappearance after restarting statins is very suggestive of the symptoms being due to true statin intolerance. An on-line tool (htpp://tools.acc.org/statinintolerance/#!/) and an app produced by the ACC/AHA are available. This tool characterizes patients based on 8 criteria into possible vs. unlikely to have statin induced muscle symptoms (table 20)
|
Table 18. Characteristic Findings with Statin Induced Myalgia
|
|
Symmetric
|
|
Proximal muscles
|
|
Muscle pain, tenderness, weakness, cramps
|
|
Symptom onset < 4 weeks after starting statin or dose increase
|
|
Improves within 2-4 weeks of stopping statin
|
|
Cramping is unilateral and involves small muscles of hands and feet
|
|
Same symptoms occur with re-challenge within 4 weeks
|
|
Table 19. Symptoms Atypical in Statin Induced Myalgia
|
|
Unilateral
|
|
Asymmetric
|
|
Small muscles
|
|
Joint or tendon pain
|
|
Shooting pain, muscle twitching or tingling
|
|
Symptom onset > 12 weeks
|
|
No improvement after discontinuing statin
|
|
Table 20. Diagnosis of Statin Associated Muscle Symptoms
|
|
Symptom timing
|
|
Symptom type
|
|
Symptom location
|
|
Sex
|
|
Age
|
|
Race/ethnicity
|
|
CK elevation > 5 times the upper limit of normal
|
|
Known risk factors for statin induced muscle symptoms and non-statin causes of muscle symptoms
|
One should also check a CK level but this is almost always in the normal range. If the CK is not elevated and the symptoms do not suggest a statin induced myalgia one can often reassure the patient and continue statin therapy. This is often successful and studies have shown that many patients that stop taking statins due to “statin induced myalgia” can be successfully treated with a statin. If the CK is elevated it should be repeated after instructing the patient to avoid exercise for 48 hours. Also, the CK levels should be compared to CK levels prior to starting therapy. If the CK remains elevated (3x upper limit of normal) the statin should be discontinued. Similarly, if the CK is normal but the symptoms are suggestive of a statin induced myalgia the statin should also be discontinued. The next step is to determine if one can identify reversible factors that could be increasing statin toxicity (hypothyroidism, drug interactions). If none are identified the next step after the myalgias have resolved is to try a low dose of a different statin that is metabolized by a different pathway (for example instead of atorvastatin, which is metabolized by the CYP3A4 pathway, rosuvastatin, which has a different pathway of metabolism). Because statin side effects are dose related, a low dose of a statin may often be tolerated. One can also try several different statins as sometimes a patient may tolerate one statin and not others. A meta-analysis has shown that every other day administration of statins is as effective as daily administration in lowering lipid levels and therefore is a very reasonable strategy (275). In some instances, using a long-acting statin (rosuvastatin or atorvastatin) 1-3 times per week can work (we usually start with once per week and then slowly increase frequency as tolerated) (276). In these circumstances (low doses or 1-3 times per week) the reduction in LDL-C may not be sufficient but one can use combination therapy with other drugs such as ezetimibe, bempedoic acid, bile acid sequestrants, or PCSK9 inhibitors to achieve LDL target goals.
Many providers have combined Coenzyme Q10 with statins to prevent statin induced myalgias. However, randomized trials with Coenzyme Q10 supplementation have not consistently shown benefit (277-282). A trial, which carefully screened patients to make sure they actually had statin induced myalgias, failed to show a benefit from Coenzyme Q10 supplementation (101). It has also been recommended that vitamin D supplementation be used to prevent statin induced myalgias but a large randomized trial failed to show a reduction in muscle symptoms with vitamin D therapy (283).
If after trying various approaches a patient still has myalgias and is unable to tolerate statin therapy one needs to utilize other approaches to lower LDL levels. Similarly, if there are other reasons why a patient cannot take a statin, such as developing muscle pathology, one will also need to utilize other approaches to lower LDL levels. These patients can be treated with ezetimibe, bempedoic acid, bile acid sequestrants, or PCSK 9 inhibitors either as monotherapy or in combination to achieve LDL goals.
There are patients who will refuse statins and other drug therapy because they do not believe in taking pharmaceuticals but will take natural products. In these patients we have employed red yeast rice, which decreases LDL-C because it contains a form of lovastatin (284,285). It is effective but one should recognize that the quality control is not similar to the standards of pharmaceutical products and that there can be batch to batch variations. Furthermore, there is a risk of drug-drug interactions if used with inhibitors of CYP3A4. However, in this particular patient population, who refuses to take statins or other drugs, this can be a reasonable alternative. If a patient just refuses statins (usually based on a belief that statins are toxic) we will employ other cholesterol lowering drugs.
CONCLUSIONS
With currently available drugs to lower LDL-C levels we are now able to markedly reduce LDL-C levels and achieve our LDL-C goals in the vast majority of patients and thereby reduce the risk of cardiovascular disease. Patients with Homozygous Familial Hypercholesterolemia and some patients with Heterozygous Familial Hypercholesterolemia still present major clinical challenges and it can be very difficult in these patients to achieve LDL-C goals.
ACKNOWLEDGEMENTS
This work was supported by grants from the Northern California Institute for Research and Education.
REFERENCES
- Feingold KR. The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- Feingold KR. Approach to the Patient with Dyslipidemia. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Grundy SM, Feingold KR. Guidelines for the Management of High Blood Cholesterol. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2022.
- Endo A. A gift from nature: the birth of the statins. Nat Med 2008; 14:1050-1052
- Alberts AW. Discovery, biochemistry and biology of lovastatin. Am J Cardiol 1988; 62:10J-15J
- Ballantyne CM, Andrews TC, Hsia JA, Kramer JH, Shear C, Efficacy ASGACC, Safety S. Correlation of non-high-density lipoprotein cholesterol with apolipoprotein B: effect of 5 hydroxymethylglutaryl coenzyme A reductase inhibitors on non-high-density lipoprotein cholesterol levels. Am J Cardiol 2001; 88:265-269
- Jones PH, Hunninghake DB, Ferdinand KC, Stein EA, Gold A, Caplan RJ, Blasetto JW. Effects of rosuvastatin versus atorvastatin, simvastatin, and pravastatin on non-high-density lipoprotein cholesterol, apolipoproteins, and lipid ratios in patients with hypercholesterolemia: additional results from the STELLAR trial. Clin Ther 2004; 26:1388-1399
- Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E, Cain VA, Blasetto JW. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol 2003; 92:152-160
- Stein EA, Lane M, Laskarzewski P. Comparison of statins in hypertriglyceridemia. Am J Cardiol 1998; 81:66B-69B
- Bos S, Yayha R, van Lennep JE. Latest developments in the treatment of lipoprotein (a). Curr Opin Lipidol 2014; 25:452-460
- van Capelleveen JC, van der Valk FM, Stroes ES. Current therapies for lowering lipoprotein (a). J Lipid Res 2016; 57:1612-1618
- Adams SP, Tsang M, Wright JM. Lipid-lowering efficacy of atorvastatin. Cochrane Database Syst Rev 2015; 3:CD008226
- Adams SP, Sekhon SS, Wright JM. Lipid-lowering efficacy of rosuvastatin. Cochrane Database Syst Rev 2014; 11:CD010254
- Liao JK. Clinical implications for statin pleiotropy. Curr Opin Lipidol 2005; 16:624-629
- Joshi PH, Jacobson TA. Therapeutic options to further lower C-reactive protein for patients on statin treatment. Curr Atheroscler Rep 2010; 12:34-42
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell 2015; 161:161-172
- Huff MW, Burnett JR. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and hepatic apolipoprotein B secretion. Curr Opin Lipidol 1997; 8:138-145
- Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J 2020; 41:2275-2284
- Causevic-Ramosevac A, Semiz S. Drug interactions with statins. Acta Pharm 2013; 63:277-293
- Hu M, Tomlinson B. Evaluation of the pharmacokinetics and drug interactions of the two recently developed statins, rosuvastatin and pitavastatin. Expert Opin Drug Metab Toxicol 2014; 10:51-65
