Archives
PEDIATRIC LIPID DISORDERS-
Thyroid Regulation and Dysfunction in the Pregnant Patient
ABSTRACT
Thyroid disease in pregnancy is a common clinical problem. During the past 2 years significant clinical and scientific advances have occurred in the field. This chapter reviews the physiology of thyroid and pregnancy focusing on iodine requirements and advances in placental function. There follows discussion on thyroid function tests in pregnancy and their interpretation noting ethnic variation in pregnancy range. Sections on iodine nutrition, thyroid autoantibodies and pregnancy complications, thyroid considerations in infertile women, hypothyroidism in pregnancy, thyrotoxicosis in pregnancy, thyroid nodules and cancer in pregnant women, fetal and neonatal considerations, thyroid disease and lactation, screening for thyroid dysfunction in pregnancy will inform the reader of the current information on these areas. Postpartum thyroid disease is also discussed. Current topical fields of importance include the role of isolated hypothyroxinemia on obstetric outcomes and neurodevelopment, the influence of thyroid autoantibodies on the same parameters and the effect of recent data on malformations associated with antithyroid drug therapy on management guidelines for thyrotoxicosis in pregnancy. It also seems as if pregnancy may have a deleterious effect on the progression differentiated thyroid cancer in pregnancy; this requires more confirmation. The intense debate on whether to screen for thyroid function in all pregnant women continues. Although the few randomised trials which have been performed are negative several areas of the world and some clinics in USA recommend screening. In general recent guidelines from USA and Europe find no evidence to support routine screening.
INTRODUCTION
Pregnancy may affect the course of thyroid disorders and, conversely, thyroid diseases may affect the course of pregnancy. Moreover, thyroid disorders (and their management) may affect both the pregnant woman and the developing fetus
MATERNAL THYROID PHYSIOLOGY
Numerous hormonal changes and metabolic demands occur during pregnancy, resulting in profound and complex effects on thyroid function Table 14-1 summarizes the main physiologic changes that occur during a normal pregnancy, and which relate to thyroid function or thyroid function testing. These changes are discussed below.
| Table 14-1. Factors affecting Thyroid Physiology during normal Pregnancy | |
| Physiologic Change | Thyroid-related consequences |
| Increased renal I- clearance | Increased 24-hr RAIU |
| Decreased plasma I- and placental I- transport to the fetus | In I- deficient women, decreased T4, increased TSH, and goiter formation |
| Increased O2 consumption by fetoplacental unit, gravid uterus and mother | Increased BMR |
| First-trimester increase in hCG | Increased free T4 and T3 Decreased basal TSH (partial blunting of the pituitary-thyroid axis) |
| Increased serum TBG | Increased total T4 and T3 |
| Increased plasma volume | Increased T4 and T3 pool size |
| Inner-ring deiodination of T4 and T3 by placenta | Accelerated rates of T4 and T3 degradation and production |
Iodine and Pregnancy
Physiologic adaptation of the thyroidal economy associated with normal pregnancy is replaced by pathologic changes when pregnancy takes place in conditions with iodine deficiency or even only mild iodine restriction. Globally, the changes in maternal thyroid function that occur during gestation can be viewed as a mathematical fraction, with hormone requirements in the numerator and the availability of iodine in the denominator. When availability of iodine becomes deficient during gestation, at a time when thyroid hormone requirements are increased, this situation presents an additional challenge to the maternal thyroid 1,2. Figure 14-1 illustrates the steps through which pregnancy induces a specific challenge for the thyroid gland and the profound difference between glandular adaptation in conditions with iodine sufficiency or deficiency.
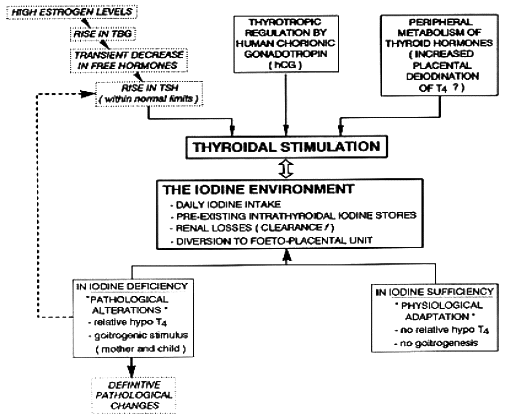
Figure 14- 1 From physiological adaptation to pathological alterations of the thyroidal economy during pregnancy. The scheme illustrates the sequence of events occurring for the maternal thyroid gland, emphasizing the role of iodine deficiency to stimulate the thyroidal machinery (from Glinoer, Ref 1).
Early in pregnancy there is an increase in renal blood flow and glomerular filtration which lead to an increase in iodide clearance from plasma (1,16). This results in a fall in plasma iodine concentrations and an increase in iodide requirements from the diet . In women with iodine sufficiency there is little thyroid impact of the obligatory increase in renal iodine losses, because the intrathyroidal iodine stores are plentiful at the time of conception and they remain unaltered throughout gestation. Pregnancy does not have a major influence on circulating iodine concentrations in iodine-sufficient regions. It should be noted, however, that the iodine excretion levels were unusually high in this study, ranging between 459-786 µg/day (17).
In regions where the iodine supply is borderline or low, the situation is clearly different and significant changes occur during pregnancy (1). Historic studies of radioiodine uptake have shown an increase (18). In addition, there is a further increment in iodine requirements, due to transplacental iodide transport necessary for iodothyronine synthesis by the fetal thyroid gland (19), which becomes progressively functional after the first trimester. When pregnancy takes place in conditions with borderline iodine availability, significant increments in both maternal and fetal thyroid volume occur, if no supplemental iodine is given during early pregnancy (20).
Thus during pregnancy, the physiologic changes that take place in maternal thyroid economy lead to an increase in thyroid hormone production of ~50% above preconception baseline hormone production. In order to achieve the necessary increment in hormone production, the iodine intake needs to be increased during early pregnancy.
Iodine deficiency present at critical stages during pregnancy and early childhood results in impaired development of the brain and consequently in impaired mental function (8,21). Iodine deficiency worldwide is a major cause of neurointellectual impairment and is discussed in detail in chapter 20.Although a variety of methods exists for the correction of iodine deficiency, the most commonly accepted and applied method is universal salt iodization (USI), i.e., the addition of suitable amounts of potassium iodide (or iodate) to all salt for human and livestock consumption. A WHO committee recommended appropriate iodine intakes for pregnant and lactating women as well as for children (Table 14-2) (22)
Table14- 2. Recommended iodine intake during pregnancy and lactation and categorization of iodine nutrition adequacy based on urinary iodine excretion
| Population Group | Median Urinary Iodine conc. | Category of Iodine intake |
| Pregnant women | 250 μg/d | |
| Lactating women | 250 μg/d | |
| Pregnant women | < 150 μg/L | Insufficient |
| 150 – 249 μg/L | Adequate | |
| 250 – 499 μg/L | More than adequate | |
| > 500 μg/L | Excessive | |
| Lactating women | < 100 μg/L | Insufficient |
| > 100 μg/L | Adequate (but see below) |
Patients with known or underlying autoimmune thyroid disorders or autonomous thyroid tissue may have side effects from excessive iodine intake, There is no clear evidence to define “how much more iodine may become too much iodine.” A recommendation was adopted to indicate that there is no proven further benefit in providing pregnant women with more than twice the daily RNI (recommended nutritional intake).
During breast-feeding, thyroid hormone production and urinary iodine excretion return to normal, but iodine is efficiently concentrated by the mammary gland. Since breast milk provides approximately 100 µg/d of iodine to the infant, it is recommended that the breast-feeding mother should continue to take 250 µg per day of iodine (see Table 14-2).
Although substantial progress has been made in the worldwide correction of iodine deficiency mainly by increasing the universal salt iodisation Nevertheless there have been many studies and reports from different world regions demonstrating the resurgence of iodine deficiency in pregnant women despite previous successful public health strategies to correct population deficiencies of the element. Therefore iodine deficiency requires constant monitoring, even after the implementation of iodine supplementation in pregnant women. Recently, iodine deficiency has re emerged in Australia and the UK and even in USA there are groups of the population with suboptimal iodine levels (24-26). The importance of iodine deficiency in pregnancy on childhood IQ has been emphasized (27). In addition there is increasing evidence of the benficial effect of iodine supplementation before and during pregnancy in ameliorating this problem (27)
Metabolism Of Iodine During Normal Pregnancy
After reduction to iodide, dietary iodine is rapidly absorbed from the gut. Then, iodide of dietary origin mixes rapidly with iodide resulting from the peripheral catabolism of thyroid hormones and iodothyronines by deiodination, and together they constitute the extra-thyroidal pool of inorganic iodide (PII). This pool is in a dynamic equilibrium with two main organs, the thyroid gland and the kidneys. Figure 14-2 schematically compares the kinetics of iodide in non-pregnant healthy adults with two different intake levels [a) adequate = 150 µg/day; and b) restricted = 70 µg/day] to the pregnancy situation with a comparable iodine intake of 70 µg/day. A normal adult utilizes ~80 µg of iodide to produce thyroid hormones (TH) and the system is balanced to fulfill these daily needs. When the iodine intake is adequate (150 µg/day, the average situation in the U.S., for instance) in non-pregnant conditions, a kinetic balance is achieved with a 35 % uptake of the available iodine by the thyroid (Figure 14-2; panel A). From the 80 µg of hormonal iodide produced each day by TH catabolism, 15 µg of iodide is lost in the feces, leaving 65 µg to be redistributed between the thyroid compartment (hence, providing 25 µg for daily TH production) and irreversible urinary losses. In such conditions, the metabolic balance is in equilibrium, with 150 µg of iodide ‘in’ & the same amount ‘out’, and 80 µg available for daily hormone production. Thus, with an iodine intake level of 150 µg/day (or above) in non-pregnant healthy adults, the system is able to maintain plentiful intra-thyroidal stores, in the order of 15-20 mg of iodine. In contrast, when the iodine intake is restricted to only 70 µg/day (a situation still seen in parts of Western Europe), the system must up-regulate the glandular iodide trapping mechanisms and increase the relative iodine intake to 50 (Figure 14-2; panel B). The higher uptake allows to recover 35 µg of iodine from dietary intake and 33 µg from TH catabolism but, in these conditions in a non-pregnant healthy adult, this is no longer strictly sufficient to sustain requirements for the production of TH, since 80 µg of iodide is still required daily. To compensate for the missing amount (i.e. ~10-12 µg), the system must use the iodine that is stored in the gland, which therefore becomes progressively depleted to lower levels (~2-5 mg of stable iodine). Over time, if the nutritional situation remains unchanged and despite some adaptation of urinary iodine losses, the metabolic balance becomes negative. The thyroid gland tries to adapt by an increased uptake, glandular hypertrophy, and a higher setting of the pituitary thyrostat.
During pregnancy, two fundamental changes take place. There is a significant increase in the renal iodide clearance (by ~1.3- to ~1.5-fold) and, concomitantly, a sustained increment in TH production requirements (by ~1.5-fold), corresponding to increased iodine requirements, from 80 to 120 µg iodide/day. Since the renal iodide clearance already increases in the first weeks of gestation and persists thereafter, this constitutes a non-avoidable urinary iodine loss, which tends to lower circulating PII levels and, in turn, induce a compensatory increase in the thyroidal clearance of iodide. These mechanisms underline the increased physiologic thyroidal activity during pregnancy. Panel C in Figure 9 indicates that when the daily iodine intake is only 70 µg during pregnancy, despite an increase in glandular uptake to 60 %, the equilibrium becomes more or less rapidly unbalanced, since the iodide entry resulting from both uptake and recycling is insufficient to fulfill the increased requirements for TH production.
Calculations show that, in such conditions, ~20 µg of iodine are missing daily and, in order to sustain TH production, the glandular machinery must draw from already depleted intra-thyroidal iodine stores. Thus in about one trimester after conception, the already low intra-thyroidal iodine stores become even more depleted and, when iodine deprivation prevails during the first half, it tends to become more severe with the progression of gestation to its final stages. A second mechanism of iodine deprivation for the mother occurs later in gestation, from the passage of a part of the available iodine from maternal circulation to the fetal-placental unit. The extent of iodine passage has not yet been precisely established. At mid-gestation, the fetal thyroid gland has already started to produce TH, indispensable for the adequate development of the fetus. In summary, augmentation of iodide trapping is the fundamental mechanism by which the thyroid adapts to changes in the iodine supply, and such mechanism is the key to understanding thyroidal adaptation to iodine deficiency. During pregnancy, increased hormone requirements and iodine losses alter the preconception steady-state. When the iodine supply is restricted (or more severely deficient), pregnancy triggers a vicious circle that leads to excessive glandular stimulation (27).
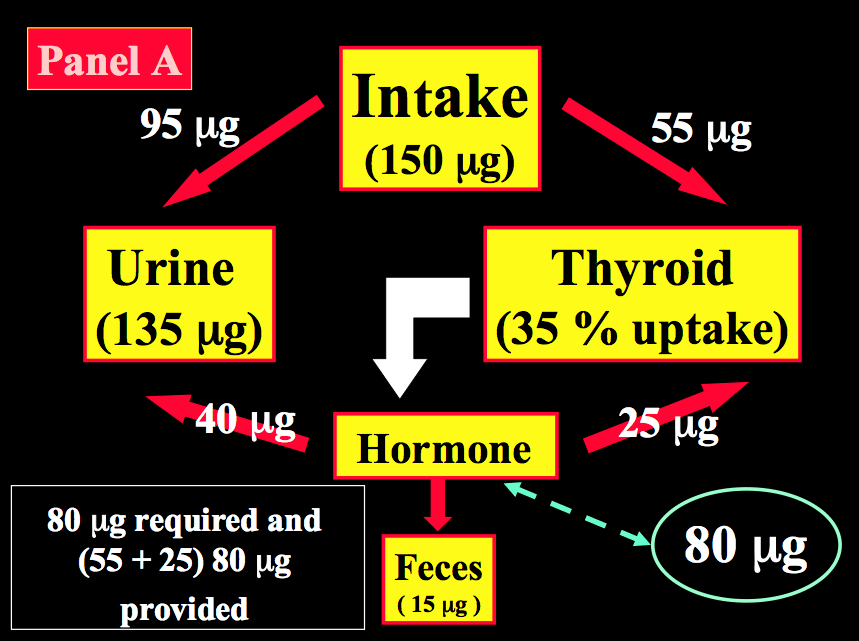
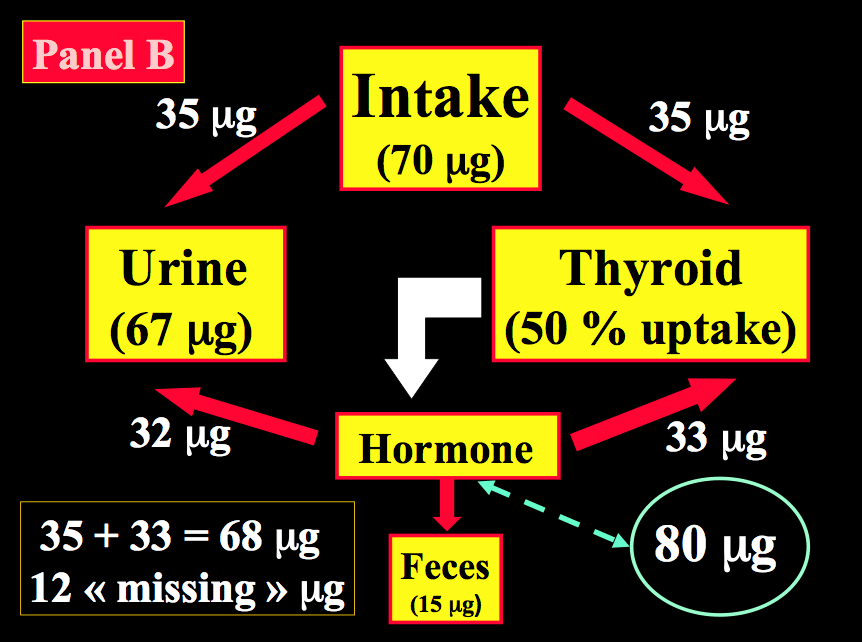
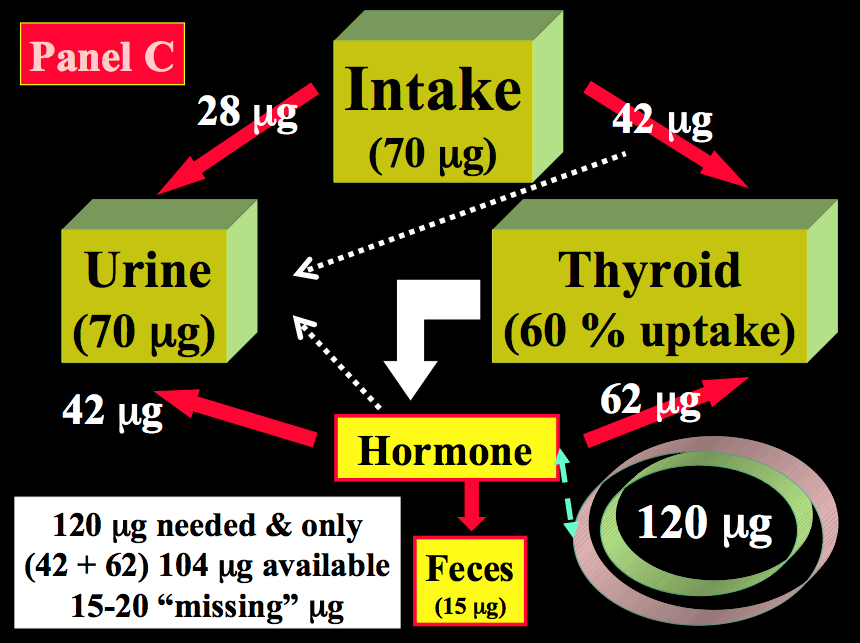
Figure 14-2 Schematic representation of the kinetics of iodide in healthy non-pregnant and pregnant adults. Panel A: non-pregnant adult with an adequate iodine intake of 150 µg/day. Panel B: non-pregnant adult with a restricted iodine intake, corresponding to 70 µg/day. Panel C: the latter condition is compared with an identically restricted level of iodine intake (i.e. 70 µg/day) in a pregnant woman. Daily TH production was set at 80 µg of iodine/day (in non-pregnant) and increased by 1.5-fold to 120 µg/day during pregnancy (from Glinoer, Ref 27).
Goiter Formation In Mother And Progeny
Iodine deficiency during pregnancy, even when considered to be only mild, results in prolonged enhanced thyroidal stimulation and leads to goitrogenesis in both mother and fetus (1). Pregnancy may therefore be considered as an ‘environmental’ factor to induce thyroid pathology in areas with a marginally reduced iodine intake. While goiter formation is not observed in pregnant women who reside in iodine-sufficient regions such as in the USA, several studies from Europe have shown that the thyroid volume (TV) increases significantly during pregnancy (1). In European regions with a sufficient iodine intake, changes in TV remain minimal (10-15% on the average), consistent mainly with vascular thyroid swelling during pregnancy (28). In other European regions with a lower iodine intake, observed changes were much larger, with TV increments ranging between 20-35% on the average, and many women exhibiting a doubling in thyroid size between 1st trimester and term (29,30). In Brussels for instance before iodine supplementation was systematically prescribed, almost 10% of women developed a goiter during pregnancy, which was only partially reversible after parturition (31). In fact there is a high prevalence of thyroid disorders in this region associated with mild iodine deficiency (32). Precise measurements of TV in newborns of these mothers indicated that TVs were 40% larger in newborns from non supplemented mothers (compared with newborns from iodine-supplemented mothers), and thyroid hyperplasia already present in 10% of these infants soon after birth (compared with none in newborns from the iodine-receiving mothers) (33).
Goitrogenesis associated with pregnancy may be one of the environmental factors explaining the preponderance of goiters in the female population. There is an association between parity and thyroid volume in an iodine deficient area (34) and this may be accentuated by active smoking(35) Rarely, a pre-existing goiter may increase in size abruptly during gestation, leading to tracheal compression and respiratory symptoms due partly to intrathyroidal hemorrhage (36,37).
The biochemical markers of enhanced thyroidal stimulation during an otherwise normal pregnancy, when iodine deficiency is present. are firstly relative hypothyroxinemia ( serum free T4 concentrations near (or below) the lower limit of the gestational reference range); Preferential T3 secretion (reflected by an elevated total T3/T4 molar ratio) and a progressive rise in TSH to reach levels that are twice (or even higher) the preconception serum TSH levels (38). In mild to moderate iodine deficiency conditions, serum thyroglobulin (Tg) increases progressively during gestation, so that at delivery, two thirds of women may have supra-normal Tg concentrations. Tg increments correlate well with gestational goitrogenesis, and hence constitute a useful prognostic marker of goiter formation, and its prevention by iodine supplementation (33). The best single parameter to evaluate the adequacy of iodine nutrition in a population is provided by measurements of the urinary iodine excretion (UIE) levels in a representative sample of the population. Although UIE is highly useful for public health estimations of iodine intake in populations, UIE alone is not a valid diagnostic criterion in individuals. Therefore, in the individual at risk of iodine deficiency the markers of thyroid stimulation already described are the best indicators of thyroid stress.
Treatment and Prevention of Maternal Goiter in Pregnancy
In countries with a longstanding and well-established USI program, pregnant women are not at risk of having iodine deficiency. Therefore, no systematic dietary fortification needs to be organized in the population. It should, however, be recommended individually to women to use vitamin/mineral tablets specifically prepared for pregnancy requirements and containing iodine supplements. In countries without an efficient USI program, or with an established USI program where the coverage is known to be only recent or partial, complementary approaches are required to reach the RNI for iodine. Such approaches include the use of iodine supplements in the form of potassium iodide (100-200 µg/day) or the inclusion of KI (125-150 µg/day) in vitamin/mineral preparations manufactured for pregnancy requirements. Finally in those areas with severe iodine deficiency and, in general, no accessible USI program and difficult socioeconomic conditions, it is recommended to administer iodized oil orally as early during gestation as possible. he importance of continuing monitoring of iodine status in the population cannot be overemphasized and has been discussed above.
To prevent gestational goitrogenesis, women should ideally be provided with an adequate level of iodine intake (~150 µg/day) already long before conception. Only then can a long term steady-state be achieved with sufficient intra-thyroidal iodine stores (10-20 mg), thus avoiding triggering of the thyroid machinery that occurs once gestation begins. To achieve such goal, public health authorities ought to implement dietary iodine supplementation national programs in the population. Correcting this public health problem has been the aim of a massive global campaign that was undertaken 15-20 years ago worldwide, based on universal salt iodization (USI), and that has shown remarkable progress so far (33). However, data demonstrate that silent iodine prophylaxis is not sufficient to restore an adequate iodine balance, and that more stringent prophylactic measures need to be taken by public health authorities.
How much supplemental iodine should be given to prevent goiter formation remains a matter of local appreciation and depends primarily on the extent of pre-existing iodine deprivation Since the ultimate goal is to restore and maintain a balanced iodine status in expecting mothers, this can be achieved in most instances with supplements of 100-200 µg of iodine per day given during pregnancy [Fig 14-3] In practice, this requires the administration of multivitamin pills designed specifically for pregnancy purposes and containing iodine supplements. It should be remembered that, because of the longstanding restriction in dietary iodine before the onset of a pregnancy, a lag period of approximately one trimester is inevitable before the benefits of iodine supplementation to improve thyroid function can be observed (33). Even then, despite iodine supplementation, iodine sufficiency may not be attained by all pregnant women (40). Because of the advocacy for salt restriction to reduce cardiovascular mortality a debate has ensued whereby use of iodinated salt seemed to be at odds with this strategy. However, simply increasing the iodine concentration in the salt can accommodate both the reduction in salt intake and the requirement to provide iodine in this way. This strategy has recently been endorsed by WHO (41).
Importantly, it has also emerged that insufficient iodine status is associated with poorer neurocognitive outcome in the offspring. While this has been accepted for many decades in relation to severe iodine (42) deficiency it is now seen to be the case in areas of mild iodine deficiency (43-45). The is accruing evidence that iodine supplementation in pregnancy even in women with mild iodine deficiency is beneficial in improving neurocognitive outcome in the child (46).
Finally, caution is needed to avoid iodine excess to the fetal (47) as well as the maternal (48) thyroid. The fetal thyroid gland is exquisitely sensitive to the inhibitory effects of high iodine concentrations, and a recent study showed that inhibitory effects of high iodine loads could lead to opposite variations in maternal and neonatal thyroid function, i.e. with facilitation of thyroid function in the mother but aggravation in the neonate (49).
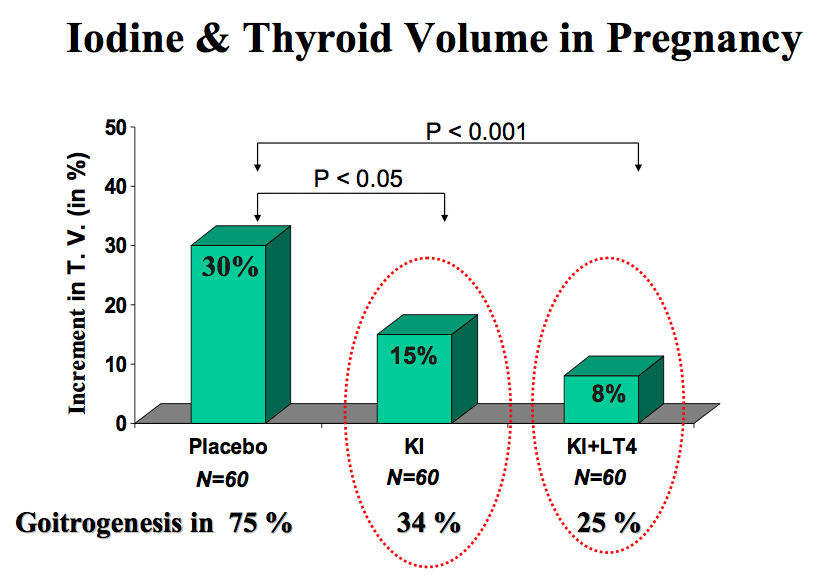
Figure 14-3: Randomized clinical trial with placebo versus KI (100 µg iodine/day) or KI + l-T4 (100 µg iodine/day and 100 µg T4/day) given during pregnancy in women with moderate iodine deficiency and laboratory features of thyroidal stimulation. In the placebo-treated group, TV increased by a mean 30% and goiter formation occurred in 75% of the women. In both actively-treated groups, the increments in TV were significantly reduced (to only 15% and 8%), as was goiter formation (from Glinoer, Ref 33).
In areas with severe iodine deficiency, iodine supplements have been administered to pregnant women using iodized salt, potassium iodide drops and iodized oil (given intramuscularly or orally), as emergency prophylactic and therapeutic approaches to avoid endemic cretinism. Several such programs have conclusively demonstrated their remarkable efficiency to prevent and treat endemic goiter, as well as to eradicate endemic cretinism (8). The results of such studies have indicated that pregnant women who reside in severely iodine-deficient regions can adequately be managed with iodine supplementation. However, except for emergency situations, there is presumably no need to use supra-physiologic amounts of iodine to normalize thyroid function parameters. Although it has not been possible, thus far, in the setting of difficult field studies to evaluate quantitatively the reduction in goiter size or goiter prevalence associated with the clear improvement in thyroid function, goiter reduction is undoubtedly a side benefit of the overall improvement in the iodine nutritional status (50, 51).
In summary, pregnancy is a strong goitrogenic stimulus for both the mother and fetus, even in areas with only a moderate iodine restriction or deficiency. Maternal goiter formation can be directly correlated with the degree of prolonged glandular stimulation that takes place during gestation. Goiters formed during gestation only partially regress after parturition, and pregnancy therefore constitutes one of the environmental factors that may help explain the higher prevalence of goiter and thyroid disorders in women, compared with men. Most importantly, goiter formation also takes place in the progeny, emphasizing the exquisite sensitivity of the fetal thyroid to the consequences of maternal iodine deprivation, and also indicating that the process of goiter formation already starts during the earliest stages of the development of the fetal thyroid gland. Iodine prophylaxis is best achieved using iodised salt. An equal if not more important benefit of using salt supplementation in gestation is the demonstrable positive effect on neurocognition in the child in areas of iodine deficiency or any degree. Monitoring of the population with urinary iodine measurements is essential.
Effects Of human Chorionic Gonadotrophin On Thyroid Function
Human chorionic Gonadotrophin (hCG) is a member of the glycoprotein hormone family that is composed of a common α-subunit and a non-covalently associated, hormone-specific β-subunit. The α-subunit of hCG consists of a polypeptide chain of 92 amino acid residues containing two N-linked oligosaccharide side-chains. The β-subunit of hCG consists of 145 residues with two N-linked and four O-linked oligosaccharide side-chains. The β-subunit of TSH is composed of 112 residues and one N-linked oligosaccharide. The β-subunits of both molecules possess 12 half-cysteine residues at highly conserved positions. Three disulfide bonds form a cystine knot structure, which is identical in both TSH and hCG and is essential for binding to their receptor (LH and hCG bind to the same receptor, the LHCG receptor). A single gene on chromosome 6 encodes for the common αsubunit, while the genes that encode for the β-subunits are clustered on chromosome 19, with seven genes (but only three actively transcribed) coding for β-hCG (52).
The structural homology between hCG and TSH provides already an indication that hCG may act as a thyrotropic agonist, by overlap of their natural functions. Human CG possesses an intrinsic (albeit weak) thyroid-stimulating activity and perhaps even a direct thyroid-growth-promoting activity (52). During normal pregnancy, the direct stimulatory effect of hCG on thyrocytes induces a small and transient increase in free thyroxine levels near the end of the 1st trimester (peak circulating hCG) and, in turn, a partial TSH suppression (1,52.) When tested in bioassays, hCG is only about 1/104 as potent as TSH during normal pregnancy. This weak thyrotropic activity explains why, in normal conditions, the effects of hCG remain largely unnoticed and thyroid function tests mostly unaltered.
The thyrotropic role of hCG in normal pregnancy is illustrated in fig 14- 4. The figure shows the inverse relationship between serum hCG and TSH concentrations, with a mirror image between the nadir of serum TSH and peak hCG levels at the end of the first trimester. The inset in the figure shows that the rise in serum free T4 is proportional to peak hCG values. At this period during gestation, 1/5th of otherwise euthyroid pregnant women have a transiently lowered serum TSH, even below the lower limit of the normal non pregnant reference range (53)
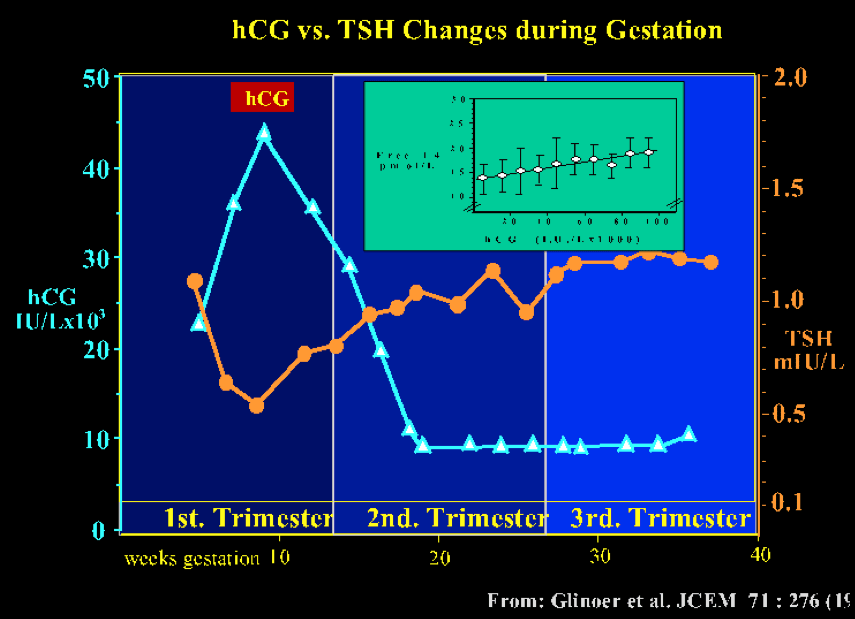
Figure 14-4 The pattern of serum TSH and hCG changes are shown as a function of gestation age in 606 healthy pregnant women. Between 8 and 14 weeks gestation, changes in serum hCG and TSH are mirror images of each other, and there is a significant negative correlation between the individual TSH (nadir) and peak hCG levels (P<0.001) (hCG: ▲-----▲ ; TSH: ●-----●). The inset shows a scattergram of serum free T4 levels in the same women plotted in relation to circulating hCG concentrations (by 10.000 IU/L increment) during the first half of gestation. The figure shows the direct relationship between free T4 and hCG, with progressively increasing free T4 levels (from Glinoer, Ref 53).
Experimental studies with desialylated and deglycosylated hCG, using T3 secretion as the response parameter (in a serum-free culture system with human thyroid follicles), have shown that removal of sialic acid or carbohydrate residues from native hCG transformed such hCG variants into thyroid stimulating super-agonists 54. Further evidence to support the patho-physiological role of hCG to stimulate excessively the human thyroid gland is can be found in studies of patients with hydatidiform mole and choriocarcinoma (see Chapter 13). In these conditions, clinical and biochemical manifestations of hyperthyroidism often occur and, as expected, the abnormal stimulation of the thyroid is rapidly relieved after appropriate surgical treatment (55).
Changes In Circulating Thyroid Hormone Binding Proteins
The increase in total serum T4 and T3 that occurs during pregnancy is due to an increase in serum thyroxine binding globulin (TBG) concentrations. Changes in TBG happen early and, by 16-20 weeks of gestation, TBG concentrations have doubled (1). The cause of the marked increase in serum TBG is probably multifactorial. TBG biosynthesis was increased, after estradiol priming, in primary cultures of hepatocytes from Rhesus monkeys (56) and changes in the glycosylation patterns of TBG, induced by estrogen, have indicated
that the increase in circulating levels of TBG was due in large part to a reduction of its plasma clearance (57). However, the lack of increase in other binding proteins (CBG & SHBG) by estrogen in HEP-G2 cells raised the possibility that other factors might be operative in the pregnant state. (57). Sera of pregnant or estrogen-treated individuals show a marked increase in the more heavily sialylated fractions of TBG. This increase in the sialic acid content of TBG inhibits the uptake of the protein by specific asialylo-glycoprotein receptors on hepatocytes, and the more heavily sialylated proteins from pregnant sera have therefore a longer plasma half-life (58). Such alterations in sialylation are not found in TBG isolated from patients with congenital TBG elevation, the latter being due to a true over-production of the protein (59) Thus, in addition to the stimulatory estrogen effects of estrogen on TBG synthesis, a major contribution to the increased TBG concentration during pregnancy is the reduced clearance of the protein. Delivery leads to a rapid reversal of this process and serum TBG concentrations return to normal within 4-6 weeks. Serum T4 and T3 also return to pregestational serum levels. In addition to the 2 to 3-fold increase in serum TBG, modest decreases in both serum transthyretin (TTR) and albumin are commonly found in pregnancy, but the physiological impact of these changes, if any, is unknown.
In a 42-year-old woman who had both established hypothyroidism and inherited TBG deficiency, the baseline TBG level was 70% below the average baseline level of non-TBG-deficient women (60).. During her pregnancies, serum TBG levels rose, although remaining at only one half the usual increment in TBG associated with normal pregnancy. Despite the patient’s low baseline TBG level and blunted pregnancy-associated TBG rise, she required an increase in her thyroxine replacement doses that mirrored those observed in hypothyroid, but non-TBG-deficient pregnant women. It was suggested therefore that an increase in TBG concentration was not the key determinant for the increase in thyroxine requirement in pregnancy. However, an alternative explanation was proposed (61). In the normal situation before pregnancy, the homeostasis of thyroid function is ensured by the equilibrium between a serum total T4 of ~100 nmol/L and a TBG concentration of ~260 nmol/L. This equilibrium implies, in turn, that ~75 % of the circulating T4 is bound to TBG and that ~35-40 % of circulating TBG is saturated by T4. During a normal pregnancy, the extracellular TBG pool expands from ~3,000 to ~7,000 nmol/L. Thus, for the homeostasis of free thyroid hormones to be maintained, the extra-thyroidal total thyroxine pool must parallel this expansion, and this can only be achieved by the thyroid gland filling up the progressively the increased hormonal pool during the first half of pregnancy (see Figure 14-5). In the exceptional case of Zigman, when this partially TBG-deficient patient was not pregnant, her serum total T4 was ~70 nmol/L and TBG ~80 nmol/L, indicating that her circulating TBG was almost completely saturated by T4, because of her severe restriction in the TBG binding capacity. However in the non pregnant condition, only a relatively small fraction of the patient’s circulating T4 could be bound to TBG: ~50%. When the patient became pregnant, her TBG deficiency was still partially responsive to estrogen induction and TBG increased 3-fold to ~240 nmol/L and total T4 to ~90 nmol/L. In other words, her total T4 concentrations had to be raised by ~30% (via an increase in thyroxine replacement), hence allowing to restore a TBG binding saturation level by T4 of ~35%, equivalent to what is observed at the onset of pregnancy in non-TBG-deficient women. Thus, the increment required in l-T4 dosage was precisely of the same proportion than that anticipated from the partial rise in serum TBG during pregnancy.


Figure 14-5 The upper panel illustrates the rapid changes that occur in serum total binding capacity of TBG during the first half of gestation under the influence of elevated estrogen levels. The lower panel shows that, in order to maintain unaltered free T4 levels, the markedly increased TBG extra-cellular pool must steadily be filled with increasing amounts of T4, until a new equilibrium is reached. This is achieved during pregnancy via an overall ~50% increase in thyroid hormone production.
Increased Plasma Volume
The increased plasma concentration of TBG, together with the increased plasma volume, results in a corresponding increase in the total T4 pool during pregnancy. While the changes in TBG are most dramatic during the first trimester, the increase in plasma volume continues until delivery. Thus, for free T4 concentration to remain unaltered, the T4 production rate must increase (or its degradation rate decrease) to allow for additional T4 to accumulate. One would predict that in a situation where the T4 input was constant, there would be an iterative increment in T4 as TBG increases, due to reduced T4 availability to degradation enzymes. The evidence that thyroxine requirements are markedly enhanced during pregnancy in hypothyroid treated women (see section on maternal hypothyroidism) strongly suggests that not only T4 degradation is decreased in early pregnancy but also that an increased T4 production occurs throughout gestation to maintain the homeostasis of free T4 concentrations (1)
Thyroxine Production Rate
The only direct measurements of T4 turnover rates in pregnancy were obtained nearly 40 years ago (62). In eight pregnant subjects (4 in 1st half & 4 in 2nd half of gestation), T4 turnover rates were estimated not to be significantly different from those of non-pregnant subjects. However, based on several considerations discussed above from more recent work, it can now be concluded that the T4 production rate is enhanced during pregnancy. Globally, it is accepted that there is a ~50 % increase in the production of T4 during gestation (1)
The Placenta
During the first trimester the human conceptus is surrounded by the placenta, which acts as an exchange unit for nutrients and waste products. The primary barrier to exchange between mother and fetus is the syncytiotrophoblast layer of the placental chorionic villi which has effective tight junctions and prevents the free diffusion of thyroid hormones across it.
The human placenta in addition to this cellular barrier also regulates the amounts of thyroid hormones passing from the mother to the fetus through its expression of placental thyroid hormone transporters, thyroid hormone binding proteins, iodothyronine deiodinases, sulfotransferases and sulfatases (63,64). The transport of iodine through the placenta is also important as the organ has shown to actively concentrate the anion (65). Oxytocin and hCG may also promote placental iodide uptake helping to protect against fetal iodine deficiency(66) Placental NIS protein levels are significantly correlated with gestational age during early pregnancy and increase with increased placental vascularization. This would lead to increased iodide supply to meet increased fetal requirements for thyroid hormone synthesis as the pregnancy progresses (67) The precise details of placental iodide concentration are unclear. It is interesting that in mothers who smoke placental iodide transport seemed unaffected despite high thiocyanate levels, suggesting that thiocyanate-insensitive iodide transporters alternative to NIS are active or that NIS in the placenta is autoregulated to keep iodide transport unaltered.(68).
Fetal circulating concentrations of total T3 are at least 10 fold lower than total T4. Unlike adults, the proportion of free unbound T4 is also higher than bound T4 in early gestation. Free T4 levels are determined by the fetal concentrations of the thyroid hormone binding proteins in the circulation and coelomic cavity and the amount of maternal T4 crossing the placenta. The concentration of free T4 in the coelomic fluid in the first trimester is approximately 50% of that found in the maternal circulation and could therefore exert biological effects in fetal tissues.
The human placenta expresses iodothyronine deiodinases type II (D2) (which activates T4 to T3) and type III (D3) (which inactivates T4 and T3). The principle subtype in the placenta is D3, having 200 times the activity of D2. D3 effectively metabolises most of the maternal T4 presented to the placenta.: still a physiologically relevant amount of T4 is transferred to the fetus. Both D2 and D3 activity per gram of placenta decrease with advancing gestation.( 64). Decidualization, which is a characteristic of the endometrium of the pregnant uterus and a response of maternal cells to the hormone progesterone, is also dependent on the strong expression and tight control of the type 3 deiodinase (69) to regulate the local T3 concentration.
A range of thyroid hormone transporters including monocarboxylate transporters (MCT) 8 and 10, system-L amnio acid transporters (LAT1 and LAT2) and organic anion transporting polypeptides (OATP) 1A2, 4A1 and Oatp1c1 have been located at the apical and basolateral membranes of the syncytiotrophoblasts (70,71). These transporters may facilitate thyroid hormone transfer across the cell barrier from the mother to the fetus ( Figure 14-6). In fact, it seems that the syncytiotrophoblast may control the quantity and forms of thyroid hormones taken up by the human placenta so that this this could be critical in regulating transplacental thyroid hormone supply from the mother to the fetus (72). Studies in the mouse with human placental tissue indicate that MCT8 makes a significant contribution to T3 uptake into human trophoblast cells and has a role in modulating human trophoblast cell invasion and viability (73). Transthyretin (TTR), a serum thyroid hormone binding protein, appears to play an important role in the delivery of maternal thyroid hormone to the developing fetus. (74).The human placenta secretes TTR into maternal and fetal circulations and the placental TTR secreted into the maternal placental circulation can be taken up by trophoblasts and translocated to the fetal circulation, forming a TTR shuttle system. This may have important implications for materno-fetal transfer of thyroid hormones (75).
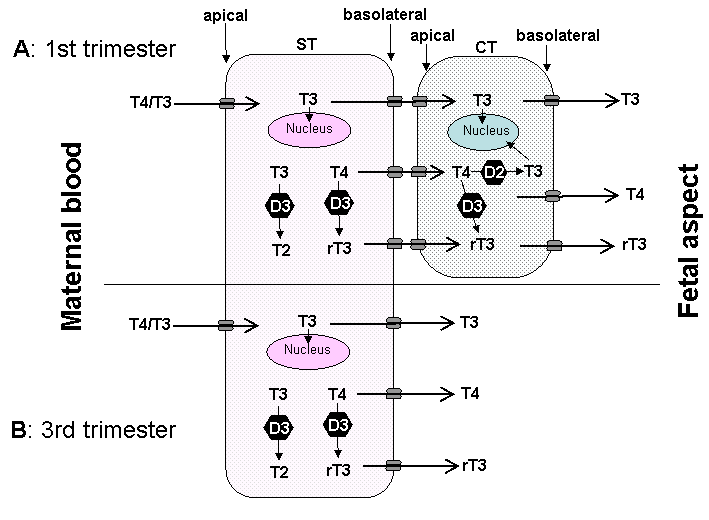
Fig 14-6 The passage of T4 and T3 from the maternal to fetal circulation requires negotiation through the apical membrane (maternal-facing) and the basolateral membrane (fetal-facing) of syncytiotrophoblasts (ST), and in the first half of pregnancy (A) through the plasma membranes of cytotrophoblasts (CT) as well. The localisation and function of the six different TH transporters ( ) present in the placenta may differ. These include monocarboxylate transporters (MCT) 8 and 10, system-L amnio acid transporters (LAT1 and LAT2) and organic anion transporting polypeptides (OATP) 1A2 and 4A1.There may also be other yet to be identified TH transporters. In addition, T4 and T3 are subject to metabolism by deioidnase type 2 (D2) and type 3 (D3) as they pass through the trophoblasts. (From 63)
In addition to the regulation of transplacental thyroid hormone transfer for fetal development, human placental development is itself is responsive to thyroid hormone from early in gestation with evidence of trophoblastic expression of thyroid hormone receptors. T3 has been shown to promote proliferation, invasion and production of epidermal growth factor by 1st trimester primary trophoblast cultures. In humans T3 has been shown to suppress apoptosis and down regulate Fas and Fas-ligand expression (76). It has been postulated that abnormal thyroid hormone levels could give rise to malplacentation which underlie the association between maternal thyroid dysfunction and adverse obstetric outcome.
The inner-ring deiodination of T4 catalyzed by the type 3 deiodinase enzyme is the source of high concentrations of reverse T3 present in amniotic fluid, and the reverse T3 levels parallel maternal serum T4 concentrations (77). This enzyme may function to reduce the concentrations of T3 and T4 in the fetal circulation (the latter being still contributed by 20-30 % from thyroid hormones of maternal origin at the time of parturition), although fetal tissue T3 levels can reach adult levels due to the local activity of the Type 2 deiodinase (see Chapter 15). Type 3 deiodinase may also indirectly provide a source of iodide to the fetus via iodothyronine deiodination. Despite the presence of placental Type 3 deiodinase, in circumstances in which fetal T4 production is reduced or maternal free T4 markedly increased, transplacental passage occurs and fetal serum T4 levels are about one third of normal.(78). Thyroxine can be detected in amniotic fluid prior to the onset of fetal thyroid function, indicating its maternal origin by transplacental transfer (79).. Figure 14-7 depicts the steep maternal to fetal gradient of total T4 concentrations in early pregnancy stages. Between 6-12 weeks gestation, if maternal total T4 concentration is set to represent 100%, the total T4 concentration in the coelomic fluid would represent 0.07% and T4 in the amniotic cavity as little as 0.0003-0.0013% of maternal total T4 concentrations. (Because of low levels of binding proteins in the amniotic cavity, the ratio of amniotic/serum FT4 is much higher.) Thus, the placental barrier to maternal iodothyronines is not impermeable to the transplacental passage of thyroid hormones of maternal origin, even in the 3rd trimester (63). Even though very small quantitatively, such concentrations may qualitatively represent an extremely important source of thyroid hormones to ensure the adequate development of the feto-maternal unit.

Figure14- 7 Steep gradient between maternal concentrations of thyroid hormones (Total T4) and those measured in the coelomic fluid and amniotic cavity with the developing embryo, during early stages of gestation.
A review concluded that a local action of thyroid hormones on female reproductive organs and embryo seemed to be crucial for a successful pregnancy and alterations of the highly regulated local activity of thyroid hormones may play an important, previously underestimated role in early pregnancy and pregnancy loss (80). Furthermore, studies in rats suggest that transcriptomic profiling of the utero-placental compartments, in addition to analysis of mRNA expression of key thyroid hormone placental signaling genes, may predict offspring obesity (81) It is important to note that there is increasing evidence that placental and angiogenic factors are affected by thyroid hormones (82). In isolated human decidual cells T3 regulates angiogenic growth factor and cytokine secretion in a cell-type specific and gestational age specific manner(83). In a large number of women from the Generation R study it was found that high levels of pro- and anti-angiogenic factors may be a risk factor for adverse pregnancy outcomes through their effects on maternal thyroid function (84). Overall thyroid hormones modulate inflammatory processes and are implicated in placental development and disease (85).
Immunological and hormonal aspects of normal pregnancy (Table 14-3)
Pregnancy has a significant effect on the immune system, in order to maintain the fetal-maternal allograft, which is not rejected despite displaying paternal histocompatability antigens. While there is no overall immunosuppression during pregnancy, clinical improvement usually occurs in patients with immunological disorders such as rheumatoid arthritis (RA) when they become pregnant. Clinical improvement occurs as well in psoriatic arthritis and Graves' disease. On the other hand, systemic lupus erythematosus (SLE) may flare during pregnancy.
Table 14-3 Immunological and Hormonal Features of Pregnancy
Clinical: Improvement in Graves’ hyperthyroidism
Rheumatoid arthritis
Psoriatic arthritis and other autoimmune diseases
Trophoblast: HLA G expression
Fas ligand expression
Lymphocytes: Th2 response
Th2 cytokines produced by the fetal/placental unit
Critical role of Treg cells (CD4+CD25+) in maternal tolerance
Possible role of Th17 lymphocyte subset
Hormones: Progesterone increase – reduction in B cell activity
Oestrogen increase – Fall in autoantibody levels
Cortisol,1,25 vitamin D and norepinephrine all affect the immune response
Other Galectin-1
The trophoblast does not express the classical major histocompatibility complex (MHC) class Ia or II which are needed to present antigenic peptides to cytotoxic cells and T helper cells respectively. Instead HLA-G, a non-classical MHC Ib molecule is expressed which may be a ligand for the natural killer (NK) cell receptor so protecting the fetus from NK cell damage ; it may also activate CD8+ T-cells that may have a suppressor function. Human trophoblasts also express the Fas ligand abundantly, thereby contributing to the immune privilege in this unique environment possibly by mediating apoptosis of activated Fas expressing lymphocytes of maternal origin.
T-cell subset studies in pregnancy are discrepant, as peripheral blood CD4+ and CD8+ cell levels have been variously reported to decline, remain unchanged and increase during pregnancy. Although, the distinction between Th1 (T cell helper 1) and Th2 (T cell helper 2) immune responses in humans remains less clear than in the mouse the general agreement is that in pregnancy there is a bias towards a Th2 response (3) This seems to be achieved by the fetal/ placental unit producing Th2 cytokines, which inhibit Th1. Th1 cytokines are potentially harmful to the fetus as, for example, interferon alpha (IFNα) is a known abortifacient. The characterisation of regulatory T cells (CD4+CD25+), a T cell subset that can prevent experimental autoimmune disease, has improved the understanding of the immunological maintenance of pregnancy. It is now thought that these cells are one of the main groups of T cell subsets which allow tolerance of the fetal semi allograft. They may be found in the decidua as well as in the maternal circulation and regulate autoimmune responses.(3)
Assessment of Thyroid Function in Pregnancy
As there is significant overlap between the symptoms experienced by normal euthyroid pregnant women and those with thyroid dysfunction clinical diagnosis is not always straightforward. Because thyroid physiology is altered in pregnancy it has become clear during the past decade that normative gestational reference ranges for thyroid hormone analytes are necessary. Most clinical laboratory reports only provide non-pregnant reference intervals for the interpretation of laboratory results...
The range of normal serum total T4 is modified during pregnancy under the influence of a rapid increase in serum TBG levels. The TBG plateau is reached at mid-gestation (see Figure 14-8, upper left panel) . If one uses total T4 to estimate thyroid function, the non pregnant reference range (5-12 µg/dl; 50-150 nmol/L) can be multiplied t by 1.5 during pregnancy. However, it should be noted that since total T4 values only reach a plateau around mid-gestation, such adaptation is only fully valid during the 2nd half of gestation (see Figure 14-8, upper right panel) . Thus, the use of total T4 does not provide an accurate estimate of thyroid function during early gestation.. However, the free thyroxine index (“adjusted T4“) appears to be a reliable assay during pregnancy (87).
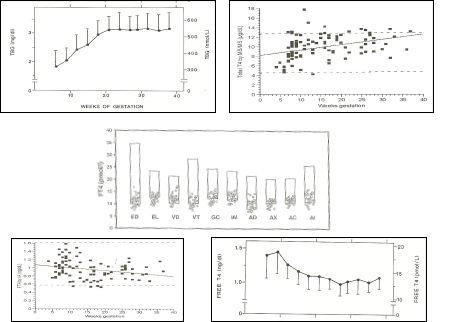
Figure 14-8
Upper left panel: pattern of changes in serum TBG concentrations (mean + sd) in 606 normal pregnant women ( Ref 1). Upper right panel: pattern of changes in serum total T4 concentrations (individual results) in 98 normal pregnancies ( Ref 86). Middle panel: free T4 measurements in 29 women in the 9th month of gestation, using equilibrium dialysis (ED), and 9 different immunoassays (EL: Elecsys; VD: Vidas; VT: Vitros ECi; GC: Gamma-Coat; IM: Immunotech; AD: Advantage; AX: AxSYM; AC: ACS: 180; AI: AIA Pack). The boxes show the non-pregnant upper and lower reference intervals. The percentages given in the upper part of the figure show the mean decrement (in percent) of serum free T4 values compared with the mean free T4 reference value for non-pregnant subjects, provided by the manufacturer. It can be seen that free T4 values were decreased by 40% when measured by ED, and by 17-34% depending on the immunoassay employed . Lower left panel: pattern of changes in serum free T4 concentrations (individual results) in 98 normal pregnancies in the USA, with an adequate iodine intake (Ref 86). Lower right panel: pattern of changes in serum free T4
Although gestation specific reference intervals for thyroid function tests are not currently in routine use in most laboratories there has been intense activity world wide in the development of such ranges (4). . Irrespective of the techniques used to measure free T4 during pregnancy, there is a characteristic pattern of serum free T4 changes during normal pregnancy. This pattern includes a slight and temporary rise in free T4 during the first trimester (due to the thyrotropic effect of hCG) and a tendency for serum free T4 values to decrease progressively during later gestational stages (88). In iodine-sufficient conditions, the physiologic free T4 decrement that is observed during the second and third trimester remains minimal (~10%), while it is enhanced (~20-25%) in iodine-deficient nutritional conditions (see Figure 14-4, lower left and right panels, respectively).
Unfortunately, few if any FT4 immunoassay manufacturers provide appropriate normal pregnancy-related reference intervals that are method-specific (specific for the method used for hormone analysis). It is therefore imperative that method- and gestation-specific reference intervals for FT4 are derived in the appropriate reference populations to prevent misinterpretation of thyroid status in pregnant women. While ‘gold standard methodology’ (e.g. tandem mass spectrometry) is useful for accurate standardisation of values (89), in practice the use of kit assays for free thyroid hormones as well as routine estimation of total bound hormones are used. These are based on analogue methods that rely on the concentrations of binding-proteins, are method-dependent and may give misleading FT4 and FT3 values in pregnancy. For example a commercially available FT4 assay has shown that it correlates more closely to total T4 assays than to FT4 measured following physical separation from binding proteins (90). A comparison of 5 different commercial assays for FT4 and FT3 showed significant interassay variation underlining the necessity for individual laboratory based reference ranges (91). Reference values for FT4 were different when measured by 7 different kits(92).Even in the same region, the use of gestational age specific reference ranges from different laboratories led to misclassification (93). FT4 assays are considered to be flawed and unreliable during pregnancy (87) but there are data showing that, despite susceptibility towards binding protein alterations, these assays may indeed reflect the gold standard assays (94). A mathematical analysis of measurement of total T4 or Free T4 in pregnancy concluded that free hormone measurement is indeed as good as the total assay (95). Gestational reference ranges for theses hormones as well as TSH should be available in every hospital dealing with pregnancy. (4). Table 14-4 shows selected reference ranges for FT4, FT3 and TSH published up to 2008. Since 2008 further country data has been documented (103-107). with emphasis being placed on obtaining first trimester ranges . Concern has been noted with regard to previous suggestions that the upper limit for TSH should be 2.5mIU/L in the first trimester(108) because of ethnic variation(109). A more realistic figure may be 3.0-4.0 mIU/L(110).
Table 14-4
Selected Trimester-Specific, Method-Specific FT4, FT3 and TSH Medians (±SD) or Means* (±SE) and Reference Intervals
| Country (ref) |
Gestation (n=) |
FT4 Median (Reference Interval) or Mean (±SEM)* |
TSH Median (Reference Interval) or Mean (±SEM) |
FT3 Median (Reference Interval) or Mean (±SEM) |
FT4/FT3 Instrument |
| pmol/L | mIU/L | pmol/L | |||
|
Australia 2008 (96) Means* |
T11 (1,817) | 13.5 (10.4-17.8) | 0.74 (0.02-2.15) | 4.35 (3.3-5.7) | Abbott Architect i |
| Non-pregnant (100) | (9.0-19.0) | (0.40-4.00) | (3.0-5.5) | ||
| Canada2 2008 (97) | T1 (224) | 15.0 (11.0-19.0) |
Roche Cobas e601/E-170 |
||
| T2 (240) | 13.5 (9.7-17.5) | ||||
| T3 (211) | 11.7 (8.1-15.3) | ||||
|
India 2008 (98) ID2 |
T1 (107) |
14.46 (12.00-19.45) 4 | 2.1 (0.60-5.00) 4 | 4.4 (1.92-5.86)4 |
Roche Cobas e411/Elecsys |
| T2 (137) | 13.4 (9.48-19.58) 4 | 2.4 (0.40-5.78) 4 | 4.3 (3.20-5.70)4 | ||
| T3 (87) | 13.28 (11.30-17.71) 4 | 2.1 (0.74-5.70) 4 | 4.1 (3.30-5.18)4 | ||
|
Switzerland 2007 (99) ID2 |
T1 (783) |
13.79 (10.53-18.28) TT45 110.64 (72.27-171.18) nmol/L |
1.04 (0.88-2.83) |
4.67 (3.52-6.22) TT3 1.78 (1.25-2.72) |
Abbott Architect i2000SR |
| T2 (528) |
12.17 (9.53-15.68) TT43 134.84 (94.77- 182.51) nmol/L |
1.02 (0.20-2.79) |
4.47 (3.41-5.78) TT3 2.15 (1.43-3.16) |
||
| T3 (598) |
11.08 (8.63-13.61) TT43 136.65 (94.88- 193.35) nmol/L |
1.13 (0.31-2.90) |
4.27 (3.33-5.59) TT3 2.19 (1.40-3.16) |
||
| USA 2008 (100) | T1 (585) |
9.9 (6.8-13.0) FT4 pmol/L |
1.1 (0.04-3.60) | Siemens Immulite 2000 | |
|
USA 2008 (101) |
T1 (9,562) | 1.13 (1.00-1.20) FT4 ng/dL | 1.05 (0.63-1.66) |
Siemens Immulite 2000
|
|
| T2 (9,562) | 1.013 (0.92-1.11) ng/dL | 1.23 (0.82-1.78) | |||
|
USA 2007 [NHANES III] (88) Means |
T1 (71) |
TT43 141.35 (3.07) nmol/L (123.64-158.29) |
0.91 (0.17) 0.28-1.06 |
Roche
|
|
| T2 (83) | TT43 152.95 (2.17) nmol/L (146.36-165.13) |
1.03 (0.20) 0.57-1.28 |
|||
| T3 (62) | TT43 142.64 (3.73) nmol/L (126.46-160.69) |
1.32 (0.27) 0.69-2.87 |
|||
|
USA (86) |
T1 (59) |
FT4 1.13 (0.23) ng/dL TT43 114.29 (34.36) nmol/L |
1.13 (0.69)
|
LC/MS/MS API 4000
|
|
| T2 (35) |
FT4 0.92 (0.30) ng/dL TT43 137.32 (24.97) nmol/L |
1.13 (0.54) | |||
| T3 (26) |
FT4 0.86 (0.21) ng/dL TT43 138.48 (25.74) nmol/L |
1.04 (0.61)
|
|||
| Non-pregnant (26) |
FT4 0.93 (0.25) ng/dL TT43 91.63 (10.17) nmol/L |
1.73 (1.13) | |||
|
USA 2007 (102)
|
T2 (2,551)
|
FT4 12.0 (9.3-15.2) TT4 128 (89.0-176.0) |
1.14 (0.15-3.11) |
FT3 4.85 (3.82-5.96) TT3 2.62 (1.82-3.68) |
Abbot Architect i2000SR |
|
* Means as marked. All are geometric means (±Standard error of the mean, SEM). 1 T 1=First trimester, Gestation weeks (GW) 1-14; T2=Second trimester, GW 15-28 ; T3=Third trimester, GW 29-40 2 Iodine nutrition status was not assessed in this study; iodine deficiency has not been ruled out. 3 To convert to SI units use www.unc.edu/~rowlett/units/scales/clinical_data.html To convert to SI units: T4 μg/dL x 12.87 to nmol/L; T3 ng/dL x 0.0154 to nmol/L; FT4 ng/dL x 12.87 to pmol/L. 4Reference interval is 90% 5 IQR=interquartile range Adapted from ref 4
|
|||||
In general, serum TSH concentrations provide the first clinical indicator for thyroid dysfunction. Due to the log-linear relationship between TSH and FT4, very small changes in T4 concentrations will provoke very large changes in serum TSH. However, in pregnancy, thyroid and pituitary functions are less stable. During early gestation, TSH is suppressed by 20-50% by week 10 due to the steep increase in hCG concentrations. Therefore, maternal serum TSH does not provide a good indicator for the control of treatment of thyroid dysfunction in the first trimester unless trimester specific ranges are available. False readings can lead to maternal under-replacement with LT4, or overtreatment with anti-thyroid drugs both of which can result in both maternal hypothyroidism and an increased risk for adverse fetal brain development. TSH is however the best measure of thyroid function during the 2nd and 3rd trimesters
Reliable trimester-specific (or gestation-specific) reference intervals for TSH are also now available, being based on an adequate sample size comprised singleton pregnancies in an iodine sufficient, antibody-free population ( see fig 14-9) The importance of the reference range is shown by the fact that 28% of singleton pregnancies with a serum TSH greater than 2 standard deviations above the mean would not have been identified when using the nonpregnant serum TSH range. The individual genetic set-points of a population may result in an intra-individual variability of the thyroid hormone levels, reflected by the reference intervals (112). Also, Afro Americans in USA have lower TSH values in gestation (113), and data from The Netherlands has also documented ethnic differences in thyroid function tests in pregnancy (114). These should be recognized when deriving normative reference ranges.
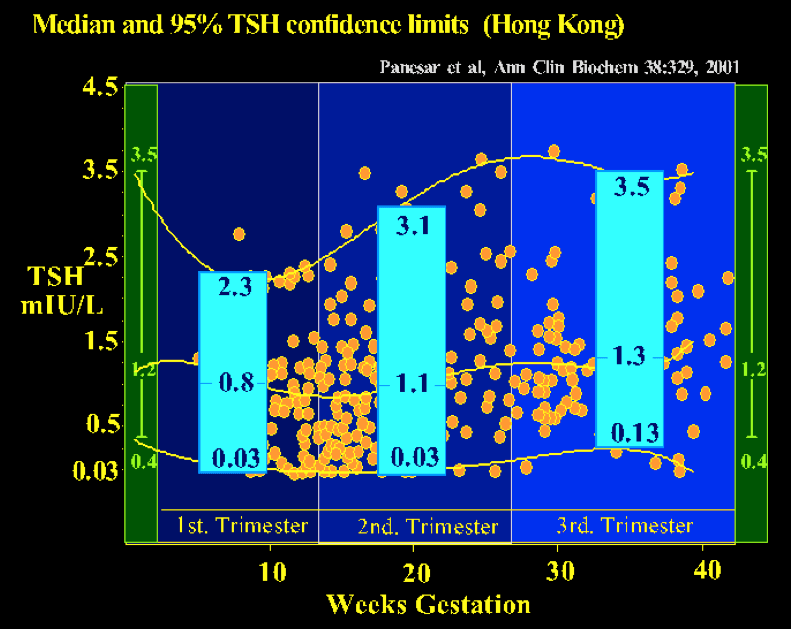
Figure 14-9 Gestation-related reference intervals for serum TSH in a Chinese population (343 healthy pregnant women & 63 non-pregnant controls). The median, 2.5th and 97.5th percentiles for serum TSH values are shown in the blue boxes for each trimester. Gestation-specific reference intervals for TSH should alleviate the potential risk of misinterpretation of thyroid function tests in pregnancy (from Panesar, Ref 111).
In summary, TSH levels may be misleading in the first trimester and T4 values either total or free will give a more accurate estimate of clinical status. Later in gestation TSH levels are reliable whereas T4 may fall especially in the 3rd trimester but this does not indicate hypothyroidism. In some cases, serum anti-TPO antibodies, anti-Tg and/or TSH receptor antibody levels can provide other information; TPO antibodies can predict the risk of hypothyroidism. Ethnic differences in trimester specific reference ranges should be noted. For example the upper limit of TSH in the first trimester was much higher than 2.5 mIU/L in Chinese pregnant women(108). Large differences in thyroid function reference intervals between different populations of pregnant women are seen due to assay variation as well as ethnicity and body mass index (115,116) In pregnant women with low TSH hyperthyroidism, TSH receptor antibodies are observed in 60–70% of the cases.
Hyperemesis Gravidarum
Vomiting occurs in normal pregnancy during the 1st trimester and ceases usually by the 15th week. Prolonged nausea and severe vomiting in early pregnancy that causes greater than a 5% weight loss, dehydration and ketonuria is defined as Hyperemesis Gravidarum (HG) and occurs in 0.5-10 cases per 1,000 pregnancies (117). Hyperemesis is associated with high hCG levels occurring at this time, but the exact cause remains uncertain. For unknown reasons, HG is more prevalent in Asian than Caucasian women. Norwegian data from 1967 to 2005 showed a prevalence of 0.9% but it affected 2.2% of Pakistani women; 1.9% of Turkish women and 0.5% of Norwegian women (118); a familial aggregation suggesting strong evidence for a genetic component of HG. has been suggested (119) When the charge-isoforms profiles of circulating hCG were compared in HG women with different ethnic backgrounds (Samoan vs. European). an increase in total serum hCG concentrations as well as an increase in the proportion of acidic hCG variants in the women suffering from HG, compared with matched control subjects was noted (120). The same study also confirmed the known association between hCG concentrations in early pregnancy and elevations in thyroid hormone levels .While there was no major association between HG and ethnic background, the authors observed a high prevalence of recurrent HG and a familial predisposition for this condition, suggesting that either long-term environmental factors or genetic factors may play a crucial role in the pathogenesis of HG and gestational transient non autoimmune thyrotoxicosis (121)
Thirty to sixty percent of patients with HG have elevations of serum free thyroid hormone concentrations with a suppressed TSH . Women with hyperemesis and elevated thyroid hormone levels most commonly do not have other clinical evidence of primary thyroid disease, such as Graves’ disease. A minor proportion of these patients may have clinical hyperthyroidism, termed ‘gestational hyperthyroidism’ or ‘gestational transient thyrotoxicosis’ (GTT). Graves’ disease can also occur coincident with hyperemesis . Many common signs and symptoms of hyperthyroidism may be mimicked by a normal pregnancy. The clinical challenge is therefore to differentiate between these two disorders
The etiology of excessive thyroid stimulation is considered to be hCG itself (or derivatives of hCG) via a direct stimulation of the thyroid cells through binding of hCG to the TSH receptor (52).. A case of severe HG was reported where the gestational thyrotoxicosis associated with HG was due to a mutation of the TSH receptor, providing hypersensitivity to hCG (122 ). Only one other similar case has since been reported world wide (123). In virtually all patients with gestational hyperthyroidism, appropriate fluid replacement will lead to resolution of the clinical symptoms. As gestation proceeds and hCG levels progressively fall, normal thyroid function is resumed. In severe (but rare) cases, antithyroid drug treatment may be required (described in more detail below). Several investigators have observed that there may even be more subtle form of hyperthyroidism associated with morning sickness (124) Severity of emesis was correlated with serum free T4 and hCG levels and inversely with the degree of TSH suppression (124), suggesting strongly that HG may reflect the extreme end of the spectrum of physiological changes that occur at this time in normal pregnancy (Fig 14-10). It is possible that high hCG levels cause both an increased estrogen secretion as well as thyroid hyperfunction, and in turn explain the coexistence of nausea and vomiting with hyperthyroidism.
