ABSTRACT
Complementary and Integrative Health (CIH) approaches, otherwise known as non-mainstream practices or Complementary and Alternative Medicine (CAM), are commonly used by patients with diabetes. Natural products, including dietary supplements, are the most frequently used complementary approach by patients with diabetes. While popular, there are regulatory, safety, and efficacy concerns regarding natural products. Commonly used dietary supplements for diabetes can be categorized as hypoglycemic agents, carbohydrate absorption inhibitors, and insulin sensitizers. Hypoglycemic agents of interest include banaba, bitter melon, fenugreek, and gymnema. American ginseng, banaba, berberine, chromium, cinnamon, gymnema, milk thistle, prickly pear cactus, soy, and vanadium are insulin sensitizers that have been studied in patients with diabetes. The carbohydrate absorption inhibitors aloe vera gel, fenugreek, flaxseed, prickly pear cactus, soy, and turmeric may be used in patients with diabetes. Mind body therapies including yoga, massage, and Tai Chi have preliminary evidence to support use in patients with diabetes. Deceptive marketing tactics may be employed by sellers of natural products. Consumers and clinicians must be aware of potential risks and make informed choices. Resources such as the Food and Drug Administration’s (FDA’s) MedWatch may be helpful. The FDA’s online health fraud website informs consumers on various types of fraud and how to avoid them.
COMPLEMEMTARY AND INTEGRATIVE HEALTH APPROACHES BACKGROUND
This chapter reviews information regarding Complementary and Integrative Health (CIH) approaches used to treat diabetes. First, background information on complementary health approaches (sometimes referred to as Complementary and Alternative Medicine or CAM) will be presented. This will be followed by a description of non-mainstream practices used by patients with diabetes. An evidence-based description of specific natural products used to treat diabetes will be next. Mind body practices will be addressed. The chapter will conclude with specific ways clinicians can assist patients in choosing safe natural products. Please note information regarding therapies to treat comorbidities of diabetes are covered elsewhere in this text.
To help clarify the contents of this chapter, nomenclature definitions relevant to CIH will be provided as defined by the National Institute of Health (NIH) National Center for Complementary and Integrative Health (NCCIH). Table 1 provides definitions of commonly used terms. Complementary medicine is defined as non-mainstream practices that are used together with conventional medicine. In contrast, alternative medicine describes non-mainstream practices used instead of conventional medicine. Complementary health approaches are non-mainstream practices. The term integrative medicine refers to medicine that brings complementary and conventional health approaches together in a coordinated manner. Integrative health describes complementary approaches that are incorporated into mainstream healthcare. Lastly, the term natural products refer to herbs, vitamins, minerals, and probiotics (1).
|
Table 1. Definitions of Terms Relevant to Complementary and Alternative Medicine (1)
|
|
Term
|
Definition
|
|
Alternative Medicine
|
Non-mainstream practice used instead of conventional medicine
|
|
Complementary Health Approaches
|
Non-mainstream practices
|
|
Complementary Medicine
|
Non-mainstream practice used together with conventional medicine
|
|
Integrative Health
|
Complementary approaches being incorporated into mainstream healthcare
|
|
Integrative Medicine
|
Medicine that brings complementary and conventional health approaches together in a coordinated fashion
|
|
Natural Products
|
Herbs, vitamins, minerals, and probiotics
|
“Complementary health approaches” is an umbrella term for non-mainstream practices. Complementary health approaches can be classified by their primary therapeutic input, or method of delivery (Table 2). A graphic representation of the examples of complementary health approaches and their categorization is provided in Figure 1 (1).
|
Table 2. Classification of Complementary Health Approaches (1)
|
|
Primary Therapeutic Input
|
Examples
|
|
Nutritional
|
Special diets, dietary supplements, herbs, and probiotics
|
|
Psychological
|
Mindfulness
|
|
Physical
|
Osteopathic manipulative therapy, chiropractic spinal manipulation, massage therapy, physical therapy
|
|
Combinations
|
Psychological and physical, e.g., yoga, Tai Chi, acupuncture, dance or art therapies, or psychological and nutritional, e.g., mindful eating
|
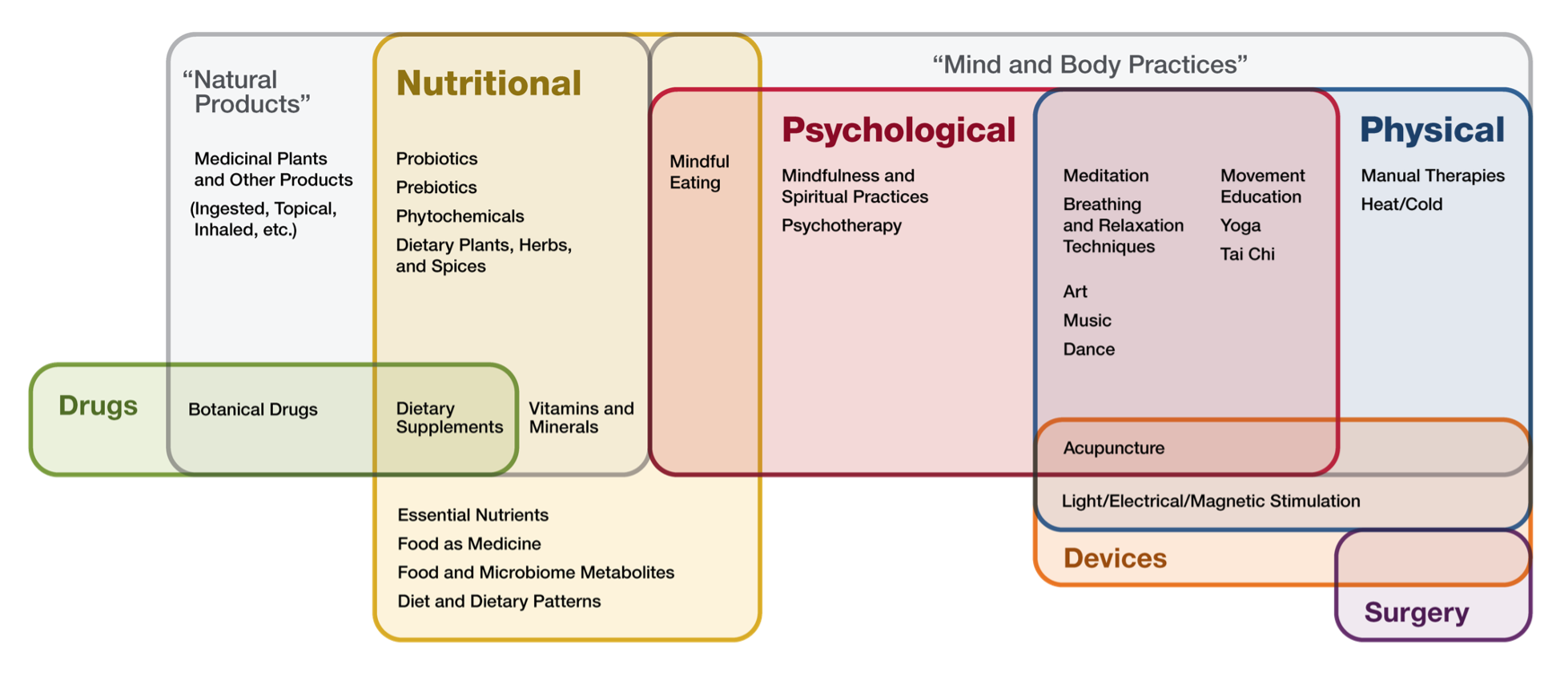
FIGURE 1. COMPLEMENTARY HEALTH APPROACH. IMAGE SOURCE: https://files.nccih.nih.gov/nutritional-psychological-physical-venn-diagram-08-01-crop.png.
The complementary health approach classification system has its limitations and, therefore, a five-domain system has been proposed to organize CIH. The domains include: (1) biologically based therapies; (2) mind-body interventions; (3) manipulative and body-based therapies; (4) alternative or whole medical systems; and (5) energy therapies. Examples of each domain are provided in Table 3 (1, 2).
|
Table 3. Domains to Classify Complementary and Integrative Health
|
|
Domain
|
Examples
|
|
Biologically based therapies
|
Dietary interventions, vitamins, minerals, supplements, herbal/botanical medicines
|
|
Mind–body interventions
|
Meditation, relaxation and breathing techniques, guided imagery, hypnosis, biofeedback, yoga, Tai Chi, qigong, expressive arts therapies, spiritual practices, and other forms of “directed” attention
|
|
Manipulative and body-based methods
|
Osteopathic manipulative therapy, chiropractic spinal manipulation, massage therapy, physical therapy
|
|
Alternative or whole medical systems
|
Traditional Chinese medicine, ayurveda, naturopathic medicine, homeopathy, Polynesian medicine, Unani-Tibb medicine, traditional African medicine, traditional Mayan medicine
|
|
Energy therapies
|
Acupuncture, Tai Chi, qigong, reiki, therapeutic or healing touch, bioenergetic therapy, and other methods that affect the body’s “bioelectric” field
|
REGULATIONS OF DIETARY SUPPLEMENTS AND NATURAL PRODUCTS
The Dietary Supplement Health and Education Act (DSHEA) was passed in 1994. This legislation created the category of dietary supplements. Prior to DSHEA, natural products (herbs, vitamins, minerals, probiotics) were classified as either food or drug. Even though natural products are biologically active, they are considered food products and are exempt from the same approval process as drugs (3).
Under DSHEA, natural product manufacturers are not allowed to sell any adulterated or misbranded product. Manufacturers are expected to ensure natural products are effective and safe. However, manufacturers are not required to provide proof of efficacy or safety before marketing and selling a particular product (3).
When DSHEA was passed, it required that good manufacturing practices (GMPs) be established. Several years later the Food and Drug Administration (FDA) provided these standards. The standards say products must be labeled correctly and be free of impurities or adulterants (4).
Labeling requirements for dietary supplements exist. Products sold as dietary supplements must contain certain informational pieces on their labels. Items such as the product name, the word “supplement”, the net content quantity, the name and place of business of the manufacturer/packer/distributor, directions for use, a “Supplement Facts” panel, and a listing of all nondietary ingredients must be included. Table 4 reviews labeling requirements and Figure 2 is an example “Supplement Facts” panel (5).
|
Table 4. Information Required to Appear on Dietary Supplement Labels (5)
|
|
Dietary Supplement Labeling Requirements
|
|
Product Name
|
|
The word “supplement” or a statement the product is a supplement
|
|
Net content quantity
|
|
Manufacturer’s, packer’s, or distributor’s name
|
|
Manufacturer’s, packer’s, or distributor’s place of business
|
|
Directions for use
|
|
“Supplement Facts” panel listing serving size, dietary ingredients, amount per serving size, and percent daily value (if established)
|
|
Nondietary ingredients such as fillers, artificial colors, sweeteners, binders,
|
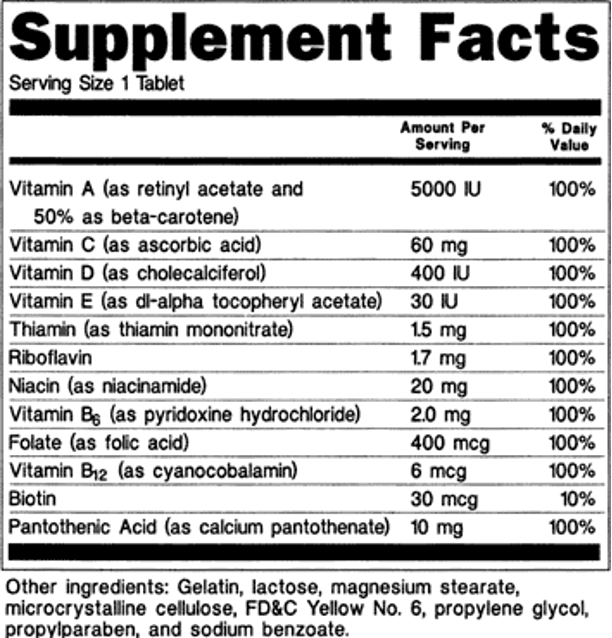
Figure 2. Example Supplement Facts Label. IMAGE SOURCE: https://www.fda.gov/food/guidanceregulation/guidancedocumentsregulatoryinformation/dietarysupplements/ucm070597.htm#4-59.
Dietary supplement products are allowed to make claims about maintaining structure or function of the body. However, products are not allowed to make claims about diagnosis, treatment, cure, or prevention of a disease. For example, a product may claim to “maintain a healthy pancreas.” Conversely, a product may not claim to “treat diabetes.” If a product does make a health maintenance claim, the label must include the following statement: “This statement has not been evaluated by the Food and Drug Administration (FDA). This product is not intended to diagnose, treat, cure, or prevent any disease” (5).
Reporting of Adverse Events
The Dietary Supplement and Nonprescription Drug Consumer Protection Act was signed into law in 2006. This act required manufacturers to report adverse events for dietary supplements and nonprescription drugs. In addition, individuals are encouraged to report supplement and nonprescription drug adverse events to the FDA (6). Despite the Dietary Supplement and Nonprescription Drug Consumer Protection Act, there is concern of underreporting of adverse events.
Safety Concerns
Safety surrounding dietary supplements and natural products is a concern for clinicians and consumers alike. Despite DSHEA and the Dietary Supplement and Nonprescription Drug Consumer Protection Act, adverse events and safety issues regarding natural products abound. In fact, an article published in 2015 in The New England Journal of Medicineestimated 23,005 emergency room visits annually were a result of adverse effects related to dietary supplements (7).
The FDA publishes recalls for prescription drugs, nonprescription drugs, and dietary supplements. The most serious recalls are classified as Class I. A Class I drug recall is one where the product in question has “reasonable probability that the use of or exposure to . . . will cause serious adverse health consequences or death” (8). From 2008 to 2012, half of all Class I drug recalls were from dietary supplements (9).
INTEGRATIVE HEALTH USE IN THE UNITED STATES
According to the Council for Responsible Nutrition (CRN), Americans spend an estimated $35 billion on dietary supplements each year. The market appears to be growing each year. CRN estimates 74% of adults in the United States use supplements (10).
Most users of natural products take a multivitamin. Other commonly used supplements include specific vitamins (D, C, and B), calcium, omega-3 fatty acids/fish oil, probiotics, green tea, protein bars, whey protein powders, and energy drinks. Mass merchandizers and pharmacies are the most common places where dietary supplements are purchased (10,11).
Reasons for dietary supplement use vary. The most popular was to support overall health and wellness. Other popular reasons include filling nutrient gaps, heart health, healthy aging, immune health, energy, bone health, preventing illness, and joint health (11).
Integrative Health Use Amongst Patients with Diabetes
According to a meta-analysis published in 2021, 51% of patients with diabetes globally use some form of CAM. Use prevalence was highest in Europe, where 76% of patients with diabetes in France use CAM; prevalence was lowest in North America, where 45% of patients used CAM (12). Reasons for CAM use specific to the United States (US) include overall wellness (28% of users), treatment of diabetes (15%), or a combination of the two (57%) (13). Figure 3 illustrates the reasons for CAM use in patients with diabetes.
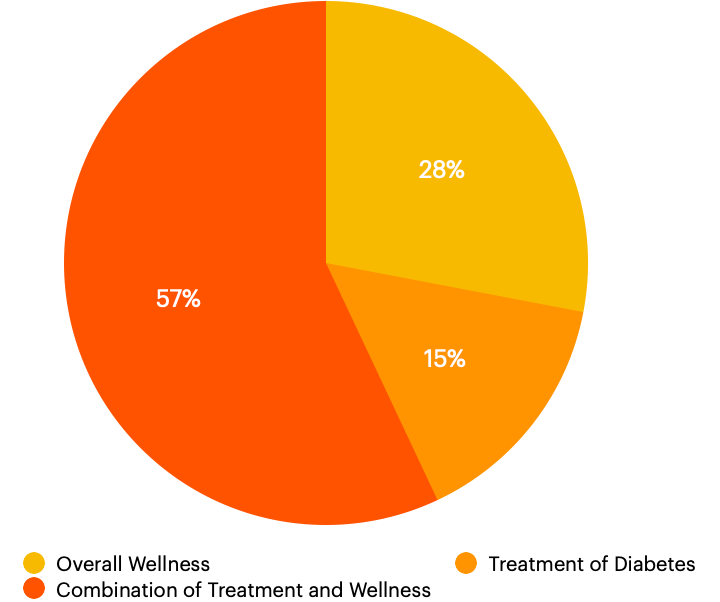
Figure 3. Reasons for CAM Use in Patients with Diabetes in the United States. Data Source (13).
Overall, the most common forms of CAM used in US patients with diabetes were herbal therapies (56.7% of users), chiropractic (25.3%), and massage (20.2%). See Figure 4 for an illustration. For those citing treatment alone as their reason for complementary health approach use, the most common types were chiropractic, herbal therapies, and massage. Those that cited wellness alone as their reason, the most common types of CAM utilized were herbal therapies, massage, and chiropractic (13).

Figure 4. Most Common Types of CAM Used in Patients with Diabetes. Data Source (13).
Sociodemographically, there are several differences in US CAM users with diabetes. The racial/ethnic group most likely to utilize complementary health approaches were non-Hispanic Whites. Those employed and with higher education attainment were also more likely to use CAM (13).
Due to the high usage, clinicians should gather comprehensive complementary health approach use histories from patients. This may prevent dangerous CAM-herb and CAM-disease interactions.
Older Adults
In particular, older adults with diabetes that utilize CAM can present unique challenges. Older adults tend to have more chronic medical conditions and diabetes complications. Additionally, older adults tend to use more medications compared to the general population.
One-quarter of older adults with diabetes utilize complementary, alternative, or integrative medicine (14). Of these older adults, 62.8% utilize herbal therapies specifically. Chiropractic (23.9%), massage (14.7%), acupuncture (10.2%), and yoga (5.2%) were the other most popular therapies used (14).
Clinicians should query older adult patients with diabetes on active CAM use. This may prevent dangerous CAM-herb and CAM-disease interactions.
NATURAL PRODUCTS
Natural products have been used for thousands of years. Natural products were depicted on clay tablets in ancient Mesopotamia (2600 BC). An ancient Egyptian pharmaceutical record, the Ebers Papyrus, dates to 2900 BC and documents hundreds of natural therapies. Documented records of natural product use have also been found in China (the Chinese Materia Medica) and Greece (from the physician Dioscorides) (15).
This section will discuss natural products that are commonly used in patients with type 2 diabetes mellitus (T2DM) for glycemic control. A brief description of each product will be followed by an overview of proposed mechanisms of action. Next, currently available evidence for the product will be reviewed. A brief discussion of adverse effects and drug interactions will also be included.