- Sirtori CR. The pharmacology of statins. Pharmacol Res 2014; 88:3-11
- Bellosta S, Corsini A. Statin drug interactions and related adverse reactions: an update. Expert Opin Drug Saf 2018; 17:25-37
- Awad K, Serban MC, Penson P, Mikhailidis DP, Toth PP, Jones SR, Rizzo M, Howard G, Lip GYH, Banach M. Effects of morning vs evening statin administration on lipid profile: A systematic review and meta-analysis. J Clin Lipidol 2017; 11:972-985 e979
- Kellick KA, Bottorff M, Toth PP, The National Lipid Association's Safety Task Force. A clinician's guide to statin drug-drug interactions. J Clin Lipidol 2014; 8:S30-46
- Lee JW, Morris JK, Wald NJ. Grapefruit Juice and Statins. Am J Med 2016; 129:26-29
- Holdaas H, Fellstrom B, Jardine AG, Holme I, Nyberg G, Fauchald P, Gronhagen-Riska C, Madsen S, Neumayer HH, Cole E, Maes B, Ambuhl P, Olsson AG, Hartmann A, Solbu DO, Pedersen TR. Effect of fluvastatin on cardiac outcomes in renal transplant recipients: a multicentre, randomised, placebo-controlled trial. Lancet 2003; 361:2024-2031
- ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr., Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563-1574
- Busti AJ, Bain AM, Hall RG, 2nd, Bedimo RG, Leff RD, Meek C, Mehvar R. Effects of atazanavir/ritonavir or fosamprenavir/ritonavir on the pharmacokinetics of rosuvastatin. J Cardiovasc Pharmacol 2008; 51:605-610
- Gervasoni C, Riva A, Rizzardini G, Clementi E, Galli M, Cattaneo D. Potential association between rosuvastatin use and high atazanavir trough concentrations in ritonavir-treated HIV-infected patients. Antivir Ther 2015; 20:449-451
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/Drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570-578
- Pham PA, la Porte CJ, Lee LS, van Heeswijk R, Sabo JP, Elgadi MM, Piliero PJ, Barditch-Crovo P, Fuchs E, Flexner C, Cameron DW. Differential effects of tipranavir plus ritonavir on atorvastatin or rosuvastatin pharmacokinetics in healthy volunteers. Antimicrob Agents Chemother 2009; 53:4385-4392
- van der Lee M, Sankatsing R, Schippers E, Vogel M, Fatkenheuer G, van der Ven A, Kroon F, Rockstroh J, Wyen C, Baumer A, de Groot E, Koopmans P, Stroes E, Reiss P, Burger D. Pharmacokinetics and pharmacodynamics of combined use of lopinavir/ritonavir and rosuvastatin in HIV-infected patients. Antivir Ther 2007; 12:1127-1132
- Sarkar S, Brown TT. Lipid Disorders in People with HIV. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, Cholesterol Treatment Trialists Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267-1278
- Cholesterol Treatment Trialists Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670-1681
- Cholesterol Treatment Trialists Collaboration, Fulcher J, O'Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, Simes J, Collins R, Kirby A, Colhoun H, Braunwald E, La Rosa J, Pedersen TR, Tonkin A, Davis B, Sleight P, Franzosi MG, Baigent C, Keech A. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015; 385:1397-1405
- Cholesterol Treatment Trialists Collaboration, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581-590
- Taylor F, Huffman MD, Macedo AF, Moore TH, Burke M, Davey Smith G, Ward K, Ebrahim S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev 2013; 1:CD004816
- Yusuf S, Bosch J, Dagenais G, Zhu J, Xavier D, Liu L, Pais P, Lopez-Jaramillo P, Leiter LA, Dans A, Avezum A, Piegas LS, Parkhomenko A, Keltai K, Keltai M, Sliwa K, Peters RJ, Held C, Chazova I, Yusoff K, Lewis BS, Jansky P, Khunti K, Toff WD, Reid CM, Varigos J, Sanchez-Vallejo G, McKelvie R, Pogue J, Jung H, Gao P, Diaz R, Lonn E. Cholesterol Lowering in Intermediate-Risk Persons without Cardiovascular Disease. N Engl J Med 2016;
- Ference BA. Mendelian randomization studies: using naturally randomized genetic data to fill evidence gaps. Curr Opin Lipidol 2015; 26:566-571
- Brown MS, Goldstein JL. Biomedicine. Lowering LDL--not only how low, but how long? Science 2006; 311:1721-1723
- Feingold KR. Maximizing the benefits of cholesterol-lowering drugs. Curr Opin Lipidol 2019; 30:388-394
- Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, Macfarlane PW, Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ, Twomey C, Westendorp RG. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623-1630
- Cholesterol Treatment Trialists Collaboration. Efficacy and safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 2019; 393:407-415
- Joseph J, Pajewski NM, Dolor RJ, Sellers MA, Perdue LH, Peeples SR, Henrie AM, Woolard N, Jones WS, Benziger CP, Orkaby AR, Mixon AS, VanWormer JJ, Shapiro MD, Kistler CE, Polonsky TS, Chatterjee R, Chamberlain AM, Forman DE, Knowlton KU, Gill TM, Newby LK, Hammill BG, Cicek MS, Williams NA, Decker JE, Ou J, Rubinstein J, Choudhary G, Gazmuri RJ, Schmader KE, Roumie CL, Vaughan CP, Effron MB, Cooper-DeHoff RM, Supiano MA, Shah RC, Whittle JC, Hernandez AF, Ambrosius WT, Williamson JD, Alexander KP. Pragmatic evaluation of events and benefits of lipid lowering in older adults (PREVENTABLE): Trial design and rationale. J Am Geriatr Soc 2023; 71:1701-1713
- Zoungas S, Curtis A, Spark S, Wolfe R, McNeil JJ, Beilin L, Chong TT, Cloud G, Hopper I, Kost A, Nelson M, Nicholls SJ, Reid CM, Ryan J, Tonkin A, Ward SA, Wierzbicki A. Statins for extension of disability-free survival and primary prevention of cardiovascular events among older people: protocol for a randomised controlled trial in primary care (STAREE trial). BMJ Open 2023; 13:e069915
- Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol 2007; 99:410-414
- Gupta M, Braga MF, Teoh H, Tsigoulis M, Verma S. Statin effects on LDL and HDL cholesterol in South Asian and white populations. J Clin Pharmacol 2009; 49:831-837
- Sakamoto T, Kojima S, Ogawa H, Shimomura H, Kimura K, Ogata Y, Sakaino N, Kitagawa A. Effects of early statin treatment on symptomatic heart failure and ischemic events after acute myocardial infarction in Japanese. Am J Cardiol 2006; 97:1165-1171
- Pais P, Jung H, Dans A, Zhu J, Liu L, Kamath D, Bosch J, Lonn E, Yusuf S. Impact of blood pressure lowering, cholesterol lowering and their combination in Asians and non-Asians in those without cardiovascular disease: an analysis of the HOPE 3 study. Eur J Prev Cardiol 2019; 26:681-697
- Feingold KR. Dyslipidemia in Patients with Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Cholesterol Treatment Trialists Collaboration, Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Peto R, Armitage J, Baigent C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117-125
- Cholesterol Treatment Trialists Collaboration , Herrington WG, Emberson J, Mihaylova B, Blackwell L, Reith C, Solbu MD, Mark PB, Fellstrom B, Jardine AG, Wanner C, Holdaas H, Fulcher J, Haynes R, Landray MJ, Keech A, Simes J, Collins R, Baigent C. Impact of renal function on the effects of LDL cholesterol lowering with statin-based regimens: a meta-analysis of individual participant data from 28 randomised trials. Lancet Diabetes Endocrinol 2016; 4:829-839
- Su X, Zhang L, Lv J, Wang J, Hou W, Xie X, Zhang H. Effect of Statins on Kidney Disease Outcomes: A Systematic Review and Meta-analysis. Am J Kidney Dis 2016;
- Wanner C, Krane V, Marz W, Olschewski M, Mann JF, Ruf G, Ritz E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238-248
- Fellstrom BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, Chae DW, Chevaile A, Cobbe SM, Gronhagen-Riska C, De Lima JJ, Lins R, Mayer G, McMahon AW, Parving HH, Remuzzi G, Samuelsson O, Sonkodi S, Sci D, Suleymanlar G, Tsakiris D, Tesar V, Todorov V, Wiecek A, Wuthrich RP, Gottlow M, Johnsson E, Zannad F. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med 2009; 360:1395-1407
- Rosenstein K, Tannock LR. Dyslipidemia in Chronic Kidney Disease. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2022.
- Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH, Dunselman P, Fonseca C, Goudev A, Grande P, Gullestad L, Hjalmarson A, Hradec J, Janosi A, Kamensky G, Komajda M, Korewicki J, Kuusi T, Mach F, Mareev V, McMurray JJ, Ranjith N, Schaufelberger M, Vanhaecke J, van Veldhuisen DJ, Waagstein F, Wedel H, Wikstrand J. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248-2261
- Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, Lucci D, Nicolosi GL, Porcu M, Tognoni G. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231-1239
- Arvind A, Osganian SA, Cohen DE, Corey KE. Lipid and Lipoprotein Metabolism in Liver Disease. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA) 2019.