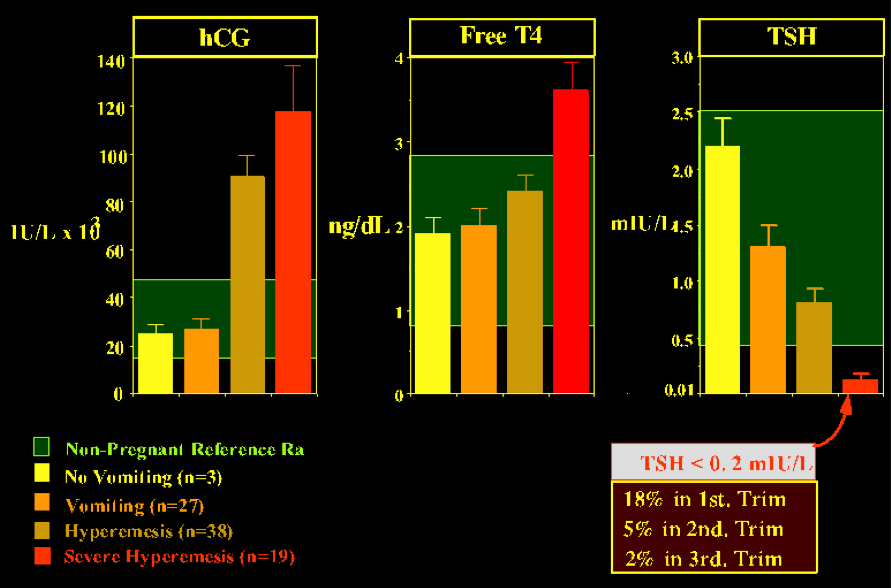
Figure 14-10 Relationship between the severity of vomiting and the mean (with SE) serum concentrations of hCG, free T4, and TSH. The inset in the lower right part of the figure shows the prevalence of suppressed TSH levels, for each trimester of gestation, in a cohort of normal pregnant women. The data were graphically adapted by Carole Spencer (thanks to Carole for allowing me to borrow the slide). The figures are based on studies by Goodwin (Ref 118)
AUTOIMMUNE THYROID DISEASE AND PREGNANCY
The whole spectrum of autoimmune thyroid disease occurs in pregnancy and the postpartum period (see table 14-5). These conditions and their relation to pregnancy are discussed in the rest of this chapter
| Table 14-5. Autoimmune Thyroid Disease During Pregnancy and the Postpartum Period |
| 1. Asymptomatic autoimmune disease a) Thyroid antibody positiive (TPOAb and/TgAb) :euthyroid b) Subclinical hypothyroidism2 Primary hypothyroidism a) Thyroid destruction (Hashimoto's disease) b) Circulating TSH-receptor-blocking antibody 3. . Graves' Diseasea)Euthyroidb) Hyperthyroid 4 Postpartum Thyroid Disease |
The prevalence of AITD in the pregnant population is comparable to that found in the general female population with a similar age range, i.e. between 5-15% (125). Careful study of women with thyroid antibodies during pregnancy has shown that despite the expected decrease in antibody titers during gestation, thyroid function gradually deteriorated towards hypothyroidism in a significant fraction of such women (Fig 14-11 a,b,c).
In the 1st trimester, serum TSH (albeit within the normal range) was already significantly shifted to higher values in women with AITD, compared with normal pregnant controls. Serum TSH remained higher throughout gestation and at parturition 40% of AITD-positive women had a serum TSH >3 mU/L, with almost one-half of them above 4 mU/L. Thus, while women with AITD were able to maintain a normal thyroid function in early gestation (due to sustained thyrotropic stimulation), their mean serum free T4 levels were significantly reduced to (or below) the lower limit of the normal reference range at delivery. Average reduction in serum free T4 reached 30% and almost one half of these women had free T4 values in the hypothyroid range by the time of delivery, confirming that these women have a reduced functional thyroid reserve. The risk of progression to hypothyroidism could be predicted from serum TSH levels and TPO-Ab titers measured in early pregnancy. When serum TSH was already above 2.5 mU/L and/or TPO-Ab titers above 1,250 U/mL before 20 weeks, these markers were predictive for the development of i hypothyroidism by the end of pregnancy. Practical use of these markers in early gestation can therefore identify those women who carry the highest risk.A Chinese study has confirmed this approach noting that between 7 and 12 weeks gestation the titers of TPOAb and TSH correlate positively and negatively with FT4 respectively (127). Preventive thyroxine treatment administered to avoid the potential deleterious effects of hypothyroxinemia and possibly thyroid antibodies on both maternal and fetal outcomes may then be considered. . There is also evidence from retrospective and some prospective studies that positive thyroid antibodies impacts adversely upon the course of pregnancy in several ways.
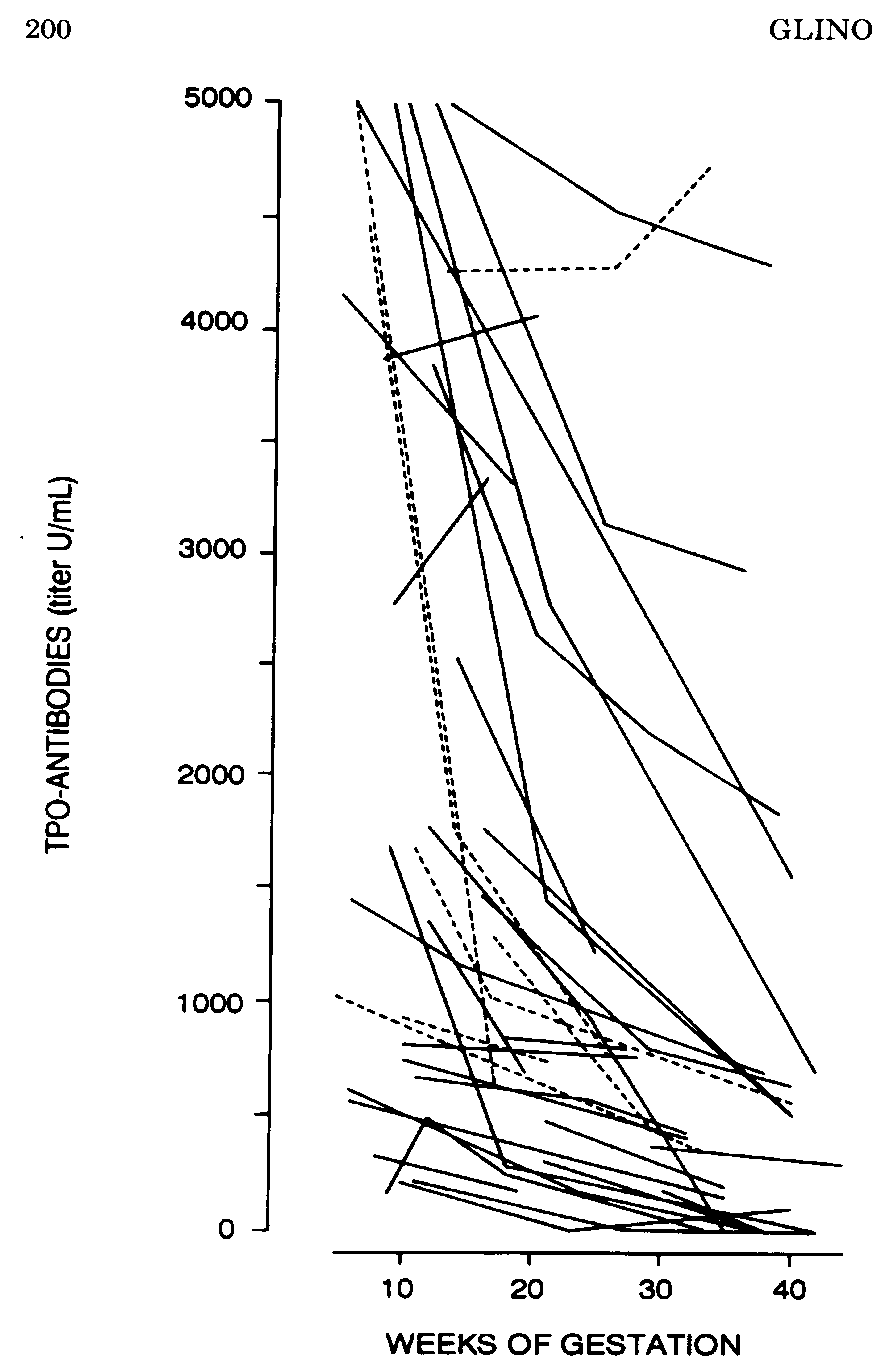
Figure 14-11a: Changes in TPO-Ab in pregnant women with AITD. There was a marked reduction in antibody titers, by 50-60% on the average (solid lines represent asymptomatic euthyroid women; dotted lines women with known hypothyroidism) (from 126).
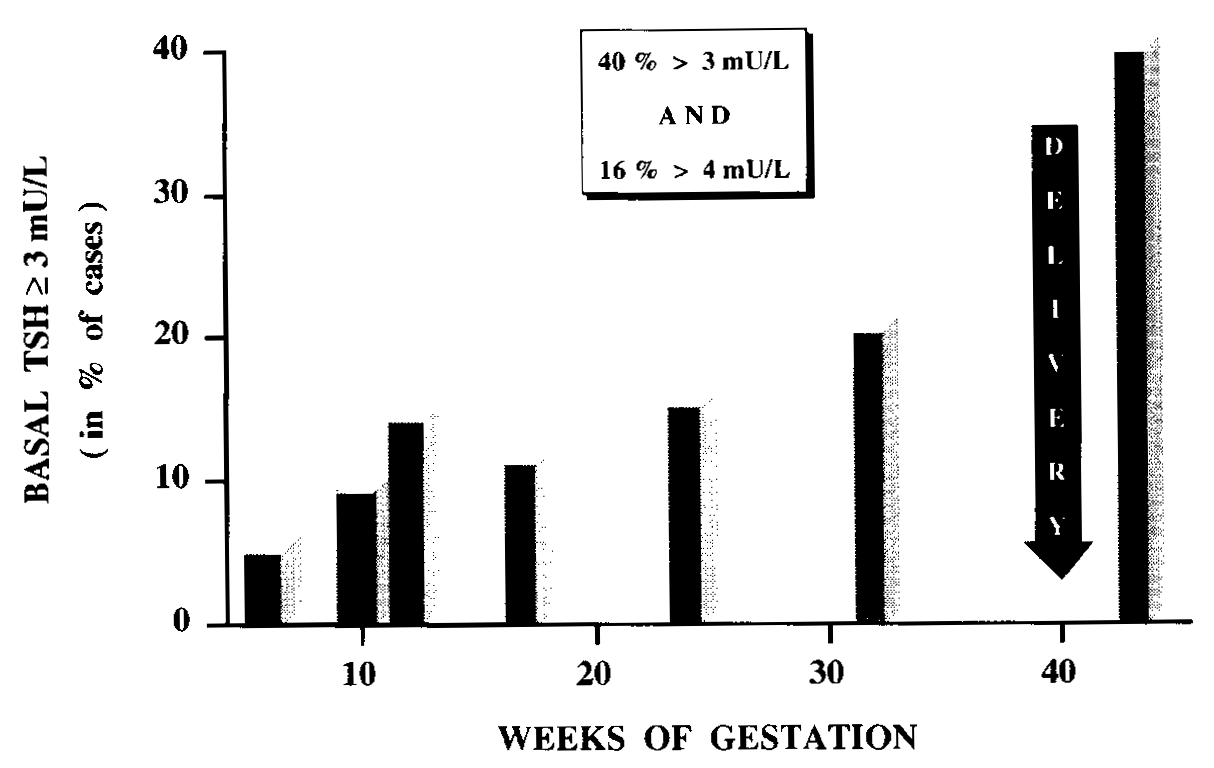
Figure 14-11b: Among women with thyroid antibodies, a progressively increasing fraction developed biochemical hypothyroidism, with 10% of them having a basal serum TSH >3 mU/L in 1st trimester, 20% in 2nd & 3rd trimesters, and finally ~40% at delivery (from 126).
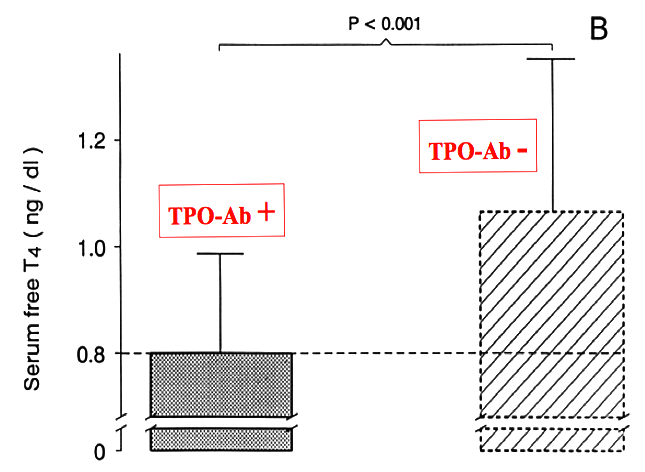
Figure14- 11c : Mean serum free T4 concentrations at delivery in women with and without thyroid immunity. In women with AITD, mean serum free T4 was not only significantly lower than in controls, but in addition, was at the lower limit of normality (from 126).
THYROID AUTOIMMUNITY AND DISORDERS OF FEMALE REPRODUCTION
Infertility
Infertility is defined as the absolute inability to conceive after one year of regular intercourse without contraception. The overall prevalence of infertility is estimated to range from 10% to 15% and has remained stable over the past few decades. The work up of infertile women usually identifies different causal factors, including male-factor infertility in 30%, female causes of infertility in 35%, a combination of both male and female infertility in 20%, and idiopathic infertility in 15%. Female causes of infertility comprise endometriosis, tubal occlusion and ovulation dysfunction. Among the factors that may negatively influence normal fertility, immunologic factors are known to play an important role in the reproduction processes of fertilization, implantation and early development of the embryo. Different investigations support the association between reproductive failure and abnormal immunological test results, including anti-phospholipid, anti-nuclear antibodies and organ specific autoimmunity, among which the presence of antithyroid antibodies (127-129). However, In women with reproductive failure the presence of autoantibodies does not appear to affect the numbers of K cells in the endometrium around the time of implantation (130). In women with repeated implantation failure the percentage of cytotoxic T cells was increase in those with thyroid autoimmunity compared to those without (131).
With regard to thyroid dysfunction, clinical hypothyroidism is clearly associated with female infertility and, in women in the reproductive age, autoimmune thyroid disease (AITD) is the most common cause of hypothyroidism (132,133). Although many of the studies relating to the association of thyroid antibodies and infertility are subject to selection bias, retrospective analysis, and different causes of infertility, they broadly confirm the association. Analysis of a large Danish population (11254 women) has shown that impaired fertility is associated with TSH, TPOAb and subclinical hypothyroidism (134). A previous large study employing control women study has documented an OR of 2.1 (1.7-2.6), p< 0.0001, in favour of the association of infertility and thyroid antibodies (135). Medically-assisted conception and onset of gestation is not hampered by AITD, but a successful outcome of the ongoing pregnancies is significantly reduced in those women with AITD due to greater early pregnancy loss (see Figure 14-23) (136)
The mechanism of the association between thyroid antibodies and infertility is not clear. A review has noted that thyroid hormone disorders and TPOAb are associated with disturbed folliculogenesis,spermatogenesis, fertilization and embryogenesis but the pathogenesis of TPOAb and reproduction is not well understood (137). It is of interest that there is an increase in infertility in women with endometriosis [RR 3.57] (138) which is known to have immune cell depression (NK cells), as well as decreased activity and cytotoxicity against autologous endometrium (139). The importance of NK cells has been emphasised (139) and impaired cellular and humoral response in women with unexplained infertility has been shown (140). The demonstration of antithyroid antibodies in ovarian follicles (141) may also suggest a critical role in infertility associated with autoimmune thyroid disease. These conclusions are strengthened by a study in mice in which it was noted that the anti TPO antibody may affect post-implantation embryo development leading to fetal loss (142). Lack of vitamin D was suggested as a predisposing factor to autoimmune diseases, and was shown to be reduced in patients with thyroid autoimmunity. In turn, its deficiency is also linked to infertility and pregnancy loss, suggesting a potential interplay with thyroid autoimmunity in the context of infertility (143)
The main practical question is whether one should give the benefit of thyroxine administration to infertile women who have positive thyroid antibodies with variable degrees of thyroid insufficiency.Screening for thyroid function in infertile women should be routinely performed (144,145) Obviously, overt thyroid dysfunction should be treated before conception or planned ART. Since SCH has a negative impact on the outcome of pregnancy after ART, thyroxine treatment should also be advised (146). It should be noted that in a study of 21 thyroxine treated women compared to 219 euthyroid women, women with hypothyroidism had a significantly decreased chance of achieving a pregnancy following IVF compared to euthyroid patients (147). The reasons are unknown and more data are required. Controlled ovarian hyperstimulation studied in 57 women led to significant elevations in TSH, often above pregnancy appropriate targets. These findings were particularly evident in women with preexisting hypothyroidism and may have important clinical implications for screening and thyroid hormone supplementation (148) Evidence on the treatment of isolated autoimmune features, but without thyroid dysfunction, was insufficiently documented until recently to advise prompt action (see later section on medical interventions).
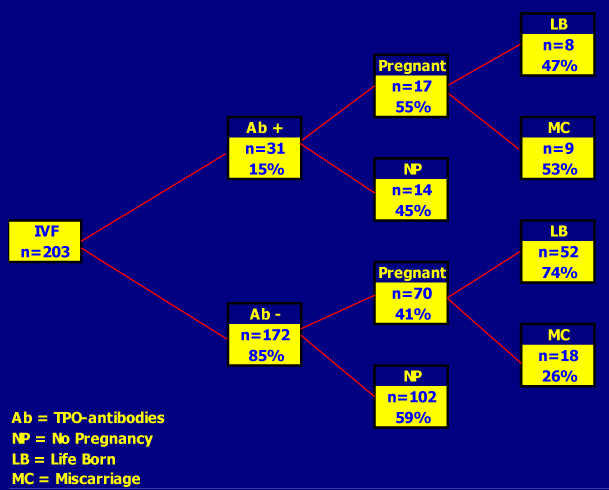
Figure 14-12: Outcome of Assisted Reproduction (IVF) in 203 women with (15%) and without (85%) thyroid autoimmunity (TAI). The rate of successfully-induced pregnancies was not decreased in TAI positive women (~50%), but miscarriages occurred twice more frequently in them (53 versus 26%; O.R for miscarriage in TAI positive cases = 3.77) (from Poppe, Ref 136).
In males, hyperthyroidism causes alterations in spermatogenesis and fertility, and most studies show that hyperthyroid male patients have abnormalities in seminal parameters, mainly sperm motility. These abnormalities tend to improve and normalize when euthyroidism is restored by treatment. Concerning hypothyroidism in males, severe and prolonged thyroid insufficiency may impair reproductive function, particularly when its onset occurs in childhood. Severe juvenile hypothyroidism may also be associated with precocious puberty. Finally, patho-zoospermia and astheno-zoospermia seem more prevalent in infertile males who present features of AITD(2). . Among 71 men with thyroid dysfunction (1/3rd with hyperthyroidism and 2/3rd with hypothyroidism), the authors found an elevated frequency of erectile dysfunction (56/71; 79%). Moreover, the restoration of a euthyroid status by thyroid treatment also restored a normal (or significantly improved) erectile function.(149)
Miscarriage
Thirty-one percent of all pregnancies end in miscarriage. Generally, women who experience a single pregnancy loss do not routinely undergo an evaluation for the cause of miscarriage. Women who experience recurrent miscarriages (i.e. 0.3%-5% of women), which is defined as three or more spontaneous miscarriages without an intervening live birth, should thoroughly be evaluated for an underlying etiology (such as infections, auto-immune disorders, exposure to drugs, etc.) (150).
Stagnaro-Green (151) reported a doubling of the spontaneous miscarriage rate in women who were Ab+ve compared with an Ab-ve cohort (17 vs 8.4%; p = 0.001). Subsequent meta analyses confirmed these associations (152,153). In a further 22 studies up to 2007 with only 6 showed no statistical correlation between the presence of antibodies and miscarriage (154). High TSH levels in women without overt thyroid dysfunction are associated with miscarriage but maternal FT4 levels and child loss were not associated (154). In 101 women with a TSH level more than 20mIU/L treated with T4 adverse pregnancy outcomes occurred no more frequently than in a control group of 205 euthyroid women. However the TSH level during pregnancy was correlated with the rate of abortion and premature delivery (155). In 216 women known to have had a miscarriage before 12 weeks gestation autoimmunity was independently associated (156). A meta analysis of 21 studies (13 cohort and 8 case control) showed a pooled odds ratio of 2.55 [CI 1.42-4.57 p=0.002] (157). A large study in which 17,298 women were screened for thyroid autoimmunity (158) showed a 3 fold increase in placental abruption in the 6% who were antibody positive (OR 3.4 CI 1.7-6.7). This 3 fold risk of placental abruption has been confirmed by a further meta analysis of 31 studies (159) involving more than 12000 women. Meta analysis of both the cohort (n=19) and case-control studies showed a positive association of thyroid antibodies with pregnancy loss (OR 3.9 CI: 2.48-6.12 p<0.001) for cohort studies and 1.8 (1.25-2.6 p,0.002) for case-control studies. A similar OR was found by a Dutch review (3.73 95% CI 1.8-7.6) (160). The association between AITD and miscarriages does not imply a causal relationship, as underlying causal mechanisms might also be attributable to a combination of factors that would potentially lead to miscarriage by themselves. In contrast an observational study of 220 women with recurrent miscarriage with TPOAbs compared to 496 women with miscarriage but no antibodies it was found that the prevalence of TPOAb in women with unexplained RM was not higher than in the general population, TPOAb-positive status did not have a prognostic value regarding the outcome of a subsequent pregnancy, and empirical thyroxine therapy in those who tested positive did not seem to improve outcome (161).However a systematic review has suggested that L Thyroxine does indeed reduce miscarriage rates (162). The American Society of Reproductive medicine asserts that there is fair evidence that thyroid autoimmunity is associated with miscarriage and that L-Thyroxine may improve pregnancy outcomes especially if TSH is > 2.5mIU/L (145).Miscarriage may be linked to a generalized immune imbalance. Women who have had multiple miscarriages have an increased number of CD5/20+ B cells compared with women who have had one or none (163).. Aberrant immune recognition of thyroglobulin (Tg) and placental antigens by antibodies to Tg has been demonstrated in mice immunized with human Tg, and resulted in decreased fetal and placental weights (164).However, evaluation of thyroglobulin expression in reproductive organs of mice showed no message in placenta, decidua or ovary suggesting that antithyroglobulin antibodies have no direct detrimental effect on such organs in patients with thyroid autoimmunity suffering from recurrent abortion (165).On the other hand Ticconi et al (166) in a case control study of 160 women with recurrent miscarriage (RM) found both TPO and Tg antibodies to be more frequently present than in 100 healthy pregnant women. Importantly, more than 90% of the RM women had evidence of other autoantibodies suggesting a more general maternal autoimmune defect in RM.
AITD may be associated with inappropriate low levels of thyroid hormones for the given gestational period, despite apparent biological euthyroidism. Only women with AITD and who experienced a miscarriage showed a difference in median serum levels of TSH and T4 compared to women without AITD (167). Women with AITD are generally older than healthy controls and increased age is an independent risk factor for miscarriage. AITD could act therefore by delaying the occurrence of conception because of its known association with infertility. Thyroid antibody-positive women would tend to become pregnant only at an older age (3-4 years older, on the average) and be more prone to pregnancy loss. There are no clear answers to the problem of thyroid autoimmunity and miscarriage and the subject has been reviewed (168).
Women Undergoing IVF
Different regimes of IVF are now frequently employed in infertile women and new approaches to ovarian stimulation are being implemented (169). A meta analysis of 4 studies on 1098 subfertile women (170) with thyroid autoimmunity and 957 controls showed an RR for miscarriage of 1.99 (CI 1.416-2.793, p<0.001) [ Fig 14-24].
Therefore, on current evidence, it does appear that the presence of thyroid autoimmunity is associated with an increased risk for spontaneous miscarriage in subfertile women achieving a pregnancy through an IVF procedure.(136,170-175); however the clinical picture is not clear cut .Negro et al. (176) found that pregnancy rates were not affected by the presence of TPOAb in euthyroid women undergoing assisted reproductive technology (ART). Intracytoplasmic sperm injection is a relatively new method of ferilisation. Studies of patients with thyroid autoimmunity and thyroid antibodies (anti TPO) concluded that these abnormalities did not affect cumulative delivery rates, fertilization, pregnancy rates, live birth rates or miscarriage rate compared to women without thyroid autoimmunity (177-179). Successful modulation of the immune system with beneficial pregnancy outcome has been reported in patients with AITD who received immunoglobulins with (or without) additional heparin or aspirin (180-182). However, the sudies were not adequately controlled, only comprising small numbers of patients who also had othe, auto-antibodies other than thyroid antibodies.
Fig 14-24
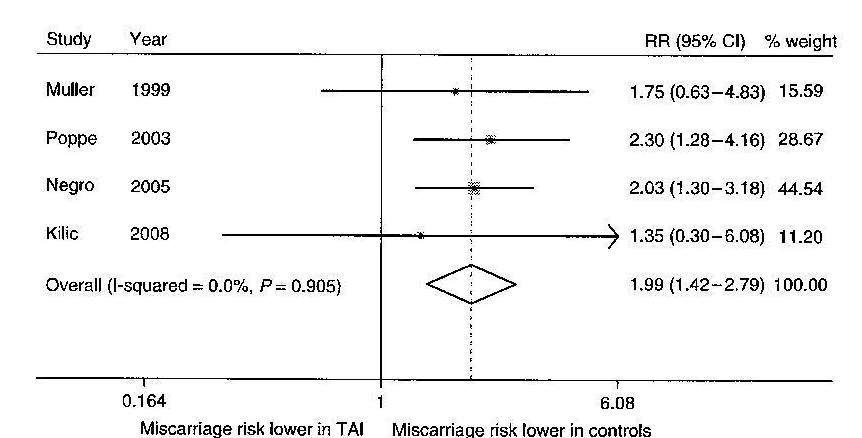
Miscarriage risk in euthyroid women with thyroid autoimmunity undergoing IVF. % weight refers to the emphasis placed on each of the 4 studies used in the analysis. From (170)
It is possible that TPOAb+ve women could have a better outcome with IVF if they also received LT4 as well as aspirin and prednisone (183) When 50 micrograms of LT4 was administered to women with subclinical hypothyroidism undergoing IVF showed an improvement in embryo quality and pregnancy outcome (184). A retrospective cohort study has confirmed that the vast majority of hypothyroid treated women who achieve pregnancy through IVF require an increase in the L-T4 dose during gestation (185). This is a similar situation to hypothyroid pregnant women not having IFV (vide infra).
These findings have implications for screening and medical intervention. For instance, if delayed conception plays a significant role to explain decreased fertility in women with AITD, it would certainly constitute an argument for screening systematically infertile women for the presence of mild thyroid underfunction that is so frequently associated with thyroid antibodies, particularly when women seek medical advice before IVF procedures. There is a high prevalence of women with elevated serum TSH levels, an association between oligo-amenorrhea and abnormally elevated serum TSH values and an overall improvement in the success rate of induced pregnancies after thyroxine administration (186). A recent study of 50 patients confirmed the link between thyroid function, forecast of conception and pregnancy, but noted that there is no recommendation on the TSH target level in patients undergoing assisted reproduction (187). Finally, women with AITD could be advised to plan for a pregnancy at a younger age, although this type of medical advice is more easily said than applicable in practice.
Although a clear association exists between thyroid autoimmunity and pregnancy loss, systematic screening cannot be universally recommended at present time, at least until adequately designed therapeutic trials will demonstrate beyond doubt a clear reduction in the rate of miscarriage with thyroxine treatment. However, many centers, in Europe and elsewhere, already routinely screen women with infertility and/or miscarriage for the presence of thyroid autoimmunity and dysfunction.
Preterm Birth
Preterm delivery (PTD) , that is birth occurring at or before 37 weeks gestation is a major cause of perinatal morbidity and mortality. It is reported to have an incidence of 12.7% (188) and an association with thyroid abnormalities was suggested (189). A subsequent review (190) concluded that autoimmune thyroid disease (positive thyroid antibodies in a euthyroid woman) is a risk factor for PTD and cited studies from Belgium, Pakistan and Italy in which PTD was observed in 16- 26.8% of TPOAb+ve women compared to 8-8.2% of antibody negative women ( all statistically significant). However, the incidence of PTD was only 4% in TPOAb+ve versus 3% in antibody negative women (p=ns) (191). Other groups have also failed to find an association between PTD and thyroid autoimmunity (192- 194). However, an increase in very preterm birth (before 34 weeks) was found in women who were TPOAb positive in the first trimester (192,195). A meta analysis of the studies defining PTD at 37 weeks showed an OR for the association of thyroid antibodies in 5 studies to be 2.07 [CI 1.17-3.68. p=0.01](159).A further meta-analysis(196) reviewed 11 prospective cohort studies involving 35, 467 participants and showed the combined RR of preterm delivery for pregnant women with thyroid antibodies compared with the reference group was 1.41 (95% CI 1.08-1.84, P=0.011). Other studies have also strengthened the association between PTD and thyroid autoimmunity (197-199). Although methodology and number of women studied varies in the different reports, current evidence suggests that the presence of TPO-Ab in pregnant women significantly increases the risk of preterm delivery. Further studies are required to evaluate other factors (eg ethnicity) associated with these findings. For example Interleukin-6 levels may also be an important factor (200). Thyroid disease is associated with systemic lupus erythematosus and pregnant patients with this disorder also have an increase in PTD (201).
Implications for Therapy
In the conditions referred to above the patients are all euthyroid. Although there may be a tendency in TPOAb+ve women to develop a raised TSH later on in pregnancy this only occurs in the minority. The Generaation R study indicated that hypothyroxinemia (RR about 3.5) as well as TPO-Antibody positivity (RR about 2.0) are risk factors for premature delivery (202) It has been suggested that L-thyroxine treatment may correct any slight deficiency in this clinical situation as well as influencing the systemic immune disturbance and the placental-decidual environment (159). Two prospective randomised trials by Negro and colleagues (176,203) support this view. In the first L-thyroxine (1mcg/kg/day)was given to women scheduled to have IVF treatment; this resulted in a 36% reduction in miscarriage rate. The later study used a mean L-T4 dose of 49.7mcg/day in women with positive antiTPOAb and noted a 75% reduction in miscarriage as well as a 69% reduction in pre-term births. More trials are awaited before a firm recommendation can be made. A systematic review (162) stated that for subclinical hypothyroidism and thyroid autoimmunity, evidence is insufficient to recommend treatment with levothyroxine. However a Cochrane review (204) stated that a reduction in preterm birth and a trend towards miscarriage with L-T4 was shown.The lack of prospective randomized controlled trials in this area of practice is currently impeding progress in high quality evidence based clinical decision making.
PRIMARY HYPOTHYROIDISM
Clinical Epidemiology
The prevalence of overt and subclinical hypothyroidism in pregnancy is estimated at 0.3-0.5% and 2-3% (or even up to 5%) respectively (205,206). Endemic iodine deficiency is the most common cause of hypothyroidism seen in pregnant women worldwide. Even mild iodine deficiency can be associated with a high prevalence of thyroid autoimmunity in the first trimester (32), and inadequate iodine status has been accompanied by a high prevalence of hypothyroidism in pregnancy (207). However the main cause of hypothyroidism in iodine-replete populations is chronic autoimmune thyroiditis (208). Other causes include post-surgical, post-radioiodine ablation and hypothyroidism secondary to pituitary disease which, although rare, can include lymphocytic hypophysitis occurring during pregnancy or postpartum (209). (Table 14-6)
Table 14-6
Etiology and Diagnosis of Hypothyroidism in Pregnancy
| Cause |
Diagnostic Feature
|
| Autoimmune thyroiditis |
Positive thyroid antibody test (TPOAb TSH Receptor blocking antibodies |
| Iodine deficiency | Low urinary iodine. Goiter |
| Post surgical |
History of Graves’ disease or toxic nodular goiter , Thyroid cancer, Benign goiter |
| Pituitary disease | Features of hypopituitarism |
Overt hypothyroidism in pregnancy may present classically but is oftentimes subtle and difficult to distinguish from the symptoms of normal pregnancy. A high index of suspicion is therefore required especially in women with a predisposition to thyroid disease such as a personal or family history of thyroid disease, the presence of goitre or the co-existence of other autoimmune disorders like type 1 diabetes.(210) which are all predictive factors of high risk of autoimmune thyroid disease. Thyroid antibodies are found in 5-15% of normal women in the childbearing age and, when the iodine nutrition status is adequate, the main cause of hypothyroidism during pregnancy is chronic autoimmune thyroiditis. An earlier review concluded that the overall prevalence of hypothyroidism was 2.2% to 3.4%, and the prevalence of thyroid antibodies ranged from 25% to 77% of hypothyroid pregnant women, with a mean prevalence of 46%. Thyroid autoimmunity was 5.2-fold more frequent in women with a diagnosis of hypothyroidism, compared with euthyroid controls (mean of 48.5% versus 9.2%) ( 211).
Clinical and Diagnostic Features
Symptoms and signs may raise clinical suspicion of hypothyroidism during pregnancy (weight increase, sensitivity to cold, dry skin, etc.) but others may go unnoticed (asthenia, drowsiness, constipation, etc.). Because many women remain asymptomatic, particular attention is required from the obstetrical care providers for this condition to be diagnosed and to evaluate more systematically thyroid function when women attend the prenatal clinic for the first time. Only thyroid function tests confirm the diagnosis. A serum TSH elevation suggests primary hypothyroidism and measurement of serum free T4 levels further distinguish between subclinical hypothyroidism (SCH) and overt hypothyroidism (OH), depending on whether free T4 is normal or clearly below normal for gestational age. Determination of thyroid antibodies, thyroperoxidase (TPO-Ab) and thyroglobulin (TG-Ab) antibodies, confirms the autoimmune origin of the disorder (212). Recently there have been a number of studies examining the relationship between ovarian reserve and thyroid autoimmunity (TA) using anti Mullerian hormone (AMH) as a marker for the former. Women with TA have a reduced ovarian follicular reserve (213). There is evidence that AMH levels are inversely correlated with TSH levels in infertile women of reproductive age (214). Although the probability of a poor response to controlled ovarian hyperstimulation (COH) is high and independent of autoimmune thyroid disease (AITD) in women with low serum AMH levels, in those women with good ovarian reserve (high AMH) the presence of AITD impairs the outcome of COH (215). In a large cross sectional retrospective study (216) it was found that a) serum thyroid hormone levels, anti-TPOAb and prevalence of subclinical hypothyroidism were no different in different ovarian reserve categories, b) a higher prevalence of subclinical and overt hypothyroidism was seen in women with a genetic cause for low ovarian reserve compared to those with unexplained cause. The relationship is clearly complex and further work is required.
Effect of Hypothyroidism on the Outcome of Pregnancy
Despite the known association between decreased fertility and hypothyroidism, the latter condition does not preclude the possibility to conceive. Gestational hypothyroidism, and particularly subclinical hypothyroidism is not rare. Abalovich at al, showed that 34% of hypothyroid women became pregnant without thyroxine treatment: 11% of them had OH and 89% SCH (217).. When hypothyroid women become pregnant and maintain the pregnancy, they carry an increased risk for early and late obstetrical complications (Table 14-7). An analysis of 223,512 singleton pregnancies from a retrospective US cohort showed that , thyroid diseases were associated with obstetrical, labor, and delivery complications (218). Unfortunately these authors had no access to treatment details. Furthermore, in a study of 92 women on T4 replacement therapy the occurrence of maternal or fetal/neonatal complications could not be predicted by maternal TSH/fT4 through pregnancy, presence of thyroid autoimmunity or dose of LT4 replacement (219).
Table 14-7
Adverse Outcomes of Hypothyroidism
MOTHER
Infertility
Miscarriage
Preterm Delivery
Anemia in pregnancy
Preeclampsia
Abruptio placenta
Postpartum haemorrhage
BABY
Increased fetal death rate
Preterm birth
Intra Uterine Growth Retardation
Low birth weight
Increased neonatal respiratory distress
Impaired neurointellectual child development
Attention Deficit Hyperactivity Disorder
Autism
The frequency of these complications depends on whether they are associated with overt (high TSH associated with low FT4) or subclinical hypothyroidism (high TSH associated with FT4 within the reference range). With regard to fetal death rates Benhadi et al (154) noted that the risk of child loss increased with higher levels of maternal TSH although maternal FT4 concentrations and child loss were not associated. Ashoor et al (220-222) however, have observed that fetal loss was associated with both an increase in TSH and a decrement in FT4 although the presence of thyroid antibodies did not affect these results. They have also shown that impaired thyroid function may predispose to the development of late pre eclampsia (221) but they found no evidence of thyroid dysfunction or maternal thyroid antibodies to be related to preterm birth (222). A definitive study by Casey et al.(223) found that subclinical hypothyroidism in pregnancy has a relative risk of 1.8 for premature birth/low birth weight.Interestingly,maternal high-normal FT4 levels in early pregnancy were associated with lower birth weight and small for gestational age in the Generation R study of more than 4000 women (224). The continuous reciprocal relationship between maternal weight and FT4 has also been noted in the 2nd trimester in more than 9000 women (225) An overview is given by Negro (226).
While the prevalence of hypothyroidism and subclinical hypothyroidism has been mentioned it should be noted that the state of isolated hypothyroxinemia (IH) occurs in around 2.5 to 10% depending on the definition employed (227). There has been controversy as to whether IH is a real entity and if so whether there are any adverse effects of the condition in gestation. The cause(s) of IH are not clear but iodine deficiency may be an important factor. IH does have adverse obstetric effects, although early studies of first trimester IH showed no adverse outcomes (228,229). Subsequent studies have documented an increase in preterm labour and macrosomia as well as gestational diabetes and increased placental abruption(230). Breech presentation,,larger fetal and infant head size and fetal distress have also been associated with IH (230-234). Despite these data guideline committees have concluded that there is not enough evidence to recommend L-T4 treatment in women with IH (13-15).
A review of the treatment of subclinical hypothyroidism SCH) in pregnancy concluded that while SCH is associated with multiple maternal and neonatal outcomes the value of L-T4 treatment remains uncertain (235). Nevertheless,adequate thyroxine treatment is critical to the outcome independent of the type of hypothyroidism (OH/SCH) A retrospective study of 150 pregnancies noted that adequate treatment of overt and SCH minimised the risks of abortion and premature delivery regardless of initial thyroid status,whereas inadequate therapy resulted in an increased rate of abortion and preterm deliveries (218) rate. A prospective randomised intervention trial also showed that even in euthyroid thyroid antibody positive pregnant women who were treated with thyroxine the rates of miscarriage and pre-term delivery were lower than euthyroid antibody positive women who did not receive thyroxine treatment (203). A retrospective study of women with SCH who were or were not prescribed L-T4 during pregnancy showed that the L-T4 group had fewer loss of pregnancies, and fewer low birth weight infants as well as better APGAR scores in the infants (236). From a practical point of view adherence to L-T4 therapy is critical and was noted to be low among 17% of women prescribed this drug in a large survey (237). Appropriate counselling is recommended.
FETAL-NEONATAL CONSEQUENCES OF MATERNAL HYPOTHYROIDISM
Role of Thyroid Hormone During Fetal Brain Development
Thyroid hormones are major factors for the normal development of the brain. Extensive animal experiments reported by Teng’s group in China have shown neurodevelopmental impairment in subclinically hypothyroid rats due to alteration of the CREB signaling pathway (238) Marginal iodine deficiency affects dendritic spine development (239) and hypothyroxinemia also inhibits brain development (240,241). Hippocampal structure and function is affected in humans and rats resulting from thyroid hormone deficiency (242,243). The mechanisms of actions of thyroid hormones in the developing brain are mainly mediated through two ligand activated thyroid hormone receptor isoforms (244). Physiological amounts of free T4 are present in coelomic and amniotic fluids surrounding the developing embryo already in first trimester. Also, specific nuclear receptors are present in fetal brain as early as ~8 weeks post-conception (245). It is known that thyroid hormone deficiency may cause severe neurological disorders resulting from the deficit of neuronal cell differentiation and migration, axonal and dendritic outgrowth, myelin formation and synaptogenesis (246). This is the situation well documented in iodine deficient areas where the maternal circulating thyroxine concentrations are too low to provide adequate fetal levels particularly in the first trimester. Even in an iodine sufficient area maternal thyroid dysfunction (hypothyroidism, subclinical hypothyroidism or hypothyroxinemia) during pregnancy results in neuro-intellectual impairment of the child; hence maternal thyroid hormones are required through gestation for proper brain development and specific effects will depend on when maternal hormone deficiency occurs during pregnancy (247) The neurobiology of fetal brain development depends on many factors including the availability of thyroxine (T4) delivery to the fetal neurones (248) . There is also an important role for the thyroid hormone transporters in one or more of these processes (249). While MCT8 facilitates thyroid hormone transport to the neurone, OATP1C1 appears to be related to thyroid hormone transport into the astrocyte. At this stage it favours the transport of T4 more than T3 but as the deiodinase II is within the astrocyte this enables conversion to occur and then allows T3 to be transferred into the neurone. Other thyroid hormone transporters are probably regulating thyroid hormone transport into the oligodendrocyte. These processes depend on maternal iodide supply, maternal T4 synthesis, maternal T4 placental transport and the conversion of T4 to T3 in the fetus by the Type II deiodinase. The discovery that children born with the Allan-Herndon-Dudely syndrome have a mutation in the thyroid transporter monocarboxylate 8 (MCT8) (250 )has accentuated the interest in many of the transporters (251).Thyroid hormone receptor development in brain occurs very early in gestation, certainly before the fetal thyroid begins to function which is around sixteen to eighteen weeks (5). In early gestation thyroid hormone effects on genes related to neurodevelopment, for example, myelin, can be recorded.
Clinical Studies on the Role of Maternal Hypothyroidism for the Psycho-Neurological Outcome in the Progeny
Man et al(252) first noted that children of mothers with inadequately treated hypothyroidism had significantly lower IQs than those born to adequately treated patients or normal controls. These pioneering data did not gain much clinical attention, probably because the prevailing dogma, at that time, was that maternal TH did not cross the placenta .
Impaired intellectual development has been reported in children born to women with non-iodine deficient hypothyroidism during pregnancy (253-255) as well as in children from hypothyroxinemic mothers (256- 261). Attention deficit disorder (262,263), autistic symptoms in offspring (264) and schizophrenia in later life (265) have been associated with maternal hypothyroxinemia. Attention deficit disorder was previously noted in offspring from mothers with thyroid autoimmunity (266). Children from mothers with anti thyroid peroxidase antibodies have been found to have intellectual impairment in early infancy (267) and a reduced childhood cognitive performance at age 4 and 7 and sensineural hearing loss at both ages (268).. Other studies have also shown suboptimal development in children exposed to hypothyroidism during pregnancy (269-271). If maternal T4 concentrations are corrected by the 20th week (272) or prior to the 3rd trimester (273,274) many of these adverse effects can be prevented..In addition, isolated hypothyroxinemia in the 2nd trimester is not associated with impaired cognitive, language and motor scores at age 2 (275). These studies emphasise the temporal nature of fetal brain development (276) and underpin the notion that women should not have an abortion if hypothyroidism is found and treated in the first trimester The seminal study of Haddow et al (253) is worthy of further comment. They found that the full IQ scores of children whose mothers had a high TSH during gestation were 7 points lower than controls (p<0.005) and that 19% of them had scores of less than 85 compared to 5% of controls (p<0.007). However there was no IQ decrement noted in the prospective double blind randomized controlled antenatal thyroid screening study (CATS) study in children of both hypothyroxinemic and high TSH mothers studied at 3 years of age who received levothyroxine therapy during pregnancy compared to children whose mothers were not treated with levothyroxine (276). As mentioned above it is possible that the timing of thyroxine administration in gestation is an important factor (241).Indeed, a more extensive replication of the CATS study has reported no difference in IQ measurements in children up to the age of 5 whose mothers received or did not receive L-T4 during gestation, but the drug was not commenced till late in the 2nd trimester (277). In a Chinese prospective population-based development study of 1017 women with singleton pregnancies clinical hypothyroidism was associated with increased fetal loss, low birth weight, and congenital circulation system malformations. Subclinical hypothyroidism was associated with increased fetal distress, preterm delivery, poor vision development, and neurodevelopmental delay. Isolated hypothyroxinemia was related to fetal distress, small for gestational age, and musculoskeletal malformations as well as spontaneous abortion (234). An important association study from the population based prospective study from Rotterdam (Generation R), which included MRI scans on the children, reported that maternal FT4 showed an inverted U shaped association with child IQ, child grey matter volume and cortex volume(278). This suggests that optimal T4 concentrations during gestation might require to be in a narrower range than previously thought. Brain morphology studies have also shown abnormal corpus callosum development in children born to women treated for hypothyroidism(279) and this maternal condition may also contribute to abnormal cortical morphology in the offspring (280). Maternal and/ neonatal thyroid function at delivery in children born at or over 37 weeks' gestation was not associated with impaired neurodevelopment at 5.5 years (281) although. lower levels of cord T4 were associated with increments in the McCarthy scales at this age. In premature infants (<34 weeks gestation) higher maternal levels of TSH at delivery were associated with significantly lower scores on the general cognitive index at 5.5 yr.(282)The neurodevelopmental impairment is similar to that seen in iodine deficient areas (see chapter on iodine deficiency) and implies that iodine status should be normalised in regions of deficiency. However, much of the USA and parts of Europe are not iodine deficient which raises the question of routine screening of thyroid function during early pregnancy or even at preconception which will be discussed below. In summary, the current weight of evidence suggests that hypothyroidism and subclinical hypothyroidism and hypothyroxinemia all have an adverse effect on neurodevelopmental outcome in the progeny. It is however the case that not all the evidence shows this and much of the evidence relates to association studies, Despite this there is a reasonable case for treatment of the woman with subclinical hypothyroidism in pregnancy to prevent these outcomes. However,treatment should be carefully monitored.
Management and Therapy of Gestational Hypothyroidism
Administration of L-thyroxine is the treatment of choice for maternal hypothyroidism, when the iodine nutrition status is adequate.
A number of studies have indicated that during pregnancy thyroxine requirements increase during gestation (283-286). The increase is due to the rapid rise in TBG levels resulting from the physiological rise in estrogen concentrations, the increased distribution volume of thyroid hormones (vascular, hepatic, and the fetal-placental unit), and finally the increased placental transport and metabolism of maternal T4(208). . If a pregnancy is planned, patients should have thyroid function tests measured soon after the missed menstrual period. If serum TSH is not increased at that time, tests should be repeated at 8-12 weeks and then again at 20 weeks, as the increase in hormone requirements may not become apparent until later during gestation. In women not receiving T4 who may have risk factors for thyroid disease (eg positive family history or other autoimmune disorder) thyroid function should be measured pre conception. If the TSH is less than 2.5mIU/L no action is required. If it is more than 3.5mIU/L thyroxine therapy may be indicated, especially if thyroid antibodies are present. If TSH is between 2.5 and 3.5mIU/L it would be prudent to check again in 4 weeks if possible. Treatment should be initiated with a dose of 100-150 µg/day or titrated according to body weight. In non pregnant women, the full replacement thyroxine dose is 1.7-2.0 µg/kg bw/day. During pregnancy, because of the increased requirements, the full replacement thyroxine dose should be increased to 2.0-2.4 mg/kg bw/day (208,287)
Women who already take thyroxine before pregnancy usually need to increase their daily dosage by 30-50%, on average, above preconception dosage; appropriate dose increments must be made once pregnancy is confirmed t The preconception thyroxine dose should be adjusted aiming to maintain serum TSH near the low-normal range (288) which should be within the trimester specific reference range ( ie approx. 2.5 mIU/L) It has been suggested that if SCH is newly diagnosed in pregnancy a T4 dose of 1.20µg/kg/day is appropriate to achieve a TSH less than 4.2 mU/L(289).
In general women who receive T4 because of previous ablative treatment (eg for thyroid cancer) require a greater increase than those receiving the drug because of Hashimoto’s thyroiditis where there still may be some reserve thyroid tissue. Therefore patients with Hashimoto’s disease require a lesser increase in T4 dose. Before pregnancy thyroid function should be checked and T4 dose adjusted to achieve a serum TSH of at least less than 2.5mIU/L. Indeed, a retrospective study has suggested that in women on T4 for hypothyroidism who are planning to become pregnant should have TSH levels not greater than about 1.2mIU/L (290) although the guidelines are not so stringent in their recommendations (10-15). The woman should be advised to increase the dose of T4 by 30-50% once pregnancy is confirmed. A convenient method for achieving this for some women would be to take 2 extra tablets of T4 per week (284). Thyroid function (T4 and TSH) should then be checked every 4 weeks as dose requirements may change during the course of gestation. It has also been suggested that The increment in thyroxine can be based on the initial degree of TSH elevation; women with a serum TSH between 5-10 mU/L, the average increment in T4 dosage is 25-50 µg/day; for those with a serum TSH between 10-20 mU/L, 50-75 µg/day; and for those with a serum TSH >20 mU/L, 75-100 µg/day (208).. The aim should be to keep the TSH around 2.5mIU/L or less remembering of course that TSH levels are difficult to interpret in the first trimester because of rising hCG concentrations. In the postpartum period most women should take their prepregnancy dose of T4. However some women with Hashimoto’s thyroiditis may require more than the pre-pregnancy dose because of possible postpartum progression of autoimmune thyroiditis (286). The question of compliance with therapy and the efforts of the physician to maintain the appropriate levels of thyroid hormone are important considerations particularly in the context of pregnancy (237). In 389 women, in the USA (185) 43% of serum TSH levels measured in 1st trimester were at or above 2.5 mU/L; In 2nd trimester, 33% of serum TSH measurements were at or above 3.0 mU/L (291). Even when the upper limit of TSH was defined as a serum TSH value above the 98th percentile of normal, 20% of values in 1st trimester and 23% in 2nd trimester were above this limit. In the UK , even in 18-45 year old pregnant women already on L-Thyroxine ,46% had a TSH level greater than 2.5 mU/L (292). These data suggest that the optimum care of pregnant women on L-thyroxine is not present despite evidence for its effect in reducing preterm birth and miscarriage (290,292). American Endocrine Society Guidelines recommend that as the potential benefits outweigh the the potential risks, women with SCH should receive T4 treatment (14,15). Women with autoimmunity (ie positive thyroid antibodies) should be carefully monitored during gestation as there is a tendency for a rise in TSH. If this occurs T4 therapy should be given.
THYROTOXICOSIS
Hyperthyroidism during pregnancy is relatively uncommon, with a prevalence estimated to range between 0.1% and 1% (0.4% clinical & 0.6% subclinical) (292). However, a population study of more than 400,000 births showed an incidence of 0.9% (293)Causes of thyrotoxicosis include those found in the general population, as well as others that occur specifically during pregnancy(294,295). While the commonest cause is Graves’ disease etiologies such as single toxic adenoma, toxic multinodular goiter, subacute or silent thyroiditis, iodide-induced thyrotoxicosis, and thyrotoxicosis factitia can occur but are uncommon during pregnancy. Molar disease should always be considered as it can potentially lead to severe thyrotoxicosis, particularly in pregnant women with a pre-existing autonomous or nodular goiter. However, since uncomplicated hydatidiform mole is easily diagnosed in early gestation, it rarely leads to severe thyrotoxicosis (52).. Other extremely rare causes of hyperthyroidism (described recently as isolated cases) include hyperplacentosis and struma ovarii (296).
In women in the childbearing age, the most common cause of hyperthyroidism is GD, as this etiology accounts for 85% of clinical hyperthyroidism in pregnancy. Another cause of hyperthyroidism is hyperemesis gravidarum. This is common and requires differentiation from Graves’ disease (2) (see section on Hyperemesis gravidarum below).
Clinical Diagnosis of Hyperthyroidism in Pregnancy
Even though the historical clues and physical findings are the same in pregnant and non pregnant patients, the diagnosis of thyrotoxicosis may be difficult to make clinically during pregnancy. Nonspecific symptoms such as fatigue, anxiety, tachycardia, heat intolerance, warm moist skin, tremor and systolic murmur may be mimicked by normal pregnancy. Alternatively, presence of goiter, ophthalmopathy and pretibial myxoedema obviously points to the suspicion of GD (see Table 14-8). A useful symptom of hyperthyroidism is that, instead of the customary weight gain, patients may report weight loss or, even more frequently perhaps, absence of weight gain despite an increased appetite (unless there is also excessive vomiting). Nausea (morning sickness) occurs frequently during normal pregnancy. However, the occurrence of hyperemesis gravidarum accompanied by weight loss must always raise the suspicion of hCG-induced hyperthyroidism.
| Table 14-8 Clinical features suggesting the possibility of hyperthyroidism due to Graves' disease in a pregnant patient |
| Historical 1. Prior history of hyperthyroidism or autoimmune thyroid disease in the patient or her family. 2. Presence of typical symptoms of hyperthyroidism including weight loss (or failure to gain weight), palpitations, proximal muscle weakness, or emotional lability. 3. Symptoms suggesting Graves' disease such as ophthalmopathy or pretibial myxedema. 4. Thyroid enlargement. 5. Accentuation of normal symptoms of pregnancy such as heat intolerance, diaphoresis, and fatigue. 6. Pruritus.Physical examination 1. Pulse rate > 100. 2. Widened pulse pressure. 3. Eye signs of Graves' disease or pretibial myxedema. 4. Thyroid enlargement especially in iodine sufficient geographical areas. 5. Onycholysis. |
Laboratory Diagnosis
Patients suspected of having hyperthyroidism require measurement of serum TSH, T4 and T3 levels, and anti-TSH receptor antibodies (TRAb). Virtually all patients with significant symptoms have a serum TSH <0.1 mU/L, as well as concurrent elevations in serum free T4 and T3 levels. However, interpretation of thyroid function tests must take into account the hCG-mediated decrease in serum TSH that occurs during pregnancy. Near the end of 1st trimester, at the time of peak hCG values, serum TSH levels may be transiently lowered to values below 0.4 mU/L in ~20% of euthyroid women (297,298). Thus, the degree and duration of TSH suppression (mainly but not only) in 1st trimester must be considered in making the differential diagnosis. Concerning T4 and T3 levels, the pitfalls and necessary caution in the interpretation of serum free T4 and T3 have been discussed earlier (see the section on thyroid function parameters in normal pregnancy).
Patients with GD usually have positive thyroid antibodies (TG-Ab and TPO-Ab) and, therefore, antibody presence should alert the clinician to the possibility that autoimmune thyroid disease is the cause of symptoms evoking hyperthyroidism. Most patients with GD have detectable TRAb. Since TRAb production tends to undergo immunologic remission during the second half of pregnancy, detection of TRAb may depend upon gestational age at determination (299). Presence of TRAb in 1st trimester is highly useful in helping make the differential diagnosis between GD and other causes of gestational hyperthyroidism.
Clinical Aspects of the Management of Graves’ Disease in Pregnancy
Prepregnancy Counseling
Prepregnancy counseling plays a very important role in the care of young women with Graves’ disease. All women of childbearing age affected with Graves’ hyperthyroidism should be strongly advised to seek contraception counseling, in order to avoid pregnancy while hyperthyroid (2,295). A discussion of the different hyperthyroid therapeutic choices is important for those women planning a pregnancy: ablative therapy, by 131I or surgery, or medical therapy. Before commencing specific treatment for a woman with GH in pregnancy it is essential to provide appropriate counselling advice [300). Risks to mother, fetus, and neonate from untreated GH during gestation are compelling reasons for recommending preconception counselling (PC). PC should include discussion as to the optimum treatment of GH in women wishing to become pregnant
- If ablative therapy is chosen the following recommendations are suggested::
- Pregnancy test prior to ablation,
- Delay of conception on average of 6 months following therapy in order to adjust LT4 doses to target values for pregnancy (serum TSH 0.3-2.5).
- Determination of TSH-receptor antibody (TRAb); the gradual disappearance from the circulation post therapy depends on the type of treatment chosen; following thyroidectomy there is a gradual disappearance of TRAb titers, while following 131I therapy there is an increase in TRAb titers that may last for 12 months followed by a gradual fall in titers (301). Therefore, in patients with high TRAb titers surgery appears to be the therapy of choice in women contemplating pregnancy (302).
- B) For women on antithyroid drugs the following discussion with the patient and her family is recommended:
- a) If still in need to ATD for more than 2 years, the possibilities of remission during pregnancy are very low, the patient, most likely, will need ATD therapy through pregnancy (see section below on management of Graves’ disease in pregnancy).
- b) PTU may induce liver toxicity with potential liver failure requiring liver transplantation; therefore recommendations by the American Thyroid Association (13) limited the use of PTU to those patients allergic to MMI, in the treatment of thyroid storm and in the first trimester of pregnancy because of the rare instances of methimazole embryopathy. In a woman on MMI therapy at the time of conception or planning a pregnancy it is advisable to switch to PTU if it is deemed that ATD are required at that time and resume MMI after the first trimester.
- c) Close follow-up throughout the pregnancy for frequent blood tests and adjustment of ATD dose, since the need for dose adjustments is common.
- d) Possibility of disease aggravation in the first trimester and recurrence in the postpartum period, due to postpartum thyroiditis or recurrence of Graves’ (303).
- e) Recommendations regarding breastfeeding while on ATDs.
- f) TRAb determination for prediction of potential fetal or neonatal complications
Complications of Hyperthyroidism and Pregnancy
These are listed in table 14-9. Untreated hyperthyroidism carries a high risk of complications (295,296) The risk of complications for mother and child , is related to the duration and adequate control of maternal hyperthyroidism. In women with unrecognized maternal GD infants showed severe prematurity (mean gestational age of 30 weeks at delivery) associated with very low birth weight (<2 Kg) and neonatal hyperthyroidism requiring treatment with ATD (308). In contrast, for those patients in whom the diagnosis was made early and treatment started promptly, the outcome was excellent. In 230 pregnant women with GD in Japan, no adverse impact on the outcome of pregnancy in patients with adequately treated Graves' disease was observed (309). Rare causes of thyrotoxicosis (eg due to an activating TSH receptor gene mutation) may also result in premature delivery and low birth weight (310)
Table 14-9 Complications of Graves’ Hyperthyroidism in Pegnancy
| Category | Item | Reference |
| Maternal | Adverse drug effects | |
| Left ventricular dysfunction | 304 | |
| Thyroid storm | 305 | |
|
Obstetric Antepartum
|
Miscarriage | |
| Preeclampsia | 306 | |
| Preterm labor [PTL] | ||
| Stillbirth | ||
| Gestational Hypertension | ||
| Fetal thyroid dysfunction | ||
| Obstetric Intrapartum | Fetal distress | |
| Preterm deliveries | ||
| Primary Cesarean Section | ||
| Placental abruption | ||
| Postpartum Hemorrhage | ||
|
Neonatal Primary Outcome
|
Birth weight < 2500 g | |
| Macrosomia >4000 g | ||
| Apgar scores | ||
| NCU admission | ||
| Respiratory Distress Syndrome (RDS) | ||
| Congenital abnormalities | ||
| Thyroid dysfunction | 307 |
Fetal & Neonatal Adverse Effects of Maternal Hyperthyroidism
Fetal Hyperthyroidism
Although rare, this is a preventable complication with potential severe sequelae, including death (311) Clinical evaluation at the time of maternal hyperthyroidism diagnosis, will indicate the few women at risk, namely those with: a) active Graves’ hyperthyroidism, b) previous history of Graves' disease treated with ablation therapy, either surgery or 131I, and c) mothers with active Graves’ hyperthyroidism undergoing therapeutic thyroidectomy in the second trimester of pregnancy . A determination of TRAb titer should be obtained between 22 and 26 weeks gestation, a value of 3-5 times above normal is an indication for fetal evaluation for detection of potential fetal thyrotoxicosis. The fetal thyroid TSH receptor starts responding to Thyroid Stimulating Immunoglobulin (TSI) stimulation during the second trimester. The placental transfer of IgG from mother to fetus increases by the end of the second trimester, reaching a level in the fetus similar to that of the mother around 30 weeks' gestation (2). Therefore, the symptoms of fetal hyperthyroidism are usually not evident until 22 to 26 weeks of gestation.
Evaluation of fetal hyperthyroidism can be assessed a) by fetal ultrasonographic data, showing the presence of fetal goiter, tachycardia (persistent fetal heart rate of >160 bpm);, b) fetal heart monitor tracing showing a sustained baseline of 170 to 180 beats per minute with moderate variability that exhibits acceleration with a lack of deceleration,[unique to fetal thyrotoxicosis (312)] c) growth retardation, increased fetal motility. Other signs developing later are intrauterine growth restriction (IUGR), oligohydramnios or hydrops, and accelerated bone maturation (311). This last sign is diagnosed by the presence of distal femoral ossification center before 31 weeks gestation (313) and is highly predictive of the disease. Serial cordocentesis for diagnosis and monitoring drug therapy has been proposed, but its value has been questioned, restricted to centers with expertise (314) because of a significant risk to the fetus (complications and/or fetal loss in 1% of cases) . It is generally recommended only if the information to be gained will change therapy.
When fetal hyperthyroidism is suspected in utero, it is reasonable to initiate ATD treatment with MMI (20 mg/day) combined with thyroxine administration, when required, to maintain maternal euthyroidism.
Fetal Hypothyroidism
Inhibitory TRAb production has been shown to cause hypothyroidism transiently in neonates born to mothers with GD (315)
Administration of ATD to treat maternal GD may induce fetal hypothyroidism that clearly should be avoided by maintaining maternal circulating thyroid hormone levels in the upper quartile of the normality range (295,296). Radioactive iodine has unintentionally been administered (in exceptional cases) to GD women who were unaware that they were pregnant and who decided, nevertheless, to maintain the pregnancy. Despite the risks of performing cordocentesis, it has been shown to be useful to predict the fetal outcome (316).
Fetal Goiter in Mothers with Graves’ Disease
The treatment of maternal hyperthyroidism may be associated with the presence of fetal goiter, thus raising clinical concern with regard to its etiology and management. Fetal goiter may result directly from the placental transfer of thyroid growth-stimulating effects of maternal TRAb, as well as from the inhibitory effect of ATD on the fetal gland inducing fetal hypothyroidism (317). The spectrum of neonatal thyroid dysfunction in pregnant women with GD receiving ATD can range from frank hypothyroidism (secondary to the exposure to MMI and maternal blocking TRAb) to neonatal Graves’ thyrotoxicosis (secondary to exposure to maternal stimulating TRAb) thus making the prenatal diagnosis extremely difficult ..
Of 72 mothers with past or present GD all infants from 31 pregnancies with no detectable TRAb and mothers without ATD treatment, were normal at birth. In the remaining 41 pregnancies, 30 women had positive TRAb and/or a treatment with ATD: fetal thyroid ultrasound was normal (32 wks gestation) and there was almost no evidence of fetal thyroid dysfunction. However,11 fetuses were found to have a goiter, of which 7 were hypothyroid and 4 hyperthyroid. The main risk factors for fetal hyperthyroidism were poorly controlled maternal hyperthyroidism and elevated TRAb. The risk factor for hypothyroidism was mothers being treated with ATD and having a serum T4 in the normal range (rather than upper limit of normal). The authors recommended TRAb measurement in women with current or past GD at the beginning of pregnancy, and close observation of those pregnancies with elevated TRAb or ATD treatment by performing monthly fetal ultrasonography after 20 weeks of gestation (313,318).
Neonatal Thyrotoxicosis
One to 5% of neonates of mothers with GD have hyperthyroidism (neonatal GD) due to the trans-placental passage of stimulating maternal TRAb. The overall incidence is low because of the balance between stimulatory and inhibitory antibodies, and also maternal treatment with ATD (311). The incidence of neonatal GD is not directly related to maternal thyroid function. Risk factors for neonatal thyroid dysfunction include history of a previously affected baby, prior radioiodine ablative treatment, and elevated TRAb titers at delivery (317). A higher TRAb value is associated with a higher risk of neonatal thyroid dysfunction (309).
Undetected fetal thyrotoxicosis may be followed by thyrotoxicosis at birth. Neonatal thyrotoxicosis is considered to be uncommon, occurring in ~1% of pregnancies in patients with Graves’ disease (319) . Risks appear highest in the offspring of women with not-well-controlled GD, as well as in women with the highest TRAb titers. Mothers with a prior history of bearing infants with neonatal GD are also at high risk of repeated episodes (296). Neonatal GD is usually diagnosed at or shortly following birth, after maternal ATD has been cleared from neonatal serum and thyroid gland. Signs of neonatal thyrotoxicosis include congestive heart failure, goiter, proptosis, jaundice, hyperirritability, failure to thrive, and tachycardia.. Cord serum free T4 and TSH determinations should be performed in all deliveries of mothers with a history of GD. Treatment should be initiated in conjunction with the neonatologist, and may include iodide, ATD, glucocorticoids, digoxin, and beta-adrenergic blocking agents, depending on the cardiovascular status. Neonatal hyperthyroidism may have a delayed onset in some infants, particularly those in whom both anti-TSH receptor blocking and stimulating antibodies coexist. Thus, the pediatrician should be alerted to measure serum free T4 if symptoms suggesting thyrotoxicosis appear during the first 6-8 weeks of life, even if cord serum results were normal, and especially when cord serum TSH was suppressed (318).
Sporadic cases of neonatal hyperthyroidism without evidence of the presence of circulating TSI in mother or infant are due to activating of mutations in the TSH receptor molecule (320). It is inherited as an autosomal dominant trait and, in contrast to Graves' neonatal hyperthyroidism, the condition persists indefinitely. Treatment with antithyroid medications followed by thyroid ablation therapy will eventually be needed in addition to genetic counseling.
Neonatal Central Hypothyroidism
Infants born to mothers with uncontrolled hyperthyroidism due to GD may present with central congenital hypothyroidism (296). High maternal serum T4 levels, during a prolonged period of time, cross the placental barrier leading to suppression of fetal TSH by pituitary feedback. In most cases, the diagnosis is made at birth or shortly thereafter, on the basis of a low neonatal serum total T4 contrasting with an inappropriately low serum TSH. In the majority of these infants, there is a return to euthyroidism within a few weeks or months; rarely, this condition may be due to mutation of the TSH receptor and result in a problem with neonatal screening. (321) There may also be a risk of thyroid ‘disintegration’ (i.e. abnormal ultrasound patterns found during childhood), possibly as the result of prolonged central hypothyroidism (322)...
Management of Graves’ Disease in Pregnancy
Graves' hyperthyroidism (GH) usually tends to improve gradually during gestation, although exacerbations can be observed in the first weeks. The spontaneous improvement: may be due to the partial immunosuppressive state of pregnancy (progressive decrease in TRAb production; changes in cytokine production) the rise in maternal serum TBG levels that tends to reduce serum free T4 & T3 fractions and obligatory iodine losses specific for pregnancy that may, paradoxically, constitute an advantage for women with GD. There is dispute as to whether the balance between blocking and stimulating TRAb activity may be modified in pregnancy (299). The exacerbation of thyrotoxicosis in women with GD during early pregnancy may be due in part to the stimulatory effect of high hCG levels (vide infra).
Antithyroid Drugs
Although antithyroid drugs (ATD) are the main treatment for GD during pregnancy (319,294) recent developments relating to the adverse effects of ATD in pregnancy have led to more caution in their use (323) Recommendations for use of PTU in the first trimester, and MMI later, are discussed below. The overall goal of therapy is to control maternal disease by maintaining the patient at a high euthyroid level, while minimizing the risk of fetal hyperthyroidism or hypothyroidism by using the smallest possible dose of ATD.
The initial recommended dose of PTU is 100 to 450 mg/day in 3 divided doses or MMI 10 to 20 mg/day; very seldom a larger initial dose is required. In patients with minimum symptoms, an initial dose of 10 mg of MMI daily or PTU 50 mg two or three times a day may be initiated. In most patients, clinical improvement is seen in 2 to 6 weeks, and improvement in thyroid tests occurs within the first 2 weeks of therapy, with normalization to chemical euthyroidism in 3 to 8 weeks in over 50% of patients (324). Resistance to drug therapy is unusual, most likely due to poor patient compliance. With clinical and thyroid test improvement, the dose of antithyroid medication should be reduced by half of the initial dose. The daily dose is adjusted every two to four weeks according to the results of thyroid tests. Serum TSH may remain suppressed despite the normalization of thyroid hormone levels for many weeks, frequently through pregnancy . The ATD dosage should be maintained at a minimum and should indeed be continued, in low dose if necessary, to the end of gestation, although there are differing opinions concerning this strategy. Patients should be assessed at regular intervals, every 2 to 4 weeks at the onset of treatment and every four weeks thereafter, to allow for proper medication adjustments to keep the FT4 or FT4I within target goals. Clinical clues of good therapeutic response are improvement in symptoms and weight gain. High FT4 levels (even in the mildly thyrotoxic range) and the presence of TRAb antibodies are useful indices of the fetal need for antithyroid treatment to prevent fetal goitre and maintenance of fetal euthyroid state (325) Continuing ATD to the end of gestation will prevent hyperthyroidism in labor which is undesirable and, if carefully monitored, should be a safe strategy. There is evidence from a retrospective non- randomized trial that continuing ATD throughout pregnancy substantially prevents postpartum recurrence of Graves’ hyperthyroidism without adverse effects on the fetus (326). However, postpartum recurrence of Graves’ hyperthyroidism has been documented more frequently (84 %) in patients previously treated for Graves’ disease with ATD before a successful pregnancy compared to a rate of 56 % in women similarly treated but not having a pregnancy (327). The case for postpartum monitoring is, therefore, very strong.
Combined administration of ATD and thyroxine to the mother should be avoided, since trans-placental passage of ATD is high while negligible for thyroid hormones and, hence, thyroxine will not protect the fetus from ATD-induced hypothyroidism.