While botanical products are often hypothesized to work by multiple mechanisms, each product is categorized below by the major mechanism thought to exert its effect. For your convenience, an alphabetized table of botanical products is also presented.
Hypoglycemic Agents
The natural products covered in this section are all agents that theoretically lower blood glucose. Each individual product may have additional mechanisms of action, which are also covered.
BANABA (LAGERSTROEMIA SPECIOSA)
Banaba is a crepe myrtle species indigenous to Southeast Asia. The first published report of banaba use is from 1940. Banaba is used for diabetes and weight loss. See Figure 5 for an illustration of the banaba plant. The banaba leaf is the portion thought to exert beneficial effects. It is thought the active constituents of the banaba leaf are corosolic acid and ellagitannins (lagerstroemin, flosin B, and reginin A) (16-18).

Figure 5. Banaba Plant Image Source: https://commons.wikimedia.org/wiki/File:Inflorescence_of_Lagerstroemia_speciosa.JPG.
Mechanism of Action
It is hypothesized that banaba lowers blood glucose by increasing insulin secretion and stimulating glucose uptake of cells (insulin-like effect). Additional proposed mechanisms include alpha-glucosidase inhibition and subsequent reduction in nutrient load; increasing insulin sensitivity via increased expression of liver peroxisome proliferator‐activated receptor‐α (PPAR‐α) mRNA and adipose tissue peroxisome proliferator‐activated receptor‐γ (PPAR‐γ) mRNA; decreased gluconeogenesis; and increased glycolysis by increasing glucokinase activity (17-21).
Evidence
A randomized controlled trial studied the activity of 1% corosolic acid (an active constituent of banaba) on glucose control in patients with type 2 diabetes. This was a dose-response study of 10 subjects aged 55-70 years. Subjects did not use any oral hypoglycemic medications for 45 days prior to the clinical trial. Five subjects in each group received either a hard or soft gelatin capsule containing 1% corosolic acid of 16, 32, and 48 mg doses (equivalent to 0.16 mg, 0.32 mg, and 0.48 mg of corosolic acid). Doses were given sequentially with a 10-day wash-out period between dose escalation. Blood glucose levels were measured via finger-prick sample. Compared to control blood glucose levels, 1% corosolic acid from a soft gelatin capsule resulted in a statistically significant percent reduction in blood glucose levels at the 32 mg (10.7% ± 1.4) and 48 mg (30.0% ± 3.4) doses (p <0.05). The hard gel formulation resulted in a significant (p <0.05) percentage reduction in blood glucose for only the 48 mg dose (20.2% ± 1.29). Results are summarized in Table 5 (18).
|
Table 5. Percent Reduction in Basal Blood Glucose Levels in Patients with Type 2 Diabetes After 15 Days of Treatment with Different Doses of 1% Corosolic Acid (18)
|
|
Dosage Form
|
Dose of 1% Corosolic Acid (Equivalent Corosolic Acid Dose)
|
Percent Reduction in Blood Glucose Levels (± SD)
|
p-value
|
|
|
32 mg (0.32 mg)
|
10.7 ± 1.4
|
≤0.01
|
|
48 mg (0.48 mg)
|
30.0 ± 3.4
|
≤0.002
|
|
32 mg (0.32 mg)
|
6.5 ± 1.13
|
≤0.09
|
|
48 mg (0.48 mg)
|
20.2 ± 1.29
|
≤0.001
|
Fukushima and colleagues performed a double-blind cross-over design trial in 31 subjects with type 2 diabetes. In this study, subjects were not randomized. Subjects received oral supplementation with a 10 mg corosolic acid (an active constituent of banaba) capsule or placebo five minutes prior to a 75-g oral glucose tolerance test. The majority of the subjects had type 2 diabetes (n=19) while seven had impaired glucose tolerance, one had impaired fasting glucose, and four had normal glucose according to 1998 WHO criteria. Subjects with diagnosed hypertension, hepatic, or renal disease; engaged in heavy exercise; or took any medication were excluded. Thirty minutes after the oral glucose tolerance test, there were no differences in plasma glucose levels. The corosolic acid treatment group showed lower glucose levels from 60 minutes until 120 minutes after glucose administration. Statistical significance was reached at 90 minutes (p<0.05) (22).
Adverse Effects and Warnings
Banaba extract appears to be well tolerated when used orally. Dizziness, headache, tremor, weakness, diaphoresis, and nausea have been reported (23).
Interactions
As banaba lowers blood glucose, it should be used with caution in those using other hypoglycemic agents. Banaba may also lower blood pressure and may have an additive effect with other antihypertensives (18, 23).
Summary
Banaba is possibly effective for the treatment of type 2 diabetes. Data from human studies investigating banaba demonstrate corosolic acid’s potential to lower blood glucose. Small sample size and short study duration limit applicability of results.
BITTER MELON (MOMORDICA CHARANTIA)
Bitter melon is a plant cultivated in India, Asia, South America, and the Caribbean. Local nomenclature for Bitter melon varies – in India it is known as karela, bitter melon, and bitter gourd. It can also be known as wild cucumber, ampalaya, and cundeamor (24, 25). A member of the melon family, bitter melon is consumed in Asian cuisine and used both orally and topically. Uses for bitter melon include diabetes, cancer, and HIV (26). See Figure 6 for an image of the bitter melon plant.

Figure 6. Bitter Melon Plant Image Source: https://commons.wikimedia.org/wiki/File:Momordica_charantia_(Bitter_melon)_leaves_and_a_flower.jpg.
Mechanism of Action
It is thought bitter melon has insulin-like properties. More specifically, it is hypothesized bitter melon: 1) inhibits mitogen-activated protein kinases (MAPKs) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in pancreatic cells; 2) promotes glucose and fatty acid catabolism; 3) stimulates fatty acid absorption; 4) induces insulin production; 5) improves insulin resistance; 6) stimulates activated protein kinases (AMPK); and 6) inhibits fructose-1,6-bisphosphate and glucose-6-phosphatase (27, 28).
Evidence
In 2024, a meta-analysis was published that aimed to examine the impact of bitter melon on glycemic control and lipid profiles in individuals with T2DM. Eight randomized controlled trials were included (n=423). The authors found bitter melon was associated with significant reductions in fasting blood glucose (weighted mean distribution [WMD] of -15.3 mg/dL; 95% CI: -25.9 to -4.7; p = 0.005; I2= 73.4 %); postprandial glucose (WMD: -41.0 mg/dL; 95% CI: -60.3 to -21.8; p = 0.000; I2 = 66.9 %); and glycosylated hemoglobin A1c (HbA1c) (WMD: -0.38%; 95% CI: -0.53 to -0.23; p = 0.000; I2= 37.6 %). Total cholesterol was also significantly reduced (WMD: -6.8 mg/dL; 95% CI: -12.6 to -1.3; p = 0.017; I2 = 63.6 %). No significant differences were observed in terms for triglycerides (TG), high-density lipoproteins (HDL), and low-density lipoprotein (LDL) (29).
A 2019 meta-analysis was conducted to evaluate the efficacy of bitter melon in lowering elevated plasma glucose levels in patients with prediabetes and T2DM. Ten studies were included in the meta-analysis and ranged from 4 to 16 weeks in follow up. Overall, bitter melon lowered fasting plasma glucose by 13 mg/dL (95% CI: -23.9 to -2.2). Postprandial glucose decreased by 25.7 mg/dL (95% CI: -39.2 to -12.1) and HbA1c decreased by 0.26% (95% CI, -0.49 to 0.03). Of note, the studies had overall bias risk of moderate to high. The study authors rated the evidence quality low due to risk of bias and inadequate sample size (30).
A 2012 Cochrane Review of four randomized controlled trials found no statistically significant difference in glycemic control with bitter melon compared to placebo. Additionally, there was no significant change in glycemic control compared to metformin and sulfonylureas. Of note, no significant interactions were noted (31).
Adverse Effects and Warnings
Adverse effects reported include abdominal discomfort, pain, and diarrhea (32).
Bitter melon has been used as an abortifacient agent. Animal research confirms two proteins isolated from the plant possess abortifacient properties and may decrease fertility (33).
Interactions
As bitter melon may lower blood glucose, it should be used with caution in those using other hypoglycemic agents.
Bitter melon may increase levels of drugs that are P-glycoprotein substrates. For example, levels of apixaban, cimetidine, corticosteroids, diltiazem, erythromycin, fexofenadine, linagliptin, rivaroxaban, and verapamil may be increased with concurrent bitter melon use.
Those with G6PD deficiency should avoid use due to risk of developing favism (27).
Summary
Bitter melon is commonly used medicinally and in cuisine. There is mixed regarding bitter melon’s blood glucose lowering efficacy in patients with type 2 diabetes. Bitter melon has abortifacient properties and should be avoided in pregnancy.
FENUGREEK (TRIGONELLA FOENUM-GRAECUM)
Fenugreek, a member of the Fabaceae family, is an aromatic herb native to the Mediterranean region, northern Africa, southern Europe, and western Asia. The plant appears clover-like and its leaves are used in southeast Asian cuisine (see Figure 7 for an illustration). The seed is considered to be the pharmaceutically active portion of Trigonella foenum-graecum. If consuming fenugreek seeds, one must crush them to release the viscous gel fiber to increase efficacy. The flavor and fragrance of the seed is similar to maple syrup. Fenugreek seeds are approximately 50% fiber (30% soluble and 20% insoluble fiber) (21, 34).
Figure 7. Fenugreek Plant Image Source: https://en.wikipedia.org/wiki/Fenugreek.
Mechanism of Action
Fenugreek is thought to lower blood glucose via multiple mechanisms. As fenugreek seeds are fiber rich, it is thought this may slow postprandial glucose absorption (34, 35). Bioactive compounds in fenugreek include 4-hydroxyisoleucine (which accounts for the majority), saponins, alkaloids, and coumarins (36). 4-hydroxyisoleucine increases glucose-dependent insulin secretion in human beta-islet cells (21, 37). Fenugreek may play a role in regulating glucagon-like peptide-1 (GLP-1) through the action of an active compound N55, which is thought to bind to GLP-1 and enhance its potency in stimulating GLP-1 receptor signaling (35). Fenugreek, along with other herbal products used for the treatment of type 2 diabetes, contains biguanide-related compounds. In fact, the history of metformin can be traced back to the use of French lilac as herbal medicine in medieval Europe (38).
Evidence
A systematic review and meta-analysis published in 2023 evaluated fourteen trials (n=894) on the effect of fenugreek on hyperglycemia. Twelve of the trials included patients with T2DM; the two remaining trials included patients with pre-diabetes and that were classified as overweight, respectively. The meta-analysis demonstrated a non-significant reduction in fasting blood glucose levels (WMD -3.70 mg/dL; 95% CI: −27.02 to 19.62; p = 0.76) and postprandial blood glucose (WMD of −10.61 mg/dL; 95% CI: −68.48 to 47.26; p = 0.72). A significant reduction in HbA1c was seen (WMD of −0.88%; 95% CI: −1.49 to −0.27; p < 0.05) with fenugreek consumption. See Table 6 for a summary of the results of the meta-analysis (39).
|
Table 6. Fenugreek Meta-Analysis Results (39)
|
|
Parameter
|
Weighted Mean Difference
(95% CI)
|
p-value
Weighted Mean Difference
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
|
HbA1c
|
-0.88% (-1.49 to -0.27)
|
<0.05
|
74.9%
|
< 0.01
|
|
Fasting blood glucose
|
-3.70 mg/dL (−27.02 to 19.62)
|
0.76
|
99.4%
|
< 0.01
|
|
Postprandial glucose
|
−10.61 mg/dL (−68.48 to 47.26)
|
0.72
|
99.1%
|
< 0.01
|
An earlier meta-analysis evaluated ten trials of fenugreek found both fasting blood glucose and HbA1c significantly decreased with fenugreek compared to placebo. The weighted mean difference in HbA1c was -0.58% (95% CI: -0.99 to -0.17; p<0.05). The same analysis found a difference in fasting blood glucose of – 12.94 mg/dL (95% CI: -21.39 to -4.49; p<0.05) (40).
A study evaluating the effect of fenugreek on glycemic control enrolled 25 patients with T2DM. Baseline fasting glucose was less than 200 mg/dL in all study participants. Half of patients received 1 g of hydroalcoholic extract of fenugreek seeds while the other half received placebo. At the end of two months, the fenugreek group’s fasting and two hour post prandial glucose levels were not different from placebo. There was, however, a difference favoring fenugreek in area under the curve of blood glucose (2375 +/- 574 vs 27597 +/- 274) as well as insulin (2492 +/- 2536 vs. 5631 +/- 2428) (p < 0.001). Homeostatic model assessment for insulin resistance (HOMA-IR) was lower in the fenugreek group (86.3 +/- 32 vs. 70.1 +/- 52) and insulin sensitivity increased (112.9 +/- 67 vs 92.2 +/- 57) (p < 0.05) (41).
Adverse Effects and Warnings
Adverse effects of fenugreek include diarrhea, heartburn, and flatulence (34).
Fenugreek is aromatic and smells similar to maple syrup. Consumption prior to delivery may cause the neonate to have this odor, which may lead to confusion with maple syrup urine disease (42).
Patients with chickpea allergies should use fenugreek with caution as there is potential for cross-reactivity. Furthermore, fenugreek may cause uterine contractions and should be avoided in pregnancy (34).
Interactions
Fenugreek preparations may contain coumarin and increase the risk of bleeding with anticoagulants. Theophylline levels may be decreased with concomitant use. As fenugreek may lower blood glucose levels, it should be used cautiously with other agents that decrease glucose (34).
Summary
Fenugreek is a commonly used herb that is possibly effective for the treatment of hyperglycemia in patients with type 2 diabetes. Due to the risk of uterine contractions, fenugreek should not be used in pregnancy. Fenugreek may contain coumarin and should be used cautiously with anticoagulants.
GYMNEMA (GYMNEMA SYLVESTRE)
Gymnema is a plant native to tropical and subtropical regions of Asia, Africa, and Australia. Ayurvedic medicine has long utilized gymnema and its Hindi name is gurmar, which means “destroyer of sugar” (43). The pharmaceutically active parts of the gymnema plant are the leaves and roots. See Figure 8 for an image of the gymnema plant (44).

FIGURE 8. GYMNEMA PLANT IMAGE SOURCE: https://commons.wikimedia.org/wiki/File:Gymnema_sylvestre_R.Br_-_Flickr_-_lalithamba.jpg.
Mechanism of Action
The active constituents of gymnema appear to be gymnemosides, saponins, stigmasterol, and various amino acid derivatives. In terms of glucose lowering, it appears gymnema reduces intestinal absorption of glucose. It may also increase enzymes that promote cellular glucose update. It is hypothesized gymnema impacts beta cells – particularly by increasing cell quantity and by stimulation of insulin release (45-47).
Evidence
In 2023, a systematic review and meta-analysis of six studies sought to determine the impact of gymnema on various cardiometabolic risk factors, including glycemic control. Participants had T2DM, metabolic syndrome, and/or impaired glucose tolerance. Fasting blood glucose significantly changed by -4.96 mg/dL (CI 95%: -7.65 to -2.27, p<0.001). HbA1c had a non-significant change of -1.78% (95% CI: -4.09 to 0.53, p=0.131). Results are shown in Table 7 (48).
|
Table 7. 2023 Gymnema Systematic Review and Meta-Analysis Results (48)
|
|
Parameter
|
Weighted Mean Difference
(95% CI)
|
p-value
Weighted Mean Difference
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
|
HbA1c
|
-1.78% (-4.09 to 0.53
|
0.131
|
98.9%
|
< 0.01
|
|
Fasting blood glucose
|
-4.96 mg/dL (-7.65 to -2.27)
|
<0.001
|
97.0%
|
< 0.01
|
An earlier systematic review and meta-analysis of ten studies (n=419) aimed to determine the effect of gymnema on glycemic control in individuals with T2DM. The study found that gymnema significantly reduced fasting blood glucose by 1.57 mg/dl (CI 95%: -2.22 to -0.93, p < .0001) and postprandial blood glucose by 1.04 mg/dl (CI 95%: -1.53 to -0.54, p < .0001). HbA1c was reduced by 3.91% (CI 95%: -7.35 to -0.16%, p < .0001), however heterogeneity did not show significance (see Table 8) (49).
|
Table 8. 2021 Gymnema Systematic Review and Meta-Analysis Results (49)
|
|
Parameter
|
Weighted Mean Difference
(95% CI)
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
|
HbA1c
|
-3.91% (-7.35 to -0.16%,)
|
99%
|
>0.05
|
|
Fasting blood glucose
|
-1.57 mg/dl (-2.22 to -0.93
|
90%
|
< 0.01
|
|
Postprandial glucose
|
−1.04 mg/dl (-1.53 to -0.54)
|
80%
|
< 0.01
|
Gymnema was studied in 64 patients with type 1 diabetes in an open-label, non-randomized, controlled trial. The study group received 200 mg twice daily of a water-soluble extract of gymnema by mouth for 6 to 60 months. All participants remained on insulin throughout the duration of the study. Insulin requirements were reduced by 50% in those in the gymnema group. Additionally, blood glucose and HbA1c levels were reduced (50).
An open-label and non-randomized trial studied gymnema in 47 patients with T2DM. In addition to conventional oral hypoglycemic agents, 400 mg of a water-soluble extract of gymnema was administered for 18 to 20 months to 22 of the participants. At the conclusion of the study, the gymnema group had significantly lower fasting glucose (29% reduction, p<0.001). HbA1c levels were also significantly (p<0.001) lower in the gymnema group (baseline 11.9% to 8.48%). Results are summarized in Table 9 (51).
|
Table 9. Effect of Gymnema on Patients with Type 2 Diabetes Mellitus (51)
|
|
|
Gymnema Group
|
Control Group
|
|
|
Baseline
|
18-20 months
|
p-value
|
Baseline
|
18-20 months
|
p-value
|
|
Fasting blood glucose (mg/dL)
|
174 ± 7
|
124 ± 5
|
<0.001
|
150 ± 4
|
157 ± 4
|
>0.05
|
|
HbA1c (%)
|
11.91 ± 0.30
|
8.48 ± 0.13
|
<0.001
|
10.24 ± 0.15
|
10.47 ± 0.14
|
>0.05
|
Adverse Effects and Warnings
A case of acute hepatitis secondary to gymnema use has been reported (43).
Interactions
Gymnema may potentiate the effects of other agents that lower glucose. There is evidence to suggest gymnema can inhibit and induce certain liver enzymes. For example, gymnema may inhibit cytochrome P450 (CYP) 1A2 and increase circulating levels of medications such as clozapine, cyclobenzaprine, mexiletine, olanzapine, propranolol, theophylline, and zolmitriptan. Gymnema may induce CYP 2C9 and decrease levels of concurrently used medications such as nonsteroidal anti-inflammatory drugs, glipizide, losartan, and warfarin (51).