- Bays H, Cohen DE, Chalasani N, Harrison SA, The National Lipid Association's Statin Safety Task Force. An assessment by the Statin Liver Safety Task Force: 2014 update. J Clin Lipidol 2014; 8:S47-57
- Herrick C, Bahrainy S, Gill EA. Statins and the Liver. Endocrinol Metab Clin North Am 2016; 45:117-128
- Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, Pagourelias ED, Theocharidou E, Karagiannis A, Mikhailidis DP. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet 2010; 376:1916-1922
- Cohen DE, Corey KE. Lipid and Lipoprotein Metabolism in Liver Disease. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, eds. Endotext. South Dartmouth (MA) 2019.
- Grinspoon SK, Fitch KV, Zanni MV, Fichtenbaum CJ, Umbleja T, Aberg JA, Overton ET, Malvestutto CD, Bloomfield GS, Currier JS, Martinez E, Roa JC, Diggs MR, Fulda ES, Paradis K, Wiviott SD, Foldyna B, Looby SE, Desvigne-Nickens P, Alston-Smith B, Leon-Cruz J, McCallum S, Hoffmann U, Lu MT, Ribaudo HJ, Douglas PS. Pitavastatin to Prevent Cardiovascular Disease in HIV Infection. N Engl J Med 2023; 389:687-699
- He Y, Li X, Gasevic D, Brunt E, McLachlan F, Millenson M, Timofeeva M, Ioannidis JPA, Campbell H, Theodoratou E. Statins and Multiple Noncardiovascular Outcomes: Umbrella Review of Meta-analyses of Observational Studies and Randomized Controlled Trials. Ann Intern Med 2018; 169:543-553
- Newman CB, Preiss D, Tobert JA, Jacobson TA, Page RL, 2nd, Goldstein LB, Chin C, Tannock LR, Miller M, Raghuveer G, Duell PB, Brinton EA, Pollak A, Braun LT, Welty FK. Statin Safety and Associated Adverse Events: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol 2019; 39:e38-e81
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735-742
- Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N, Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 2011; 305:2556-2564
- Feingold KR. Dyslipidemia in Diabetes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA) 2020.
- Erqou S, Lee CC, Adler AI. Statins and glycaemic control in individuals with diabetes: a systematic review and meta-analysis. Diabetologia 2014; 57:2444-2452
- Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685-696
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005-2016
- Sattar N. Statins and diabetes: What are the connections? Best Pract Res Clin Endocrinol Metab 2023; 37:101749
- Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, Sofat R, Stender S, Johnson PC, Scott RA, Leusink M, Verweij N, Sharp SJ, Guo Y, Giambartolomei C, Chung C, Peasey A, Amuzu A, Li K, Palmen J, Howard P, Cooper JA, Drenos F, Li YR, Lowe G, Gallacher J, Stewart MC, Tzoulaki I, Buxbaum SG, van der AD, Forouhi NG, Onland-Moret NC, van der Schouw YT, Schnabel RB, Hubacek JA, Kubinova R, Baceviciene M, Tamosiunas A, Pajak A, Topor-Madry R, Stepaniak U, Malyutina S, Baldassarre D, Sennblad B, Tremoli E, de Faire U, Veglia F, Ford I, Jukema JW, Westendorp RG, de Borst GJ, de Jong PA, Algra A, Spiering W, Maitland-van der Zee AH, Klungel OH, de Boer A, Doevendans PA, Eaton CB, Robinson JG, Duggan D, Consortium D, Consortium M, InterAct C, Kjekshus J, Downs JR, Gotto AM, Keech AC, Marchioli R, Tognoni G, Sever PS, Poulter NR, Waters DD, Pedersen TR, Amarenco P, Nakamura H, McMurray JJ, Lewsey JD, Chasman DI, Ridker PM, Maggioni AP, Tavazzi L, Ray KK, Seshasai SR, Manson JE, Price JF, Whincup PH, Morris RW, Lawlor DA, Smith GD, Ben-Shlomo Y, Schreiner PJ, Fornage M, Siscovick DS, Cushman M, Kumari M, Wareham NJ, Verschuren WM, Redline S, Patel SR, Whittaker JC, Hamsten A, Delaney JA, Dale C, Gaunt TR, Wong A, Kuh D, Hardy R, Kathiresan S, Castillo BA, van der Harst P, Brunner EJ, Tybjaerg-Hansen A, Marmot MG, Krauss RM, Tsai M, Coresh J, Hoogeveen RC, Psaty BM, Lange LA, Hakonarson H, Dudbridge F, Humphries SE, Talmud PJ, Kivimaki M, Timpson NJ, Langenberg C, Asselbergs FW, Voevoda M, Bobak M, Pikhart H, Wilson JG, Reiner AP, Keating BJ, Hingorani AD, Sattar N. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet 2015; 385:351-361
- Sugiyama T, Tsugawa Y, Tseng CH, Kobayashi Y, Shapiro MF. Different time trends of caloric and fat intake between statin users and nonusers among US adults: gluttony in the time of statins? JAMA Intern Med 2014; 174:1038-1045
- Higuchi S, Izquierdo MC, Haeusler RA. Unexplained reciprocal regulation of diabetes and lipoproteins. Curr Opin Lipidol 2018; 29:186-193
- Rojas-Fernandez C, Hudani Z, Bittner V. Statins and cognitive side effects: what cardiologists need to know. Cardiol Clin 2015; 33:245-256
- Rojas-Fernandez CH, Goldstein LB, Levey AI, Taylor BA, Bittner V, The National Lipid Association's Safety Task Force. An assessment by the Statin Cognitive Safety Task Force: 2014 update. J Clin Lipidol 2014; 8:S5-16
- Richardson K, Schoen M, French B, Umscheid CA, Mitchell MD, Arnold SE, Heidenreich PA, Rader DJ, deGoma EM. Statins and cognitive function: a systematic review. Ann Intern Med 2013; 159:688-697
- Trompet S, van Vliet P, de Craen AJ, Jolles J, Buckley BM, Murphy MB, Ford I, Macfarlane PW, Sattar N, Packard CJ, Stott DJ, Shepherd J, Bollen EL, Blauw GJ, Jukema JW, Westendorp RG. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol 2010; 257:85-90
- Collins R, Armitage J, Parish S, Sleight P, Peto R. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20536 people with cerebrovascular disease or other high-risk conditions. Lancet 2004; 363:757-767
- McGuinness B, Craig D, Bullock R, Malouf R, Passmore P. Statins for the treatment of dementia. Cochrane Database Syst Rev 2014; 7:CD007514
- Kashani A, Phillips CO, Foody JM, Wang Y, Mangalmurti S, Ko DT, Krumholz HM. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation 2006; 114:2788-2797
- de Denus S, Spinler SA, Miller K, Peterson AM. Statins and liver toxicity: a meta-analysis. Pharmacotherapy 2004; 24:584-591
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C-60C
- Alsheikh-Ali AA, Maddukuri PV, Han H, Karas RH. Effect of the magnitude of lipid lowering on risk of elevated liver enzymes, rhabdomyolysis, and cancer: insights from large randomized statin trials. J Am Coll Cardiol 2007; 50:409-418
- Russo MW, Scobey M, Bonkovsky HL. Drug-induced liver injury associated with statins. Semin Liver Dis 2009; 29:412-422
- Tolman KG. Defining patient risks from expanded preventive therapies. Am J Cardiol 2000; 85:15E-19E
- Boutari C, Pappas PD, Anastasilakis D, Mantzoros CS. Statins' efficacy in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin Nutr 2022; 41:2195-2206
- Rosenson RS, Baker SK, Jacobson TA, Kopecky SL, Parker BA, The National Lipid Association's Muscle Safety Expert Panel. An assessment by the Statin Muscle Safety Task Force: 2014 update. J Clin Lipidol 2014; 8:S58-71
- Stroes ES, Thompson PD, Corsini A, Vladutiu GD, Raal FJ, Ray KK, Roden M, Stein E, Tokgozoglu L, Nordestgaard BG, Bruckert E, De Backer G, Krauss RM, Laufs U, Santos RD, Hegele RA, Hovingh GK, Leiter LA, Mach F, Marz W, Newman CB, Wiklund O, Jacobson TA, Catapano AL, Chapman MJ, Ginsberg HN, European Atherosclerosis Society Consensus Panel. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J 2015; 36:1012-1022
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681-1690
- Cholesterol Treatment Trialists Collaboration. Effect of statin therapy on muscle symptoms: an individual participant data meta-analysis of large-scale, randomised, double-blind trials. Lancet 2022; 400:832-845
- Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403-414
- Buettner C, Davis RB, Leveille SG, Mittleman MA, Mukamal KJ. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182-1186
- Cohen JD, Brinton EA, Ito MK, Jacobson TA. Understanding Statin Use in America and Gaps in Patient Education (USAGE): an internet-based survey of 10,138 current and former statin users. J Clin Lipidol 2012; 6:208-215