Assessing TRAb concentration is essential in the management. Titers of TRAb measured after 22 weeks gestation may be slightly elevated suggesting a very low probability for the fetus to develop hyperthyroidism, and a good indicator for using lower doses of ATD. The classical course of Graves’ disease during pregnancy frequently encompasses exacerbation of hyperthyroidism during 1st trimester and a gradual improvement in the 2nd half of gestation. Maternal production of TRAb may remain elevated after thyroid ablation using radioiodine or even after a prior thyroidectomy or the apparent cure of the disease by antithyroid drug (ATD) therapy given several years before pregnancy. In euthyroid pregnant women who have previously received ATD for GD but who are currently not receiving ATD treatment, the risk of fetal/neonatal thyrotoxicosis is negligible and, therefore, systematic measurement of TRAb is not mandatory. For a euthyroid pregnant woman (with or without thyroid hormone replacement therapy) who has previously been treated with radioiodine or undergone thyroid surgery for GD, the risk of fetal/neonatal thyrotoxicosis depends upon the level of TRAb produced by the mother. As a result, TRAb should be measured in early pregnancy to evaluate this risk. If significantly elevated TRAb is detected at weeks 18-22 or the mother is taking ATD in the third trimester, a TRAb measurement should again be performed in late pregnancy (weeks 30-34) to evaluate the need for neonatal and postnatal monitoring It should be remembered that the standard TRAb assays measure displacement of binding by TSH to the TSH receptor and do not distinguish between stimulating and blocking TRAbs. Assays that do distinguish are usually only available in research settings.
PTU, MMI and CBZ (converted to MMI by the liver), are equally effective in controlling the disease (213). The risk of hepatic toxicity due to PTU has been emphasized due to the number of cases requiring liver transplantation and as a cause of death (329). MMI can also induce a milder cholestatic liver toxicity not associated with liver failure (330). While PTU can rarely cause antineutrophil cytoplasmic antibody-associated vasculitis (331) agranulocytosis and liver failure were very rare in a large population survey in pregnancy of PTU and MMI (332); birth defects were the dominant side effect in pregnancy, their relative incidence being re-evaluated recently by the late Professor Laurberg and his Danish colleagues. A detailed literature analysis concluded that both MMI and PTU use in early pregnancy may result in birth defects in 2-3% of exposed children and that the highest risk was in gestational weeks 6-10 (ie during organogenesis)(333). A meta analysis concurred with this view (334). This has therapeutic implications (vide infra). It is claimed that studies which have not found ATD associated birth defects were either not sufficiently powered or did not study outcomes at optimal ages (335). Aplasia cutis, occurred in a small group of infants born of mothers on MMI therapy (336),. It has been reported in infants from mothers receiving PTU but much less commonly (337). ”Methimazole embryopathy” includes choanal atresia and/or esophageal atresia, minor dysmorphic features and development delay An OR (odds ratio) of 18 (95% Cl 3-121) for choanal atresia among infants whose mothers received MMI in the first trimester compared to the general population was noted (338); PTU did not seem to be a major human teratogen in one study (339) but 3/47 PTU 1st trimester exposed mothers had children with congenital abnormalities (229) A retrospective review (340) of the pregnancy outcomes of 6744 pregnant women with Graves’ disease in relation to all observed congenital anomalies showed a significantly higher rate of major anomalies in the MMI group of babies (4.1%) compared to those seen in the PTU group (2.1%) [p=0.002]. However, examination of Danish records of more than 817,000 infants showed that birth defects (in the neck and face and urinary system) due to PTU do indeed occur but are generally less severe than in children exposed to MMI or CBZ; but these children did require surgical correction (341). In line with these data a meta-analysis indicated that PTU was a safer choice for treatment according to the risk of birth defects but that a shift between MMI and PTU failed to provide protection against birth defects. That is to say both drugs can be associated with birth defects (342).
An advisory committee recommended limiting the use of PTU to the first trimester of pregnancy (343). Exceptions to this are patients with MMI allergy or those with thyroid storm. It is accepted that when PTU is not available MMI can be used in the first trimester.
PTU and MMI are equipotent in the management of hyperthyroidism in pregnancy, both drugs having similar placental transfer kinetics (344). Furthermore, when the efficacies of both drugs have been compared in pregnant women, euthyroidism was achieved equally with equivalent amounts of drugs and at the same weeks of treatment (345). Obstetric and neonatal outcomes were no different in both groups.
In view of the recent information on teratogenic effects of thionamide drugs in pregnancy revised management guidelines have been suggested (13).
Women taking MMI or PTU should be instructed to confirm potential pregnancy as soon as possible and contact their physician immediately pregnancy is diagnosed. If she is on low dose ATD the physician should consider discontinuing ATDs (depending on clinical disease status) because of potential teratogenic effects. Clinical and laboratory testing should occur every 2 weeks or with longer intervals if euthyroidism persists. If ATD are required PTU should be used through 16 weeks of pregnancy. Pregnant women receiving MMI who are in need of continuing therapy during pregnancy should be switched to PTU as early as possible;
a dose ratio of approximately 1:20 should be used (e.g. methimazole 5 mg daily = PTU 100 mg twice daily). If ATD therapy is required after 16 weeks gestation, it remains unclear whether PTU should be continued or therapy changed to methimazole as both medications are associated with potential adverse effects and shifting potentially may lead to a period of less-tight control. General treatment guidelines are shown in table 14-10
| Table 14-10. Treatment guidelines for Graves' disease during pregnancy | |
| 1. | Monitor pulse, weight gain, thyroid size, serum free T4 and T3, and TSH every 2-4 weeks, and titrate ATD as necessary. |
| 2. | At pregnancy diagnosis see above paragraph.PTU recommended for 1st trimester. Then switch to MMI. |
| 3. | Use the lowest dosage of ATD that maintains the patient in a euthyroid or mildly hyperthyroid state. The ATD dose can usually be adjusted downward after 1st trimester and often (but not always) discontinued during last trimester. |
| 4. | Do not attempt to normalize serum TSH. Serum TSH concentrations between 0.1 & 0.4 mU/L are generally appropriate, but lower levels are acceptable if the patient is clinically satisfactory. |
| 5. | While as little as 100-200 mg PTU/day may affect fetal thyroid function, dosages as high as 400 mg PTU (~30 mg MMI) have been used. |
| 6. | Communicate regularly with obstetric care providers, especially with respect to fetal pulse and growth in the 2nd half of gestation. |
| 7. | Consider thyroidectomy if persistently high doses of ATD are required (PTU >600 mg/d or MMI >40 mg/d), or if the patient is not compliant or cannot tolerate the administration of ATD. |
| 8. | Beta-adrenergic blocking agents and low doses of iodine may be used peri-operatively to control hyperthyroidism. |
| 9. | ATD will often need to be reinstituted or increased after delivery. |
Beta-Adrenergic Blocking Agents
Propranolol may be used transiently to control symptoms of acute hyperthyroid disease and for pre-operative preparation, and there are no significant teratogenic effects of propranolol reported in humans or animals. If a patient requires long-term propranolol administration, careful monitoring of fetal growth is advised, because of a possible association with intrauterine growth restriction (346)
Iodides
Iodine crosses the placenta. If given in large amounts and for prolonged periods, it may induce fetal goiter and hypothyroidism. However, iodine has been used in small amounts, 6 to 40 mg/day in a group of pregnant Japanese women with mild hyperthyroidism (347). Elevation in serum TSH was observed in 2 of 35 newborns, and the mothers were slightly hyperthyroid at the time of delivery. In general however, iodine therapy is not routinely indicated in the treatment of hyperthyroidism in pregnancy.
Radioactive Iodine Administration
Radioactive iodine administration is contraindicated during pregnancy. In case of inadvertent radioiodine administration, the fetus is exposed to radiation from mother’s blood (approximately 0.5-1.0 Rad per mci administered). Since fetal thyroid uptake of radioiodine commences after the 12th week, exposure to maternal radioiodine prior to this time is not associated with fetal thyroid dysfunction (348). However, treatment with radioiodine after 12 weeks leads to significant radiation effects on the fetal thyroid. Multiple incidents of inadvertent exposure to radioiodine have been reported, causing fetal thyroid destruction, in utero hypothyroidism, and subsequent neural damage (349).
Surgery
Subtotal thyroidectomy in pregnancy is effective in managing the disease and usually should be performed in the second trimester. Unacceptable side effects of ATDs,poor patient compliance and very large goiter with potential obstruction are indications for surgery as well as patient preference .The mother should be prepared with β-blocking agents to rendered her hemodynamicaly stable and with Lugol’s solution for at least 10 days to reduce thyroid gland vascularity. A TRAb assay should also be performed as the fetus is at risk of hyperthyroidism (350).
Breast Feeding in Mothers with Treated Graves’ Disease
Lactation during ATD therapy has been discussed (351,). PTU and MMI are secreted in human milk, although PTU less so because of more extensive binding to albumin. With moderate doses of MMI or PTU (MMI: <20 mg/d; PTU: <250-300 mg/d), the risk to the infant is practically negligible.. The drug should be taken by the mother after a feeding but there is no need to monitor infant thyroid function. There is also a possibility that allergic reactions associated with ATD (agranulocytosis or rash) may occur in the infant. Wide experience has confirmed that the use of ATD in lactatin mothers does not pose a risk to the neonate and appears to be safe.
Gestational Non Autoimmune Hyperthyroidism
Thyrotoxicosis and hCG
Non autoimmune gestational hyperthyroidism or gestational transient thyrotoxicosis “GTT” is characterized by elevated serum free T4 and T3 levels, suppressed TSH, variable clinical evidence of hyperthyroidism, usually minimal thyroid enlargement, and absence of thyroid auto-antibodies and ophthalmopathy. The syndrome occurs transiently near the end of the 1st trimester of gestation, usually in hitherto healthy women who have otherwise a normal pregnancy, and it is frequently associated with excessive vomiting (296). GTT occurs in women without past history of GD and absence of TRAb. GTT is not always clinically apparent, due to its transient nature but is common in hyperemesis gravidarum (HG) up to 45%). The severity of GTT is related to serum hCG levels which are elevated. In patients with HG and GTT, thyroid function normalized by the second trimester without antithyroid treatment. GTT does not affect pregnancy outcomes (352). The prevalence of GTT is highly variable, being as low as 0.3% (Japan) or as high as 11% (Hong Kong) (353,354). A figure of around 2-3% of normal pregnancies is the usual case in Europe. (2). GTT is always transient; elevated serum free T4 values revert gradually to normal in parallel with the decrease in hCG concentrations. Serum TSH often remains partially (or totally) suppressed for several weeks after free T4 reverted to normal, i.e. until after mid-gestation (355).
Twin pregnancy is associated with sustained and high hCG concentrations Peak hCG values are significantly higher (almost double) and of a much longer duration in women with a twin pregnancy (up to 6 weeks compared to a few days in singleton pregnancy).. While peak hCG values lasted only for a few days in singleton pregnancy, peak hCG levels (>100,000 UI/L) lasted for up to six weeks in twin pregnancies (356). Hence intense vomiting is more frequently noted in women with a twin pregnancy.
Treatment
In most cases, no specific treatment is required and symptoms can be relieved by administration of beta-adrenergic blocking agents for a short period, while waiting for the spontaneous recovery of elevated thyroid hormones to occur. Hydration and antiemetics may be needed. In patients with a severe clinical presentation (clear symptomatic hyperthyroidism) it is important to rule out the presence of Graves’ disease by measurement of TRAb. Therapy with PTU for a few weeks has been suggested by some.
Pathogenic Mechanisms in GTT
The etiology of the syndrome is due to hCG itself or derivatives of hCG (52). Based on the example of GTT associated with twin pregnancy, a direct quantitative effect of elevated hCG concentrations to stimulate the thyroid gland is probably sufficient to explain hyperthyroidism in most pregnant women, provided that hCG remains above 75,000-100,000 UI/L for a sufficient period of time. Thus, GTT is directly related to both the amplitude and duration of peak hCG values (357). Human CG acts as a weak TSH agonist to increase cAMP production, iodide transport and cell growth in thyrocytes (52). It remains possible that abnormal h CG molecular variants, with a prolonged half life, are produced in these situations explaining sustained prolonged high circulating hCG levels (52). hCG molecular variants with a more potent thyrotropic activity have been detected, although these variants are more usually found in women with hydatidiform mole or choriocarcinoma (358). The hCG stimulation of the thyroid is related to the marked homology between hCG and TSH molecules, as well as between LH/CG and TSH receptors (259). Gestational non autoimmune hyperthyroidism can be considered an example of an endocrine ‘spill-over’ syndrome, a concept based on molecular mimicry between hormone ligands and their receptors (52).
TSH Receptor Mutations Hypersensitive to hCG
The thyrocyte may be a passive bystander of abnormal thyrotropic activity of hCG in GTT, or it may play an active role in its response to hCG through variable degrees of sensitivity of the TSH receptor. A woman with recurrent gestational hyperthyroidism was reported (122) who, after two miscarriages, presented with overt hyperthyroidism and hyperemesis early in pregnancy,. During her next pregnancy, she experienced a relapse of the same situation. The patient’s mother had also been diagnosed with hyperthyroidism during her 2nd and 3rd gestations, mistaken for GD. Study of the TSH receptor of this patient disclosed a single mutation in the extracellular domain of the TSH receptor (K183R), rendering the mutant receptor highly sensitive to hCG, and accounting for recurrent thyrotoxicosis during pregnancies in the presence of normal hCG levels. This finding remained unique until the same group described another case in 2016 (123). It is possible that some women who develop GTT may have an abnormality at the level of the TSH receptor, but perhaps not the same mutation as described (see Figure 14-14).
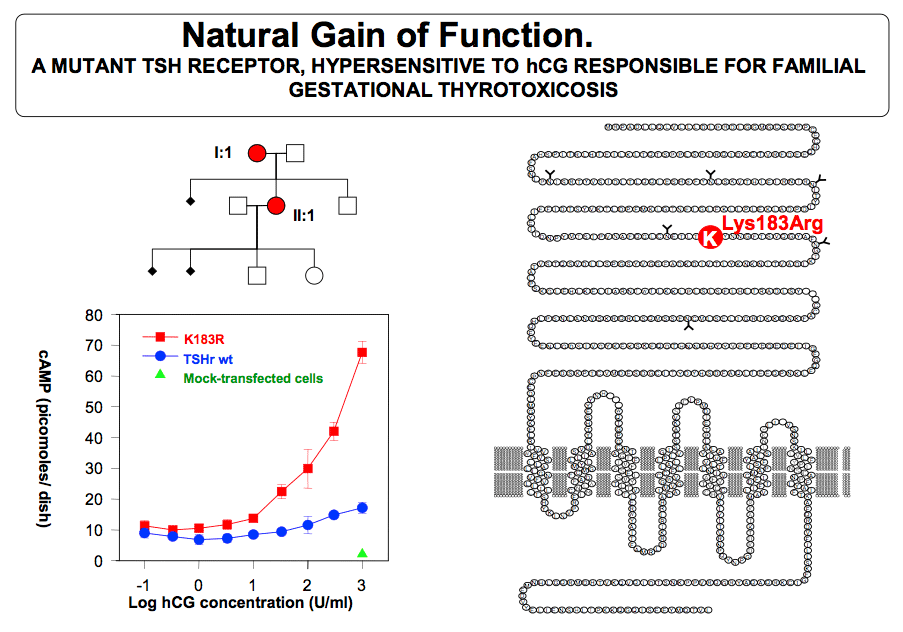
Figure14- 14: TSH receptor mutation, with a Lysine to Arginine mutation in position 183 of the ecto-domain. The graph on the left shows that the mutation confers high sensitivity to hCG (red curve), compared with wild type TSH receptor (blue curve). The family tree (upper right) shows the pedigree of the patient. (from 122).
Hyperemesis Gravidarum and Gestational Hyperthyroidism
Hyperemesis Gravidarum (HG) is reported to occur in 0.3 to 1.0% of pregnancies; it is defined as persistent nausea and vomiting in the first trimester of pregnancy, resulting in greater than 5% weight loss, ketonuria, dehydration liver and electrolyte abnormalities (hypokalemia, metabolic alkalosis, hyponatremia, hypochloremia) in severe cases (359). The onset of nausea is at about 4 to 6 week’s gestation, with worsening by 7- 9 weeks gestation, resolution by the end of the first trimester in 60% of cases, and complete resolution by 20 weeks in the vast majority of women.
The incidence of hyperthyroidism in women with HG depends on the severity of symptoms, ethnic background, perhaps dietary iodine intake, interpretation of thyroid tests and other unknown factors. The diagnosis of HG is based on the presence of clinical and physical clues: lack of hyperthyroid symptoms before conception, similar symptoms in previous pregnancies, and absence of goiter or Graves’ ophthalmopathy. Serum FT4 or Free Thyroxine Index (FT4I) are above the reference range, serum TSH is suppressed or undetectable and markers of thyroid autoimmunity (TPOAb and TRAb) are absent. In less than 20 % of affected women, serum TT3 is slightly elevated.
Ethnic variation in the incidence of HG suggesting strong evidence for a genetic component of HG. has been noted in a Norwegian study which showed 2.2% of Pakistani women; 1.9% of Turkish women and 0.5% of Norwegian women (360). A subsequent study found that, sisters and mothers were more affected than controls (361), An association between HG, hyperthyroidism and hydatidiform mole, has been documented (39). From the clinical aspect the most severely affected women have the lowest TSH and the highest FT4 and FT3 (many in the thyrotoxic range) (362)..
Antithyroid medications are not required in the vast majority of cases. In one series in which antithyroid medication was used, pregnancy outcome was not significantly different to a similar group of patients receiving no therapy (363). Occasionally, severe vomiting and hyperthyroidism may require parenteral nutrition
The differential diagnosis from Graves’ hyperthyroidism may be difficult, as vomiting may also be a presenting symptom of hyperthyroidism of Graves' disease. The diagnosis of transient hyperthyroidism of hyperemesis gravidarum should be considered in women with severe vomiting, no clinical manifestations of Graves' disease, and biochemical evidence of hyperthyroidism. Vomiting should be persistent and severe with a significant weight loss, since most women with morning sickness of pregnancy have normal thyroid function tests . Thyroid gland color flow Doppler sonography may be helpful in the diagnosis (364). Hyperemesis Gravidarum may also occur in women with Graves’ hyperthyroidism and in those with a previous history of Graves’ hyperthyroidism in remission; this is explained by the thyrotrophic action of hCG early in gestation. The differential diagnosis between the two entities may be difficult, the presence of TRAb favoring the diagnosis of Graves’ hyperthyroidism.
Trophoblastic diseases, partial and complete hydatidiform moles, and choriocarcinoma are other causes of hyperthyroidism early in pregnancy. Patients may present without symptoms in spite of chemical hyperthyroidism, or with various degrees of severity, including congestive heart failure. Evacuation of the mole eliminates the source of the excessive hCG and reverses the clinical and biochemical features of hyperthyroidism. Treatment with β-adrenergic blocking agents is effective in controlling the symptoms.
NODULAR THYROID DISEASE
Thyroid Nodule Growth During Pregnancy
Thyroid nodules can be detected in up to 10% of pregnant women. In an iodine deficient area there was no correlation between pregnancy and nodular thyroid disease (365). In a Chinese population pregnancy is associated with an increase in preexisting thyroid nodules as well as new thyroid nodule formation (366). In a retrospective study from the California Cancer Registry from 1991 to 1999, 129 cases of thyroid cancer were diagnosed during pregnancy: 3.3/100,000 diagnosed before pregnancy; 0.3/100,000 at the time of delivery and 10.8/100,000 within one year after delivery (367).
Diagnostic Evaluation and Management of a Thyroid Nodule in Pregnancy
Fine needle aspiration biopsy is the first investigation of choice and in one report yielded a malignancy/suspicious result in 35%. (368). In the presence of a single thyroid nodule detected on physical examination, or a dominant nodule in a multinodular gland, confirmed by ultrasonography, the following approach is suggested :
a) solid lesion <1cm, follow up in the postpartum;
b) nodules >1-1.5 cm, should be considered for FNA if there are suspicious findings on ultrasound,
c) in the presence of tracheal obstruction, immediate surgery;
d) if the FNA is diagnostic of malignancy or it is a suspicious lesion, some authors recommend that surgery may be postponed until after delivery, unless there are lymph node metastases or the lesion is a large primary or there is extensive lymph node involvement in a medullary cancer,
e) surgery and FNAB could both be postponed until after delivery with probable safety,
f) a woman with a malignant lesion or rapid growth should be offered surgery in the second trimester of gestation;
g) Some authors recommend that women with follicular lesions or early stage papillary carcinoma may postpone the surgery until postpartum, since these lesions are not expected to progress rapidly (369,370).
In a retrospective study of 61 women pregnant at the time of the diagnosis of differentiated thyroid carcinoma (papillary cancer in 87%, follicular cancer 13%) 14 were operated on during pregnancy, the other 47 women undergoing surgery 1 to 84 months after delivery (371). The outcome was compared with a group of 598 nonpregnant women matched for age and similar follow up (median 22.4 years and 19.5 years respectively). Treatment and outcome were similar in those operated in the postpartum period. It was concluded that both diagnostic studies and initial therapy might be delayed until after delivery in most patients. Cancer registry data compared disease-related survival in 6505 women diagnosed with thyroid cancer during pregnancy or 1 year post delivery and noted no significant difference in outcome up to 11 years compared to an age-matched non-pregnant cohort (372). The impact of pregnancy on thyroid cancer had been considered to be minimal in that there is no difference in rates of metastases or recurrence compared to non-pregnant women with the same disease.
However, an Italian study of 123 women with differentiated thyroid cancer in relation to pregnancy concluded that pregnancy had a negative impact on the outcome, by showing a poorer prognosis compared to those women diagnosed in nongravid periods (373). The role of estrogen and estrogen receptor status in this regard is still not clear as there are data showing a proliferative effect on thyroid cancer cell lines(374) but there are other reports of estrogen only stimulating adenomatous tissue but not neoplastic thyroid (375). In addition recent studies on the effect of pregnancy on prognosis of differentiated thyroid cancer (DTC) have stated that persistence /recurrence of DTC is significantly higher in pregnant patients (376) and that pregnancy was associated with increase in size of papillary microcarcinoma (377), More studies are required on follow up and mechanisms for these observations.
Pregnancy and Co-existing Thyroid Malignancy
Whether women already treated for thyroid malignancy should become pregnant is of concern, but current evidence suggests that treated differentiated thyroid cancer without evidence of residual disease should not inhibit an intended pregnancy. A meta analysis of the association of thyroid carcinoma with pregnancy concluded that multiple pregnancies and a <5 year interval were indetified as high risk factors for thyroid carcinoma but thyroid carcinoma during pregnancy was not associated with a significant risk of lymphatic and distant metastases (378). In a retrospective analysis on 36 women who became pregnant a median of 4.3 years after initial therapy for differentiated thyroid carcinoma, and were evaluated a median of 4 months after delivery (0.1-1.7 years), total thyroidectomy was performed in 80% and lobectomy in 20% (379). From the clinical progression and serum thyroglobulin (Tg) values it was concluded that pregnancy “is probably a mild stimulus to cancer growth as evidence by minor disease progression in some patients with known structural disease before pregnancy”. Hirsch et al (380) evaluated 63 consecutive women (90 births), followed from 1992 to 2009 who had delivered at least once after total thyroidectomy plus 131-Iodine (in 58 of them) for papillary thyroid cancer. Serum thyroglobulin (Tg) values and neck ultrasound were compared before and after pregnancy. Six women out of the 63 showed disease progression during the first pregnancy and two had disease progression only during the second pregnancy. Serum TSH levels during pregnancy correlated with disease persistence before pregnancy and disease progression during pregnancy. An interesting finding was that a non-suppressed TSH level during pregnancy did not stimulate disease progression during pregnancy; They concluded that pregnancy does not cause thyroid cancer recurrence in PTC survivors who have no structural or biochemical evidence of disease persistence at the time of conception. A study of 24 patients with papillary cancer suggested that PTC during pregnancy may be more locoregionally aggressive but no difference in survival or recurrence was demonstrated compared to non pregnant women (381). The conclusion from these studies indicate no progression of the disease in women free of disease before pregnancy, however there is a possibility of progression in those patients with evidence of residual cancer at the time of conception. Further studies are needed before a firm recommendation could be offered to patients
In patients who are clinically and biochemically free of disease but who present with a high risk tumor, TSH suppression should be maintained with serum TSH levels between 0.1 – 0.5 mU/L. In low-risk patients free of disease, TSH may be kept within the low normal range (0.3–1.5 mU/L). Finally, in patients who have not undergone remnant ablation, who are clinically free of disease and have undetectable suppressed serum Tg and normal neck US, the serum TSH may be allowed to remain in the low normal range (0.3–1.5 mU/L) (371). The recent ATA guidelines(13) suggest that PTC detected in early pregnancy should be monitored sonographically. If it grows substantially before 24-26 weeks gestation, or if cytologically malignant cervical lymph nodes are present, surgery should be considered during pregnancy. However, if the disease remains stable by midgestation, or if it is diagnosed in the second half of pregnancy, surgery may be deferred until after delivery.The impact of pregnancy on women with newly-diagnosed medullary carcinoma or anaplastic cancer is unknown. However, a delay in treatment is likely to adversely impact outcome. Therefore, surgery should be strongly considered, following assessment of all clinical factors.
Effect of Previous 131 Iodine Therapy
Previous 131I therapy does not result in demonstrable adverse events in subsequent pregnancies (382). One of the most common concerns in young people, male and female, is the potential side effects on future fertility and pregnancies following Iodine 131 therapy, both for Graves’ therapy and thyroid cancer. The majority of studies (383-385) concluded that in young men, following 131Iodine doses of up to 3.7 GBq, serum FSH and LH increases with some oligospermia, with normalization within 18 months following treatment. There are few cases of permanent damage, related to patient age, therefore it is recommended to perform a semen analysis before treatment. The risk of testicular dysfunction is increased after repeated or high cumulative radioiodine activity (386). No effects on the progeny have been reported. The effect on women is very consistent in the many reported series. Women may have irregular menses in the first twelve months following therapy, with restitution of normal cycling and fertility thereafter. In a review of 54 studies (387) there was no increase in miscarriages, congenital malformations, or prematurity compared to previous treatment. Early onset of menopause has been reported (387). Overall doses up to 3.7 GBq resulted only in transient menstrual cycle abnormalities lasting up to one year, but no permanent ovarian failure. Although there are no specific studies assessing the risk of pregnancies within 12 months of 131Iodine treatment, the overall consensus by experts in the field is to postpone pregnancy for one year after ablation therapy. The importance of achieving a serum TSH within target should be emphasized. Determination of serum hCG in addition to the pregnancy test on the day of radioactive therapy is recommended, since several cases of false negative pregnancy tests have been reported.
Thyroid hormone administration is justified to achieve a slightly suppressed (but detectable) serum TSH in pregnant women with an FNAB positive for or suspicious for cancer and who elect to delay surgical treatment until postpartum.
POSTPARTUM THYROID DYSFUNCTION
In addition to changes in circulating thyroid hormone concentrations observed during gestation (1), pronounced alterations in the immune system are evident (388). The cellular changes consist of a change from the so-called Th1 state to a predominance of cytokines such as IL-4 consistent with a Th2 status. On the humoral side the titre of anti thyroid peroxidase antibodies (anti TPOAb), found in 10% of pregnant women at 14-16 weeks gestation, decreases markedly during the 2nd and 3rd trimesters. At birth the Th2 status abruptly reverts back to the non pregnant Th1 position and this is accompanied by a dramatic rebound in the titre of antiTPOAb which reaches a maximum between 3 and 6 months postpartum (‘immune rebound phenomenon’). If thyrotropin receptor stimulating antibodies (TRAb) are present in early pregnancy they behave in a similar manner through gestation and the postpartum period. These immunological changes at delivery and the postpartum set the scene for the development of postpartum thyroid dysfunction. The changes in postpartum thyroid dysfunction may be transient or permanent and may be due to destructive or stimulating disease (Fig 14-15).
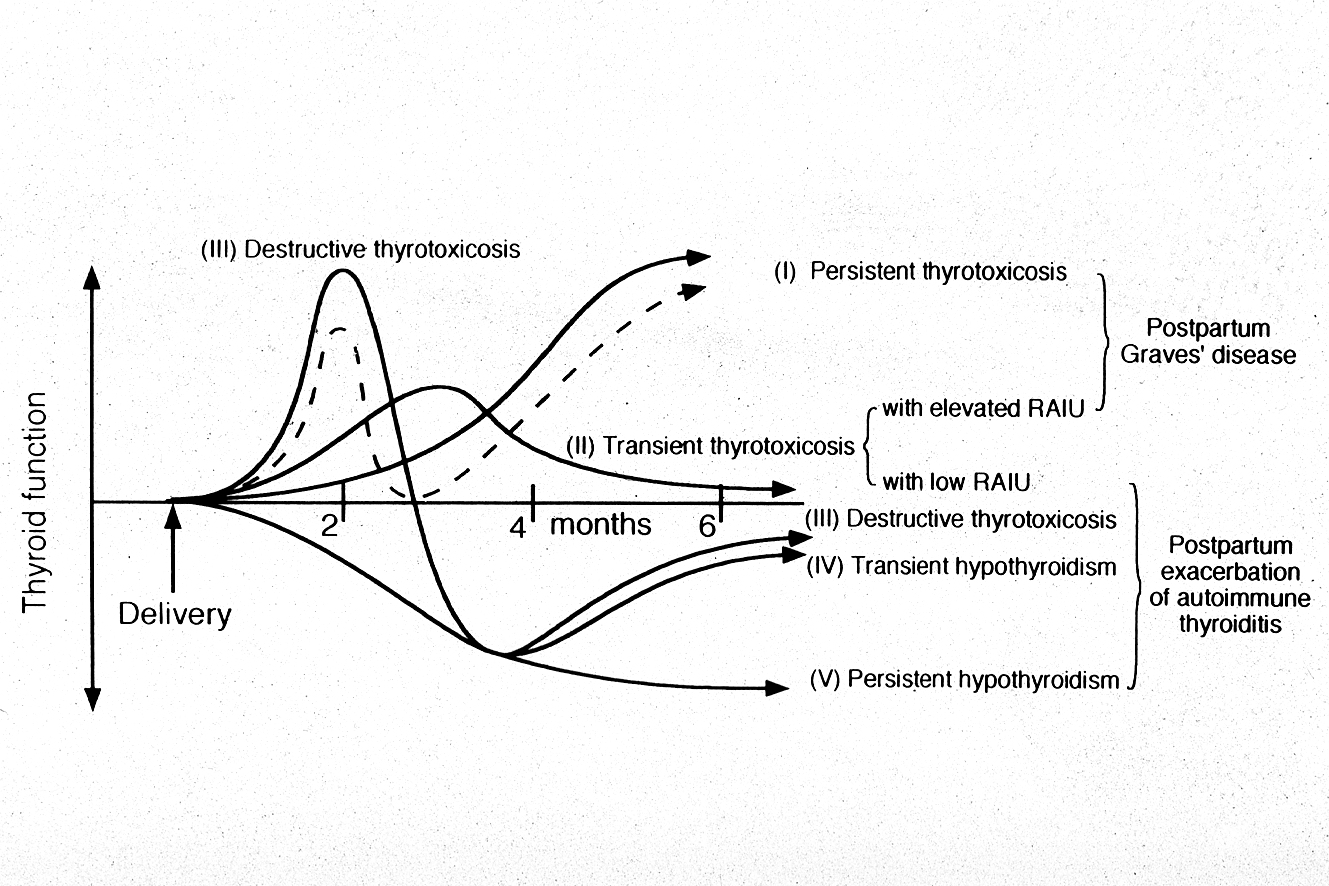
Figure 14-15 (from 389) Patterns of Postpartum Thyroid Dysfunction
POSTPARTUM GRAVES’ DISEASE
Individual patients at high risk of postpartum onset of Graves' disease can be found in early pregnancy by the detection of TRAb. Up to 40% of women with Graves’ hyperthyroidism have been found to occur after a recent pregnancy (390) and The PP period is significantly associated with a relapse of hyperthyroidism in GD patients being in remission after ATD (391).
It is important to differentiate postpartum Graves' disease with accompanying hyperthyroidism from the other causes of postpartum hyperthyroidism. The presence of circulating TRAb , radioiodine uptake, together with clinical examination and thyroid scintiscanning will usually resolve any diagnostic difficulty. A combination of positive TRAb and high thyroid blood flow suggests the presence of Graves’ disease (392). Spectral Doppler sonography may also be useful at this time (393). However silent thyroiditis (i.e. postpartum thyroiditis) commonly develops concomitantly with the activation of Graves’ disease and may delay or mask the development of Graves’ hyperthyroidism .The serum thyroglobulin concentration, which is raised in postpartum destructive thyroiditis with hyperthyroidism, has also been shown to be useful for the differentiation of this condition from Graves’ hyperthyroidism following delivery. Therapy of postpartum Graves’ hyperthyroidism should be carried out by the usual methods remembering that radioiodine is contraindicated during breast feeding. In addition, radiation safety requirements may make it very difficult for the mother to care for her new born child. Ideally another carer should look after the child for at least a week if an activity of 400-600 MBq (app. 10-16 mci) is administered. Alternatively the patient may be treated with antithyroid drugs at this stage. Clearly, prevention of postpartum patients Graves’ hyperthyroidism may be achieved by adequate treatment of the condition before the onset of gestation (394). Screening for TRAb during pregnancy may detect patients at risk of postpartum relapse
POSTPARTUM THYROIDITIS
Postpartum patients with lassitude and other symptoms of hypothyroidism were described in 1948 and these complaints were treated successfully with thyroid extract (395). The syndrome remained generally unrecognised until the 1970s when reports from Japan and Canada rediscovered the existence of postpartum thyroid disease and established the nature of the condition to be related to the immune rebound phenomenon (see above). Several reviews are available (396-399).
Incidence
A variable incidence (from 3-17%) has been reported worldwide because of wide variations in the number of women studied, the frequency of thyroid assessment postpartum, diagnostic criteria employed and differences in hormone assay methodology. However there is a general consensus that the disease occurs in 5-9% of unselected postpartum women. Women with Type 1 diabetes have a three-fold incidence of PPT compared to non-diabetics. In these cases there is a strong association with thyroid antibodies. PPT is also more likely to occur in women who has had a previous episode.
Clinical Spectrum
PPTD is a destructive thyroiditis characterized by transient hyperthyroidism (median time of onset 13 weeks) and/or hypothyroidism (median time of onset 19 weeks) which may occur up to 9 months following delivery (400). The distribution of clinical presentation is approximately 19% hyperthyroid alone, 49% hypothyroid alone and the remaining 32% hyper followed by hypothyroidism. Although the clinical manifestations of the hyperthyroid state are not usually severe in PPT, lack of energy and irritability are particularly prominent even in antibody +ve women who do not develop thyroid dysfunction. In contrast the symptomatology of the hypothyroid phase may be profound. Raised levels of circulating thyroid peroxidase autoantibody (TPOAb) are detected in 10% of pregnant women at 16 weeks Of these, 50% develop PPTD during the first six months of the postpartum period (401) Permanent hypothyroidism is reported in as many as 30% of these cases after 3 years, and in 50 % at 7 - 10 years .An association with depression has been observed with PPTD (402); depression is also associated with thyroid antibody positive women irrespective of thyroid status (403,404). However, a study in an Australian population found no association between PPTD or thyroid antibody status with depression (405). TPOAb positivity has also been associated with dysphoric mood during and after pregnancy (406).
Diagnosis
Hyperthyroidism in the postpartum period may be due to a recurrence or the development of new Graves' disease or to the hyperthyroid phase of postpartum thyroiditis. Symptoms of hyperthyroidism are much more evident in Graves' disease .As postpartum hyperthyroidism is a destructive process radioiodine uptake will be very low at early and late times after isotope administration. TSH receptor antibodies are not seen unless there is coexisting Graves' disease. Hyperthyroidism due to postpartum thyroiditis is diagnosed by a suppressed TSH together with an elevated FT4 or FT3, or elevated FT3 and FT4, with either set of criteria occurring on more than one occasion. A report (407) of PPT with hyperthyroidism in a patient with thyroid hormone resistance noted the suppression of TSH in this state which is unusual in RTH. If possible a normal range of thyroid hormone concentrations should be derived in the postpartum period as they fall into a narrower range than the general population. Antibodies to thyroxine and T3, which may occur in autoimmune thyroid disease, may cause confusion in diagnosis but they are infrequent.
Hypothyroidism in the postpartum period is diagnosed when either TSH >3.6 mU/L together with FT4<8 pmol/l or FT3 <4.2 pmol/l or TSH 10mU/l on one or more occasion is present. The most frequent symptoms have been found to be lack of energy, aches and pains, poor memory, dry skin and cold intolerance. Thyroid ultrasonography has demonstrated diffuse or multifocal echogenicity reflecting the abnormal thyroid morphology and consistent with the known lymphocytic infiltration of the thyroid .The destructive nature of the thyroiditis is also shown by the increase in urinary iodine excretion in the hyperthyroid as well as the hypothyroid phase of the syndrome. In addition there is evidence that an early rise in serum thyroglobulin (a further indicator of thyroid destruction) may help in the identification of those women at risk of PPT.
Management of PPT
The hyperthyroid phase is relatively asymptomatic and usually requires no specific therapy. If symptoms of tachycardia and palpitations are troublesome of if other symptoms of hyperthyroidism such as sweating or anxiety are present then beta adrenoreceptor blocking agents may be used. Propanolol is the drug of choice but if nightmares develop a more cardioselective β blocker may be used. If this class of drug is contraindicated verapamil may be effective for cardiac symptoms. Antithyroid drugs are not indicated as the condition is a destructive thyroiditis. In contrast, patients experience persistent and troublesome symptoms related to the hypothyroid period and treatment with L-Thyroxine should be given starting with 0.1mg per day increasing as necessary. At this stage it will not be clear whether the patient has developed transient or permanent hypothyroidism. In this instance it is reasonable to treat with thyroxine for one year and then review the patient to determine the thyroxine requirement. This will normally mean that the patient should stop the therapy for 4-6 weeks and then have a thyroid function test. Patients who have been known to have transient thyroid dysfunction postpartum should be checked at least annually as 50% of them will develop hypothyroidism after 7 years. This is in contrast to TPOAb +ve women who have not experienced any thyroid dysfunction postpartum whose rate of hypothyroidism at 7 years follow up is only 5%. Clearly these patients require less intensive surveillance.
Follow Up and Course of the Syndrome
While the hyperthyroidism of PPT always resolves several long term studies of the hypothyroid phase have documented persistence of hypothyroidism in 20-30% of cases (397). Follow-up assessment of antiTPO positive women (at 16 weeks gestation) 9 years later has shown that the rate of development of hypothyroidism was significantly greater (48% vs 8% ) in those who had had PPT compared to those who were euthyroid antibody positive (408).. Recurrence of transient PPT has been observed by several workers and there is a 70% chance of developing recurrent PPT after a first attack and a 25% risk even if the woman was only anti- TPO positive without thyroid dysfunction during the first postpartum period.
The Etiology and Nature of PPT
Factors which predispose towards the development of PPT include the presence of thyroid antibodies (usually TPO but occasionally Tg), a previous episode, type 1 diabetes mellitus and a positive family history of thyroid disease. There is no influence of breast feeding, cigarette smoking, parity or baby gender on the development of PPT. PPT is usually only seen in women who were known to have positive TPO antibodies as determined at 16 weeks gestation although other groups have described the condition in women without thyroid antibodies and with no discernible immune abnormality (401). The destructive nature of the thyroiditis has been noted above. There is no evidence that ambient iodine concentrations affect the incidence of the disease and iodine administration to marginally iodine deficient pregnant women will not prevent the onset of PPTD.
PPTD is an organ specific syndrome which has been regarded as a model of aggravation of the autoimmune state. Abnormal thyroid morphology has been shown by multifocal echogenicity and lymphocytic infiltration and follicles suggestive of Hashimoto’s disease in thyroid biopsies. .The rapid perturbation of the immune system in the post partum year contrasts with the relatively stable situation seen in chronic Hashimoto's thyroiditis. Although the antibody response is dramatic,its precise role in the immunopathogenesis of the condition remains to be determined. Probably, the antibody titre is merely a marker of disease and the immunological damage is mediated by lymphocyte and complement associated mechanisms. A prospective study of lymphocyte sub populations in anti TPO +ve pregnant women and antibody negative controls showed a significant fall in the CD4+/CD8+ ratio in late pregnancy and into the postpartum period in the controls (409). In contrast, women who subsequently developed PPT had a significantly higher CD4+/CD8+ ratio and T cell activation than in normal TPO-ve women. In addition, a particular lymphocyte subset ( CD45RA+ T cells), was also significantly elevated in those women destined to develop PPT and it is possible that this subset serves as a marker in this respect. It has also been suggested that the immunological determinants of postpartum thyroid dysfunction may in part occur antenatally (410). HLA haplotype restriction of the type commonly seen in autoimmune thyroid disease is seen in PPT.
Quantitative examination of complement C3b in PPT patients has shown that, not only is there activation of the complement system by thyroid directed autoantibodies, but that complement activation is related to the extent of the thyroiditis and correlates with the severity of the thyroid dysfunction .
Finally an exciting development has been reported by Negro et al who showed that the administration of selenium (as sodium selenite ) to TPOAb positive women through gestation led to a reduction in the postpartum TPOAb rise and also a significant reduction in postpartum hypothyroidism (411). These data still require confirmation.
SCREENING FOR THYROID DISEASE IN PREGNANCY
Screening for Disease
Medical screening is the systematic application of a test or inquiry to identify individuals at sufficient risk of a specific disorder to benefit from further investigation or direct preventive action (412). The requirements for a justifiable screening test are shown in table 14-11.
Table 14-11
Justification for Screening Test (adapted from 412)
- Well defined disorder with known incidence/prevalence
- Medically important disorder
- Screening test simple and safe with established cut off values
- Effective treatment available
- Cost of test relative to benefit should be known
- Adequate logistics for the testing and follow up
- Patient and management acceptability
It will be apparent that screening a population must be considered very carefully in respect of the condition being screened for, the effectiveness (and safety) of any intervention and the potential anxiety of the patient. If the effectiveness is not known with certainty then evidence should be sought, usually in the form of a randomised trial. The criteria listed in table 1 will now be discussed. Some data relevant to the debate has been mentioned in this chapter.
Does Thyroid Screening in Pregnancy Meet the Above Criteria for Screening?
The prevalence of Graves’ disease is approximately 3.0/1000 with an
incidence of about 0.5/1000/year. The prevalence and incidence in women
during child bearing years is not known but thyrotoxicosis is said to occur in 2
/1000 pregnancies and Graves’ disease would be expected to account for at
least 80% of these cases. While these figures are low, Graves’ hyperthyroidism
can have a dramatic effect on the mother as well as the fetus. There are significant maternal, fetal and neonatal complications (see section on Graves’ disease). Subclinical hyperthyroidism (i.e. normal circulating concentrations of T4 and T3 but subnormal TSH levels) occurs in approximately 1.7% of pregnant women and is not associated with adverse pregnancy outcomes (413). Screening for this condition is clearly not warranted, although if a low TSH is found the establishment of the cause will improve obstetric outcome in a number of women (414)
In contrast to hyperthyroidism, hypothyroidism is quite common in pregnancy . The incidence of subclinical hypothyroidism (raised TSH and normal or low normal T4) is at least 2.5% and these women have no clinical features and are often asymptomatic, but 50–60% will have evidence of autoimmune thyroid disease (positive TPOAbs and or thyroglobulin antibodies, TgAbs) in iodine-sufficient areas. It should be noted however that endemic iodine deficiency is the most common cause of hypothyroidism seen in pregnant women worldwide. Overt hypothyroidism occurs in only about 5% of all women who have a high TSH. During the last decade, it has become apparent that untreated maternal hypothyroidism and subclinical hypothyroidism in pregnancy is associated with adverse fetal and obstetric outcomes discussed in this chapter. There is a greater prevalence of subclinical hypothyroidism in women with delivery before 32 weeks and there is even an association between thyroid autoimmunity and adverse obstetric outcome, which is independent of thyroid function.(5). Higher maternal TSH levels even within the normal reference range are associated with an increased risk of miscarriages, fetal and neonatal distress (154) as well as preterm delivery.(18) In a prospective study, euthyroid TPOAb+ve women who received interventional L-thyroxine in early pregnancy had a reduced miscarriage rate and less preterm delivery.(203). Further prospective randomised trials are required to confirm these interesting data. Of equal or even greater importance than the above is the detrimental effect of hypothyroidism during pregnancy on fetal brain development. The neurodevelopmental impairment is similar to that seen in iodine deficient areas and implies that iodine status should be normalised in regions of deficiency. However, much of the USA is not iodine deficient which raises the question of routine screening of thyroid function during early pregnancy or even at preconception. In contrast gestational iodine deficiency in Europe is not uncommon (415) and decrements in mentation can be seen in iodine deficient areas as well as iodine sufficient ones. It should be noted however that the only randomized prospective trial adequately powered to answer the question as to whether thyroxine therapy to hypothyroid or hypothyroxinemic mothers resulted in benefit to the IQ of their children was negative (276). While there may be reasons for this result, further high quality evidence based studies must be performed to assess the situation such as the one in abstract (277).
As discussed in this chapter, isolated hypothyroxinaemia (IH) (low FT4 and normal TSH) either due to iodine deficiency or autoimmune thyroid disease has been shown to result in lower IQ in infants and young children in retrospective and prospective studies. IH Although it has been found to be associated with adverse perinatal outcomes (227) and is associated with reduced motor and intelligence performance in neonates (416) and in children aged 25–30 months in a Chinese population (255). While treatment of overt hypothyroidism has been shown to prevent the obstetric and neonatal complications the evidence for treatment of subclinical hypothyroidism in prevention is less secure. However a recent screening study where women were characterised as high risk or low risk in terms of the chance of adverse obstetric outcome there was a significant reduction in these outcomes even in low risk women who were screened for subclinical hypothyroidism (417).
Evidence for Intervention in High Risk Clinical Situations
The strength of evidence relating maternal hypothyroidism to low IQ in children suggests strongly that screening thyroid function in early gestation with l-thyroxine intervention in appropriate women would be beneficial. In addition there is evidence that such a strategy would be cost-effective. A study by Thung et al (418) compared the cost effectiveness of no screening versus routine screening for subclinical hypothyroidism in pregnancy.. The decision model demonstrated a saving of approximately $8.3 million per 100,000 women screened with an increment of 589.3 quality adjusted life years. Similar results were obtained by Dosiu et al (419) using a different screening model.
The lack of sufficient class A evidence has added to the controversy. Recent studies of targeted screening have concluded that targeted screening is unsatisfactory and that the data support the case for universal screening although more studies are required (420-422). The recommendation for universal screening has been made for pregnant women in Egypt (423), and pregnant women in China (424,425). An Asian survey found variable practice in relation to screening (426). Meanwhile other workers have recommended against screening (427,428). A recent opinion stated that testing maternal TSH as part of first trimester screening does not predict adverse pregnancy outcomes because mainly mild abnormalities in thyroid function are detected (429,430).
Several organisations have issued guidelines on whether to adopt a screening strategy for thyroid function in early pregnancy (11-15), the latest being the revised version of The American Thyroid Association guidelines (13). Generally, because of the lack of class A evidence (randomized controlled trial data) the recommendations from these guidelines) do not endorse universal screening. Instead, a targeted approach was suggested in specific clinical situations, although as noted below, this approach has limitations. The published Endocrine society of America guidelines (14) included a split recommendation, some members favoring universal screening, and others favoring targeted screening.A comparison of the 2 sets of recommendations is shown in table 14-12.
Table 14-12
Recommended patient profiles for targeted thyroid disease case finding in women seeking pregnancy, or newly pregnant:
ATA guidelines
History of thyroid dysfunction or prior thyroid surgery
Age >30 years
Symptoms of thyroid dysfunction or the presence of goiter
TPOAb positivity
Type 1 diabetes or other autoimmune disorders
History of miscarriage or preterm delivery
History of head or neck radiation
Family history of thyroid dysfunction
Morbid obesity (BMI ≥40 kg/m2)
Use of amiodarone or lithium, or recent administration of iodinated radiologic contrast
Infertility
Residing in an area of known moderate to severe iodine insufficiency
Endocrine Society Guidelines
Women over age 30 years ( ? Valid see 310a)
family history of autoimmune thyroid disease or hypothyroidism
goiter
thyroid antibodies, primarily thyroid peroxidase antibodies
symptoms or clinical signs suggestive of thyroid hypofunction
type 1 diabetes mellitus, or other autoimmune disorders
infertility
prior history of preterm delivery
prior therapeutic head or neck irradiation or prior thyroid surgery
currently receiving levothyroxine replacement
A comparison between the first ATA guidelines (12) and those of The Endocrine Society (14) concluded that the data available at that time are neither for or against universal screening (431).
Althoughtargeted screening might seem a reasonable approach in relation to economic and logistic factors, there has been accruing evidence that a substantial number of women with thyroid dysfunction would not be diagnosed in these circumstances. Vaidya (432) found that targeted testing of a previously defined high risk group who had a personal history of thyroid or other autoimmune disorders or a family history of thyroid disease (413 women) failed to detect 28% of pregnant women with a TSH>4.2mIU/L. Li et al (255) found that this strategy missed 36% of women with TSH>4.0mIU/L. Overall , targeted screening may miss 33-88% of women with a thyroid abnormality (420,423,424,432-435, ). For example, screening ‘low risk women’ identified 28% with thyroid dysfunction excluding those with just positive thyroid antibodies (420). The variability seen in these data may relate to different definitions of thyroid dysfunction and different ethnicity of the populations studied. A meta analysis performed by obstetricians in The Netherlands concluded that he overall lack of evidence precludes a recommendation for universal screening and is only justified in a research setting (162). Despite a lack of consensus among professional organisation many areas of the world are in fact performing routine screening (436-438).In the USA 74% of respondents at an ATA meeting advocated universal thyroid screening with TSH (439). To date there is an ongoing disussion relating to the evidence that levothyroxine treatment of pregnant women with subclinical hypothyroidism, isolated hypothyroxinemia, or thyroid autoimmunity is beneficial (439,,13,15). Therefore, there is ongoing debate regarding the need for universal screening for thyroid dysfunction during pregnancy . Efforts are still required to provide more high quality evidence to justify screening. There is some evidence that screening (with thyroxine intervention therapy) may at least prevent or reduce some obstetric complications associated with SCH in pregnancy (440,441). Meanwhile, optimal cooperation and communication between endocrinologists and obstetricians is also necessary.
Conclusion
The screening criteria for subclinical hypothyroidism in pregnancy are largely met. The condition is not rare and several retrospective studies imply adverse obstetric and child neurodevelopmental outcomes. However there are few prospective randomized trials to substantiate the benefit of screening and the CATS study did not show a benefit in child IQ at age 3 years (276). Nevertheless there seems to be a case for screening to prevent adverse obstetric outcomes. From the child cognitive function aspect there should be further studies where intervention is initiated early in the first trimester during the course of brain development. Unfortunately the recent NIH study reported in abstract indicated that recruitment of mothers was at least midway in the second trimester (277)
From the forgoing discussion this author believes that the lack of high quality clinical epidemiological evidence base probably does not justify universal screening at the present time. Other authors note that the data on the beneficial effects of treatment for subclinical hypothyroidism remain uncertain, but that the other established benefits justify universal screening at this time. However, it is likely that more evidence will be produced which may alter this view in the future. Other authors have suggested that screening may be worthwhile rather than searching for minor abnormalities in thyroid function tests (442) and that screening could be introduced on the premise that overt hypothyroidism is the target condition and that there is agreement with regard to treatment ( Meanwhile it must be admitted that screening is occurring round the world in a pragmatic fashion.
FINAL CONCLUSIONS
Pregnancy has profound effects on the regulation of thyroid function in healthy women and patients with thyroid disorders. These effects need to be recognized, precisely assessed, clearly interpreted, and correctly managed. For healthy pregnant women who reside in areas with a restricted iodine intake, relative hypothyroxinemia & goitrogenesis occur frequently, indicating that pregnancy constitutes a challenge for the thyroidal economy.
Overt thyroid dysfunction occurs in 2-3% of pregnancies, but subclinical thyroid dysfunction (both hyper- & hypothyroidism) is probably more prevalent and frequently remains undiagnosed, unless specific screening programs are initiated to disclose thyroid function abnormalities in early gestation. Maternal alterations of thyroid function due to iodine deficiency, hypothyroidism and hyperthyroidism have important implications for fetal/neonatal outcome. In recent years, particular attention has been focused on potential developmental risks for the fetuses of women with subclinical hypothyroidism during early gestation. These include obstetric problems and the possibility of impaired neurodevelopment.
Pregnancy increases the metabolic rate, blood flow, heart rate, and cardiac output, and various subjective sensations such as fatigue and heat intolerance that may suggest the possibility of coexistent thyrotoxicosis. Other metabolic changes which also impact the hypothalamic pituitary thyroid system are the potential direct stimulation of the maternal thyroid by hCG, as well as the accelerated metabolism of thyroxine, presumably due to increased placental deiodination enzymes.
In patients with hypothyroidism, it is important to recognize that therapeutic requirements for exogenous thyroxine are increased by 50% on average during pregnancy. This should be taken into account in the management of such patients.
Main causes of thyrotoxicosis in pregnancy include Graves' disease (uncommon, but potentially pregnancy-threatening) and gestational non autoimmune transient hyperthyroidism (more common, but remaining mild usually). The natural history of Graves' disease is altered during pregnancy, with a tendency for exacerbation in 1st trimester, amelioration during 2nd & 3rd trimesters, and typically a rebound during the postpartum period. These changes are the consequences of partial immune suppression during gestation with a rebound during the postpartum period. This must be kept in mind when treating thyrotoxic patients, since all ATD cross the placenta and may affect fetal thyroid function. PTU is now recommended only for 1st trimester and MMI for the rest of pregnancy.
Fetal and neonatal hyperthyroidism is due to the transplacental transfer of maternal stimulating TSH-receptor antibodies (TRAb). The diagnosis of fetal (and neonatal) hyperthyroidism is usually made on the basis of fetal tachycardia, accelerated bone age, and intrauterine growth retardation. It may occur in infants born to women with active Graves' disease, but also to women who have had prior definitive cure of their disease by surgery or radioactive iodine, but maintain high titers of TRAb. The proper management of pregnant patients with Graves' disease remains a difficult challenge in clinical endocrinology.
Thyroid nodules discovered during pregnancy should be aspirated for cytological diagnosis. If a malignancy is diagnosed, surgery should be performed during pregnancy or shortly thereafter. Pregnancy by itself usually does not adversely affect the natural history of differentiated thyroid carcinoma.
During the postpartum period, particular attention should be given to women with thyroid autoimmunity, since hypothyroidism and hyperthyroidism are frequently exacerbated in the months following the delivery.
Antenatal screening for thyroid dysfunction is being actively discussed by the thyroid community. At present evidence based studies are very limited and do not support this strategy. However many clinics worldwide are currently screening. Dialogue between endocrinologist and obstetrician is important in this regard The results of further randomized trials are awaited.
REFERENCES
1 Glinoer D. The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endocr Rev 1997;18: 404-33
2 KrassasGE, PoppeK, GlinoerD
Thyroid function and human reproductive health. Endocr Rev 2010;31:702-55
3 Weetman AP. Immunity, thyroid function and pregnancy: molecular mechanisms Nat Rev Endocrinol. 2010;6: 311-18.
4 Lazarus JH, Soldin O, Evans C. Assessing thyroid function in pregnancy. In: Brent G, ed. Thyroid Function Testing. New York : Springer, 2010: 209-33
5 John H. Lazarus Thyroid function in pregnancy British Medical Bulletin 2011; 97:137-48
6 Spencer L, Bubner T, Bain E, Middleton P Screening and subsequent management for thyroid dysfunction pre-pregnancy and during pregnancy for improving maternal and infant health. Cochrane Database Syst Rev. 2015 21:CD011263. doi: 10.1002/14651858.
7 Okosieme OE, Lazarus JH Important considerations in the management of Graves' disease in pregnant women. Expert Rev Clin Immunol. 2015;11:947-57. doi:10.1586/1744666X.2015.1054375.
8 Zimmermann MB. Iodine deficiency. Endocr Rev. 2009; 30:376-408
9 .Moog NK, Entringer S, Heim C, Wadhwa PD, Kathmann N, Buss C Influence of maternal thyroid hormones during gestation on fetal brain development.Neuroscience. 2015 Oct 3. pii: S0306-4522(15)00897-0. doi: 10.1016/j.neuroscience.2015.09.070.
10 Pearce EN Thyroid disorders during pregnancy and postpartum..Best Pract Res Clin Obstet Gynaecol. 2015 ;29:700-6. doi: 10.1016/j.bpobgyn.2015.04.007
11 Abalovich M, Amino N, Barbour LA, Cobin RH, De Groot L, Glinoer D, Mandel SJ, Stagnaro-Green A: Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2007; 92 (Supplement): S1-S47),
12 Stagnaro-Green A, Abalovich M, Alexander E, Azizi F, Mestman J, Negro R, Nixon A, Pearce EN, Soldin OP, Sullivan S, Wiersinga W; American Thyroid Association Taskforce on Thyroid Disease During Pregnancy and Postpartum. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum.Thyroid. 2011;21:1081-125
13 Erik K. Alexander, Elizabeth N. Pearce , Gregory A. Brent, Rosalind S. Brown, Herbert Chen,Chrysoula Dosiou, William A. Grobman , Peter Laurberg , John H. Lazarus, Susan J. Mandel , Robin P. Peeters , and Scott Sullivan 2016 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease during Pregnancy and the Postpartum. Thyoid 2016 in press
14 De Groot L, Abalovich M, Alexander EK, Amino N, Barbour L, Cobin RH, Eastman CJ, Lazarus JH, Luton D, Mandel SJ, Mestman J, Rovet J, Sullivan S. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline.J Clin Endocrinol Metab. 2012;97:2543-65
15 Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaidya B. 2014 European Thyroid Association Guidelines for the Management of Subclinical Hypothyroidism in Pregnancy and in Children Eur Thyroid J 2014;3:76-94 (DOI: 10.1159/000362597)
- Glinoer D: Pregnancy and iodine. Thyroid 2001;11:471-81
17 Liberman CS, Pino SC, Lieh Fang S, Braverman LE, Emerson CH: Circulating iodide concentrations during and after pregnancy. J Clin Endocrinol Metab 1998 ; 83:3545-9
- Halnan KE: The radioiodine uptake of the human thyroid in pregnancy. Clin Sci 1958 17:281-90,
19 Burns R, Azizi F, Hedayati M, Mirmiran P, O'Herlihy C, Smyth PPIs placental iodine content related to dietary iodine intake? Clin Endocrinol (Oxf). 2011; 75 :261-4.
- Crooks J, Tulloch MI, Turnbull AC, et al: Comparative incidence of goitre in pregnancy in Iceland and Scotland. Lancet 1969; 2:625-7,
21, Farebrother J, Naude CE, Nicol L, Andersson M, Zimmermann MB.Iodised salt and iodine supplements for prenatal and postnatal growth: a rapid scoping of existing systematic reviews. Nutr J. 2015 2;14:89. doi: 10.1186/s12937-015-0079-z.
22 WHO Secretariat, Andersson M, de Benoist B, Delange F, Zupan J. Prevention and control of iodine deficiency in pregnant and lactating women and in children less than 2-years-old: conclusions and recommendations of the Technical Consultation.Public Health Nutr. 2007; 10(12A):1606-11
23 Charlton K, Land MA, Ma G, Yeatman H, Houweling F Iodine status similarly suboptimal in Australian women who have desirable salt intakes compared to those with excessive intakes..Nutrition. 2014;30: 234-5. doi: 10.1016/j.nut.2013.05.009
24 Vanderpump MP, Lazarus JH, Smyth PP, Laurberg P, Holder RL, Boelaert K, Franklyn JA; British Thyroid Association UK Iodine Survey Group Iodine status of UK schoolgirls: a cross-sectional survey.Lancet. 2011;377: 2007-12.
- Lee KW, Cho MS, Shin D, Song WO. Changes in iodine status among US adults, 2001-2012. Int J Food Sci Nutr. 2016 ;67:184-94. doi: 10.3109/09637486.2016.1144717
26 Zimmermann MB The effects of iodine deficiency in pregnancy and infancy.
Paediatr Perinat Epidemiol. 2012 ;26 Suppl 1:108-17.
- Glinoer D: The regulation of thyroid function during normal pregnancy: importance of the iodine nutrition status. In: Best Practice & Research in Clinical Endocrinology and Metabolism: The Thyroid and Pregnancy (Editor: Glinoer D) 2004; 18:133-52
- Smyth PP, Hetherton AM, Smith DF, Radcliff M, O'Herlihy C: Maternal iodine status and thyroid volume during pregnancy: correlation with neonatal iodine intake. J Clin Endocrinol Metab 1997; 82:2840-3
29 Pedersen KM, Laurberg P, Iversen E, Knudsen PR, Gregersen HE, Rasmussen OS, Larsen KR, Eriksen GM, Johannesen PL. Amelioration of some pregnancy-associated variations in thyroid function induced by iodine supplementation. J Clin Endocrinol Metab 1993; 77:1078-83
- Romano R, Jannini EA, Pepe M, Grimaldi A, Olivieri M, Spennati P, Cappa F, D'Armiento M. The effects of iodoprophylaxis on thyroid size during pregnancy. Am J Obstet Gynecol 1991;164:482-5,
31 Glinoer D, Lemone M, Bourdoux P, De Nayer P, DeLange F, Kinthaert J, LeJeune B. Partial reversibility during late postpartum of thyroid abnormalities associated with pregnancy. J Clin Endocrinol Metab1992; 74:453-7
32 Moreno-Reyes R, Glinoer D, Van Oyen H, Vandevijvere S.High prevalence of thyroid disorders in pregnant women in a mildly iodine-deficient country: a population-based study.
J Clin Endocrinol Metab. 2013;98:3694-701
- Glinoer D, De Nayer P, Delange F, Lemone M, Toppet V, Spehl M, Grün JP, Kinthaert J, Lejeune B. A randomized trial for the treatment of mild iodine deficiency during pregnancy: maternal and neonatal effects. J Clin Endocrinol Metab 1995; 80:258-69
- Rotondi M, Amato G, Biondi B, Mazziotti G, Del Buono A, Rotonda Nicchio M, Balzano S, Bellastella A, Glinoer D, Carella C. Parity as a thyroid size-determining factor in areas with moderate iodine deficiency. J Clin Endocrinol Metab 2000; 85:4534-37
- Knudsen N, Bülow I, Laurberg P, Ovesen L, Perrild H, Jørgensen T. Parity is associated with increased thyroid volume solely among smokers in an area with moderate to mild iodine deficiency. Europ J Endocrinology 2002;146:39-43
36 Glinoer D, Demeester R, Lemone M, Larsimont D, Andry G. Acute increase in goiter size during a normal pregnancy: an exceptional case report. Thyroid 2003;13:881-884
37.Aloumanis K, Mavroudis K, Vassiliou I, Arkadopoulos N, et al: Urgent thyroidectomy for acute airway obstruction caused by a goiter in a euthyroid pregnant woman. Thyroid 2006; 16:85-88,
- Vermiglio F, Lo Presti VP, Castagna MG, et al: Increased risk of maternal thyroid failure with pregnancy progression in an iodine deficient area with major iodine deficiency disorders. Thyroid 1999;9:19-24
39 Report of a WHO Technical Consultation on prevention and control of iodine deficiency in pregnancy, lactation, and in children less than 2 years of age (Geneva, 24-26 January 2005) (de Benoist B, Delange F, Editors). Public Health Nutrition (Special Issue) 10 (12A):1527-1611, 2007
40 Vandevijvere S, Amsalkhir S, Mourri AB, Van Oyen H, Moreno-Reyes R.
Br J Nutr. 2013: 28;109:2276-84.
41 Technical Consultation: Salt reduction and iodine fortification strategies in public health co-sponsored by the George Institute for Global Health, AustraliaIn collaboration with the International Council for the Control of Iodine Deficiency Disorders Global Network 25 - 27 March 2013Sydney, Australia
42 The effect of iodine prophylaxis on the incidence of endemic cretinism.
Pharoah PO, Buttfield IH, Hetzel BS.Adv Exp Med Biol. 1972;30:201-21
43 Melse-Boonstra A, Gowachirapant S, Jaiswal N, Winichagoon P, Srinivasan K, Zimmermann MB. Iodine supplementation in pregnancy and its effect on child cognition.
J Trace Elem Med Biol. 2012 ;26:134-6
44 van Mil NH, Tiemeier H, Bongers-Schokking JJ, Ghassabian A, Hofman A, Hooijkaas H, Jaddoe VW, de Muinck Keizer-Schrama SM, Steegers EA, Visser TJ, Visser W, Ross HA, Verhulst FC, de Rijke YB, Steegers-Theunissen RP. Low urinary iodine excretion during early pregnancy is associated with alterations in executive functioning in children.
J Nutr. 2012;142:2167-74.
45 Bath SC, Steer CD, Golding J, Emmett P, Rayman MP.
Lancet. 2013 ;382:331-7.
46 Taylor PN, Okosieme OE, Dayan CM, Lazarus JH. Impact of iodine supplementation in mild-to-moderate iodine deficiency: systematic review and meta-analysis.
Eur J Endocrinol. 2013 2;170: R1-R15. PMID:24088547
47 Fuse Y, Ohashi T, Yamaguchi S, Yamaguchi M, Shishiba Y, Irie M Iodine status of pregnant and postpartum Japanese women: effect of iodine intake on maternal and neonatal thyroid function in an iodine-sufficient area. J Clin Endocrinol Metab. 2011;96:3846-54
48 Sang Z, Wei W, Zhao N, Zhang G, Chen W, Liu H, Shen J, Liu J, Yan Y, Zhang W.
Thyroid dysfunction during late gestation is associated with excessive iodine intake in pregnant women.J Clin Endocrinol Metab. 2012;97: E1363-9.
49 Nohr SB, Laurberg P: Opposite variations in maternal and neonatal thyroid function induced by iodine supplementation during pregnancy. J Clin Endocrinol Metab 2000; 85:623-627
50 Zimmermann MB, Aeberli I, Torresani T, Bürgi H: Increasing the iodine concentration in the Swiss iodized salt program markedly improved iodine status in pregnant women and children: a 5-y prospective national study Am J Clin Nutrition 2005; 82:388-92
- Andersson M, Takkouche B, Egli I, et al: Current global iodine status and progress over the last decade towards the elimination of iodine deficiency. Bull WHO 2005; 83:518-25,
- Hershman JM: Physiological and pathological aspects of the effect of human chorionic gonadotropin on the thyroid. In: Best Practice & Research in Clinical Endocrinology and Metabolism: The Thyroid and Pregnancy (Editor: Glinoer D) 2004 ; 18:249-264,
53 . Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S.: Serum levels of intact human chorionic gonadotropin (hCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881-8
54 Kraiem Z, Lahat N, Sadeh O, Blithe DL, Nisula BC: Desialylated and deglycosylated human chorionic gonadotropin are superagonists of native human chorionic gonadotropin in human thyroid follicles. Thyroid 1997;7:783-8
55 Walkington L, Webster J, Hancock BW, Everard J, Coleman RE.Hyperthyroidism and human chorionic gonadotrophin production in gestational trophoblastic disease.Br J Cancer. 2011;104:1665-9
- Glinoer D, Gershengorn MC, Dubois A, Robbins J.: Stimulation of thyroxine-binding globulin synthesis by isolated rhesus monkey hepatocytes after in vivo -estradiol administration. Endocrinology 1977;100:807-13
- Ain KB, Mori Y, Refetoff S: Reduced clearance rate of thyroxine-binding globulin (TBG) with increased sialylation: a mechanism for estrogen-induced elevation of serum TBG concentration. J Clin Endocrinol Metab1987; 65:689-96
58 Ain KB, Refetoff S: Relationship of oligosaccharide modification to the cause of serum thyroxine-binding globulin excess. J Clin Endocrinol Metab 1988;66:1037-43
- Refetoff S: Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev 1989; 10:275-93,
- Zigman JM, Cohen SE, Garber JR: Impact of thyroxine-binding globulin on thyroid hormone economy during pregnancy. Thyroid 2003; 13:1169-75
61 Glinoer D: Increased TBG during pregnancy and increased hormonal requirements. Thyroid (letter to the Editor) 14:179, 2004
62 Dowling JT, Appleton WG, Nicoloff JT: Thyroxine turnover during human pregnancy. J Clin Endocrinol Metab 1964;27:1749-50
63 Chan SY, Vasilopoulou E, Kilby MD. The role of the placenta in thyroid hormone
delivery to the fetus. Nat Clin Pract Endocrinol Metab 2009; 5:45-54.
64 Patel J, Landers K, Li H, Mortimer RH, Richard K. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol Metab. 2011;22:164-70.
65 Burns R, O'Herlihy C, Smyth PP. The Placenta as a Compensatory Iodine Storage Organ.Thyroid. 2011, 21: 541-46
66 Burns R, O'Herlihy C, Smyth PP. Regulation of iodide uptake in placental primary cultures.
Eur Thyroid J. 2013;2:243-51. doi: 10.1159/000356847
67 Li H, Patel J, Mortimer RH, Richard K Ontogenic changes in human placental sodium iodide symporter expression. Placenta. 2012 ;33:946-8.