Summary
Gymnema has been long used in Ayurvedic medicine. It is thought to lower blood glucose via multiple mechanisms. There are minimal human studies evaluating gymnema’s efficacy, but preliminary evidence shows promise.
Insulin Sensitizers
The natural products contained in this section are known to be insulin sensitizers. This means they improve the sensitivity of cells to the effects of insulin. Products may also employ other mechanisms of action, which are addressed.
AMERICAN GINSENG (PANAX QUINQUEFOLIUS)
American ginseng is from the Panax genus. While named similarly, American ginseng (Panax quinquefolius) is different from Asian ginseng (Panax ginseng). American ginseng, as its name suggests, is found mostly in North America and is considered endangered in some areas (53). Currently in the United States, only eighteen states allow for the harvesting of American ginseng. See Figure 9 for an image of the American ginseng plant (54).

FIGURE 9. AMERICAN GINSENG PLANT IMAGE SOURCE: https://commons.wikimedia.org/wiki/File:American-ginseng-with-fruit.jpg.
Mechanism of Action
The root of the American ginseng plant is the portion that exerts a pharmaceutical effect. Saponins, specifically ginsenosides, are hypothesized to reduce glucose levels 55-57). The primary mechanism is thought to be insulin sensitization, but increased insulin secretion and prevention of beta-cell loss may also play a role (56, 57).
Evidence
Vuksan and colleagues performed a randomized, double-blind, cross-over trial to evaluate the efficacy and safety of American ginseng as complementary therapy in patients with T2DM using conventional therapy. Participants (n=24) received either 1 g of American ginseng extract or placebo for 8-weeks while continuing their original conventional therapies. After a 4-week washout period, participants were crossed over to the opposite 8-week treatment arm. American ginseng reduced HbA1c by 0.29% (p = 0.041) and plasma blood glucose by 12.8 mg/mL (p=0.008). The safety parameters studied, liver and kidney function, were not affected (58).
Vuksan and colleagues performed a randomized, single-blind, controlled study in ten patients to evaluate the efficacy of various doses of American ginseng. Participants received either placebo or 3, 6, or 9 g of American ginseng prior to a 25-g oral glucose challenge. Capillary glucose was measured during the study duration. Compared to placebo, American ginseng significantly decreased glucose (p<0.05) for the 3, 6, and 9 g doses. Glucose area under the curve was also reduced (19.7% for the 3 g dose, 15.3% for the 6 g dose, and 15.9% for the 9 g dose). There was no difference between the various American ginseng doses (56).
Due to concern over varying ginsenoside concentrations in different products, a follow up study was completed. The objective of this study was to determine the efficacy of American ginseng with a different ginsenoside profile. Twelve participants received 6 g of American ginseng or placebo after a 75-g oral glucose load. There was no significant difference in venous blood glucose levels at -40, 0, 15, 30, 45, 60, 90, and 120 minutes between the groups. There was also no difference in plasma insulin levels. The American ginseng used in this study contained 1.66% total ginsenosides with the breakdown being 0.90% protopanaxodiol ginsenosides (PPD) and 0.75% protopanaxatriol ginsenosides (PPT) (59).
Adverse Effects and Warnings
Headache is the most common side effect reported with American ginseng use (60).
American ginseng should not be confused for Asian ginseng (Panax ginseng), Siberian ginseng (Eleutherococcus senticosus) (53).
Interactions
American ginseng may stimulate immune function and may theoretically decrease the effect of immunosuppressants (61).
American ginseng can decrease the efficacy of warfarin. Concomitant use is not advised (62).
As American ginseng can lower glucose, it should be used cautiously with other glucose lowering agents (56).
Summary
Limited trials suggest American ginseng (Panax quinquefolius) may lower blood glucose. However, variability of product ginsenoside profile can impact efficacy. Concomitant warfarin use is not recommended due to decreased warfarin effectiveness. American ginseng is often confused with Asian (Panax ginseng) or Siberian ginseng (Eleutherococcus senticosus).
BANABA (LAGERSTROEMIA SPECIOSA)
Banaba is a natural product with multiple mechanisms. It was previously covered in the “Hypoglycemia Agents” section.
BERBERINE
Berberine is a bitter tasting plant alkaloid extracted from various plants. Goldenseal, goldthread, Oregon grape, European barberry, phellodendron, and tree turmeric are all sources. See Figure 10 for an image of European barberry, a source of berberine. Berberine is used for glucose lowering, dyslipidemia, hypertension, and infections (63, 64).

FIGURE 10. EUROPEAN BARBERRY (BERBERIS VULGARIS) – BERBERINE IMAGE SOURCE: https://commons.wikimedia.org/wiki/File:Berberis_vulgaris_%27Atropurpurea%27_003.JPG.
Mechanism of Action
Berberine’s glucose lowering properties are thought to be from increased insulin secretion, increased glycolysis, increased levels of GLUT-4 and GLP-1, activation of PPAR gamma receptors, and alpha-glucosidase inhibition (65-68).
Evidence
The effect of berberine on metabolic profiles in patients with T2DM was studied in a systematic review and meta-analysis. Thirty-seven randomized controlled trials with 3,048 patients were included. with at least 60 participants over the age of 18 were included. Compared to the control group, berberine was associated with a significant reduction in fasting plasma glucose (-14.8 mg/dL; 95% CI: -17.1 to -0.12.6; p<0.05), HbA1c (-0.63%; 95% CI: -0.72 to -0.53; p<0.05), and two-hour post prandial blood glucose (-20.9 mg/dL; 95% CI: -24.5 to -17.3; p<0.05). There was significant heterogeneity with each of the three outcomes. Adverse effects were included in 14 of the included studies. Analysis revealed incidence of adverse effects was lower with berberine compared to the control groups. Hypoglycemia was reported in nine studies with no significant difference between the berberine and control groups (fixed effects model; RR = 0.48; 95% CI: 0.21 to 1.08; p = 0.08). Results are presented in Table 10 (69).
|
Table 9. Berberine Systematic Review and Meta-Analysis Results (69)
|
|
Parameter
|
Weighted Mean Difference
(95% CI)
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
|
HbA1c
|
-0.63% (-0.72 to -0.53)
|
52%
|
< 0.001
|
|
Fasting blood glucose
|
14.8 mg/dL (-17.1 to -0.12.6)
|
60%
|
< 0.00001
|
|
Postprandial glucose
|
-20.9 mg/dL (-24.5 to -17.3)
|
68%
|
< 0.001
|
The analysis also concluded that berberine alone or in combination with oral hypoglycemic agents did not significantly increase the incidence of total adverse events (RR = 0.73; 95% CI: 0.55 to 0.97; p = 0.03) and the risk of hypoglycemia (RR = 0.48, 95% CI 0.21 to 1.08; p = 0.08).
Berberine has also been studied in a randomized controlled trial compared to metformin, a medication in the biguanide class. In this study, 36 patients that were recently diagnosed with T2DM were randomized to either berberine 500 mg three times daily or metformin 500 mg three times daily for three months. In the berberine group, HbA1c decreased from 9.5 ± 0.5% to 7.5 ± 0.4% (p<0.01). Fasting blood glucose changed from 191 ± 16 mg/dL to 124 ± 9 mg/dL (p<0.01). Postprandial glucose also decreased from 357 ± 31 mg/dL to 214 ± 16 mg/dL (p<0.01). These differences were similar to metformin. At the end of the trial, the HbA1c lowering effect of berberine was similar to metformin (70).
Adverse Effects and Warnings
Gastrointestinal side effects are most common with berberine (diarrhea, constipation, flatulence, abdominal pain, and vomiting).
Uterine contractions are an adverse effect of berberine. Berberine is also thought to cross the placenta and neonatal kernicterus may result when ingested during pregnancy. Use in pregnancy is not recommended. Berberine can be transferred through breastmilk (71, 72).
Interactions
Berberine may inhibit cytochrome P450 3A4, 2D6, and 2C9 and should be used cautiously with other agents that are substrates, inhibitors, or inducers of these hepatic enzymes. Of note, cyclosporine levels can be increased and concomitant use is not advised.
As berberine lowers glucose, caution should be exercised when used with other agents that lower glucose. Berberine may increase the risk of bleeding when used with anticoagulants (64).
Summary
Berberine is an alkaloid extract derived from various plants. There is evidence to suggest berberine lowers fasting glucose, postprandial glucose, and HbA1c. Berberine may cause uterine contractions and kernicterus so should be avoided during pregnancy. There is concern that berberine inhibits several CYP enzymes and may contribute to multiple drug interactions.
CHROMIUM
Chromium is a mineral essential to humans. It is found naturally in brewer’s yeast (where it was first discovered), oysters, mushrooms, liver, potatoes, beef, cheese, and fresh vegetables. Chromium exists in two valences – trivalent and hexavalent. Trivalent chromium (Cr+3 or Cr III) is the biologically active form found in food and supplements. Hexavalent chromium (Cr+6 of Cr VI) is a toxic manufacturing byproduct and may cause lung cancer, dermatologic issues, and perforated nasal septum with chronic exposure (73, 74). Chromium in this section will refer to the commercially available trivalent chromium.
Chromium may be referred to as glucose tolerance factor (74, 75). However, glucose tolerance factor is a complex that contains, amongst other molecules, chromium. There is an apparent association between low chromium levels and impaired glycemic control (76).
The Food and Nutrition Board of the Institute of Medicine determined there was not sufficient evidence to set an Estimated Average Requirement for chromium consumption. However, they did suggest Adequate Intake (AI) levels. For adults, the AI is 35 mcg per day for men and 25 mcg per day for women. Due to the fact few serious adverse effects are associated with excess chromium from food, there is no designated Tolerable Upper Intake Level (74).
Chromium is typically found in the chloride, nicotinate, and picolinate salt forms. It is thought the picolinate salt is absorbed by humans best (73, 77).
Mechanism of Action
The exact mechanism of chromium has not been elucidated. Chromium has an insulin sensitizing effect by reducing the content and activity of the tyrosine phosphatase PTP-1B (78). Alternatively, chromium might act directly on the insulin receptor (79, 80).
Evidence
Despite plausible mechanisms of action, there is mixed evidence surrounding chromium for the treatment of diabetes (75, 81).
A systematic review and meta-analysis of ten studies (n=509) was published in 2021 to determine the effect of chromium supplementation on blood glucose and lipid levels in patients with T2DM. The weighted mean difference in HbA1c indicated a significant reduction in HbA1c (-0.54%; 95% CI: -0.98 to -0.09; p = 0.02). No difference in fasting plasma glucose was found (-29.65 mg/dL; 95% CI: -68.62 to 9.31; p=0.14). There was no difference found in triglycerides, total cholesterol, low-density lipoproteins, and high-density lipoproteins. Results are summarized in Table 10 (82).
|
Table 10. 2021 Meta-Analysis Results of the Effect of Chromium on Glycemic Control in Patients with Diabetes (82)
|
|
Parameter
|
Weighted Mean Difference
(95% CI)
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
|
HbA1c
|
-0.54% (-0.98 to -0.09)
|
84%
|
< 0.01
|
|
Fasting blood glucose
|
-29.65 mg/dL (-68.62 to 9.31)
|
97%
|
< 0.00001
|
A meta-analysis of 28 randomized controlled studies aimed to investigate the effect of chromium on glycemic control in patients with T2DM was published in 2020. The revealed significant reductions in HbA1c (-0.71%; p = 0.004) and fasting blood glucose (-19.0 mg/dL; p = 0.030) with chromium use. Insulin levels (-12.35 pmol/L, p <0.001) and homeostatic model assessment for insulin resistance (HOMA-IR) were also significantly lower with chromium. HOMA-IR levels decreased by 1.53 (p <0.001). There was significant heterogeneity between studies for HbA1c, fasting blood glucose, insulin, and HOMA-IR. Results are shown in Table 11 (83).
|
Table 11. 2020 Meta-Analysis Results of the Effect of Chromium on Glycemic Control in Patients with Diabetes (83)
|
|
Parameter
|
Weighted Mean Distribution
(95% CI)
|
p-value Weighted Mean Distribution
|
Heterogeneity
(I2)
|
p-value Heterogeneity
|
p-value Begg’s test
|
|
HbA1c (%)
|
-0.71
(-1.19 to -0.23)
|
0.004
|
99.2%
|
<0.001
|
0.143
|
|
Fasting blood glucose (mg/dL)
|
-19.0
(-36.15 to -1.85)
|
0.030
|
99.8%
|
<0.001
|
0.086
|
|
Insulin level (pmol/L)
|
-12.35
(-17.86 to -6.83)
|
<0.001
|
98.1%
|
<0.001
|
0.363
|
|
HOMA-IR
|
-1.53
(-2.35 to -0.72)
|
<0.001
|
89.9%
|
<0.001
|
0.466
|
Another meta-analysis of 25 randomized controlled trials evaluating the efficacy of chromium supplementation was published in 2014. Of these trials, 22 studied chromium monosupplementation, while two trials studied chromium in combination with biotin and one trial studied chromium with vitamins C and E. Trial duration varied from four to 24 weeks. In the 14 included trials that assessed HbA1c, there was a statistically significant change of -0.55% (95% CI: -0.88 to -0.22). Twenty-four studies evaluated fasting glucose and the pooled mean change was -20.7 mg/dL (95% CI: -33.1 to -8.5). Monosupplementation with chromium significantly decreased triglycerides (-26.6 mg/dL; p=0.002) and increased high density lipoprotein concentration (4.6 mg/dL; p=0.01). There was no change in total cholesterol or low-density lipoprotein concentrations. The meta-analysis authors concluded chromium supplementation had favorable effects on HbA1c and fasting glucose in patients with diabetes (75). Results from this meta-analysis are presented in Table 12.
|
Table 12. Meta-Analysis Results of the Effect of Chromium on Glycemic Control in Patients with Diabetes (75)
|
|
Parameter
|
Pooled Mean Difference (95% CI)
|
Heterogeneity P-Value
|
|
HbA1c (%)
|
-0.55% (-0.88 to -0.22)
|
<0.00001
|
|
Fasting glucose (mg/dL)
|
-20.7 mg/dL (-33.1 to -8.5)
|
<0.00001
|
A meta-analysis from 2002 of 15 randomized controlled trials was conducted to determine the efficacy of chromium on glycemic control. Doses in the included trials ranged from 10 to 1,000 micrograms of chromium daily and varied in terms of source (brewer’s yeast, chromium chloride, chromium nicotinate, chromium picolinate, or chromium-niacin). Study duration ranged from one to 16 months. In terms of fasting glucose, 14 studies and 463 patients were included (n=38 with diabetes and n=425 without diabetes). For all included patients, the fasting glucose pooled mean difference was 0.5 mg/dL (95% CI: -1.6 to 2.7) with no evidence of heterogeneity (p=0.97). The effect of chromium supplementation on two-hour OGTT results were included in five of the 14 trials. The majority of patients did not have a diabetes diagnosis (n=133 versus 8 with a diabetes diagnosis). The pooled mean difference was 4.7 mg/dL (95% CI, -4.3 to 13.7) with no evidence of heterogeneity (p=0.98). Fasting insulin levels were recorded in 10 of the studies (8 patients with diabetes and 326 without diabetes). The pooled mean difference in fasting insulin with chromium use was 0.28 pmol/L (95% CI, -7.0 to 7.5, heterogeneity p=0.097). Three of the trials assessed HbA1c (33 healthy subjects, 24 with glucose intolerance, and 155 with diabetes). There was no association between chromium supplementation and HbA1c in the study of healthy subjects. The single study that included subjects with glucose intolerance showed chromium supplementation was associated with a nonsignificant reduction in HbA1c (mean difference -0.30%; 95% CI: -0.86 to 0.25). The study that included subjects with diabetes showed a reduction in HbA1c for different chromium doses (mean difference for 1000 micrograms: -1.90%; 95% CI: -2.34 to -1.46; mean difference for 200 micrograms: -1.00%; 95% CI: -1.55 to -0.45). Data from the meta-analysis is presented in the following table. The authors of the meta-analysis concluded there was no effect of chromium on glucose or insulin concentrations in subjects without diabetes. The data for those with diabetes was inconclusive. Table 13 summarizes these results (81).
|
Table 13. Meta-Analysis Results of the Impact of Chromium on Glycemic Control (81)
|
|
Parameter
|
Number of Studies
|
N Patients with Diabetes
|
N Patients without Diabetes
|
Chromium Supplementation Pooled Mean Difference (95% CI)
|
Heterogeneity p Value
|
|
Fasting glucose (mg/dL)
|
14
|
38
|
425
|
0.5 (-1.6 to 2.7)
|
0.97
|
|
2-hour glucose tolerance test (mg/dL)
|
5
|
8
|
133
|
4.7 (-4.3 to 13.7)
|
0.98
|
|
Fasting insulin (pmol/L)
|
10
|
8
|
326
|
0.28 (-7.0 to 7.5)
|
0.097
|
Adverse Effects and Warnings
Trivalent chromium has demonstrated safety in large doses (74, 75). The picolinate form may cause cognitive, perceptual, and moto dysfunction (84).
Hexavalent chromium is toxic and is listed as a known carcinogen (74).
Interactions
There is a theoretical interaction between chromium and iron (74).
Summary
Chromium is a mineral essential to humans and may be referred to as glucose tolerance factor. There is conflicting evidence in terms of efficacy. Meta-analyses published in 2020 and 2014 suggested chromium decreased HbA1c and fasting glucose in patients with diabetes. Another meta-analysis published in 2002 found chromium to have no impact on glycemic control in those without diabetes.
CINNAMON (CINNAMOMUM AROMATICUM, CINNAMOMUM CASSIA)
Cinnamon is a natural product derived from the dried inner bark of the evergreen tree. It is commonly used in many cuisines. Cinnamon commonly found in grocery stores for culinary purposes is usually Cinnamomum cassia, but may be Ceylon cinnamon (85, 86).
Mechanism of Action
Procyanidin polymers appear to be responsible for cinnamon’s insulin sensitizing actions. It is hypothesized that these compounds increase phosphorylation of the insulin receptor, therefore, increasing sensitivity to insulin. Cinnamon may also stimulate insulin release and increase GLP-1 and GLUT-4 levels. Evidence also suggests cinnamon increases cellular glucose uptake (87-90).
Evidence
Cinnamon has shown mixed results in various trials in patients with diabetes (91-94).