- Parker BA, Capizzi JA, Grimaldi AS, Clarkson PM, Cole SM, Keadle J, Chipkin S, Pescatello LS, Simpson K, White CM, Thompson PD. Effect of statins on skeletal muscle function. Circulation 2013; 127:96-103
- Gupta A, Thompson D, Whitehouse A, Collier T, Dahlof B, Poulter N, Collins R, Sever P. Adverse events associated with unblinded, but not with blinded, statin therapy in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-Lowering Arm (ASCOT-LLA): a randomised double-blind placebo-controlled trial and its non-randomised non-blind extension phase. Lancet 2017; 389:2473-2481
- Joy TR, Monjed A, Zou GY, Hegele RA, McDonald CG, Mahon JL. N-of-1 (single-patient) trials for statin-related myalgia. Ann Intern Med 2014; 160:301-310
- Taylor BA, Lorson L, White CM, Thompson PD. A randomized trial of coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis 2015; 238:329-335
- Nissen SE, Stroes E, Dent-Acosta RE, Rosenson RS, Lehman SJ, Sattar N, Preiss D, Bruckert E, Ceska R, Lepor N, Ballantyne CM, Gouni-Berthold I, Elliott M, Brennan DM, Wasserman SM, Somaratne R, Scott R, Stein EA. Efficacy and Tolerability of Evolocumab vs Ezetimibe in Patients With Muscle-Related Statin Intolerance: The GAUSS-3 Randomized Clinical Trial. JAMA 2016; 315:1580-1590
- Herrett E, Williamson E, Brack K, Beaumont D, Perkins A, Thayne A, Shakur-Still H, Roberts I, Prowse D, Goldacre B, van Staa T, MacDonald TM, Armitage J, Wimborne J, Melrose P, Singh J, Brooks L, Moore M, Hoffman M, Smeeth L. Statin treatment and muscle symptoms: series of randomised, placebo controlled n-of-1 trials. BMJ 2021; 372:n135
- Howard JP, Wood FA, Finegold JA, Nowbar AN, Thompson DM, Arnold AD, Rajkumar CA, Connolly S, Cegla J, Stride C, Sever P, Norton C, Thom SAM, Shun-Shin MJ, Francis DP. Side Effect Patterns in a Crossover Trial of Statin, Placebo, and No Treatment. J Am Coll Cardiol 2021; 78:1210-1222
- Nielsen SF, Nordestgaard BG. Negative statin-related news stories decrease statin persistence and increase myocardial infarction and cardiovascular mortality: a nationwide prospective cohort study. Eur Heart J 2016; 37:908-916
- De Vera MA, Bhole V, Burns LC, Lacaille D. Impact of statin adherence on cardiovascular disease and mortality outcomes: a systematic review. Br J Clin Pharmacol 2014; 78:684-698
- Cziraky MJ, Willey VJ, McKenney JM, Kamat SA, Fisher MD, Guyton JR, Jacobson TA, Davidson MH. Risk of hospitalized rhabdomyolysis associated with lipid-lowering drugs in a real-world clinical setting. J Clin Lipidol 2013; 7:102-108
- Davidson MH, Stein EA, Dujovne CA, Hunninghake DB, Weiss SR, Knopp RH, Illingworth DR, Mitchel YB, Melino MR, Zupkis RV, Dobrinska MR, Amin RD, Tobert JA. The efficacy and six-week tolerability of simvastatin 80 and 160 mg/day. Am J Cardiol 1997; 79:38-42
- Rosenson RS, Bays HE. Results of two clinical trials on the safety and efficacy of pravastatin 80 and 160 mg per day. Am J Cardiol 2003; 91:878-881
- Search Collaborative Group, Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 2008; 359:789-799
- Hopewell JC, Reith C, Armitage J. Pharmacogenomics of statin therapy: any new insights in efficacy or safety? Curr Opin Lipidol 2014; 25:438-445
- Mammen AL. Statin-Associated Autoimmune Myopathy. N Engl J Med 2016; 374:664-669
- Mohassel P, Mammen AL. Anti-HMGCR Myopathy. J Neuromuscul Dis 2018; 5:11-20
- Wild R, Feingold KR. Effect of Pregnancy on Lipid Metabolism and Lipoprotein Levels. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Bruckert E, Giral P, Tellier P. Perspectives in cholesterol-lowering therapy: the role of ezetimibe, a new selective inhibitor of intestinal cholesterol absorption. Circulation 2003; 107:3124-3128
- Liebeskind A, Peterson AL, Wilson D. Sitosterolemia. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Pandor A, Ara RM, Tumur I, Wilkinson AJ, Paisley S, Duenas A, Durrington PN, Chilcott J. Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med 2009; 265:568-580
- Morrone D, Weintraub WS, Toth PP, Hanson ME, Lowe RS, Lin J, Shah AK, Tershakovec AM. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: a pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis 2012; 223:251-261
- Ballantyne CM, Weiss R, Moccetti T, Vogt A, Eber B, Sosef F, Duffield E. Efficacy and safety of rosuvastatin 40 mg alone or in combination with ezetimibe in patients at high risk of cardiovascular disease (results from the EXPLORER study). Am J Cardiol 2007; 99:673-680
- Sahebkar A, Simental-Mendia LE, Pirro M, Banach M, Watts GF, Sirtori C, Al-Rasadi K, Atkin SL. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: a systematic review and meta-analysis of randomized controlled trials. Sci Rep 2018; 8:17887
- Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med 2008; 358:1431-1443
- Sager PT, Capece R, Lipka L, Strony J, Yang B, Suresh R, Mitchel Y, Veltri E. Effects of ezetimibe coadministered with simvastatin on C-reactive protein in a large cohort of hypercholesterolemic patients. Atherosclerosis 2005; 179:361-367
- Ballantyne CM, Houri J, Notarbartolo A, Melani L, Lipka LJ, Suresh R, Sun S, LeBeaut AP, Sager PT, Veltri EP. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Circulation 2003; 107:2409-2415
- Yu L. The structure and function of Niemann-Pick C1-like 1 protein. Curr Opin Lipidol 2008; 19:263-269
- Turley SD, Dietschy JM. Sterol absorption by the small intestine. Curr Opin Lipidol 2003; 14:233-240
- Telford DE, Sutherland BG, Edwards JY, Andrews JD, Barrett PH, Huff MW. The molecular mechanisms underlying the reduction of LDL apoB-100 by ezetimibe plus simvastatin. J Lipid Res 2007; 48:699-708
- Pramfalk C, Jiang ZY, Parini P. Hepatic Niemann-Pick C1-like 1. Curr Opin Lipidol 2011; 22:225-230
- Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 2008; 359:1343-1356
- Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, Neal B, Jiang L, Hooi LS, Levin A, Agodoa L, Gaziano M, Kasiske B, Walker R, Massy ZA, Feldt-Rasmussen B, Krairittichai U, Ophascharoensuk V, Fellstrom B, Holdaas H, Tesar V, Wiecek A, Grobbee D, de Zeeuw D, Gronhagen-Riska C, Dasgupta T, Lewis D, Herrington W, Mafham M, Majoni W, Wallendszus K, Grimm R, Pedersen T, Tobert J, Armitage J, Baxter A, Bray C, Chen Y, Chen Z, Hill M, Knott C, Parish S, Simpson D, Sleight P, Young A, Collins R. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181-2192
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med 2015; 372:2387-2397
- Bohula EA, Wiviott SD, Giugliano RP, Blazing MA, Park JG, Murphy SA, White JA, Mach F, Van de Werf F, Dalby AJ, White HD, Tershakovec AM, Cannon CP, Braunwald E. Prevention of Stroke with the Addition of Ezetimibe to Statin Therapy in Patients With Acute Coronary Syndrome in IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2017; 136:2440-2450
- Giugliano RP, Cannon CP, Blazing MA, Nicolau JC, Corbalan R, Spinar J, Park JG, White JA, Bohula EA, Braunwald E. Benefit of Adding Ezetimibe to Statin Therapy on Cardiovascular Outcomes and Safety in Patients With Versus Without Diabetes Mellitus: Results From IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2018; 137:1571-1582
- Bonaca MP, Gutierrez JA, Cannon C, Giugliano R, Blazing M, Park JG, White J, Tershakovec A, Braunwald E. Polyvascular disease, type 2 diabetes, and long-term vascular risk: a secondary analysis of the IMPROVE-IT trial. Lancet Diabetes Endocrinol 2018; 6:934-943
- Ouchi Y, Sasaki J, Arai H, Yokote K, Harada K, Katayama Y, Urabe T, Uchida Y, Hayashi M, Yokota N, Nishida H, Otonari T, Arai T, Sakuma I, Sakabe K, Yamamoto M, Kobayashi T, Oikawa S, Yamashita S, Rakugi H, Imai T, Tanaka S, Ohashi Y, Kuwabara M, Ito H. Ezetimibe Lipid-Lowering Trial on Prevention of Atherosclerotic Cardiovascular Disease in 75 or Older (EWTOPIA 75): A Randomized, Controlled Trial. Circulation 2019; 140:992-1003
- Kim BK, Hong SJ, Lee YJ, Hong SJ, Yun KH, Hong BK, Heo JH, Rha SW, Cho YH, Lee SJ, Ahn CM, Kim JS, Ko YG, Choi D, Jang Y, Hong MK. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet 2022; 400:380-390