68 Andersen SL, Nøhr SB, Wu CS, Olsen J, Pedersen KM, Laurberg P
Thyroglobulin in smoking mothers and their newborns at delivery suggests autoregulation of placental iodide transport overcoming thiocyanate inhibition. Eur J Endocrinol. 2013;15:168:723-31.
69 Deng WB, Liang XH, Liu JL, Yang ZM. Regulation and function of deiodinases during decidualization in female mice.Endocrinology. 2014;155:2704-17. doi: 10.1210/en.2014-1015
70 Loubiere LS, Vasilopoulou E, Bulmer JN, Taylor PM, Stieger B, Verrey F, McCabe CJ, Franklyn JA, Kilby MD, Chan SY. Expression of thyroid hormone transporters in the human placenta and changes associated with intrauterine growth restriction. Placenta 2010; 31:295-304.
71 Sun YN, Liu YJ, Zhang L, Ye Y, Lin LX, Li YM, Yan YQ, Chen ZP. Expression of organic anion transporting polypeptide 1c1 and monocarboxylate transporter 8 in the rat placental barrier and the compensatory response to thyroid dysfunction.PLoS One. 2014 24;9:e96047. doi: 10.1371/journal.pone.0096047.
72 Loubière LS, Vasilopoulou E, Glazier JD, Taylor PM, Franklyn JA, Kilby MD, Chan SY.
Expression and function of thyroid hormone transporters in the microvillous plasma membrane of human term placental syncytiotrophoblast.Endocrinology. 2012 ;153:6126-35.
73 Vasilopoulou E, Loubière LS, Heuer H, Trajkovic-Arsic M, Darras VM, Visser TJ, Lash GE, Whitley GS, McCabe CJ, Franklyn JA, Kilby MD, Chan SY Monocarboxylate transporter 8 modulates the viability and invasive capacity of human placental cells and fetoplacental growth in mice. PLoS One. 2013 12;8:e65402. doi: 10.1371/journal.pone.0065402.
74 Landers KA, Mortimer RH, Richard K. Transthyretin and the human placenta.
Placenta. 2013 ;34:513-7
75 Mortimer RH, Landers KA, Balakrishnan B, Li H, Mitchell MD, Patel J, Richard K. Secretion and transfer of the thyroid hormone binding protein transthyretin by human placenta.Placenta. 2012 ;33:252-6
76 Laoag-Fernandez JB, Matsuo H, Murakoshi H, Hamada AL, Tsang BK, Maruo T. 3,5,3 '-triiodothyroninedown-regulates Fas and Fas ligand expression andsuppresses caspase-3 and poly (adenosine 5 '-diphosphate-ribose) polymerase cleavage and apoptosis in early placental extravillous trophoblasts in vitro. Journal of Clinical Endocrinology and Metabolism 2004;89:4069-4077.
77 Roti E, Gnudi A, Braverman LE: The placental transport, synthesis and metabolism of hormones and drugs which affect thyroid function. Endocr Rev1983; 4:131-149
78 Vulsma T, Gons MH, De Vijlder JMM: Maternal fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect of thyroid dysgenesis. N Engl J Med , 1989;321:13-6
79 Contempre B, Jauniaux E, Calvo R, Jurkovic D, Campbell S, de Escobar GM : Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J Clin Endocrinol Metab1993; 77:1719-22
80 Colicchia M, Campagnolo L, Baldini E, Ulisse S, Valensise H, Moretti C Molecular basis of thyrotropin and thyroid hormone action during implantation and early development..Hum Reprod Update. 2014;20: 884-904. doi: 10.1093/humupd/dmu028
81 Saben J, Kang P, Zhong Y, Thakali KM, Gomez-Acevedo H, Borengasser SJ, Andres A, Badger TM, Shankar K. RNA-seq analysis of the rat placentation site reveals maternal obesity-associated changes in placental and offspring thyroid hormone signaling. Placenta. 2014;35:1013-20. doi: 10.1016/j.placenta.2014.09.015.
82 Silva JF, Ocarino NM, Serakides R.Placental angiogenic and hormonal factors are affected by thyroid hormones in rats. Pathol Res Pract. 2015;211:226-34. doi: 10.1016/j.prp.2014.11.003.
83 Vasilopoulou E, Loubière LS, Lash GE, Ohizua O, McCabe CJ, Franklyn JA, Kilby MD, Chan SY.Triiodothyronine regulates angiogenic growth factor and cytokine secretion by isolated human decidual cells in a cell-type specific and gestational age-dependent manner.Hum Reprod. 2014;29:1161-72. doi: 10.1093/humrep/deu046.
84 Korevaar TI, Steegers EA, de Rijke YB, Visser WE, Jaddoe VW, Visser TJ, Medici M, Peeters RP Placental Angiogenic Factors Are Associated With Maternal Thyroid Function and Modify hCG-Mediated FT4 Stimulation. J Clin Endocrinol Metab. 2015;100:E1328-34. doi: 10.1210/jc.2015-2553
85 Chen CY, Chen CP, Lin KH. Biological functions of thyroid hormone in placenta. Int J Mol Sci. 2015 16;16:4161-79. doi: 10.3390/ijms16024161.
86 Kahric-Janicic N, Soldin SJ, Soldin OP, West T, Gu J, Jonklaas J: Tandem mass spectrometry improves the accuracy of free thyroxine measurements during pregnancy. Thyroid 17:303-11, 2007
87 Lee RH, Spencer CA, Mestman JH, Miller EA, Petrovic I, Braverman LE, Goodwin TM. Free T4 immunoassays are flawed during pregnancy. Am J Obstet Gynecol. 2009 ;200:260.e 1-6 doi: 10.1016/j.ajog.2008.10.042
88 Soldin, O. P. Soldin D, Sastoque M. Gestation-specific thyroxine and thyroid stimulating hormone levels in the United States and worldwide. Ther Drug Monit 2007; 29: 553-9.
89 van Deventer HE, Soldin SJ.The expanding role of tandem mass spectrometry in optimizing diagnosis and treatment of thyroid disease. Adv Clin Chem. 2013; 61:127-52
90 Fritz, K. S., R. B. Wilcox, Nelson JC. A direct free thyroxine (T4)
immunoassay with the characteristics of a total T4 immunoassay. Clin Chem 2007;
53: 911-5
91 Berta E, Samson L, Lenkey A, Erdei A, Cseke B, Jenei K, Major T, Jakab A, Jenei Z, Paragh G, Nagy EV, Bodor M. Evaluation of the thyroid function of healthy pregnant women by five different hormone assays.Pharmazie 2010 ;65:436-39.
92 Springer D, Bartos V, Zima T. Reference intervals for thyroid markers in early pregnancy determined by 7 different analytical systems. Scand J Clin Lab Invest. 2014 ;74:95-01. doi: 10.3109/00365513.2013.860617.
93 Bliddal S, Feldt-Rasmussen U, Boas M, Faber J, Juul A, Larsen T, Precht DH. Gestational age-specific reference ranges from different laboratories misclassify pregnant women's thyroid status: comparison of two longitudinal prospective cohort studies. Eur J Endocrinol. 2013 27;170:329-9. doi: 10.1530/EJE-13-0672
94 Anckaert E, Poppe K, Van Uytfanghe K, Schiettecatte J, Foulon W, Thienpont LM. FT4 immunoassays may display a pattern during pregnancy similar to the equilibrium dialysis ID-LC/tandem MS candidate reference measurement procedure in spite of susceptibility towards binding protein alterations Clin Chim Acta. 2010; 41:1348-53
95 Midgley JE, Hoermann R.Measurement of total rather than free thyroxine in pregnancy: the diagnostic implications.Thyroid. 2013 ;23:259-61
96 Gilbert, R. M., N. C. Hadlow, Walsh JP, Fletcher SJ, Brown SJ, Stuckey BG, Lim EM . Assessment of thyroid function during pregnancy: first-trimester (weeks 9-13) reference intervals derived from Western Australian women. Med J Aust 2008;189: 250-3.
97 Gong, Y. and B. R. Hoffman . Free thyroxine reference interval in each trimester of pregnancy determined with the Roche Modular E-170 electrochemiluminescent immunoassay. Clin Biochem 2008;41: 902-6.
98 Marwaha, R. K., S. Chopra, Gopalakrishnan S, Sharma B, Kanwar RS, Sastry A, Singh S. Establishment of reference range for thyroid hormones in normal pregnant Indian women. BJOG 2008; 115: 602-6.
99 Stricker, R. Echenard M, Eberhart R, Chevailler MC, Perez V, Quinn FA, Stricker R. et al. (2007). Evaluation of maternal thyroid function during pregnancy: the importance of using gestational age-specific reference intervals. Eur J Endocrinol 157: 509-14.
100 Pearce, E. N. Oken E, Gillman MW, Lee SL, Magnani B, Platek D, Braverman LE. . Association of first-trimester thyroid function test values with thyroperoxidase antibody status, smoking, and multivitamin use. Endocr Pract 2008;14: 33-9.
101 Lambert-Messerlian, G., M. McClain, Haddow JE, Palomaki GE, Canick JA, Cleary-Goldman J, Malone FD, Porter TF, Nyberg DA, Bernstein P, D'Alton ME. First- and second-trimester thyroid hormone reference data in pregnant women: a FaSTER (First- and Second-Trimester Evaluation of Risk for aneuploidy) Research Consortium study. Am J Obstet Gynecol 2008;199: 62. e1-6. doi: 10.1016/j.ajog.2007.12.003
102 La'ulu, S. L. and W. L. Roberts . Second-trimester reference intervals for thyroid tests: the role of ethnicity. Clin Chem 2007;53:1658-64.
103 Thevarajah M, Chew YY, Lim SC, Sabir N, Sickan J. Determination of trimester specific reference intervals for thyroid hormones during pregnancy in Malaysian women.Malays J Pathol. 2009; 31:23-7.
104 Vila L, Serra-Prat M, Palomera E, et al. Reference values for thyroid function tests in pregnant women living in Catalonia, Spain. Thyroid; 2010 :20:221-5.
105 Ashoor G, Kametas NA, Akolekar R, Guisado J, Nicolaides KH. Maternal thyroid function at 11-13 weeks of gestation.Fetal Diagn Ther. 2010; 27:156-163
106 Azizi F, Ladan M, Amouzegar A, Hossein D, Maryam T, Sahar A, Hedayati M.
Establishment of the trimester-specific reference range for free thyroxine index. Thyroid. 2013 ;23:354-9.
107 Xing J, Yuan E, Li J, Zhang Y, Meng X, Zhang X, Rong S, Lv Z, Tian Y, Jia L.Trimester- and Assay-Specific Thyroid Reference Intervals for Pregnant Women in China.
Int J Endocrinol. 2016;2016:3754213. doi: 10.1155/2016/3754213.
108 Li C, Shan Z, Mao J, Wang W, Xie X, Zhou W, Li C, Xu B, Bi L, Meng T, Du J, Zhang S, Gao Z, Zhang X, Yang L, Fan C, Teng W. Assessment of thyroid function during first-trimester pregnancy: what is the rational upper limit of serum TSH during the first trimester in Chinese pregnant women? J Clin Endocrinol Metab. 2014; 99:73-9. doi: 10.1210/jc.2013-1674
109 Medici M, Korevaar TI, Visser WE, Visser TJ, Peeters RP.Thyroid function in pregnancy: what is normal? Clin Chem. 2015;61:704-13. doi: 10.1373/clinchem.2014.236646
110 McNeil AR, Stanford PE. Reporting Thyroid Function Tests in Pregnancy.
Clin Biochem Rev. 2015;36:109-26
111 Panesar, NS, Li CY, Rogers MS. Reference intervals for thyroid hormones in pregnant Chinese women. Ann Clin Biochem 2001; 38: 329-32.
112 Boas M, Forman JL, Juul A, Feldt-Rasmussen U, Skakkebaek NE, Hilsted L, Chellakooty M, Larsen T, Larsen JF, Petersen JH, Main KM. Narrow intra-individual variation of maternal thyroid function in pregnancy based on a longitudinal study on 132 women. Eur J Endocrinol. 2009;161:903-10.
113 Walker JA, Illions EH, Huddleston JF, Smallridge RC.Racial comparisons of thyroid function and autoimmunity during pregnancy and the postpartum period. Obstet Gynecol. 2005 ;106:1365-71.
114 Korevaar TI, Medici M, de Rijke YB, Visser W, de Muinck Keizer-Schrama SM, Jaddoe VW, Hofman A, Ross HA, Visser WE, Hooijkaas H, Steegers EA, Tiemeier H, Bongers-Schokking JJ, Visser TJ, Peeters RP. Ethnic differences in maternal thyroid parameters during pregnancy: the Generation R study.J Clin Endocrinol Metab. 2013 ;98:3678-86.
115 Han C, Li C, Mao J, Wang W, Xie X, Zhou W, Li C, Xu B, Bi L, Meng T, Du J, Zhang S, Gao Z, Zhang X, Yang L, Fan C, Teng W, Shan Z. High Body Mass Index Is an Indicator of Maternal Hypothyroidism, Hypothyroxinemia, and Thyroid-Peroxidase Antibody Positivity during Early Pregnancy. Biomed Res Int. 2015;2015:351831. doi: 10.1155/2015/351831
116 Haddow JE, Neveux LM, Palomaki GE, Lambert-Messerlian G, Malone FD, D'Alton ME. An Inverse Relationship Between Weight and Free Thyroxine During Early Gestation Among Women Treated for Hypothyroidism.Thyroid. 2015;25:949-53. Doi
10.1089/thy.2015.0085.
117 Verberg MF, Gillott DJ, Al-Fardan N, Grudzinskas JG: Hyperemesis gravidarum, a literature review. Hum Reprod Update 2005;11:527- 39,
118 Grjibovski AM, Vikanes A; Stoltenberg C Magnus P. Consanguinity and the risk of hyperemesis gravidarum in Norway. Acta Obstetr Gynecol Scand 2008; 87:20-5.
119 Zhang Y, Cantor RM, MacGibbon K, Romero R, Goodwin TM, Mullin PM, Fejzo MS. Familial aggregation of hyperemesis gravidarum. Am J Obstet Gynecol 2011; 204:230.e1-7 doi: 10.1016/j.ajog.2010.09.018
120.Jordan V, Grebe SK, Cooke RR, Ford HC, Larsen PD, Stone PR, Salmond CE. Acidic forms of chorionic gonadotrophin in European and Samoan women are associated with hyperemesis gravidarum and may be thyrotrophic. Clin Endocrinol1999; 50:619-27,
121 Poppe K, Glinoer D, Tournaye H, Devroey P, van Steirteghem A, Kaufman L, Velkeniers B. Assisted reproduction and thyroid immunity: an unfortunate combination? J Clin Endocrinol Metab 2003; 88:4149-52
122 Rodien P, Brémont C, Sanson ML, Parma J, Van Sande J, Costagliola S, Luton JP, Vassart G, Duprez L.Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. N EnglJ Med.1998; 17;339:1823-6.
123 Coulon AL, Savagner F, Briet C, Vernin M, Munier M, Chabre O, Rodien P. Prolonged and Severe Gestational Thyrotoxicosis Due to Enhanced hCG Sensitivity of a Mutant Thyrotropin Receptor. JClin Endocrinol Metab. 2016 ;101:10-1. doi: 10.1210/jc.2015-3670.
124 Goodwin TM, Montoro M, Mestman JH: The role of chorionic gonadotropin in transient hyperthyroidism of hyperemesis gravidarum. J Clin Endocrinol Metab 1992; 75: 1333-37.
125. Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, Grimley Evans J, Hasan DM, Rodgers H, Tunbridge F, et al.The incidence of thyroid disorders in a community: a twenty-year follow-up of the Whickam survey. Clin Endocrinol 1995;43:55-68,
126 Glinoer D, Rihai M, Grün JP, Kinthaert J: Risk of subclinical hypothyroidism in pregnant women with autoimmune thyroid disorders. J Clin Endocrinol Metab1994; 79:197-04,
127 Lin L, Zhang XL, Long Y.Analysis of thyroid peroxidase antibody in early pregnancy. Genet Mol Res. 2014;13: 5107-14. doi: 10.4238/2014.
128 Van Steirteghem A Current status of assisted reproductive technology (ART) 30 years after the first IVF birth .in The Thyroid and Reproduction Ed John Lazarus, Valdis Pirags and Sigrid Butz 2009 Thieme p 7-14
129 Dosiou C, Giudice LC. Natural killer cells in pregnancy and recurrent pregnancy loss: endocrine and immunologic perspectives. Endocrine Rev 2005; 26:44-62
130 Mariee NG, Tuckerman E, Laird S, Li TC. The correlation of autoantibodies and uNK cells in women with reproductive failure. J Reprod Immunol. 2012 ;95:59-66
131 Huang C, Liang P, Diao L, Liu C, Chen X, Li G, Chen C, Zeng Y. Thyroid Autoimmunity is Associated with Decreased Cytotoxicity T Cells in Women with Repeated Implantation Failure.
Int J Environ Res Public Health. 2015 25;12:10352-61. doi: 10.3390/ijerph120910352.
132 Medenica S, Nedeljkovic O, Radojevic N, Stojkovic M, Trbojevic B, Pajovic B.Thyroid dysfunction and thyroid autoimmunity in euthyroid women in achieving fertility.Eur Rev Med Pharmacol Sci. 2015;19:977-87
133 Cho MK.Thyroid dysfunction and subfertility.Clin Exp Reprod Med. 2015;42:131-5. doi: 10.5653/cerm.2015.42.4.131
134 Feldthusen AD, Pedersen PL, Larsen J, Toft Kristensen T, Ellervik C, Kvetny J. Impaired Fertility Associated with Subclinical Hypothyroidism and Thyroid Autoimmunity: The Danish General Suburban Population Study.J Pregnancy. 2015;2015:132718. doi: 10.1155/2015/132718.
135 Poppe K, Velkeniers B, Glinoer D. Thyroid disease and female reproduction.Clin Endocrinol 2007;66:309-21
136 Poppe K, Glinoer D, Tournaye H, Devroey P, van Steirteghem A, Kaufman L, Velkeniers B. Assisted reproduction and thyroid autoimmunity: an unfortunate combination?J Clin Endocrinol Metab. 2003;88: 4149-52
137 Vissenberg R, Manders VD, Mastenbroek S, Fliers E, Afink GB, Ris-Stalpers C, Goddijn M, Bisschop PH.Pathophysiological aspects of thyroid hormone disorders/thyroid peroxidase autoantibodies and reproduction. Hum Reprod Update. 2015;21:378-87.
doi:10.1093/humupd/dmv004
138 Matarese G, De Placido G, Nikas Y, Alviggi C. Pathogenesis of endometriosis: natural immunity dysfunction or autoimmune disease? Trends Mol Med. 2003; 9 :223-8.
139 Konova E. The role of NK cells in the autoimmune thyroid disease-associated pregnancy loss. Clin Rev Allergy Immunol. 2010; 39:176-84
140 Kim NY, Cho HJ, Kim HY, Thyroid autoimmunity and its association with cellular and humoral immunity in women with reproductive failures. Am J Reprod Immunol. 2011; 65: 78-87:
141 Monteleone P, Parrini D, Faviana P, Carletti E, Casarosa E, Uccelli A, Cela V, Genazzani AR, Artini PG. Female Infertility Related to Thyroid Autoimmunity: The Ovarian Follicle Hypothesis.Am J Reprod Immunol. 2011; 66:108-14 doi: 10.1111/j.1600-0897.2010.00961.
142 Lee YL, Ng HP, Lau KS, Liu WM, O WS, Yeung WS, Kung AW. Increased fetal abortion rate in autoimmune thyroid disease is related to circulating TPO autoantibodies in an autoimmune thyroiditis animal model. Fertil Steril. 2009;91: 2104-9 doi: 10.1016/j.fertnstert.2008.07.1704.
143 Twig G, Shina A, Amital H, Shoenfeld Y. Pathogenesis of infertility and recurrent pregnancy loss in thyroid autoimmunity. J Autoimmun. 2012;38:275-81 doi: 10.1016/j.jaut.2011.11.014.
144 Artini PG, Uccelli A, Papini F, Simi G, Di Berardino OM, Ruggiero M, Cela V. Infertility and pregnancy loss in euthyroid women with thyroid autoimmunity.
Gynecol Endocrinol. 2013;291:36-41.
145 Practice Committee of the American Society for Reproductive Medicine Subclinical hypothyroidism in the infertile female population: a guideline.Fertil Steril. 2015 ;104:545-53. doi: 10.1016/j.fertnstert.2015.05.028.
146 Yoshioka W, Amino N, Ide A, Kang S, Kudo T, Nishihara E, Ito M, Nakamura H, Miyauchi A.Thyroxine treatment may be useful for subclinical hypothyroidism in patients with female infertility. Endocr J. 2015; 62:87-92. doi: 10.1507/endocrj.EJ14-0300.
147 Scoccia B, Demir H, Kang Y, Fierro MA, Winston NJ. In vitro fertilization pregnancy rates in levothyroxine-treated women with hypothyroidism compared to women without thyroid dysfunction disorders. Thyroid 2012;226:631-6.
148 Gracia CR, Morse CB, Chan G, Schilling S, Prewitt M, Sammel MD, Mandel SJ.
Thyroid function during controlled ovarian hyperstimulation as part of in vitro fertilization. Fertil Steril. 2012 ;9: 585-91.
- Krassas GE, Tziomalos K, Papadopoulou F, Pontikides N, Perros P: Erectile dysfunction in patients with hyper- and hypothyroidism: how common and should we treat? J Clin Endocrinol Metab 2008; 93:1815 -9. doi: 10.1210/jc.2007-2259
- Li TC, Makris M, Tomsu M, Tuckerman E, Laird S .Recurrent miscarriage: aetiology, management and prognosis. Hum Reprod Update. 2002;8:463-81
151 Stagnaro-Green A, Roman SH, Cobin RH, el-Harazy E, Alvarez-Marfany M, Davies TF. Detection of at-risk pregnancy by means of highly sensitive assays for thyroid autoantibodies. JAMA. 1990; 19:1422-5.
152 Prummel MF, Wiersinga WM. Thyroid autoimmunity and miscarriage.Eur J Endocrinol. 2004;150: 751-5
153 Krassas GE Perros P Kaprara A Thyroid autoimmunity, infertility and miscarriage Expert Rev. Endocrinol Metab 2008 3 127-36
154 Benhadi N, Wiersinga WM, Reitsma JB, Vrijkotte TG, Bonsel GJ. Higher maternal TSH levels in pregnancy are associated with increased risk for miscarriage, fetal or neonatal death. Eur J Endocrinol. 2009;160:985-91
155 Hirsch D, Levy S, Nadler V, Kopel V, Shainberg B, Toledano Y Pregnancy outcomes in women with severe hypothyroidism. Eur J Endocrinol. 2013 29;169: 313-20.
156 De Vivo A, Mancuso A, Giacobbe A, Moleti M, Maggio Savasta L, De Dominici R, Priolo AM, Vermiglio F. Thyroid function in women found to have early pregnancy loss. Thyroid 2010; 20:633-37.
157 Chen L, Hu R. Thyroid autoimmunity and miscarriage: a meta-analysis.Clin Endocrinol . 2011;74:513-19
158 Abbassi-Ghanavati M, Casey BM, Spong CY, McIntire DD, Halvorson LM, Cunningham FG. . Pregnancy outcomes in women with thyroid peroxidase antibodies. Obstet Gynecol. 2010; 116 381-86.
159 Thangaratinam S, Tan A, Knox E, Kilby MD, Franklyn J, Coomarasamy A. Association between thyroid autoantibodies and miscarriage and preterm birth: meta-analysis of evidence. BMJ. 2011;9: 342:d2616. doi: 10.1136 /bmj.d2616
160 van den Boogaard E, Vissenberg R, Land JA, van Wely M, van der Post JA, Goddijn M, Bisschop PH Significance of (sub)clinical thyroid dysfunction and thyroid autoimmunity before conception and in early pregnancy: a systematic review. Hum Reprod Update. 2011;17:605-19. doi: 10.1093/humupd/dmr024.
161 Yan J, Sripada S, Saravelos SH, Chen ZJ, Egner W, Li TC Thyroid peroxidase antibody in women with unexplained recurrent miscarriage: prevalence, prognostic value, and response to empirical thyroxine therapy Fertil Steril. 2012 ; 98:378-82.
162 Vissenberg R, van den Boogaard E, van Wely M, van der Post JA, Fliers E, Bisschop PH, Goddijn M. Treatment of thyroid disorders before conception and in early pregnancy: a systematic review. Hum Reprod Update. 2012;18: 360-73.
163 Roberts J, Jenkins C, Wilson R, Pearson C, Franklin IA, MacLean MA, McKillop JH, Walker JJ.: Recurrent miscarriage is associated with increased numbers of CD5/20 positive lymphocytes and an increased incidence of thyroid antibodies. Eur J Endocrinol 1996;134:84-6
164 Matalon ST, Blank M, Levy Y, Carp HJ, Arad A, Burek L, Grunebaum E, Sherer Y, Ornoy A, Refetoff S, Weiss RE, Rose NR, Shoenfeld Y. The pathogenic role of anti-thyroglobulin antibody on pregnancy: evidence from an active immunization model in mice. Hum Reprod 2003;18:1094-9
165 Moravej A, Jeddi-Tehrani M, Salek-Moghaddam AR, Dokouhaki P, Ghods R, Rabbani H, Kazemi-Sefat GE, Shahbazi M, Zarnani AH..Evaluation of thyroglobulin expression in murine reproductive organs during pregnancy.Am J Reprod Immunol. 2010; 1;64:97-103. doi: 10.1111/j.1600-0897.2010.00827.x
166 Ticconi C, Giuliani E, Veglia M, Pietropolli A, Piccione E, Di Simone N. Thyroid autoimmunity and recurrent miscarriage.Am J Reprod Immunol. 2011;66: 452-9.
167 Bagis T, Gokcel A, Saygili ES: Autoimmune thyroid disease in pregnancy and the postpartum period: relationship to spontaneous abortion. Thyroid 2001; 11:1049-53
168 Velkeniers B, Van Meerhaeghe A, Poppe K, Unuane D, Tournaye H, Haentjens P. Levothyroxine treatment and pregnancy outcome in women with subclinical hypothyroidism undergoing assisted reproduction technologies: systematic review and meta-analysis of RCTs. Hum Reprod Update. 2013;19:251-8. doi: 10.1093/humupd/dms052
169 Macklon NS, New Approaches to Ovarian Stimulation, In: John Lazarus Valdis Pirags Sigrid Butz, Eds. The Thyroid and Reproduction, Stuttgart: Georg Thieme Verlag, 2009:15-26
170 Toulis KA, Goulis DG, Venetis CA, Kolibianakis EM, Negro R, Tarlatzis BC, Papadimas I. Risk of spontaneous miscarriage in euthyroid women with thyroid autoimmunity undergoing IVF: a meta-analysis. Eur J Endocrinol. 2010 ;162:643-52. doi: 10.1530/EJE-09-0850.
171 Muller AF, Verhoeff A, Mantel MJ, Berghout A. Thyroid autoimmunity and abortion: a prospective study in women undergoing in vitro fertilization. Fertil Steril.
1999;71: 30-4
172 Negro R, Mangieri T, Coppola L Presicce G, Casavola EC, Gismondi R, Locorotondo G, Caroli P, Pezzarossa A, Dazzi D, Hassan H.Levothyroxine treatment in thyroid peroxidase antibody-positive women undergoing assisted reproduction technologies: a prospective study. Hum Reprod. 2005; 20:1529-33
173 Shoenfeld Y, Carp HJ, Molina V, Blank M, Cervera R, Balasch J, Tincani A, Faden D, Lojacono A, Doria A, Konova E, Meroni PL. Autoantibodies and prediction of reproductive failure. Am J Reprod Immunol. 2006;56: 337-44.
174 Kilic S, Tasdemir N, Yilmaz N, Yuksel B, Gul A, Batioglu S. The effect of anti-thyroid antibodies on endometrial volume, embryo grade and IVF outcome.Gynecol Endocrinol. 2008 ;24:649-55
175 Zhong YP, Ying Y, Wu HT, Zhou CQ, Xu YW, Wang Q, Li J, Shen XT, Li J.
Relationship between antithyroid antibody and pregnancy outcome following in vitro fertilization and embryo transfer. Int J Med Sci. 2012;9:121-5.
176 Negro R, Formoso G, Coppola L Presicce G, Mangieri T, Pezzarossa A, Dazzi D. Euthyroid women with autoimmune disease undergoing assisted reproduction technologies: the role of autoimmunity and thyroid function.J Endocrinol Invest. 2007 ;30:3-8.
177 Tan S, Dieterle S, Pechlavanis S, Janssen OE, Fuhrer D. Thyroid autoantibodies per se do not impair intracytoplasmic sperm injection outcome in euthyroid healthy women. Eur J Endocrinol. 2014; 8;170:495-500. doi: 10.1530/EJE-13-0790.
178 Łukaszuk K, Kunicki M, Kulwikowska P, Liss J, Pastuszek E, Jaszczołt M, Męczekalski B, Skowroński K. The impact of the presence of antithyroid antibodies on pregnancy outcome following intracytoplasmatic sperm injection-ICSI and embryo transfer in women with normal thyreotropine levels.J Endocrinol Invest. 2015 ;38:1335-43
179 Unuane D, Velkeniers B, Deridder S, Bravenboer B, Tournaye H, De Brucker M. Impact of thyroid autoimmunity on cumulative delivery rates in in vitro fertilization/intracytoplasmic sperm injection patients.Fertil Steril. 2016;106:144-50. doi: 10.1016/j
180 Kiprov DD, Nachtigall RD, Weaver RC, Jacobson A, Main EK, Garovoy MR. The use of intravenous immunoglobulin in recurrent pregnancy loss associated with combined alloimmune and autoimmune abnormalities. Am J Reprod Immunol 1996; 36:228-34,
- Sher G, Maassarani G, Zouves C, Feinman M, Sohn S, Matzner W, Chong P, Ching W. The use of combined heparin/aspirin and immunoglobulin G therapy in the treatment of in vitro fertilization patients with antithyroid antibodies. Am J Reprod Immunol 1998; 39:223-5
- Stricker RB, Steinleitner A, Bookoff CN, Weckstein LN, Winger EE. Successful treatment for immunological abortion with low-dose intravenous immunoglobulin. Fertil Steril 2000;73:536-40,
183 Revelli A, Casano S, Piane LD et al. A retrospective study on IVF outcome in euthyroid patients with anti-thyroid antibodies: effects of levothyroxine, acetyl-salicylic acid and prednisolone adjuvant treatments. Reprod Biol Endocrinol. 2009; 27:7:137. doi:10.1186/1477-7827-7-137
184 Kim CH, Ahn JW, Kang SP, Kim SH, Chae HD, Kang BM Effect of levothyroxine treatment on in vitro fertilization and pregnancy outcome in infertile women with subclinical hypothyroidism undergoing in vitro fertilization/intracytoplasmic sperm injection. Fertil Steril. 2011;95:1650-4.
185 Busnelli A, Vannucchi G, Paffoni A, Faulisi S, Fugazzola L, Fedele L, Somigliana E Levothyroxine dose adjustment in hypothyroid women achieving pregnancy through IVF.
Eur J Endocrinol. 2015;173:417-24. doi: 10.1530/EJE-15-0151.
186 Arojoki M, Jokimaa V, Juuti A, Juuti A, Koskinen P, Irjala K, Anttila L. Hypothyroidism among infertile women in Finland. Gynecol Endocrinol 14:127-31, 2000
187 Alnot-Burette J, Nakib I, Lipere A, Delemer B, Graesslin O. Thyroid function for infertile women during ovarian hyperstimulation as part of IVF. Gynecol Obstet Fertil. 2016 ;44:156-62. doi: 10.1016/j.gyobfe.2016.01.007.
188 Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;5: 371:75-84. doi: 10.2217/17455057.4.6.625
189 Stagnaro-Green A, Chen X, Bogden JD, Davies TF, Scholl TO. The thyroid and pregnancy: a novel risk factor for very preterm delivery. Thyroid. 2005;15: 351-57
190 Stagnaro-Green A. Maternal thyroid disease and preterm delivery. J Clin Endocrinol Metab. 2009; 94: 21-25.
191 Tierney K, Delpachitra P, Grossmann M Onwude J, Sikaris K, Wallace EM, Hamilton EJ, Tong S . Thyroid function and autoantibody status among women who spontaneously deliver under 35 weeks of gestation.Clin Endocrinol. 2009 ; 71: 892-95.
192 Haddow JE, Cleary-Goldman J, McClain MR. Palomaki GE, Neveux LM, Lambert-Messerlian G, Canick JA, Malone FD, Porter TF, Nyberg DA, Bernstein PS, D'Alton ME. First- and Second-Trimester Risk of Aneuploidy (FaSTER) Research Consortium. Thyroperoxidase and thyroglobulin antibodies in early pregnancy and preterm delivery. Obstet Gynecol. 2010 ;116:58-62.
193.Lata K, Dutta P, Sridhar S, Rohilla M, Srinivasan A, Prashad GR, Shah VN, Bhansali A. Thyroid autoimmunity and obstetric outcomes in women with recurrent miscarriage: a case-control study Endocr Connect. 2013; 22;2:118-24. doi: 10.1530/EC-13-0012.
194 .Bliddal S, Boas M, Hilsted L, Friis-Hansen L, Tabor A, Feldt-Rasmussen Thyroid function and autoimmunity in Danish pregnant women after an iodine fortification program and associations with obstetric outcomes U. Eur J Endocrinol. 2015;173:709-18.
195 Negro R, Schwartz A, Gismondi R,Tinelli A, Mangieri T, Stagnaro-Green A. Thyroid antibody positivity in the first trimester of pregnancy is associated with negative pregnancy outcomes. J Clin Endocrinol Metab. 2011;96:E920-24
196 He X, Wang P, Wang Z, He X, Xu D, Wang B.
Thyroid antibodies and risk of preterm delivery: a meta-analysis of prospective cohort studies. Eur J Endocrinol. 2012;167:455-64.
197 Karakosta P, Alegakis D, Georgiou V, Roumeliotaki T, Fthenou E, Vassilaki M, Boumpas D, Castanas E, Kogevinas M, Chatzi L.Thyroid dysfunction and autoantibodies in early pregnancy are associated with increased risk of gestational diabetes and adverse birth outcomes. J Clin Endocrinol Metab. 2012;97:4464-72. doi: 10.1210/jc.2012-2540
198 Saki F, Dabbaghmanesh MH, Ghaemi SZ, Forouhari S, Omrani GR, Bakhshayeshkaram M. Thyroid autoimmunity in pregnancy and its influences on maternal and fetal outcome in Iran (a prospective study).Endocr Res. 2015;40:139-45. Doi:10.3109/07435800.2014.966384
199 Kumru P, Erdogdu E, Arisoy R, Demirci O, Ozkoral A, Ardic C, Ertekin AA, Erdogan S, Ozdemir NN. Effect of thyroid dysfunction and autoimmunity on pregnancy outcomes in low risk population.Arch Gynecol Obstet. 2015;291:1047-54. doi: 10.1007/s00404-014-3533-9.
200 Oztas E, Erkenekli K, Ozler S, Aktas A, Buyukkagnıcı U, Uygur D, Danisman N. First trimester interleukin-6 levels help to predict adverse pregnancy outcomes in both thyroid autoantibody positive and negative patients. J Obstet Gynaecol Res. 2015; 41:1700-7. doi: 10.1111/jog.12799.
201 Stagnaro-Green A, Akhter E, Yim C, Davies TF, Magder LS, Petri M. Thyroid disease in pregnant women with systemic lupus erythematosus: increased preterm delivery. Lupus 2011; 20:690-99
202 Korevaar TI, Schalekamp-Timmermans S, de Rijke YB, Visser WE, Visser W, de Muinck Keizer-Schrama SM, Hofman A, Ross HA, Hooijkaas H, Tiemeier H, Bongers-Schokking JJ, Jaddoe VW, Visser TJ, Steegers EA, Medici M, Peeters RP.Hypothyroxinemia and TPO-Antibody Positivity Are Risk Factors for Premature Delivery: The Generation R Study. J Clin Endocrinol Metab. 2013;98:4382-90.
203 Negro R, Formoso G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H.Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroiddisease: effects on obstetrical complications. J Clin Endocrinol Metab 2006;91:2587-91.
204 Reid SM, Middleton P, Cossich MC, Crowther CA, Bain E. Interventions for clinical and subclinical hypothyroidism pre-pregnancy and during pregnancy..Cochrane Database Syst Rev. 2013; 31;:CD007752. doi: 10.1002/14651858.CD007752.pub3.
- Klein RZ, Haddow JE, Faix JD, Brown RS, Hermos RJ, Pulkkinen A. Prevalence of thyroid deficiency in pregnant women. Clin Endocrinol 35:41-6 1991
206 Teng W, Shan Z, Patil-Sisodia K, Cooper DS. Hypothyroidism in pregnancy.Lancet Diabetes Endocrinol. 2013 ;1:228-37. doi: 10.1016/S2213-8587(13)70109-8
207 Jaiswal N, Melse-Boonstra A, Thomas T, Basavaraj C, Sharma SK, Srinivasan K, Zimmermann MB. High prevalence of maternal hypothyroidism despite adequate iodine status in Indian pregnant women in the first trimester..Thyroid. 2014;24:1419-29. doi: 10.1089/thy.2014.0071.
208 Mandel SJ. Hypothyroidism and chronic autoimmune thyroiditis in the pregnant state: maternal aspects. Best Pract Res Clin Endocrinol Metab. 2004;18: 213-24.
209 Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev 26:599-614 2005
210 . Gallas PR, Stolk RP, Bakker K, Endert E, Wiersinga WM. Thyroid dysfunction during pregnancy and in the first postpartum year in women with diabetes mellitus type 1. Eur J Endocrinol 2002; 147:443-51
211 Glinoer D: Thyroidal and immune adaptation to pregnancy: focus on maternal hypo- and hyperthyroidism. In: Pirags V, Lazarus J, Butz S (Eds) The Thyroid and Autoimmunity. Georg Thieme Verlag; Stuttgart-New 46-58, 2008
- Krassas GE: Thyroid disease and female reproduction. Fertil Steril 2000; 74:1063-70,
213 Saglam F, Onal ED, Ersoy R, Koca C, Ergin M, Erel O, Cakir B.Anti-Müllerian hormone as a marker of premature ovarian aging in autoimmune thyroid disease. Gynecol Endocrinol. 2015;31:165-8. doi: 10.3109/09513590.2014.973391
214 Kuroda K, Uchida T, Nagai S, Ozaki R, Yamaguchi T, Sato Y, Brosens JJ, Takeda S Elevated serum thyroid-stimulating hormone is associated with decreased anti-Müllerian hormone in infertile women of reproductive age. J Assist Reprod Genet. 2015;32:243-7. doi: 10.1007/s10815-014-0397-7.
215 Magri F, Schena L, Capelli V, Gaiti M, Zerbini F, Brambilla E, Rotondi M, De Amici M, Spinillo A, Nappi RE, Chiovato L. Anti-Mullerian hormone as a predictor of ovarian reserve in ART protocols: the hidden role of thyroid autoimmunity Reprod Biol Endocrinol. 2015 21;13:106. doi: 10.1186/s12958-015-0103-3.
.216 Polyzos NP, Sakkas E, Vaiarelli A, Poppe K, Camus M, Tournaye H. Thyroid autoimmunity, hypothyroidism and ovarian reserve: a cross-sectional study of 5000 women based on age-specific AMH values. Hum Reprod. 2015;30:1690-6. doi: 10.1093/humrep/dev089.
217 Abalovich M, Gutierrez S, Alcaraz G, Maccallini G, Garcia A, Levalle O. Overt and subclinical hypothyroidism complicating pregnancy. Thyroid 2002;12:63-8
218 Männistö T, Mendola P, Grewal J, Xie Y, Chen Z, Laughon SK Thyroid diseases and adverse pregnancy outcomes in a contemporary US cohort. J Clin Endocrinol Metab. 2013 ;98:2725-33.
219Poulasouchidou MK, Goulis DG, Poulakos P, Mintziori G, Athanasiadis A, Grimbizis G, Tarlatzis BC Prediction of maternal and neonatal adverse outcomes in pregnant women treated for hypothyroidism. Hormones (Athens). 2012;11:468-76.
220 Ashoor G, Maiz N, Rotas M, Jawdat F, Nicolaides KH. Maternal thyroid function at 11 to 13 weeks of gestation and subsequent fetal death.Thyroid. 2010;20: 989-93
221 Ashoor G, Maiz N, Rotas M, Kametas NA, Nicolaides KH Maternal thyroid function at 11 to 13 weeks of gestation and subsequent development of preeclampsia .Prenat Diagn. 2010 ;30:1032-8.
222 Ashoor G, Maiz N, Rotas M, Jawdat F, Nicolaides KH. Maternal thyroid function at 11-13 weeks of gestation and spontaneous preterm delivery. Obstet Gynecol. 2011;117:293-8.
223 Casey BM, Dashe JS, Wells CE, McIntire DD, Byrd W, Leveno KJ. Subclinical hypothyroidism and pregnancy outcome. Obstet Gynecol 2005; 105:239-45
224 Medici M, Timmermans S, Visser W, de Muinck Keizer-Schrama SM, Jaddoe VW, Hofman A, Hooijkaas H, de Rijke YB, Tiemeier H, Bongers-Schokking JJ, Visser TJ, Peeters RP, Steegers EA. Maternal thyroid hormone parameters during early pregnancy and birth weight: the Generation R Study. J Clin Endocrinol Metab. 2013;98:59-66.
225 Haddow JE, Craig WY, Palomaki GE, Neveux LM, Lambert-Messerlian G, Canick JA, Malone FD, D'Alton ME; First And Second Trimester Risk Of Aneuploidy Faster Research Consortium. Impact of adjusting for the reciprocal relationship between maternal weight and free thyroxine during early pregnancy. Thyroid. 2013 ;23: 225-30.
226 Negro R. Thyroid insufficiency during pregnancy: complications and implications for screening Expert Rev. Endocrinol. Metab. 2008: 32; 1-10
227 Furnica RM, Lazarus JH, Gruson D, Daumerie C. Update on a new controversy in endocrinology: isolated maternal hypothyroxinemia. J Endocrinol Invest. 2015 ;38:117-23. doi: 10.1007/s40618-014-0203-5.
228 Casey BM, Dashe JS, Spong CY, McIntire DD, Leveno KJ, Cunningham GF.Perinatal significance of isolated maternal hypothyroxinemia identified in the first half of pregnancy. Obstet Gynecol 2007;109:1129-35
229 Hamm MP, Cherry NM, Martin JW, Bamforth F, Burstyn I. The impact of isolated maternal hypothyroxinemia on perinatal morbidity. J Obstet Gynaecol Can. 2009 ; 31:1015-21.
230 Cleary-Goldman J, Malone FD, Lambert-Messerlian G, Sullivan L, Canick J, Porter TF, Luthy D, Gross S, Bianchi DW, D'Alton ME. Maternal thyroid hypofunction and pregnancy outcome.Obstet Gynecol. 2008 ;112:85-92. doi: 10.1097/AOG.0b013e3181788dd7
231 Pop VJ, Brouwers EP, Wijnen H, Oei G, Essed GG, Vader HL. Low concentrations of maternal thyroxin during early gestation: a risk factor of breech presentation? BJOG. 2004 ;111:925-30
232 Mil NH, Steegers-Theunissen RP, Bongers-Schokking JJ, El Marroun H, Ghassabian A, Hofman A, Jaddoe VW, Visser TJ, Verhulst FC, de Rijke YB, Steegers EA, Tiemeier H.Maternal hypothyroxinemia during pregnancy and growth of the fetal and infant head.van Reprod Sci. 2012;19:1315-22. doi: 10.1177/1933719112450338
233 Korevaar TI, Schalekamp-Timmermans S, de Rijke YB, Visser WE, Visser W, de Muinck Keizer-Schrama SM, Hofman A, Ross HA, Hooijkaas H, Tiemeier H, Bongers-Schokking JJ, Jaddoe VW, Visser TJ, Steegers EA, Medici M, Peeters RP. Hypothyroxinemia and TPO-antibody positivity are risk factors for premature delivery: the generation R study. J Clin Endocrinol Metab. 2013;98:4382-90. doi: 10.1210/jc.2013-2855.
234 Su PY, Huang K, Hao JH, Xu YQ, Yan SQ, Li T, Xu YH, Tao FB.Maternal thyroid function in the first twenty weeks of pregnancy and subsequent fetal and infant development: a prospective population-based cohort study in China. J Clin Endocrinol Metab. 2011;;96:3234-41. doi: 10.1210/jc.2011-0274
235 Maraka S, Singh Ospina NM, O'Keeffe DT, Rodriguez-Gutierrez R, Espinosa De Ycaza AE, Wi CI, Juhn YJ, Coddington CC 3rd, Montori VM, Stan MN. Effects of Levothyroxine Therapy on Pregnancy Outcomes in Women with Subclinical Hypothyroidism.Thyroid. 2016 May 16. [Epub ahead of print
236 Maraka S, Ospina NM, O'Keeffe DT, Espinosa De Ycaza AE, Gionfriddo MR, Erwin PJ, Coddington CC 3rd, Stan MN, Murad MH, Montori VM.Subclinical Hypothyroidism in Pregnancy: A Systematic Review and Meta-Analysis. Thyroid. 2016;26:580-90. doi: 10.1089/thy.2015.0418.
237 Juch H, Lupattelli A, Ystrom E, Verheyen S, Nordeng H Medication adherence among pregnant women with hypothyroidism-missed opportunities to improve reproductive health? A cross-sectional, web-based study. Patient Educ Couns. 2016 11. pii: S0738-3991(16)30158-6. doi: 10.1016/j.pec.2016.04.006.
238 Zhang Y, Fan Y, Yu X, Wang X, Bao S, Li J, Fan C, Shan Z, Teng W. Maternal Subclinical Hypothyroidism Impairs Neurodevelopment in Rat Offspring by Inhibiting the CREB Signaling Pathway.Mol Neurobiol. 2015 ;52:432-41. doi: 10.1007/s12035-014-8855-x.
239 Min H, Dong J, Wang Y, Wang Y, Yu Y, Shan Z, Xi Q, Teng W, Chen J. Marginal Iodine Deficiency Affects Dendritic Spine Development by Disturbing the Function of Rac1 Signaling Pathway on Cytoskeleton. Mol Neurobiol. 2016 Jan 7
240 Hu X Wang R, Shan Z, Dong Y, Zheng H, Jesse FF, Rao E, Takahashi E, Li W1, Teng W, Teng X. Perinatal Iron Deficiency-Induced Hypothyroxinemia Impairs Early Brain Development Regardless of Normal Iron Levels in the Neonatal Brain.Thyroid. 2016 May 27. [Epub ahead of print]
241 Moog NK, Entringer S, Heim C, Wadhwa PD, Kathmann N, Buss C. Influence of maternal thyroid hormones during gestation on fetal brain development.Neuroscience. 2015 Oct 3. pii: S0306-4522(15)00897-0. doi: 10.1016/j.neuroscience.2015.09.070
242 Willoughby KA, McAndrews MP, Rovet JF. Effects of maternal hypothyroidism on offspring hippocampus and memory. Thyroid. 2014 ;24:576-84. doi: 10.1089/thy.2013.0215.
243 Gilbert ME, Sanchez-Huerta K, Wood C. Mild Thyroid Hormone Insufficiency During Development Compromises Activity-Dependent Neuroplasticity in the Hippocampus of Adult Male Rats. Endocrinology. 2016 ;157:774-87. doi: 10.1210/en.2015-1643
244 Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone.J Neuroendocrinol. 2008 ;20: 784-94.
245 Morreale de Escobar G, Obregon MJ, Escobar del Rey F: Maternal thyroid hormones early in pregnancy and fetal brain development. In: Best Practice & Research in Clinical Endocrinology and Metabolism: The Thyroid and Pregnancy (Editor: Glinoer D) 18:225, 2004
246 Bernal J.Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab. 2007;3: 249-59.
247 Rovet JF The role of thyroid hormones for brain development and cognitive function..Endocr Dev. 2014;26:26-43. doi: 10.1159/000363153
248 Schroeder AC, Privalsky ML. Thyroid hormones, t3 and t4, in the brain. Front Endocrinol (Lausanne). 2014 Mar 31;5:40. doi: 10.3389/fendo.2014.00040.
249 Bernal J, Guadaño-Ferraz A, Morte B. Thyroid hormone transporters-functions and clinical implications. Nat Rev Endocrinol. 2015;11:406-17.
250 Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005 ;77:41-53.
251 Wirth EK, Schweizer U, Köhrle J.Transport of thyroid hormone in brain.Front Endocrinol (Lausanne). 2014; 24;5:98. doi: 10.3389/fendo.2014.00098.
252 Man EB, Jones WS, Holden RH, Mellits ED: Thyroid function in human pregnancy. VIII. Retardation of progeny aged 7 years; relationships to maternal age and maternal thyroid function. Amer J Obstet Gynecol 1971;111:905-16
253 Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O'Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, Faix JD, Klein RZ.Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med.1999 19;341:549-55.
254 Smit BJ, Kok JH, Vulsma T, Briët JM, Boer K, Wiersinga WM.
Neurologic development of the newborn and young child in relation to maternal thyroid function.Acta Paediatr. 2000;89:291-5
255 Li Y, Shan Z, Teng W, Yu X, Li Y, Fan C, Teng X, Guo R, Wang H, Li J, Chen Y, Wang W, Chawinga M, Zhang L, Yang L, Zhao Y, Hua T. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months.Clin Endocrinol. 2010;72:825-9 doi: 10.1111/j.1365-2265.2009.03743.x
256 Pop VJ, Kuijpens JL, van Baar AL, Verkerk G, van Son MM, de Vijlder JJ, Vulsma T, Wiersinga WM, Drexhage HA, Vader HL. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy.Clin Endocrinol (Oxf). 1999;50:149-55.
257 Pop VJ, Brouwers EP, Vader H, Vulsma T, van Baar AL, de Vijlder JJ. Maternal hypothyroxinaemia during early pregnancy and subsequent child development: a 3-year follow-up study. Clin Endocrinol (Oxf). 2003;59:282-8.
258 Henrichs J, Bongers-Schokking JJ, Schenk JJ, Ghassabian A, Schmidt HG, Visser TJ, Hooijkaas H, de Muinck Keizer-Schrama SM, Hofman A, Jaddoe VV, Visser W, Steegers EA, Verhulst FC, de Rijke YB, Tiemeier H. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab. 2010 ;95:4227-34.
259 Suárez-Rodríguez M, Azcona-San Julián C, Alzina de Aguilar V.
Hypothyroxinemia during pregnancy: the effect on neurodevelopment in the child.
Int J Dev Neurosci. 2012 ;30:435-8.
260 Finken MJ, van Eijsden M, Loomans EM, Vrijkotte TG, Rotteveel J.
J Clin Endocrinol Metab. 2013 ;98:1417-26.
261 Julvez J, Alvarez-Pedrerol M, Rebagliato M, Murcia M, Forns J, Garcia-Esteban R, Lertxundi N, Espada M, Tardón A, Riaño Galán I, Sunyer J. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology. 2013 ;24:150-7.
262 Vermiglio F, Lo Presti VP, Moleti M, Sidoti M, Tortorella G,Scaffidi G, Castagna MG, Mattina F, Violi MA, Crisà A, ArtemisiaA, Trimarchi F.Attention deficit and hyperactivity disorders in theoffspring of mothers exposed to mild-moderate iodine deficiency:a possible novel iodine deficiency disorder in developed countries.J Clin Endocrinol Metab. 2004;89:6054-60.
- Modesto T, Tiemeier H, Peeters RP, Jaddoe VW, HofmanA, Verhulst FC, Ghassabian A. Maternal Mild Thyroid HormoneInsufficiency in Early Pregnancy and Attention-Deficit/
Hyperactivity Disorder Symptoms in Children. JAMA Pediatr.2015;169:838-45.
264 Román GC, Ghassabian A, Bongers-Schokking JJ, Jaddoe VW,Hofman A, de Rijke YB, Verhulst FC, Tiemeier H. Association of gestational maternal hypothyroxinemia and increased autism risk.Ann Neurol. 2013;74:733-42.
265 Gyllenberg D, Sourander A, Surcel HM, Hinkka-Yli-SalomäkiS, McKeague IW, Brown AS. Hypothyroxinemia During Gestation and Offspring Schizophrenia in a National Birth Cohort. BiolPsychiatry. 2015 19. pii: S0006-3223(15)00520-X.
266 Ghassabian A, Bongers-Schokking JJ, de Rijke YB, van Mil N, Jaddoe VW, de Muinck Keizer-Schrama SM, Hooijkaas H, Hofman A, Visser W, Roman GC, Visser TJ, Verhulst FC, Tiemeier H. Maternal thyroid autoimmunity during pregnancy and the risk of attention deficit/hyperactivity problems in children: the Generation R Study.Thyroid. 2012; 22:178-86
267 Pop VJ, de Vries E, van Baar AL, Waelkens JJ, de Rooy HA, Horsten M, Donkers MM, Komproe IH, van Son MM,Vader HL. Maternal thyroid peroxidase antibodies during pregnancy: a marker of impaired child development? J Clin Endocrinol Metab. 1995; 80:3561-6.
268 Wasserman EE, Pillion JP, Duggan A, Nelson K, Rohde C, Seaberg EC, Talor MV, Yolken RH, Rose NR.Childhood IQ, hearing loss, and maternal thyroid autoimmunity in the Baltimore Collaborative Perinatal Project. Pediatr Res. 2012 ;72:525-30.
269 Kooistra L, Crawford S, van Baar AL, Brouwers EP, Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy.Pediatrics. 2006;117:161-7
270 Mirabella G, Westall CA, Asztalos E, Perlman K, Koren G, Rovet J. Development of contrast sensitivity in infants with prenatal and neonatal thyroid hormone insufficiencies.Pediatr Res. 2005 ;57:902-7.
271 Ghassabian A, Bongers-Schokking JJ, Henrichs J, Jaddoe VW, Visser TJ, Visser W, de Muinck Keizer-Schrama SM, Hooijkaas H, Steegers EA, Hofman A, Verhulst FC, van der Ende J, de Rijke YB, Tiemeier H. Maternal thyroid function during pregnancy and behavioral problems in the offspring: the generation R study.Pediatr Res. 2011;69:454-9.
272 Momotani N, Iwama S, Momotani K. Neurodevelopment in children born to hypothyroid mothers restored to normal thyroxine (T₄) concentration by late pregnancy in Japan: no apparent influence of maternal T₄ deficiency. J Clin Endocrinol Metab. 2012;97:1104-8.
273 Downing S, Halpern L, Carswell J, Brown RS Severe maternal hypothyroidism corrected prior to the third trimester is associated with normal cognitive outcome in the offspring. Thyroid. 2012 ;22:625-30.
274 Behrooz HG, Tohidi M, Mehrabi Y, Behrooz EG, Tehranidoost M, Azizi F Subclinical hypothyroidism in pregnancy: intellectual development of offspring Thyroid. 2011;2:1143-7.
275 Craig WY, Allan WC, Kloza EM, Pulkkinen AJ, Waisbren S, Spratt DI, Palomaki GE, Neveux LM, Haddow JEMid-gestational maternal free thyroxine concentration and offspring neurocognitive development at age two years. J Clin Endocrinol Metab. 2012 ;97:E22-8.
276 Lazarus JH, Bestwick JP, Channon S, Paradice R, Maina A, Rees R, Chiusano E, John R, Guaraldo V, George LM, Perona M, Dall'Amico D, Parkes AB, Joomun M, Wald NJ. Antenatal thyroid screening and childhood cognitive function. N Engl J Med. 2012 9;366:493-501
277 Brian Casey Effect of treatment of maternal subclinicalhypothyroidism or hypothyroxinemia on IQ in offspring American Journal of Obstetrics & Gynecology Supplement to JANUARY 2016 Abstr 2 S2
278 Korevaar TI, Muetzel R, Medici M, Chaker L, Jaddoe VW, de Rijke YB, Steegers EA, Visser TJ, White T, Tiemeier H, Peeters RP. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study.Lancet Diabetes Endocrinol. 2016 ;4:35-43. doi: 10.1016/S2213-8587(15)00327-7
279 Samadi A, Skocic J, Rovet JF. Children born to women treated for hypothyroidism during pregnancy show abnormal corpus callosum development.Thyroid. 2015;25:494-502. doi: 10.1089/thy.2014.0548
280 Lischinsky JE, Skocic J, Clairman H, Rovet J. Preliminary Findings Show Maternal Hypothyroidism May Contribute to Abnormal Cortical Morphology in Offspring. Front Endocrinol (Lausanne). 2016; 25;7:16. doi: 10.3389/fendo.2016.00016.
281 Williams FL, Watson J, Ogston SA, Visser TJ, Hume R, Willatts P. Maternal and umbilical cord levels of T4, FT4, TSH, TPOAb, and TgAb in term infants and neurodevelopmental outcome at 5.5 years. J Clin Endocrinol Metab. 2013;98:829-38.
282 Williams F, Watson J, Ogston S, Hume R, Willatts P, Visser T; Scottish Preterm Thyroid Group. Mild maternal thyroid dysfunction at delivery of infants born ≤34 weeks and neurodevelopmental outcome at 5.5 years. J Clin Endocrinol Metab. 2012;97: 1977-85
283 Mandel SJ, Larsen PR, Seely EW, Brent GA. Increased need for thyroxine during
pregnancy in women with primary hypothyroidism.N Engl J Med.1990;12: 323:91-6
284 Yassa L, Marqusee E, Fawcett R, Alexander EK. Thyroid hormone early adjustment in pregnancy (the THERAPY) trial. J Clin Endocrinol Metab. 2010;95: 3234-41
285 Loh JA, Wartofsky L, Jonklaas J, Burman KD. The magnitude of increased levothyroxine requirements in hypothyroid pregnant women depends upon the etiology of the hypothyroidism. Thyroid. 2009;19:269-75
286 Galofré JC, Haber RS, Mitchell AA, Pessah R, Davies TF. Increased postpartum thyroxine replacement in Hashimoto's thyroiditis. Thyroid. 2010; 20: 901-08.
287 . Arafah BM. Increased need for thyroxine in women with hypothyroidism during estrogen therapy. New Engl J Med 2001; 344:1743-9,
288 Rotondi M, Precerutti S, Chiovato L: Maternal hypothyroidism during pregnancy: possible preventive strategies. Clin Endocrinol 2006; 64:599-601
289 Abalovich M, Vázquez A, Alcaraz G, Kitaigrodsky A, Szuman G, Calabrese C, Astarita G, Frydman M, Gutiérrez S. Adequate levothyroxine doses for the treatment of hypothyroidism newly discovered during pregnancyThyroid. 2013;23:1479-83.
290 Abalovich M, Alcaraz G, Kleiman-Rubinsztein Jet al. The relationship of preconception thyrotropin levels to requirements for increasing the levothyroxine dose during pregnancy in women with primary hypothyroidism. Thyroid. 2010;20: 1175-8.
291 McClain MR, Lambert-Messerlian G, Haddow JE, Palomaki GE, Canick JA, Cleary-Goldman J, Malone FD, Porter TF, Nyberg DA, Bernstein P, D'Alton ME. Sequential first- and second-trimester TSH, free thyroxine, and thyroid antibody measurements in women with known hypothyroidism: a FaSTER trial study. Am J Obstet Gynecol 2008; 199(2): el-6
292 Taylor PN, Minassian C, Rehman A, Iqbal A, Draman MS, Hamilton W, Dunlop D, Robinson A, Vaidya B, Lazarus JH, Thomas S, Dayan CM, Okosieme OE.TSH levels and risk of miscarriage in women on long-term levothyroxine: a community-based study.J Clin Endocrinol Metab. 2014;99: 3895-902. doi: 10.1210/jc.2014-1954
293 Andersen SL, Olsen J, Carlé A, Laurberg P. Hyperthyroidism incidence fluctuates widely in and around pregnancy and is at variance with some other autoimmune diseases: a Danish population-based study. J Clin Endocrinol Metab. 2015;100:1164-71. doi: 10.1210/jc.2014-3588.
294 Okosieme OE, Lazarus JH Important considerations in the management of Graves' disease in pregnant women. Expert Rev Clin Immunol. 2015;11:947-57. doi: 10.1586/1744666X.2015.1054375
295 Marx H, Amin P, Lazarus JH: Hyperthyroidism and pregnancy. Brit Med J 336:663-67, 2008
296 Mestman JH Hyperthyroidism in pregnancy.Curr Opin Endocrinol Diabetes Obes. 2012;19: 394-401
297 Haddow JE, Knight GJ, Palomaki GE, McClain MR, Pulkkinen AJ. The reference range and within-person variability of thyroid stimulating hormone during the first and second trimesters of pregnancy. J Med Screen 11:170-4 2004
298 Dorizzi RM, Ozzola G, Sommella C, Catania F, Lelli F, Migali E, Polverini G. An approach to establish reference intervals for thyrotropin in pregnancy using the ADVIA Centaur analyzer. Clin Lab. 2010; 56:417-25.
299 Barbesino G, Tomer Y. Clinical review: Clinical utility of TSH receptor antibodies.J Clin Endocrinol Metab. 2013; 98: 2247-55. doi: 10.1210/jc.2012-4309.
300 Lazarus JH Pre-conception counselling in Graves’ disease Eur Thyroid J 2012;1:24-9
301 Laurberg P, Wallin G, Tallstedt L, Abraham-Nordling M, Lundell G, Torring O: TSH receptor autoimmunity in Graves’ disease after therapy with antithyroid drugs, surgery, or radioiodine: a 5 year prospective randomized study. Eur J Endocrinol 2008; 158:69-75.
302 Lauberg P, Bournaud C, Karmmisholt J, Orgiazzi J: Management of Graves’ hyperthyroidism in pregnancy: focus on both maternal and foetal thyroid function and caution against surgical thyroidectomy in pregnancy. European Journal of Endocrinology 2009; 160:1-8.
303 Tagami T, Hagiwara H, Kimura T, Usui T, Shimatsu A, Naruse M. The incidence of gestational hyperthyroidism and postpartum thyroiditis in treated patients with Graves’ disease. Thyroid 2007; 17:767-72.
304 Sheffield JS, Cunningham FG: Thyrotoxicosis and heart failure that complicate pregnancy. Am J Obstet Gynecol 2004; 190:211-17
305 Chonh HW, See KC, Phua J: Thyroid storm with multiorgan failure. Thyroid 2010; 20:333-6.
306 Millar LK, Wing DA, Leung AS, et al: Low birth weight and preeclampsia in pregnancies complicated by hyperthyroidism Obstet Gynecol 1994; 84: 946-9
307 Kempers MJE, van Trotsenburg ASP, van Rijn RR, et al: Loss of integrity of thyroid morphology and function in children born to mothers with inadequately treated Graves’ disease. J Clin Endocrinol Metab 2007; 92:2984-91.
308 Smith C, Thomsett M, Choong C, Rodda C, McIntyre HD, Cotterill AM Congenital thyrotoxicosis in premature infants. Clin Endocrinol 2001; 54:371-6
309 Mitsuda N, Tamaki H, Amino N, Hosono T, Miyai K, Tanizawa O. Risk factors for developmental disorders in infants born to women with Graves' disease. Obstet Gynecol 1992; 80:359-64
310 Vaidya B, Campbell V, Tripp JH, Spyer G, Hattersley AT, Ellard S. Premature birth and low birth weight associated with non-autoimmune hyperthyroidism due to an activating thyrotropin receptor gene mutation. Clin Endocrinol 2004; 60:711-8
311 Polak M, Luton D. Fetal thyroïdology.Best Pract Res Clin Endocrinol Metab. 2014 ;28:161-73. doi: 10.1016/j.beem.2013.04.013.
312 Towers CV, Thomas S, Steiger RM: The fetal heart monitor tracing in pregnancies complicated by fetal thyrotoxicosis. Amer J Perinatal 2009; 26:373-7.
313 Luton D, Le Gac I, Vullard E, Castanet M, Guibourdenche J, Noel M, Toubert ME, Léger J, Boissinot C, Schlageter MH, Garel C, Tébeka B, Oury JF, Czernichow P, Polak M. Management of Graves’ disease during pregnancy: the key role of fetal thyroid gland monitoring. J Clin Endocrinol Metab 2005;90:6093-98.
314 Kilpatrick S: Umbilical cord sampling in women with thyroid disease in pregnancy: is it necessary? Am J Obstet Gynecol 2003:;189:1-2
315 Evans C, Gregory JW, Barton J, Bidder C, Gibbs J, Pryce R, Al-Muzaffar I, Ludgate M, Warner J, John R, Moat SJ Transient congenital hypothyroidism due to thyroid-stimulating hormone receptor blocking antibodies: a case series.Ann Clin Biochem. 2011;48:386-90. doi: 10.1258/acb.2011.011007
316 Calderwood C, Williams H, Campbell IW, Toft AD, Cameron A. Cordocentesis to predict fetal outcome after administration of radio-active iodine for Graves’ disease. J Obstet Gynaecol 2002; 22:217-8
317 Polak M, Le Gac I, Vuillard E, Guibourdenche J, Leger J, Toubert ME, Madec AM, Oury JF, Czernichow P, Luton D. Fetal and neonatal thyroid function in relation to maternal Graves' disease.Best Pract Res Clin Endocrinol Metab. 2004;18:289-302.
318 Luton D, Le Gac I, Noel M, Guibourdenche J, Polak M: Thyroid function during pregnancy in women with past Graves’ disease. Brit J Obstet Gynaecol 2005;112:1565-7
319 Cooper DS, Laurberg P. Hyperthyroidism in pregnancy.Lancet Diabetes Endocrinol. 2013 ;1:238-49. doi: 10.1016/S2213-8587(13)70086-X.
320 Chester J, Rotenstein D, Ringkananont U, Steuer G, Carlin B, Stewart L, Grasberger H, Refetoff S.t al.Congenital neonatal hyperthyroidism caused by germline mutations in the TSH receptor gene.J Pediatr Endocrinol Metab. 2008;21:479-86.
321 Nicholas AK, Jaleel S, Lyons G, Schoenmakers E, Dattani MT, Crowne E, Bernhard B, Kirk J, Roche EF, Chatterjee VK, Schoenmakers N. Molecular spectrum of TSHβ subunit gene defects in central hypothyroidism in the UK and Ireland. Clin Endocrinol (Oxf). 2016; 30. doi: 10.1111/cen.13149
322 . Kempers MJ, Van Trotsenburg AS, Van Rijn RR, Smets AM, Smit BJ, de Vijlder JJ, Vulsma T. Loss of integrity of thyroid morphology and function in children born to mothers with inadequately treated Graves’ disease. J Clin Endocrinol Metab 2007 92:2984-91,
323 Napier C, Pearce SH. Rethinking antithyroid drugs in pregnancy. Clin Endocrinol (Oxf). 2015 ;82:475-7. doi: 10.1111/cen.12577.
324 Wing DA, Mmiller LK, Koonings PP: A comparison of propylthiouracil versus methimazole in the treatment of hyperthyroidism in pregnancy. Am J Obstet Gynecol 1994; 170: 90-95
325 Momotani N, Noh J, Oyangi H, Ishikawa N, Ito K. Antithyroid during therapy for Graves’ disease during pregnancy: optimal regimen for fetal thyroid status. N Engl J Med 1986;315:24-8.
326 Y. Nakagawa, K. Mori, S. Hoshikawa, M. Yamamoto, S. Ito, K. Yoshida, Postpartum recurrence of Graves hyperthyroidism can be prevented by the continuation of antithyroid drugs during pregnancy. Clin Endocrinol (Oxf). 2002; 57: 467–71
327 M. Rotondi, C. Cappelli, B. Pirali, I. Pirola, F. Magri, R. Fonte, M. Castellano, E.A. Rosei, L. Chiovato, The effect of pregnancy on subsequent relapse from Graves’ disease after a successful course of antithyroid drug therapy. J. Clin. Endocrinol. Metab 2008; 93: 3985–88
328 Mandel SJ, Cooper DS: The use of antithyroid drugs in pregnancy and lactation. J Clin Endocrinol Metab 2001; 86:2354-9
329 Rivkees SA, Szarfman A: Dissimilar hepatotoxicity profiles of propylthiouracil and methimazole in children. J Clin Endocrinol Metab 2010;95: 3260-67.
330 Akmal A, Kung J. Propylthiouracil, and methimazole, and carbimazole-related hepatotoxicity. Expert Opin DrugSaf.2014;13:1397-406. doi: 10.1517/14740338.2014.953796.
331 Kimura M, Seki T, Ozawa H, Ishihara T, Komatsu M, Tajiri S, Yanagi H, Nishina M, Noh JY, Fukagawa M, Takagi A.The onset of antineutrophil cytoplasmic antibody-associated vasculitis immediately after methimazole was switched to propylthiouracil in a woman with Graves' disease who wished to become pregnant.Endocr J. 2013;60:383-8.