In 2012, a Cochrane Review was published evaluating the effectiveness of cinnamon in patients with T2DM. The primary outcomes included fasting glucose, postprandial glucose, and adverse effects. Change in HbA1c was a secondary outcome. There was no change in fasting glucose (-1.4 mg/dL; 95% CI: -6.1 to 3.2), post-prandial glucose (-7.0; 95% CI: -14.9 to 0.9), or HbA1c (-0.06%; 95% CI: -0.29 to 0.18]. There was no difference in adverse effects between users and non-users of cinnamon (OR 0.83; 95% CI: 0.22 to 3.07), p = 0.77; n = 264; 4 trials). Table 14 presents the results from this review (95).
|
Table 14. Cochrane Review Results of the Effect of Cinnamon on Glycemic Control in Patients with Diabetes (95)
|
|
Parameter
|
Weighted Mean Distribution
(95% CI)
|
p-value Weighted Mean Distribution
|
Heterogeneity
(I2)
|
Number of Trials
|
|
HbA1c (%)
|
-0.06%
(-0.29 to 0.18)
|
0.63
|
0%
|
6
|
|
Fasting blood glucose (mg/dL)
|
-1.4
(-6.1 to 3.2)
|
0.06
|
0%
|
8
|
|
Postprandial glucose (mg/dL)
|
-7.0
(-14.9 to 0.9)
|
0.08
|
n/a
|
1
|
More recently, a systematic review and meta-analysis by Moridpour and colleagues was published in 2024 that aimed to assess the effects of cinnamon supplementation in managing glycemic control in patients with T2DM. Twenty-four randomized controlled trials were included. The pooled results indicated cinnamon had a statistically significant (p<0.05) reduction in fasting blood glucose (-1.32 mg/dL; 95 % CI: -1.77 to -0.87; p<0.001), HbA1c (-0.67%; 95 % CI: -1.18 to -0.15; p=0.011), and HOMA-IR (-0.44; 95 % CI: -0.77 to -0.10; p<0.001). Results are summarized in Table 15 (94).
|
Table 15. Systematic Review and Meta-Analysis of the Effect of Cinnamon on Glycemic Control in Patients with Diabetes (94)
|
|
Parameter
|
Weighted Mean Distribution
(95% CI)
|
p-value Weighted Mean Distribution
|
Heterogeneity
(I2)
|
p-value
Heterogeneity
|
Number of Trials
|
|
Fasting blood glucose (mg/dL)
|
-1.32
(-1.77 to -0.87)
|
<0.001
|
94.0%
|
<0.001
|
23
|
|
HbA1c (%)
|
-0.67%
(-1.18 to -0.15)
|
0.011
|
94.7%
|
<0.001
|
18
|
|
HOMA-IR
|
-0.44
(-0.77 to -0.10)
|
<0.001
|
79.1%
|
<0.001
|
8
|
A systematic review and meta-analysis by Deyno and colleagues was published to evaluate the efficacy of cinnamon in patients with type 2 diabetes mellitus and pre-diabetes. Sixteen randomized controlled studies were included in the meta-analysis. There was no significant change in weighted mean difference of HbA1c and lipid profiles. There was, however, a statistically significant difference in fasting blood glucose and HOMA-IR. High heterogeneity was observed in the included studies and cinnamon doses ranged from 1 g to 14.4 g a day. Results can be found in Table 16 (96).
|
Table 16. Meta-Analysis Results for the Effect of Cinnamon on Glycemia and Lipoprotein Levels (96)
|
|
Parameter
|
Weighted Mean Difference (95% CI)
|
Heterogeneity
(I2)
|
|
Glycemic
|
|
|
Fasting plasma glucose (mg/dL)
|
-9.8 (-16.4 to -3.2)
|
83.6%
|
|
HbA1c (%)
|
-0.104 (-0.138 to 0.110)
|
69.6%
|
|
HOMA-IR
|
-0.714 (-1.388 to -0.04)
|
84.4%
|
|
Lipoprotein
|
|
|
Total Cholesterol (mg/dL)
|
-3.6 (-7.3 to 0.2)
|
86.4%,
|
|
Low density lipoprotein concentration (mg/dL)
|
-2.1 (-4.9 to 0.7)
|
86.0%
|
|
High density lipoprotein concentration (mg/dL)
|
-0.1 (-1.1 to 0.9)
|
81.0%
|
|
Triglycerides (mg/dL)
|
-1.8 (-4.0 to 0.4)
|
69.0%
|
A randomized, double-blind, placebo-controlled trial evaluated the effect of cinnamon on 66 Chinese patients with type 2 diabetes (HbA1c greater than 7% and fasting glucose greater than 144 mg/dL). Patients were not receiving insulin or other glucose-lowering agents aside from glicazide, a sulfonylurea, of which all participants were taking 30 mg daily. Patients were randomized to 120 mg daily of cinnamon, 360 mg daily of cinnamon, or placebo for 12 weeks. Both the 120 mg and 360 mg cinnamon groups experienced significantly lower HbA1c and fasting plasma glucose measurements. There was no significant change in either parameter for the placebo group. Results are provided in Table 17 (97).
|
Table 17. Effect of Various Cinnamon Doses on Glycemic Parameters (97)
|
|
|
Cinnamon 120 mg Daily Group
|
Cinnamon 360 mg Daily Group
|
Placebo Group
|
|
Change in HbA1c (%) (95% CI)
|
-0.67 (-1.1 to -0.25)
|
-0.93 (-1.38 to -0.47)
|
0.00 (-0.61 to 0.61)
|
|
Change in fasting plasma glucose (mg/dL) (95% CI)
|
-18.4 (-29.0 to -7.57)
|
-29.2 (-41.8 to -16.8)
|
-3.96 (-24 to 16)
|
Adverse Effects and Warnings
Cinnamon is typically tolerated well (98-100).
Cinnamon is a natural source of coumarin and use therefore presents a theoretical risk of hepatic injury (101).
Interactions
Due to concerns of hepatic injury when large doses of cinnamon are used, use cautiously with other hepatotoxic agents.
Cinnamon may decrease glucose levels and should be used cautiously with other agents that lower glucose.
Summary
Cinnamon is derived from the dried inner bark of evergreen trees and is commonly used as a spice in cuisine. In terms of glycemic lowering, cinnamon studies have shown varying results. A 2012 Cochrane review found cinnamon to be no more effective than placebo; a 2024 meta-analysis found cinnamon to have statistically significant improvements on fasting plasma glucose and HbA1c. Cinnamon is typically well tolerated.
GYMNEMA (GYMNEMA SYLVESTRE)
Gymnema has multiple mechanisms of action is addressed under the “Hypoglycemic Agents” section.
MILK THISTLE (SILYBUM MARIANUM)
Milk thistle (Silybum marianum) is a member of the aster family which also includes daisies and thistles (102). The plant itself is edible and was native to Europe before introduction to North America. Currently, milk thistle is found in Europe, North America, India, China, South America, Africa, and Australia (103).
Milk thistle has a long history of medicinal use. Use dates back to the time of ancient Greece. Milk thistle is used for diabetes, liver support, and menstrual support. An image of the milk thistle plant can be found in Figure 11 (103, 104).

FIGURE 11. MILK THISTLE PLANT IMAGE SOURCE: https://commons.wikimedia.org/wiki/File:(1)_Milk_thistle.jpg.
Mechanism of Action
The mechanism of action of milk thistle for glycemic control is not fully understood. The pharmaceutically active portions of the plant are the seeds and the above ground portions. Milk thistle seed extract is comprised primarily (up to 80%) of silymarin. Silymarin contains various flavonolignans, the most active being silybin or silibinin (102, 103, 105, 106).
Silymarin decreases insulin resistance and may have a protective pancreatic effect through a mechanism thought to involve antioxidant properties (107, 108). Some studies suggest that silymarin may also regenerate pancreatic beta cells and enhance insulin sensitivity of liver and muscle cells (106). Carbohydrate-induced glycolysis is decreased by silibinin through pyruvate kinase inhibition (103, 109).
Evidence
Milk thistle has been studied in randomized controlled trials. Soleymani and colleagues published a meta-analysis of 30 studies to determine the effects of milk thistle on cardiometabolic syndrome. Adults (with and without diabetes) were included. The study demonstrated that milk thistle significantly reduced the levels of fasting plasma glucose (WMD: -17.96 mg/dL; 95% CI: -32.91 to -3.02); HbA1c (WMD: -1.25%; 95% CI: -2.34 to -0.16); total cholesterol (WMD: -17.46 mg/dL; 95% CI: -30.98 to -3.95); triglycerides (WMD: -25.70 mg/dL; 95% CI: -47.23 to -4.17); low-density lipoproteins (WMD: -10.53 mg/dL; 95% CI: -19.12 to -1.94). High-density lipoprotein levels increased (WMD: 3.36 mg/dL; 95% CI: 0.88 to 5.84). There was no difference in BMI. The majority of patients included in the meta-analysis had type 2 diabetes mellitus. Results are summarized in Table 18 (106).
|
Table 18. Effect of Various Cinnamon Doses on Glycemic Parameters (106)
|
|
Parameter
|
Weighted Mean Difference (95% CI)
|
p-value
Weighted Mean Difference
|
Heterogeneity
(I2)
|
p-value
Heterogeneity
|
|
Glycemic
|
|
Fasting plasma glucose (mg/dL)
|
-17.96
(-32.91 to -3.02)
|
<0.05
|
82.4%
|
<0.001
|
|
HbA1c (%)
|
-1.25% (-2.34 to -0.16)
|
<0.05
|
92.9%
|
<0.001
|
|
Lipoprotein
|
|
Total Cholesterol (mg/dL)
|
-17.46 (-30.98 to -3.95)
|
<0.05
|
62.9%
|
0.006
|
|
Low density lipoprotein concentration (mg/dL)
|
-10.53 (-19.12 to -1.94)
|
<0.05
|
37.5%
|
0.119
|
|
High density lipoprotein concentration (mg/dL)
|
3.36 (0.88 to 5.84)
|
<0.05
|
81.0%
|
|
|
Triglycerides (mg/dL)
|
-25.70 (-47.23 to -4.17)
|
<0.05
|
54.3%
|
0.025
|
|
Body Mass Index (kg/m2)
|
0.07 kg (-0.7, 0.83)
|
>0.05
|
0%
|
0.94
|
In 2020, a meta-analysis was published to evaluate the efficacy and safety of milk thistle in patients with glucose or lipid metabolic dysfunction. Sixteen studies (n=1358) were included in the analysis. Fasting blood glucose levels and HbA1c were reduced significantly in milk thistle users compared to placebo. There was no difference between the groups in liver enzymes, creatinine phosphokinase, or creatinine. Results are shown in Table 19 (110).
|
Table 19. Effect of Silymarin on Glucose and Lipid Parameters (110)
|
|
Parameter
|
Weighted Mean Difference (95% CI)
|
p-value
|
|
Glycemic
|
|
Fasting plasma glucose (mg/dL)
|
-22.9 mg/dL (-32 to -13.7)
|
<0.001
|
|
HbA1c (%)
|
-1.88 (-2.57 to -1.20)
|
<0.001
|
A meta-analysis of trials was published in 2011 that evaluated the impact of milk thistle on glycemic control in patients with type 2 diabetes. Two studies (n=89) were identified that met analysis criteria. The mean pooled difference in fasting glucose was -38.1 mg/dL (95% CI: -66.6 to -9.5). The mean pooled difference in HbA1c was -1.92% (95% CI: -3.32 to -0.51). Heterogeneity for both had p-values of less than 0.05. The authors concluded milk thistle may improve glycemic control in patients with type 2 diabetes.
The two studies included in the aforementioned meta-analysis each individually showed statistically significant change in fasting glucose as well as HbA1c (107, 111). Results are shown in the Tables 20 and 21.
|
Table 20. The Effect of Milk Thistle on Glycemic Control (107)
|
|
Parameter
|
Milk Thistle Treatment (SD)
|
Control (SD)
|
P-Value
|
|
Fasting glucose (mg/dL)
|
133 (39)
|
188 (48)
|
0.001
|
|
HbA1c (%)
|
6.8 (1.1)
|
9.5 (2.2)
|
0.001
|
|
Table 21. The Effect of Milk Thistle on Fasting Glucose and HbA1c (111)
|
|
Parameter
|
Milk Thistle Treatment (SD)
|
Control (SD)
|
P-Value
|
|
Fasting glucose (mg/dL)
|
167.58 (9.9)
|
193.14 (16.1)
|
<0.01
|
|
HbA1c (%)
|
7.45 (0.8)
|
8.71 (0.63)
|
<0.05
|
Milk thistle has also been studied in combination with berberine. The combination of the two was more effective than berberine alone in reducing HbA1c in type 2 diabetes patients (112).
Adverse Effects and Warnings
Milk thistle is typically well tolerated. Side effects include nausea, diarrhea, and abdominal bloating (113).
As milk thistle is a member of the aster family, cross reactivity may exist with other plants. Those with a daisy or ragweed allergy may experience a cross reaction with milk thistle use (102).
Interactions
Milk thistle may inhibit certain cytochrome P450 isoenzymes. The isoenzymes 2C8, 2C9, 2D6, 3A4, and 3A5 may all be inhibited with concomitant use (114, 115).
As milk thistle may lower glucose levels, it should be used cautiously with other hypoglycemia agents. Increased warfarin levels may occur with concomitant use (116).
Summary
Milk thistle is a member of the aster family and is used for lowering glucose, liver support, and menstrual support. There is modest evidence to suggest milk thistle may lower glucose in patients with diabetes.
PRICKLY PEAR CACTUS (OPUNTIA FICUS-INDICA AND OTHER OPUNTIA SPECIES), NOPAL
Prickly pear cactus is native to Mexico and found widely in the southwestern United States, Africa, Australia, and the Mediterranean. The berries of the cactus are oval, edible, and may vary in color (117, 118). Prickly pear cactus has been used historically in Mexican cultures as a treatment for type 2 diabetes. Figure 12 illustrates the prickly pear cactus plant (119).

FIGURE 12. PRICKLY PEAR CACTUS PLANT IMAGE SOURCE: https://commons.wikimedia.org/wiki/File:Prickly_pear_cactus_in_Texas.jpg.
Mechanism of Action
Much of the prickly pear cactus plant is pharmaceutically active – the leaves, flowers, stems, and fruit are all thought to exert an effect. The plant contains carbohydrate, protein, fat, and fiber (120). It is thought prickly pear cactus lowers glucose by acting as an insulin sensitizer and by slowing carbohydrate absorption (117, 120, 121).
Evidence
A systematic review that included 20 articles investigated the effects of various parts of the prickly pear cactus plant on glucose and insulin. Studies that used prickly pear cactus fruit generally did not impact serum glucose or insulin. Studies that used the cladode portion of the plant (flat, leaf-like stem) predominantly demonstrated reductions in glucose and insulin (122).
A randomized, double-blind, placebo-controlled study was conducted to evaluate and effect of prickly pear cactus in obese patients with pre-diabetes. Patients received either 200 mg of a proprietary prickly pear product (n=15) or placebo (n=14). Patients underwent two different oral glucose tolerance tests – one without prickly pear cactus to determine baseline values and one half an hour after prickly pear cactus ingestion. There was a significant difference (p<0.05) in plasma glucose concentrations at 60, 90, and 120 minutes following the glucose tolerance test for the prickly pear cactus group. There was no difference in HbA1c, insulin levels, high sensitivity C-Reactive Protein, body weight, or fat mass. There was also no difference in comprehensive metabolic profile parameters (123).
Most prickly pear cactus trials were published in Spanish only with abstracts available in English. Two trials showed a decrease in postprandial glucose from prickly pear administration (124, 125). Another trial showed when added to a high-carbohydrate or high-soy-protein breakfast, prickly pear cactus decreased glucose area under the curve (126).
Adverse Effects and Warnings
Prickly pear cactus is generally tolerated well when used orally. Side effects include nausea, diarrhea, and headache (120).
Interactions
Prickly pear cactus may lower glucose levels and should be used cautiously with other agents that impact glycemic control (123, 126).
Summary
Prickly pear cactus was used historically in Mexican cultures and is gaining popularity. There is preliminary data to suggest prickly pear cactus may be effective in lowering glucose. Some parts of the plant, i.e., the cladode, may be more efficacious than others, i.e., fruit. However, more studies are needed to determine efficacy. Prickly pear cactus is usually well tolerated.
SOY (GLYCINE MAX)
Soy comes from the soybean, a legume originating from Asia. In fact, prior to the 1950s soybean was seldom grown outside of the region. Now soybeans are grown in other regions such as North and South America. Soybeans are used in various food preparations such as edamame, tofu, and soymilk. An image of a soybean plant can be found in Figure 13 (127.

FIGURE 13. SOYBEAN PLANT IMAGE SOURCE: https://pixabay.com/en/soy-soybean-nature-green-998566/.
Mechanism of Action
The portion of soy that is pharmaceutically active is the bean. Soybeans are protein-rich and contain calcium, iron, potassium, amino acids, vitamins, and fiber (128). Soybeans contain phytoestrogens (isoflavones and lignans) and phytosterols, which are biologically active (129. 130).
Soy works via various mechanisms. Soy has insulin sensitizing properties and may slow carbohydrate absorption due to its fiber content. It is hypothesized that the fiber content of soy helps reduce glucose levels (128, 131).
Evidence
Results on the efficacy of soy in type 2 diabetes are conflicting. A systematic review and meta-analysis summarizing the association of soy intake and the risk of type 2 diabetes was published in 2020. Fifteen studies were included (n = 565,810) and multivariable-adjusted relative risks were determined. The relative risk of incidence of type 2 diabetes was 0.83 (95% CI: 0.68 to 1.01; p>0.05) for total soy. The relative risk for soy milk was 0.89 (95% CI: 0.71 to 1.11; p>0.05); tofu was 0.92 (95% CI: 0.84 to 0.99; p<0.05); soy protein was 0.84 (95% CI: 0.75 to 0.95; p<0.05); and soy isoflavones was 0.88 (95% CI: 0.81 to 0.96; p<0.05). High heterogeneity was observed in the total soy (I2 = 90.8%) and soy milk (I2= 91.7%) categories. Inverse linear associations were observed for the tofu, soy protein, and soy isoflavone groups. The quality of evidence was rated as low for the total soy, soy milk, tofu, soy protein, and soy isoflavone groups. The study authors suggested dietary intake of tofu, soy protein, and soy isoflavones are inversely associated with type 2 diabetes incidence. They found no association between total soy intake and incidence of type 2 diabetes. The authors cautioned that the overall quality of evidence was low (132).