- Lee B, Hong SJ, Rha SW, Heo JH, Hur SH, Choi HH, Kim KJ, Kim JH, Kim HK, Kim U, Choi YJ, Lee YJ, Lee SJ, Ahn CM, Ko YG, Kim BK, Choi D, Hong MK, Jang Y, Kim JS. Moderate-intensity statin plus ezetimibe vs high-intensity statin according to baseline LDL-C in the treatment of atherosclerotic cardiovascular disease: A post-hoc analysis of the RACING randomized trial. Atherosclerosis 2023; 386:117373
- Lee SH, Lee YJ, Heo JH, Hur SH, Choi HH, Kim KJ, Kim JH, Park KH, Lee JH, Choi YJ, Lee SJ, Hong SJ, Ahn CM, Kim BK, Ko YG, Choi D, Hong MK, Jang Y, Kim JS. Combination Moderate-Intensity Statin and Ezetimibe Therapy for Elderly Patients With Atherosclerosis. J Am Coll Cardiol 2023; 81:1339-1349
- Toth PP, Morrone D, Weintraub WS, Hanson ME, Lowe RS, Lin J, Shah AK, Tershakovec AM. Safety profile of statins alone or combined with ezetimibe: a pooled analysis of 27 studies including over 22,000 patients treated for 6-24 weeks. Int J Clin Pract 2012; 66:800-812
- Luo L, Yuan X, Huang W, Ren F, Zhu H, Zheng Y, Tang L. Safety of coadministration of ezetimibe and statins in patients with hypercholesterolaemia: a meta-analysis. Intern Med J 2015; 45:546-557
- Kashani A, Sallam T, Bheemreddy S, Mann DL, Wang Y, Foody JM. Review of side-effect profile of combination ezetimibe and statin therapy in randomized clinical trials. Am J Cardiol 2008; 101:1606-1613
- Savarese G, De Ferrari GM, Rosano GM, Perrone-Filardi P. Safety and efficacy of ezetimibe: A meta-analysis. Int J Cardiol 2015; 201:247-252
- Peto R, Emberson J, Landray M, Baigent C, Collins R, Clare R, Califf R. Analyses of cancer data from three ezetimibe trials. N Engl J Med 2008; 359:1357-1366
- Wu H, Shang H, Wu J. Effect of ezetimibe on glycemic control: a systematic review and meta-analysis of randomized controlled trials. Endocrine 2018; 60:229-239
- Aldridge MA, Ito MK. Colesevelam hydrochloride: a novel bile acid-binding resin. Ann Pharmacother 2001; 35:898-907
- Heel RC, Brogden RN, Pakes GE, Speight TM, Avery GS. Colestipol: a review of its pharmacological properties and therapeutic efficacy in patients with hypercholesterolaemia. Drugs 1980; 19:161-180
- Insull W, Jr. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J 2006; 99:257-273
- Huijgen R, Abbink EJ, Bruckert E, Stalenhoef AF, Imholz BP, Durrington PN, Trip MD, Eriksson M, Visseren FL, Schaefer JR, Kastelein JJ. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial hypercholesterolemia: a 12-week, multicenter, randomized, double-blind, controlled trial. Clin Ther 2010; 32:615-625
- Zema MJ. Colesevelam HCl and ezetimibe combination therapy provides effective lipid-lowering in difficult-to-treat patients with hypercholesterolemia. Am J Ther 2005; 12:306-310
- Bays H, Rhyne J, Abby S, Lai YL, Jones M. Lipid-lowering effects of colesevelam HCl in combination with ezetimibe. Curr Med Res Opin 2006; 22:2191-2200
- Fonseca VA, Handelsman Y, Staels B. Colesevelam lowers glucose and lipid levels in type 2 diabetes: the clinical evidence. Diabetes Obes Metab 2010; 12:384-392
- Devaraj S, Autret B, Jialal I. Effects of colesevelam hydrochloride (WelChol) on biomarkers of inflammation in patients with mild hypercholesterolemia. Am J Cardiol 2006; 98:641-643
- Bays HE, Davidson M, Jones MR, Abby SL. Effects of colesevelam hydrochloride on low-density lipoprotein cholesterol and high-sensitivity C-reactive protein when added to statins in patients with hypercholesterolemia. Am J Cardiol 2006; 97:1198-1205
- Einarsson K, Ericsson S, Ewerth S, Reihner E, Rudling M, Stahlberg D, Angelin B. Bile acid sequestrants: mechanisms of action on bile acid and cholesterol metabolism. Eur J Clin Pharmacol 1991; 40 Suppl 1:S53-58
- Kliewer SA, Mangelsdorf DJ. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig Dis 2015; 33:327-331
- Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009; 50:1955-1966
- Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res 2012; 53:1723-1737
- Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res 2002; 43:2-12
- Bronden A, Knop FK. Gluco-Metabolic Effects of Pharmacotherapy-Induced Modulation of Bile Acid Physiology. J Clin Endocrinol Metab 2020; 105
- The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA 1984; 251:351-364
- The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 1984; 251:365-374
- Levy RI, Brensike JF, Epstein SE, Kelsey SF, Passamani ER, Richardson JM, Loh IK, Stone NJ, Aldrich RF, Battaglini JW, et al. The influence of changes in lipid values induced by cholestyramine and diet on progression of coronary artery disease: results of NHLBI Type II Coronary Intervention Study. Circulation 1984; 69:325-337
- Watts GF, Lewis B, Brunt JN, Lewis ES, Coltart DJ, Smith LD, Mann JI, Swan AV. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas' Atherosclerosis Regression Study (STARS). Lancet 1992; 339:563-569
- Blankenhorn DH, Nessim SA, Johnson RL, Sanmarco ME, Azen SP, Cashin-Hemphill L. Beneficial effects of combined colestipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts. JAMA 1987; 257:3233-3240
- Brown G, Albers JJ, Fisher LD, Schaefer SM, Lin JT, Kaplan C, Zhao XQ, Bisson BD, Fitzpatrick VF, Dodge HT. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med 1990; 323:1289-1298
- Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ. Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimens. JAMA 1990; 264:3007-3012
- Ito MK, McGowan MP, Moriarty PM, National Lipid Association Expert Panel on Familial Hypercholesterolemia. Management of familial hypercholesterolemias in adult patients: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011; 5:S38-45
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the Next Breakthrough in the Cardiovascular Field? J Am Coll Cardiol 2015; 65:2638-2651
- McKenney JM. Understanding PCSK9 and anti-PCSK9 therapies. J Clin Lipidol 2015; 9:170-186
- Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, Brockmeyer M, Kandzari DE, Kubica JM, D'Agostino RB, Sr., Kubica J, Volpe M, Agewall S, Kereiakes DJ, Kelm M. Effects of Proprotein Convertase Subtilisin/Kexin Type 9 Antibodies in Adults With Hypercholesterolemia: A Systematic Review and Meta-analysis. Ann Intern Med 2015; 163:40-51
- O'Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, Im K, Lira Pineda A, Wasserman SM, Ceska R, Ezhov MV, Jukema JW, Jensen HK, Tokgozoglu SL, Mach F, Huber K, Sever PS, Keech AC, Pedersen TR, Sabatine MS. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019; 139:1483-1492
- Sahebkar A, Di Giosia P, Stamerra CA, Grassi D, Pedone C, Ferretti G, Bacchetti T, Ferri C, Giorgini P. Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: a meta-analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol 2016; 81:1175-1190
- Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2531-2540
- Roth EM, Taskinen MR, Ginsberg HN, Kastelein JJ, Colhoun HM, Robinson JG, Merlet L, Pordy R, Baccara-Dinet MT. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol 2014; 176:55-61
- Kereiakes DJ, Robinson JG, Cannon CP, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: The ODYSSEY COMBO I study. Am Heart J 2015; 169:906-915 e913
- Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J 2015; 36:1186-1194
- Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, Somaratne R, Legg JC, Nelson P, Scott R, Wasserman SM, Weiss R. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014; 311:1870-1882
- Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L, Ceska R, Roth E, Koren MJ, Ballantyne CM, Monsalvo ML, Tsirtsonis K, Kim JB, Scott R, Wasserman SM, Stein EA. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370:1809-1819
- Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, Hovingh GK, Cariou B, Gouni-Berthold I, Somaratne R, Bridges I, Scott R, Wasserman SM, Gaudet D. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 385:331-340
- Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, Zhao J, Pordy R, Baccara-Dinet MT, Gipe DA, Geiger MJ, Farnier M. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36:2996-3003
- Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 385:341-350
- Raal FJ, Hovingh GK, Blom D, Santos RD, Harada-Shiba M, Bruckert E, Couture P, Soran H, Watts GF, Kurtz C, Honarpour N, Tang L, Kasichayanula S, Wasserman SM, Stein EA. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol 2017; 5:280-290
- Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation 2013; 128:2113-2120
- Blom DJ, Harada-Shiba M, Rubba P, Gaudet D, Kastelein JJP, Charng MJ, Pordy R, Donahue S, Ali S, Dong Y, Khilla N, Banerjee P, Baccara-Dinet M, Rosenson RS. Efficacy and Safety of Alirocumab in Adults With Homozygous Familial Hypercholesterolemia: The ODYSSEY HoFH Trial. J Am Coll Cardiol 2020; 76:131-142
- Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, Bruckert E, Cho L, Dent R, Knusel B, Xue A, Scott R, Wasserman SM, Rocco M. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014; 63:2541-2548
- Moriarty PM, Thompson PD, Cannon CP, Guyton JR, Bergeron J, Zieve FJ, Bruckert E, Jacobson TA, Kopecky SL, Baccara-Dinet MT, Du Y, Pordy R, Gipe DA. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol 2015; 9:758-769
- Sattar N, Preiss D, Robinson JG, Djedjos CS, Elliott M, Somaratne R, Wasserman SM, Raal FJ. Lipid-lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: a meta-analysis of individual patient data. Lancet Diabetes Endocrinol 2016; 4:403-410
- Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154-156
- Warden BA, Fazio S, Shapiro MD. Familial Hypercholesterolemia: Genes and Beyond. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005; 37:161-165
- Cohen JC, Boerwinkle E, Mosley TH, Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264-1272
- Feingold KR. Introduction to Lipids and Lipoproteins. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 2009; 50 Suppl:S172-177
- Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res 2012; 53:2515-2524
- Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike J, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK, Ginsberg HN. Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation 2017; 135:352-362
- Watts GF, Chan DC, Dent R, Somaratne R, Wasserman SM, Scott R, Burrows S, Barrett PHR. Factorial Effects of Evolocumab and Atorvastatin on Lipoprotein Metabolism. Circulation 2017; 135:338-351
- Watts GF, Chan DC, Pang J, Ma L, Ying Q, Aggarwal S, Marcovina SM, Barrett PHR. PCSK9 Inhibition with alirocumab increases the catabolism of lipoprotein(a) particles in statin-treated patients with elevated lipoprotein(a). Metabolism 2020; 107:154221
- Ying Q, Chan DC, Pang J, Marcovina SM, Barrett PHR, Watts GF. PCSK9 inhibition with alirocumab decreases plasma lipoprotein(a) concentration by a dual mechanism of action in statin-treated patients with very high apolipoprotein(a) concentration. J Intern Med 2022; 291:870-876
- Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Seidah NG, Honarpour N, Lira A, Xue A, Chirovolu P, Jackson S, Di M, Peach M, Somaratne R, Wasserman SM, Scott R, Stein EA. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the role of the LDL receptor. J Lipid Res 2016;
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA, Open-Label Study of Long-Term Evaluation against LDLCI. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500-1509
- Sabatine MS, Leiter LA, Wiviott SD, Giugliano RP, Deedwania P, De Ferrari GM, Murphy SA, Kuder JF, Gouni-Berthold I, Lewis BS, Handelsman Y, Pineda AL, Honarpour N, Keech AC, Sever PS, Pedersen TR. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol 2017; 5:941-950
- Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, Kuder J, Murphy SA, Jukema JW, Lewis BS, Tokgozoglu L, Somaratne R, Sever PS, Pedersen TR, Sabatine MS. Low-Density Lipoprotein Cholesterol Lowering With Evolocumab and Outcomes in Patients With Peripheral Artery Disease: Insights From the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk). Circulation 2018; 137:338-350
- Giugliano RP, Keech A, Murphy SA, Huber K, Tokgozoglu SL, Lewis BS, Ferreira J, Pineda AL, Somaratne R, Sever PS, Pedersen TR, Sabatine MS. Clinical Efficacy and Safety of Evolocumab in High-Risk Patients Receiving a Statin: Secondary Analysis of Patients With Low LDL Cholesterol Levels and in Those Already Receiving a Maximal-Potency Statin in a Randomized Clinical Trial. JAMA Cardiol 2017; 2:1385-1391
- Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni-Berthold I, Lopez-Miranda J, Schiele F, Mach F, Ott BR, Kanevsky E, Pineda AL, Somaratne R, Wasserman SM, Keech AC, Sever PS, Sabatine MS. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet 2017; 390:1962-1971
- Sabatine MS, De Ferrari GM, Giugliano RP, Huber K, Lewis BS, Ferreira J, Kuder JF, Murphy SA, Wiviott SD, Kurtz CE, Honarpour N, Keech AC, Sever PS, Pedersen TR. Clinical Benefit of Evolocumab by Severity and Extent of Coronary Artery Disease. Circulation 2018; 138:756-766
- Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, Jukema JW, Lecorps G, Mahaffey KW, Moryusef A, Pordy R, Quintero K, Roe MT, Sasiela WJ, Tamby JF, Tricoci P, White HD, Zeiher AM. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med 2018; 379:2097-2107
- Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016; 316:2373-2384
- Raber L, Ueki Y, Otsuka T, Losdat S, Haner JD, Lonborg J, Fahrni G, Iglesias JF, van Geuns RJ, Ondracek AS, Radu Juul Jensen MD, Zanchin C, Stortecky S, Spirk D, Siontis GCM, Saleh L, Matter CM, Daemen J, Mach F, Heg D, Windecker S, Engstrom T, Lang IM, Koskinas KC. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients With Acute Myocardial Infarction: The PACMAN-AMI Randomized Clinical Trial. JAMA 2022; 327:1771-1781
- Perez de Isla L, Diaz-Diaz JL, Romero MJ, Muniz-Grijalvo O, Mediavilla JD, Argueso R, Sanchez Munoz-Torrero JF, Rubio P, Alvarez-Banos P, Ponte P, Manas D, Suarez Gutierrez L, Cepeda JM, Casanas M, Fuentes F, Guijarro C, Angel Barba M, Saltijeral Cerezo A, Padro T, Mata P. Alirocumab and Coronary Atherosclerosis in Asymptomatic Patients with Familial Hypercholesterolemia: The ARCHITECT Study. Circulation 2023; 147:1436-1443
- Marston NA, Gurmu Y, Melloni GEM, Bonaca M, Gencer B, Sever PS, Pedersen TR, Keech AC, Roselli C, Lubitz SA, Ellinor PT, O'Donoghue ML, Giugliano RP, Ruff CT, Sabatine MS. The Effect of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Inhibition on the Risk of Venous Thromboembolism. Circulation 2020; 141:1600-1607
- Schwartz GG, Steg PG, Szarek M, Bittner VA, Diaz R, Goodman SG, Kim YU, Jukema JW, Pordy R, Roe MT, White HD, Bhatt DL. Peripheral Artery Disease and Venous Thromboembolic Events After Acute Coronary Syndrome: Role of Lipoprotein(a) and Modification by Alirocumab: Prespecified Analysis of the ODYSSEY OUTCOMES Randomized Clinical Trial. Circulation 2020; 141:1608-1617
- de Carvalho LSF, Campos AM, Sposito AC. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibitors and Incident Type 2 Diabetes: A Systematic Review and Meta-analysis With Over 96,000 Patient-Years. Diabetes Care 2018; 41:364-367
- Giugliano RP, Mach F, Zavitz K, Kurtz C, Im K, Kanevsky E, Schneider J, Wang H, Keech A, Pedersen TR, Sabatine MS, Sever PS, Robinson JG, Honarpour N, Wasserman SM, Ott BR. Cognitive Function in a Randomized Trial of Evolocumab. N Engl J Med 2017; 377:633-643
- Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, Civeira F, Somaratne R, Nelson P, Liu T, Scott R, Wasserman SM, Sabatine MS. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation 2014; 129:234-243
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1489-1499
- Shapiro MD, Feingold KR. Monogenic Disorders Causing Hypobetalipoproteinemia. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2024.