332 Andersen SL, Olsen J, Laurberg P.Antithyroid Drug Side Effects in the Population and in Pregnancy. J Clin Endocrinol Metab. 2016;101:1606-14. doi: 10.1210/jc.2015-4274.
333 Therapy of endocrine disease: antithyroid drug use in early pregnancy and birth defects: time windows of relative safety and high risk?Laurberg P, Andersen SL.Eur J Endocrinol. 2014 ;171:R13-20. doi: 10.1530/EJE-14-0135
334 Li H, Zheng J, Luo J, Zeng R, Feng N, Zhu N, Feng Q Congenital anomalies in children exposed to antithyroid drugs in-utero: a meta-analysis of cohort studies. PLoS One. 2015 14;10(5):e0126610. doi: 10.1371/journal.pone.0126610
335 Laurberg P, Andersen SL. Antithyroid Drug Use in Pregnancy and Birth Defects: Why Some Studies Find Clear Associations, and Some Studies Report None.Thyroid. 2015 ;25:1185-90. doi: 10.1089/thy.2015.0182.
336 Mandel S, Brent GA, Larsen PR: Review of antithyroid drugs use during pregnancy and report of a case of aplasia cutis. Thyroid 1994; 4: 129-133.
337 Löllgen RM, Calza AM, Schwitzgebel VM, Pfister RE.Aplasia cutis congenita in surviving co-twin after propylthiouracil exposure in utero.J Pediatr Endocrinol Metab. 2011;24:215-8.
338 Barbero P, Valdez R, Rodriguez H, Tiscornia C, Mansilla E, Allons A, Coll S, Liascovich R. Choanal atresia associated with maternal hyperthyroidism treated with methimazole: A case control study. Am J Med Genet 2008;146A: 2390-95.
339 Rosenfeld H. Ornoy A, Shchtman S, Diav-Citrin O: Pregnancy outcome, thyroid dysfunction and fetal goiter after in utero exposure to PTU: a controlled cohort study. B J Clin Pharmacol 2008;68: 609-17.
340 Yoshihara A, Noh J, Yamaguchi T, Ohye H, Sato S, Sekiya K, Kosuga Y, Suzuki M, Matsumoto M, Kunii Y, Watanabe N, Mukasa K, Ito K, Ito K. Treatment of graves' disease with antithyroid drugs in the first trimester of pregnancy and the prevalence of congenital malfor mation.J Clin Endocrinol Metab. 2012;97:2396-403.
341 Andersen SL, Olsen J, Wu CS, Laurberg P. Severity of birth defects after propylthiouracil exposure in early pregnancy Thyroid. 2014 ;24:1533-40. doi: 10.1089/thy.2014.0150
342 Li X, Liu GY, Ma JL, Zhou L. Risk of congenital anomalies associated with antithyroid treatment during pregnancy: a meta-analysis.Clinics (Sao Paulo). 2015;70:453-9. doi: 10.6061/clinics/2015(06)12
343 Bahn RS, Burch HS, Cooper DS, Garber JR, Greenlee CM, Klein IL, Laurberg P, McDougall IR, Rivkees SA, Ross D, Sosa JA, Stan MN The role of propylthiouracil in the management of Graves' disease in adults: report of a meeting jointly sponsored by the American Thyroid Association and the Food and Drug Administration. Thyroid 2009;19:673–74.
344 Mortimer RH, Cannell GR, Addison RS: Methimazole and propylthiouracil equally cross the perfused human term placenta lobule? J Clin Endocrinol Metab 1997;82: 3099-102.
345 Gianetti E, Russo L, Orlandi F, Chiovato L, Giusti M, Benvenga S, Moleti M, Vermiglio F, Macchia PE, Vitale M, Regalbuto C, Centanni M, Martino E, Vitti P, Tonacchera M. Pregnancy outcome in women treated with methimazole or propylthiouracil during pregnancy. J Endocrinol Invest. 2015;38:977-85. doi: 10.1007/s40618-015-0281-z
346 Redmond GP: Propranolol and fetal growth retardation. Semin Perinatol 1982; 6:142-7
347 Momotani N, Hisaoka T, Noh J, Ishikawa N, Ito K. Effects of iodine on thyroid status of fetus versus mother in treatment of Graves’ disease complicated by pregnancy. J Clin Endocrinol Metab 1992; 75:738-44
348 Zanzonico PB: Radiation dose to patients and relatives incident to 131-I therapy. Thyroid 1997; 7:199-204
- Berg GE, Nystrom EH, Jacobsson L Lindberg S, Lindstedt RG, Mattsson S, Niklasson CA, Norén AH, Westphal OG.Radioiodine treatment of hyperthyroidism in a pregnant woman. J Nucl Med 1998; 39:357-61
350 Abeillon-du Payrat J, Chikh K, Bossard N, Bretones P, Gaucherand P, Claris O, Charrié A, Raverot V, Orgiazzi J, Borson-Chazot F, Bournaud C.Predictive value of maternal second-generation thyroid-binding inhibitory immunoglobulin assay for neonatal autoimmune hyperthyroidism.Eur J Endocrinol. 2014;171:451-60. doi: 10.1530/EJE-14-0254
351 Karras S, Tzotzas T, Kaltsas T, Krassas GE: Pharmacological treatment of hyperthyroidism during lactation: review of the literature and novel data. Pediatr Endocrinol Rev 2010; 8: 25-33
352 Sun S, Qiu X, Zhou J. Clinical analysis of 65 cases of hyperemesis gravidarum with gestational transient thyrotoxicosis. J Obstet Gynaecol Res. 2014 ;40:1567-72. doi: 10.1111/jog.12372.
353 Tanaka S, Yamada H, Kato EH, Furuta I, Fukushi M, Takasugi N, Fujimoto S.Gestational transient hyperthyroxinaemia (GTH): screening for thyroid function in 23163 pregnant women using dried blood spots. Clin Endocrinol 1998; 49:325-9
354 Yeo CP, Khoo DH, Eng PH, Tan HK, Yo SL, Jacob E. Prevalence of gestational thyrotoxicosis in Asian women evaluated in the 8th to 14th weeks of pregnancy: correlations with total and free beta human chorionic gonadotrophin. Clin Endocrinol 2001; 55:391-8
355 Utiger RD: Some women with hyperemesis gravidarum have transient hyperthyroidism. Clinical Thyroidology 2002;14:56,
356 Grün JP, Meuris S, De Nayer P, Glinoer D: The thyrotropic role of human chorionic gonadotropin (hCG) in the early stages of twin (versus single) pregnancy. Clin Endocrinol 1997;46:719-25
357 Yoshikawa N, Nishikawa M, Horimoto M,et al. Thyroid-stimulating activity in sera of normal pregnant women.J Clin Endocrinol Metab. 1989 ;69:891-5
358 Talbot JA, Lambert A, Anobile CJ, McLoughlin JD, Price A, Weetman AP, Robertson WR. The nature of human chorionic gonadotrophin glycoforms in gestational thyrotoxicosis. Clin Endocrinol 2001; 55:33-9
359 Vassart G, Dumont JE: The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr Rev 1992;13: 596-611
360 Grjibovski AM, Vikanes A; Stoltenberg C Magnus P. Consanguinity and the risk of hyperemesis gravidarum in Norway: Acta Obstetr Gynecol Scan 2007; 87: 20-5
361.Zhang Y, Cantor RM, MacGibbon K, Romero R, Goodwin TM, Mullin PM, Fejzo MS.Familial aggression of hyperemesis gravidarum. Am J Obstet Gynecol 2011; 204:230.e1-230.e7.
362 Goodwin TM. Hyperemesis gravidarum. Obstet Gynecol Clin North Am. 2008;35:401-17, viii. doi: 10.1016/j.ogc.2008.04.002
363 Bober SA, McGill AC, Tunbridge WMG: Thyroid function in hyperemesis gravidarum. Acta Endocrinologica 1986; 111:404-10.
364 Kari Kumar KVS, Vamsikrishna P, Verma A, Muthukrishnan J, Meena U, Modi KD: Evaluation of thyrotoxicosis during pregnancy with color doppler sonography. International Journal of Gynecology and Obstetrics 2008; 102:152-5.
365 Karger S, Schötz S, Stumvoll M, Berger F, Führer D. Impact of pregnancy on prevalence of goitre and nodular thyroid disease in women living in a region of borderline sufficient iodine supply. Horm Metab Res. 2010;42:137-42
366 Kung AW, Chau MT, Lao TT, Tam SC, Low LC The effect of pregnancy on thyroid nodule formationJ Clin Endocrinol Metab. 2002;87:1010-114.
- Smith LH, Danielsen B, Allen ME, Cress R. Cancer associated with obstetric delivery: Results of linkage with California cancer registry. Am J Obstet Gynecol 2003;189:1128-35
- Hay I. Nodular thyroid disease diagnosed during pregnancy: How and when to treat. Thyroid 1999;9:667-70.
369 Pacini F, Schlumberger M, Dralle H Ilisea R, Smith Y, Wiersinga W. European consensus of the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur J Endocrinol 2006; 154:787-803
370 Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009;19:1167-1214.
371 Moosa M, Mazzaferri EL Outcome of differentiated thyroid cancer diagnosed in pregnant women. J Clin Endocrinol Metab1997; 82:2862-6
372 Yasmeen S, Cress R, Romano PS, Xing G, Berger-Chen S, Danielsen B, Smith LH Thyroid cancer in pregnancy. Int J Gynaecol Obstet 2005; 91:15-20
373 Vannucchi G, Perrino M, Rossi S, Colombo C, Vicentini L, Dazzi D, Beck-Peccoz P, Fugazzola L. Clinical and molecular features of differentiated thyroid cancer diagnosed during pregnancy.Eur J Endocrinol. 2010;162:145-51.
374 Lee ML, Chen GG, Vlantis AC, Tse GM, Leung BC, van Hasselt CA. Induction of thyroid papillary carcinoma cell proliferation by estrogen is associated with an altered expression of Bcl-xL. Cancer J. 2005;11:113-21.
375 Hishinuma A, Yamanaka T, Kasai K, So S, Tseng CC, Bamba N, Ohtake H, Shimoda S.Different growth control of the two human thyroid cell lines of adenomatous goiter and papillary carcinoma.Thyroid. 1995;5:41-6.
376 Messuti I, Corvisieri S, Bardesono F, Rapa I, Giorcelli J, Pellerito R, Volante M, Orlandi F.Impact of pregnancy on prognosis of differentiated thyroid cancer: clinical and molecular features.Eur J Endocrinol. 2014;10:170:659-66. doi: 10.1530/EJE-13-090311;doi:10.4061/2011/549609
377 Ito Y, Miyauchi A, Kudo T, Ota H, Yoshioka K, Oda H, Sasai H, Nakayama A, Yabuta T, Masuoka H, Fukushima M, Higashiyama T, Kihara M, Kobayashi K, Miya A. Effects of Pregnancy on Papillary Microcarcinomas of the Thyroid Re-Evaluated in the Entire Patient Series at Kuma Hospital. Thyroid. 2016 ;26:156-60. doi: 10.1089/thy.2015.0393
378 Zhou YQ, Zhou Z, Qian MF, Gong T, Wang JD. Association of thyroid carcinoma with pregnancy: A meta-analysis. Mol Clin Oncol. 2015 ;3:341-6
379 .Leboeuf R, Emerick LE, Martorella AJ, Tuttle RM Impact of pregnancy on serum thyroglobulin and detection of recurrent disease shortly after delivery in thyroid cancer survivors. Thyroid 2007;17:543-7
380 Hirsch D, Levy S, Tsvetov G, Weinstein R, Lifshitz A, Singer J, Shraga-Slutzky I, Grozinski-Glasberg S, Shimon I, Benbassat C.Impact of pregnancy on outcome and prognosis of survivors of papillary thyroid cancer. Thyroid 2010; 20:1179- 85. doi:10.1089/thy.2010.0081.
381 Lee JC, Zhao JT, Clifton-Bligh RJ, Gill AJ, Gundara JS, Ip J, Sywak MS, Delbridge LW, Robinson BG, Sidhu SB Papillary thyroid carcinoma in pregnancy: a variant of the disease? Ann Surg Oncol. 2012;19: 4210-6
382 Lin JD, Wang HS, Weng HF, Kao PF. Outcome of pregnancy after radioactive iodine treatment for well differentiated thyroid carcinomas. J Endoc Invest 1998;21 662-7.
383 Sawka AM, Lakra DC, Lea J Alshehri B, Tsang RW, Brierley JD, Straus S, Thabane L, Gafni A, Ezzat S, George SR, Goldstein DP. 2008 A systematic review examining the effects of therapeutic radioactive iodine on ovarian function and future pregnancy in female thyroid cancer survivors. Clin Endocrinol (Oxf) 2008:69:479-90
384 Garsi JP, Schlumberger M, Rubino C Ricard M, Labbé M, Ceccarelli C, Schvartz C, Henri-Amar M, Bardet S, de Vathaire F. Therapeutic administration of 131I for differentiated thyroid cancer: radiation dose to ovaries and outcome of pregnancies.J Nucl Med. 2008;49: 845-52.
385 Sioka C, Fotopoulos A: Effects of I-131 therapy on gonads and pregnancy outcome in patients with thyroid cancer. Fertility Sterility 2011; 95: 1552-9
386 Sawka AM, Lea J, Alshehis B, Straus S, Tsang RW, Brierley JD, Thabane L, Rotstein L, Gafni A, Ezzat S, Goldstein DP. A systemic review of the gonadal effects of therapeutic radioactive iodine in male thyroid cancer survivors. Clin Endocrinol 2008: 68:610-17
387 Cecarelli C, Bencivelli W, Morciano D, Pinchera A, Pacini F. I 131 therapy for differentiated thyroid cancer leads to an earlier onset of menopause: results of a retrospective study. J Clin Endocr Metab 2001; 86: 3512-15
388 Weetman AP.Immunity, thyroid function and pregnancy: molecular mechanisms.Nat Rev Endocrinol. 2010;6:311-8
389 Amino N, Tada H. Hidaka Y. Thyroid disease after pregnancy: postpartum thyroiditis. In: Wass JAH, Shalet SM (eds), Oxford textbook of Endocrinology and Diabetes. Oxford, UK: Oxford University press, 2002: 527-32
390 Tada H, Hidaka Y, Tsuruta E, Kashiwai T, Tamaki H, Iwatani Y, Amino N. Prevalence of postpartum onset of disease within patients with Graves’ disease of child-bearing age. Endocr J 1994; 41: 325-27
391 Rotondi M, Cappelli C, Pirali B,et al. The effect of pregnancy on subsequent relapse from Graves' disease after a successful course of antithyroid drug therapy Rotondi M, Cappelli C, Pirali B, Pirola I, Magri F, Fonte R, Castellano M, Rosei EA, Chiovato LJ Clin Endocrinol Metab. 2008;93:3985-8
392 Ide A, Amino N, Kang S, Yoshioka W, Kudo T, Nishihara E, Ito M, Nakamura H, Miyauchi A. Differentiation of postpartum Graves' thyrotoxicosis from postpartum destructive thyrotoxicosis using antithyrotropin receptor antibodies and thyroid blood flow.Thyroid. 2014 ;24:1027-31. doi: 10.1089/thy.2013.0585.
393 Gaberšček S, Osolnik J, Zaletel K, Pirnat E, Hojker S. An Advantageous Role of Spectral Doppler Sonography in the Evaluation of Thyroid Dysfunction During the Postpartum Period.
J Ultrasound Med. 2016 ;35: 1429-36. doi: 10.7863/ultra.15.07033
394 Lazarus JH, Ludgate ME. Prevention and treatment of postpartum Graves’ disease. In: Bailliere’s Clinical Endocrinology and Metabolism. New Aspects of Clinical Graves’ Disease. Bailliere Tindall, London. Vol 11(3): pp 549-560; 1997
395 Roberton HEW: Lassitude, coldness and hair changes following pregnancy, and their treatment with thyroid extract. BMJ ii: 2275, 1948
396 Stagnaro-Green A Postpartum thyroiditis..Best Pract Res Clin Endocrinol Metab. 2004 ;18:303-16
397 Lazarus JH, Postpartum Thyroid Disease in The Thyroid and Reproduction Eds John Lazarus Valdis Pirags Sigrid Butz. Georg Thieme Verlag, Stuttgart 2009 p 105-113
398 Landek-Salgado MA, Gutenberg A, Lupi I, Kimura H, Mariotti S, Rose NR, Caturegli P.Pregnancy, postpartum autoimmune thyroiditis, and autoimmune hypophysitis: intimate relationships.Autoimmun Rev. 2010 ;9:153-7.
399 Pearce EN.Thyroid disorders during pregnancy and postpartum.Best Pract Res Clin Obstet Gynaecol. 2015;29:700-6. doi: 10.1016/j.bpobgyn.2015.04.007
400 Muller AF, Drexhage HA, Berghout A. Postpartum thyroiditis and autoimmune thyroiditis in women of childbearing age: recent insights and consequences for antenatal and postnatal care. Endocr Rev. 2001;22:605-30.
401 Lazarus JH, Hall R, Othman S Parkes AB, Richards CJ, McCulloch B, Harris B. The clinical spectrum of postpartum thyroid disease. QJM 1996; 89:429-35.
402 Harris B, Fung H, Johns S Kologlu M, Bhatti R, McGregor AM, Richards CJ, Hall R. Transient post-partum thyroid dysfunction and postnatal depression. J Affect Disord. 1989; 17: 243-9.
403 Harris B, Othman S, Davies JA Weppner GJ, Richards CJ, Newcombe RG, Lazarus JH, Parkes AB, Hall R, Phillips DI. Association between postpartum thyroid dysfunction and thyroid antibodies and depression. BMJ. 1992; 305:152-6.
404 Pop VJ, de Rooy HA, Vader HL van der Heide D, van Son M, Komproe IH, Essed GG, de Geus CA. Postpartum thyroid dysfunction and depression in an unselected population. N Engl J Med. 1991; 324:1815–6.
405 Kent GN, Stuckey BG, Allen JR Lambert T, Gee V. Postpartum thyroid dysfunction: clinical assessment and relationship to psychiatric affective morbidity. Clin Endocrinol (Oxf). 1999; 51: 429-38.
406 Groer MW, Vaughan JH Positive thyroid peroxidase antibody titer is associated with dysphoric moods during pregnancy and postpartum J Obstet Gynecol Neonatal Nurs. 2013 ;42:E 26-32.
407 Paragliola RM, Concolino P, De Rosa A, Mello E, Zuppi C, Pontecorvi A, Capoluongo E, Corsello SM The first case of association between postpartum thyroiditis and thyroid hormone resistance in an Italian patient showing a novel p.V283A THRB mutation.
Thyroid. 2013; 23: 506-10.
408 Premawardhana LDKE, Parkes AB, Ammari F, John R, Darke C, Adams H, Lazarus JH. Postpartum thyroiditis and long-term thyroid status: prognostic influence of thyroid peroxidase antibodies and ultrasound echogenicity. J Clin Endocrinol Metab 2000; 85:71-5.
409 Stagnaro-Green A Roman SH, Cobin RH, el-Harazy E, Wallenstein S, Davies TF. A prospective study of lymphocyte-initiated immunosuppression in normal pregnancy: evidence of a T-cell etiology for postpartum thyroid dysfunction. J Clin Endocrinol Metab1992:;74: 645-53
410 Kokandi AA Parkes AB, Premawardhana LD, John R, Lazarus JH. Association of postpartum thyroid dysfunction with antepartum hormonal and immunological changes. J Clin Endocrinol Metab 2003; 88:1126-32
411 Negro R, Greco G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H.The influence of selenium supplementation on postpartum thyroid status in pregnant women with thyroid peroxidase autoantibodies.J Clin Endocrinol Metab. 2007;92:1263-8.
412 Wald N, Law M Medical Screening in Oxford Textbook of Medicine 2010 5th Edition Eds Warrell DA, Cox TM and Firth JD. Oxford University Press, Oxford, New York Vol 1 pp94 – 108
413 Casey BM, Dashe JS, Wells CE, McIntire DD, Leveno KJ, Cunningham FG. Subclinical hyperthyroidism and pregnancy outcomes. Obstet Gynecol 2006; 107:337-41
414 Lazarus JH, Kaklamanou M. Significance of low thyroid-stimulating hormone in pregnancy. Curr Opin Endocrinol Diabetes Obes. 2007;14: 389-92
415 Lazarus JH. Iodine status in Europe in 2014. Eur Thyroid J. 2014; 3:3-6. doi: 10.1159/000358873.
416 Kooistra L, Crawford S, van Baar AL, Brouwers EP, Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy. Pediatrics.2006; 117:161-7.
417 Negro R, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Stagnaro-Green A.Universal screening versus case finding for detection and treatment of thyroid hormonal dysfunction during pregnancy.J Clin Endocrinol Metab. 2010; 95: 1699-707
418 Thung SF, Funai EF, Grobman WA. The cost-effectiveness of universal screening in pregnancy for subclinical hypothyroidism. Am J Obstet Gynecol. 2009; 200: 267.e1-7.
419 Dosiou C, Sanders GD, Araki SS, Crapo LM. Screening pregnant women for autoimmune thyroid disease: a cost-effectiveness analysis. Eur J Endocrinol. 2008; 158: 841-51
420 Granfors M, Åkerud H, Skogö J, Stridsberg M, Wikström AK, Sundström-Poromaa I. Targeted thyroid testing during pregnancy in clinical practice. Obstet Gynecol. 2014;124:10-5. doi: 10.1097/AOG.
421 Nazarpour S, Tehrani FR, Simbar M, Tohidi M, AlaviMajd H, Azizi F. Comparison of universal screening with targeted high-risk case finding for diagnosis of thyroid disorders.
Eur J Endocrinol. 2016 ;174:77-83. doi: 10.1530/EJE-15-0750.
422 Jouyandeh Z, Hasani-Ranjbar S, Qorbani M, Larijani B. Universal screening versus selective case-based screening for thyroid disorders in pregnancy.Endocrine. 2015;48:116-23. doi: 10.1007/s12020-014-0385-9.
423 Ahmed IZ, Eid YM, El Orabi H, Ibrahim HR. Comparison of universal and targeted screening for thyroid dysfunction in pregnant Egyptian women.Eur J Endocrinol. 2014 ;171: 285-91. doi: 10.1530/EJE-14-0100..
424 Yang H, Shao M, Chen L, Chen Q, Yu L, Cai L, Lin Z, Zhang C, Lu X. Screening strategies for thyroid disorders in the first and second trimester of pregnancy in China.
PLoS One. 2014 12;9:e99611. doi: 10.1371/journal.pone.0099611
425 Ma L, Qi H, Chai X, Jiang F, Mao S, Liu J, Zhang S, Lian X, Sun X, Wang D, Ren J, Yan Q. The effects of screening and intervention of subclinical hypothyroidism on pregnancy outcomes: a prospective multicenter single-blind, randomized, controlled study of thyroid function screening test during pregnancy. J Matern Fetal Neonatal Med. 2016; 29:1391-4. doi: 10.3109/14767058.2015.1049150.
426 Azizi F, Amouzegar A, Mehran L, Alamdari S, Subekti I, Vaidya B, Poppe K, San Luis T Jr, Akamizu T.Screening and management of hypothyroidism in pregnancy: results of an Asian survey.Endocr J. 2014; 61:697-704.
427 Casey B1, de Veciana M2. Thyroid screening in pregnancy.Am J Obstet Gynecol. 2014 ;211:351-3.e1. doi: 10.1016/j.ajog.2014.08.013.
428 Spencer L1, Bubner T, Bain E, Middleton P. Screening and subsequent management for thyroid dysfunction pre-pregnancy and during pregnancy for improving maternal and infant health. Cochrane Database Syst Rev. 2015 21;:CD011263. doi:
10.1002/14651858.CD011263.pub2.
429 Ong GS, Hadlow NC, Brown SJ, Lim EM, Walsh JP. Does the thyroid-stimulating hormone measured concurrently with first trimester biochemical screening tests predict adverse pregnancy outcomes occurring after 20 weeks gestation?J Clin Endocrinol Metab. 2014;99:E2668-72. doi: 10.1210/jc.2014-1918.
430 Taylor PN, Okosieme OE, Premawardhana L, Lazarus JH. Should all women be screened for thyroid dysfunction in pregnancy? Womens Health (Lond). 2015;11:295-307. doi: 10.2217/whe.15.7
431 Amouzegar A, Mehran L, Sarvghadi F, Delshad H, Azizi F, Lazarus JH.
Comparison of the American Thyroid Association with the Endocrine Society practice guidelines for the screening and treatment of hypothyroidism during pregnancy.
Hormones (Athens). 2014;13:307-13. doi: 10.14310/horm.2002.1486
432 Vaidya B, Anthony S, Bilous M et al. Detection of thyroid dysfunction in early pregnancy: Universal screening or targeted high-risk case finding J Clin Endocrinol Metab. 2007;92:203-07.
433 Horacek J, Spitalnikova S, Dlabalova B, Malirova E, Vizda J, Svilias I, Cepkova J, Mc Grath C, Maly J. Universal screening detects two-times more thyroid disorders in early pregnancy than targeted high-risk case finding. Eur. J. Endocrinol. 2010; 163:645-65
434 Jiskra J, Bartáková J, Holinka Š, Límanová Z, Springer D, Antošová M, Telička Z, Potluková Low prevalence of clinically high-risk women and pathological thyroid ultrasound among pregnant women positive in universal screening for thyroid disorders. E.Exp Clin Endocrinol Diabetes. 2011;119:530-5.
435 .Wang W, Teng W, Shan Z, Wang S, Li J, Zhu L, Zhou J, Mao J, Yu X, Li J, Chen Y, Xue H, Fan C, Wang H, Zhang H, Li C, Zhou W, Gao B, Shang T, Zhou J, Ding B, Ma Y, Wu Y, Xu H, Liu W. The prevalence of thyroid disorders during early pregnancy in China: the benefits of universal screening in the first trimester of pregnancy Eur J Endocrinol. 2011;164: 263-8.
436 Haddow JE, McClain MR, Palomaki GE, Kloza EM, Williams J. Screening for thyroid disorders during pregnancy: results of a survey in Maine. American Journal of Obstetrics and Gynecology. 2006; 194: 471-4.
437 Vaidya B, Hubalewska-Dydejczyk A, Laurberg P, Negro R, Vermiglio F, Poppe K. Treatment and screening of hypothyroidism in pregnancy: results of a European survey. European Journal of Endocrinology. 2012 ;166: 49-54
438 Vila L, Velasco I, González S, Morales F, Sánchez E, Torrejón S, Soldevila B, Stagnaro-Green A, Puig-Domingo M.L Vila, Barcelona, Spain Controversies in Endocrinology: On the need of universal thyroid screening in pregnant women. Eur J Endocrinol. 2013; 29;170(:R17-30. doi: 10.1530/EJE-13-0561
439 Srimatkandada P, Stagnaro-Green A, Pearce EN. Attitudes of ATA survey responden.ts toward screening and treatment of hypothyroidism in pregnancy.Thyroid. 2015;25:368-9. doi: 10.1089/thy.2014.0322.
440 Reid SM, Middleton P, Cossich MC, Crowther CA, Bain E.
Interventions for clinical and subclinical hypothyroidism pre-pregnancy and during pregnancy. Cochrane Database Syst Rev. 2013 May 31;5:CD007752. doi: 10.1002/14651858.
441Taylor PN, Lacey A, Thayer D, Yusof M, Tabasum A, Muller I, Marsh L, Ludgate M Rees A, Boelaert K, Chan S, Nelson S, Rees A Lazarus JH, Dayan CM, Vaidya B, Okosieme O.Controlled Antenatal Thyroid Study: Obstetric Outcomes.British Thyroid Association 2016; Abstr. No 01 Newcastle-on-Tyne.
442 Laurberg P, Andersen SL, Pedersen IB, Andersen S, Carlé A.Screening for overt thyroid disease in early pregnancy may be preferable to searching for small aberrations in thyroid function tests.Clin Endocrinol (Oxf). 2013 ;79:297-304. doi: 10.1111/cen.12232
443 Pop VJ Pregnancy, postpartum and the thyroid: isn't it time to offer women optimal care?
Facts Views Vis Obgyn. 2014;6:166-70.
Thyroid Nodules
ABSTRACT
Thyroid cancer accounts for only 0.4% of all cancer deaths, with an incidence of 11 cases and about 0.5-0.6 deaths per 100,000 population in the United States each year,according to the November 2011 SEER report (http://seer.cancer.gov/statfacts/html/thyro.html). Its clinical importance, by contrast, is out of all proportion to its incidence, because cancers of the thyroid must be differentiated from the much more frequent benign adenomas and multinodular goiters. The latter, depending on the criteria employed, occur in up to 4% of the population, and thyroid nodules may be present in 20% or more of adults subjected to routine thyroid echography. The differential diagnosis of thyroid nodules is now easily accomplished by fine needle aspiration cytology in 60-90% of the cases, allowing a significant reduction in the number of thyroid surgeries performed for thyroid nodules. This chapter is concerned with the clinical and pathological description of benign and malignant thyroid nodules and with the diagnostic and therapeutic approach to them. What will be said applies also to nodules found within a multinodular goiter, although as a separate entity this disease is discussed in Chapter 17.
INTRODUCTION
DEFINITION
The thyroid adenoma is a benign neoplastic growth contained within a capsule. The term adenoma and nodule are often used interchangeably in the literature. This practice is imprecise because adenoma implies a specific benign new tissue growth with a glandlike cellular structure, whereas a nodule could as well be a cyst, carcinoma, lobule of normal tissue, or other focal lesion different from the normal gland. In the following section, the term nodule appears frequently when there is need for a nonspecific term.
Table 18-1. Neoplasms of the Thyroid
|
| I. Adenomas (Fig. 18-1, below) A. Follicular 1. Colloid variant 2. Embryonal 3. Fetal 4. Hurthle cell variant B. Papillary (probably malignant) C. TeratomaII. Malignant Tumors A. Differentiated 1. Papillary adenocarcinoma a. Pure papillary adenocarcinoma b.Mixed papillary and follicular carcinoma (variants including tall cell, follicular, oxyphyl, solid) 2. Follicular adenocarcinomas (variants: "malignant adenoma", Hurthle cell carcinoma or oxyphil carcinoma, clear-cell carcinoma, insular carcinoma) B. Medullary carcinoma- (not a tumor of follicular cells) C. Undifferentiated 1. Small cell (to be differentiated from lymphoma) 2. Giant cell 3. Carcinosarcoma D. Miscellaneous 1. Lymphoma, sarcoma 2. Squamous cell epidermoid carcinoma 3. Fibrosarcoma 4. Mucoepithelial carcinoma 5. Metastatic tumor |
Figure 18-1A) Follicular and microfollicular adenoma. The nodule shows microfollicles, is sharply circumscribed by a delicate even fibrous capsule, and there is no invasion of the capsule or blood vessels by the tumor.
Figure 18-1B) The central area of a microfollicular adenoma displays regular nuclei and some interfollicular edema.
Figure 18-1C) Hurthle (oxyphile) cell tumor, lower half of photomicrograph, with well circumscribed margin established by an intact delicate fibrous capsule. This is a Hurthle cell tumor of low malignant potential (an adenoma).
Figure 18-1D) High power view of a Hurthle cell tumor made up of microfollicles lined by large acidophilic cells, the cytoplasm of which is granular and filled with mitochondria.
PATHOLOGY OF NODULES
Thyroid nodules are not expression of a single disease but are the clinical manifestation of a wide range of different diseases. Non-neoplastic nodules are the result of glandular hyperplasia arising spontaneously or following partial thyroidectomy; rarely, thyroid hemiagenesia may present as hyperplasia of the existing lobe, mimicking a thyroid nodule. Non-neoplastic thyroid diseases, such as Hashimoto’s thyroiditis or subacute thyroiditis may appear as thyroid lumps which are not true nodules but just the expression of the underlying thyroid disease.
Benign neoplastic nodules are divided into embryonal, fetal, follicular, Hurthle, and possibly papillary adenomas on the basis of their characteristic pattern 12. Examples appear in Figure 18-1 (above). The adenomas usually exhibit a uniform orderly architecture and few mitoses, and show no lymphatic or blood vessel invasion. They are characteristically enveloped by a discrete fibrous capsule or a thin zone of compressed surrounding thyroid tissue. All types of nodules may become partially cystic, presumably through necrosis of a portion of the growth. Cyst formation is very common in colloid nodules.
Whether papillary adenoma is a real entity is debatable; most observers believe that all papillary tumors should be considered as carcinomas. Others consider that some papillary tumors are benign adenomas. It is our impression that papillary tumors are best thought of as carcinomatous, although the degree of invasive potential may be very slight in some instances. The same confusion extends to Hurthle cell adenomas. Many pathologists consider all of these tumors as low-grade carcinomas in view of their frequent late recurrences. For this reason, the nondefinitive term Hurthle cell tumor is commonly used. Pathologists usually grade them on the probability of being malignant, based on factors such as invasion of the thyroid tumor capsule or blood vessels, without differentiation into benign and malignant. Hurthle cell tumors are found on electron microscopy to be packed with mitochondria, which accounts for their special eosinophilic staining quality.
Nearly half of all single nodules have on gross inspection a gelatinous appearance, are composed of large colloid-filled follicles, and are not completely surrounded by a well-defined fibrous capsule. These nodules are listed as colloid variants of follicular adenomas in our classification. Many pathologists report these as colloid nodules, and suggest that each is a focal process perhaps related to multinodular goiter rather than a true adenoma. These tumors are usually not surrounded by a capsule of compressed normal tissue, and often show degeneration of parenchyma, hemosiderosis, and colloid phagocytosis (Fig. 18-2). Recent studies indicate that most adenomas, as well as carcinomas, are truly clonal -- derived from one cell -- whereas colloid nodules, at least in multinodular goiters, tend to be polyclonal (2).
Figure 18-2A) "Colloid nodules" display macrofollicles lined by flattened thyroid epithelial cells. The nodules are circumscribed and do not have a fibrous capsule.
Figure 18-2B) Possible evolution of a "colloid nodule". An area of nodular hyperplasia on left, and a "developing" colloid nodule on right, with macrofollicles and some remaining focal hyperplasia.
Figure 18-3. Fine needle aspiration cytology specimens.
CAUSE OF NODULES
Thyroid adenomas are monoclonal "new growths" that are formed in response to the same sort of stimuli as are carcinomas. Heredity does not appear to play a major role in their appearance. One clue to their origin is that they are four times more frequent in women than in men, although no definitive relation of estrogen to cell growth has been demonstrated. Thyroid radiation, chronic TSH stimulation, and oncogenes believed to be related to the origin of these lesions are discussed below in the section on thyroid cancer. Of specific interest in relation to benign nodules is the remarkable observation by Vassart and colleagues that activating mutations of the TSH receptor are the specific cause of most autonomously functioning thyroid nodules (3) including those found in the context of a mulitnodular goiter (4). Please see the discussion on causation under "Carcinoma".
COURSE AND SYMPTOMS OF NODULES
Thyroid adenomas grow slowly and may remain dormant for years. This is presumably related to the fact that adult thyroid cells normally divide once in eight years. (5) Pregnancy tends to make nodules increase in size, and to cause development of new nodules. An adenoma may first come to attention because the patient accidentally finds a lump in the neck or because a physician discovers it upon routine examination. Rarely, symptoms such as dysphagia, dysphonia, or stridor may develop, but it is unusual for these tumors to attain sufficient size to cause significant symptoms in the neck. Typically, they are entirely asymptomatic. Occasionally there is bleeding into the tumor, causing a sudden increase in size and local pain and tenderness. After bleeding into an adenoma, transient symptoms of thyrotoxicosis may appear with elevated serum T4 levels, and suppression of thyroidal RAIU. Spontaneous regression of adenomas can occur.
M.G., 47-Year-Old Woman: Hemorrhage into a Nodule
When the patient was first examined, enlargement of the thyroid had been known for at least 6 years. A scan showed a cold nodule in the left lobe. The patient was thought to have a thyroid nodule and to be euthyroid. There was on exam a normal right thyroid lobe, and a 4 x 5 cm soft mass occupying the position of the left lobe. The impression at this time was that she had an adenoma that might be cystic. Antithyroid antibodies were not detectable.
The patient was examined by several observers who palpated the thyroid. One-half hour after leaving the clinic, the patient's neck gradually began to enlarge, and she developed pain in the area of the thyroid and a rasping hoarseness. She had no difficulty in swallowing or breathing. The pain was significant enough to keep her awake that night, and she returned to the hospital the next day. The patient was very anxious, and there was a 10 x 12-cm tender fluctuant swelling occupying the area of the thyroid. Inspiratory stridor was present, and there were a few rhonchi in the lungs.
During the subsequent 3 days the pain in the neck gradually diminished, but the size of the mass remained more or less the same. Chest x-ray films revealed marked deviation of the trachea to the right. Operation was elected. A greatly enlarged left lobe of the thyroid was found, with hemorrhage into an adenoma. The encapsulated mass measured 6.5 x 5.5 cm and was smooth and cystic. There was a large multiloculated hematoma and considerable necrotic tissue. A left lobectomy was performed. Microscopic examination showed a microfollicular thyroid adenoma with recent hemorrhage and necrosis. The postoperative course was unremarkable. The patient remained well after surgery without further difficulty.
Usually hemorrhage occurs without known provocation, but occasionally is seen after trauma to the neck. In this instance, palpation may have been sufficient to induce bleeding.
Does an adenoma ever develop into a carcinoma? At the practical level, thyroid adenomas appear to be benign from the start and most thyroid carcinomas are likewise malignant from their inception, and do not appear on pathological examination to originate in an adenoma. Perhaps this has to do with the specific discrete mutational event causing their development. However, in animals chronically given 131-I and antithyroid drugs, a gradual progression of types of lesions from adenomas to carcinomas is seen. Pathologic examination occasionally gives evidence for conversion of an adenoma to a carcinoma. Transformation of hyperplastic thyroid tissue into invasive cancer occurs in occasional patients with congenital goitrous hypothyroidism, and occasionally cancers are seen inside an adenoma or in a gland that was known to have harbored a nodule for many years. Occasionally a patient develops metastatic cancer years after resection of an embryonal or Hurthle cell adenoma. Rearrangement of the RET gene, the genetic abnormality responsible for a subset of papillary thyroid carcinoma, is frequently found in microcarcinoma, suggesting that this lesion is malignant from the beginning without the need for accumulating several genetic lesions (6). Furthermore, no case of RET/PTC positive anaplastic cancer has been found up to now, again suggesting that there is no transition from papillary, at least RET/PTC positive, carcinoma to anaplastic cancer (6). All of these points suggest that transformation of an adenoma into a carcinoma occurs occasionally, but it appears to be an unusual sequence of events.
HOT NODULES
About 10% of thyroid follicular adenomas are functional enough (are "hot" on scan) to produce overt thyrotoxicosis or subclinical hyperthyroidism (suppressed TSH being the only abnormality) and account for perhaps 2% of all thyrotoxic patients. Another 10% may be borderline in function and are classified as warm or "hot" (or hyperfunctioning, in comparison to the remainder of the thyroid gland) on isotopic scans. Although hyperfunctioning nodules may remain unchanged for years, some gradually develop into toxic nodules, especially if their diameter exceeds 3 cm (7). Others undergo spontaneous necrosis with a return of function in the formerly suppressed normal gland. Patients with functioning autonomous nodules may be overtly thyrotoxic; more commonly, however, the nodule functions enough to suppress the remainder of the gland, but not enough to produce clinical hyperthyroidism (8). In such patients, T3 levels may be slightly elevated, serum TSH below normal, and the pituitary response to TRH is typically suppressed (9). If the nodule is resected, the gland resumes normal function, and serum TSH and the TRH response is normalized (see also Chapter 13).
In certain areas such as Switzerland, up to one-third of all thyrotoxic patients have hyperfunctioning adenomas (10), largely in multinodular glands. Perhaps this situation is generally true in endemic goiter areas.
Activating TSH receptor mutations have been found by Vassart and co-workers (11) to be the cause of most hyperfunctional nodules, and are now known to be common in "hot" nodules in patients with multi-nodular goiter.. These mutations generally involve the extracellular loops of the transmembrane domain and the transmembrane segments, and are proven to induce hyperfunction by transfection studies. Mutations of the stimulatory GTP binding protein subunit are also present in some patients with hyperfunctioning thyroid adenomas (12).
METABOLIC FUNCTION OF NODULES
The biochemical defect responsible for diminished iodine metabolism in the "cold" (i.e., inactive) nodule can result from deletion of specific metabolic processes required for hormone synthesis. Slices of cold nodules incubated in vitro were unable to accumulate iodide against a concentration gradient, although peroxidase and iodide organification activities were present (13). This finding was consistent with a specific defect of the iodide transport process. Others have also observed this phenomenon and have shown that TSH can bind to the membranes of the cells and activate adenyl cyclase as usual, but that subsequent metabolic steps are not induced (14). Activity of the sodium-potassium-activated ATPase, thought to be related to iodide transport, is intact, and ATP levels are normal, even though iodide transport is inoperative. Other nodules appear to be cold because they lack peroxidase (15). These nodules can be "hot" when scanned with 99mTcO4 due to active transport of the isotope, but relatively cold on scanning at 24 hours after 131-I is given, since iodide binding is poor (16). The adenyl cyclase system in the plasma membrane of some hyperfunctioning nodules has been found to be hyper-responsive to TSH in some studies (17) but not in others (18). As noted above, most hyperfunctional nodules are associated with- presumably caused by- activating mutations of the TSH receptor. All of the foregoing reports suggest that adenoma formation is associated with mutational events that cause loss or dysfunction of normal metabolic activities The recent cloning of the sodium/iodide synporter gene (NIS) (19) has allowed the study of its expression in thyroid nodules. NIS expression is increased, with respect to normal thyroid tissue, in hyperfunctioning nodules and low or absent in cold nodules both benign and malignant (20).
CLINICAL EVALUATION AND MANAGEMENT OF NODULES
Table 18-2. Differential Diagnosis of the Thyroid Nodule |
| Adenoma Cyst Carcinoma Multinodular goiter Hashimoto's thyroiditis Subacute thyroiditis Effect of prior operation or 131I therapy Thyroid hemiagenesis Metastasis Parathyroid cyst or adenoma Thyroglossal cyst Nonthyroidal lesions Inflammatory or neoplastic nodes Cystic hygroma Aneurysm Bronchocele Laryngocele |
HISTORY OF NODULES
Conditions to be considered in the differential diagnosis are listed in Table 18-2. They include adenoma, cyst, multinodular goiter, a prominent area of thyroiditis, an irregular regrowth of tissue if surgery has been performed, thyroid hemiagenesis, and of course, thyroid cancer. Hashimoto's thyroiditis offten presents with a lumpy gland on physical exam, and a nodular or pseudo-nodular appearance on ultrasound is frequent. In some patients Hurthle cell rich nodular areas develop, and of course some patients have coexistent but (presumably) etiologically distinct adenomas or cancers.
Factors that must be considered in reaching a decision for management include the history of the lesion, age, sex, and family history of the patient, physical characteristics of the gland, local symptoms, and laboratory evaluation. The age of the patient is an important consideration since the ratio of malignant to benign nodules is higher in youth and lower in older age. Male sex carries a similar importance (21). Nodules are less frequent in men, and a greater proportion are malignant.
Rarely, the family history may be helpful in the decision regarding surgery. Patients with the hereditable multiple endocrine neoplasia syndrome (MEN), type I, may have thyroid adenomas, parathyroid adenomas, islet cell tumors, and adrenal tumors, whereas patients with MEN types II and III, have pheochromocytomas, medullary thyroid carcinomas, hyperparathyroidism, and mucosal neuromas (22-24) (vi). Further, we have observed that 6% of our patients with thyroid carcinoma have a history of malignant thyroid neoplasm in other family members, and familial medullary cancer (without MEN) is well known. Familial thyroid tumors occur in Cowden's disease, Gardner's syndrome, and familial polyposis coli (vi.)
A most important piece of information regarding a nodule is a history of prior neck irradiation. Any irradiation above 50 rads (50 cGrays) to the thyroid during childhood should be viewed with concern. Exposure to 100-700 rads during the first 3 or 4 years of life has been associated with a 1-7% incidence of thyroid cancer occurring 10-30 years later (25-30). Radiation exposure during adolescence or early adulthood for acne or for other reasons has also been identified as a cause of this disease. Although this association was known by 1950, patients were still being seen with radiation-related tumors who received x-ray treatment as late as 1959. Radiation therapy for other benign or malignant lesions in the neck is still in use in selected patients; such exposure will thus continue to be a relevant part of the history. Because of the high prevalence (20-40%) of carcinoma in nodules resected from irradiated glands, the finding of one or more clear-cut nodules in a radiated gland, or a cold area on scan, must be viewed with alarm and requires consideration for removal, as indicated below. In this case, multiple nodules do not indicate that the lesions are benign. In contrast, prior exposure to internal radiation from 131-I for diagnostic or therapeutic purposes has not to date been associated with an increased risk of developing thyroid carcinoma.
The history of the neck lump itself is important. Recent onset, growth, hoarseness, pain, nodes in the supraclavicular fossae, symptoms of brachial plexus irritation, and local tenderness all suggest malignancy, but of course do not prove it. The usual cause of sudden swelling and tenderness in a nodule is hemorrhage into a benign lesion. Although the presence of a nodule for many years suggests a benign process, some cancers grow slowly. In our series, the average time from recognition of a nodule to diagnosis of cancer was 3 years. A history of residence in an endemic goiter zone during the first decades of life is also relevant and must raise the possibility of multinodular goiter as the true diagnosis.
PHYSICAL FINDINGS
The adenoma is typically felt as a discrete lump in an otherwise normal gland, and it moves with the thyroid. Enlarged lymph nodes should be carefully sought, particularly in the area above the isthmus, in the cervical chains, and in the supra- clavicular areas. Their presence suggests malignant disease unless a good alternative diagnosis is apparent, such as recent oro-pharyngeal sepsis or viral infection. Fixation of the nodule to strap muscles or the trachea is alarming. Characteristically a benign thyroid adenoma is part of the thyroid and moves with deglutition, but can be moved in relation to strap muscles and within the gland substance to some extent. Pain, tenderness, or sudden swelling of the nodule usually indicates hemorrhage into the nodule but can also indicate an invasive malignancy. Hoarseness may arise from pressure or by infiltration of a recurrent laryngeal nerve by a neoplasm. Obviously the presence of a firm, fixed lesion, associated with pain, hoarseness, or any one of these features, should signal some degree of alarm. The converse situation, the absence of such characteristics, suggests but does not prove benignity. Fluctuance in the lesion suggest the presence of a cyst that is, usually, benign.
The presence of a diffusely multinodular gland, ascertained on the basis of palpation, US, or scanning, has in past years been interpreted as a sign of safety. Multinodular goiters coming to surgery have a significant prevalence of carcinoma (4-17%), but this finding was believed to be due largely to selection of patients for surgery, and not to be typical of multinodular goiters in the general population (31,32). However, in an era of generalized iodine sufficiency, when multinodular goiters are less common, this opinion needs re-evaluation. Frates et al (32.1) evaluated outcome in 1985 patients who underwent 3483 FNAs. Solitary nodules had the expected higher incidence of cancer than non-solitary nodules. However on a "per patient" basis, patients with MNG had the same incidence of diagnosed cancer (14.9%) as did patients with a solitary nodule (14.8%). Male sex, non-cystic nature, hypo-echogenicity, and stippled calcifications were associated with increased risk of malignancy per nodule. Usually FNA has been recommended for dominant or growing nodules. Frates et al recommend that for exclusion of malignancy in a thyroid with multiple nodules larger than 10mm, up to four nodules should be considered for FNA, and that the risk factors noted above may guide selection of nodules for biopsy.
Occasionally the gland has, in addition to a nodule, the diffuse enlargement and firm consistency of chronic thyroiditis, a palpable pyramidal lobe, and antibody test results that may be positive. These findings strongly suggest thyroiditis but do not disclose the nature of the nodule, which must be evaluated independently. It should be remembered that 14 - 20% (30, 33) of thyroid cancer specimens contain diffuse or focal thyroiditis. In addition, a positive association of thyroid cancer and Hashimoto's thyroiditis has been reported, but is not proven (v.i.).
Thyroid function tests
The patient is usually euthyroid, and this impression is supported by normal values for the serum FTI and T3 levels. Thyrotoxicity produced by an adenoma is discussed below. Low free thyroid hormones or elevated TSH results should raise the question of thyroiditis.
Curiously, it has been observed and confirmed that there is a direct correlation between TSH levels in patients with nodules and the risk of thyroid malignancy, and also with the degree of aggressiveness of the tumor(33.1)
The serum TG concentration may be elevated, as in all other goitrous conditions, and therefore is not a valuable tool in differential diagnosis. Calcitonin assay is indicated in the presence of a suggestive family history or of coincident features of the MEN-II syndromes. A chest x-ray should be taken if a normal film has not been reported in the prior 6 months. Soft tissue x-ray films of the neck may disclose indentation or deviation of the trachea if the tumor is more than 3 or 4 cm in diameter. Fine, stippled calcifications through the tumor (psammoma bodies) are virtually pathognomonic of papillary cancer. Patchy or "signet ring" calcification occurs in old cysts and degenerating adenomas, and has no such connotation.
Calcitonin assays
Although MTC constitutes a small fraction of thyroid malignancies, and an even smaller proportion of thyroid nodules, several reports suggest that routine screening of nodular goiters by CT assay is an appropriate approach (34-37). Such screening offers the possibility of finding tumors before they have metastasized, and MTC is rarely found on FNA. Whether the considerable expense is justified is yet indeterminate. Routine screening has been adopted as a standard procedure for evaluation of nodules in many clinics in Europe, but not in the USA. While calcitonin levels above 60 typically signal the presence of Medullary cancer, abnormal levels between 10 and 60 may be present with C-cell hyperplasia or no objective abnormality, and may spontaneously normalize(37.1).
FINE NEEDLE ASPIRATION CYTOLOGY
For many years core needle biopsy of the thyroid was employed successfully in some clinics to provide a histologic diagnosis on which to base therapy (38). Difficulties in acceptance of the procedure by surgeons, patients, and pathologists prevented its widespread application. As an alternative technique, thin needle aspiration cytologic examination has been widely adopted after favorable reports by Walfish et al (39) and Gershengorn et al (40). The procedure is technically simple and acceptable to patients, but requires an experienced operator and collaboration with a skilled cytopathologist (Figure 18-1). Two to four aspirations of the nodule, in different areas of the nodule, are recommended by many expert cytologists, particularly when the nodule is large enough (41). One common technique for sampling the whole nodule, is to introduce the needle in the center of the nodule, aspirate and then move the needle in another direction and aspirate again. The technique of FNA, and US-guided FNA, is described above in Chapter 6.
Fig 18-1a. Benign colloid nodule, showing scattered bits of coloid, few RBCs, and normal follicular cells
Figure 18-1B Epithelial cells in a follicular arrangement suggesting adenoma, but which could be from a follicular carcinoma.
Figure 18-1C Epithelial cells in a papillary formation from a papillary thyroid carcinoma. Nuclear grooves are also apparent. (Courtesy of Dr. Richard DeMay, University of Chicago/Chicago Lying-in Hospital)
Significant complications such as bleeding, infection, induced necrosis, or cyst formation are rare. Surprisingly the release of thyroglobulin into the blood stream appears to induce development of anti-thyroxine antibodies, of IgG or IgM class, in some (2-20%) patients, especially those with prior evidence of autoimmune thyroid disease (42). Adequate specimens can be obtained in over 90% of patients. False-negative and false-positive diagnoses do of course occur, but are each under 5% with experienced hands. Willems and Lowhagen (43), in reviewing a collected series of nearly 4,000 surgically proven fine needle aspiration studies, found that 11.8% were considered to be malignant lesions. False-negative diagnoses of cancer were made in 6.6-27.5% and false-positive diagnoses in only 0-2%. Currently the results of FNA are viewed as the "gold standard" for diagnosis in most cases, and play a crucial role in the selection of patients for operation. Gharib and co-workers recently analyzed data on 10,000 FNAs, and found the procedure to be the preferred first step in diagnosis (44). The diagnostic accuracy was nearly 98%, with under two percent false positives and false negatives. Miller et al.52 compared fine needle aspiration, large needle aspiration, and cutting needle biopsy. They found fine needle aspiration cytologic examination was able to detect almost all carcinomas, but believe that cutting needle biopsy is a useful additional procedure, especially in larger (over 2-3 cm) nodules. A word of caution comes from a study by Tee et al, who reviewed published studies and concluded that FNA may miss up to a third of all thyroid malignancies (44.1)
A particular problem is posed when cytology discloses a follicular proliferation or a Hurthle celll proliferation. FNA cannot differentiate follicular adenoma from follicular carcinoma, since this distinction can only be based on the presence of capsular or vascular invasion, which cannot be detected on a cytologic smear. In these cases, the histological verification of the lesion is common, even though only 10-20% of nodules with follicular histology are proven to be malignant. An additional indication for FNA is the diagnostic evaluation of extra-thyroidal neck masses, especially lymph nodes, both at presentation and when the diagnosis of thyroid carcinoma has already been established. In these cases FNA may be integrated with the measurement of thyroglobulin content in the liquid recovered after washing the needle. If the lesion is metastatic from a differentiated thyroid cancer, thyroglobulin concentrations are very high (45).
In our practice, 5-8% of aspirates are found diagnostic of malignancy, 10-20% are considered suspicious (including those with follicular proliferation) but not diagnostic, 2-5% fail to provide an adequate specimen, and the remainder are considered benign, usually suggestive of a "colloid nodule" or thyroiditis. An inadequate specimen should lead to reaspiration. In a recent review of 153 patients with nondiagnostic FNAs, 60 patients had reaspiration. Of these 38% remain nondiagnostic. Of the group that went to operation, 37% had a malignancy. The authors conclude that nondiagnostic FNAs should not be considered benign, and that reaspiration, if uninformative, should be followed by selective surgical treatment (46). Non-palpable nodules can be biopsied under ultrasound guidance. Non-palpable thyroid nodules, typically < 1 cm in size, are usually non-malignant. It is uncertain whether ultrasound guided fine needle aspiration biopsy is appropriate in these individuals. However, considering that 4% are reported to be papillary cancers when diagnosed under ultrasound guidance, probably most careful physicians will elect to do such a biopsy, if possible (47). Of course a positive diagnosis of cancer leads to surgery. We, as others, tend to operate on patients with suspicious FNA histology, since about 25% prove at surgery to be malignant. In the remainder, continued observation and replacement thyroxine therapy are offered (v.i.). Patients who are not operated are seen at 6 or 12 month intervals, and examined for any sign such as pain, growth, hoarseness, or nodes that might indicate a change in the character of the tumor. Patients are usually re-biopsied after 2-3 years and possibly again after 5-8 years to document the benign nature of the lesion. The outcome of reaspiration of benign nodular thyroid disease was investigated by Erdogan et al in studies on more than 200 patients (48). Three of 216 patients had a diagnosis changed from benign to papillary carcinoma at the time of the second biopsy. The authors conclude that a second aspiration of clinically suspicious nodules can correct some initial false negative results, but routine reaspiration was not useful in clinically stable disease. A variety of techniques have been applied to improve accuracy of interpretation of FNA cytology or histology. Staining with antibodies to TPO and TG, or to T cell antigens, is fairly routine.
GENETIC DIAGNOSIS
Thanks to great technological advances, our understanding of the genomics of thyroid cancer has dramatically improved in last years. A number of mutations have been demonstrated in differentiated thyroid cancers and some of them are considered driver mutations for a specific cancer histotypes, often configuring a genotype-phenotype relationship. The MUC1 gene, and telomerase activity, are highly expressed in carcinomas rather than in adenomas in operative specimens (49, 49a). Overexpression of cyclin D1 and underexpression of p27 predict metastatic behavior in papillary nodules (49b). Expression of c-MET, galectin, VEGF, cathepsin B, thymosin, and HMG1 has been correlated with increased probability of malignancy. Dipeptidyl aminopeptidase IV activity is almost universally present in follicular carcinomas, and usually negative in adenomas (49c).
Published studies have clearly demonstrated that the most important driver genetic events of papillary and follicular thyroid cancer, respectively, occur in the MAPK and PI3K–AKT pathways. In this view, several reports have shown the validity of using a panel of oncogenes including at least BRAF, RET/PTC, PAX8/PPAR, RAS isoforms and hTERT mutations with possible addition of TRK rearrangement (50-50c). The main effect was due to determination of BRAF V600E mutation, which is present in 50-80% of papillary cancers. However cytologic diagnosis of papillary cancer is itself very accurate and BRAF identification would primarily help in a few uncertain cases. Nikiforev et al (50) found that in each of the indeterminate categories (using the Bethesda classification), atypia of undetermined significance/follicular lesion of undetermined significance, follicular or oncocytic Hurthle cell neoplasm, and suspicious for malignant cells, addition of the mutation screening changed diagnostic accuracy from 14-54%, up to 88-95%. A more comprehensive analysis is offered by the next-generation sequencing (NGS) approach which is able to interrogate multiple genes simultaneously in a cost-effective way with high sensitivity and working with low input of starting material (50d-50h).
Diagnosis by determining expression of relevant genes using gene microarray technology logically should be informative, since expression profiles of hundreds of genes can be analyzed at one time. Studies using the methodology on samples derived from FNA are currently being reported, although none has as yet reached practical clinical utility. Durand et al analyzed the level of expression of 200 potentially informative genes in 56 thyroid tissue samples (benign or malignant tumors and paired normal tissue) using nylon microarrays. Expression patterns of a series of 19 genes allowed discrimination between follicular adenomas+normal tissue, from follicular thyroid and papillary thyroid carcinomas. The procedure is believed applicable to the material collected by FNAB, but needs testing in a prospective study (51).
Several studies (51a-51g) addressed the issue whether the search of miRNAs in cytological material can refine FNAC diagnosis specially in indeterminate lesions since miRNAs can be easily extracted from fresh FNAC sample or from residual cells left within the needle cup. Generally, all the studies concluded that a set of miRNAs seems to be more sensitive than single miRNA and show consistent results in terms of diagnostic odd ratio (mean 20.3) but inconsistent data in terms of set of miRNA proposed. Future prospective and retrospective research are recommended on a large cohort of indeterminate lesions to validate the diagnostic value of miRNA in FNAC.Basically, we still wait a proven genetic diagnostic technique that can be applied to FNA samples with near-perfect reliability and accuracy. However, screening for mutated BRAF, RAS, RET/PTC and PAX8/PPRgamma, has already successfully become a part of routine examination of thyroid nodule cytology in some clinics.
Nishino (51h ) has recently provided an excellent description and comparison of the current commercially- available methods for classifying thyroid fine-needle aspirates by molecular techniques, especially those in the category of “atypia of undetermined significance/follicular lesion of undetermined significance” (AUS/FLUS) with a 5% to 15% risk of malignancy, and the “follicular neoplasm/suspicious for follicular neoplasm” (FN/SFN) category typically associated with a 15% to 30% risk of malignancy. The Afirma Gene Expression Classifier (GEC) and Afirma BRAF use microarray technology to assess the mRNA expression profiles of cytologically indeterminate thyroid nodules.The test is designed to “rule out” malignancy with a high PPV and low NPV, and is most clearly applicable to aspirates in the FN/SFN category, and may be less useful for AUS/FLUS aspirates. ThyGenX test is designed to “rule in” a possible diagnosis of malignancy by assaying FNA sample DNA for the most common oncogenic mutations in BRAF, KRAS, HRAS, NRAS, and chromosomal translocations resulting in RET/PTC1, RET/PTC3, and PAX8/PPARc fusions. Positive assay for BRAFV600E mutations and Ret/ PTC rearrangements carry a very high PPV. ThyroSeq V2 is similarly directed to “rule in” diagnosis of a malignancy. It uses parallel “next generation sequencing” targeted multiple portions of the genome in parallel searching for gene mutations in PIK3CA, PTEN, TP53, TSHR, CTNNB1, RET, AKT1, and TERT, as well as gene fusions involving RET, BRAF, NTRK1, NTRK3, AKT, PPARc, and THADA. It is also probably most useful on samples in the FN/SFN category, and less reliable in the AUS/FLUS category. Please consult this reference for extensive data on test performance.
Guidelines published by AACE and AME (51i) in January 2016 on management of thyroid nodules offers a wealth of carefully researched information. The authors advise that Molecular Testing should be considered to complement cytologic evaluation, and is appropriate if the results are expected to change therapy.The studies should include the detection of BRAF and RET/PTC and, possibly, PAX8/PPARG and RAS mutations if such detection is available. This group also concluded that there is “insufficient evidence to recommend use of gene expression classifiers (GECs) for decisions on cytologically indeterminate nodules”, that with the exception of mutations such as BRAFV600E, molecular evidence is insufficient to determine the extent of surgery , and that patients with nodules that are negative on mutation testing should still be monitored by close follow-up. Please consult this reference for a full description of their views,
THYROID ULTRASOUND
It is accepted practice to perform ultrasound examination of all thyroid nodules as the first study, or in conjunction with FNA. Good technique demonstrates nodules if more than 3 mm in size, indicates cystic areas, may demonstrate a capsule around the nodule, and the size of the lobes. (Fig 18-2) It often displays multiple nodules when only one is noted clinically. The technique is more sensitive than scintiscanning, is noninvasive, involves less time, allows serial exams, and is usually less expensive. From 3-20% of lesions are found to be totally or partially cystic. Purely cystic lesions are reported to have a lower incidence of malignancy than solid tumors (3% versus 10%), and diagnosis of a cyst raises the possibility of aspiration therapy (52). Mixed solid and cystic lesions allegedly have a higher frequency of malignant change than either pure cysts or solid lesions. While US can not diagnose malignancy, certain features such as irregular borders of the nodule, lack of a "halo", hypo-echogenicity, evidence of calcium flakes, marginal nodules in a cyst, increased blood flow, and growth on serial ultrasounds, are suggestive signs. Schlumberger and coworkers found that cystic appearance, hyperechoic punctuations, loss of hilum, and peripheral vascularization were major ultrasound criteria of lymph node malignancy. LNs with hyperechoic punctuations are highly suspicious of malignancy. LNs with a hyperechoic hilum should be considered benign. Peripheral vascularization has the best sensitivity-specificity compromise. Round shape, hypoechogenicity, and the loss of hilum taken as single criteria are not specific enough to suspect malignancy (52.1). Ultrasound gives also valuable information on the extranodular thyroid tissue, that may be useful for the differential diagnosis: a typical pattern of diffuse hypoechogeneity is almost synonymous with autoimmune thyroiditis (53).
Figure 18-2.Ultrasooigraphic examination by transverse image of the thyroid containing a solid nodule in the left lobe and a homogeneous appearance of the right lobe. The structure peripheral to the nodule is the internal jugular vein.
A current problem is the proper way to manage thyroid nodules found incidentally on ultrasounds or CAT scans done for other purposes. As noted above, US-detectable nodules are present in a large proportion of all adults- perhaps 20 % as an average figure. If these are actually palpable, they fit into the schema just outlined. Whether to do US guided FNA on all non-palpable nodules, those generally smaller than 1cm and often as small as 3-5mm, is a question not clearly answered. Many patients request biopsy because they are anxious about the problem. Some studies have indicated that the incidence of carcinoma in these clinically non-detectable nodules is effectively the same as in larger nodules. At present our operational approach is to attempt biopsy in nodules 5-10 mm in size, and base treatment on the results. In smaller nodules or those judged very difficult to sample, the patient is advised to have repeated follow-up by exam and US at 6-12 month intervals. In a study of this problem by Papini et al 7% of nodules under 1 cm in size were found to harbor carcinomas. These authors believe that FNA is generally indicated, and especially if the nodule is solid and hypo-echogenic, has irregular margins, intranodular vascular spots, or microcalcifications.
ISOTOPE SCANS
The scintiscan received much attention in the past as an aid in the differential diagnosis of thyroid lesions (Chapter 6). The scan can provide evidence for a diagnosis in a multinodular goiter, in Hashimoto's thyroiditis, and rarely in thyroid cancer when functioning cervical metastases are seen. If the scan demonstrates a hyperfunctioning nodule suppressing the remainder of the gland, and the patient is thyrotoxic as demonstrated by an elevated serum FT4 or FT3 level, or suppressed sTSH, the chance of malignancy is very low. Tumors that accumulate RAI in a concentration equal to or greater than that of the surrounding normal thyroid tissue, but that do not produce thyrotoxicosis, are also typically benign (54, 55). In fact, some observers insist that functioning nodules cannot be malignant (55), in spite of reports of malignant change in occasional warm or hot nodules (55-59). Malignant tumors usually fail to accumulate iodide to a degree equal to that of the normal gland. However, most cold nodules turn out to be benign adenomas and cysts, not cancers. (Figure 18-3, below) The reported incidence of cancer in cold nodules is highly variable; a review of 400 cases found 10% to be cancer (60), and this experience is typical. Tumors smaller than 1 cm in size are below the discriminating power of most of the available scanning devices. Thus, a nodule 1 cm or less in diameter that fails to collect RAI (cold nodule) might not be delineated at all on the scintiscan. Further, many nodules turn out to be neither cold nor hot (preferential isotope accumulation); rather, they accumulate RAI in approximately the same concentration as the surrounding thyroid tissue. Normal tissue in front of or behind the nodule may also accumulate isotope and in this way obscure a deficit in collection within the lesion itself. For all of these reasons, it is our impression that the thyroid scintiscan has value, but except for the clearly toxic nodule, does not form an absolute predictor as to whether a palpable nodule is malignant or benign. Usually pertechnetate scanning provides the same information as RAI scanning, but exceptions occur. The first-line diagnostic procedures consist of ultrasound and TSH measurements. Thyroid scan is applied to autonomously functioning nodules and FNA to all the other nodules either cystic or solid or mixed.
| Fig. 18-3. Scintiscans of thyroid. The scan on the left is normal. A typical scan of a "cold" thyroid nodule failing to accumulate iodide isotope is shown on the right. Incidentally, a pyramidal lobe is also seen on this scan, which might suggest the presence of Hashimoto's Thyroiditis. |
Several alternative isotopes, such as selenomethionine, radioactive phosphate, gallium, and technetium-labeled bleomycin, have been introduced for scanning, but none has proven to give clear cut evidence of malignancy or to be diagnostically superior to FNA. The same is true for other imaging techniques such as thyroid thermography, CT scan or MRI. All these techniques should not be used in daily clinical practice. Interestingly, fluorodeoxyglucose-PET scanning for other purposes occasionally turns up a hot spot in the thyroid.. On examination these thyroid nodules have a high rate of malignancy (61).
While FNA clearly has reduced the initial incidence of surgery in the management of nodules, the long term effect is less clear. Over three years of follow-up, at least 30% of our patients, who are believed to have a benign nodule, eventually undergo surgery. This occurs because they, or their physicians, remain concerned about the nodule, or the nodule is painful or grows, or is cosmetically unsatisfactory. But in most cases operation probably occurs because the patient cannot be entirely reassured by the physician that the lesion is safe.
THE DECISION FOR SURGERY
All thyroid nodules once discovered should undergo a complete diagnostic evaluation, regardless of the presenting manifestation or size. This allows the selection of patients with nodules that are malignant or suspicious of malignancy and therefore eligible for surgery. In addition, surgical treatment may be needed for some benign nodules, either single or associated with multinodular goiter, when they are large or associated with signs and symptoms of compression, discomfort, or for cosmetic concerns. All the other nodules are candidates either for medical therapy or simply follow-up with no therapy. A simple and practical flow-chart for the management of thyroid nodules, based on the results of the diagnostic evaluation is offered in Fig.18-4.
What is the prognosis, with or without treatment, for these thyroid malignancies? The majority are papillary or mixed papillary-follicular tumors, with fewer pure follicular and rare solid or anaplastic carcinomas. In general, the death rate due to thyroid carcinoma, 5-6 per 10-6 persons per year in the USA in reported by the SEER program and possibly 6/million/year indicated recently, is approximately 5% of the incidence rate of 110 per million people each year (62, 63, http://seer.cancer.gov/statfacts/html/thyro.html.). (Incidence rates are 160/10-6 women and 56/10-6 men) Although it would be comforting to believe that the difference between the incidence and death rates is due to the effectiveness of surgical and medical therapy, it also reflects the remarkable benignity of many of these tumors. Some patients carry them throughout their lives and die from other causes. Although no controlled series is available, it seems obvious that some carcinomas that occur first as nodules will cause death. About all that can be stated with certainty is that some patients do die from thyroid carcinoma, and that if the surgeon removes a nodule that is really an invasive tumor before it has metastasized, or while it is still under the control of the defense mechanisms of the body, a cure is effected.