In 2020, Zuo and colleagues conducted a systematic review and meta-analysis regarding the intake of soy and the association with type 2 diabetes mellitus and cardiovascular disease events. The review included 29 studies with 16,521 individuals with T2DM and 54,213 individuals with cardiovascular disease events. The follow up duration of the studies ranged from 2.5 to 24 years. In groups with the highest soy consumption compared to the lowest soy consumption, there was a significant reduction in the risk of T2DM (17%; total relative risk [TRR] = 0.83; 95% CI: 0.74 to 0.93); cardiovascular disease events (13%; TRR = 0.87; 95% CI: 0.81 to 0.94); coronary heart disease (21%; TRR = 0.79; 95% CI: 0.71 to 0.88); and stroke (12%; TRR = 0.88; 95% CI: 0.79 to 0.99). Daily tofu intake of 26.7 g of reduced cardiovascular disease event risk by 18% (TRR = 0.82; 95% CI: 0.74 to 0.92) and daily intake of 11.1 g of natto lowered cardiovascular disease event risk by 17% (TRR = 0.83; 95% CI: 0.78 to 0.89). The authors concluded that soy consumption was negatively associated with the risk of developing T2DM and cardiovascular disease events (133).
In 2018, a meta-analysis was published that aimed to evaluate the efficacy of soy in preventing diabetes. Eight studies were included in the meta-analysis. Soy intake decreased the risk of type 2 diabetes with an overall risk reduction of 0.77 (95% CI: 0.66 to 0.97). Soy protein and isoflavone intake lowered the risk of diabetes with risk reduction of 0.77 (95% CI: 0.80 to 0.97). A subgroup analysis looked at the relationship of soy intake in women and Asian populations. Women had a risk reduction of 0.65 (95% CI: 0.49 to 0.87) and Asian populations had a risk reduction of 0.73 (95% CI: 0.61 to 0.88). The study authors concluded soy intake may be associated with a decreased risk of type 2 diabetes (134).
Yang and colleagues performed a meta-analysis evaluating the impact of soy on glycemic control and lipoproteins in patients with type 2 diabetes. Eight studies were included and found there was no association between soy consumption and fasting glucose and HbA1c. There was, however, a significant reduction in serum cholesterol, triacylglycerol, and LDL-C, associated with soy use (p<0.001 for all). The authors concluded there was no significant effect of soy on fasting glucose, insulin, or HbA1c, but there was a favorable effect on serum lipids (135).
Another meta-analysis was published in 2011 examining the impact of soy intake on glycemic control. Twenty-four trials were included (n=1,518). The pooled mean change in fasting glucose was -0.69 mg/dL (95% CI, -1.65 to 0.27). Fasting insulin concentrations decreased by 0.18 mg/dL (95% CI, -0.70 to 0.34). The authors concluded there was no significant overall effect of soy on fasting glucose and insulin, but there was a favorable change in studies that used whole soy foods or soy diet (136).
Adverse Effects and Warnings
Soy is generally well tolerated with side effects being nausea, diarrhea, and bloating. There is concern that soy may alter thyroid function, but this appears to occur in those with iodine deficiency (137-139).
Interactions
Fermented soy products such as tofu may contain small amounts of tyramine. Tyramine should be avoided in those using monoamine oxidase inhibitors (140).
Summary
Soy comes from the soybean plant and contains phytoestrogens and phytosterols. There is some data to suggest soy consumption may decrease the risk of type 2 diabetes. Two meta-analyses showed soy did not significantly decrease fasting glucose of HbA1c in patients with type 2 diabetes.
VANADIUM
Vanadium is a mineral found in food sources such as mushrooms, shellfish, black pepper, parsley, dill seed, and certain prepared foods. Beer and wine are also sources. Grains account for 13 to 30 percent of vanadium in adult diets (74).
Mechanism of Action
Vanadium increases sensitivity to insulin and may mimic insulin’s actions. It may stimulate glucose oxidation and transport, stimulate hepatic glycogen synthesis, inhibit hepatic gluconeogenesis, and increase glucose uptake. Vanadium inhibits phosphotyrosine phosphatase enzymes which impact the insulin receptor (141, 142).
Evidence
A systematic review of five trials (n=48) evaluated vanadium’s impact in glycemic control. Doses varied from 50 mg to 300 mg of vanadium over three to six weeks. All trials reported reductions in fasting glucose values. However, none of the trials included controls (85).
A study of vanadium’s role in glycemic control enrolled 11 patients with type 2 diabetes. Patients were treated with 150 mg of vanadyl sulfate (a salt form of vanadium) for 6 weeks. Treatment with vanadyl sulfate decreased fasting glucose from 194 mg/dL ± 16 to 155 mg/dL ± 15. There was no change in body weight. Patients had an increased rate of hepatic glucose production (HGP) compared with controls (4.1 ± 0.2 vs. 2.7 ± 0.2 mg/kg lean body mass/min; p< 0.001), which was closely correlated with fasting glucose (r = 0.56; p< 0.006). Vanadyl sulfate reduced HGP by about 20% (P < 0.01), and the decline in HGP was correlated with the reduction in FPG (r = 0.60; p<0.05). Vanadyl sulfate also caused a modest increase in insulin-mediated glucose disposal (from 4.3 ± 0.4 to 5.1 ± 0.6 mg/kg lean body mass/min; p< 0.03), although the improvement in insulin sensitivity did not correlate with the decline in fasting glucose after treatment (r = -0.16; p>0.05). Thus, vanadyl sulfate at a dose of 150 mg/day for 6 weeks improves hepatic and muscle insulin sensitivity in patients with type 2 diabetes. The glucose-lowering effect of vanadyl sulfate correlated well with the reduction in HGP, but not with insulin-mediated glucose disposal, suggesting that liver, rather than muscle, is the primary target of vanadyl sulfate action at therapeutic doses (143).
Li and colleagues conducted a case-control study to explore the association of plasma vanadium with gestational diabetes mellitus. They included 252 newly diagnosed gestational diabetes mellitus cases with 252 controls matched by age, parity, and gestational age. Plasma concentrations of vanadium were significantly lower in the gestational diabetes mellitus group compared to the control group (p<0.001). The authors concluded there was an inverse association between plasma vanadium and gestational diabetes mellitus (144).
Adverse Effects and Warnings
Acute vanadium toxicity does not appear to be a common concern. Mild gastrointestinal effects such as abdominal cramps and loose stools may occur. Animal studies suggest vanadium may cause anemia. However, this has not been shown in humans (74).
Interactions
There is theoretical concern that vanadium may increase the risk of bleeding in anticoagulant agents (145).
Summary
Vanadium is a mineral that may increase sensitivity to insulin. There is a lack of human data to support the use of vanadium as a glucose lowering agent, but there is promise.
Carbohydrate Absorption Inhibitors
The natural products contained in this section are known to be carbohydrate absorption inhibitors. This means they theoretically lower plasma glucose by preventing the absorption of ingested carbohydrate. Products may also employ other mechanisms of action, which are addressed.
ALOE VERA GEL
Aloe is a desert plant that appears similar to a cactus and typically grows in hot and dry climates. See Figure 14 for an image of the aloe plant. Aloe byproducts are commonly used in cosmetics and medicine. The byproducts aloe vera gel and aloe latex are common (146).

FIGURE 14. ALOE VERA PLANT IMAGE SOURCE: https://en.wikipedia.org/wiki/File:Aloe_aristata.jpg.
Mechanism of Action
The portion of the aloe extract that is thought to be pharmaceutically active is the leaf. Aloe gel is clear and can be extracted from the leaf (147, 148). Monosaccharides, polysaccharides, tannins, sterols, enzymes, amino acids, salicylic acid, arachidonic acid, lipids, vitamins, and minerals are all found in aloe vera gel (148). Studies in mice suggest aloe gel may stimulate beta-cells, while human studies show conflicting evidence. Aloe latex contains anthraquinones and may be toxic. Aloe latex should not be confused with aloe gel (149, 150).
Evidence
In 2021, an analysis of four systematic reviews analyzing the metabolic effects of aloe vera in individuals with type 2 diabetes and pre-diabetes was published. Fasting blood glucose was significantly lower in the aloe vera group compared to placebo (-5.61 mg/dL; 95% CI: -7.94 to -3.28; p<0.001); HbA1c was also lower in the aloe vera group (-0.95%; 95% CI: -1.76 to -0.14; p=0.02). There was no significant change in total cholesterol, low-density lipoproteins, triglycerides, or high-density lipoproteins. Results are summarized in Table 22 (151).
|
Table 22. Effect of Aloe Vera on Metabolic Parameters (151)
|
|
Parameter
|
Pooled Mean Difference (95% CI)
|
p-value
Pooled Mean Difference
|
Heterogeneity
(I2)
|
p-value
Heterogeneity
|
|
Glycemic
|
|
Fasting plasma glucose (mg/dL)
|
-5.61 (-7.94 to -3.28)
|
<0.001
|
98%
|
<0.001
|
|
HbA1c (%)
|
-0.95% (-1.76 to -0.14)
|
0.02
|
78%
|
0.004
|
|
Lipoprotein
|
|
Total Cholesterol (mg/dL)
|
-6.79 (-44.07 to 30.48)
|
0.72
|
96%
|
<0.001
|
|
Low density lipoprotein concentration (mg/dL)
|
24.49 (-13.70 to 62.67)
|
0.21
|
99%
|
<0.00001
|
|
High density lipoprotein concentration (mg/dL)
|
4.44 (-4.24 to 13.12)
|
0.32
|
98%
|
<0.00001
|
|
Triglycerides (mg/dL)
|
28.04 (-41.90 to 97.98)
|
0.43
|
100
|
<0.001
|
A meta-analysis was published in 2016 evaluating the impact of aloe vera on fasting glucose and HbA1c in patients with type 2 diabetes. Nine studies were included in the fasting glucose analysis. Aloe vera use decreased fasting glucose by 26.6 mg/dL (p<0.001). Five studies were included in the HbA1c analysis. HbA1c decreased by 1.05% in aloe vera treated individuals (p<0.004). Results suggested patients with higher fasting glucose levels may benefit more from aloe vera use (these patients demonstrated a decrease of 109.9 mg/dL; p<0.01). The authors concluded their results support the use of aloe vera for decreasing fasting glucose and HbA1c in patients with diabetes (152).
Aloe vera has also been studied in pre-diabetes. Alinejad-Mofrad and colleagues compared aloe vera to placebo in patients with prediabetes (n=72). Patients were randomized to three groups: aloe vera 300 mg daily, aloe vera 500 mg daily, and placebo for eight weeks. Treatment with aloe vera (both 300 mg and 500 mg daily) decreased fasting glucose and HbA1c compared to placebo. Results are presented in Table 23 (153).
|
Table 23. Comparison Fasting Glucose and HbA1c with use of Aloe Vera in Patients with Prediabetes (153)
|
|
Parameter
|
Time
|
Placebo
|
Within Group p-Value
|
Aloe Vera 300 mg Group
|
Within Group p-Value
|
Aloe Vera 500 mg Group
|
Within Group p-Value
|
Between Group p-value
|
|
Fasting glucose (mg/dL)
|
Baseline
|
110 ± 3.91
|
n/a
|
112 ± 2.5
|
n/a
|
111 ± 4.1
|
n/a
|
0.69
|
|
8 weeks
|
110 ± 4.22
|
0.19
|
108 ± 2.78*
|
0.001
|
104 ± 4.2*
|
<0.001
|
0.001*
|
|
HbA1c (%)
|
Baseline
|
6.01 ± 0.16
|
n/a
|
6 ± 0.24
|
n/a
|
6 ± 0.23
|
n/a
|
0.37
|
|
8 weeks
|
6.03 ± 0.14
|
0.059
|
5.8 ± 0.21*
|
0.042
|
5.6 ± 0.33*
|
0.011
|
0.04*
|
* indicates statistical significance compared to placebo (p<0.05)
A meta-analysis and systematic review of randomized controlled trials evaluated aloe vera in patients with type 2 diabetes and prediabetes. Eight trials were included (five enrolled patients with diabetes and three enrolled patients with prediabetes). In patients with diabetes, both fasting glucose (-21 mg/dL; p<0.05) and HbA1c (-1%; p=0.01) decreased significantly. In patients with prediabetes, fasting glucose decreased statistically significantly, but in a very small amount (-4 mg/dL; p<0.0001). There was no change in HbA1c in the prediabetes patients (154).
Adverse Effects and Warnings
Aloe vera gel, when used orally, is well tolerated. Aloe latex, however, can cause abdominal pain and cramps. Unlike aloe vera gel, aloe latex contains anthraquinones which may be toxic (148).
Interactions
Aloe vera gel may decrease glucose and should be used cautiously with other products with the same effect. It may also increase the risk of bleeding when used with anticoagulant or antiplatelet drugs (148).
Summary
Aloe vera gel is derived from the leaf of the aloe plant. There is preliminary evidence to suggest it lowers glucose in those with diabetes and prediabetes. Aloe vera gel is typically well tolerated.
FENUGREEK (TRIGONELLA FOENUM-GRAECUM)
Fenugreek has multiple mechanisms of action is addressed under the “Hypoglycemic Agents” section.
FLAXSEED (LINUM USITATISSIMUM)
Flaxseed is a grain that is native to Europe, Asia, and the Mediterranean. Flax is a blue flowering crop and the seeds exist in brown, yellow, and green colors (see Figure 15). Whole flaxseeds primarily contain fat (41%), dietary fiber (28%), and protein (21%). The oil contained in flaxseeds is particularly rich in polyunsaturated fat (73%) and lower in monounsaturated (18%) and saturated (9%) fats. Flaxseed oil is a rich source of the omega-3 fatty acid alpha linolenic acid (ALA) (155).

FIGURE 15. BROWN FLAXSEEDS IMAGE SOURCE: https://en.wikipedia.org/wiki/Flax#/media/File:Brown_Flax_Seeds.jpg.
Mechanism of Action
The pharmaceutically active portion of flaxseed is the seed and the oil. The high soluble fiber content is thought to decrease carbohydrate absorption. The high omega-3 fatty acid content (ALA) is also thought to play a crucial role as these acids have been shown improve insulin sensitivity and glycemic control. ALA has been shown to increase plasma GLP-1 levels (156). Flaxseeds contain lignans, which have been proven to have antioxidant effects (157-159).
Evidence
A systematic review and meta-analysis of randomized controlled trials aimed at determining the impact of flaxseed supplementation in patients with type 2 diabetes mellitus was published in 2023. Thirteen studies were included in the analysis. HbA1c was significantly reduced in the flaxseed supplementation group (-0.19%; 95% CI: -0.38 to 0.00; p=0.045). The authors found no significant change in fasting plasma glucose, insulin, HOMA-IR, lipid parameters, body weight, BMI, or blood pressure (160).
A single-blinded, randomized, controlled trial evaluated the impact of flaxseed in 53 patients with type 2 diabetes that also had constipation. Patients either received cookies with flaxseed twice a day or cookies free of flaxseed twice daily for 12 weeks. Constipation scores, weight (-3.8 kg), and fasting plasma glucose (-26.7 mg/dL) all decreased from baseline in the flaxseed group (p<0.05). Constipation scores, weight (-3.8 versus 0 kg), fasting plasma glucose (-26.7 versus 1.9 mg/dL), and HbA1c (-0.8% versus 1.0%) were significantly different in the flaxseed group compared to placebo (p<0.05) (161).
A placebo-controlled, crossover study evaluated the impact of flaxseed on diabetes. Seventy-three patients with type 2 diabetes took either placebo or 360 mg daily of flaxseed for 12 weeks. After an eight-week washout period, patients crossed over to the other group. HbA1c decreased a small, but statistically significant, amount (0.1%; p=0.01). There was no significant change in fasting plasma glucose or lipoprotein levels (162).
An open-label study evaluated the effect of flaxseed in patients with type 2 diabetes (n=29). Patients received 10 g of flaxseed daily (n=18) or placebo (n=11). Fasting glucose decreased 28.9 mg/dL in the flaxseed group (p=0.02) and slightly increased in the placebo group. HbA1c decreased 0.59% (from 8.75% to 8.16%) in the flaxseed group (p=0.009) and increased 0.1% in the placebo group. Triglycerides and LDL-C also decreased significantly in the flaxseed group (1630.
Flaxseed has also been studied in patients with prediabetes. Hutchins and colleagues randomized 25 patients with prediabetes to take 26 g of flaxseed, 13 g of flaxseed, or placebo for 12 weeks. After a 2-week washout period, patients were crossed over to another group. Fasting glucose levels did not decrease significantly in the 26 g group compared to placebo. However, fasting glucose decreased significantly in the 13 g group compared to placebo (-2 mg/dL, p=0.036). Insulin levels also decreased significantly in the 13 g group (p=0.021) (164).
Adverse Effects and Warnings
Few adverse effects are reported with flaxseed use.161 Gastrointestinal side effects are the most commonly reported (155).
Interactions
Oil from flaxseeds is shown to decrease platelet aggregation and may, therefore, increase the risk of bleeding in users of anticoagulants and antiplatelets (165).
Theoretically, flaxseed may decrease the absorption of acetaminophen and ketoprofen. However, this interaction has not been shown in human studies (166).
Summary
Flaxseed is rich in fat (primarily omega-3 fatty acids) and fiber. There are conflicting results regarding flaxseed’s efficacy in glycemic control. Most individuals tolerate flaxseed well.
PRICKLY PEAR CACTUS (OPUNTIA FICUS-INDICA), NOPAL
Prickly pear cactus has multiple mechanisms of action is addressed under the “Insulin Sensitizers” section.
SOY (GLYCINE MAX)
Soy has multiple mechanisms of action is addressed under the “Insulin Sensitizers” section.
TURMERIC (CURCUMA LONGA, CURCUMA DOMESTICA, CURCUMA AROMATIC)
Turmeric is a member if the Zingiberaceae (ginger) family (167). Turmeric has a long history of use in Ayurvedic and Chinese medicine (see Figure 16). Curcumin is considered the active constituent of turmeric and is yellow-colored and fragrant. Curcumin is typically what is used as a flavoring and coloring agent in turmeric-containing products (168).

FIGURE 16. TURMERIC IMAGE SOURCE: https://en.wikipedia.org/wiki/Wikipedia:Featured_picture_candidates/Turmeric.
Mechanism of Action
Turmeric has multiple proposed mechanisms of action relating to glycemia. Turmeric can induce peroxisome proliferator-activated receptor-gamma activation. It may also activate hepatic enzymes associated with glycolysis and gluconeogenesis. Turmeric may also enhance tumor necrosis factor alpha (168).
Evidence
In 2023, a systematic review and network meta-analysis of randomized controlled trials was published that compared the effectiveness of different herbs in the management of T2DM. Turmeric (curcumin) was one of the herbs studied. Forty-four trials were included in the review. Compared to controls, turmeric use was correlated with a statistically significant reduction in fasting plasma glucose (-13.15 mg/dL; 95% CI: -23.64 to -2.66; p<0.05). The authors found no change in HbA1c associated with turmeric use (169).