- LaRosa JC, Grundy SM, Kastelein JJ, Kostis JB, Greten H, Treating to New Targets Steering C, Investigators. Safety and efficacy of Atorvastatin-induced very low-density lipoprotein cholesterol levels in Patients with coronary heart disease (a post hoc analysis of the treating to new targets [TNT] study). Am J Cardiol 2007; 100:747-752
- Wiviott SD, Cannon CP, Morrow DA, Ray KK, Pfeffer MA, Braunwald E. Can low-density lipoprotein be too low? The safety and efficacy of achieving very low low-density lipoprotein with intensive statin therapy: a PROVE IT-TIMI 22 substudy. J Am Coll Cardiol 2005; 46:1411-1416
- Everett BM, Mora S, Glynn RJ, MacFadyen J, Ridker PM. Safety profile of subjects treated to very low low-density lipoprotein cholesterol levels (<30 mg/dl) with rosuvastatin 20 mg daily (from JUPITER). Am J Cardiol 2014; 114:1682-1689
- Sinning D, Landmesser U. Low-density Lipoprotein-Cholesterol Lowering Strategies for Prevention of Atherosclerotic Cardiovascular Disease: Focus on siRNA Treatment Targeting PCSK9 (Inclisiran). Curr Cardiol Rep 2020; 22:176
- Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, Kastelein JJP. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med 2020; 382:1507-1519
- Wright RS, Collins MG, Stoekenbroek RM, Robson R, Wijngaard PLJ, Landmesser U, Leiter LA, Kastelein JJP, Ray KK, Kallend D. Effects of Renal Impairment on the Pharmacokinetics, Efficacy, and Safety of Inclisiran: An Analysis of the ORION-7 and ORION-1 Studies. Mayo Clin Proc 2020; 95:77-89
- Ray KK, Troquay RPT, Visseren FLJ, Leiter LA, Scott Wright R, Vikarunnessa S, Talloczy Z, Zang X, Maheux P, Lesogor A, Landmesser U. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol 2023; 11:109-119
- Raal FJ, Kallend D, Ray KK, Turner T, Koenig W, Wright RS, Wijngaard PLJ, Curcio D, Jaros MJ, Leiter LA, Kastelein JJP. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N Engl J Med 2020; 382:1520-1530
- Hovingh GK, Lepor NE, Kallend D, Stoekenbroek RM, Wijngaard PLJ, Raal FJ. Inclisiran Durably Lowers Low-Density Lipoprotein Cholesterol and Proprotein Convertase Subtilisin/Kexin Type 9 Expression in Homozygous Familial Hypercholesterolemia: The ORION-2 Pilot Study. Circulation 2020; 141:1829-1831
- Raal F, Durst R, Bi R, Talloczy Z, Maheux P, Lesogor A, Kastelein JJP. Efficacy, Safety, and Tolerability of Inclisiran in Patients With Homozygous Familial Hypercholesterolemia: Results From the ORION-5 Randomized Clinical Trial. Circulation 2024; 149:354-362
- Wright RS, Koenig W, Landmesser U, Leiter LA, Raal FJ, Schwartz GG, Lesogor A, Maheux P, Stratz C, Zang X, Ray KK. Safety and Tolerability of Inclisiran for Treatment of Hypercholesterolemia in 7 Clinical Trials. J Am Coll Cardiol 2023; 82:2251-2261
- Laufs U, Banach M, Mancini GBJ, Gaudet D, Bloedon LT, Sterling LR, Kelly S, Stroes ESG. Efficacy and Safety of Bempedoic Acid in Patients With Hypercholesterolemia and Statin Intolerance. J Am Heart Assoc 2019; 8:e011662
- Goldberg AC, Leiter LA, Stroes ESG, Baum SJ, Hanselman JC, Bloedon LT, Lalwani ND, Patel PM, Zhao X, Duell PB. Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial. JAMA 2019; 322:1780-1788
- Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, Robinson PL, Ballantyne CM. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N Engl J Med 2019; 380:1022-1032
- Lalwani ND, Hanselman JC, MacDougall DE, Sterling LR, Cramer CT. Complementary low-density lipoprotein-cholesterol lowering and pharmacokinetics of adding bempedoic acid (ETC-1002) to high-dose atorvastatin background therapy in hypercholesterolemic patients: A randomized placebo-controlled trial. J Clin Lipidol 2019; 13:568-579
- Ballantyne CM, Laufs U, Ray KK, Leiter LA, Bays HE, Goldberg AC, Stroes ES, MacDougall D, Zhao X, Catapano AL. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur J Prev Cardiol 2019:2047487319864671
- Ballantyne CM, Banach M, Mancini GBJ, Lepor NE, Hanselman JC, Zhao X, Leiter LA. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: A randomized, placebo-controlled study. Atherosclerosis 2018; 277:195-203
- Ridker PM, Lei L, Ray KK, Ballantyne CM, Bradwin G, Rifai N. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: A secondary analysis of the CLEAR harmony trial. J Clin Lipidol 2023; 17:297-302
- Ray KK, Nicholls SJ, Li N, Louie MJ, Brennan D, Lincoff AM, Nissen SE. Efficacy and safety of bempedoic acid among patients with and without diabetes: prespecified analysis of the CLEAR Outcomes randomised trial. Lancet Diabetes Endocrinol 2024; 12:19-28
- Nissen SE, Nicholls SJ, Lincoff AM. Bempedoic Acid for Primary Prevention of Cardiovascular Events-Reply. JAMA 2023; 330:1696-1697
- Bays HE, Bloedon LT, Lin G, Powell HA, Louie MJ, Nicholls SJ, Lincoff AM, Nissen S. Safety of bempedoic acid in patients at high cardiovascular risk and with statin intolerance. J Clin Lipidol 2023;
- Burke AC, Telford DE, Huff MW. Bempedoic acid: effects on lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol 2019; 30:1-9
- Ference BA, Ray KK, Catapano AL, Ference TB, Burgess S, Neff DR, Oliver-Williams C, Wood AM, Butterworth AS, Di Angelantonio E, Danesh J, Kastelein JJP, Nicholls SJ. Mendelian Randomization Study of ACLY and Cardiovascular Disease. N Engl J Med 2019; 380:1033-1042
- Nissen SE, Lincoff AM, Brennan D, Ray KK, Mason D, Kastelein JJP, Thompson PD, Libby P, Cho L, Plutzky J, Bays HE, Moriarty PM, Menon V, Grobbee DE, Louie MJ, Chen CF, Li N, Bloedon L, Robinson P, Horner M, Sasiela WJ, McCluskey J, Davey D, Fajardo-Campos P, Petrovic P, Fedacko J, Zmuda W, Lukyanov Y, Nicholls SJ. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med 2023; 388:1353-1364
- Neef D, Berthold HK, Gouni-Berthold I. Lomitapide for use in patients with homozygous familial hypercholesterolemia: a narrative review. Expert Rev Clin Pharmacol 2016; 9:655-663
- Gouni-Berthold I, Berthold HK. Mipomersen and lomitapide: Two new drugs for the treatment of homozygous familial hypercholesterolemia. Atheroscler Suppl 2015; 18:28-34
- Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014; 129:1022-1032
- Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ, Phase 3 Ho FHLSi. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet 2013; 381:40-46
- Blom DJ, Averna MR, Meagher EA, du Toit Theron H, Sirtori CR, Hegele RA, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Larrey D, Bloedon LT, Foulds P, Rader DJ, Cuchel M. Long-Term Efficacy and Safety of the Microsomal Triglyceride Transfer Protein Inhibitor Lomitapide in Patients With Homozygous Familial Hypercholesterolemia. Circulation 2017; 136:332-335
- Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med 2008; 5:497-505
- Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res 2003; 44:22-32
- Blom DJ, Cuchel M, Ager M, Phillips H. Target achievement and cardiovascular event rates with Lomitapide in homozygous Familial Hypercholesterolaemia. Orphanet J Rare Dis 2018; 13:96
- Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years' treatment with lomitapide. JAMA Intern Med 2014; 174:443-447
- Underberg JA, Cannon CP, Larrey D, Makris L, Blom D, Phillips H. Long-term safety and efficacy of lomitapide in patients with homozygous familial hypercholesterolemia: Five-year data from the Lomitapide Observational Worldwide Evaluation Registry (LOWER). J Clin Lipidol 2020; 14:807-817
- Larrey D, D'Erasmo L, O'Brien S, Arca M. Long-term hepatic safety of lomitapide in homozygous familial hypercholesterolaemia. Liver Int 2023; 43:413-423
- Agarwala A, Jones P, Nambi V. The role of antisense oligonucleotide therapy in patients with familial hypercholesterolemia: risks, benefits, and management recommendations. Curr Atheroscler Rep 2015; 17:467
- Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL, Flaim JD, Crooke ST. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 2010; 375:998-1006
- Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, Chin W, Tribble DL, McGowan M. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 2012; 126:2283-2292
- Santos RD, Duell PB, East C, Guyton JR, Moriarty PM, Chin W, Mittleman RS. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J 2015; 36:566-575
- Panta R, Dahal K, Kunwar S. Efficacy and safety of mipomersen in treatment of dyslipidemia: a meta-analysis of randomized controlled trials. J Clin Lipidol 2015; 9:217-225
- Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol 2016; 10:1011-1021
- Hashemi N, Odze RD, McGowan MP, Santos RD, Stroes ES, Cohen DE. Liver histology during Mipomersen therapy for severe hypercholesterolemia. J Clin Lipidol 2014; 8:606-611
- Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, Ali S, Banerjee P, Chan KC, Gipe DA, Khilla N, Pordy R, Weinreich DM, Yancopoulos GD, Zhang Y, Gaudet D. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med 2020; 383:711-720
- Rosenson RS, Burgess LJ, Ebenbichler CF, Baum SJ, Stroes ESG, Ali S, Khilla N, Hamlin R, Pordy R, Dong Y, Son V, Gaudet D. Evinacumab in Patients with Refractory Hypercholesterolemia. N Engl J Med 2020; 383:2307-2319
- Rosenson RS, Burgess LJ, Ebenbichler CF, Baum SJ, Stroes ESG, Ali S, Khilla N, McGinniss J, Gaudet D, Pordy R. Longer-Term Efficacy and Safety of Evinacumab in Patients With Refractory Hypercholesterolemia. JAMA Cardiol 2023; 8:1070-1076
- Feingold KR. Triglyceride Lowering Drugs. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2024.