THERAPY FOR NODULES (Table 18-3,18-4) (Figure 18-4)
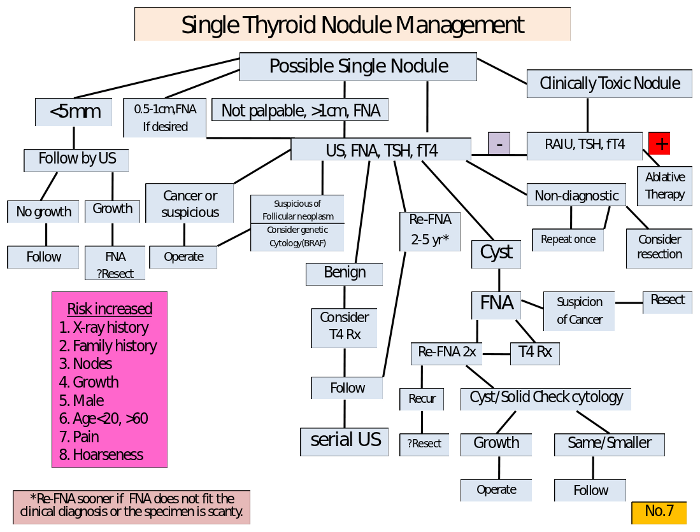 |
| Figure 18-4.Diagnostic sequence and therapeutic decisions in managing a patient with an apparent single nodule of the thyroid. |
Toxic Nodules
Three therapeutic options are available for toxic nodules: surgery, 131-I therapy and ethanol injection. Antithyroid drugs can be used if necessary prior to definitive therapy, for example during pregnancy. Radioiodine is a very effective therapy and is becoming the treatment of choice in most patients over 21 years of age and particularly in older patients and those with coincident serious illness, because of its ease and convenience, slightly lower expense, avoidance of a scar, and avoidance of hospitalization. The activity of 131-I to be administered will depend on the size of the nodule and usually ranges between 185 and 740 MBq (5-20 mCi). Euthyroidism and a variable shrinkage of the nodule are obtained in most patients. When one single dose is ineffective, the procedure may be repeated. With time, hypothyroidism may develop in up to 30-40% of the patients, since the remainder of the gland receives 1,000-8,000 rads (64). Hypothyroidism is more frequent in patients with positive anti-thyroid autoantibodies prior to therapy (59a). Further, the patient receives 30-60 rads of whole body irradiation (65). Although, in theory, this radiation could induce cancer formation, this has not been reported.
Surgery is indicated for large nodules, particularly when they have a large cystic component, in very young patients (although rare) and in those refusing radioiodine therapy. Surgery consists of a total lobectomy and must be performed after restoration of a normal thyroid function by antithyroid drugs. Also after surgery, late hypothyroidism is common (30-40% in our experience), while the occurrence of surgical complications is nearly absent in the hands of experienced surgeons.
The third option for the treatment of toxic or pre-toxic nodules, ethanol injections, has been proposed by Italian authors (66, 67). The procedure consists in percutaneous intra-nodular ethanol injection, which induces cellular dehydration followed by coagulative necrosis and vascular thrombosis and occlusion. Volumes of .4 - 2 ml are injected, and patients may receive up to 9 or more treatments at intervals of several days. The technique requires a well-trained staff. Transient, sometimes severe, local pain is the most frequent side effect, followed by transient fever, and occasionally transient dysphonia. Long term follow-up studies have shown that the rate of recurrence is limited to a few patients, and almost no patient developed hypothyroidism (68). However, in our opinion, this therapeutic option should be limited to highly selected cases, such as small nodules, well accessible to palpation, in patients at surgical risk or refusing radioiodine. Small autonomous functioning thyroid nodules, without thyrotoxicosis, can be left untreated and followed. Nearly 30-40% will eventually evolve into toxic nodules (69), but many may stay as they are or even undergo spontaneous cystic degeneration.
Cysts
Cystic lesions are aspirated and often reaspirated one or more times. Possibly long-term replacement with thyroid hormone tends to prevent recurrence, although this outcome is uncertain (70). If, after repeated aspiration, the lesion is still clearly evident, it must be considered a mixed solid/cystic lesion and probably should be resected. Cytologic examination of the aspirated fluid should be done, but the specimens are often not satisfactory for diagnosis. Some physicians attempt to sclerose cysts by aspirating fluid, and then reinjecting one-half volume of a 10/1 mixture of saline and an injectable form of tetracycline containing 100 mg/ml of the drug. Care must be taken to avoid subcutaneous leakage which is very painful. The technique is not widely used, but is reported to be effective (71). Sclerotherapy by ethanol injection is an other promising method of treatment for thyroid cysts. A major reduction in cyst volume, with very low rate of recurrence has been reported in two publications (72, 73). Large cysts (over 40 ml volume) can also be treated by ethanol injection in several sessions with > 50% reduction in size in most cases (74).
Solid nodules
Solid, mixed, functioning, or "cold" nodules constitute the remaining group and indeed the majority of cases. Here major reliance is placed on aspiration cytology. If the results are positive for carcinoma, resection is offered. If no specimen is obtained, reaspiration is performed. If the specimen is suspicious, reaspiration or resection is mandatory. Usually "suspicious" cytology and follicular proliferation leads to resection, since about 25% of nodules with this cytological picture are carcinomas at final pathology
Table 18-3. Factors to be Considered in Management of Cold Nodules
History |
Physical Examination |
Laboratory tests |
Radiation |
Size |
FTI |
Age |
Fixation |
Antithyroid antibodies |
Sex |
Cystic nature |
TG |
Duration |
Tenderness |
Chest Xray film |
Local Sysmptoms |
Adenopathy |
Fine needle aspiration |
Growth |
Diffuse/local process |
Ultrasound scan |
MEN Syndrome |
Vocal cord paralysis |
?) Isotope scan |
Thyrotoxicity |
Single vs multiple |
?) Xray soft tissues of neck |
Geographic residence |
||
Family History |
THYROXINE TREATMENT
If a benign result is obtained on cytologic examination, the patient is followed intermittently and may, or may not, be given mildly "suppressive" doses of thyroxine. There is no unanimity on the value of this treatment, and many endocrinologists do not believe thyroid therapy is useful. However solid evidence for the value (modest) of the treatment, and lack of problems, is gradually accumulating. Patients are given thyroxine in a dose adjusted to keep TSH in the minimally depressed range -- e.g. 0.1-0.3 µU/ml. (Fig. 18-5, below) It is recognized that the efficacy of this treatment to shrink nodules is modest, but it does appear to reduce the size of 10-20% of the lesions (75-79), may prevent further growth, and keeps patients under obseervation.. Meta-analyses of studies on thyroid hormone suppressive therapy for solitary thyroid nodules were presented by Castro et al (80) and Zelmanovitz et al (81). A 50% reduction in nodule volume was found in 17% more of T4-treated patients than those left untreated, and nodule volume increased more than 50% in a larger proportion of untreated patients. Nodules which are recent, small, colloid or showing degenerative changes at cytology are those more prone to respond to thyroxin treatment (82,83). An almost identical result (84) was found in a randomized, double-blind, placebo controlled study done by a group of French clinicians, who also noted that thyroxin treatment dramatically reduced the number of newly recognized nodules during follow-up. Sdano et al (83a) did a meta-analysis of 9 randomized studies, with 609 subjects.Subjects were 88% more likely to experience >50% nodule volume reduction with THST than placebo or no treatment (relative risk = 1.88; 95% CI = 1.18-3.01; P = 0.008). However studies with follow-up after thyroxine withdrawal demonstrate rapid increase in thyroid nodule and goiter volumes, and these authors did not advise routine use for benign nodules. Age is also important in the selection process. We prefer to treat young adult patient up to 60 years of age. In older patients therapy must be considered on an individual basis, after excluding other underlying chronic diseases, such as heart problems. If a patient is already on L-T4 and has a good compliance with no side effects, treatment may be continued after 60 years, slightly reducing the daily dose. In case of multinodular goiter, a condition frequently associated with intra-glandular areas of functional autonomy, particular attention should be paid to the TSH pre-treatment level. If this is already in the low-normal range, as frequently seen, it is advisable to start l-T4 at very low daily doses (25 µg/day), increasing the dose gradually according to the TSH modifications, to avoid iatrogenic thyrotoxicosis. The level of hormone replacement is chosen in the belief that it is unlikely to cause symptoms of hyperthyroidism, such as palpitations, and unlikely to cause osteoporosis, even with prolonged use, while presumably does reduce growth stimulation to the nodule.The usual l-T4 dose is 1 - 2.0 µg/Kg/day, to be administered in the morning and while fasting. The appropriate dose should be checked by measuring FT3 and TSH, 3-4 months after its institution. Normal free T3 values exclude significant over-treatment, even when FT4 is in the upper limit of normal.
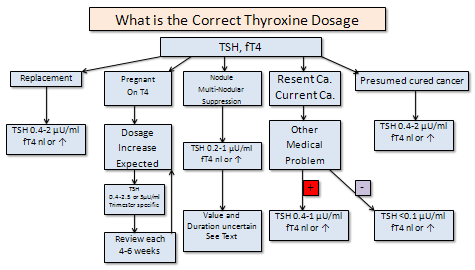 |
Fig. 18-5-Correct thyroxine treatment
It is not appropriate to maintain postmenopausal women, on doses of hormone that produce even mild hyperthyroidism for a long period, in view of the potential induction of osteoporosis and arrhythmias. However it should be noted that prolonged suppression of TSH to about 0.1uU/ml by T4 doses just above physiological in premenopausal women does not appear to induce bone loss, nor is it certain that this therapy increases fracture risk in older women (85). Serum TG is usually measured at each visit. Suppression of TG to a normal level by T4 therapy is a gratifying and reassuring response, and is correlated with nodule shrinkage (86). Progressive elevation suggests resection should be considered.
Patients receiving thyroxin therapy, or not, are followed indefinitely at 6- to 12-month intervals, and future management varies with the patient's course. Adverse factors to be noted include growth, development of local symptoms, or adenopathy. In all patients under age 25, all men, and those with a history of neck irradiation, any change usually constitutes grounds for resection. In women 25 years of age and up, the situation should be carefully reviewed and reaspiration performed. If the changes can be explained in the context of a benign process and the reaspirate is benign, continued careful medical follow-up is acceptable, but operation will often be preferred by the patient or physician. The desired course in follow-up is for a gradual reduction and disappearance of the offending lesion. Although this result occasionally is seen, most often the lump remains the same or a bit smaller and persists year after year.
Table 18 - 4. Management of Nodules
| Category | Aspiration Data | Management |
| Thyrotoxic hot nodule | Lobectomy, or 131I therapy, or (?) sclerotherapy | |
| "Mainly cystic | Aspirate for Dx and therapy; reaspirate as needed | T4 therapy; resection; (?) sclerotherapy |
| Other nodules | Probable cancer | operate |
| Inadequate specimen | reaspirate | |
| Benign | follow with/with out T4 therapy and periodic exam , US, and FNA | |
| Suspicious or hypercellular | Probably operate |
SURGICAL RESECTION
The most important requirement for surgery is the selection of an experienced surgeon in an institution with an adequate Department of Pathology. The patient is occasionally pretreated for several weeks with of thyroid hormone to suppress the normal thyroid and thereby better delineate the nodule from the normal gland. The surgeon should be prepared to do a lobectomy if the lesion is benign, or a more extensive operation and appropriate lymph node removal if, from the operative findings and examination of frozen sections, it is malignant. If the lesion is described as a hypercellular follicular adenoma, we feel it is best to do a lobectomy and contralateral subtotal lobectomy. Many of these lesions turn out on final pathology to be malignant, and reoperation is then avoided.
The first operation is the time for definitive surgery. Although a surgeon with limited experience in neck surgery can remove a thyroid nodule, an adequate near-total thyroidectomy and modified radical neck dissection requires experience, if damage to the recurrent laryngeal nerves or induction of hypoparathyroidism is to be avoided. In this day of specialization, patients deserve a surgeon who has more than a casual interest in the field. Indeed, in the absence of such surgical skill, medical therapy may offer significantly fewer problems for certain patients with nodules than those arising out of inadequate surgery.
Lobectomy for benign solitary adenomas is a relatively harmless procedure. The incidence of death, recurrent laryngeal nerve paralysis, or permanent hypoparathyrodism should be zero. It is less of a procedure than is subtotal thyroidectomy for Graves' disease. Only 1-2 days are required in the hospital, and currently some surgeons do thyroid lobectomy as an “outpatient” proceedure.. Post-operative morbidity is slight and transient, and the cosmetic appearance is almost always satisfactory. Complications increase if a carcinoma is discovered, and thyroidectomy and node dissection are required. However, this risk is more than balanced by the benefits (87), since long-term survival after surgery for intrathyroidal or locally metastatic thyroid carcinoma is almost equal to that of the "normal" population.
A satisfactory outcome often depends on a pathologist competent in thyroid histopathology. Occasionally the difficulty of interpreting frozen sections will lead to thyroidectomy for a tumor that is ultimately classified as benign. Performance of an occasional unnecessary thyroidectomy is not a serious problem in the hands of a surgeon who has few operative complications. On the other hand, reoperation at a later date for cancer erroneously diagnosed at initial surgery as benign is all too often required, and does not offer the patient the best chance of operative cure and freedom from operative complications.
After operation many thyroidologists favor treating patients with long-term replacement therapy with thyroid hormone. Although the recurrence of "solitary" nodules is infrequent, replacement therapy is safe, inexpensive, and probably provides some protection. There is no general agreement on this point. We note that thyroxin treatment has been shown to decrease recurrence of nodules in patients operated for thyroid disease induced by childhood irradiation (88).
Occasionally, when the patient is recuperating from lobectomy for a presumed benign lesion, the permanent tissue sections indicate to the pathologist that the diagnosis is actually carcinoma of the follicular or papillary type. If the lesion is over 1 cm in size, there is a history of irradiation, or the lesion is follicular, completion of the thyroidectomy is advisable, as discussed below, since up to 30% of such re-operations disclose residual tumor. This is certainly appropriate if there is nodularity in the residual thyroid on US exam. An alternative approach is to ablate the residual lobe with 30 mCi of RAI, which is effective in eliminating most of the tissue. Although easy for the patient, and certainly free of surgical complications, whether this provides the same protection as re-operation is not known.
SCLEROTHERAPY
Bennedbaek and Hegedus have carefully evaluated percutaneous ethanol injection therapy in benign solitary solid cold thyroid nodules. Ninety-eight percent ethanol (25 – 50% of nodule volume) was injected under ultrasound guidance intending to achieve uniform spread throughout the nodule. The overall reduction in nodule volume at six months was 46 – 51%. While the majority of patients benefited from reduction in nodule size, significant side effects included transient thyrotoxicosis, permanent facial dysesthesia, paranodular fibrosis, pain, and tenderness. They conclude that the optimum strategy is yet uncertain and that the procedure is limited, especially by local pain and the significant number of side effects, so that caution is advisable in routine use (89).
Dossing et al treated 78 euthyroid outpatients with a benign solitary solid and scintigraphically cold thyroid nodule causing local discomfort, with Interstitial Laser Photocoagulation (ILP). using one laser fiber under ultrasound (US) guidance and an output power of 1.5-3.5 W. The overall median nodule volume decreased from 8.2 ml (range 2.0-25.9) to 4.1 ml (range 0.6-33.0; P<0.001) at the final evaluation. After 12--96 months (median 38 months) of ILP, 21 patients (29%) had thyroid surgery because of an unsatisfactory result..US-guided ILP results in a satisfactory long-term clinical response in the majority of patients with a benign solitary solid cold thyroid nodule, but further large-scale studies are needed (89.1).
THYROID NODULES IN CHILDREN
In areas of iodide deficiency diffuse thyroid enlargement is found during pre-teen years, and multi-nodularity commonly occurs by teenage. In contrast, in an iodide-replete country, discovery of a nodule in the thyroid of a child is (fortunately) uncommon, and always raises alarm because of the risk of neoplasia. There is a +/- 5% incidence of cancer in "single nodules" in adults, but up to 18% of those found in children are malignant. This high incidence raises the issue whether all such nodules should be immediately resected, or if it is legitimate to employ the available diagnostic tests to select those for whom surgery should be advised?
Nodules in children are infrequently cystic, perhaps because cystic nodules represent an end stage in some long standing benign growths. Of course cystic lesions can be malignant. We have observed that a significant proportion of enlarging thyroid masses in children produce hyperthyroidism and turn out to be "hyperplastic colloid nodules" on pathological exam, although there are reports that up to 1/4 of "hot nodules" in children are papillary cancers. The majority are inactive on isotope scanning, solid on ultrasound, do not produce hyperthyroidism, and are painless.
If there is rapid growth, if possibly related nodes are present or if there is any other suggestion of malignancy, or if the nodule is the apparent cause of hyperthyroidism, resection is inevitable. For the remaining patients, a question is whether to do a fine needle aspiration cytological exam, or offer resection without this examination. Many thyroidologists caring for pediatric patients would suggest resection directly, but the alternative position is to do an FNA, be guided by the results, and to follow the patient closely with resection in mind if any unfavorable sign occurs (90). Our attitude is to perform FNA in virtually all cases, the reason being not only the selection of therapy, but, even if surgery is already planned, to provide the surgeon with the most likely diagnosis, thus allowing better planning of the surgical procedure. Also, this approach is taken in view of the occasional false positive or false negative diagnosis by intra-operative pathological examination. Since the natural history of such nodules is to enlarge, many will ultimately come to operation. In these patients thyroxine treatment may have value as reported by Renshaw et al(90.1). These authors found in a study of 78 euthyroid children with benign nodules, that T4 threatment reduced size up to 50%, while untreated nodules typically enlarged.
The operative approach is comparable to that used in adults. If the nodule is definitively benign at surgical pathological exam and the remainder of the thyroid is normal, lobectomy or more limited resection is performed. If the lesion is a hypercellular follicular adenoma or if there is uncertainly in the exam, or if other nodules are found, a lobectomy and contra-lateral sub-total resection is often performed. Near-total thyroidectomy with/without node resection is done for malignancy.
INCIDENTALOMAS
Wide spread use of ultrasound for exam of any neck pathology has resulted in frequent recognition of thyroid nodules that are too small to be palpated on clinical examination. Usually such nodules are < 1cm in largest diameter, typically they are asymptomatic, and are not associated with lymph nodes or other suggestions of malignancy. Often incidentally found, such nodules produce a problem because of the difficulty in achieving a specific diagnosis, which is desired by the patient.
The usual differential diagnostic possibilities, described above, are present. The probability of malignancy is lower than in larger lesions, although exactly how much so is uncertain. In a recent meta-nalysis by Tan and Gharib (91), the risk for malignancy in incidentalomas ranged betwen 0.45% and 13%.Tiny cystic lesions are especially unlikely to harbor a malignancy. Presence of neck adenopathy, local symptoms such as pain or dysphonia, growth under observation, or a history of external radiation to the neck, signal concern and suggest that resection is the proper course.
If the lesion can be palpated it is appropriate to offer FNA cytological exam and proceed as for management of larger lesions. More typically the nodule can not be precisely demarcated on exam. Ultrasound guided FNA is possible for lesions closer to 1 cm in size, and in patients who clearly want every diagnostic assurance available. The smaller lesions are difficult to aspirate with certainty even under ultrasonic guidance. (See discussion above) Considering the probable benign nature of most such lesions, the slow growth and spread of differentiated thyroid cancers, and our ability to offer close surveillance via yearly (or more frequent) ultrasound exams, a common alternative course is "observe" tiny lesions periodically and reserve resection for those that grow or produce other symptoms.
Suppressive doses of thyroxin can be offered to patients being followed medically. It is clear that shrinkage can be anticipated in only a small fraction of lesions, but this treatment may help reduce future growth, provides a reason for the patient to remain under medical observation, and may, by shrinking the normal tissue, make the nodule palpable and thus more easily examined. Growth under observation and in the absence of cytological diagnosis, or development of nodes or local symptoms, indicate the need for resection.
REFERENCES
- Hedinger, C, Dillwyn Williams, E, Sobin, L 1989. The WHO histological classification of thyroid tumors. A commentary on the second edition. Cancer 63:908-911
2.Namba, H, Matsuo, K, Fagin, J 1990 Clonal composition of benign and malignant human thyroid tumors. J Clin In 86:.
3.Parma, J, Duprez, L, Van Sande, J, et al. 1993. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651
4.Tonacchera, M, Chiovato, L, Pinchera, A, et al. 1998 Hyperfunctioning thyroid nodules in toxic multinodular goiter share activating thyrotropin receptor mutations with solitary toxic adenoma. J Clin Endocrinol Metab 83:492-8
5. Coclet, J, Foureau, F, Ketelbant, P, Galand, P, Dumont, J 1989. Cell population kinetics in dog and human adult thyroid. Clinical Endocrinol 31:655-665
6. Viglietto, G, Chiappetta, G, Martinez-Tello, F J, et al. 1995 RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 11:1207-10
7. Hamburger, J I 1980; Evolution of toxicity in solitary nontoxic autonomously functioning thyroid nodules. J Clin Endocrinol Metab 50:1089-1093
8. Silverstein, G, Burke, G, Cogan, R 1967. The natural history of the auto-nomous hyperfunctioning thyroid nodule. Ann Intern Med 67:539
9. Evered, D, Clark, F, Peterson, V 1974. Thyroid function in euthyroid subjects with autonomous thyroid nodules. Clin Endocrinol 3:149
10. Horst, W, Rosler, H, Schneider, C, Labhart, A 1967. 306 cases of toxic adenoma. Clinical aspects; findings in radioiodine diagnostics; radiochromatography and histology; results of 131I and surgical treatment. J Nucl Med 8:515
11.Parma, J, Duprez, L, Van Sande, J, et al. 1993. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649-651
12.Suarez, H, du Villard, J, Caillou, B, Schlumberger, M, Parmentier, C, Monier, M 1991. Gsp mutations in human thyroid tumors. Oncogene 6:677-679
13. see 10
13a. DeGroot, L 1970. Lack of iodide trapping in "cold" thyroid nodules. Acta Endocrinol Panam 1:27
14. Field, J, Larsen, P, Yamashita, K, Mashiter, K, Dekke, A 1973. Demonstration of iodide transport defect but normal iodide organification in nonfunctioning nodules of human thyroid glands. J Clin Invest 52:2404
15. Fragu, P, Nataf, B 1977. Human thyroid peroxidase activity in benign and malign thyroid disorders. J Clin Endocrinol Metab 45:1089
16. Demeester-Mirkine, N, Van Sande, J, Corvilain, H, Dumont, J 1975. Benign thyroid nodule with normal iodide trap and defective organification. J Clin Endocrinol Metab 41:1169
17. Burke, G, Szabo, M 1972. Dissociation of in vivo and in vitro "autonomy" in hyperfunctioning thyroid nodules. J Clin Endocrinol Metab 35:199
18. Sande Van, J, Mockel, J, Boeynaems, J, Dor, P, Andry, G, Dumont, J 1980. Regulation of cyclic nucleotide and prostaglandin formation in normal human thyroid tissue and in autonomous nodules. J Clin Endocrinol Metab 50:776
19. Smanik, P, Ryu, K-Y, Thel, K, Mazzaferri, E, Jhiang, S 1997. Expression; exon-intron organization; and chromosome mapping of the human sodium iodide symporter. Endocrinology 138:3555-8
20.Arturi, F, Russo, D, Schlumberger, M, et al. 1998. Iodine symporter gene expression in human thyroid tumors. J Clin Endocrinol Metab 83:2493-96
21. Thomas, C J, Buckwalter, J, Staab, E, Kerr, C 1976. Evaluation of dominant thyroid masses. Ann Surg 183:464
22. Sipple, J H 1984 Multiple endocrine neoplasia type 2 syndromes: historical perspectives. Henry Ford Hosp Med J 32:219-21
23.Schimke, R, Hartmann, W, Prout, T, Rimoin, D 1968. Syndrome of bilateral pheochromocytoma; medullary thyroid carcinoma; and multiple neuromas. N Engl J Med 279:1
24.Sapira, J, Altman, M, Vandyk, K, Shapiro, A 1965. Bilateral adrenal pheo-chromocytoma and medullary thyroid carcinoma. N Engl J Med 273:140
25. Duffy, B J, Fitzgerald, P 1950. Cancer of the thyroid in children. A report of twenty-eight cases. J Clin Endocrinol 10:1296
26. Clark, D 1955. Association of irradiation with cancer of the thyroid in children and adolescents. JAMA 159:1007
27. Modan, B, Ron, E, Werner, A 1977. Thyroid cancer following scalp irradiation. Therapeutic Radiology 123:741
28 DeGroot, L, Frohman, L, Kaplan, E, Refetoff, S e 1977. Radiation-Associated Thyroid Carcinoma. New York Grune & Stratton:539 pages
29. Refetoff, S, Harrison, J, Karanfilski, B, Kaplan, E, DeGroot, L, Bekerman, C 1975. Continuing occurrence of thyroid carcinoma after irradiation to the neck in infancy and childhood. N Engl J Med 292:171
30 DeGroot, L, Paloyan, E 1973. Thyroid carcinoma and radiation. A Chicago endemic. JAMA 225:487
31. Sokal, J 1959. The problem of malignancy in nodular goiter -- recapitulation and a challenge. JAMA 170:61
32. Veith, F, Brooks, J, Grigsby, W, Selenkow, H 1964. The nodular thyroid gland and cancer. N Engl J Med 270:431
32.1 Frates MC, Benson CB, Doubilet PM, Kunreuther E, Contreras M, Cibas ES, Orcutt J, Moore FD Jr, Larsen PR, Marqusee E, Alexander EK. Prevalence and distribution of carcinoma in patients with solitary and multiple thyroid nodules on sonography. J Clin Endocrinol Metab. 2006 Sep;91(9):3411-7.
33. Hoffman, G, Thompson, N, Heffron, C 1972. The solitary thyroid nodule. Arch Surg 105:379
33.1 Haymart MR, Repplinger DJ, Leverson GE, Elson DF, Sippel RS, Jaume JC, Chen H.Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage.J Clin Endocrinol Metab. 2008 Mar;93(3):809-14.
34. Pacini, F, Fontanelli, M, Fugazzola, L, et al. 1994. Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosisof unsuspected sporadic medullary thyroid carcinoma. J Clin Endocrinol Mertab 78:826-9
35. Rieu, M, Lame, M, Richard, A, et al. 1995. Prevalence of sporadic medullary thyroid carcinoma. the importance of routine measurement of serum calcitonin in the diagnostic evaluation of thyroid nodules. Clin Endocrinol (Oxf) 42:453-7
36. Niccoli, P, Wion-Barbot, N, Caron, P, et al. 1997. Interest of routine measurement of serum calcitonin. Study in a large series of thyroidectomized patients. J Clin Endocrinol Metab 82:338-341
37. Gagel, R 1997. The goitrous patient with an elevated serum calcitonin -- what to do? J Clin Endocrinol Metab 82:335
37.1 Cherenko M, Slotema E, Sebag F, De Micco C, Henry JF. Mild hypercalcitoninaemia and sporadic thyroid disease. Br J Surg 2010 May:97 (5) :684-90 - Hamlin, E, Vickery, A 1956. Needle biopsy of the thyroid gland. N Engl J Med 254:742
39. Walfish, P, Hazani, E, Strawbridge, H, Miskin, M, Rosen, B 1977. Combined ultrasound and needle aspiration cytology in the assessment and management of hypofunctioning thyroid nodule. Ann Intern Med 87:270
40. Gershengorn, M, McClung, M, Chu, W, Hanson, T, Weintraub, B, Robbins, J 1977. Fine-needle aspiration cytology in the preoperative diagnosis of thyroid nodules. Ann Intern Med 87:265
41.Baloch, Z, Sack, M, Livolsi, V, Gupta, P 1998. Fine-needle aspiration of thyroid. an institutional experience. Thyroid 8:565-9
42. Benvenga, S, Bartolone, L, Squadrito, S, Trimarchi, F 1997. Thyroid hormone autoantibodies elicited by diagnostic fine needle biopsy. J Clin Endocrinol Metab 82:4217-4223
43 Wilems, J-S, Lowhagen, T 1981. Fine needle aspiration cytology in thyroid disease. . Clin Endocrinol Metab 2:247
44 Gharib, H, Goellner, J, Johnson, D 1993. Fine needle aspiration cytology of the thyroid. A 12-year experience with 11;000 biopsies. Clin Lab Med 13:699-709
44.1 Tee YY, Lowe AJ, Brand CA, Judson RT.Fine-needle aspiration may miss a third of all malignancy in palpable thyroid nodules: a comprehensive literature review.Ann Surg. 2007 Nov;246(5):714-20.
45. Pacini, F, Fugazzola, L, Lippi, F, et al. 1992 Detection of thyroglobulin in fine needle aspirates of nonthyroidal neck masses. a clue to the diagnosis of metastatic thyroid cancer. J Clin Endocrinol Metab 74:1401-4
46. Chow, L S, Gharib, H, Goellner, J R, van Heerden, J A 2001 Nondiagnostic thyroid fine-needle aspiration cytology: management dilemmas. Thyroid 11:1147-51
47. Khurana, K, Richards, V, Chopra, P, Izquierdo, R, Rubens, D, Mesonero, C 1998. The role of ultrasonography-guided fine-needle aspiration biopsy in the management of nonpalpable and palpable thyroid nodules. Thyroid 8:511
48. Erdogan, M, Kamel, N, Aras, D, Akdogan, A, Baskal, N, Erdogan, G 1998. Value of reaspirations in benign nodular thyroid disease. Thyroid 8:1087
49. Bieche, I, Ruffet, E, Zweibaum, A, Vilde, F, Lidereau, R, Franc, B 1997. MUC1 Mucin gene; transcripts; and protein in adenomas and papillary carcinomas of the thyroid. Thyroid 7:725
49a Brousset, P, Chaouche, N, Leprat, F, et al. 1997. Telomerase activity in human thyroid carcinomas originating from the follicular cells. J Clin Endocrinol Metab 82:4214-4216
49b Khoo, M L, Beasley, N J, Ezzat, S, Freeman, J L, Asa, S L 2002 Overexpression of cyclin D1 and underexpression of p27 predict lymph node metastases in papillary thyroid carcinoma. J Clin Endocrinol Metab 87:1814-8
49c. Maruta, J; Hashimoto, H; Yamashita, H; Yamashita, H; Noguchi, S. Diagnostic applicability of dipeptidyl aminopeptidase IV activity in cytological samples for differentiating follicular thyroid carcinoma from follicular adenoma. Arch Surg 139 83-88 2004.
50. Nikiforov YE, Ohori NP, Hodak SP, Carty SE, Lebeau SO, Ferris RL, Yip L, Seethala RR, Tublin ME, Stang MT, Coyne C, Johnson JT, Stewart AF, Nikiforova MN. Impact of Mutational Testing on the Diagnosis and Management of Patients with Cytologically Indeterminate Thyroid Nodules: A Prospective Analysis of 1056 FNA Samples. J Clin Endocrinol Metab. 2011;96:3390-7.
50a. Ferraz C, Eszlinger M, Paschke R.Current state and future perspective of molecular diagnosis of fine-needle aspiration biopsy of thyroid nodules. J Clin Endocrinol Metab. 2011;96:2016-26.
50b. Nikiforov YE, Steward DL, Robinson-Smith TM, Haugen BR, Klopper JP, Zhu Z, et al. Molecular testing for mutations in improving the fine-needle aspiration diagnosis of thyroid nodules. J Clin Endocrinol Metab, 94, 2092-2098 2009.
50c. Cantara S, Capezzone M, Marchisotta S, Capuano S, Busonero G, Toti P, et al. Impact of proto-oncogene mutation detection in cytological specimens from thyroid nodules improves the diagnostic accuracy of cytology. J Clin Endocrinol Metab;95: 1365-1369, 2010.
50d. Le Mercier M, D'Haene N, De Nève N, Blanchard O, Degand C, Rorive S, Salmon I. Next-generation sequencing improves the diagnosis of thyroid FNA specimens with indeterminate cytology. Histopathology. 2015;66:215-24.
50e. Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98:E1852-60.
50f. Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, Sanford T, Buddavarapu K, Krosting J, Garmire L, Wylie D, Shinde R, Beaudenon S, Alexander EK, Mambo E, Adai AT, Latham GJ. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15:234-47.
50g. Kanagal-Shamanna R, Portier BP, Singh RR, Routbort MJ, Aldape KD, Handal BA, Rahimi H, Reddy NG, Barkoh BA, Mishra BM, Paladugu AV, Manekia JH, Kalhor N, Chowdhuri SR, Staerkel GA, Medeiros LJ, Luthra R, Patel KP. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: promises and challenges for routine clinical diagnostics. Mod Pathol. 2014;27:314-27.
50h. Nikiforov YE, Carty SE, Chiosea SI, Coyne C, Duvvuri U, Ferris RL, Gooding WE, LeBeau SO, Ohori NP, Seethala RR, Tublin ME, Yip L, Nikiforova MN. Impact of the Multi-Gene ThyroSeq Next-Generation Sequencing Assay on Cancer Diagnosis in Thyroid Nodules with Atypia of Undetermined Significance/Follicular Lesion of Undetermined Significance Cytology. Thyroid. 2015;25:1217-23.
- Durand S, Ferraro-Peyret C, Selmi-Ruby S, Paulin C, El Atifi M, Berger F, Berger-Dutrieux N, Decaussin M, Peix JL, Bournaud C, Orgiazzi J, Borson-Chazot F, Rousset B Evaluation of gene expression profiles in thyroid nodule biopsy material to diagnose thyroid cancer. J Clin Endocrinol Metab. 2008 Apr;93(4):1195-202.
51a. Mazeh H, Levy Y, Mizrahi I, Appelbaum L, Ilyayev N, Halle D, Freund HR, Nissan A. Differentiating benign from malignant thyroid nodules using micro ribonucleic acid amplification in residual cells obtained by fine needle aspiration biopsy. J Surg Res. 2013 Apr;180:216-21.
51b. Agretti P1, Ferrarini E, Rago T, Candelieri A, De Marco G, Dimida A, Niccolai F, Molinaro A, Di Coscio G, Pinchera A, Vitti P, Tonacchera M. MicroRNA expression profile helps to distinguish benign nodules from papillary thyroid carcinomas starting from cells of fine-needle aspiration. Eur J Endocrinol. 2012;167:393-400.
51c. Wei WJ, Shen CT, Song HJ, Qiu ZL, Luo QY. MicroRNAs as a potential tool in the differential diagnosis of thyroid cancer: a systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014.
51d. Kitano M, Rahbari R, Patterson EE, Steinberg SM, Prasad NB, Wang Y, Zeiger MA, Kebebew E. Evaluation of candidate diagnostic microRNAs in thyroid fine-needle aspiration biopsy samples. Thyroid. 2012;22:285-91.
51e. Keutgen XM, Filicori F, Crowley MJ, Wang Y, Scognamiglio T, Hoda R, Buitrago D, Cooper D, Zeiger MA, Zarnegar R, Elemento O, Fahey TJ 3rd. A panel of four miRNAs accurately differentiates malignant from benign indeterminate thyroid lesions on fine needle aspiration. Clin Cancer Res. 2012;18(7):2032-8.
51f. Mazeh H, Mizrahi I, Halle D, Ilyayev N, Stojadinovic A, Trink B, Mitrani-Rosenbaum S, Roistacher M, Ariel I, Eid A, Freund HR, Nissan A. Development of a microRNA-based molecular assay for the detection of papillary thyroid carcinoma in aspiration biopsy samples. Thyroid. 2011;21(2):111-8.
51g. Kitano M1, Rahbari R, Patterson EE, Steinberg SM, Prasad NB, Wang Y, Zeiger MA, Kebebew E. Evaluation of candidate diagnostic microRNAs in thyroid fine-needle aspiration biopsy samples. Thyroid. 2012;22:285-91.
51h. Nishino M Molecular cytopathology for thyroid nodules: A review of methodology and test performance. Cancer Cytopathol. 2016 Jan;124(1):14-27. doi: 10.1002/cncy.21612. Epub 2015 Sep 8.
51i. Gharib H, Papini E, Garber JR, Duick DS, Harrell RM, Hegedüs L, Paschke R, Valcavi R, Vitti P; American Association Of Clinical Endocrinologists, American College Of Endocrinology, And Associazione Medici Endocrinologi Medical Guidelines For Clinical Practice For The Diagnosis And Management Of Thyroid Nodules - 2016 UPDATE. Endocr Pract. 2016 May;22(5):622-39. doi: 10.4158/EP161208.GL.
52 Clark, O, Okerlund, M, Cavalieri, R, Greenspan, F 1979. Diagnosis and treatment of thyroid; parathyroid; and thyroglossal duct cysts. J Clin Endocrinol Metab 48:983
52.1 Leboulleux S, Girard E, Rose M, Travagli JP, Sabbah N, Caillou B, Hartl DM, Lassau N, Baudin E, Schlumberger M Ultrasound criteria of malignancy for cervical lymph nodes in patients followed up for differentiated thyroid cancer. J Clin Endocrinol Metab. 2007 Sep;92(9):3590-4
53. Marcocci, C, Vitti, P, Cetani, F, Catalano, F, Concetti, R, Pinchera, A 1991. Thyroid ultrasonography helps to identify patients with diffuse lymphocytic thyroiditis who are prone to develop hypothyroidism. J Clin Endocrinol Metab 72:209-13
54. Kendall, L, Condon, R 1969. Prediction of malignancy in solitary thyroid nodules. Lancet 1:1071
55. Miller, J, Hamburger, J 1965.. The thyroid scintigram. 1. The hot nodule. Radiology 84:66
56. Attie, J 1960. The use of radioactive iodine in the evaluation of thyroid nodules. Surgery 47:611
57. Dische, S 1964. The radioisotope scan applied to the detection of carcinoma in thyroid swellings. Cancer 17:473
58. Fujimoto, Y, Oka, A, Nagataki, S 1972. Occurrence of papillary carcinoma in hyperfunctioning thyroid nodule. Report of a case. Endocrinol Jpn 19:371
59. Scott, M, Crawford, J 1976. Solitary thyroid nodules in childhood. Is the incidence of thyroid carcinoma declining? Pediatrics 58:521
60.Messaris, G, Evangelou, G, Tountas, C 1973. Incidence of carcinoma in cold nodules of the thyroid gland. Surgery 74:447
61. Van den Bruel, A, Maes, A, De Potter, T, et al. 2002 Clinical relevance of thyroid fluorodeoxyglucose-whole body positron emission tomography incidentaloma. J Clin Endocrinol Metab 87:1517-20
62. Hakama, M Berlin; Springer-Verlag Different world thyroid cancer rates; in Hedinger CE (ed). Thyroid Cancer. International Union Against Cancer Monograph Series:Vol 12.
63. Young, J J, Percy, C, Asire, A e 1082 pages Surveillance; Epidemiology; and End Results. Incidence and Mortality Data; 1973-77. National Cancer Institute Monograph 57 NIH Publication No. 81:2330
64. Goldstein, R, Hart, I 1983. Follow-up of solitary autonomous thyroid nodules treated with 131I. New Engl J Med 309:1473
65. Gorman, C, Robertson, J 1978. Radiation dose in the selection of 131I or surgical treatment for toxic thyroid adenoma. Ann Intern Med 89:85
66. Paracchi, A, Ferrari, C, Livraghi, T, et al. 1992. Percutaneous intranodular ethanol injection. A new treatment for autonomous thyroid adenoma. J Endocrinol Invest 15:353-362
67. Lippi, F, Ferrari, C, Manetti, L, et al. 1996. Treatment of solitary autonomous thyroid nodules by percutaneous ethanol injection. results of an Italian multicenter study. The Multicenter Study Group. J Clin Endocrinol Metab 81:3261-3264
68. Monzani, F, Caraccio, N, Goletti, O, et al. S54-8 Treatment of hyperfunctioning thyroid nodules with percutaneous ethanol injection. Eight years' experience. Exp Clin Endocrinol Diabetes 106: Suppl 4
69. Hamburger, J I 1980; Evolution of toxicity in solitary nontoxic autonomously functioning thyroid nodules. J Clin Endocrinol Metab 50:1089-1093
70. McCowen, K, Reed, J, BL., F 1980. The role of thyroid therapy in patients with thyroid cysts. Amer J Med 68:853
71. Treece, G, Georgitis, W, Hofeldt, F 1983 Resolution of recurrent thyroid cysts with tetracycline instillation. Arch Int Med 143: 2285.
72. Monzani, F, Lippi, F, Goletti, O, et al. 1994. Percutaneous aspiration and ethanol sclero-therapy for thyroid cysts. J Clin Endocrinol Metab 78:800-802
73. Zingrillo, M, Torlontano, M, Ghiggi, M, et al. 1996. Percutaneous ethanol injection of large thyroid cystic nodules. Thyroid 6:403-8
74.Del Prete, S, Caraglia, M, Russo, D, et al. 2002 Percutaneous ethanol injection efficacy in the treatment of large symptomatic thyroid cystic nodules: ten-year follow-up of a large series. Thyroid 12:815-21
75. see 70
76. Papini, E, Bacci, V, Panunzi, C, et al. 1993. A prospective randomized trial of levothyroxine suppressive therapy for solitary thyroid nodules. Clin Endocrinol 38:507-513
77. Burch, H 1995. Evaluation and management of the solid thyroid nodule. Endocrinol Metab Clin N Am 24:663-710
78. Mainini, E, Martinelli, I, Morandi, G, Villa, S, Stefani, I, Mazzi, C 1995. Levothyroxine suppressive therapy for solitary thyroid nodule. J Endocrinol Invest 18:796-799
79. Cooper, D 1995. Clinical review 66. Thyroxine suppression therapy for benign nodular disease. J Clin Endocrinol Metab 80:331-334
80. Castro, M R, Caraballo, P J, Morris, J C 2002 Effectiveness of thyroid hormone suppressive therapy in benign solitary thyroid nodules: a meta-analysis. J Clin Endocrinol Metab 87:4154-9
81 Zelmanovits, F, Genro, S, Gross, J 1998. Suppressive therapy with levothyroxine for solitary thyroid nodules. A double-blind controlled clinical study and cumulative meta-analyses. J Clin Endocrinol Metab 83:3881-3885
82. Herms, A, Huysmans, D 1998. Treatment of benign nodular thyroid disease. N Engl J Med 312:601-4
83. Larosa, G, Ippolito, A, Luppo, L, et al. 1996. Cold thyroid nodule reduction with l-thyroxine can be predicted by initial nodule volume and cytological characteristics. J Clin Endocrinol Metab; 81:4385-7
83.a. Sdano MT, Falciglia M, Welge JA, Steward DL. Efficacy of thyroid hormone suppression for benign thyroid nodules: meta-analysis of randomized trials Otolaryngol Head Neck Surg. 2005 Sep;133(3):391-6
84. Wemeau, J L, Caron, P, Schvartz, C, et al. 2002 Effects of thyroid-stimulating hormone suppression with levothyroxine in reducing the volume of solitary thyroid nodules and improving extranodular nonpalpable changes: a randomized, double-blind, placebo-controlled trial by the French Thyroid Research Group. J Clin Endocrinol Metab 87:4928-34
85 see 77
86 Morita, T, Tamai, H, Ohshima, A, et al. 1989. Changes in serum thyroid hormone; thyrotropin and thyroglobulin concentrations during thyroxine therapy in patients with solitary thyroid nodules. J Clin Endocrinol Metab 69:227
87 Woolner, L, Beahrs, O, Black, B, McConahey, W, Keating, F J vol 12 Long term survival rates. in Hedinger Chr E (ed). Thyroid Cancer. International Union Against Cancer: Monograph Series
88. Fogelfeld, L, Wiviott, M, Shore-Freedman, E, et al. 1989. Recurrence of thyroid nodules after surgical removal in patients irradiated in childhood for benign conditions. N Engl J Med 320:835-840
89. Bennedbaek, F, Hegedus, L 1999. Percutaneous ethanol injection therapy in benign solitary solid cold thyroid nodules. A randomized trial comparing one injection with three injections. Thyroid 9:225-233
89.1 Døssing H, Bennedbæk FN, Hegedüs L. Long-term outcome following interstitial laser photocoagulation of benign cold thyroid nodules. Eur J Endocrinol. 2011 Jul;165(1):123-8.
90. Khurana, K, Labrador, E, Izquierdo, R, Mesonero, C, Pisharodi, L 1999. The role of fine-needle aspiration biopsy in the management of thyroid nodules in children; adolescents; and young adults. A multi-institutional study. Thyroid 9:383
91.Tan, G, Gharib, H 1997. Thyroid incidentalomas. management approaches to nonpalpable nodules discovered incidentally on thyroid imaging. Ann Intern Med 126:226-31
Cellular Uptake of Thyroid Hormones
ABSTRACT
The biological activity of thyroid hormone (TH) is regulated at the target tissue level by two important processes, i.e. deiodination and plasma membrane transport. The first process involves the expression of the deiodinase D2, which converts the prohormone T4 to bioactive T3, and/or of the deiodinase D3 which converts both T4 and T3 to inactive metabolites. Intracellular metabolism and action of TH in target cells as well as transcellular TH transport across, for instance, the blood-brain barrier (BBB) and the intestinal wall depends on the expression of transporters facilitating uptake and/or efflux of iodothyronines.
Recently, several important TH transporters have been identified, including monocarboxylate transporter 8 (MCT8), MCT10 and organic anion transporting polypeptide 1C1 (OATP1C1). The physiological relevance of MCT8 has been demonstrated in studies of male patients with the Allan-Herndon-Dudley syndrome, characterized by severe psychomotor retardation and abnormal TH levels caused by hemizygous mutations of the X-linked MCT8 gene. In human brain, MCT8 appears to be important in particular for T4 and T3 transport across the BBB and for T3 uptake in neurons, whereas OATP1C1 is predominantly involved in T4 uptake in astrocytes to allow its conversion to T3 by D2 also expressed in these cells. MCT10 also transports aromatic amino acids and its physiological role in tissue TH transport remains to be established. For complete coverage of this and related areas of Endocrinology, visit our free web-book, www.thyroidmanager.org .
INTRODUCTION
It was thought for a long time that thyroid hormone (TH) crosses the plasma membrane of tissue cells by simple diffusion since iodothyronines are lipophilic compounds which would easily pass the lipid bilayer of the plasma membrane. However, it has become increasingly clear that diffusion plays a minor role, if any, in TH transport across the plasma membrane. Rather, TH is transported into cells by specific carrier-mediated mechanisms. In the second half of the 20th century, many studies have been published about the biochemical characterization of TH transport mechanisms in a variety of cell types. In general, these studies have indicated that cellular TH transport is a saturable process which in liver cells may be Na+ dependent and in other cell types may be inhibited by aromatic and/or aliphatic amino acids. Early studies of cellular TH transport have been extensively reviewed in 2001 (1) and only some of them will be mentioned here.
About 80% of circulating T3 is produced outside the thyroid gland by peripheral conversion of T4, and only 20% is directly secreted by the thyroid gland (2). T3 is considered to be the major bioactive TH, whereas T4 is mainly a prohormone that becomes activated upon its conversion to T3 (2). Most TH actions are initiated by binding of T3 to its nuclear receptors in target cells (3,4). Therefore, the biological activity of TH is determined largely by the intracellular T3 concentration, which depends on a) the circulating concentration of T3 and its precursor T4; b) the activities of deiodinases that catalyze the production (D1, D2) or degradation (D1,D3) of T3; and c) the activities of transporters which mediate the cellular uptake or efflux of T3 and T4 (Fig. 1). It should be noted that TH bioactivity may be regulated in an autocrine fashion as shown in Fig. 1, or in a paracrine fashion, where T3 production and action take place in the same cell or in separate cells, respectively.
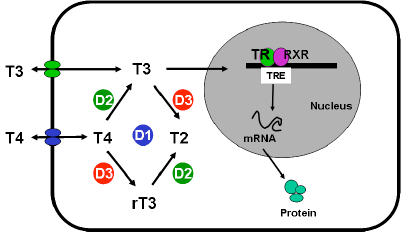
Fig. 1. Thyroid hormone transport, metabolism and action in a target cell.
Recently, three relatively specific TH transporters have been identified. OATP1C1 is a member of the organic anion transporting polypeptide family, which shows preference for T4 above T3, and is expressed almost exclusively in brain (5). In human brain, OATP1C1 is importantly expressed in astrocytes, where it facilitates entry of T4 allowing its conversion to T3 also expressed in these cells (6). MCT8 and MCT10 are members of the monocarboxylate transporter family. While MCT10 is also known to transport aromatic amino acids, no other substrates have been identified for MCT8 than iodothyronines and iodotyrosines (7-9). MCT8 and MCT10 are expressed in various tissues (10). In human brain, MCT8 is importantly expressed in endothelial cells of the blood-brain barrier (BBB) as well as in neurons (6). Mutations in the MCT8 transporter have been identified in male patients with the Allan-Herndon-Dudley syndrome, characterized by severe X-linked psychomotor retardation and elevated serum T3 levels (11-14).
THYROID HORMONE TRANSPORTERS
Organic Anion Transporters
Organic anion transporting polypeptides (OATPs) represent a large family of homologous proteins, many of which have been shown to transport different iodothyronines and their sulfate conjugates (Table 1) (15-17). The genes coding for these transporters are now referred as the SLCO family. The OATPs accept a wide range of substrates, not only anionic but also neutral and sometimes even cationic compounds. Some members are expressed in a single tissue, whereas others have a wider tissue distribution. The SLCO1A2, 1B1, 1B3 and 1C1 genes are clustered together with a related pseudogene on human chromosome 12p12 (17). The encoded OATPs have all been shown to transport iodothyronines (18). Of these, OATP1B1 and 1B3 are expressed specifically in the liver, OATP1C1 is expressed only in brain and testis, and OATP1A2 is expressed in brain, liver and kidney. In terms of TH transport, OATP1C1 is the most intriguing as it shows a high specificity and affinity towards T4 and rT3. In mouse brain, Oatp1c1 is localized both in capillary, the choroid plexus, and astrocytes, but in human brain localization of OATP1C1 in endothelial cells is negligible (5,19-22). Therefore, the primary role of OATP1C1 in human brain appears to be the transport of T4 into astrocytes to allow its conversion to T3 by D2 that is also expressed in astrocytes.
Table 1. Characteristics of human thyroid hormone transporters
| Gene | Protein |
Chr |
Tissue distribution | Ref. |
| SLC10A1 | NTCP | 14q24.1 | Liver | (26) |
| SLCO1A2 | OATP1A2 | 12p12 | Brain, kidney, liver | (18,111,112) |
| SLCO1B1 | OATP1B1 | 12p12 | Liver | (112-114) |
| SLCO1B3 | OATP1B3 | 12p12 | Liver | (18,112) |
| SLCO1C1 | OATP1C1 | 12p12 | Brain, cochlea, testis | (5,115) |
| SLCO3A1 | OATP3A1 | 15q26 | Brain, testis | (116) |
| SLCO4A1 | OATP4A1 | 20q13.33 | Multiple | (111) |
| SLCO4C1 | OATP4C1 | 5q21.2 | Kidney, other | (117) |
| SLC7A5 | LAT1 | 16q24.3 | Multiple, tumors | (41) |
| SLC7A8 | LAT2 | 14q11.2 | Multiple, tumors | (41) |
| SLC16A2 | MCT8 | Xq13.2 | Brain, liver, kidney, heart, thyroid etc | (8,9) |
| SLC10A10 | MCT10 | 16q21-q22 | Multiple. | (7) |
It should be realized that the organization of the OATP1 subfamily is very different in humans than in mice and rats (Fig. 2) (23). Although human, mouse and rat OATP1C1 are clearly orthologues, the OATP1A branch has only 1 member in humans (1A2) but 4 members in mice (1A1,4-6) and 5 members in rats (1A1,3-6), whereas the OATP1B branch has 2 members in humans (1B1,3) and one member in rats and mice (1B2). Therefore, mice and rats are not good animal models for TH transport by members of the OATP1A/B subfamily. Considering the different cellular distribution of OATP1C1 in human and mouse brain, the same precaution may also apply to this transporter.
OATPs transport their substrates in a Na-independent manner. The solute carrier family 22 (SLC22) also contains many organic anion transporters (OATs) and organic cation transporters (OCTs) (24), but information about the possible transport of TH by any of these transporters has not been published. We have demonstrated that the Na-taurocholate co-transporting polypeptide (NTCP, SLC10A1) facilitates uptake of the different iodothyronines as well as their sulfates (25,26). The SLC10 family contains 7 members of which NTCP is expressed exclusively in liver (27-29). SLC10A2 is another bile acid transporter expressed in the intestine and kidney. SLC10A6 transports different organic anions but substrates for the other SLC10 transporters have not yet been identified. NTCP is the only SLC10 family member capable of transporting TH (26).
Interestingly, NTCP has recently been identified as the receptor involved in the infection of liver cells by hepatitis B virus (HBV) and hepatitis D virus (HDV) (30). This NTCP-mediated internalization of HBV and HBD is specifically inhibited by the novel drug Myrcludex B, a synthetic peptide derived from the HBV/HBD surface protein preS1 (30). Myrcludex B also inhibits bile acid transport by NTCP (31,32) and it would be interesting to know if it affects TH transport into the liver. Clinical trials are now conducted with this drug in hepatitis B and D patients (33).
Amino Acid Transporters
Iodothyronines are a particular class of amino acids built from two tyrosine residues. Therefore, it is no surprise that amino acid transporters, in particular the L and T-type amino acid transporters, are involved in TH uptake into several tissues (34-38). L-type transporters mediate uptake of large neutral, branched-chain and aromatic amino acids, whereas T-type transporters are specific for the aromatic amino acids Phe, Tyr and Trp.
Four L-type transporters (LAT1-4) have been identified, two of which (LAT1,2) belong to the heterodimeric amino acid transporter family. These transporters consist of a heavy chain and a light chain, linked through a disulfide bond (39). There are 2 possible heavy chains, SLC3A1 (rBAT) and SLC3A2 (4F2hc or CD98), and in humans there are 13 possible light chains belonging to the SLC7 gene family. The 4F2 or CD98 cell surface antigen is expressed in many tissues, especially on activated lymphocytes and tumor cells. 4F2hc is a glycosylated protein with a single transmembrane domain, whereas the light chains are not glycosylated and have 12 transmembrane domains (40). LAT1 and LAT2 consist of the SLC3A2 heavy chain in combination with the SLC7A5 and SLC7A8 light chain, respectively. They are obligate exchangers, implying that the cellular uptake of extracellular substrates is tightly coupled to the efflux of intracellular substrates.
Significant Na-independent transport of iodothyronines has been observed in Xenopus oocytes expressing heterodimeric transporters consisting of human SLC3A2 and either human SLC7A5 (LAT1) or mouse SLC7A8 (LAT2) (Table 1) (41). The rate of iodothyronine uptake by the 4F2hc/LAT1 transporter decreased in the order 3,3’-T2 > T3 ~ rT3 > T4. Apparent Km values were found to be in the micromolar range, being lowest for T3 (1.5 µM) (41).
Ritchie et al. have reported on the stimulation of T3 transport in oocytes injected with mRNA for 4F2hc and for the IU12 Xenopus LAT1 homolog (42). They have also shown that overexpression of the heterodimeric L type transporter in cells results in increased intracellular T3 availability and, thus, augmented T3 action (43). Furthermore, they demonstrated T3 uptake via the 4F2hc/LAT1 transporter into human BeWo placental choriocarcinoma cells, suggesting that this transporter plays a role in the trans-placental transfer of maternal TH to the fetus (44). Indeed both LAT1 and LAT2 have been localized in the human placenta, in particular in cytotrophoblasts (45-48).
TH transport by LAT1 and LAT2 has recently been characterized in more detail, and a structural model of LAT2 has been obtained (49-51). LAT1 facilitates cellular uptake of all iodothyronines tested with a clear preference for 3,3’-T2 and rT3. LAT2 also shows significant cellular uptake of T3 and, in particular, 3,3’-T2. Both LAT1 and LAT2 also markedly facilitate cellular entry of 3-iodotyrosine (MIT) (51).
In addition to LAT1 and LAT2, two other L-type amino acid transporters have recently been identified in the SLC43 family. LAT3 (SLC43A1) and LAT4 (SLC43A2) are monomeric proteins containing 12 trans-membrane domains, which apparently do not require ancillary proteins for proper expression in the plasma membrane (52). Both LAT3 and LAT4 especially facilitate the cellular efflux of their substrates, including 3,3’-T2 and MIT (51). The function of the third member of this small family (SLC43A3) is unknown.
A T-type amino acid transporter (TAT1) has been cloned from rats and humans, showing transport of Phe, Tyr and Trp (53,54). This protein is a member of the monocarboxylate transporter family, and is also called MCT10 (SLC16A10). The MCT family consists of 14 members, and earned its name because MCT1-4 have been characterized as monocarboxylate transporters (55). Endogenous substrates are being recognized for other MCT family members, such as β-hydroxybutyrate for MCT7 (56), carnitine for MCT9 (57), and carnitine for MCT12 (58) The degree of homology between MCT proteins is especially high for MCT1-4 and for MCT8/10. In contrast to the initial failure to demonstrate TH transport by MCT10, we have demonstrated that both MCT8 and MCT10 are highly effective iodothyronine transporters (Table 1) (7-9).
MCT8 and MCT10
MCT8 and MCT10 have identical gene structures; both consist of 6 exons and 5 introns, with a particularly long first intron (~100 kb). The MCT10 gene is located on human chromosome 16q21-q22 and codes for a protein of 515 amino acids. The MCT8 gene is located on human chromosome Xq13.2 and has 2 possible translation start sites, coding for proteins of 613 or 539 amino acids (Fig. 2). The significance of the N-terminal extension of long versus short human MCT8 (indicated in yellow in Fig. 2) remains to be investigated. A patient has been reported with psychomotor retardation associated with a Met to Leu mutation (M1L) at the upstream translation start site. However this mutation was also identified in a healthy relative (59), suggesting that the long MCT8 protein does not have a crucial physiological function. Moreover, a recent study indicated that if the long MCT8 protein is generated it undergoes effective ubiquitination and proteasomal degradation (60). Most species, including mice and rats, only express the short MCT8 protein as they lack the first translation start site. Functional studies of human MCT8 have been carried out so far by expression of the short protein.
Both MCT8 and MCT10 proteins have 12 putative transmembrane domains, with both N- and C-terminal ends located on the inside of the plasma membrane. The common amino acids in MCT8 and MCT10 are indicated in green in Fig. 2, showing a high degree of homology in particular in the transmembrane domains. Unique to the MCT8 structure is the presence of PEST domains in the N-terminal intracellular part of the protein, rich in Pro (P), Glu (E), Ser (S) and Thr (T) residues. The function of these domains is unknown. MCT8 is expressed in many tissues, including human liver, kidney, heart, brain, placenta, adrenal gland, skeletal muscle, and thyroid. MCT10 also shows a wide tissue distribution, with particularly high expression in human skeletal muscle, intestine, kidney and pancreas (10).
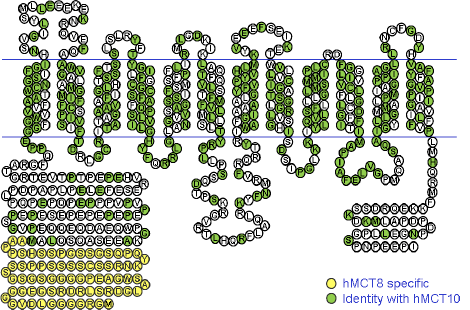
Fig. 2. Structure of the human MCT8 protein with 12 transmembrane domains. The blue lines represent the plasma membrane. The N- and C-terminal domains are located in the cytoplasm. The N-terminal extension of long vs. short MCT8 protein generated using the first or the second translation start site is indicated in yellow. The amino acid identity with human MCT10 is indicated in green.
After the cloning of MCT8 in 1994 (61), no reports on the biological function or the transported substrates have been published until Friesema et al. identified rat MCT8 as a specific TH transporter (8). Expression of rat Mct8 in Xenopus oocytes induced a ~10-fold increase in iodothyronine uptake, much greater than that induced by any other transporter, including rat NTCP, rat OATP1A1 and human LAT1 (8). Although rat MCT8 does not discriminate between T4, T3, rT3 and 3,3’-T2, it does not transport iodothyronine sulfates, the amino acids Phe, Tyr, Trp and Leu, or the monocarboxylates lactate and pyruvate. Apparent Km values amount to 2-5 µM for the different iodothyronines in the absence of protein in the medium. T4 and T3 transport are largely Na+ independent (8).
Subsequent studies in mammalian cells transfected with human MCT8 or MCT10 cDNA have demonstrated that both transporters effectively facilitate transmembrane TH transport (7,9). Co-transfection of the high-affinity cytoplasmic TH-binding protein mu-crystallin (CRYM) strongly augments the cellular accumulation of iodothyronines compared with cells transfected with MCT8 or MCT10 alone. These and other findings suggest that MCT8 and MCT10 facilitate both cellular uptake and efflux of T4 and T3. MCT10 appears to transport T3 better than MCT8 whereas the opposite is true for T4. Transfection of MCT8 or MCT10 into cells that express D1, D2 or D3 results in a marked increase in the intracellular metabolism of different iodothyronine substrates (7,9).
CLINICAL RELEVANCE OF MCT8
Worldwide, over 100 families have been reported where males are affected by severe psychomotor retardation associated with a particular combination of abnormal serum TH levels. A large family with this X-linked mental retardation (XLMR) syndrome was first reported in 1944 by Allan, Herndon and Dudley (62,63). Since then, this disorder is usually referred to as the Allan-Herndon-Dudley syndrome (AHDS). Only 60 years later it was realized that patients with AHDS also have abnormal TH levels (11,12,64).
Usually, patients with AHDS are born at term following an uncomplicated pregnancy with a normal birthweight, body length and head circumference. During the first 6 months a general hypotonia is noticed. During development the truncal hypotonia remains, whereas the distal hypotonia progresses into dystonia and spasticity. The truncal hypotonia results in poor head control. Growth is relatively normal, but final body length is reduced and body weight is usually extremely low with obvious signs of muscle wasting. There is also progressive microcephaly. In the first 2 years of life, brain MRI shows clearly delayed myelination. Although myelination improves in subsequent years, it never really normalizes. This is supported by a recent study of post-mortem brains from a fetal and a 11-year old AHDS patient (65). Based on observations of delayed myelination, AHDS has also been referred to as a Pelizaeus-Merzbacher-like disorder (PMLD) (66).
Although in some families the clinical phenotype is somewhat milder, AHDS patients are usually incapable of sitting, standing or walking independently, and do not develop any speech. They are severly mentally retarded with IQ values <40. Feeding is a problem in AHDS patients as they have difficulties swallowing; aspiration is a frequent cause of pneumonia. For a detailed description of the clinical features of patients with AHDS, the reader is referred to recent literature (66-69).
AHDS patients have a characteristic combination of abnormal serum TH levels (68). Both T4 and FT4 levels are low-normal to clearly reduced, whereas serum T3 and FT3 are markedly elevated. Serum rT3 is always low. Consequently, the serum T3/T4 and T3/rT3 ratios are strongly elevated. Serum TSH is usually within the normal range, but the mean serum TSH level in AHDS patients is about twice that in healthy controls. Serum SHBG levels are markedly elevated, and several studies have reported on elevated serum lactate levels in young patients (70,71).
In 2004, it was demonstrated by the group of Refetoff and by our group that AHDS represents a TH resistance syndrome caused by a defect in TH uptake in target cells due to mutations in MCT8. Since then, MCT8 mutations have been identified in over 100 families with AHDS. These mutations include 1) deletions affecting one or more exons, 2) frame-shift mutations resulting in scrambled and often truncated proteins, 3) splice site mutations, 4) nonsense mutations resulting in truncated proteins; 5) deletions or insertions of one or more codons and, thus, amino acids, 6) missense mutations asociated with single amino acid substitutions. A list of missense mutations is provided in Table 2.
Table 2. Missense MCT8 mutations in AHDS patients
| Exon 1 | Exon 2 | Exon 3 | Exon 4 | Exon 5 | Exon 6 |
|
c.563T>A p.I188N (118) |
c.661G>A p.G221R (64,66,119) |
c.812G>A p.R271H (72,120-124) |
c.1298T>A p. L433H (NP) |
c.1412T>C p.L471P (11,125,126) |
c.1658C>A p.A553D (NP) |
|
c.575A>G p.H192R (66) |
c.670G>A p.A224T (120,127) |
c.826G>A p.G276R (128) |
c.1301T>G p.L434W (64) |
c.1475T>C p.L492P (80) |
c.1673G>A p.G558D (59) |
|
c.575A>C p.H192P (2,129) |
c.671C>T p.A224V (11,72,118,130,131) |
c.844G>T p.G282C (132) |
c.1333C>T p.R445C (79,133) |
c.1481T>C p.L494P (65) |
c.1690G>A p.G564R (75,134) |
|
c.581C>T p.S194F (64,73) |
c.671C>A p.A224E (2) |
c.869C>T p.S290F (81,135) |
c.1333C>A p.R445S (136) |
c.1484G>C p.G495A (137) |
c.1691G>A p.G564E (NP) |
|
c.587G>A p.G196E (138) |
c.703G>A p.V235M (64,73) |
c.872T>G p.L291R (139) |
c.1358A>T p.D453V (NP) |
c.1492G>A p.D498N (140) |
c.1703T>C p.L568P (64,73) |
|
c.911T>C p.L304P (141) |
c.1535T>C p.L512P (12) |
||||
|
c.962C>T p.P321L (66,142) |
c.1610C>T p.P537L (143) |
||||
|
c.1061A>G Y354C (144) |
c.1621G>T p.G541C (145) |
||||
|
c.1163G>A p.R388Q (146) |
|||||
|
c.1201G>A p.G401R (147) |
The larger deletions, frame shift mutations and nonsense mutations are obviously deleterious for MCT8 function. The functional consequences of single amino acid substitutions, deletions or insertions have been investigated in cells transfected with wild-type or mutated MCT8. Most mutations were found to result in an amost complete loss of TH transport by MCT8. However, the extent to which these mutations affect MCT8 function depends on the type of cell used for transfection for reasons which need to be fully explored (72-75). Studies of the localization of wild-type and mutant MCT8 protein have indicated two distinct pathogenic mechanisms, in that certain mutations interfere with the trafficking of the transporter to the plasma membrane, while other mutations allow proper routing of MCT8 but interfere with the substrate translocation process (73,76,77). The functional consequences of MCT8 mutations have also been demonstrated using fibroblasts cultured from skin biopsies, showing that T4 and T3 uptake by cells from AHDS patients is markedly reduced compared with cells cultured from healthy controls (75,78-82).
ANIMAL STUDIES
Studies in humans and animals have indicated that MCT8 is expressed in a variety of tissues, including brain. The distribution of MCT8 expression in mouse brain has been studied in detail by Heuer et al (83). These studies have demonstrated that MCT8 is predominantly expressed in neurons in different brain areas, including hippocampus, cerebral cortex, striatum, hypothalamus and cerebellum. In addition, MCT8 is importantly expressed in capillary endothelial cells, the choroid plexus, and tanycytes which line the third ventricle (83,84). MCT8 expression in neurons coincides with expression of D3. D2 is largely expressed in adjacent astrocytes. In mouse brain, the T4 transporter OATP1C1 is expressed in capillaries, in the choroid plexus and in astrocytes (83-86). However, in primate brain localization of OATP1C1 in capillaries appears to be negligible (22,87).
MCT8 (KO) mice have been studied in the laboratories of Heuer (88-90), Refetoff (91-94), Bernal (95-97) and Schweizer (98-100). In contrast to the severe neurological phenotype in male patients with MCT8 mutations, neither hemizygous MCT8 KO male mice nor homozygous MCT8 KO female mice show an obvious phenotype. However, they show the same abnormal serum thyroid parameters as patients with MCT8 mutations, i.e. a large decrease in T4, a large increase in serum T3, and slightly elevated TSH levels. In addition, MCT8 KO mice show the following features: 1) normal brain T4 uptake but impaired brain T3 uptake, 2) decreased brain T4 and T3 contents, 3) increased D2 and decreased D3 activities in brain, 4) normal liver T4 and T3 uptake, 5) increased kidney T4 and T3 uptake, 6) increased kidney T4 and T3 contents, and 7) increased D1 activity in both liver and kidney (90).
The paradoxical increase in renal T4 and T3 uptake in MCT8 KO mice is unexplained, but the increase in renal T4 content in combination with the increased D1 expression may account for an enhanced renal T4 to T3 conversion, and thus contribute to the decrease in serum T4 and increase in serum T3 (90). In addition, there is evidence suggesting that thyroidal hormone secretion is affected by MCT8 inactivation, perhaps leading to preferential T3 secretion (89,92). Since MCT8 is expressed in the hypothalamus, inactivation of MCT8 is associated with an impaired feedback of TH at the hypothalamic level, contributing to the slightly increased serum TSH level (88,101).
In addition to MCT8 KO mice, a number of other interesting mouse models have been generated which in addition to MCT8 are also deficient in other TH-related genes. Liao et al (102) have studied the effects of the deletion of D1 and/or D2 on serum and brain TH levels in MCT8 KO and WT mice. The results indicate that D1 plays an important role in the altered TH homeostasis in MCT8 KO mice, probably involving and increased T4 to T3 conversion in the thyroid and the kidneys (89,90,92,103). In a recent study, the effect of MCT8 deletion was investigated in D3 deficient mice (104). Inactivation of D3 in mice is associated with marked morbidity and mortality, which is largely prevented by deletion of MCT8. The mechanism by which MCT8 deletion improves the phenotype of D3 deficient mice is unknown.