A systematic review and meta-analysis published in 2021 was conducted to evaluate the effect of curcumin on glycemic and lipid profiles in patients with type 2 diabetes. There was a statistically significant difference in HbA1c in turmeric users (-0.42%, 95% CI -0.72 to -0.11; p< 0.05). There was non-significant (p = 0.107) moderate heterogeneity (I2 = 42.42) (170).
The effect of turmeric in delaying the development of type 2 diabetes was studied in patients with prediabetes in a randomized, double-blinded, placebo-controlled trial. Subjects (n=240) were randomly assigned to curcumin capsules or placebo for nine months. After nine months, 16.4% of subjects in the placebo group were diagnosed with type 2 diabetes. No subjects in the curcumin group were diagnosed in this timeframe (p<0.05). Markers of insulin sensitivity also showed favor for the curcumin group (higher HOMA-beta and lower HOMA-IR; p<0.05) (171).
Adverse Effects and Warnings
Turmeric is usually well tolerated. Itching, constipation, and vertigo have been reported with use (171).
Interactions
There is a risk of increased bleeding when turmeric is combined with anticoagulants (172).
Summary
Turmeric and its active constituent, curcumin, have been long used medicinally. There is limited evidence in humans to suggest curcumin use may delay the onset of type 2 diabetes in patients with prediabetes. Turmeric is typically well tolerated.
Summary of Natural Products
A summary of the natural products is presented alphabetically in Table 24.
|
Table 24. Summary of Natural Products Used for Diabetes Listed Alphabetically
|
|
Natural Product
|
Adverse Effects
|
Interactions
|
|
Aloe Vera Gel
|
Aloe vera gel is typically well tolerated
Aloe latex (which contains anthraquinones) can cause abdominal pain and cramps
|
May decrease glucose and should be used cautiously with other products with the same effect
|
|
Banaba (Lagerstroemia speciosa)
|
Dizziness, headache, tremor, weakness, diaphoresis, nausea
|
Additive effect with antihypertensives
Use with caution in those using hypoglycemic agents
|
|
Berberine
|
Diarrhea, constipation, flatulence, abdominal pain, and vomiting
Uterine contractions
May cross the placenta and result in neonatal kernicterus when ingested during pregnancy
|
May inhibit cytochrome P450 3A4 and should be used cautiously with other agents that are substrates, inhibitors, or inducers of this hepatic enzyme
As berberine lowers glucose, caution should be exercised when used with other agents that lower glucose
|
|
Bitter melon
|
Abdominal discomfort, pain, and diarrhea
May contain abortifacient proteins
|
Use with caution in those using other hypoglycemic agents
Those with G6PD deficiency should avoid use due to risk of developing favism
|
|
Chromium
|
Chromium picolinate may cause cognitive, perceptual, and motor dysfunction
|
There is a theoretical interaction between chromium and iron
|
|
Cinnamon
|
Cinnamon is typically tolerated well
Cinnamon is a natural source of coumarin and use, therefore, presents a theoretical risk of hepatic injury
|
May contain coumarin and increase the risk of bleeding with anticoagulants
Use cautiously with other hepatotoxic agents due to concerns of hepatic injury when large doses are used
May decrease glucose levels and should be used cautiously with other agents that lower glucose
|
|
Fenugreek (Trigonellafoenum-graecum)
|
Diarrhea, heartburn, and flatulence
Fenugreek smells similar to maple syrup; consumption prior to delivery may cause the neonate to have this odor, which may lead to confusion with maple syrup urine disease
Patients with chickpea allergies should use fenugreek with caution as there is potential for cross-reactivity
Fenugreek may cause uterine contractions and should be avoided in pregnancy
|
May contain coumarin and increase the risk of bleeding with anticoagulants
Theophylline levels may be decreased with concomitant use
Use cautiously with other agents that decrease glucose
|
|
Flaxseed (Linumusitatissimum)
|
Gastrointestinal side effects are the most commonly reported
|
Flaxseed may decrease the absorption of acetaminophen and ketoprofen
Flaxseed oil may increase the risk of bleeding with antiplatelets and anticoagulants
|
|
American Ginseng(Panax quinquefolius)
|
Headache
|
May stimulate immune function and theoretically decrease the effect of immunosuppressants
Can decrease the efficacy of warfarin; concomitant use is not advised
Use cautiously with other glucose lowering agents
|
|
Gymnema (Gymnemasylvestre)
|
A case of acute hepatitis secondary to gymnema use has been reported
|
May potentiate the effects of other agents that lower glucose
|
|
Milk thistle (Silybummarianum)
|
Nausea, diarrhea, and abdominal bloating may occur with use
Those with a daisy or ragweed allergy may experience a cross reaction with milk thistle use
|
May inhibit certain cytochrome P450 isoenzymes; the isoenzymes 2C8, 2C9, 2D6, 3A4, and 3A5 may all be inhibited with concomitant use
Increased warfarin levels may occur with concomitant use
May lower glucose levels and should be used cautiously with other hypoglycemia agents
|
|
Prickly Pear Cactus(Opuntia ficus-indica andother Opuntia species),Nopal
|
Prickly pear cactus is generally tolerated well when used orally
Side effects include nausea, diarrhea, and headache
|
Prickly pear cactus may lower glucose levels and should be used cautiously with other agents that impact glycemic control
|
|
Soy
|
Soy is generally well tolerated
May cause nausea, diarrhea, and bloating
Soy may alter thyroid function, but this appears to occur in those with iodine deficiency
|
Fermented soy products such as tofu may contain small amounts of tyramine; tyramine should be avoided in those using monoamine oxidase inhibitors
|
|
Turmeric (Curcuma longa, Curcuma domestica, Curcuma aromatic)
|
Itching, constipation, and vertigo have been reported with use
|
There is a risk of increased bleeding when turmeric is combined with anticoagulants
|
|
Vanadium
|
Mild gastrointestinal effects such as abdominal cramps and loose stools may occur
Animal studies suggest vanadium may cause anemia
|
Theoretical concern that vanadium may increase the risk of bleeding in anticoagulant agents
|
MIND BODY PRACTICES FOR TYPE 2 DIABETES
According to the National Institute of Heath, mind body practices include, but are not limited to, yoga, chiropractic and osteopathic manipulation, meditation, massage, acupuncture, Tai Chi, healing touch, hypnotherapy, and movement manipulation (1). Mind body practices are used for overall health and to help with specific disease states. There is recent interest in studying the impact of mind body practices on type 2 diabetes.
Yoga has been studied in patients with type 2 diabetes. Preliminary studies indicate yoga may reduce BMI, improve glycemic control, improve lipid levels, and improve body composition. Yoga may also decrease blood pressure (173-177).
The impact of massage on glycemic control in patients with diabetes has been studied. One study showed parent-provided full body massage at bedtime improved serum glucose levels and decreased anxiety of both the massage giver and receiver (178). Another study compared changes in metabolic parameters of individuals with type 2 diabetes between a control group, a routine massage group, and an abdominal massage group. The control group had no significant change in fasting plasma glucose, postprandial plasma glucose, or HbA1c. The routine massage and abdominal massage groups had significant reductions in fasting plasma glucose, postprandial plasma glucose, and HbA1c (69).
A meta-analysis exploring the impact of Tai Chi on glucose and lipid metabolism in patients with T2DM was conducted and included 16 randomized controlled trials. Tai Chi significantly reduced fasting plasma glucose and HbA1c (179). Individual studies on Tai Chi have shown it may decrease fasting glucose values in patients with diabetes. However, Tai Chi does not appear to reduce glucose more than other types of gentle exercise (180-182).
RELIABLE RESOURCES FOR PROVIDERS AND PATIENTS
Clinicians need to be aware of the deceptive marketing tactics employed by natural product manufacturers. Patients and clinicians need reliable and dependable resources regarding integrative medicine.
MedWatch is a website developed and updated by the FDA. MedWatch provides timely safety information on dietary supplements as well as medications and cosmetics. MedWatch can be accessed at: https://www.fda.gov/Safety/MedWatch/.
The FDA also has a health fraud online resource. The website describes health fraud and provides actionable ways patients can protect themselves. The FDA’s health fraud website can be accessed at: https://www.fda.gov/consumers/health-fraud-scams
In terms of efficacy, primary literature is a reasonable resource. The US National Library of Medicine has an online database that can be searched free of change. The database can be accessed at: https://www.ncbi.nlm.nih.gov/pubmed.
Additional online resources are provided in Table 25.
|
Table 25. Complementary Health Approach Online Resources for Patients and Clinicians
|
|
Source
|
Description
|
Website Address
|
|
American Botanical Council
|
Non-profit, international member-based organization providing education using evidence-based and traditional information to promote the responsible use of herbal medicine
|
http://abc.herbalgram.org/site/PageServer
|
|
ConsumerLab.com
|
Independent testing site that reviews natural products and specific manufacturers
|
http://www.consumerlab.com
|
|
National Center for Complementary and Integrative Health (NCCIH)
|
National (US) center that supports and disseminates research results on complementary health approaches
|
https://www.nccih.nih.gov
|
|
National Institute of Health Office of Dietary Supplements (ODS)
|
National (US) center that supports and disseminates research results on dietary supplements
|
http://ods.od.nih.gov/index.aspx
|
|
Natural Medicines
|
A scientifically-based and practical database on natural medicines (Subscription required)
|
https://naturalmedicines.therapeuticresearch.com
|
|
The Cochrane Library
|
An electronic database designed to provide high quality scientific evidence
|
https://www.cochranelibrary.com
|
|
United States Food and Drug Administration Health Fraud Website
|
The website describes health fraud and provides actionable ways patients can protect themselves.
|
https://www.fda.gov/consumers/health-fraud-scams
|
|
United States Food and Drug Administration MedWatch
|
MedWatch provides timely safety information on dietary supplements as well as medications and cosmetics.
|
https://www.fda.gov/Safety/MedWatch/
|
|
United States Food and Drug Administration Office of Nutritional Products, Labeling, and Dietary Supplements
|
FDA office responsible for developing policy and regulations for dietary supplements, medical foods, and related areas, as well as for their scientific evaluation
|
http://www.fda.gov/Food/DietarySupplements/default.htm
|
|
WebMD Health
|
WebMD provides comprehensive health information and tools for managing health care for health care professionals and their patients
|
http://diabetes.webmd.com/default.htm
|
IDENTIFYING POTENTIALLY HARMFUL PRODUCTS
According to the FDA’s health fraud website, there are certain red flags that can alert a consumer to potentially harmful natural products, including dietary supplements. Red flags to be wary of include: claims that a product is a cure-all for a wide variety of ailments; suggestions that a product can treat or cure diseases; promotions using words such as "scientific breakthrough" or "miraculous cure"; undocumented testimonies by consumers or doctors claiming amazing results; limited availability and advance payment requirements; promises of no-risk, money-back guarantees; promises of an "easy" fix; and claims that the product is "natural" or "non-toxic" (which doesn't necessarily mean safe) (183). These safety alerts are provided in Table 26. Additionally, the FDA fraud website warms to avoid websites that fail to list the company’s name, physical address, phone number, or other contact information.
|
Table 26. FDA Health Fraud Indicators a Natural Product May Be Ineffective or Unsafe (183)
|
|
Claims that a product is a quick, effective cure-all or a diagnostic tool for a wide variety of ailments
|
|
Suggests that a product can treat or cure diseases
|
|
Promotions using words such as "scientific breakthrough," "miraculous cure," "secret ingredient," and "ancient remedy"
|
|
Text with impressive-sounding terms such as: "hunger stimulation point" and "thermogenesis" for a weight loss product
|
|
Undocumented case histories by consumers or doctors claiming amazing results
|
|
Limited availability and advance payment requirements
|
|
Promises of no-risk, money-back guarantees
|
|
Promises of an "easy" fix
|
|
Claims that the product is "natural" or "non-toxic" (which doesn't necessarily mean safe)
|
REFERENCES
- Health NCfCaI. Complementary, Alternative, or Integrative Health: What’s In a Name? National Institute of Health. Accessed June 26, 2024. https://nccih.nih.gov/health/integrative-health
- Chan S, Hecht FM. Integrative Medicine. In: Feldman MD, Christensen JF, Satterfield JM, Laponis R, eds. Behavioral Medicine: A Guide for Clinical Practice, 5e. McGraw-Hill Education; 2019.
- Dietary Supplements (2024).
- Administration UFaD. Current Good Manufacturing Practices (CGMPs) for Dietary Supplements. 2024 June 11, Updated 2023 Dec 12. https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations
- National Institutes of Health OoDS. Dietary supplements: Background information. 2019 May 13, Updated 2020 March 11. Accessed 2024 June 11, https://ods.od.nih.gov/factsheets/DietarySupplements-Consumer/
- Dietary Supplement and Non-Prescription Drug Consumer Protection Act. Pub L. No. 109-462. (2006).
- Geller AI, Shehab N, Weidle NJ, et al. Emergency Department Visits for Adverse Events Related to Dietary Supplements. N Engl J Med. Oct 2015;373(16):1531-40. doi:10.1056/NEJMsa1504267
- Drug Recalls (2018).
- Harel Z HS, Waid R, Mamdani M, Bell CM. The frequency and characteristics of dietary supplement recalls in the United States. JAMA Intern med. 2013;173:926-8.
- Nutrition CfR. Consumer Survey on Dietary Supplements. 2023 Oct 5 2023;
- Nutrition CfR. Consumer Survey on Dietary Supplements. 30 Sept 2019;
- Alzahrani AS, Price MJ, Greenfield SM, Paudyal V. Global prevalence and types of complementary and alternative medicines use amongst adults with diabetes: systematic review and meta-analysis. Eur J Clin Pharmacol. Mar 8 2021;doi:10.1007/s00228-021-03097-x
- Rhee TG, Westberg SM, Harris IM. Complementary and alternative medicine in US adults with diabetes: Reasons for use and perceived benefits. J Diabetes. Apr 2018;10(4):310-319. doi:10.1111/1753-0407.12607
- Rhee TG, Westberg SM, Harris IM. Use of Complementary and Alternative Medicine in Older Adults With Diabetes. Diabetes Care. Apr 2018;doi:10.2337/dc17-0682
- Dias DA, Urban S, Roessner U. A historical overview of natural products in drug discovery. Metabolites. Apr 2012;2(2):303-36. doi:10.3390/metabo2020303
- Kakuda T, Sakane I, Takihara T, Ozaki Y, Takeuchi H, Kuroyanagi M. Hypoglycemic effect of extracts from Lagerstroemia speciosa L. leaves in genetically diabetic KK-AY mice. Biosci Biotechnol Biochem. Feb 1996;60(2):204-8.
- Hayashi T, Maruyama H, Kasai R, et al. Ellagitannins from Lagerstroemia speciosa as activators of glucose transport in fat cells. Planta Med. Feb 2002;68(2):173-5. doi:10.1055/s-2002-20251
- Judy WV, Hari SP, Stogsdill WW, Judy JS, Naguib YM, Passwater R. Antidiabetic activity of a standardized extract (Glucosol) from Lagerstroemia speciosa leaves in Type II diabetics. A dose-dependence study. J Ethnopharmacol. Jul 2003;87(1):115-7.
- Hattori K, Sukenobu N, Sasaki T, et al. Activation of insulin receptors by lagerstroemin. J Pharmacol Sci. Sep 2003;93(1):69-73.
- López-Murillo LD, González-Ortiz M, Martínez-Abundis E, Cortez-Navarrete M, Pérez-Rubio KG. Effect of Banaba (Lagerstroemia speciosa) on Metabolic Syndrome, Insulin Sensitivity, and Insulin Secretion. J Med Food. Feb 2022;25(2):177-182. doi:10.1089/jmf.2021.0039
- Derosa G, D'Angelo A, Maffioli P. The role of selected nutraceuticals in management of prediabetes and diabetes: An updated review of the literature. Phytother Res. Oct 2022;36(10):3709-3765. doi:10.1002/ptr.7564
- Fukushima M, Matsuyama F, Ueda N, et al. Effect of corosolic acid on postchallenge plasma glucose levels. Diabetes Res Clin Pract. Aug 2006;73(2):174-7. doi:10.1016/j.diabres.2006.01.010
- Center TR. Banaba. 2024 March 8. https://naturalmedicines.therapeuticresearch.com
- Grover JK, Yadav S, Vats V. Medicinal plants of India with anti-diabetic potential. J Ethnopharmacol. Jun 2002;81(1):81-100.
- Berman BM, Swyers JP, Kaczmarczyk J. Complementary and alternative medicine: herbal therapies for diabetes. J Assoc Acad Minor Phys. 1999;10(1):10-4.
- Center TR. Bitter melon. 2024 Feb 12. https://naturalmedicines.therapeuticresearch.com
- Basch E, Gabardi S, Ulbricht C. Bitter melon (Momordica charantia): a review of efficacy and safety. Am J Health Syst Pharm. Feb 2003;60(4):356-9.
- Oyelere SF, Ajayi OH, Ayoade TE, et al. A detailed review on the phytochemical profiles and anti-diabetic mechanisms of Momordica charantia. Heliyon. Apr 2022;8(4):e09253. doi:10.1016/j.heliyon.2022.e09253
- Zhang X, Zhao Y, Song Y, Miao M. Effects of Momordica charantia L. supplementation on glycemic control and lipid profile in type 2 diabetes mellitus patients: A systematic review and meta-analysis of randomized controlled trials. Heliyon. May 30 2024;10(10):e31126. doi:10.1016/j.heliyon.2024.e31126
- Peter EL, Kasali FM, Deyno S, et al. Momordica charantia L. lowers elevated glycaemia in type 2 diabetes mellitus patients: Systematic review and meta-analysis. J Ethnopharmacol. Mar 1 2019;231:311-324. doi:10.1016/j.jep.2018.10.033
- Ooi CP, Yassin Z, Hamid TA. Momordica charantia for type 2 diabetes mellitus. Cochrane Database Syst Rev. Aug 2012;(8):CD007845. doi:10.1002/14651858.CD007845.pub3
- Dans AM, Villarruz MV, Jimeno CA, et al. The effect of Momordica charantia capsule preparation on glycemic control in type 2 diabetes mellitus needs further studies. J Clin Epidemiol. Jun 2007;60(6):554-9. doi:10.1016/j.jclinepi.2006.07.009
- Leung SO, Yeung HW, Leung KN. The immunosuppressive activities of two abortifacient proteins isolated from the seeds of bitter melon (Momordica charantia). Immunopharmacology. Jun 1987;13(3):159-71.