- Mattijssen F, Kersten S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim Biophys Acta 2012; 1821:782-789
- Kersten S. New insights into angiopoietin-like proteins in lipid metabolism and cardiovascular disease risk. Curr Opin Lipidol 2019; 30:205-211
- Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet 1999; 21:424-428
- Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD, Quertermous T. Endothelial lipase is a major determinant of HDL level. J Clin Invest 2003; 111:347-355
- Shimamura M, Matsuda M, Yasumo H, Okazaki M, Fujimoto K, Kono K, Shimizugawa T, Ando Y, Koishi R, Kohama T, Sakai N, Kotani K, Komuro R, Ishida T, Hirata K, Yamashita S, Furukawa H, Shimomura I. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol 2007; 27:366-372
- Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, Hamon SC, Kim HI, Cohen JC, Hobbs HH, Van Hout C, Gromada J, Murphy AJ, Yancopoulos GD, Sleeman MW, Gusarova V. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res 2020; 61:1271-1286
- Reeskamp LF, Millar JS, Wu L, Jansen H, van Harskamp D, Schierbeek H, Gipe DA, Rader DJ, Dallinga-Thie GM, Hovingh GK, Cuchel M. ANGPTL3 Inhibition With Evinacumab Results in Faster Clearance of IDL and LDL apoB in Patients With Homozygous Familial Hypercholesterolemia. Arterioscler Thromb Vasc Biol 2021:ATVBAHA120315204
- Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J Lipid Res 2015; 56:1296-1307
- Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, Fennell T, Banks E, Ambrogio L, Cibulskis K, Kernytsky A, Gonzalez E, Rudzicz N, Engert JC, DePristo MA, Daly MJ, Cohen JC, Hobbs HH, Altshuler D, Schonfeld G, Gabriel SB, Yue P, Kathiresan S. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 2010; 363:2220-2227
- Dewey FE, Gusarova V, Dunbar RL, O'Dushlaine C, Schurmann C, Gottesman O, McCarthy S, Van Hout CV, Bruse S, Dansky HM, Leader JB, Murray MF, Ritchie MD, Kirchner HL, Habegger L, Lopez A, Penn J, Zhao A, Shao W, Stahl N, Murphy AJ, Hamon S, Bouzelmat A, Zhang R, Shumel B, Pordy R, Gipe D, Herman GA, Sheu WHH, Lee IT, Liang KW, Guo X, Rotter JI, Chen YI, Kraus WE, Shah SH, Damrauer S, Small A, Rader DJ, Wulff AB, Nordestgaard BG, Tybjaerg-Hansen A, van den Hoek AM, Princen HMG, Ledbetter DH, Carey DJ, Overton JD, Reid JG, Sasiela WJ, Banerjee P, Shuldiner AR, Borecki IB, Teslovich TM, Yancopoulos GD, Mellis SJ, Gromada J, Baras A. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med 2017; 377:211-221
- Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, Nomura A, Erdmann J, Schunkert H, Samani NJ, Kraus WE, Shah SH, Yu B, Boerwinkle E, Rader DJ, Gupta N, Frossard PM, Rasheed A, Danesh J, Lander ES, Gabriel S, Saleheen D, Musunuru K, Kathiresan S. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol 2017; 69:2054-2063
- Ahmad Z, Banerjee P, Hamon S, Chan KC, Bouzelmat A, Sasiela WJ, Pordy R, Mellis S, Dansky H, Gipe DA, Dunbar RL. Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation 2019; 140:470-486
- Feingold KR. Lipoprotein Apheresis. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Awad K, Mikhailidis DP, Toth PP, Jones SR, Moriarty P, Lip GYH, Muntner P, Catapano AL, Pencina MJ, Rosenson RS, Rysz J, Banach M. Efficacy and Safety of Alternate-Day Versus Daily Dosing of Statins: a Systematic Review and Meta-Analysis. Cardiovasc Drugs Ther 2017; 31:419-431
- Kennedy SP, Barnas GP, Schmidt MJ, Glisczinski MS, Paniagua AC. Efficacy and tolerability of once-weekly rosuvastatin in patients with previous statin intolerance. J Clin Lipidol 2011; 5:308-315
- Banach M, Serban C, Sahebkar A, Ursoniu S, Rysz J, Muntner P, Toth PP, Jones SR, Rizzo M, Glasser SP, Lip GY, Dragan S, Mikhailidis DP, Lipid, Blood Pressure Meta-analysis Collaboration G. Effects of coenzyme Q10 on statin-induced myopathy: a meta-analysis of randomized controlled trials. Mayo Clin Proc 2015; 90:24-34
- Schaars CF, Stalenhoef AF. Effects of ubiquinone (coenzyme Q10) on myopathy in statin users. Curr Opin Lipidol 2008; 19:553-557
- Skarlovnik A, Janic M, Lunder M, Turk M, Sabovic M. Coenzyme Q10 supplementation decreases statin-related mild-to-moderate muscle symptoms: a randomized clinical study. Med Sci Monit 2014; 20:2183-2188
- Bogsrud MP, Langslet G, Ose L, Arnesen KE, Sm Stuen MC, Malt UF, Woldseth B, Retterstol K. No effect of combined coenzyme Q10 and selenium supplementation on atorvastatin-induced myopathy. Scand Cardiovasc J 2013; 47:80-87
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409-1412
- Fedacko J, Pella D, Fedackova P, Hanninen O, Tuomainen P, Jarcuska P, Lopuchovsky T, Jedlickova L, Merkovska L, Littarru GP. Coenzyme Q(10) and selenium in statin-associated myopathy treatment. Can J Physiol Pharmacol 2013; 91:165-170
- Hlatky MA, Gonzalez PE, Manson JE, Buring JE, Lee IM, Cook NR, Mora S, Bubes V, Stone NJ. Statin-Associated Muscle Symptoms Among New Statin Users Randomly Assigned to Vitamin D or Placebo. JAMA Cardiol 2023; 8:74-80
- Becker DJ, Gordon RY, Halbert SC, French B, Morris PB, Rader DJ. Red yeast rice for dyslipidemia in statin-intolerant patients: a randomized trial. Ann Intern Med 2009; 150:830-839, W147-839
- Halbert SC, French B, Gordon RY, Farrar JT, Schmitz K, Morris PB, Thompson PD, Rader DJ, Becker DJ. Tolerability of red yeast rice (2,400 mg twice daily) versus pravastatin (20 mg twice daily) in patients with previous statin intolerance. Am J Cardiol 2010; 105:198-204