Of special interest are studies using mice which in addition to MCT8 are also deficient in other TH transporters, such as MCT8/MCT10 (105) and MCT8/LAT2 (106) double knockout (DKO) mice. The additional deficiency of LAT2 or MCT10 results in interesting changes in TH levels compared with MCT8 only KO mice, although deletion of MCT10 or LAT2 alone have little effect on TH homeostasis (105-107). Perhaps most interesting are the findings obtained in MCT8/OATP1C1 DKO mice (21). In contrast to the only mild reduction in brain T3 levels and the lack of an obvious phenotype in mice deficient in MCT8 alone or OATP1C1 alone (108), MCT8/OATP1C1 DKO mice show a dramatic decrease in brain T3 content associated with a markedly impaired neurodevelopment (21). These findings suggest overlapping activities of MCT8 and OATP1C1 in TH transport in the brain, as they are both capable of transporting T4 across the BBB. Apparently, development of the human brain is more vulnerable to mutations in MCT8, since OATP1C1 is not significantly expressed in the human BBB and thus cannot compensate for the loss of MCT8.
Figure 3 shows a schematic of the regulation of T3 supply to neuronal target cells, based largely on studies by Heuer et al (21) and Bernal et al (109). The steps involved in this process include 1) TH transport across the BBB by both OATP1C1 and MCT8 in mice and by MCT8 alone in humans, 2) uptake of T4 in astrocytes by OATP1C1, 3) conversion of T4 to T3 by D2 in astrocytes, 4) release of T3 from the astrocytes by an unidentified transporter, 5) uptake of T3 in neurons by MCT8. These neurons may also express D3 for termination of T3 activity. MCT8 may also be involved in T3 uptake by oligodendrocytes, but this remains to be established. This schema is an oversimplification as, for instance, it ignores the importance of TH transport across the blood-CSF barrier by MCT8 and OATP1C1.
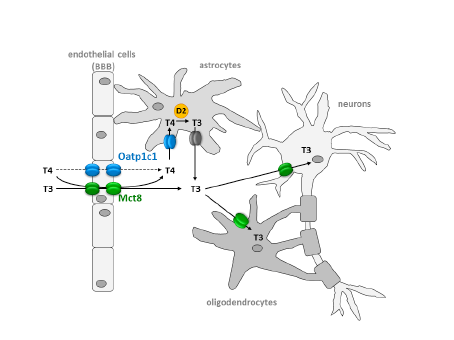
Fig. 3. Schematic of steps involved in the supply of bioactive T3 to target cells in the brain. In contrast to the mouse brain, OATP1C1 does not seem to play an important role in T4 transport across the blood-brain barrier. (Courtesy of Drs. Steffen Mayerl and Heike Heuer).
PATHOGENESIS OF AHDS BY MCT8 MUTATIONS
TH plays an essential role in brain development. This requires optimal spatio-temporal regulation of T3 supply to brain target cells, in particular neurons. MCT8 is supposed to be crucial for T4 and T3 transport across the BBB and may also play an important role in T3 uptake by neurons. Inactivation of MCT8 results in an impaired development of the central nervous system and thus in severe psychomotor retardation. It is also possibe that MCT8 is more important for T3 uptake in certain subsets of neurons than for others. This may result in a dysbalance of T3 supply to different neuronal populations and thus in a defect in the coordinated development of neuronal networks in the brain.
In addition to the TH dysregulation in the brain, the effects of MCT8 mutations on the thyroid state of peripheral tissues should also be considered. Usually, the heart appears to function normally in MCT8 patients despite exposure to highly elevated serum T3 levels. This suggest a partially impaired cardiac T3 upake in case of a MCT8 mutation, implying the involvement of additional TH transporters in the heart. MCT8 patients show extensive muscle wasting and increased serum SHBG levels, which likely reflect a hyperthyroid state of the skeletal muscles and liver, respectively (hepatic SHBG production is increased by TH). This would indicate that MCT8 inactivation does not impair muscle and liver T3 uptake, suggesting a more important role of other transporters. Finally, findings in MCT8 KO mice suggest that the kidneys are also in a hyperthyroid state in MCT8 patients, but there is no direct evidence for this assumption.
CONCLUSIONS AND PERSPECTIVES
Much progress has been made in recent years with the identification of TH transporters and their role in the tissue-specific regulation of TH bioactivity in health and disease. However, it is likely that important TH transporters still remain to be discovered. For instance, none of the TH transporters characterized recently at the molecular level have the properties of transporters involved in TH uptake in liver cells, such as nanomolar affinities, ATP and Na+ dependence, as determined in previous studies (1). Further, the physiological relevance of OATP1C1 and MCT10 need to be demonstrated. Although it has been demonstrated that mutations in MCT8 cause severe psychomotor retardation, the pathogenic mechanism has not been established. For this it is essential to know exactly where MCT8 is expressed in the human brain and other tissues. A beginning has been made with the treatment of AHDS patients with T3 analogues which do not require MCT8 for cellular uptake, such as DITPA (110) and Triac (https://clinicaltrials.gov/ct2/show/NCT02060474), but further work is needed to develop an optimal therapy for these patients.
REFERENCES
- Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 2001; 22:451-476
- Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev 2008; 29:898-938
- Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev 2001; 81:1097-1142
- Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev 2014; 94:355-382
- Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol 2002; 16:2283-2296
- Bernal J. Thyroid hormones in brain development and function. 2015;
- Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol 2008; 22:1357-1369
- Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 2003; 278:40128-40135
- Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 2006; 20:2761-2772
- Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet 2008; 23:22-44
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004; 364:1435-1437
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 2004; 74:168-175
- Dumitrescu AM, Refetoff S. Impaired Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism and Action. 2000;
- Bernal J. Thyroid Hormones in Brain Development and Function. 2000;
- Hagenbuch B. Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab 2007; 21:209-221
- Suzuki T, Abe T. Thyroid hormone transporters in the brain. Cerebellum 2008; 7:75-83
- Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch 2004; 447:653-665
- van der Deure WM, Peeters RP, Visser TJ. Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters. J Mol Endocrinol 2010; 44:1-11
- Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, Sugiyama Y. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem 2003; 278:43489-43495
- Tohyama K, Kusuhara H, Sugiyama Y. Involvement of multispecific organic anion transporter, Oatp14 (Slc21a14), in the transport of thyroxine across the blood-brain barrier. Endocrinology 2004;
- Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, Buder K, Boelen A, Visser TJ, Heuer H. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest 2014; 124:1987-1999
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008; 149:6251-6261
- Hagenbuch B, Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med 2013; 34:396-412
- Koepsell H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 2013; 34:413-435
- Friesema EC, Docter R, Moerings EP, Stieger B, Hagenbuch B, Meier PJ, Krenning EP, Hennemann G, Visser TJ. Identification of thyroid hormone transporters. Biochem Biophys Res Commun 1999; 254:497-501
- Visser WE, Wong WS, van Mullem AA, Friesema EC, Geyer J, Visser TJ. Study of the transport of thyroid hormone by transporters of the SLC10 family. Mol Cell Endocrinol 2010; 315:138-145
- Doring B, Lutteke T, Geyer J, Petzinger E. The SLC10 carrier family: transport functions and molecular structure. Curr Top Membr 2012; 70:105-168
- Claro da Silva T, Polli JE, Swaan PW. The solute carrier family 10 (SLC10): beyond bile acid transport. Mol Aspects Med 2013; 34:252-269
- Anwer MS, Stieger B. Sodium-dependent bile salt transporters of the SLC10A transporter family: more than solute transporters. Pflugers Arch 2014; 466:77-89
- Li W, Urban S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J Hepatol 2016; 64:S32-40
- Haag M, Hofmann U, Murdter TE, Heinkele G, Leuthold P, Blank A, Haefeli WE, Alexandrov A, Urban S, Schwab M. Quantitative bile acid profiling by liquid chromatography quadrupole time-of-flight mass spectrometry: monitoring hepatitis B therapy by a novel Na(+)-taurocholate cotransporting polypeptide inhibitor. Anal Bioanal Chem 2015; 407:6815-6825
- Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, de Waart DR, Elferink RP, Mier W, Stieger B, Beuers U, Urban S, van de Graaf SF. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology 2015; 62:207-219
- Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, Lehr T, Lempp FA, Wedemeyer H, Haag M, Schwab M, Haefeli WE, Blank A, Urban S. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B - first results of a Phase Ib/IIa study. J Hepatol 2016;
- Lakshmanan M, Goncalves E, Lessly G, Foti D, Robbins J. The transport of thyroxine into mouse neuroblastoma cells, NB41A3: the effect of L-system amino acids. Endocrinology 1990; 126:3245-3250
- Blondeau JP, Beslin A, Chantoux F, Francon J. Triiodothyronine is a high-affinity inhibitor of amino acid transport system L1 in cultured astrocytes. J Neurochem 1993; 60:1407-1413
- Zhou Y, Samson M, Osty J, Francon J, Blondeau JP. Evidence for a close link between the thyroid hormone transport system and the aromatic amino acid transport system T in erythrocytes. J Biol Chem 1990; 265:17000-17004
- Zhou Y, Samson M, Francon J, Blondeau JP. Thyroid hormone concentrative uptake in rat erythrocytes. Involvement of the tryptophan transport system T in countertransport of tri-iodothyronine and aromatic amino acids. Biochem J 1992; 281 ( Pt 1):81-86
- Yan Z, Hinkle PM. Saturable, stereospecific transport of 3,5,3'-triiodo-L-thyronine and L-thyroxine into GH4C1 pituitary cells. J Biol Chem 1993; 268:20179-20184
- Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 2013; 34:139-158
- Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch 2004; 447:532-542
- Friesema EC, Docter R, Moerings EP, Verrey F, Krenning EP, Hennemann G, Visser TJ. Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology 2001; 142:4339-4348
- Ritchie JW, Peter GJ, Shi YB, Taylor PM. Thyroid hormone transport by 4F2hc-IU12 heterodimers expressed in Xenopus oocytes. J Endocrinol 1999; 163:R5-9
- Ritchie JW, Shi YB, Hayashi Y, Baird FE, Muchekehu RW, Christie GR, Taylor PM. A role for thyroid hormone transporters in transcriptional regulation by thyroid hormone receptors. Mol Endocrinol 2003; 17:653-661
- Ritchie JW, Taylor PM. Role of the System L permease LAT1 in amino acid and iodothyronine transport in placenta. Biochem J 2001; 356:719-725
- Loubiere LS, Vasilopoulou E, Bulmer JN, Taylor PM, Stieger B, Verrey F, McCabe CJ, Franklyn JA, Kilby MD, Chan SY. Expression of thyroid hormone transporters in the human placenta and changes associated with intrauterine growth restriction. Placenta 2010; 31:295-304
- Loubiere LS, Vasilopoulou E, Glazier JD, Taylor PM, Franklyn JA, Kilby MD, Chan SY. Expression and function of thyroid hormone transporters in the microvillous plasma membrane of human term placental syncytiotrophoblast. Endocrinology 2012; 153:6126-6135
- Vasilopoulou E, Loubiere LS, Lash GE, Ohizua O, McCabe CJ, Franklyn JA, Kilby MD, Chan SY. Triiodothyronine regulates angiogenic growth factor and cytokine secretion by isolated human decidual cells in a cell-type specific and gestational age-dependent manner. Hum Reprod 2014; 29:1161-1172
- Vasilopoulou E, Loubiere LS, Martin-Santos A, McCabe CJ, Franklyn JA, Kilby MD, Chan SY. Differential triiodothyronine responsiveness and transport by human cytotrophoblasts from normal and growth-restricted pregnancies. J Clin Endocrinol Metab 2010; 95:4762-4770
- Hinz KM, Meyer K, Kinne A, Schulein R, Kohrle J, Krause G. Structural insights into thyroid hormone transport mechanisms of the L-type amino acid transporter 2. Mol Endocrinol 2015; 29:933-942
- Kinne A, Wittner M, Wirth EK, Hinz KM, Schulein R, Kohrle J, Krause G. Involvement of the L-Type Amino Acid Transporter Lat2 in the Transport of 3,3'-Diiodothyronine across the Plasma Membrane. Eur Thyroid J 2015; 4:42-50
- Zevenbergen C, Meima ME, Lima de Souza EC, Peeters RP, Kinne A, Krause G, Visser WE, Visser TJ. Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology 2015; 156:4345-4355
- Bodoy S, Fotiadis D, Stoeger C, Kanai Y, Palacin M. The small SLC43 family: facilitator system l amino acid transporters and the orphan EEG1. Mol Aspects Med 2013; 34:638-645
- Kim DK, Kanai Y, Chairoungdua A, Matsuo H, Cha SH, Endou H. Expression cloning of a Na+-independent aromatic amino acid transporter with structural similarity to H+/monocarboxylate transporters. J Biol Chem 2001; 276:17221-17228
- Kim do K, Kanai Y, Matsuo H, Kim JY, Chairoungdua A, Kobayashi Y, Enomoto A, Cha SH, Goya T, Endou H. The human T-type amino acid transporter-1: characterization, gene organization, and chromosomal location. Genomics 2002; 79:95-103
- Halestrap AP. The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med 2013; 34:337-349
- Hugo SE, Cruz-Garcia L, Karanth S, Anderson RM, Stainier DY, Schlegel A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev 2012; 26:282-293
- Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, Altmaier E, CardioGram, Deloukas P, Erdmann J, Grundberg E, Hammond CJ, de Angelis MH, Kastenmuller G, Kottgen A, Kronenberg F, Mangino M, Meisinger C, Meitinger T, Mewes HW, Milburn MV, Prehn C, Raffler J, Ried JS, Romisch-Margl W, Samani NJ, Small KS, Wichmann HE, Zhai G, Illig T, Spector TD, Adamski J, Soranzo N, Gieger C. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011; 477:54-60
- Abplanalp J, Laczko E, Philp NJ, Neidhardt J, Zuercher J, Braun P, Schorderet DF, Munier FL, Verrey F, Berger W, Camargo SM, Kloeckener-Gruissem B. The cataract and glucosuria associated monocarboxylate transporter MCT12 is a new creatine transporter. Hum Mol Genet 2013; 22:3218-3226
- Frints SG, Lenzner S, Bauters M, Jensen LR, Van Esch H, des Portes V, Moog U, Macville MV, van Roozendaal K, Schrander-Stumpel CT, Tzschach A, Marynen P, Fryns JP, Hamel B, van Bokhoven H, Chelly J, Beldjord C, Turner G, Gecz J, Moraine C, Raynaud M, Ropers HH, Froyen G, Kuss AW. MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet 2008; 16:1029-1037
- Zwanziger D, Schmidt M, Fischer J, Kleinau G, Braun D, Schweizer U, Moeller LC, Biebermann H, Fuehrer D. The long N-terminus of the human monocarboxylate transporter 8 is a target of ubiquitin-dependent proteasomal degradation which regulates protein expression and oligomerization capacity. Mol Cell Endocrinol 2016;
- Lafreniere RG, Carrel L, Willard HF. A novel transmembrane transporter encoded by the XPCT gene in Xq13.2. Hum Mol Genet 1994; 3:1133-1139
- Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Mental Defic 1944; 48:325-334
- Schwartz CE, Ulmer J, Brown A, Pancoast I, Goodman HO, Stevenson RE. Allan-Herndon syndrome. II. Linkage to DNA markers in Xq21. Am J Hum Genet 1990; 47:454-458
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 2005; 77:41-53
- Lopez-Espindola D, Morales-Bastos C, Grijota-Martinez C, Liao XH, Lev D, Sugo E, Verge CF, Refetoff S, Bernal J, Guadano-Ferraz A. Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab 2014; 99:E2799-2804
- Vaurs-Barriere C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Vinas JM, De Michele G, Dan B, Brady AF, Boespflug-Tanguy O, Touraine R. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol 2009; 65:114-118
- Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol 2005; 252:663-666
- Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab 2006; 2:512-523
- Holden KR, Zuniga OF, May MM, Su H, Molinero MR, Rogers RC, Schwartz CE. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol 2005; 20:852-857
- Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, Ramirez P, Iorcansky S, Rivarola MA, Belgorosky A. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res 2007; 67:1-6
- Namba N, Etani Y, Kitaoka T, Nakamoto Y, Nakacho M, Bessho K, Miyoshi Y, Mushiake S, Mohri I, Arai H, Taniike M, Ozono K. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr 2008; 167:785-791
- Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Gruters A, Barrett TG, Mancilla EE, Svensson J, Wemeau JL, Busi da Silva Canalli MH, Lundgren J, McEntagart ME, Hopper N, Arts WF, Visser TJ. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab 2007; 92:2378-2381
- Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology 2008; 149:2184-2190
- Kinne A, Roth S, Biebermann H, Koehrle J, Gruters A, Schweizer U. Surface translocation and T3 uptake of mutant MCT8 proteins are cell type-dependent. Journal of molecular endocrinology 2009; 43:263-271
- Visser WE, Jansen J, Friesema EC, Kester MH, Mancilla E, Lundgren J, van der Knaap MS, Lunsing RJ, Brouwer OF, Visser TJ. Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat 2009; 30:29-38
- Kersseboom S, Kremers GJ, Friesema EC, Visser WE, Klootwijk W, Peeters RP, Visser TJ. Mutations in MCT8 in patients with Allan-Herndon-Dudley-syndrome affecting its cellular distribution. Mol Endocrinol 2013; 27:801-813
- Kinne A, Roth S, Biebermann H, Kohrle J, Gruters A, Schweizer U. Surface translocation and tri-iodothyronine uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol 2009; 43:263-271
- Visser WE, Swagemakers SM, Ozgur Z, Schot R, Verheijen FW, van Ijcken WF, van der Spek PJ, Visser TJ. Transcriptional profiling of fibroblasts from patients with mutations in MCT8 and comparative analysis with the human brain transcriptome. Hum Mol Genet 2010; 19:4189-4200
- Capri Y, Friesema EC, Kersseboom S, Touraine R, Monnier A, Eymard-Pierre E, Des Portes V, De Michele G, Brady AF, Boespflug-Tanguy O, Visser TJ, Vaurs-Barriere C. Relevance of different cellular models in determining the effects of mutations on SLC16A2/MCT8 thyroid hormone transporter function and genotype-phenotype correlation. Hum Mutat 2013; 34:1018-1025
- Visser WE, Vrijmoeth P, Visser FE, Arts WF, van Toor H, Visser TJ. Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin Endocrinol (Oxf) 2013; 78:310-315
- Armour CM, Kersseboom S, Yoon G, Visser TJ. Further insights into the Allan-Herndon-Dudley Syndrome: Clinical and functional characterization of a novel MCT8 mutation. PLOS ONE 2015;
- Kersseboom S, Horn S, Visser WE, Chen J, Friesema EC, Vaurs-Barriere C, Peeters RP, Heuer H, Visser TJ. In vitro and mouse studies support therapeutic utility of triiodothyroacetic acid in MCT8 deficiency. Mol Endocrinol 2015:me00009999
- Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 2005; 146:1701-1706
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N. Expression of the thyroid hormone transporters MCT8 (SLC16A2) and OATP14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008; 149:6251-6261
- Schnell C, Shahmoradi A, Wichert SP, Mayerl S, Hagos Y, Heuer H, Rossner MJ, Hulsmann S. The multispecific thyroid hormone transporter OATP1C1 mediates cell-specific sulforhodamine 101-labeling of hippocampal astrocytes. Brain Struct Funct 2015; 220:193-203
- Wittmann G, Szabon J, Mohacsik P, Nouriel SS, Gereben B, Fekete C, Lechan RM. Parallel regulation of thyroid hormone transporters OATP1c1 and MCT8 during and after endotoxemia at the blood-brain barrier of male rodents. Endocrinology 2015; 156:1552-1564
- Ito K, Uchida Y, Ohtsuki S, Aizawa S, Kawakami H, Katsukura Y, Kamiie J, Terasaki T. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci 2011; 100:3939-3950
- Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 2007; 117:627-635
- Trajkovic-Arsic M, Muller J, Darras VM, Groba C, Lee S, Weih D, Bauer K, Visser TJ, Heuer H. Impact of monocarboxylate transporter-8 deficiency on the hypothalamus-pituitary-thyroid axis in mice. Endocrinology 2010; 151:5053-5062
- Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, Mittag J, Bauer K, Heuer H. Consequences of monocarboxylate transporter 8 deficiency for renal transport and metabolism of thyroid hormones in mice. Endocrinology 2010; 151:802-809
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 2006; 147:4036-4043
- Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S. Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest 2010; 120:3377-3388
- Di Cosmo C, Liao XH, Ye H, Ferrara AM, Weiss RE, Refetoff S, Dumitrescu AM. Mct8-deficient mice have increased energy expenditure and reduced fat mass that is abrogated by normalization of serum T3 levels. Endocrinology 2013; 154:4885-4895
- Ferrara AM, Liao XH, Gil-Ibanez P, Marcinkowski T, Bernal J, Weiss RE, Dumitrescu AM, Refetoff S. Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology 2013; 154:2533-2541
- Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, Morte B, Bernal J. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3'-triiodo-L-thyronine. Endocrinology 2009; 150:2491-2496
- Morte B, Ceballos A, Diez D, Grijota-Martinez C, Dumitrescu AM, Di Cosmo C, Galton VA, Refetoff S, Bernal J. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology 2010; 151:2381-2387
- Rodrigues TB, Ceballos A, Grijota-Martinez C, Nunez B, Refetoff S, Cerdan S, Morte B, Bernal J. Increased oxidative metabolism and neurotransmitter cycling in the brain of mice lacking the thyroid hormone transporter SLC16A2 (MCT8). PLoS One 2013; 8:e74621
- Wirth EK, Roth S, Blechschmidt C, Holter SM, Becker L, Racz I, Zimmer A, Klopstock T, Gailus-Durner V, Fuchs H, Wurst W, Naumann T, Brauer A, de Angelis MH, Kohrle J, Gruters A, Schweizer U. Neuronal 3',3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci 2009; 29:9439-9449
- Braun D, Kinne A, Brauer AU, Sapin R, Klein MO, Kohrle J, Wirth EK, Schweizer U. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia 2011; 59:463-471
- Wirth EK, Sheu SY, Chiu-Ugalde J, Sapin R, Klein MO, Mossbrugger I, Quintanilla-Martinez L, de Angelis MH, Krude H, Riebel T, Rothe K, Kohrle J, Schmid KW, Schweizer U, Gruters A. Monocarboxylate transporter 8 deficiency: altered thyroid morphology and persistent high triiodothyronine/thyroxine ratio after thyroidectomy. Eur J Endocrinol 2011; 165:555-561
- Alkemade A, Friesema EC, Kalsbeek A, Swaab DF, Visser TJ, Fliers E. Expression of thyroid hormone transporters in the human hypothalamus. J Clin Endocrinol Metab 2011; 96:E967-971
- Liao XH, Di Cosmo C, Dumitrescu AM, Hernandez A, Van Sande J, St Germain DL, Weiss RE, Galton VA, Refetoff S. Distinct roles of deiodinases on the phenotype of Mct8 defect: a comparison of eight different mouse genotypes. Endocrinology 2011; 152:1180-1191
- Wirth EK, Rijntjes E, Meyer F, Kohrle J, Schweizer U. High T3, Low T4 Serum Levels in Mct8 Deficiency Are Not Caused by Increased Hepatic Conversion through Type I Deiodinase. Eur Thyroid J 2015; 4:87-91
- Patrizia Stohn J, Elena Martinez M, Matoin K, Morte B, Bernal J, Anne Galton V, St Germain D, Hernandez A. MCT8 Deficiency in Male Mice Mitigates the Phenotypic Abnormalities Associated with the Absence of a Functional Type 3 Deiodinase. Endocrinology 2016:en20161162
- Muller J, Mayerl S, Visser TJ, Darras VM, Boelen A, Frappart L, Mariotta L, Verrey F, Heuer H. Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology 2014; 155:315-325
- Nunez B, Martinez de Mena R, Obregon MJ, Font-Llitjos M, Nunes V, Palacin M, Dumitrescu AM, Morte B, Bernal J. Cerebral cortex hyperthyroidism of newborn mct8-deficient mice transiently suppressed by lat2 inactivation. PLoS One 2014; 9:e96915
- Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, Kohrle J, Schweizer U. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J 2011; 439:249-255
- Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology 2012; 153:1528-1537
- Bernal J, Guadano-Ferraz A, Morte B. Thyroid hormone transporters--functions and clinical implications. Nat Rev Endocrinol 2015; 11:406-417
- Verge CF, Konrad D, Cohen M, Di Cosmo C, Dumitrescu AM, Marcinkowski T, Hameed S, Hamilton J, Weiss RE, Refetoff S. Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J Clin Endocrinol Metab 2012; 97:4515-4523
- Fujiwara K, Adachi H, Nishio T, Unno M, Tokui T, Okabe M, Onogawa T, Suzuki T, Asano N, Tanemoto M, Seki M, Shiiba K, Suzuki M, Kondo Y, Nunoki K, Shimosegawa T, Iinuma K, Ito S, Matsuno S, Abe T. Identification of thyroid hormone transporters in humans: different molecules are involved in a tissue-specific manner. Endocrinology 2001; 142:2005-2012
- Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, Fattinger K, Meier PJ, Hagenbuch B. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology 2001; 120:525-533
- Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, Nomura H, Unno M, Suzuki M, Naitoh T, Matsuno S, Yawo H. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem 1999; 274:17159-17163
- van der Deure WM, Friesema EC, de Jong FJ, de Rijke YB, de Jong FH, Uitterlinden AG, Breteler MM, Peeters RP, Visser TJ. Organic anion transporter 1B1: an important factor in hepatic thyroid hormone and estrogen transport and metabolism. Endocrinology 2008; 149:4695-4701
- van der Deure WM, Hansen PS, Peeters RP, Kyvik KO, Friesema EC, Hegedus L, Visser TJ. Thyroid hormone transport and metabolism by organic anion transporter 1C1 and consequences of genetic variation. Endocrinology 2008; 149:5307-5314
- Huber RD, Gao B, Sidler Pfandler MA, Zhang-Fu W, Leuthold S, Hagenbuch B, Folkers G, Meier PJ, Stieger B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am J Physiol Cell Physiol 2007; 292:C795-806
- Mikkaichi T, Suzuki T, Onogawa T, Tanemoto M, Mizutamari H, Okada M, Chaki T, Masuda S, Tokui T, Eto N, Abe M, Satoh F, Unno M, Hishinuma T, Inui K, Ito S, Goto J, Abe T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc Natl Acad Sci U S A 2004; 101:3569-3574
- Kim JH, Kim YM, Yum MS, Choi JH, Lee BH, Kim GH, Yoo HW. Clinical and endocrine features of two Allan-Herndon-Dudley syndrome patients with monocarboxylate transporter 8 mutations. Horm Res Paediatr 2015; 83:288-292
- Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, St-Onge J, Steinmetz A, Huet F, Chouchane M, Darmency-Stamboul V, Callier P, Thauvin-Robinet C, Faivre L, Riviere JB. Diagnostic odyssey in severe neurodevelopmental disorders: Towards clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet 2016;
- Raymond F, Whibley A, Price S, Rosser E, Rahman N, Holder S, Stewart F, Tarpey P, Futreal A, Stratton M, Gold I. Raised T3 levels and mutations in MCT8 (SLC16A2) cause X-linked cerebral palsy and mental retardation. Eur J Med Genet 2008; 14:60
- Fuchs O, Pfarr N, Pohlenz J, Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol 2009; 51:240-244
- Hu H, Wrogemann K, Kalscheuer V, Tzschach A, Richard H, Haas SA, Menzel C, Bienek M, Froyen G, Raynaud M, Van Bokhoven H, Chelly J, Ropers H, Chen W. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J 2009; 3:41-49
- Kientz C, Herbepin Granados V, Lebrun M, Touraine R. 2012 DETERMINATION D'UN RATIO DES HORMONES THYROÏDIENNES T3/T4 COMME MARQUEUR PREDICTIF DE MUTATION DU GENE MCT8. 6èmes Assises de Génétique Humaine et Médicale; 2012; Marseille.
- Riess A, Kohlhase J, Grasshoff U, Schöning M, Riess OH, Tzschach A. Allan-Herndon-Dudley syndrome: increased serum triiodothyronine (T3) is a key diagnostic marker. Eur J Hum Genet 2013; 21:161
- Redin C, Gerard B, Lauer J, Herenger Y, Muller J, Quartier A, Masurel-Paulet A, Willems M, Lesca G, El-Chehadeh S, Le Gras S, Vicaire S, Philipps M, Dumas M, Geoffroy V, Feger C, Haumesser N, Alembik Y, Barth M, Bonneau D, Colin E, Dollfus H, Doray B, Delrue MA, Drouin-Garraud V, Flori E, Fradin M, Francannet C, Goldenberg A, Lumbroso S, Mathieu-Dramard M, Martin-Coignard D, Lacombe D, Morin G, Polge A, Sukno S, Thauvin-Robinet C, Thevenon J, Doco-Fenzy M, Genevieve D, Sarda P, Edery P, Isidor B, Jost B, Olivier-Faivre L, Mandel JL, Piton A. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 2014; 51:724-736
- Tonduti D, Vanderver A, Berardinelli A, Schmidt JL, Collins CD, Novara F, Genni AD, Mita A, Triulzi F, Brunstrom-Hernandez JE, Zuffardi O, Balottin U, Orcesi S. MCT8 deficiency: extrapyramidal symptoms and delayed myelination as prominent features. J Child Neurol 2013; 28:795-800
- Boccone L, Dessi V, Meloni A, Loudianos G. Allan-Herndon-Dudley syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. Eur J Med Genet 2013; 56:207-210
- Dateki S, Haraguchi K, Sato T, Nakatomi A, Fujiwara M, Sakurai M, Namba N, Ozono K, Moriuchi H. A novel MCT8 mutation in a Japanese patient with Allan-Herndon-Dudley syndrome. Horm Res 2013; 80:360
- EmVClass. http://geneticslab.emory.edu/emvclass/emvclass.php.
- Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol 2005; 153:359-366
- Yamamoto T, Shimojima K, Umemura A, Uematsu M, Nakayama T, Inoue K. SLC16A2 mutations in two Japanese patients with Allan–Herndon–Dudley syndrome. Human Genome Variation 2014; 1
- Wood T, Hobson D, Browning B, Rogers C, Skinner C, Ardinger HH, Collins F, Aronsky A, Friez MJ, Schwartz CE. The utilization of T3/T4 screening of males with MR of unknown etiology to identify patients with Allan-Herndon-Dudley syndrome. Eur J Hum Genet 2008; 16:26
- Philips AK, Siren A, Avela K, Somer M, Peippo M, Ahvenainen M, Doagu F, Arvio M, Kaariainen H, Van Esch H, Froyen G, Haas SA, Hu H, Kalscheuer VM, Jarvela I. X-exome sequencing in Finnish families with intellectual disability--four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis 2014; 9:49
- Ramos HE, Morandini M, Carre A, Tron E, Floch C, Mandelbrot L, Neri N, De Sarcus B, Simon A, Bonnefont JP, Amiel J, Desguerre I, Valayannopoulos V, Castanet M, Polak M. Pregnancy in women heterozygous for MCT8 mutations: risk of maternal hypothyroxinemia and fetal care. Eur J Endocrinol 2011; 164:309-314
- Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, Moharir M, Siriwardena K, Weiss SK, Weksberg R, Snead OC, 3rd. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015; 56:707-716
- Ono E, Ariga M, Oshima S, Hayakawa M, Imai M, Ochiai Y, Mochizuki H, Namba N, Ozono K, Miyata I. Three novel mutations of the MCT8 (SLC16A2) gene: individual and temporal variations of endocrinological and radiological features. Clinical Pediatric Endocrinology 2016; 25:23-35
- Anik A, Kersseboom S, Demir K, Catli G, Yis U, Bober E, van Mullem A, van Herebeek RE, Hiz S, Abaci A, Visser TJ. Psychomotor retardation caused by a defective thyroid hormone transporter: report of two families with different MCT8 mutations. Horm Res Paediatr 2014; 82:261-271
- Ono E, Ariga M, Oshima S, Hayakawa M, Imai M, Ochiai Y, Matsushima S, Miyata I, Namba N, Ozono K, Ida H. Endocrinological investigations in two cases of MCT8 abnormality with novel mutations in the SLC16A2 gene. 95th Annual Meeting of The Endocrine Society2013:SUN-618.
- La Piana R, Vanasse M, Brais B, Bernard G. Myelination Delay and Allan-Herndon-Dudley Syndrome Caused by a Novel Mutation in the SLC16A2 Gene. J Child Neurol 2015; 30:1371-1374
- Ugrasbul F, H.H. A. A patient presenting with central hypothyroidism, developmental delay and poor head control. Should we be checking T3 levels? Horm Res 2009; 72:458-459
- Noguchi A, Takahashi I, Shoji Y, Oyamada M, Takahashi T. Transient acidosis in infancy with a novel variant in MCT8 (Monocarboxylate transporter 8) gene. Hormone Research 2009; 72:310-311
- Gika AD, Siddiqui A, Hulse AJ, Edward S, Fallon P, McEntagart ME, Jan W, Josifova D, Lerman-Sagie T, Drummond J, Thompson E, Refetoff S, Bonnemann CG, Jungbluth H. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol 2010; 52:475-482
- Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, Refetoff S. A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics 2008; 121:e199-202
- Langley KG, Trau S, Bean LJ, Narravula A, Schrier Vergano SA. A 7-month-old male with Allan-Herndon-Dudley syndrome and the power of T3. Am J Med Genet A 2015; 167A:1117-1120
- Kobayashi S, Onuma A, Inui T, Wakusawa K, Tanaka S, Shimojima K, Yamamoto T, Haginoya K. Clinical course and images of four familial cases of Allan-Herndon-Dudley syndrome with a novel monocarboxylate transporter 8 gene mutation. Pediatr Neurol 2014; 51:414-416
- Fu J, Refetoff S, Dumitrescu AM, Weiss RE. OR29-3: Whole-Exome Sequencing Identified a Novel MCT8 Gene Mutation in a Child with Mild Cognitive, Motor and Behavior Abnormalities. 2014;
- Faruk Aydin O, Kara C, Jones J, Wood TC, May MM, Friez MJ, Schwartz CE. Allan-Herndon-Dudley syndrome caused by a novel MCT8/SLC16A2 mutation in a Turkish family. Horm Res 2013; 80:352-353
Evaluation of Infertility, Ovulation Induction and Assisted Reproduction
ABSTRACT
Infertility is a prevalent condition among men and women and can be emotionally devastating. The cause of a couple’s infertility may be female, male, a combination, or unexplained. This chapter serves to outline the evidence-based evaluation of infertility and treatments including ovulation induction and assisted reproductive technologies.
Reproduction is a mandate of nature. The continuity of species demands promulgation. However, as humans have attempted to control nature, reproduction has evolved into an option. But, preventing conception is quite often easier than reproduction on demand. Indeed, 12 months of intercourse without contraception and without conception is defined as infertility. Although infertility has plagued humans throughout history, its face has changed throughout by economics of large families, women’s role in the workplace, smaller families born later in life and effective contraception. Advanced technology has aided the diagnosis of infertility and revolutionized the treatment. This chapter reviews the history, diagnosis and therapy of infertility. For complete coverage of this and all related areas of Endocrinology, please visit our FREE on-line web-textbook, www.endotext.org.
HISTORY
Modern gynecologists wrote about infertility in the mid-20th century as a descriptive state induced by a certain personality type of the woman (3,4). As more data become known of the physical causes of infertility, the depression and anxiety thought to be a cause of infertility evolved into an effect of the disease.
The next significant advance in the diagnosis of infertility was laparoscopy. Laparoscopy enabled tubal factor infertility and endometriosis to be diagnosed in women without major abdominal surgery. It would be much later when surgical intervention was actually introduced through the laparoscope, but, nonetheless, diagnosis became more aggressive.
The first therapeutic breakthrough was probably the introduction of clomiphene citrate in the 1960s for ovulation induction in anovulatory women (5). This was followed by gonadotropin injections.
Most recently, the birth of Louise Brown in July of 1978, the world’s first child conceived after in vitro fertilization, altered the approach to therapy once again. Because of its technologic glamour, IVF was highlighted by the media in the last quarter of the 20th century as the Mecca of infertility therapy and the prime example of interfering with the intimacy of reproduction. The biologic fidelity of gametes from the married couple resulting in the wife’s gestation and delivery of their child was broken. Gestational parent, gamete donors, and surrogate mothers became the topic of heated ethical and legal battles.
DEMOGRAPHICS
In the United States approximately 16.6% of women ages 15-44 are infertile, and 11.9% (7.3 million) women have received fertility services (6). Infertility is not considered a disease by most third party payers, and medical diagnosis and treatment usually requires the couple to provide payment. Because infertility is not a life-threatening disease, choice of therapy often rests with the couple, which is frequently a choice of cost-consideration rather than cost-effectiveness. Couples frequently request therapy which is the least costly even though the efficacy may be low. The most important role of the physician in these cases is education and barring therapy whose risk-benefit ratio is too low for safety.
Thus, the evaluation and therapy of infertile couples is often dictated by the amount of funds available. This review, however, discusses the disease in an academic manner without emphasis on practicality. Studies available to evaluate the science rather than the art of medicine will be emphasized in order to provide readers with factual information, but this should not be interpreted as minimizing the practical and sensitive side so important in the clinical practice of medicine. Rather, armed with as much information as possible, each physician may be best able to tailor the evolution and therapy to the needs and resources of the individual.
EVALUATION
The evaluation of infertility assesses each component of reproductive physiology to identify an abnormality: the cervix, the uterus, the endometrium, the semen, the ovarian function, the fallopian tubes and the peritoneum. Of course, each evaluation begins with a history and physical. Often, historical evidence directs and streamlines subsequent laboratory evaluation, frequently eschewing other tests before beginning therapy. It follows, that each abnormality found should be treated to maximize fertility.
AGE EFFECTS
Infertility increases with age. The effect of the female’s age is more dramatic and earlier than that of the male, but an age effect is seen with advancing male age also.
Figure 1:Graph of Births over Time as a Function of Maternal Age (7)
Fertility peaks when the woman is in her late teens and early twenties. It begins to decline at age thirty and drops more rapidly after age 35 years (8). It plummets after age 40 and pregnancy after age 45 is rare (7,9). This phenomenon is also reflected in the live birth rate and has been remarkably stable over time and geography as seen in figure 1. Miscarriages are also more frequent as maternal age rises (10). Although one may suggest that this is because of the well-known increase in aneuploid conceptions with maternal age (11), euploid pregnancies are also lost with higher frequency as the mother ages.
The effect of maternal age on fertility is theoretically an effect of a larger proportion of abnormal embryos with increasing maternal age. Data from in vitro fertilization in which normal appearing embryos were examined with fluorescent in situ hybridization (FISH), revealed 39% abnormal embryos from women who are greater or equal to 40 years of age compared to 5% from women who are 20 to 34 years of age (12). A more recent study utilizing comprehensive chromosomal screening through comparative genomic hybridization (CGH) illustrated a 51.3% aneuploidy rate in embryos from infertile women aged 30 – 43 years. Of these women, those with age 40 and older produced an average of 57% aneuploid embryos (13).
Frequently unaddressed, is the effect of the male’s age. Sperm count does drop with age, although the individual variation is wide as is the male’s sexual function. The decreasing fertility of the male roughly lags behind that of the female by 10 years. Significant infertility is seen after age 55 years, although this data is difficult to come by, because of the more impressive effect of maternal age. In order to gain firm figures, men at various ages older than 45, married to women younger than 30 would have to be surveyed for intercourse frequency and conception. The only data available are those from the Mormon genealogy registers (see Figure 2)(14).
Figure 2:Data from Mormon Registries: Fertility as a Function of Maternal and Paternal Age (13)
Thus, men older than 55 married to women older than 35 may have a synergistic age effect on their fertility, but one which is difficult to quantify.
In vitro fertilization outcomes echo earlier trends between female age and pregnancy. While increasing female age leads to a decrease in live birth when using autologous oocytes, donor-oocyte-IVF cycle outcomes appear unaffected by increasing recipient age (Figure 3)(15).
Figure 3: Live birth rates among IVF cycles performed in the United States in 2013 (15).
CERVICAL FACTOR
The nulliparous cervix is approximately 4 cm in length. Its columnar epithelium is pierced by the ducts of mucous secreting glands which spew forth their contents as a protective barrier preventing bacteria from entering the upper reproductive tract and as a welcome channel to lead sperm into the upper tract. Cervical mucus is comprised of hyaluronic acid micelles which are affected by the hormonal milieu exposed to the glands. Late in the follicular phase, as estrogen rises, the micelles align in a parallel arrangement forming channels to guide the sperm. Under the microscope this can be seen as the classic "fern" pattern of dried cervical mucus (Figure 4). The pH is alkaline and nourishing to the sperm. Indeed, sperm can live in normal cervical mucus for as long as 4 days. The abundant mucus frequent oozes from the cervical os into the vagina to lure the sperm from the ejaculate, while protecting them from the acidity of the vagina. At mid-cycle, just prior to and after ovulation the rising progesterone increases the salt content of the mucus, breaking the micelle channels and thickening the consistency of the mucus. The "fern" pattern is no longer seen; the mucus thickens and becomes hostile to sperm and bacteria alike.
Figure 3:The classic fern pattern of dried cervical mucous.
History
Women with a history of cervical infections, surgery or cryotherapy may have damage to the cervical glands and lack mucus. This may result in the inability of the sperm to survive the harsh vaginal acidity and not make the assent to the uterus. However, a considerable amount of cervix must be removed or damaged for a true "cervical" factor to cause infertility.
Infections rarely result in infertility. Although gonorrhea and chlamydia have often been accused, it is difficult to prove that either of these actually destroys cervical glands and are more likely to result in tubal factor infertility (16). Acute infection may alter cervical pH, killing sperm, although this is not well documented.
Cervical conization which removes the cervical glands is the most likely cause of cervical factor infertility. Cryotherapy and laser vaporization may destroy the lower canal, but glands above the point of metaplasia usually provide enough mucus to retain fertility (17).
Physical Examination
The normal squamocolumar junction with clear cervical mucus almost always rules out a cervical factor. When the cervix is scarred with a narrow external os and almost flush with the vagina, the cervical glands are frequently absent.
Diagnosis
This diagnosis of cervical factor is based heavily of the history of cervical damage by surgery or infection. Nonetheless, the classic diagnosis of cervical factor infertility has been the post coital test (aka Sims Hauser test). The postcoital test was proposed to determine the adequacy of sperm and the receptivity of cervical mucus, however it has been the subject of debate over the last 10 years. In a randomized 24 month study female patients with abnormal post-coital tests were compared to those with normal tests, and importantly there was no difference in pregnancy rates between the two groups (18). In a literature review assessing use of the post coital test, the sensitivity of the test ranged from 0.09 to 0.71, specificity from 0.62 to 1.00, predictive value of abnormal from 0.56 to 1.00, and predictive value of normal from 0.25 to 0.75 (19). In light of problems of poor validity, lack of standard methodology, lack of a uniform definition of normal, and unknown reproducibility, the postcoital test in longer considered of significant value in infertility assessments. Without a history of cervical damage by infection or surgery, the diagnosis of cervical factor is largely circumstantial.
Treatment
A true cervical factor is best treated with intrauterine insemination as described below. Briefly, washed sperm is injected into the uterus at the time of ovulation. Because the sperm survive a limited time, it is probably best to time ovulation with urinary LH to narrow the insemination-ovulation interval.
In addition, prior cervical surgery may predispose to an incompetent cervix during pregnancy (20). Thus, the patient should be closely monitored for painless dilation of the cervix during pregnancy and evaluated for cervical cerclage.
ENDOMETRIAL FACTOR
A hostile endometrial environment will impede embryonic implantation as shown by the placement of an intrauterine device into the endometrial cavity for contraception. This hostile effect is postulated to be reproduced by endometrial polyps and submucous fibroids. In addition, hormonal, immune and biochemical factors have been postulated to result in a hostile endometrial environment.
Poor progesterone effect resulting in delayed maturation of the endometrial lining, commonly referred to as the luteal phase defect, has been considered as hostile to implantation. Indeed, luteal phase defect has been demonstrated in a case of trisomy 16, suggesting that the defect is a secondary phenomenon of an primary defect, namely, genetic abnormality of an oocytes and its follicular apparatus resulting in abnormally low hormonal stimulation and finally, poor endometrial maturation (21). Teleologically, this would prevent pregnancy with abnormal embryos. Theoretically, this mechanism may go astray during genetically normal cycles and result in failed implantation, but has never been proved to be a cause of infertility, per se.
Other factors, which have been implicated in the development of a hostile environment for implantation, are autoimmune factors such as lupus anticoagulant and Integrin IIIβ (22). It has been shown that antiphospholipid antibodies are not a factor in implantation, but the concentrations of Integrin III β are yet to be determined (23).
History
Patients with endometrial polyps and submucous fibroids may have premenstrual spotting or heavy menstrual bleeding. A luteal phase defect is not detected in any medical history.
Physical Examination
Endometrial factors cannot be diagnosed by physical examination.
Diagnosis
Endometrial polyps and submucous fibroids are detected by either hysterosalpingography, a saline sonohysterogram, or hysteroscopy (Figure 5). A dilation and curettage (D&C) may be performed and histology of the curettings will confirm the diagnosis. Submucous fibroids may be felt as an irregularity while curetting the endometrial cavity.
Figure 5 :Hysteroscopic view of an endometrial polyp.
Abnormal autoimmune factors do not cause infertility and need not be diagnosed. Integrin IIIβ is still a research tool and diagnosis of an abnormal concentration should be left to research protocols.
The luteal phase defect is not a documented cause of infertility (24-26). The high prevalence of out-of-phase endometrial biopsies in fertile women makes histological dating of the endometrium of no use in the routine evaluation of infertility (26). Because its role in spontaneous abortion is not known, some physicians prefer to evaluate the endometrium in infertile patients. The diagnosis is made by performing an endometrial biopsy in the luteal phase. A histological dating lagging more than 2 days behind the actual postovulatory date diagnoses the defect.
Treatment
Endometrial polyps may be removed by operative hysteroscopy or uterine curettage. Submucous fibroids may be removed through hysteroscopic surgery. Depending upon the depth of myometrial invasion, laparoscopy or laparotomy may be necessary. Pritts et al performed a meta analysis of women undergoing hysteroscopic myomectomy and found a modest improvement in pregnancy outcome (relative risk of 1.72 with a confidence interval of 1.13-2.58)(27).
As mentioned, other causes of endometrial hostility have not been proven to be related to infertility and, thus, treatment is not warranted.
UTERINE FACTOR
In addition to the endometrium and cervix, the shape and competency of the uterine fundus must be considered. Anomalies of mullerian fusion and fibroids have been suggested to result in the inability of implantation or growth of the pregnancy. It is difficult to postulate how abnormalities of the uterus can cause infertility except through abnormal blood flow leading to poor implantation and a subsequent abnormality involving placentation. Indeed, evidence that does exist relates infertility to large fibroids suggesting tubal factor infertility. Mullerian fusion abnormalities resulting in a uterine septum or bicornuate uterus are largely associated with recurrent miscarriage rather than the ability to conceive.
History
Historical clues of an abnormal uterus are rare. Occasionally, patients with incomplete mullerian fusion may have dysmenorrhea, but the symptom itself is so nonspecific, that the physician is not likely to alter either diagnosis or therapeutic course.
Physical Examination
A bimanual pelvic examination will reveal large fibroids. The bicornuate uterus is typically unable to be palpated. A compete uterus didelphys will be revealed by a vaginal and cervical examination.
Diagnosis
Uterine fibroids may be diagnosed by ultrasound (Figure 6). Their imposition on the endometrial cavity may be illustrated by hysterosalpingogram and confirmed by hysteroscopy. Ultrasound is less valuable in diagnosing mullerian anomalies, but hysterosalpingogram will identify the abnormal uterus (Figures 7,8,9) . MRI is often helpful in identifying both fibroids and the type of mullerian anomaly found on hysterosalpingogram (Figure 10).
Figure 6: Transverse sonographic image of the uterus demonstrating overall enlargement and multiple leiomyomata.
Figure 7: Hysterosalpingogram of a normal uterus. Note the normal appearing intrauterine cavity with bilateral tubal spillage of contrast
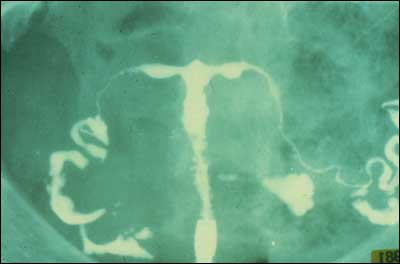
Figure 8: Hysterosalpingogram of a T-shaped uterus secondary to in utero DES exposure.
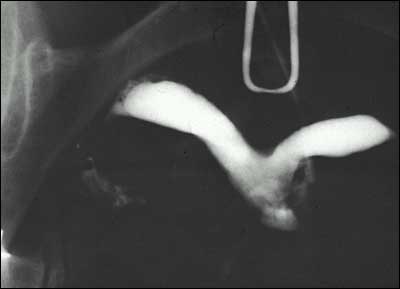
Figure 9: Hysterosalpingogram of a uterine anomaly. With HSG, a septum versus a bicornuate uterus cannot be distinguished..
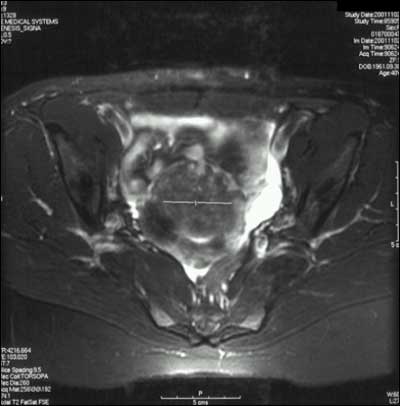
Figure 10: Transverse MRI image demonstrating multiple leiomyomata..
The septate uterus can be differentiated from the bicornuate uterus by laparoscopy and MRI. Both laparoscopy and hysteroscopy will identify fibroids and their location in the uterus and pelvis. If fibroids interfere with tubal pick-up of oocytes, mechanical infertility will result. Similarly, fibroids impinging on the endometrial cavity may interfere with implantation and placentation.
Treatment
Uterine fibroids may be treated by myomectomy. Depending on the location of the fibroid, hysteroscopic or laparoscopic removal may be possible. Otherwise laparotomy is necessary. Treatment of the fibroid with a GnRH agonist will shrink the fibroid, but as soon as the agonist is discontinued and estrogen activity resumes, the fibroid will return to at least its original size (28).
Myomectomy is indicated when the myomas result in bleeding severe enough to cause anemia, when pressure is placed on the on the bladder to cause urinary tract infections, or when pressure on the bowel results in constipation. It is less clear whether myomectomy improves fertility and, in fact, may even result in pelvic adhesions and lead to tubal factor infertility (29). Myomas which impinge or are present within the endometrial cavity may likely cause spontaneous abortion, but evidence proving this causation is difficult to find. Even harder to prove is the efficacy of myomectomy resulting in an increase in term pregnancy.
TUBAL FACTOR
The mechanical ability of the sperm to be conducted to the site of the oocyte or the fertilized oocyte to be propelled back into the uterus, demands patent fallopian tubes. Clearly, the tube must be open to allow fertility. Also important is the ability of the tube to freely move across the surface of the ovary in order to sweep the oocyte into the tube. If the tube is not able to pick up the oocyte from the surface of the ovary, no pregnancy will result.
History
Patients with tubal factor infertility often have a history of a pelvic infection, endometriosis, or previous abdominal or pelvic surgery. Frequently, however, patients are unable to clearly identify a source of their adhesions. For example, a patient who experienced a chlamydial infection may have attributed the lower abdominal pain, fever and cramping to a gastrointestinal viral infection and often cannot recollect the time of infection.
Adhesions may be asymptomatic or may result in pelvic pain. Normal movement of the bowel and ovaries across the visceral surfaces of the abdomen may be impeded by pelvic adhesions and results in these organs pulling on the abdominal wall. This may result in both pelvic and abdominal pain.
Physical Examination
Tubal disease significant enough to result in hydrosalpinges may result in adnexal masses. Similarly adhesions, which adhere the ovaries to uterus, may often be interpreted as a uterus with posterior fibroids on physical examination.
Diagnosis
Tubal adhesions or hydrosalpinges are suggested when contrast pools are seen on a hysterosalpingogram (Figure 11), but definitive diagnosis requires visualization at laparoscopy. Tubal occlusion may be diagnosed by hysterosalpingography or by laparoscopy. Most recently, fluid extravasation into the pelvis after flushing of an intrauterine cannula may be seen by an experienced ultrasonographer (30). However, this test is less reliable than the former and is dependent on the skill of the sonographer, patient’s factors such as weight and air in the bowel and resolution capabilities of the ultrasound machine.
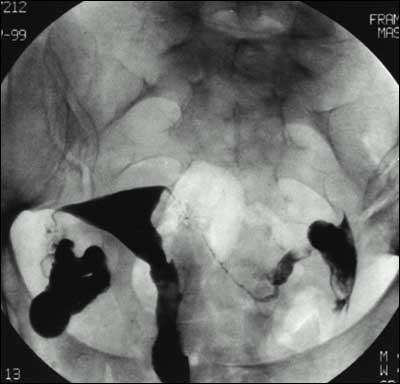
Figure 11: Hysterosalpingogram demonstrating hydrosalpinges. Note the marked dilation of the tubes and the pooling of contrast..
Treatment
In vitro fertilization results in the highest chances of pregnancy for the couple whose infertility is the result of pelvic adhesions or blocked Fallopian tubes. Surgical correction may increase the chances of fertility, however, unless the adhesions are filmy and their extent is limited, surgical lysis incurs a high likelihood of adhesion recurrence.
ENDOMETRIOSIS
Endometriosis is the abnormal development of endometrial glands and stroma outside of the uterus. It is associated with dysmenorrhea, dyspareunia, and infertility. The American Society of Reproductive Medicine (ASRM) developed a revised staging scheme in order to standardize communication between physicians regarding their patients, between investigators for research protocols and to follow effects of therapy (31).
The mechanism of action whereby endometriosis causes infertility is complex. Severe endometriosis with adhesions and adherent pelvic organs results in mechanical infertility. It is difficult to assign a mechanism to minimal and moderate endometriosis. Increased peritoneal fluid, increase peritoneal prostaglandin concentration, and interference with normal ovarian folliculogenesis have all been postulated (32). However, no direct evidence exists between mild and minimal endometriosis and resultant infertility (33). Indeed, some have postulated endometriosis is a result rather than a cause of infertility.
History
Patients with endometriosis experience a spectrum of symptoms ranging from incapacitating dysmenorrhea and severe dyspareunia to none at all. Although 30% of infertile patients have endometriosis (34), it is unknown how many patients who have endometriosis are infertile. Classically, patients have dysmenorrhea which begins during the menses and over the years extends to the prior luteal phase.
Cyclical hemoptysis and hematochezia may occur in patients with endometriosis implants in the lung; however, very few of patients with endometriosis have these remote implants.
Physical Examination
Endometriosis may be occasionally palpated on pelvic exam if there are implants on the uterosacral ligaments or the posterior surface of the uterus; however, physical examination is usually not helpful in the diagnosis of endometriosis.
Diagnosis
Endometriosis is diagnosed by visual inspection at the time of laparoscopy or laparotomy or if endometriomas are visualized on pelvic ultrasound. Endometrial implants over the pelvic organs may be biopsied for histological confirmation. Endometrial glands and stroma are seen microscopically. The American Society of Reproductive Medicine recommends using a standard staging system to document the severity of each patient’s disease. The system incorporates superficial and deep lesions, adhesions, and the location and size of each lesion. Standardized staging allows communication between physicians and evaluation of treatment efficacy.
Treatment
Endometriosis causing pain may be treated with surgical or medical therapy. Surgical extirpation of lesions and adhesions is successful in alleviating pain for endometriosis but is less effective in increasing fertility caused by endometriosis (35). Preventing menstruation with the use of continuous oral contraceptive or continuous progestin therapy may also relieve pain. Medical menopause induced by GNRH analogues has also been effective in reducing the implants of endometriosis. However, once normal cycles and hormones resume, the implants return. Medical therapy does not have much effect on the adhesions associated with endometriosis (36,37). Similarly, endometriomas of the ovary are decreased in size by medical therapy but are rarely completely treated. In vitro fertilization is more effective than either surgery or medicine in increasing fertility caused by endometriosis (32).
OVULATORY FUNCTION
Pregnancy is direct evidence of ovulation. Patients who do not ovulate cannot conceive without assisted reproductive technology. Ovulatory function requires the integration of many normally functioning systems. Normal thyroid function, normal insulin action, normal adrenal function and perhaps normal cerebral function are all required for ovulation. When these systems are disrupted, follicular function is altered and ovulation becomes disordered. This results in a series of endocrine events, which leads to elevated estrone production, elevated androgen production and altered insulin action. These events, in turn, prevent ovulation. This cycle of disordered events has become known as polycystic ovarian syndrome. This syndrome is discussed in detail in Chapter 6.
History
Normal, regular menstrual cycles usually reflect ovulatory cycles. Indeed, 95% of regular menstrual cycles are ovulatory (38). If cycles are irregular, the patient is most likely not ovulating, obviously a cause of infertility. Patients who do not ovulate and have polycystic ovarian disease may also have other associated findings of the disorder including insulin resistance, androgen excess, acanthosis nigricans and obesity.
Physical Examination
Anovulation alone is not associated with any physical findings. Patients will have abundant, thin cervical mucus most of the time. This is the result of continuous, unopposed estrogen. In those whose have elevated androgens, hirsutism may be present. Acanthosis nigricans may also be found at the neckline, in the axilla, and underneath the breasts.
Diagnosis
Anovulation is diagnosed by a serum progesterone measurement less than 4 ng/ml drawn at least 4 days prior to a menstrual bleed. Proliferative endometrium on an endometrial biopsy performed during the week prior to a menstrual cycle is also diagnostic of anovulation. Finally, a basal body temperature chart revealing no biphasic increase in temperature is diagnostic of anovulation in 80% of cases; 20% of women with a monophasic basal body temperature will actually ovulate (39,40).
Treatment
Ovulation may be stimulated by administering clomiphene citrate, letrozole, or gonadotropins. Clomiphene citrate is a serum estrogen receptor modulator, which increases pituitary secretion of LH and FSH. When given to anovulatory women, the gonadotropins stimulate follicular development. The follicle continues to develop normally and feeds back to signal the pituitary that the oocyte is mature and an LH surge is needed. The so-called "ovarian clock" is restored and ovulation may be timed by monitoring urinary LH concentrations. Clomiphene successfully induces ovulation in 85% of women, although pregnancy rates are lower than 65% (41,42). When clomiphene is unsuccessful, gonadotropins may directly stimulate the ovarian follicle to develop and chorionic gonadotropin may be administered to simulate the LH surge and result in ovulation.
Patients with polycystic ovarian disease have often been treated with insulin sensitizing agents, particularly metformin (43,44). Most recently however, a multicenter randomized placebo-controlled trial failed to show any superiority of metformin over clomid, and combining clomid with metformin was no better than clomid alone (45) Metformin use in PCOS should be restricted to women with glucose intolerance and the routine use of this drug in ovulation induction is not recommended (46,47).
Letrozole, an aromatase inhibitor, has also been used to induce ovulation in anovulatory women. The medication decreases peripheral aromatization of testosterone to estrogen leading to an increase in FSH secretion from the pituitary gland and resultant follicular maturation (48). The Reproductive Medicine Network performed a double-blind, multi-center study comparing pregnancy outcomes in women with PCOS using either clomiphene citrate or letrozole. Patients using letrozole had a higher cumulative live birth rate than those who used clomiphene citrate (27.5% vs. 19.1%, P=0.007) and a lower multiple pregnancy rate (P=0.03) (49).
Gonadotropins, FSH or a combination of FSH and LH can also be used to induce ovulation in anovulatory women. These medications are generally injected subcutaneously and lead to follicular growth. Recruitment of more than one dominant follicle is not uncommon during a gonadotropin stimulation, so patients are followed closely with ultrasound monitoring and serum estradiol levels. The treatment modality is associated with an increased risk of high-order-multiple pregnancy when compared to natural conception, clomiphene citrate, and letrozole.
CHRONIC DISEASE
History
A thorough menstrual history should be done with all infertile patients to detect chronic diseases such as severe anemia, autoimmune diseases, liver disease and renal disease. Debilitating disease and chronic malnutrition may also result in infertility (50). Chronic disease most likely interferes with ovulation by central mechanisms. Treatment of the disease rarely restores ovulation because such treatment is usually palliative.
Physical Examination
The exam findings may be subtle, and include clubbing of nailbeds, poor skin turgor, subcutaneous adiposity, conjunctival pallor, thinning of hair, jaundice, cushingoid features, recent weight loss, poor skin turgor, delayed capillary refill, gum bleeds, and cheilosis, Fatigue, exercise intolerance, dyspnea on exertion, peripheral edema, cardiac murmurs are consistent with underlying cardiac disease. Splenomegaly, dilated abdominal wall collateral vasculature, and ascites or signs of anemia may represent longstanding gastrointestinal disease. Pulmonary disease is manifested by a chronic cough, diminished breath sounds, wheezing, dyspnea or simply fatigue.
Diagnosis
In most instances, the diagnosis will already be established. Physical exam findings may indicate the patient’s overall health, and nonspecific studies may be helpful such as: CBC with peripheral smear, urine dipstick for proteinuria, hematuria, pyuria and microscopic evaluation (epithelial cells, WBC casts, etc.), liver functions, and serum chemistries to include electrolytes, creatinine, BUN, magnesium, phosphate, uric acid, and protein. A sedimentation rate is a nonspecific test of inflammation, and cardiac status may be initially assessed with an ECG, CXR, echocardiography, and stress test. Clearly, the diagnostic tests must be tailored to the individual.
Treatment
Treatment of infertility associated with chronic disease requires addressing the underlying health problem and optimizing the patient’s present state of health. This usually requires a multi-specialty effort, specific to the patient’s health needs. Special consideration should be taken to assess if a relative or absolute contraindication to pregnancy exists, as the pregnancy may well exacerbate an already tenuous medical condition. Maternal surrogacy is an option in patients who are unable to tolerate the physical stresses of pregnancy, such as patients with severe hypertension, significant structural cardiac anomalies, advanced multiple sclerosis, to name a few. If premature ovarian failure has occurred, either secondary to chronic disease, or to treatments such as chemotherapy or radiotherapy, donated oocytes are an option.
MALE FACTOR
Extensive discussion of male factor infertility is within Section: Endocrinology of the Male edited by Robert MacLachlan,M.D (www.endotext.org). A recent study performed by the Reproductive Medicine Network correlated semen parameters to fertility. The "new norms" by which fertility can now be defined are: a sperm density greater than 15 million/ml, motility greater than 32%, and a strict morphology greater than 4% (51). Abnormal semen parameters have a host of causes resulting from an absent vas deferens secondary to cystic fibrosis heterozygosity to a varicocele. Nonetheless, when the semen parameters are low, the number of competent sperm available to fertilize an oocyte is decreased and the monthly fecundity is lowered. It is more difficult to explain why male factor infertility occurs in the presence of normal semen parameters. Some have suggested that abnormal capacitation, decreased motility, abnormal morphology, chemical composition, or antibody presence compromise fertilization. These factors remain unproved.
History
Male factor infertility usually presents when the gynecologist orders a semen analysis in the initial infertility evaluation. Rarely does it present as symptoms in the male. The male partner may have a history of undescended testicles or testicular pain due to a varicocele, but this is usually elicited after the semen analysis is abnormal.
Physical Examination
Males with a varicocele will often have a palpable vessel in the scrotum. Otherwise no physical findings are abnormal in most infertile males.
Diagnosis
The diagnosis of male factor infertility begins with a semen analysis. Abnormal parameters include: sperm concentration of less than 15 x 10(6) per milliliter, less than 32% of sperm motility, with less than 4% with normal morphologic features (51). Tests such as the sperm penetration assay, antisperm antibodies, free oxygen radicals have been implicated as resulting in infertility, but the documentation is unconfirmed. Sonographic evaluation of the scrotum will confirm the presence of a varicocele.
Treatment
If the infertility is due to a mechanical issue, such as retrograde ejaculation, either pharamacologic or surgical correction of the bladder neck is feasible. In patients with neurological damage from spinal cord or pelvic trauma, multiple sclerosis, or retroperitoneal surgery, rectal electroejaculation or surgical sperm recovery may be utilized. If the male partner is azoospermic due to hypogonadotropic hypogonadism, the patient is treated with gonadotropin therapy. Obstructive azoospermia may be corrected surgically or sperm recovery techniques such as TESE (testicular sperm extraction)(Figure 12) or MESA (microsurgical epididymal sperm aspiration)(Figure13) may be used. Since the wide acceptance of ICSI (intracytoplasmic sperm injection)(Figure14), an oocyte is mechanically fertilized with a single sperm, a method which ameliorates all but the most severe male factor infertility. Clomiphene citrate is no better than placebo for increasing a low sperm count. Recently, sperm motility has been treated with carnitine supplements, but randomized trails are pending. (For a full discussion on male factor infertility, see Endocrinology of the Male edited by Robert McLachlan, M.D.at www.endotext.org).

Figure 12: Testicular sperm extraction (TESE)
Figure 13: Microsurgical epididymal sperm aspiration (MESA)
Figure 14: Intracytoplasmic sperm injection (ICSI)
UNEXPLAINED INFERTILITY
Approximately 15% of patients with infertility have an unidentifiable cause. Thus, unexplained infertility is a failure of both the couple to conceive and the physician to explain why. Perhaps even more psychologically difficult to accept than sterility, unexplained infertility poses impressive stress to the couple. Despite doing all they can correctly, the couple is unable to conceive and has no answer to their problem even after enduring expensive, painful testing. Their only recourse is empiric therapy.
History
Couples with unexplained infertility have routine intercourse, have regular menstrual cycles and normal infertility testing results.
Physical Examination
Inherent to the diagnosis, physical examination reveals no abnormalities.
Diagnosis
The diagnosis of unexplained infertility is made in couples who have intercourse regularly or well-timed intercourse and fail to conceive despite regular ovulatory cycles, a normal semen analysis, normal laparoscopy, and normal hysterosalpingography.
Treatment
Couples with unexplained infertility may conceive without therapy but it may take from 3 to 7 years (52). To decrease the waiting time, empiric therapy is instituted. Empiric therapy usually begins with ovulation induction with oral agents or gonadotropins combined with intrauterine insemination. This combined therapy increases the cyclic fecundity over either alone in couples with unexplained infertility (53, 54). After three or four unsuccessful cycles or alternatively, in vitro fertilization may be offered.
ASSISTED REPRODUCTIVE TECHNOLOGY
The birth of Louise Brown in 1978, the world’s first "test tube baby", revolutionized therapy for infertile patients. No longer were women without fallopian tubes unable to conceive and gestate. For the first time, medicine was able to view the first few human embryonic divisions and reproductive medical scientists learned better how to induce ovulation and handle gametes. Numerous "spin-off" technologies arose in the subsequent two decades: controlled ovarian hyperstimulation (COH-IUI), intracytoplasmic sperm injection (ICSI) and oocyte donation allowed pregnancy in couples without gametes. This section briefly describes the reproductive technologies available to infertile patients and referred to in treatment sections above in the chronological order of their introduction into the therapeutic armamentarium.
In Vitro Fertilization (IVF)
In Vitro Fertilization (IVF) was first successfully performed by Patrick Steptoe, MD and Robert Edwards, PhD in a woman with tubal factor infertility. The woman timed her ovulation and underwent laparotomy for oocyte retrieval. In the ensuing years, spontaneous cycles were replaced by first clomiphene citrate ovulation induction and then stimulation with gonadotropins. Ovulation stimulation allowed the retrieval of more than one mature oocyte, thus increasing the statistical chance of embryonic implantation during one cycle. The early pregnancy rates were approximately 8% per cycle and cancellation prior to oocyte retrieval was high. The introduction of GnRH analog allowed down regulation prior to ovarian stimulation and thwarted the numbers of cycles cancelled due to spontaneous premature LH surges. GnRH analog therapy was most likely the reason that IVF pregnancy rates jumped to about 15% per cycle in the late 1980s.
By the time IVF in the United States was introduced by the Jones in 1983, oocytes retrieval was primarily by laparoscopy. In 1983, Gleicher et al reported the first transvaginal ultrasound-directed oocyte aspiration, and currently, most aspirations are performed in this manner (55).
Indications
Initially, IVF was indicated in patients with tubal factor infertility. The relatively high success rates have allowed extension to couples with endometriosis, drug-resistant polycystic ovarian disease, and unexplained infertility. Additionally, IVF can be used with donor oocytes to treat women with age-related ovarian dysfunction, ovarian failure or surgically removed ovaries. Combined with ICSI (see below) IVF can also be used to treat couples with sperm disorders and immunologic infertility.