- Basch E, Ulbricht C, Kuo G, Szapary P, Smith M. Therapeutic applications of fenugreek. Altern Med Rev. Feb 2003;8(1):20-7.
- Cortez-Navarrete M, Pérez-Rubio KG, Escobedo-Gutiérrez MJ. Role of Fenugreek, Cinnamon, Curcuma longa, Berberine and Momordica charantia in Type 2 Diabetes Mellitus Treatment: A Review. Pharmaceuticals (Basel). Mar 30 2023;16(4)doi:10.3390/ph16040515
- Marles RJ, Farnsworth NR. Antidiabetic plants and their active constituents. Phytomedicine. Oct 1995;2(2):137-89. doi:10.1016/S0944-7113(11)80059-0
- Sauvaire Y, Petit P, Broca C, et al. 4-Hydroxyisoleucine: a novel amino acid potentiator of insulin secretion. Diabetes. Feb 1998;47(2):206-10.
- Perla V, Jayanty SS. Biguanide related compounds in traditional antidiabetic functional foods. Food Chem. Jun 2013;138(2-3):1574-80. doi:10.1016/j.foodchem.2012.09.125
- Shabil M, Bushi G, Bodige PK, et al. Effect of Fenugreek on Hyperglycemia: A Systematic Review and Meta-Analysis. Medicina (Kaunas). Jan 27 2023;59(2)doi:10.3390/medicina59020248
- Khodamoradi K, Khosropanah MH, Ayati Z, et al. The Effects of Fenugreek on Cardiometabolic Risk Factors in Adults: A Systematic Review and Meta-analysis. Complement Ther Med. Aug 2020;52:102416. doi:10.1016/j.ctim.2020.102416
- Gupta A, Gupta R, Lal B. Effect of Trigonella foenum-graecum (fenugreek) seeds on glycaemic control and insulin resistance in type 2 diabetes mellitus: a double blind placebo controlled study. J Assoc Physicians India. Nov 2001;49:1057-61.
- Sewell AC, Mosandl A, Böhles H. False diagnosis of maple syrup urine disease owing to ingestion of herbal tea. N Engl J Med. Sep 1999;341(10):769. doi:10.1056/NEJM199909023411020
- Shiyovich A, Sztarkier I, Nesher L. Toxic hepatitis induced by Gymnema sylvestre, a natural remedy for type 2 diabetes mellitus. Am J Med Sci. Dec 2010;340(6):514-7. doi:10.1097/MAJ.0b013e3181f41168
- Tiwari P, Mishra BN, Sangwan NS. Phytochemical and pharmacological properties of Gymnema sylvestre: an important medicinal plant. Biomed Res Int. 2014;2014:830285. doi:10.1155/2014/830285
- Persaud SJ, Al-Majed H, Raman A, Jones PM. Gymnema sylvestre stimulates insulin release in vitro by increased membrane permeability. J Endocrinol. Nov 1999;163(2):207-12.
- Kanetkar P, Singhal R, Kamat M. Gymnema sylvestre: A Memoir. J Clin Biochem Nutr. Sep 2007;41(2):77-81. doi:10.3164/jcbn.2007010
- Gymnema sylvestre. Altern Med Rev. Feb 1999;4(1):46-7.
- Zamani M, Ashtary-Larky D, Nosratabadi S, et al. The effects of Gymnema Sylvestre supplementation on lipid profile, glycemic control, blood pressure, and anthropometric indices in adults: A systematic review and meta-analysis. Phytother Res. Mar 2023;37(3):949-964. doi:10.1002/ptr.7585
- Devangan S, Varghese B, Johny E, Gurram S, Adela R. The effect of Gymnema sylvestre supplementation on glycemic control in type 2 diabetes patients: A systematic review and meta-analysis. Phytother Res. Dec 2021;35(12):6802-6812. doi:10.1002/ptr.7265
- Shanmugasundaram ER, Rajeswari G, Baskaran K, Rajesh Kumar BR, Radha Shanmugasundaram K, Kizar Ahmath B. Use of Gymnema sylvestre leaf extract in the control of blood glucose in insulin-dependent diabetes mellitus. J Ethnopharmacol. Oct 1990;30(3):281-94.
- Baskaran K, Kizar Ahamath B, Radha Shanmugasundaram K, Shanmugasundaram ER. Antidiabetic effect of a leaf extract from Gymnema sylvestre in non-insulin-dependent diabetes mellitus patients. J Ethnopharmacol. Oct 1990;30(3):295-300.
- Fabio GD, Romanucci V, De Marco A, Zarrelli A. Triterpenoids from Gymnema sylvestre and their pharmacological activities. Molecules. Jul 2014;19(8):10956-81. doi:10.3390/molecules190810956
- Mucalo I, Jovanovski E, Rahelić D, Božikov V, Romić Z, Vuksan V. Effect of American ginseng (Panax quinquefolius L.) on arterial stiffness in subjects with type-2 diabetes and concomitant hypertension. J Ethnopharmacol. Oct 2013;150(1):148-53. doi:10.1016/j.jep.2013.08.015
- Services USFaW. Li st of States and Tribes with Approved Export Programs for Furbearers, Alligators, and Ginseng. 2017.
- Lim W, Mudge KW, Vermeylen F. Effects of population, age, and cultivation methods on ginsenoside content of wild American ginseng (Panax quinquefolium). J Agric Food Chem. Nov 2005;53(22):8498-505. doi:10.1021/jf051070y
- Vuksan V, Stavro MP, Sievenpiper JL, et al. Similar postprandial glycemic reductions with escalation of dose and administration time of American ginseng in type 2 diabetes. Diabetes Care. Sep 2000;23(9):1221-6.
- Liu T, Wang D, Zhou X, et al. Study on the mechanism of American ginseng extract for treating type 2 diabetes mellitus based on metabolomics. Front Pharmacol. 2022;13:960050. doi:10.3389/fphar.2022.960050
- Vuksan V, Xu ZZ, Jovanovski E, et al. Efficacy and safety of American ginseng (Panax quinquefolius L.) extract on glycemic control and cardiovascular risk factors in individuals with type 2 diabetes: a double-blind, randomized, cross-over clinical trial. Eur J Nutr. Apr 2019;58(3):1237-1245. doi:10.1007/s00394-018-1642-0
- Sievenpiper JL, Arnason JT, Leiter LA, Vuksan V. Variable effects of American ginseng: a batch of American ginseng (Panax quinquefolius L.) with a depressed ginsenoside profile does not affect postprandial glycemia. Eur J Clin Nutr. Feb 2003;57(2):243-8. doi:10.1038/sj.ejcn.1601550
- Stavro PM, Woo M, Leiter LA, Heim TF, Sievenpiper JL, Vuksan V. Long-term intake of North American ginseng has no effect on 24-hour blood pressure and renal function. Hypertension. Apr 2006;47(4):791-6. doi:10.1161/01.HYP.0000205150.43169.2c
- McElhaney JE, Gravenstein S, Cole SK, et al. A placebo-controlled trial of a proprietary extract of North American ginseng (CVT-E002) to prevent acute respiratory illness in institutionalized older adults. J Am Geriatr Soc. Jan 2004;52(1):13-9.
- Janetzky K, Morreale AP. Probable interaction between warfarin and ginseng. Am J Health Syst Pharm. Mar 1997;54(6):692-3.
- Berberine. Altern Med Rev. Apr 2000;5(2):175-7.
- Center TR. Berberine. 1 May 2024.
- Zhou JY, Zhou SW, Zhang KB, et al. Chronic effects of berberine on blood, liver glucolipid metabolism and liver PPARs expression in diabetic hyperlipidemic rats. Biol Pharm Bull. Jun 2008;31(6):1169-76.
- Zhang H, Wei J, Xue R, et al. Berberine lowers blood glucose in type 2 diabetes mellitus patients through increasing insulin receptor expression. Metabolism. Feb 2010;59(2):285-92. doi:10.1016/j.metabol.2009.07.029
- Lu SS, Yu YL, Zhu HJ, et al. Berberine promotes glucagon-like peptide-1 (7-36) amide secretion in streptozotocin-induced diabetic rats. J Endocrinol. Feb 2009;200(2):159-65. doi:10.1677/JOE-08-0419
- Yaribeygi H, Jamialahmadi T, Moallem SA, Sahebkar A. Boosting GLP-1 by Natural Products. Adv Exp Med Biol. 2021;1328:513-522. doi:10.1007/978-3-030-73234-9_36
- Xie W, Su F, Wang G, et al. Glucose-lowering effect of berberine on type 2 diabetes: A systematic review and meta-analysis. Front Pharmacol. 2022;13:1015045. doi:10.3389/fphar.2022.1015045
- Yin J, Xing H, Ye J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism. May 2008;57(5):712-7. doi:10.1016/j.metabol.2008.01.013
- Chan E. Displacement of bilirubin from albumin by berberine. Biol Neonate. 1993;63(4):201-8. doi:10.1159/000243932
- Abascal K YE. Recent clinical advances with berberine. Altern Complement Ther. 2010;16(5):281-7.
- Balk EM, Tatsioni A, Lichtenstein AH, Lau J, Pittas AG. Effect of chromium supplementation on glucose metabolism and lipids: a systematic review of randomized controlled trials. Diabetes Care. Aug 2007;30(8):2154-63. doi:10.2337/dc06-0996
- Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. Mar 2001;101(3):294-301. doi:10.1016/S0002-8223(01)00078-5
- Suksomboon N, Poolsup N, Yuwanakorn A. Systematic review and meta-analysis of the efficacy and safety of chromium supplementation in diabetes. J Clin Pharm Ther. Jun 2014;39(3):292-306. doi:10.1111/jcpt.12147
- Cefalu WT, Hu FB. Role of chromium in human health and in diabetes. Diabetes Care. Nov 2004;27(11):2741-51.
- Evans GW, Pouchnik DJ. Composition and biological activity of chromium-pyridine carboxylate complexes. J Inorg Biochem. Feb 1993;49(3):177-87.
- Wang ZQ, Zhang XH, Russell JC, Hulver M, Cefalu WT. Chromium picolinate enhances skeletal muscle cellular insulin signaling in vivo in obese, insulin-resistant JCR:LA-cp rats. J Nutr. Feb 2006;136(2):415-20. doi:10.1093/jn/136.2.415
- Li M, Youngren JF, Dunaif A, et al. Decreased insulin receptor (IR) autophosphorylation in fibroblasts from patients with PCOS: effects of serine kinase inhibitors and IR activators. J Clin Endocrinol Metab. Sep 2002;87(9):4088-93. doi:10.1210/jc.2002-020363
- Pender C, Goldfine ID, Manchem VP, et al. Regulation of insulin receptor function by a small molecule insulin receptor activator. J Biol Chem. Nov 2002;277(46):43565-71. doi:10.1074/jbc.M202426200
- Althuis MD, Jordan NE, Ludington EA, Wittes JT. Glucose and insulin responses to dietary chromium supplements: a meta-analysis. Am J Clin Nutr. Jul 2002;76(1):148-55. doi:10.1093/ajcn/76.1.148
- Zhao F, Pan D, Wang N, et al. Effect of Chromium Supplementation on Blood Glucose and Lipid Levels in Patients with Type 2 Diabetes Mellitus: a Systematic Review and Meta-analysis. Biol Trace Elem Res. Feb 2022;200(2):516-525. doi:10.1007/s12011-021-02693-3
- Asbaghi O, Fatemeh N, Mahnaz RK, et al. Effects of chromium supplementation on glycemic control in patients with type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. Nov 2020;161:105098. doi:10.1016/j.phrs.2020.105098
- Fox GN, Sabovic Z. Chromium picolinate supplementation for diabetes mellitus. J Fam Pract. Jan 1998;46(1):83-6.
- Nahas R, Moher M. Complementary and alternative medicine for the treatment of type 2 diabetes. Can Fam Physician. Jun 2009;55(6):591-6.
- Suksomboon N, Poolsup N, Boonkaew S, Suthisisang CC. Meta-analysis of the effect of herbal supplement on glycemic control in type 2 diabetes. J Ethnopharmacol. Oct 2011;137(3):1328-33. doi:10.1016/j.jep.2011.07.059
- Anderson RA, Broadhurst CL, Polansky MM, et al. Isolation and characterization of polyphenol type-A polymers from cinnamon with insulin-like biological activity. J Agric Food Chem. Jan 2004;52(1):65-70. doi:10.1021/jf034916b
- Jarvill-Taylor KJ, Anderson RA, Graves DJ. A hydroxychalcone derived from cinnamon functions as a mimetic for insulin in 3T3-L1 adipocytes. J Am Coll Nutr. Aug 2001;20(4):327-36.
- Imparl-Radosevich J, Deas S, Polansky MM, et al. Regulation of PTP-1 and insulin receptor kinase by fractions from cinnamon: implications for cinnamon regulation of insulin signalling. Horm Res. Sep 1998;50(3):177-82. doi:10.1159/000023270
- Center TR. Cinnamon. 2024 March 25. https://naturalmedicines.therapeuticresearch.com
- Akilen R, Tsiami A, Robinson N. Efficacy and safety of 'true' cinnamon (Cinnamomum zeylanicum) as a pharmaceutical agent in diabetes: a systematic review and meta-analysis. Diabet Med. Apr 2013;30(4):505-6. doi:10.1111/dme.12068
- Allen RW, Schwartzman E, Baker WL, Coleman CI, Phung OJ. Cinnamon use in type 2 diabetes: an updated systematic review and meta-analysis. Ann Fam Med. 2013 Sep-Oct 2013;11(5):452-9. doi:10.1370/afm.1517
- Baker WL, Gutierrez-Williams G, White CM, Kluger J, Coleman CI. Effect of cinnamon on glucose control and lipid parameters. Diabetes Care. Jan 2008;31(1):41-3. doi:10.2337/dc07-1711
- Moridpour AH, Kavyani Z, Khosravi S, et al. The effect of cinnamon supplementation on glycemic control in patients with type 2 diabetes mellitus: An updated systematic review and dose-response meta-analysis of randomized controlled trials. Phytother Res. Jan 2024;38(1):117-130. doi:10.1002/ptr.8026
- Leach MJ, Kumar S. Cinnamon for diabetes mellitus. Cochrane Database Syst Rev. Sep 12 2012;2012(9):Cd007170. doi:10.1002/14651858.CD007170.pub2
- Deyno S, Eneyew K, Seyfe S, et al. Efficacy and safety of cinnamon in type 2 diabetes mellitus and pre-diabetes patients: A meta-analysis and meta-regression. Diabetes Res Clin Pract. Oct 2019;156:107815. doi:10.1016/j.diabres.2019.107815
- Lu T, Sheng H, Wu J, Cheng Y, Zhu J, Chen Y. Cinnamon extract improves fasting blood glucose and glycosylated hemoglobin level in Chinese patients with type 2 diabetes. Nutr Res. Jun 2012;32(6):408-12. doi:10.1016/j.nutres.2012.05.003
- Khan A, Safdar M, Ali Khan MM, Khattak KN, Anderson RA. Cinnamon improves glucose and lipids of people with type 2 diabetes. Diabetes Care. Dec 2003;26(12):3215-8.
- Vanschoonbeek K, Thomassen BJ, Senden JM, Wodzig WK, van Loon LJ. Cinnamon supplementation does not improve glycemic control in postmenopausal type 2 diabetes patients. J Nutr. Apr 2006;136(4):977-80. doi:10.1093/jn/136.4.977
- Crawford P. Effectiveness of cinnamon for lowering hemoglobin A1C in patients with type 2 diabetes: a randomized, controlled trial. J Am Board Fam Med. 2009 Sep-Oct 2009;22(5):507-12. doi:10.3122/jabfm.2009.05.080093
- Felter SP, Vassallo JD, Carlton BD, Daston GP. A safety assessment of coumarin taking into account species-specificity of toxicokinetics. Food Chem Toxicol. Apr 2006;44(4):462-75. doi:10.1016/j.fct.2005.08.019
- Pepping J. Milk thistle: Silybum marianum. Am J Health Syst Pharm. Jun 1999;56(12):1195-7.
- Voroneanu L, Nistor I, Dumea R, Apetrii M, Covic A. Silymarin in Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J Diabetes Res. 2016;2016:5147468. doi:10.1155/2016/5147468
- Flora K, Hahn M, Rosen H, Benner K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am J Gastroenterol. Feb 1998;93(2):139-43. doi:10.1111/j.1572-0241.1998.00139.x
- Shahbazi F, Sadighi S, Dashti-Khavidaki S, et al. Effect of Silymarin Administration on Cisplatin Nephrotoxicity: Report from A Pilot, Randomized, Double-Blinded, Placebo-Controlled Clinical Trial. Phytother Res. Jul 2015;29(7):1046-53. doi:10.1002/ptr.5345
- Soleymani S, Ayati MH, Mansourzadeh MJ, Namazi N, Zargaran A. The effects of Silymarin on the features of cardiometabolic syndrome in adults: A systematic review and meta-analysis. Phytother Res. Feb 2022;36(2):842-856. doi:10.1002/ptr.7364
- Huseini HF, Larijani B, Heshmat R, et al. The efficacy of Silybum marianum (L.) Gaertn. (silymarin) in the treatment of type II diabetes: a randomized, double-blind, placebo-controlled, clinical trial. Phytother Res. Dec 2006;20(12):1036-9. doi:10.1002/ptr.1988
- Soto C, Pérez J, García V, Uría E, Vadillo M, Raya L. Effect of silymarin on kidneys of rats suffering from alloxan-induced diabetes mellitus. Phytomedicine. Dec 2010;17(14):1090-4. doi:10.1016/j.phymed.2010.04.011
- Detaille D, Sanchez C, Sanz N, Lopez-Novoa JM, Leverve X, El-Mir MY. Interrelation between the inhibition of glycolytic flux by silibinin and the lowering of mitochondrial ROS production in perifused rat hepatocytes. Life Sci. May 2008;82(21-22):1070-6. doi:10.1016/j.lfs.2008.03.007
- Xiao F, Gao F, Zhou S, Wang L. The therapeutic effects of silymarin for patients with glucose/lipid metabolic dysfunction: A meta-analysis. Medicine (Baltimore). Oct 2 2020;99(40):e22249. doi:10.1097/md.0000000000022249
- Hussain SA. Silymarin as an adjunct to glibenclamide therapy improves long-term and postprandial glycemic control and body mass index in type 2 diabetes. J Med Food. Sep 2007;10(3):543-7. doi:10.1089/jmf.2006.089
- Di Pierro F, Putignano P, Villanova N, Montesi L, Moscatiello S, Marchesini G. Preliminary study about the possible glycemic clinical advantage in using a fixed combination of Berberis aristata and Silybum marianum standardized extracts versus only Berberis aristata in patients with type 2 diabetes. Clin Pharmacol. 2013;5:167-74. doi:10.2147/CPAA.S54308
- Mulrow C, Lawrence V, Jacobs B, et al. Milk thistle: effects on liver disease and cirrhosis and clinical adverse effects. Evid Rep Technol Assess (Summ). 2000;(21):1-3.
- Albassam AA, Frye RF, Markowitz JS. The effect of milk thistle (Silybum marianum) and its main flavonolignans on CYP2C8 enzyme activity in human liver microsomes. Chem Biol Interact. Jun 2017;271:24-29. doi:10.1016/j.cbi.2017.04.025
- Kawaguchi-Suzuki M, Frye RF, Zhu HJ, et al. The effects of milk thistle (Silybum marianum) on human cytochrome P450 activity. Drug Metab Dispos. Oct 2014;42(10):1611-6. doi:10.1124/dmd.114.057232
- Center TR. Milk thistle. 12 March 2024. https://naturalmedicines.therapeuticresearch.com
- Onakpoya IJ, O'Sullivan J, Heneghan CJ. The effect of cactus pear (Opuntia ficus-indica) on body weight and cardiovascular risk factors: a systematic review and meta-analysis of randomized clinical trials. Nutrition. May 2015;31(5):640-6. doi:10.1016/j.nut.2014.11.015
- López-Romero P, Pichardo-Ontiveros E, Avila-Nava A, et al. The effect of nopal (Opuntia ficus indica) on postprandial blood glucose, incretins, and antioxidant activity in Mexican patients with type 2 diabetes after consumption of two different composition breakfasts. J Acad Nutr Diet. Nov 2014;114(11):1811-8. doi:10.1016/j.jand.2014.06.352
- Argáez-López N, Wacher NH, Kumate-Rodríguez J, et al. The use of complementary and alternative medicine therapies in type 2 diabetic patients in Mexico. Diabetes Care. Aug 2003;26(8):2470-1.
- K R, R M, M E. Glycemic effects of various species of nopal (Opuntia sp.) in type 2 diabetes mellitus. Texas J Rural Health. 1998;26:68-76.
- RM W, H K, Y E, J S, . SH. Effect of prickly pear (Opuntia robusta) on glucose- and lipid-metabolism in non-diabetics with hyperlipidemia--a pilot study. Wien Klin Wochenschr. 2002;114(19-20):840-6.
- Gouws CA, Georgousopoulou EN, Mellor DD, McKune A, Naumovski N. Effects of the Consumption of Prickly Pear Cacti (Opuntia spp.) and its Products on Blood Glucose Levels and Insulin: A Systematic Review. Medicina (Kaunas). May 15 2019;55(5)doi:10.3390/medicina55050138
- Godard MP, Ewing BA, Pischel I, Ziegler A, Benedek B, Feistel B. Acute blood glucose lowering effects and long-term safety of OpunDia supplementation in pre-diabetic males and females. J Ethnopharmacol. Aug 9 2010;130(3):631-4. doi:10.1016/j.jep.2010.05.047
- Frati AC, Gordillo BE, Altamirano P, Ariza CR, Cortés-Franco R, Chavez-Negrete A. Acute hypoglycemic effect of Opuntia streptacantha Lemaire in NIDDM. Diabetes Care. Apr 1990;13(4):455-6.
- Frati-Munari AC, Gordillo BE, Altamirano P, Ariza CR. Hypoglycemic effect of Opuntia streptacantha Lemaire in NIDDM. Diabetes Care. Jan 1988;11(1):63-6.
- Lopez-Romero P, Pichardo-Ontiveros E, Avila-Nava A, et al. The effect of nopal (Opuntia ficus indica) on postprandial blood glucose, incretins, and antioxidant activity in Mexican patients with type 2 diabetes after consumption of two different composition breakfasts. J Acad Nutr Diet. Nov 2014;114(11):1811-8. doi:10.1016/j.jand.2014.06.352
- T. H. On the domestication of the soybean. Econ Bot. 1970;24(4):408-21.
- Erdman JW. AHA Science Advisory: Soy protein and cardiovascular disease: A statement for healthcare professionals from the Nutrition Committee of the AHA. Circulation. Nov 2000;102(20):2555-9.
- Zand RS, Jenkins DJ, Diamandis EP. Steroid hormone activity of flavonoids and related compounds. Breast Cancer Res Treat. Jul 2000;62(1):35-49.
- Tham DM, Gardner CD, Haskell WL. Clinical review 97: Potential health benefits of dietary phytoestrogens: a review of the clinical, epidemiological, and mechanistic evidence. J Clin Endocrinol Metab. Jul 1998;83(7):2223-35. doi:10.1210/jcem.83.7.4752
- Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr. Jun 2001;73(6):1131S-1141S. doi:10.1093/ajcn/73.6.1131S
- Tang J, Wan Y, Zhao M, Zhong H, Zheng JS, Feng F. Legume and soy intake and risk of type 2 diabetes: a systematic review and meta-analysis of prospective cohort studies. Am J Clin Nutr. Mar 1 2020;111(3):677-688. doi:10.1093/ajcn/nqz338
- Zuo X, Zhao R, Wu M, Wan Q, Li T. Soy Consumption and the Risk of Type 2 Diabetes and Cardiovascular Diseases: A Systematic Review and Meta-Analysis. Nutrients. Mar 10 2023;15(6)doi:10.3390/nu15061358
- Li W, Ruan W, Peng Y, Wang D. Soy and the risk of type 2 diabetes mellitus: A systematic review and meta-analysis of observational studies. Diabetes Res Clin Pract. Mar 2018;137:190-199. doi:10.1016/j.diabres.2018.01.010
- Yang B, Chen Y, Xu T, et al. Systematic review and meta-analysis of soy products consumption in patients with type 2 diabetes mellitus. Asia Pac J Clin Nutr. 2011;20(4):593-602.
- Liu ZM, Chen YM, Ho SC. Effects of soy intake on glycemic control: a meta-analysis of randomized controlled trials. Am J Clin Nutr. May 2011;93(5):1092-101. doi:10.3945/ajcn.110.007187
- Albertazzi P, Pansini F, Bonaccorsi G, Zanotti L, Forini E, De Aloysio D. The effect of dietary soy supplementation on hot flushes. Obstet Gynecol. Jan 1998;91(1):6-11.
- Messina M, Redmond G. Effects of soy protein and soybean isoflavones on thyroid function in healthy adults and hypothyroid patients: a review of the relevant literature. Thyroid. Mar 2006;16(3):249-58. doi:10.1089/thy.2006.16.249
- Divi RL, Chang HC, Doerge DR. Anti-thyroid isoflavones from soybean: isolation, characterization, and mechanisms of action. Biochem Pharmacol. Nov 1997;54(10):1087-96.
- Shulman KI, Walker SE. Refining the MAOI diet: tyramine content of pizzas and soy products. J Clin Psychiatry. Mar 1999;60(3):191-3.
- Harland BF, Harden-Williams BA. Is vanadium of human nutritional importance yet? J Am Diet Assoc. Aug 1994;94(8):891-4.
- Mukherjee B, Patra B, Mahapatra S, Banerjee P, Tiwari A, Chatterjee M. Vanadium--an element of atypical biological significance. Toxicol Lett. Apr 2004;150(2):135-43. doi:10.1016/j.toxlet.2004.01.009
- Cusi K, Cukier S, DeFronzo RA, Torres M, Puchulu FM, Redondo JC. Vanadyl sulfate improves hepatic and muscle insulin sensitivity in type 2 diabetes. J Clin Endocrinol Metab. Mar 2001;86(3):1410-7. doi:10.1210/jcem.86.3.7337
- Li X, Zhu Y, Yin J, et al. Inverse Association of Plasma Vanadium Concentrations with Gestational Diabetes Mellitus. Nutrients. Mar 29 2022;14(7)doi:10.3390/nu14071415
- Funakoshi T, Shimada H, Kojima S, et al. Anticoagulant action of vanadate. Chem Pharm Bull (Tokyo). Jan 1992;40(1):174-6.
- Panel CIRE. Final report on the safety assessment of AloeAndongensis Extract, Aloe Andongensis Leaf Juice,aloe Arborescens Leaf Extract, Aloe Arborescens Leaf Juice, Aloe Arborescens Leaf Protoplasts, Aloe Barbadensis Flower Extract, Aloe Barbadensis Leaf, Aloe Barbadensis Leaf Extract, Aloe Barbadensis Leaf Juice,aloe Barbadensis Leaf Polysaccharides, Aloe Barbadensis Leaf Water, Aloe Ferox Leaf Extract, Aloe Ferox Leaf Juice, and Aloe Ferox Leaf Juice Extract. Int J Toxicol. 2007;26 Suppl 2:1-50. doi:10.1080/10915810701351186
- Akaberi M, Sobhani Z, Javadi B, Sahebkar A, Emami SA. Therapeutic effects of Aloe spp. in traditional and modern medicine: A review. Biomed Pharmacother. Dec 2016;84:759-772. doi:10.1016/j.biopha.2016.09.096
- Vogler BK, Ernst E. Aloe vera: a systematic review of its clinical effectiveness. Br J Gen Pract. Oct 1999;49(447):823-8.
- Choi HC, Kim SJ, Son KY, Oh BJ, Cho BL. Metabolic effects of aloe vera gel complex in obese prediabetes and early non-treated diabetic patients: randomized controlled trial. Nutrition. Sep 2013;29(9):1110-4. doi:10.1016/j.nut.2013.02.015
- Beppu H, Shimpo K, Chihara T, et al. Antidiabetic effects of dietary administration of Aloe arborescens Miller components on multiple low-dose streptozotocin-induced diabetes in mice: investigation on hypoglycemic action and systemic absorption dynamics of aloe components. J Ethnopharmacol. Feb 2006;103(3):468-77. doi:10.1016/j.jep.2005.10.034
- Araya-Quintanilla F, Gutiérrez-Espinoza H, Cuyul-Vásquez I, Pavez L. Effectiveness of aloe vera in patients with type 2 Diabetes Mellitus and pre-diabetes: An overview of systematic reviews. Diabetes Metab Syndr. Nov-Dec 2021;15(6):102292. doi:10.1016/j.dsx.2021.102292
- Dick WR, Fletcher EA, Shah SA. Reduction of Fasting Blood Glucose and Hemoglobin A1c Using Oral Aloe Vera: A Meta-Analysis. J Altern Complement Med. Jun 2016;22(6):450-7. doi:10.1089/acm.2015.0122
- Alinejad-Mofrad S, Foadoddini M, Saadatjoo SA, Shayesteh M. Improvement of glucose and lipid profile status with Aloe vera in pre-diabetic subjects: a randomized controlled-trial. J Diabetes Metab Disord. 2015;14:22. doi:10.1186/s40200-015-0137-2
- Suksomboon N, Poolsup N, Punthanitisarn S. Effect of Aloe vera on glycaemic control in prediabetes and type 2 diabetes: a systematic review and meta-analysis. J Clin Pharm Ther. Apr 2016;41(2):180-8. doi:10.1111/jcpt.12382
- Bloedon LT, Szapary PO. Flaxseed and cardiovascular risk. Nutr Rev. Jan 2004;62(1):18-27.
- Adachi T, Tanaka T, Takemoto K, Koshimizu T-a, Hirasawa A, Tsujimoto G. Free fatty acids administered into the colon promote the secretion of glucagon-like peptide-1 and insulin. Biochemical and Biophysical Research Communications. 2006/02/03/ 2006;340(1):332-337. doi:https://doi.org/10.1016/j.bbrc.2005.11.162
- Javidi A, Mozaffari-Khosravi H, Nadjarzadeh A, Dehghani A, Eftekhari MH. The effect of flaxseed powder on insulin resistance indices and blood pressure in prediabetic individuals: A randomized controlled clinical trial. J Res Med Sci. 2016;21:70. doi:10.4103/1735-1995.189660
- Jenkins DJ, Kendall CW, Vidgen E, et al. Health aspects of partially defatted flaxseed, including effects on serum lipids, oxidative measures, and ex vivo androgen and progestin activity: a controlled crossover trial. Am J Clin Nutr. Mar 1999;69(3):395-402. doi:10.1093/ajcn/69.3.395
- Nettleton JA, Katz R. n-3 long-chain polyunsaturated fatty acids in type 2 diabetes: a review. J Am Diet Assoc. Mar 2005;105(3):428-40. doi:10.1016/j.jada.2004.11.029
- Xi H, Zhou W, Sohaib M, et al. Flaxseed supplementation significantly reduces hemoglobin A1c in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Nutr Res. Feb 2023;110:23-32. doi:10.1016/j.nutres.2022.12.008
- Soltanian N, Janghorbani M. A randomized trial of the effects of flaxseed to manage constipation, weight, glycemia, and lipids in constipated patients with type 2 diabetes. Nutr Metab (Lond). 2018;15:36. doi:10.1186/s12986-018-0273-z
- Pan A, Sun J, Chen Y, et al. Effects of a flaxseed-derived lignan supplement in type 2 diabetic patients: a randomized, double-blind, cross-over trial. PLoS One. Nov 2007;2(11):e1148. doi:10.1371/journal.pone.0001148
- Mani UV, Mani I, Biswas M, Kumar SN. An open-label study on the effect of flax seed powder (Linum usitatissimum) supplementation in the management of diabetes mellitus. J Diet Suppl. Sep 2011;8(3):257-65. doi:10.3109/19390211.2011.593615
- Hutchins AM, Brown BD, Cunnane SC, Domitrovich SG, Adams ER, Bobowiec CE. Daily flaxseed consumption improves glycemic control in obese men and women with pre-diabetes: a randomized study. Nutr Res. May 2013;33(5):367-75. doi:10.1016/j.nutres.2013.02.012
- Nordström DC, Honkanen VE, Nasu Y, Antila E, Friman C, Konttinen YT. Alpha-linolenic acid in the treatment of rheumatoid arthritis. A double-blind, placebo-controlled and randomized study: flaxseed vs. safflower seed. Rheumatol Int. 1995;14(6):231-4. doi:10.1007/bf00262088
- Laitinen LA, Tammela PS, Galkin A, Vuorela HJ, Marvola ML, Vuorela PM. Effects of extracts of commonly consumed food supplements and food fractions on the permeability of drugs across Caco-2 cell monolayers. Pharm Res. Oct 2004;21(10):1904-16.
- Araújo CC, Leon LL. Biological activities of Curcuma longa L. Mem Inst Oswaldo Cruz. Jul 2001;96(5):723-8.
- Zhang DW, Fu M, Gao SH, Liu JL. Curcumin and diabetes: a systematic review. Evid Based Complement Alternat Med. 2013;2013:636053. doi:10.1155/2013/636053
- Kumar S, Sharma SK, Mudgal SK, et al. Comparative effectiveness of six herbs in the management of glycemic status of type 2 diabetes mellitus patients: A systematic review and network meta-analysis of randomized controlled trials. Diabetes Metab Syndr. Aug 2023;17(8):102826. doi:10.1016/j.dsx.2023.102826
- Altobelli E, Angeletti PM, Marziliano C, Mastrodomenico M, Giuliani AR, Petrocelli R. Potential Therapeutic Effects of Curcumin on Glycemic and Lipid Profile in Uncomplicated Type 2 Diabetes-A Meta-Analysis of Randomized Controlled Trial. Nutrients. Jan 27 2021;13(2)doi:10.3390/nu13020404
- Chuengsamarn S, Rattanamongkolgul S, Phonrat B, Tungtrongchitr R, Jirawatnotai S. Reduction of atherogenic risk in patients with type 2 diabetes by curcuminoid extract: a randomized controlled trial. J Nutr Biochem. Feb 2014;25(2):144-50. doi:10.1016/j.jnutbio.2013.09.013
- Daveluy A, Géniaux H, Thibaud L, Mallaret M, Miremont-Salamé G, Haramburu F. Probable interaction between an oral vitamin K antagonist and turmeric (Curcuma longa). Therapie. 2014 Nov-Dec 2014;69(6):519-20. doi:10.2515/therapie/2014062
- Garrow D, Egede LE. Association between complementary and alternative medicine use, preventive care practices, and use of conventional medical services among adults with diabetes. Diabetes Care. Jan 2006;29(1):15-9.
- Hegde SV, Adhikari P, Kotian S, Pinto VJ, D'Souza S, D'Souza V. Effect of 3-month yoga on oxidative stress in type 2 diabetes with or without complications: a controlled clinical trial. Diabetes Care. Oct 2011;34(10):2208-10. doi:10.2337/dc10-2430
- Innes KE, Selfe TK. Yoga for Adults with Type 2 Diabetes: A Systematic Review of Controlled Trials. J Diabetes Res. 2016;2016:6979370. doi:10.1155/2016/6979370
- Wu S, Wang L, He Y, et al. Effects of different mind-body exercises on glucose and lipid metabolism in patients with type 2 diabetes: A network meta-analysis. Complement Ther Clin Pract. Nov 2023;53:101802. doi:10.1016/j.ctcp.2023.101802
- Cangelosi G, Acito M, Grappasonni I, et al. Yoga or Mindfulness on Diabetes: Scoping Review for Theoretical Experimental Framework. Ann Ig. Mar-Apr 2024;36(2):153-168. doi:10.7416/ai.2024.2600
- T F, M H-R, A L. Glucose levels decreased after giving massage therapy to children with diabetes mellitus. . Spectrum. 1997;10:23-5.
- Zhao H, Teng J, Song G, et al. The optimal exercise parameters of Tai Chi on the effect of glucose and lipid metabolism in patients with type 2 diabetes mellitus: A meta-analysis. Complement Ther Med. Dec 2023;79:102995. doi:10.1016/j.ctim.2023.102995
- Champagne CP, Gardner NJ, Roy D. Challenges in the addition of probiotic cultures to foods. Crit Rev Food Sci Nutr. 2005;45(1):61-84. doi:10.1080/10408690590900144
- Tsang T, Orr R, Lam P, Comino E, Singh MF. Effects of Tai Chi on glucose homeostasis and insulin sensitivity in older adults with type 2 diabetes: a randomised double-blind sham-exercise-controlled trial. Age Ageing. Jan 2008;37(1):64-71. doi:10.1093/ageing/afm127
- Lam P, Dennis SM, Diamond TH, Zwar N. Improving glycaemic and BP control in type 2 diabetes. The effectiveness of tai chi. Aust Fam Physician. Oct 2008;37(10):884-7.
- Administration USFaD. 6 Tip-offs to Rip-offs: Don't Fall for Health Fraud Scams. Updated 2021 March 4. Accessed June 26, 2024. https://www.fda.gov/consumers/consumer-updates/6-tip-offs-rip-offs-dont-fall-health-fraud-scams