Procedure:
IVF begins with ovulation induction. Although it may be performed in natural cycles, removal of the oocytes requires intensive hormonal monitoring and the availably of the medical team too often to be practical. In addition, pregnancy rates are lower with natural cycle IVF.
A variety of ovulation induction regimens are available. Many programs in the United States administer ovarian down regulation with a GnRH agonist in the luteal phase prior to the cycle of stimulation. Oral contraceptives are often added prior to down-regulation. After spontaneous ovarian activity is suppressed, folliculogenesis is stimulated with gonadotropins. The regimen used is particular to each program, but those that have been tested in clinical trials all seem to result in similar pregnancy and delivery rates. Choices are made based on cost, availability, route of administration, and familiarity of the physician with the regimen.
GnRH antagonists are also used to thwart spontaneous LH surges during ovulation induction. Thus far, they have not proven superior to the GnRH agonists in pregnancy outcome and are more expensive on a mg per mg basis (56) but offer the advantage of fewer total injections to the patient. Another proposed advantage of GnRH antagonist cycles is a decreased risk of moderate and severe Ovarian Hyperstimulation Syndrome (OHSS) when combining this protocol with a GnRH-agonist induction of final oocyte maturation (57-59).
During ovulation induction, the ovaries are monitored for follicular growth by frequent transvaginal ultrasound examinations and serum estradiol concentrations. When clinical parameters suggest the presence of mature oocytes, human chorionic gonadotropin (hCG) is administered to mimic the LH surge and allow further progression of the oocytes through meiosis. Some IVF programs now use a GnRH agonist to trigger ovulation, as it appears that the incidence of moderate and severe ovarian hyperstimulation syndrome (OHSS) is reduced in high-risk patients when an agonist is used. Approximately 35 hours later, the patient undergoes a follicular aspiration. In the United States, most aspirations are performed transvaginally with ultrasound direction. Conscious sedation is typically used. Regional analgesia is not usually employed because the concentration of the local anesthesia in the follicular fluid has been shown to decrease the pH and affect the fertilization of the oocyte (60). Concomitant identification of the oocyte by nearby laboratory technicians informs the physician to proceed with aspiration of each sequential follicle (Figures 15,16).
Figure 15: Human oocyte immediately following retrieval.
Figure 16: Human oocyte after the surrounding cumulus cells have been removed.
Between 4 and 6 hours after aspiration, the oocytes are mixed with 15,000-30,000 motile, previously prepared sperm. Human fertilization occurs in the next 18 hours and the first two cell divisions usually occur in the subsequent 24 hours (61). Embryos are then transferred into the uterine cavity typically three or five days later. The American Society of Reproductive Medicine (ASRM) recommends determining the number of embryos to be transferred by patient criteria, with favorable prognosis patients generally receiving no more than 1-2 embryos, and poor prognosis patients of advanced maternal age receiving no more than 5 embryos (ASRM Practice Committee Report: Guidelines on the Number of Embryos to Transfer, revised September, 2012). These guidelines were proposed to decrease the incidence of multiple pregnancies, yet still facilitate a favorable pregnancy rate.
The embryos are transferred through the vagina into the uterus. Approximately 2 weeks later a pregnancy test is performed. Because aspiration removes granulosa cells which would otherwise produce the progesterone necessary for endometrial development, many programs support the luteal phase with exogenous progesterone supplementation. This has never been shown to be necessary or effective, but is of low risk (62). If given, supplementation is continued for approximately 4 weeks after the positive pregnancy test.
Contraindications:
IVF is contraindicated in women in whom pregnancy is contraindicated.
Outcome:
Federal law requires that the outcome of all IVF cycles be reported to the national database kept by the CDC and ASRM/SART (www.cdc.gov/nccdphp/drh/art99/index99.htmwebsite and www.sart.org).
For the 87,089 IVF cycles performed nationally in 2013, there was a 40.1% live birth rate per initiated cycle for women younger than 35 years, 31.4% for women between ages 35 and 37, 21.2% for women between 38 and 40, and 11.2% for women over 40 years (SART www.sart.org).
Gamete Intrafallopian Transfer (GIFT)
Gamete Intrafallopian Transfer (GIFT) is a procedure developed in 1983 by Dr. Ricardo Asch in San Antonio (63). The procedure begins in a therapeutic manner similar to IVF, however, when the oocytes are aspirated, they are mixed with sperm and immediately replaced in the patient"s fallopian tubes. GIFT requires less laboratory services and expertise than does IVF, but does require the patient to incur the risks of general anesthesia and laparoscopy. With increasing training and availability of IVF laboratory personnel, GIFT is rarely utilized.
Indications:
GIFT requires at least one normal, patent fallopian tube. Patients with unexplained infertility are those who benefit most by this procedure. Additionally, because embryos are not formed in the laboratory, some religious groups may find GIFT more acceptable than IVF.
Procedure:
GIFT begins with ovulation induction in a manner similar as described above for IVF (see IVF). When the patient is ready for oocyte aspiration, the aspiration may be performed either transvaginally with ultrasound guidance, or transabdominally under laparoscopic guidance. The oocytes are immediately combined with previously prepared sperm and allowed to incubate for 10 minutes to allow sperm binding to the zona pellucida. Approximately 25,000 motile sperm and two oocytes are placed in each Fallopian tube through a catheter placed in the tube under laparoscopic vision. Luteal support with exogenous progesterone may be given. Two weeks after aspiration a pregnancy test is performed.
Contraindications:
Patients with abnormal Fallopian tubes are not eligible for GIFT treatment. Any contraindication to pregnancy is a contraindication to GIFT.
Outcome:
Federal law requires that the outcome of all GIFT cycles be reported to the national database kept by the CDC and ASRM/SART (www.cdc.gov/nccdphp/drh/art99/index99.htmwebsite).
Overall, GIFT was performed with less than 1% of the aspirations in the United States in 2011, and because of the small number of cases, clinic outcome data specific for GIFT is unavailable. In general, the outcome of GIFT is thought to be comparable to that of ovulation induction with gonadotropins and intrauterine insemination.
Controlled Ovarian Hyperstimulation and Intrauterine Insemination (COH-IUI)
Controlled Ovarian Hyperstimulation and Intrauterine Insemination (COH-IUI) was first introduced in 1983 by Dodson et al. as a direct spin-off of GIFT (64). These clinical investigators reasoned that if the cyclic fecundity rate may be increase by placing processed sperm into the normal fallopian tube with 4 oocytes, perhaps the physical placement can be replaced by the patient’s natural physiology. Their first series of 85 patients were stimulated with gonadotropins to develop 4 mature follicles and the normal fallopian tubes were entrusted with oocyte pick-up. After administering hCG, processed sperm were delivered into the intrauterine cavity and allowed to be propelled naturally to the site of the oocyte. Previous studies revealed that this transport occurred within two minutes. This therapy was applied to patients with unexplained infertility so rapidly that it resulted in a demand on the supply of urinary gonadotropins so large that a backlog of drugs in the United States occurred.
Indications:
Unexplained infertility was the initial indication for COH-IUI. This was later extended to include some patients with pelvic adhesions (but patent tubes) and also patients with male factor infertility.
Procedure:
Beginning early in the menstrual cycle, gonadotropins are administered daily to patients. After approximately 5 days of therapy, ultrasound folliculograms and serum estradiol measurements are performed every 1 to 2 days to determine the dose and frequency of further gonadotropin administration. When three to four mature follicles are detected, human chorionic gonadotropin (hCG) is administered to trigger ovulation. Between 24 and 48 hours after hCG administration, an intrauterine insemination (IUI) is performed.
The sperm is prepared for IUI in a manner similar to that first described for IVF. After liquification, the ejaculate is diluted with a buffered media and centrifuged. The sperm is thus separated from the seminal plasma when the supernatant is discarded and the sperm-rich pellet is re-suspended in media. Processing thereafter varies with the laboratory and sperm characteristics. The sperm suspension may be layered in a sephadex column (Percoll), recentrifuged with media, or allowed to "swim-up" into an overlying layer of media. These procedures are intended to remove non-motile sperm, debris, bacteria, and white cells before intrauterine insemination. They also remove 60-80% of the motile sperm. No technique has proved superior for pregnancy outcome.
Contraindications:
Blocked fallopian tubes are a contraindication to COH-IUI because the tubes are required to pick up the oocytes from the ovary and facilitate the mechanical apposition of sperm and oocyte.
Outcome:
Pregnancy rates and pregnancy outcome after COH-IUI depend upon diagnosis. Few randomized studies are available to assess accurate outcomes after this therapy because it is often attempted in patients who would otherwise be treated with IVF. For example, a patient with pelvic adhesions who cannot afford IVF may opt for a cycle of COH-IUI in hope that more ovulations may increase the chance of an oocyte being geographically close to the fimbrial opening and afford tubal pick-up. The more ovulated oocytes, the better statistical chance of an oocyte reaching the tubal opening. Nonetheless, patients such as this would be less likely to conceive that a patient with normal tube-egg pick-up.
Strict criteria for unexplained and male factor infertility were applied in the randomized trial conducted by the Reproductive Medicine Network (65). This study revealed that patients with truly unexplained infertility had a 33% pregnancy/cycle of COH-IUI, higher than couples treated with either COH or IUI alone, and higher than controls.
The multiple pregnancy rate was 13.4% overall and the spontaneous abortion rate was 19% overall (22.3% in the COH treated groups). Interestingly, this study treated couples with more than one motile sperm and no other infertility factors. In those couples with sperm counts less than 0.2-21.8 x 10-6, couples were found to have and higher pregnancy rate when treated with COH-IUI than either COH or IUI alone, and higher than controls.
Not all couples with unexplained infertility are optimal candidates for COH-IUI. Women with an elevated serum AMH or high basal antral follicle count may experience production of multiple dominant follicles, leading to a higher risk of high-order-multiple pregnancy and/or OHSS. These negative outcomes were noted to be more prevalent in the above study with 6 cases of OHSS, 3 sets of quadruplets, and 4 sets of triplets among the 186 total pregnancies in the COH-IUI group (65). Many argue the relatively low reported pregnancy rates in COH-IUI compared with risks do not justify its use after failure of CC-IUI since IVF success rates have improved greatly over the last decade (66).
OOCYTE AND EMBRYO DONATION
Oocytes used in IVF are not required to be from the female partner of the infertile couple. Indeed, these procedures allowed women to utilize donor gametes as their male counterparts have been doing for over a century. A fertile woman who agrees to donate her oocytes anonymously or to a known couple, undergoes ovulation induction, and the retrieved oocytes are fertilized. Of course, the sperm may also be from a sperm donor. After embryonic development, the embryos are replaced into the female partner of the infertile couple. She is treated with exogenous hormones to synchronize her cycle with the donor.
Similarly, a couple can use embryos donated from another person or couple to become pregnant. The hormonal preparation of the female partner’s uterus is similar to an oocyte-donation cycle. In the US, each state has a unique set of laws governing use of donated embryos and oocytes, so practitioners must stay updated on changes in reproductive laws in their state. Legal restrictions and variable costs associated with embryo and oocyte donation has led to reproductive “tourism” where couples travel to other states or countries for reproductive treatment.
Indications:
Oocytes or embryos obtained from a donor is indicated for patients with medical or surgical menopause or genetic disorders that may impact offspring. Oocyte donors must be physically and mentally healthy and without major genetic diseases in the family, have attained the state’s age of legal majority, and preferably be within 21-34 years of age. The donor must be screened for HIV, hepatitis B surface antigen, hepatitis C antibody, syphilis, chlamydia, and gonorrhea (American Society of Reproductive Medicine Practice Committee Report: Recommendations for gamete and embryo donation: 2012.)
Procedure:
Both oocyte donors and recipients are carefully selected, counseled and screened before being accepted into the program. Donors may be known to the patient, a volunteer that altruistically donates to an anonymous recipient (usually reimbursed for missed work and effort), or an individual that is undergoing IVF and donates her spare oocytes. Stimulation and transfer are performed as with routine IVF.
Contraindications:
Any contraindication to pregnancy, spontaneous or induced ovulation would preclude either donation of oocytes or use in an IVF cycle.
Outcome:
For the 8,921 fresh treatment donor-oocyte cycles initiated in 2013, the reported delivery rate was 49.6% per initiated cycle (www.SART.org). Neither recipient age nor diagnosis plays a substantial role in the success of oocyte donation (67).
INTRACYTOPLASMIC SPERM INJECTION (ICSI)
Advancements in IVF promulgated its extension to couples with male factor infertility since in vitro fertilization required fewer moving sperm than in vivo fertilization, even after IUI. Furthermore, in the early 1990’s the zona pellucida was incised (zona slitting) or treated with hyaluronic acid (zona drilling) to facilitate sperm transgression across it in cases of male factor infertility. Another technique injected sperm under the zona pellucida (SZI). Indeed, inadvertent penetration of the cytoplasm and injection of a sperm during a SZI procedure in resulted in a pregnancy and sired the best treatment of male factor infertility since donor sperm: Intracytoplasmic Sperm Injection (ICSI) (68).
Indications:
ICSI is indicated for male factor infertility in which the count, motility, or strict morphology is low. The definite parameters will depend upon each program; in general, a sperm density <5 x 106, motility <20% and strict morphology <5% are indications. In addition, patients with antisperm antibodies may be best treated with ICSI. For 2013, 67% of reported IVF cycles in the United States utilized ICSI as the means for fertilization (www.SART.org).
ICSI also enabled men without sperm in their ejaculate to sire a pregnancy. Sperm aspirated from the epididymis (MESA) and from the testicle (TESE) may be used for injection. It is important, however, to realize that men with azoospermia may have a genetic defect which may possibly be inherited and result in infertility in subsequent male offspring. Similarly, men with at least one vas deferens congenitally absent may be a carrier of cystic fibrosis. His wife should be screened for cystic fibrosis mutations to assure that she, too, is not a carrier, lest they have a child affected with the disease.
ICSI has been proposed in non-male-factor infertility, but current literature does not support routine use (69). A randomized, multi-center trial showed similar clinical pregnancy rates with conventional insemination and ICSI in non-male-factor patients (33% vs. 26%) and a higher rate of failed fertilization (5%) in the conventional insemination group. Based on these data, the number needed to treat (NNT) to prevent one failed fertilization with ICSI is 33 (70-71).
Procedure:
Patients treated with ICSI undergo the same procedures as with IVF except that 5 hours after aspiration, one sperm is injected into each oocyte. The sperm is pretreated to remove it from the seminal plasma and placed in poly vinyl propylpyrrhidol (PVPP) which slows its movement and allows a single sperm to be aspirated into a glass micropipette. The tail of the sperm is frequently broken off to prevent migration from the cytoplasm after injection.
Contraindications:
Contraindications to ICSI are those of IVF.
Outcome:
ICSI requires IVF and Federal law requires that the outcome of all IVF cycles be reported to the national database kept by the CDC and ASRM/SART.
PREIMPLANTATION GENETIC TESTING
Preimplantation genetic testing encompasses procedures involving the removal of polar bodies from oocytes,blastomeres from cleavage-stage embryos, and trophectoderm cells from day 5-6 embryos to test for mutations in gene sequence or chromosome number prior to embryo transfer. The term "preimplantation genetic diagnosis" (PGD) is used when one or both parents carry a specific gene mutation or chromosomal rearrangement and testing is performed to determine whether that genetic abnormality has been transmitted to the oocyte or embryo. The term "preimplantation genetic screening" (PGS) applies when the genetic parents are presumed to be chromosomally normal and their embryos are screened for
abnormalities in chromosome number.
In 1990, the first established pregnancies using this procedure were reported (72). In two couples known to be at risk of transmitting adrenoleukodystrophy and X-linked mental retardation, two female embryos were transferred after in vitro fertilization (IVF), biopsy of a single cell at the six to eight cells stage, and sexing by DNA amplification of a Y chromosome-specific repeated sequence. Both women were confirmed as carrying normal female twins.
Indication:
PGD is indicated for couples at risk for transmitting a specific genetic disease or abnormality to their offspring. Historically evidence did not currently support the routine use of PGS (aneuploidy screening) to increase live birth rates in women of advanced maternal age, with multiple miscarriages or multiple IVF failures (73), but emerging research with newer PGS techniques has shown a modest improvement in implantation rates. A meta-analysis of 11 studies illustrated a higher sustained implantation rate (IR) with pooled relative risk (RR) of 1.39 (95% CI 1.21 – 1.60) among the randomized trials and RR of 1.75 (95% CI 1.15 – 1.45) among observational studies (74). PGS technologies continue to evolve and improve, making elective single embryo transfer of the embryo with highest implantation potential more attainable.
Procedure:
Following oocyte recovery with IVF (see IVF) material from an oocyte or embryo is
extracted by creating a perforation in the zona pellucid via a laser, acid Tyrode’s
solution, or a sharpened glass needle. The polar body, blastomere(s) or trophectoderm cells are then extracted using a small suction pipette or by gently compressing the oocyte or embryo to
extrude material through the opening. To identify specific gene mutations, PGD employs techniques involving the polymerase chain reaction (PCR) to amplify a segment of the genome that contains the specific gene of interest. Fluorescence in situ hybridization (FISH) is a technique that uses DNA probes labeled with distinctly colored fluorochromes. These probes bind to specific DNA sequences unique to each chromosome and detect missing or excess chromosomal material. Where FISH limits the number of chromosomes able to be tested at once, newer technologies have combined DNA microarrays and comparative genomic hybridization (CGH) to screen more genetic loci in PGS. More recent PGS platforms utilize single-nucleotide polymorphism (SNP) microarray, quantitative polymerase chain reaction (qPCR), and next-generation sequencing (NGS) to screen day 5-6 embryos for genetic abnormalities. In order to utilize newer PGS technologies, IVF centers must have experience in trophectoderm biopsy, ability to grow embryos to the blastocyst stage, and a successful cryopreservation program.
Contraindications:
Contraindications to PGD/PGS are those of IVF.
Outcome:
The estimated risk of transferring an affected embryo mistakenly identified as normal by traditional single-gene PGD is approximately 2% for recessive disorders and 11% for dominant disorders (75). Prenatal diagnostic testing to confirm the results of PGD is encouraged strongly because
the methods used for PGD have technical limitations that include the possibility for a false negative result.
NGS involves embryonic DNA being divided into 100-200 base pair sequences and comparing these sequences to a reference genome, and both whole chromosome and segmental chromosome imbalances can be identified (76). However, with this technology, mosaic DNA will labelled “abnormal,” and many argue embryos with some mosaicism may actually be viable in vivo. NGS outcomes are being followed closely as the technology evolves.
FERTILITY PRESERVATION
An increasing number of women are delaying reproduction until their late 30s, 40s and even into their fifth decade of life. Many women understandably have an interest in preserving their fertility, and are seeking the option to cryopreserve their oocytes for later use. Oocyte cryopreservation however, is still considered an experimental procedure and data related to clinical outcomes are limited.
According to the American Society for Reproductive Medicine, the only established methods of fertility preservation for women are oocyte and embryo cryopreservation (77,78). Cryopreservation of embryos (as opposed to oocytes) is well established, with the first successful pregnancy from frozen human embryos reported in 1983. Women in committed relationships may elect to cryopreserve embryos with their partners’ sperm, and single women may elect to cryopreserve embryos utilizing donor sperm. Oocyte cryopreservation has become more prevalent in the last decade for fertility preservation in women facing cancer treatment as well as those planning to electively defer childbearing. The American Society for Reproductive Medicine (ASRM) now views elective oocyte cryopreservation as a viable, non-experimental treatment option for women (78). Currently in the US, employers such as Apple and Facebook offer to help offset costs associated with oocyte cryopreservation in attempt to help career-oriented women preserve fertility. Thawed oocytes appear to have similar fertilization and implantation rates as fresh oocytes (79) due to improved cell survival from newer vitrification techniques, and there does not appear to be an increase in congenital anomalies in babies born after this process (80).
In the past, retransplantation of cryopreserved ovarian tissue has been proposed as a mechanism for fertility preservation in cancer patients. With only a small number of live births in the United States to date, this methodology will not be discussed further (for a concise review, refer to citation 77 and 81).
Indications:
Fertility preservation is an option for women with cancer or other illnesses requiring treatment that may compromise their fertility. Fertility preservation is also an elective option for women who are interested in childbearing at a later time, but patients need to be informed of the potential benefits, limitations and risks of this developing technology.
Procedure:
Procedures for oocyte and embryo cryopreservation begin in a manner similar as described for IVF (see IVF). The oocytes are cryopreserved immediately upon
harvest, or oocytes are fertilized and the embryos are cryopreserved typically on day three, five or six of culture.
Contraindication:
Fertility preservation procedures are contraindicated in women in whom exposure to fertility medications or oocyte retrieval is contraindicated.
Outcome:
In the absence of clinic-specific out comes, there is an overall 2% live birth rate per oocyte thawed via traditional slow freeze techniques and 4% live birth rate per oocyte thawed via vitrification (82,83). For cryopreserved embryos transferred in 2013, the live birth rate was 44.4% for women under age 35, 40.6% for women ages 35-37, 36.1% for women ages 38-40, 31.6% for women 41 to 42 years (www.SART.org).
Oocyte Nuclear Transfer:
Research continues in ART among women who are carriers for mitochondrial disorders, a diverse group of maternally-derived mutations resulting in progressive and life-threatening diseases. Animal studies replacing the cytoplasm and mitochondrial DNA (mtDNA) from an affected oocyte with that of a healthy donor oocyte have shown promise. The FDA in the US currently does not support human clinical trials to confirm or refute animal studies due to unknown implications for the offspring (www.fda.gov).
Summary:
Advances in technology have dramatically altered the treatment of infertile couples and have improved pregnancy and live birth outcomes. However, the methodical approach beginning with a detailed medical history and physical examination is a mainstay. Avoiding empirical tests and treatments will afford couples the most economical and successful therapy for this emotionally devastating disease.
BIBLIOGRAPHY
- Aldrich CK. A case of recurrent pseudocyesis. Perspect Biol Med. 1972;16:11-21.
- Grossi, N. A little pregnant or not at all? Pharos Spring 1999; 16-19.
- Dunbar HF. Emotions and bodily changes. 3rd Ed. New York:Columbia University Press.1946;341.
- Hamilton GV. A research in marriage. New York: Lear Publishers Inc.1948
- Kistner, R. Induction of ovulation with clomiphene citrate. Ob Gyn Survey. 1965. Dec (20)6;873-900.
- Chandra A, Martinez GM, Mosher WD, Abma JC, Jones J. Fertility, family planning, and reproductive health: Data from the 2002 National Survey of Family Growth. Vital Health Statistics 23(25). 2005.
- Menken J, Trussell J, Larsen U. Age and infertility. Science 1986:233: 1389-1394.
- Schwartz D, Mayaux MJ. Female fecundity as a function of age: results of artificial insemination in 2193 nulliparous women with azoospermic husbands. NEJM 1982;306:404-410.
- Teitze C. Reproductive span and rate of reproduction among Hutterite women.
Fert Steril. 1957;8(1): 89-97. - Wharburton D. Reproductive loss: how much is preventable? NEJM. 1987.316:158-160.
- Hassold T, Chen N, Funkhouser J, Jooss T, Manuel B, Matsura J, Matsuyama A, Wilson C, Yamane LA, Jacobs PA. A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet London 1980;44:151-164.
- Munne S, Alikani M, Tomkin G et al.,Embryo morphology, developmental rates and maternal age are correlated with chromosomal abnormalities. Fert Steril 1995;64(2):382-391.
- Schoolcraft B, Fragouli E, Stevens S, Munne S, Katz-Jaffe M, Wells D. Clinical application of comprehensive chromosomal screening at the blastocyst stage. Fertil Steril 2010;94(5):1700-6.
- Mineau G, Trussell J. A specification of marital fertility by parents age, age at marriage and marital duration. Demography.1982;19(3):335-349.
- 2010 Assisted Reproductive Technology Report. Center for Disease Control. http://www.cdc.gov/art/ART2013/index.htm.
- Ruijs GJ, Kauer FM, Jager S, Schroder FP, Schirm J, Kremer J. Further details on sequelae at the cervical and tubal level of Chlamydia trachomatis infection in infertile women. Fert Steril. 1991.Jul;56(1):20-6.17.. Querleu D. Fertility after conization .Rev Fr Gynecol Obstet 1991 Feb 15;86(2):81-2
- Oei SG, Helmerhorst FM, Keirse MJ. When is the post-coital test normal? Hum Repro 1995;10 (7):1711-1714.
- Griffith CS, Grimes DA : The validity of the postcoital test.Am J Obstet Gynecol 1990 Mar;162(3):615-20.
- Moinian M, Andersch B. Does cervix conization increase the risk of complications in subsequent pregnancies? Acta Obstet Gynecol Scand 1982;61(2):101-321. Wentz AC, Martens P, Wilroy RS Jr. Luteal phase inadequacy and a chromosomal anomaly in recurrent abortion. : Fertil Steril 1984 Jan;41(1):142-3
- Choudhury SR, Knapp LA. Human reproductive failure I: Immunological factors. Hum Repro Update 2001 Mar-Apr;7(2):113-34.
- Eldar-Geva T, Wood C, Lolatgis N, Rombauts L, Kovacs G, Fuscaldo J, Trounson AO. Cumulative pregnancy and live birth rates in women with antiphospholipid antibodies undergoing assisted reproduction. Hum Reprod 1999 Jun;14(6):1461-6.
- Wentz AC, Kossoy LR, Parker RA. The impact of luteal phase inadequacy in an infertile population. Am J Obstet Gynecol 1990;162:937-45.
- Peters AJ, Lloyd RP, Coulam CB. Prevalence of out of phase endometrial biopsy specimens. Am J Obstet Gynecol 1992;166:1738-46.
- Coutifaris C, Myers ER, Guzick DS, Diamond MP, Carson SA, Legro RS, McGovern PG, Schlaff WD, Carr BR, Steinkampf MP, Silva S, Vogel DL, Leppert PC; NICHD National Cooperative Reproductive Medicine Network. Histological dating of timed endometrial biopsy tissue is not related to fertility status. Fertil Steril. 2004 Nov;82(5):1264-72
- Pritts EA. Fibroids and infertility:A systematic review of the evidence. Obstet Gynecol Survey.2001;56(8):483-491.
- Schlaff WD, Zerhouni EA, Huth JA, Chen J, Damewood MD, Rock JA. A placebo-controlled trial of a depot gonadotropin-releasing hormone analogue (leuprolide) in the treatment of uterine leiomyomata. : Obstet Gynecol 1989 Dec;74(6):856-62
- Vercellini P, Maddalena S, DeGiorgi O, Aimi G, Crosignani PG. Abdominal myomectomy for infertility:a comprehensive review. Hum Repro 1998;13(4):873-879.
- Sankpal RS, Confino E, Matel A, Cohen LS. Investigation of the uterine cavity and fallopian tubes using three-dimensional saline sonohysterosalpingography. Int J Gynecol Obstet 2001 (73):125-129.
- ASRM. Revised American Society for Reproductive Medicine classification of endometriosis:1996. Fert Steril. 1997;67(5):817-821.
- Adamson GD. Treatment of endometriosis-associated infertility. Seminars in Repro Endocrinol.1997;15(3)263-271.
- Olive DL, Schwartz LB. Endometriosis. NEJM.1993;328(24):1759-1769
- Witz CA, Burns WN. Endometriosis and infertility: is there a cause and effect relationship? Gynecol Obstet Invest 2002;53 Suppl 1:2-11
- Parazzini, F and the Gruppo Italiano per lo Studio dell Endometriosi. Ablation of lesion or no treatment in minimal-mild endometriosis in infertile women:a randomized trial. Hum Repro.1999;14(5):1332-1334.
- Wellbery C. Diagnosis and treatment of endometriosis. Am Fam Physician 1999;60:1753-62,1767-8.
- Fabre V, Camus M, Devroey P. Endometriosis and sterility. Rev Prat 1999;49:279-81.
- Guermandi E, Vegetti W, Bianchi M, Uglietti A, Ragni G, Crosignani P. Reliability of ovulation tests in infertile women. Obstet Gynecol.2001;97:92-6.
- Moghissi KS, Syner FN, Evans TN. A composite picture of the menstrual cycle. Am J Obstet Gynecol 1972;114:405.
- Bauman JE. Basal body temperature:Unreliable method of ovulation detection. Fert Steril 1981.36:729-733
- Gorlitsky GA, Kase Ng, Speroff L. Ovulation and pregnancy rates with clomiphene citrate. Obstet Gynecol.1978;51:265.
- Gysler M, March CM, Mishell jr DR, Bailey EJ. A decade’s experience with an individualized clomiphene citrate treatment regimen including its effect on the postcoital test. Fertil Steril. 1989;52:35.
- Nardo LG, Rai R. Metformin therapy in the management of polycystic ovary syndrome: endocrine, metabolic and reproductive effects. Gynecol Endocrinol 2001 Oct;15(5):373-80
- Heard MJ, Pierce A, Carson SA, Buster Je. Pregnancies following use of metformin for ovulation induction in patients with polycystic ovarian syndrome. Fertil Steril.2002;77(4):669-673.
- Legro RS, Barnhart HX, Schlaff WD, Carr BR, Diamond MP, Carson SA, Steinkampf MP, Coutifaris C, McGovern PG, Cataldo NA, Gosman GG, Nestler JE, Giudice LC, Leppert PC, Myers ER; Cooperative Multicenter Reproductive Medicine Network. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N Engl J Med.2007 Feb 8;356(6):551-66.
- 46. Thessaloniki ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Consensus on infertility treatment related to polycystic ovary syndrome. Hum Reprod. 2008 Mar;23(3):462-77.
- Nestler JE, Jakubowicx DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycyctic ovarian syndrome. NEJM 1998;1876-80.
- Casper RF, Mitwally MF. Review: aromatase inhibitors for ovulation induction. J Clin Endocrinol Metab 2006;91:760-771.
- Legro RS, Brzyski RG, Diamond MP, Coutifaros C, et al. Letrozole versus clomiphene for infertility in the polycystic ovary syndrome. NEJM 2014;371(2)119-29.
- Falcone T. What the internist needs to know about infertility.Cleve Clin J Med 2001 Jan;68(1):65-72; discussion 73.
- Cooper TG, Noonan E, von Eckardstein S, et al. Hum Reprod Update 2010;16(3):231-45
- Collins JA, Milner RA, Rowe TC. The effect of treatment on pregnancy among couples with unexplained infertility. Int J Fertil 1991 May-Jun;36(3):140-1, 145-52
- Serhal PF, Katz M, Little V, Woronowski H. Unexplained infertility-the value of pergonal superovulation combined with intrauterine insemination.Fertil Steril 1988;49:602-605.
- Guzick DS, Sullivan MW, Adamson GD, Cedars MI, Falk RJ, Peterson EP, Steinkampf MP. Efficacy of treatment for unexplained infertility. Fertil Steril 1998;70(2):207-13.
- Gleicher N, Friberg J, Fullan N, Giglia RV, Mayden K, Kesky T, Siegel I. EGG retrieval for in vitro fertilization by sonographically controlled vaginal culdocentesis. Lancet 1983 Aug 27; 2(8348):508-9.
- Del Gadillo JC, Siebzehnrubl E, Dittrich R, Wildt L, Lang N. Comparison of GnRH agonists and antagonists in unselected IVF/ICSI patients treated with different controlled ovarian hyperstimulation protocols: a matched study. Eur J Obstet Gynecol Reprod Biol 2002 May 10;102(2):179-83
- Pundir J, Sunkara SK, El-Toukhy T, Khalaf Y. Meta-analysis of GnRH antagonist protocols: do they reduce the risk of OHSS in PCOS? Repro Biomed Online 2012;24(1):6-22.
- Engmann L, DiLuigi A, Schmidt D, Nulsen J, Maier D, Benadiva C. The use of gonadotropin-releasing (GnRH) agonist to induce oocyte maturation after cotreatment with GnRH antagonist in high-risk patients undergoing in vitro fertilization prevents risk of ovarian hyperstimulation symdrome: a prospective randomized controlled study. Fertil Steril 2008;89(1):84-91.
- Iliodromiti S, Blockeel C, Tremellen KP, Fleming R, Tournaye H, Humaidan P, Nelson SM. Consistent high clinical pregnancy rates and low ovarian hyperstimulation syndrome rates in high-risk patients after GnRH agonist triggering and modified luteal support: a retrospective multicenter study. Hum Reprod 2013;28(9):2529-36.
- Schnell VL, Sacco AG, Savoy-Moore RT, Ataya KM, Moghissi KS. Effects of oocyte exposure to local anesthetics on in vitro fertilization and embryo development in the mouse. Reprod Toxicol 1992;6(4):323-7
- Edwards RG, Steptoe PC, Purdy JM. Fertilization and cleavage of preovulatory human oocytes. Nature. 1970. 227: 1307-9.
- Daya S. Efficacy of progesterone support in the luteal phase following in-vitro fertilization and embryo transfer: meta-analysis of clinical trials. Hum Reprod 1988 Aug;3(6):731-4
- Asch RH, Balmaceda JP, Ellsworth LR, Wong PC. Preliminary experiences with gamete intrafallopian transfer (GIFT). Fertil Steril 1986 Mar;45(3):366-71
- Dodson WC, Whitesides DB, Hughes CL, Easley HA, Haney AF. Superovulation with intrauterine insemination in the treatment of infertility:a possible alternative to gamete intrafallopian transfer and in vitro fertilization. Fertil Steril.1987;48(3):441-445.
- Guzick DS, Carson SA, Coutifaris C, Overstreet JW, Factor-Litvak P, Steinkampf MP, et al. Efficacy of superovulation and intrauterine insemination in the treatment of infertility. National Cooperative Medicine Network. NEJM 1999;340(3):177-83.
- Reindollar RH, Goldman MB. Gonadotropin therapy: a 20th century relic. Fertil Steril 2012;97(4):813-818.
- Paulson RJ, Hatch IL, Lobo RA, Sauer MV. Cumulative conception and live birth rates after oocyte donation;implications regarding endometrial receptivity. Hum Repro.1997;12:835-9.
- Van Steirteghem, A., Nagy, P., Joris, H. et al. The development of intracytoplasmic injection. Hum. Reprod.,1996. 11 (Suppl.1), 59-72
- American Society for Reproductive Medicine Practice Committee Opinion: ICSI for non-male factor infertility. 2012.
- Bukulmez O, Yarali H, Yucel A, Sari T, Gurgan T. Intracytoplasmic sperm injection versus in vitro fertilization for patients with a tubal factor as their sole cause of infertility: a prospective, randomized trial. Fertil Steril 2000;73:38-42.
- Poehl M, Holagschwandtner M, Bichler K, Krischker U, Jurgen S, Feichtinger W. IVF patients with nonmale factor “to ICSI” or “not to ICSI” that is the question. J Assist Reprod Genet 2001;18:205-8.
- A.H. Handyside, E.H. Kontogianni, K. Hardy and R.M. Winston, Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification, Nature 344 (April (6268)) 1990 68-770.
- Preimplantation genetic testing: a Practice Committee Opinion. The Practice Committee of the Society for Assisted Reproductive Technology and the Practice Committee of the American Society for Reproductive Medicine Fertil Steril 2008;90:S136-43.
- Dahdouh EM, Balayla J, Garcia-Velasco JA. Comprehensive chromosomal screening improves embryo selection: a meta-analysis. Fertil Steril 2015;104(6):1503-12.
- Lewis CM, Pinel T, Whittaker JC, Handyside AH. Controlling misdiagnosis
errors in preimplantation genetic diagnosis: a comprehensive model encompassing extrinsic and intrinsic sources of error. Hum Reprod 2001;16:43-50. - Fiorentino F, Biricik A, Bono S, Spizzichino L, Cotroneo E, et al. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil Steril 2014;101(5):1375-1382.
- Ethics Committee of the American Society for Reproductive Medicine. Fertility Preservation and reproduction in cancer patients. Fertil Steril 2005;83(6):1622-1628.
- Practice Committee of the American Society for Reproductive Medicine. Mature oocyte cryopreservation. January 2013.
- Cobo A, Mesenguer M, Remohi J, Pellicer A. Use of cryo-banked oocytes in an ovum donation programme: a prospective, randomized, controlled, clinical trial. Hum Reprod 2010;25:2239-46.
- Noyes N, Porcu E, Borini A. Over 900 oocyte cryopreservation babies born with no apparent increase in congenital anomalies. Reprod Biomed Online 2009;18(6)769-76.
- Bromer JG, Patrizio P. Fertility preservation: the rationale for cryopreservation of the whole ovary. Semin Reprod Med. 2009 Nov;27(6):465-71.
- Oktay K, Cil AP, Bang H. Efficiency of oocyte cryopreservation: a metaanalysis.Fertil Steril 2006;86:70-80.
83. Goldman KN, Noyes NL, Knopman JM, McCaffrey C, Grifo JA. Oocyte efficiency: does live birth rate differ when analyzing cryopreserved and fresh oocytes on a per-oocyte basis? Fertil Steril 2013;100(3):712-7.
Central Causes of Amenorrhea
ABSTRACT
Normal ovulatory menstrual cycles reflect a coordination of an intact hypothalamic-pituitary-ovarian unit. This process involves neuroendocrine messaging between the hypothalamus, pituitary, and gonads. Common central causes of amenorrhea include functional hypothalamic anovulation, exercise-associated anovulation, pharmacologic-induced anovulation, psychiatric associated disorders such as anorexia nervosa and bulimia, and genetic-developmental defects of the gonadotropin releasing hormone (GnRH) neuronal unit. While brain radiation, trauma or a congenital deficiency of gonadotropins can certainly result in amenorrhea, many more common subtle factors can also affect this pathway. Neuroendocrine regulation of ovulation involves the interaction between neurons secreting GABA, kisspeptin, and neurokinin which modulate the function of GnRH neurons. There is also a link between the reproductive axis, nutritional-energy balance status, and dysregulation of two key peptides, leptin and ghrelin, that occurs when there are extreme, nutritionally stressful conditions such as with anorexia nervosa, extreme exercise, or starvation. In the evaluation of patients with central amenorrhea, the history and physical are crucial to determine the laboratory evaluation and if brain imaging is required. Understanding the patient’s goals will allow an individualized treatment regimen, as it will likely differ if the patient desires children imminently. For complete analysis of this and all areas of Endocrinology, please visit our FREE web-textbook, www.endotext.org.
Introduction
The hypothalamic-pituitary-ovarian axis must be fully synchronized for normal ovulation and successful reproduction. Factors that modulate hypothalamic gonadotropin hormone releasing hormone (GnRH) include pubertal maturation, alterations in energy balance, body composition and metabolism, and stress and emotional changes. Disruption of normal menstrual cycles and normal ovulation are often associated with a variety of life style factors such as excessive exercise, nutritional deprivation, and psychological stress. In the vast majority of cases, this is associated with normal neuroanatomic findings. In a small subset, neuroendocrine abnormalities such as isolated gonadotropin deficiency (Kallmann syndrome), head trauma, radiation effects, Sheehan syndrome, and pituitary apoplexy are identified (see Table 1). Regardless of the etiology, the final common pathway is a change in the normal pattern of episodic secretion by the GnRH pulse generator resulting in disruption of ovulation and amenorrhea. These types of disorders will be discussed in this chapter.
| Functional Hypothalamic Anovulation |
|
| Physiologic Anovulation |
|
| Pharmacologic-Associated Anovulation |
|
| Psychiatric-Associated Disorders |
|
| Organic Defects of the Hypothalamic-Pituitary Unit |
|
NEUROENDOCRINE CONTROL OF GNRH SECRETION DURING THE REPRODUCTIVE CYCLE
Prior to the existence of laboratory techniques to assess gonadotropin secretion, physicians relied on clinical judgement to arrive at the diagnosis of central amenorrhea (1). The advent of the radioimmunoassay in the late 1960s paved the way for a laboratory diagnosis of this disease. The secretion of endogenous GnRH is difficult to assess in the human because this decapeptide is rapidly metabolized within 2 to 4 minutes in the peripheral circulation. Thus, it is not possible to directly assess GnRH secretion and the majority of clinical investigations utilize frequent measurements of LH concentrations as the surrogate marker for hypothalamic GnRH secretion. In women with regular menstrual cycles, clinical studies have demonstrated a characteristic pulsatile secretion of LH at a frequency of 90-120 minutes during the follicular phase and a frequency of 180-240 minutes during the luteal phase (2). Small alterations in LH pulsatile frequency and or amplitude can result in a range of disorders including luteal phase defects, oligo-ovulation and anovulation. Thus, most studies have examined for changes in LH pulsatile frequency and amplitude as the major endpoint in studies to investigate functional hypothalamic amenorrhea.
Puberty
Normal female pubertal development requires an elaborate orchestration of the hypothalamic-pituitary-gonadal axis. Prepubertal girls have low LH, FSH, and estradiol. Prepubertal girls respond with a rise in gonadotropin secretion (predominantly FSH) in response to exogenous GnRH administration (3). This response indicates that the hypothalamic GnRH pulse generator is less active during this period of development. At pubertal onset there is a distinct diurnal variation as the gonadotropins rise (4). Although the exact triggers at the time of puberty are poorly understood, there are three distinct changes that are observed in the hypothalamic-pituitary unit (5).
- A nocturnal sleep-related augmentation of pulsatile LH secretion begins as a result of the increase in GnRH pulsatility.
- The sensitivity set point of the hypothalamus to estradiol and testosterone decreases, thereby resulting in an increased basal gonadotropin secretion.
- In the female, a positive feedback capability develops, and critical levels of estradiol can trigger a large release of GnRH, and subsequently LH, leading to ovulation.
Central axis control of gonadotropins
The effect of estradiol on the CNS is generally dichotomous. While E2 sensitizes the pituitary gonadotrophs to GnRH by inducing GnRH receptor expression, it also suppresses the release of GnRH from the hypothalamus (6). This "negative feedback" is the dominant effect of estradiol for the vast majority of the ovulatory cycle. The negative control of gonadotropin release is augmented by inhibin secreted from ovarian granulosa cells. Unlike other endocrine systems, continued exposure to higher levels of estradiol (>250 pg/mL) for prolonged duration (>48hours) produced by the dominant ovarian follicle results in a sudden release of a GnRH-LH surge termed "positive feedback" that triggers ovulation at midcycle (7).
Neuroendocrine regulation of ovulation
During embryogenesis in primates, the GnRH cells migrate from the nasal placode into the bilateral arcuate nuclei in the mid-hypothalamus. The neurons of the arcuate nucleus that target the GnRH cells synapse with incoming axons from extrahypothalamic brain areas such as the amygdala, hippocampus, and cortex that target the GnRH cells. Conceptually, these interneuronal connections release excitatory (E) or inhibitory (I) neurotransmitters. Variations in the synaptic balance of these neurotransmitters exist during the ovulatory cycle. During negative feedback, the E/I ratio is low and during positive feedback the E/I ratio is high (8). Gamma amino isobutyric acid (GABA) neurotransmitters are primarily associated with inhibitory GnRH activity, while kisspeptin and neurokinin are associated with excitatory GnRH activity (9). Estradiol effects on GnRH pulsatility appears to be mediated by altering the E/I ratio through synaptogenesis and synaptolysis of neuronal pathways targeting GnRH secretion (10). During the bulk of the cycle (negative feedback) the effect of E2 is synaptogenic and induces neurotransmitters with a net inhibition of GnRH secretion. In contrast, during the pre-ovulatory phase (positive feedback), estradiol triggers synaptolysis, thereby causing the ovulatory surge.
Kisspeptins are G protein-coupled receptor ligands originally identified as human metastasis suppressor gene products that have the ability to suppress melanoma and breast cancer metastasis. These peptides have recently been found to play an important role in initiating the secretion of GnRH at puberty (11). Kisspeptin, encoded by the KISS1 gene is located on the long arm of chromosome 1 (1q32) and is a peptide consisting of 145 amino acids. In the brain, the gene is transcribed within the hippocampal dentate gyrus and activates the G protein-coupled receptor GPR54.
In 2003 the discovery of the necessary role kisspeptin has in puberty led to the understanding of the neuroendocrine effect on human reproduction (12). The GnRH neurons of primates, rodents and sheep are found in close apposition with kisspeptin neurons. GnRH neurons express the kisspeptin receptor, and when kisspeptin is incubated with hypothalamic explants, it stimulates the release of GnRH. This effect is not observed in kisspeptin receptor knockout mice. While GnRH neurons show an increase in firing rate in vitro following kisspeptin treatment, this effect is attenuated by the application of a kisspeptin receptor antagonist. The discovery of inactivating mutations in the kisspeptin/GPR54 further ascertained its role in human reproduction. In humans, inactivating GPR54 mutations are associated with normosmic hypogonadotropic hypogonadism while activation of GPR54 signaling is associated with precocious puberty. Central and peripheral administration of kisspeptin stimulates the hypothalamic-pituitary-gonadal axis while pre-administration of a GnRH antagonist abolishes this effect. Collectively, these observations strongly suggest that kisspeptin can be a primary mediator for activation of GnRH neurons.
All of the current studies initiating puberty with exogenous kisspeptin were performed on monkeys and rodents; however, one study demonstrated return of reproductive hormone production in women with hypothalamic amenorrhea who received twice-weekly kisspeptin injections for 8 weeks (13). Another potential clinical application for kisspeptin is to use it as a trigger for the LH surge during ovulation induction for in vitro fertilization (IVF). It has been shown to effectively induce oocyte maturation, and may be a more physiologic mechanism for inducing ovulation than hCG, which is currently used for ovulation induction. There is speculation that a kisspeptin trigger may also decrease the risk for ovarian hyperstimulation syndrome; however, studies comparing kisspeptin and a GnRH agonist need to be completed prior to recommending the use of kisspeptin (14). Further studies are ongoing to further establish the role of kisspeptin in restoring reproductive function in certain conditions.
Modulation of GnRH Secretion by Opioidergic, Dopaminergic, and Excitatory Amino Acids
Animal studies have shown that other neurotransmitter systems such as dopamine, norepinephrine, and serotonin can regulate GnRH or LH secretion. These studies suggest that activation of the noradrenergic system is associated with increased release of GnRH whereas dopaminergic or serotonergic activation can either inhibit or stimulate GnRH release (15-21). These observations can explain in part the CNS-associated disruption of normal menstrual cycles in patients who take phenothiazine (dopamine receptor antagonists), stimulants, antidepressants, and sedatives on a chronic basis.
Excitatory amino acids such as glutamate and aspartate have been shown to be localized to the arcuate nucleus in the media basal hypothalamus adjacent to GnRH neurons and have been implicated in a regulatory role for GnRH secretion primarily during pubertal maturation (22). These two amino acids appear to activate GnRH secretion during puberty in monkeys.
Endogenous opiates peptides such as endorphins, enkephalins, and dynorphins appear to play largely an inhibitory role in GnRH and LH secretion. The same functional neuronal network that secretes kisspeptin also co-secrete dynorphin, and collectively it is known as the kisspeptin-neurokinin B-dynorphin (KNDY) pathway (23). KNDy neurons in the infundibular/arcuate nucleus have an effect on GnRH by influencing both the GnRH cell body and the neurosecretory terminals. Given the KNDy cells express both neurokinin B receptors and the kappa opioid peptide receptor (dynorphin receptor), it is suspected that the stimulatory role of neurokinin B and the inhibitory role of dynorphin work together to cause a pulsatile release of kisspeptin, which results in a GnRH pulsatile release (24). In patients with hypothalamic amenorrhea, blockade of endogenous opiate receptors with the receptor antagonists such as naloxone or naltrexone (dynorphin antagonists) will induce an increase an increase in pulsatile release of GnRH and LH (25). Long term treatment of hypothalamic amenorrhea patients with naltrexone can result in return of normal menstrual cycles in some individuals (26). These findings indirectly suggest that endogenous opiate activity is suppressing GnRH secretion. The use of an inhibitory neurotransmitter may be beneficial in patients who require a suppression of GnRH or LH secretion. For example, patients with PCOS generally have a dysregulation of gonadotropin secretion which likely contributes to irregular ovulation may benefit from exogenous regulation of the opiate peptides (27).
LINKAGE BETWEEN NUTRITIONAL STATUS AND HYPOTHALAMIC AMENORRHEA
For many years, clinicians have sought to identify the functional link between nutritional status and reproduction. Recent identification of neuropeptides that alter feeding behaviors has provided a physiological mechanism to explain the shutdown of the H-P-O axis for individuals who experience significant changes in nutritional status (i.e. starvation and obesity). Two key peptides, leptin and ghrelin have been identified and appear to regulate feeding behavior which may mediate the dysregulation of the reproductive axis under extreme, nutritionally stressful conditions.
Leptin is a 16 Kd, 167 amino acid polypeptide that was first isolated in 1994. This peptide is a product of the ob gene. Based on phylogenetic studies, leptin is conserved and has been identified in amphibians, rat, sheep, and human (28). Leptin is primarily produced by the adipocytes but has also been shown to be synthesized in other tissues such as skeletal muscle, heart, stomach, and the placental-fetal unit. Leptin is a member of the cytokine family and is classified as an anorexigenic peptide. Other peptides in this class include pro-opiomelanocortin and the cocaine and amphetamine-regulated transcription peptides. Leptin’s action is opposed by orexigenic peptides such as neuropeptide Y and the agouti-related peptide.
Leptin is known to circulate in two forms: as a free form and as a bound form complexed to soluble leptin-binding proteins or to circulating leptin receptors. Physiological studies demonstrate that leptin is secreted in a pulsatile manner with a diurnal rhythm. Current commercial assays for leptin appear to measure total leptin in the serum compartment. Transport of leptin into the brain has been described as unidirectional through the blood brain barrier into brain tissue.
For patients with HA, most studies report a decrease in total circulating leptin with a loss of the normal diurnal rhythm (29). This lower leptin level is a common characteristic of several energy-deficient conditions and is associated with a decrease of the LH pulse frequency. There have now been six receptor isoforms described for leptin. These receptors have been localized to the brain, ventral hypothalamus, lung, kidney, pancreas, adrenals, ovaries, skeletal muscle, and hematopoietic stem cells. Within the brain, the arcuate nucleus appears to have the greatest concentrations of leptin receptors (as well as GnRH receptors).
Binding of leptin to its receptor causes functional dimers to form and activates the JAK/STAT3 intracellular signaling pathways. The effects of leptin on GnRH release may be mediated through kisspeptin. The leptin receptor (Ob-Rb) is not expressed by GnRH neurons of the hypothalamus. However Ob-Rb mRNA is found in 40% of Kiss1 mRNA-expressing cells of the arcuate nucleus (30). The expression of Kiss1 mRNA in the mutated leptin receptors is reduced in comparison with wild-type mice. Furthermore, the expression of Kiss1 mRNA has been shown to be influenced by nutritional status. In pre-pubertal rats, which have been food deprived for 72 h, the hypothalamic expression of Kiss1 mRNA is markedly reduced (31).
In contrast to leptin, ghrelin, another energy-balance peptide, was found to be elevated in individuals with HA. Ghrelin is a 28-amino acid orexigenic peptide that was first isolated in the stomach and is a potent stimulus to GH release. In the fasting state, ghrelin serves as the hunger signal from the periphery to the CNS, acting on the hypothalamic arcuate nucleus, a region which is known to control food intake. Based on these relationships, it is tempting to rationalize that leptin and ghrelin simply serve as natural on/off switches for regulation of feeding behavior.
Functional Hypothalamic Amenorrhea
A practical definition of functional hypothalamic amenorrhea (HA) is the absence of menstrual cycles for more than 6 months without evidence of anatomic or organic abnormalities (32). Three main types of functional hypothalamic amenorrhea have been recognized, associated with stress, weight loss, or exercise (33). Other more serious organic disorder can mimic HA such as isolated gonadotropin deficiency (see table 1). Thus, this diagnosis should be made after exclusion of other causes.
HA is associated with increased cortisol secretion that reflects increased activity of the hypothalamic-pituitary-adrenal (HPA) axis (34). There appears to be an increased secretion of a "stress response complex" of hormones such as CRH, ACTH, cortisol, PRL, oxytocin, vasopressin, norepinephrine, and epinephrine (TABLE 2). CRH has been shown to directly inhibit GnRH secretion in rats, monkeys, and humans at the hypothalamic level in in vitro and in vivo experimental models. This inhibition can be negated by administration of a CRH receptor antagonist or by naloxone, an opiate receptor antagonist. Taken together, these observations suggest that the inhibitory effect of CRH is mediated in part by increased opioidergic activity.
The increased secretion of ACTH at the pituitary level may also suppress pituitary response to GnRH. In addition, increased cortisol levels may also dampen pituitary response to GnRH. It must be emphasized that an acute stress response will be unlikely to alter ovulatory function since the H-P-O axis is quite resilient. On the other hand, it appears that chronic environmental stressors lead to long term activation of the HPA axis which in turn can induce ovulatory dysfunction at either the hypothalamic or the pituitary level.
Modification of the stress response can restore normal HPO function. The validation for this comes from evidence of complete resumption of normal ovulatory function in women with HA treated with cognitive behavioral therapy (CBT) (35). Treated women exhibited increased LH pulsatility and decreased cortisol levels. A similar study at the same institution found that CBT resulted in an increase in leptin and TSH levels, and improved the neuroendocrine and metabolic components of HA (36). This provides evidence that stress-reducing behavioral changes may be successful in restoring normal ovulation and metabolic function in women with HA.
A constellation of other neuroendocrine alterations occur in HA. There is a suppression of the hypothalamic-pituitary-thyroid axis with a resultant decrease in circulating thyroid hormones (37). There also may be a decrease in prolactin secretion as well as alterations in other neurohormones such as melatonin (38). There may be difficulties in recognizing the stressors that have elicited the stress response in FHA since the stressors may not be objective or quantifiable (39). Human stressors often reflect attitudes of self or society. Subjective stressors such as these may not be as easy to quantitate as stressors with objective measures such as food deprivation or outright violence.
Evaluation
The American Academy of Pediatrics and the American College of Obstetricians and Gynecologists have advocated for menstrual status to be considered a “vital sign” at routine clinical visits, given the importance of estrogen for bone and other tissues (40). Many individuals with this disorder will provide a history of normal menarche and regular menstrual cycles between 26 and 35 days in length. These women typically are intelligent, high-achievers, who are usually thin or of normal body weight. A detailed interview may identify a stressful event or emotional crisis (divorce, relation breakup, death of a friend or relative) preceding the amenorrhea. Other interpersonal and environmental stressors may also be present such as academic pressures, job stresses, or psychosexual problems. A careful review of the patient's current lifestyle including exercise intensity, dietary choices, and the use of sedatives or hypnotics may be helpful in characterizing the psychogenic stress components. The patient may have features of the female athlete triad, which consist of amenorrhea, insufficient caloric intake with or without an eating disorder, and low bone density or osteoporosis.
The physical examination should focus on identifying galactorrhea, thyroid dysfunction, and evidence of hyperandrogenemia (i.e., acne, hirsutism). The pelvic examination should be normal except for a thinned vaginal mucosa or absent cervical mucus which are characteristics of hypoestrogenism. Despite these findings, these patients do not usually experience hot flushes.
(Table 3, 4)
Laboratory evaluation should include FSH, prolactin (PRL), and TSH. Most of the other pituitary hormones should be in the normal range (Table 4). In many patients, the progestin challenge test (medroxyprogesterone acetate 10 mg for 7 days) will demonstrate an absence of withdrawal uterine bleeding or vaginal spotting. This test is a bioassay for the absence of estrogen priming of the endometrium and reflects the chronically low circulating levels of estradiol.
The basic defect in women with functional hypothalamic amenorrhea is the failure of the hypothalamus to increase GnRH output in the presence of severe hypoestrogenism. Most investigators believe that there is a slowing of the GnRH pulse generator as reflected by a decrease in peripheral pulsatile LH secretion in these women. The pattern of LH secretion may vary. During the early onset, LH pulse frequency and amplitude may be normal, and in more severe cases, regression to a prepubertal pattern may occur. During recovery from HA, sleep- associated increases may be observed (see Figure 1).
The pituitary gland is fully functional and capable of synthesizing and release of LH and FSH. However, responses to exogenous GnRH in these individuals may vary depending on the endogenous GnRH priming of the pituitary gland. LH and FSH responses to exogenous GnRH may be absent, normal, or supernormal. In these patients, after a period of priming with intravenous pulsatile GnRH (1-2 mg/90 minutes), normal levels of LH and FSH can be restored and responses to exogenous GnRH become normal. Taken together, these observations suggest that endogenous GnRH secretion is deficient and, gonadotropin secretion and ovarian function can be normalized with physiologic replacement of exogenous, pulsatile GnRH.
Magnetic resonance imaging (MRI) of the brain is not routinely needed in patients with
presumed hypothalamic amenorrhea (41). However, it is indicated in patients who have a history of severe or persistent headaches, persistent vomiting that is not self-induced, central hypothyroidism, hyperprolactinemia or galactorrhea, or a change in thirst, urination, or vision.
Figure 1
CLINICAL MANAGEMENT OF FUNCTIONAL HYPOTHALAMIC AMENORRHEA
The clinical evaluation of HA should focus on a carefully conducted history and physical examination that reviews life style variables and interpersonal relationships. Since this is a diagnosis of exclusion, significant organic diseases must be excluded (Table 1). In many patients, spontaneous recovery of menstrual function will take place following accommodation to environmental stressors or after modification of life style. Psychological counseling may also be appropriate for these individuals. Because of the functional nature of HA, an individualized and expectant management should be considered the initial approach. In those who remain amenorrheic, periodic evaluation of menstrual status every 4 to 6 months is prudent.
In the infertile patient with this diagnosis who fails to resume normal cycles, a trial of low dose clomiphene citrate (25-50 mg for 5 days) is appropriate. In these patients, higher doses of clomiphene may suppress the H-P axis due to the weak estrogenic properties of clomiphene in an already hypoestrogenic environment. For clomiphene failures, use of human menopausal gonadotropins as an alternative method is highly successful. If available, pulsatile administration of GnRH 5 mg/90 minutes intravenously using a modified insulin pump will successfully induce ovulation after a 13 to 14 day treatment period (42). This latter approach is associated with ovulation rates of greater than 90% with generation of a single dominant ovarian follicle and much lower rates of ovarian hyperstimulation. In these patients, corpus luteum support can be maintained with either GnRH or human chorionic gonadotropin 1500 units intramuscularly every three days for 4 doses.
For the woman who remains amenorrheic for more than 1 year, the long term risks of hypoestrogenism including reduced bone mineral density and osteoporosis become factors. In young women with persistent hypoestrogenism, the bone mass can decrease at a rate of 2-5% per year for the first three to five years. Dual energy X-ray absorptiometry (DEXA) is often necessary to convince patients to begin estrogen therapy. The minimal dose of estrogen necessary to conserve bone has been established in menopausal women. At least 0.3 mg of conjugated estrogen, 1 mg of micronized estradiol, or 0.025 mg of transdermal estrogen is necessary to protect against bone loss. Use of a progestin on a cyclic basis such as medroxyprogesterone acetate for 10 to 12 days each month is necessary to ensure regularly shedding of endometrium and prevent endometrial hyperplasia. Calcium and vitamin D supplements may also be taken; however, the primary therapy requires treatment of the underlying process. The use of bisphosphonates in reproductive-aged women should be carefully considered.
As previously discussed, Leptin plays a critical role in regulation of kisspeptin and in HA. A possible future treatment for HA may include leptin replacement as it has been shown to restore pulsatility of gonadotropin-releasing hormone and ovulation as well as full development of secondary sexual characteristics in females with hypogonadotropic hypogonadism (43). However, the effect of leptin therapy on bone health is unknown. Similarly possible future treatments options may include exogenous kisspeptin.
Table 2. Associated Neuroendocrine Abnormalities in Hypothalamic Amenorrhea.
| Increased daytime cortisol secretion
Increased amplitude and duration of nocturnal melatonin secretion Increased nocturnal secretion of GH Elevated CRH levels in cerebral spinal fluid Blunted elevation of PRL, ACTH, and cortisol during the noon meal |
Table 3. Common Features of Women with Psychogenic Hypothalamic Amenorrhea
| Single marital status
Obsessive-compulsive habits History of significant stressful life events History of sexual abuse History of prior irregular menstrual cycles Normal or thin habitus Tendency to use sedatives or hypnotic drugs Involved in professional occupations High intelligence |
Table 4. Expected Serum Hormonal Parameters in Functional Hypothalamic Amenorrhea
| Hormone | Expected Values |
| LH
|
Normal or low
|
| FSH | Normal or low |
| PRL | Normal or low |
| TSH | Normal |
| GH | Normal or low |
| Estradiol | Normal or low |
| ACTH | Normal |
| Cortisol | Daytime elevation with diurnal variation |
| Testosterone | Normal or low |
| T3 | Normal |
| T4 | Normal |

Figure 1. Examples of LH pulsatile secretion at various stages of the hypothalamic-pituitary activation. GnRH-LH secretion can progress from an apulsatile stage; to a high amplitude infrequent pulse stage; to a sleep-entrained pulse stage; and to regular every ninety minute pulse (1).
BULIMIA AND ANOREXIA NERVOSA
Severe eating disorders such as bulimia and anorexia nervosa are also associated with disruption of reproductive function. Bulimia is characterized by alternating episodes of consumption of large amounts of food over a short time (binge eating) followed by periods of self-induced vomiting, excessive use of laxatives or diuretics and food restriction. The incidence of bulimia is estimated to be 4.5 to 18 percent among high school and college students (44). Bulimia usually begins between the ages of 17 to 25 years.
Anorexia nervosa is a severe eating disorder characterized by extreme weight loss (greater than 25% of ideal body weight), body-image disturbance, and an intense fear of becoming obese (45). Anorexia patients are usually between the age of 12 yrs and the mid-thirties and have a bimodal age of onset at 13-14 years and 17-18 years. There is a 90 to 95 % female predominance with the majority of patients coming from Caucasian, middle-class or upper-middle class families. The overall incidence of anorexia ranges from 0.64 per 100,000 to 1.12 per 100,000. The mortality associated with anorexia has been reported to be as high as 9 percent usually secondary to cardiac arrhythmia which is precipitated by electrolyte abnormalities and/or diminished heart muscle mass. Suicide has also been more common (2 to 5%) in patients with anorexia nervosa. These statistics are sobering making it extremely important for the clinician to recognize early signs of this disorder so that appropriate intervention and treatment can be initiated.
The clinical features of bulimia and anorexia are listed in Tables 5 and 6. Due to the marked reduction in caloric intake in anorexia, basal metabolism is lowered by decreased conversion of thyroxine to triiodothyroxine to maintain homeostasis. Thyroxine is converted via an alternative pathway to reverse triiodothyroxine, a relatively inactive isoform (Table 7). This protective functional mechanism is also commonly seen in severely ill patients and during prolonged starvation. Anorexics also suffer from defects in thermoregulation and are hypothermic. Because secretion of vasopressin is impaired, anorexics also have partial diabetes insipidus and are unable to concentrate their urine.
Anorectic and bulimic patients have hyperactivation of the HPA axis (46). Studies show a persistent hypersecretion of cortisol throughout the day with an increase in 24 hour free cortisol secretion. Despite the increased cortisol production, the manifestations of hypercortisolism are not present due to a decrease in cellular glucocorticoid receptors (47). The reduced number of these receptors may also provide an explanation for the incomplete suppression of the pituitary-adrenal axis by dexamethasone. Pituitary CRH responses are also blunted in bulimics and anorectics. As with other stress response syndromes, anorexics have evidence for increased central opioid activity with reported increases in cerebrospinal fluid levels of b-endorphins (48). Hypolepitinemia is commonly seen in women with anorexia. In these patients the low levels of leptin are representative of a chronic energy deficiency (49).
Like functional hypothalamic amenorrhea, anorexics have a prepubertal pattern of LH secretion presumably due to marked decrease in GnRH secretion. With weight gain, anorexics can display transitional patterns of LH secretion and may have normal or supranormal responses to GnRH (Figure 1). In some patients, despite return to normal body weight, up to 50% remain anovulatory.
Table 5. Common Features of Bulimia
|
Irregular menstrual cycles Dental enamel erosion Acute irritation of esophageal mucosa Esophageal or gastric rupture Hypokalemia Aspiration pneumonia Ipecac poisoning |
Table 6. Common Features of Anorexia Nervosa
| Preoccupation with handling of food
Bulimic behavior Calorie counting Distortion of body self-image Hyperactivity Obsessive-compulsive personality Increased incidence of past sexual abuse
Amenorrhea Constipation Coarse, dry skin Soft, lanugo-type hair
Hypothermia with defective thermoregulation Mild bradycardia Cardiac arrhythmia Hypotension
Hypokalemia secondary to diuretic or laxative abuse Low bone mass Increased serum beta-carotene levels Anemia Leukopenia Elevated hepatic enzymes |
Table 7 Neuroendocrine Abnormalities Associated with Anorexia Nervosa
| Diminished GnRH-LH pulsatile frequency and amplitude
Low blood LH and FSH levels Impaired ACTH response to CRH stimulation testing Resistance to dexamethasone suppression Increased ACTH levels Increased 24 hour urinary free cortisol levels Low prolactin levels Low TSH levels High reverse T3 and low T3 levels Elevated GH levels Decreased IGF-1 levels Diabetes insipidus |
The success rates for treatment of anorexia nervosa and bulimia remain low. Therapeutic approaches include behavior modification, group therapy, and individual psychotherapy. Generally, a team approach consisting of a psychiatrist and a general medicine specialist with expertise in eating disorders is desirable. Because of the high mortality rate and the significant morbidity associated with anorexia, it is important to obtain psychiatric consultation and follow-up in all patients with eating disorders. For patients who fail to resume menstrual function even after restoration of body weight, estrogen replacement therapy is indicated.
EXERCISE-INDUCED HYPOTHALAMIC AMENORRHEA
With the increased participation of women in all types of recreational and competitive athletic activities, health issues related to exercised-induced amenorrhea have become common. Depending on the type of sport and competition level, the incidence of amenorrhea varies from 5 to 25%. The incidence of menstrual irregularities appears to be greatest in sports that favor a low body weight physique such as ballet dancers (6-43%) and middle and long distance runners (24-26%). The incidence appears to be less frequent in bicyclers (12%) and swimmers (12%) (50).
In those athletes with menstrual irregularity, LH pulsatility is altered and can range from a decreased frequency to a transitional pattern (Figure 1). It is well known that acute exercise leads to hyperactivation of the HPA axis. Is it the stress of exercise or the low energy availability that alters LH pulsatility in exercising women? This key question has been answered in part by controlled studies in which women undergo dietary caloric restriction imposed in the face of increasing exercise demands. It would appear that LH pulsatility is not disrupted by the stress of exercise but rather LH pulsatility is disrupted because of reduced energy availability (51).
It is important to emphasize for so many of these athletes that the “female athlete triad” amenorrhea, osteoporosis, and eating disorders coexist. Management of these patients should emphasize measurement of bone density, counseling about diets, weight changes, trying to keep weight near normal levels, and calcium intakes. Women with exercise-related amenorrhea tend to have impaired lipid and metabolic profiles including an elevated serum total cholesterol, LDL cholesterol, and triglycerides compared with healthy women (52). One goal of therapy should be to decrease the level of exercise, improve the diet, and achieve weight gain. For others, exercise may not induce amenorrhea but may be associated with longer menstrual cycles, luteal phase defects, and intermenstrual spotting. These reproductive defects may be reversible with a decrease in exercise level or intensity. Alternatively, clomiphene given at doses between 25-100 mg for 5 days may increase GnRH pulsatility sufficiently to correct this defect. If amenorrhea persists for over 6 months, it may be prudent to begin estrogen replacement therapy or oral contraceptive pills. Long term health risks in these individuals are reproductive dysfunction and skeletal abnormalities.
OTHER CAUSES OF HYPOGONADOTROPINISM
Isolated Gonadotropin Deficiency (IGD)
This disorder is characterized by a decrease or absence of endogenous GnRH secretion resulting in very low to undetectable LH and FSH levels. Individuals with this disorder have incomplete development of secondary sexual characteristics, primary amenorrhea, eunuchoid features, and in some cases a decreased sense of smell or anosmia (Kallmann syndrome) (53). This disorder can have an autosomal dominant inheritance pattern. IGD has been linked to variance in over 15 genes. Common genetic variants include KAL1, FGFR1, PROKR2, PROK2, CHD7, FGF8, KISS1, KISS1R, TAC3, TACR3, GNRHR, and GNRH1 (54). The underlying defect is due to a failure of GnRH neurons to form completely in the olfactory placode or to migrate from the olfactory bulb to the media basal hypothalamus during early embryo development. For individuals with anosmia or hypo-osmia, there is evidence of hypoplasia of the olfactory bulbs on magnetic resonance scan (55).
Baseline levels of LH and FSH may be in the prepubertal or normal range. However, levels of other pituitary hormones such as TSH, GH, PRL, and ACTH are normal. Due to the failure to increase gonadal sex steroid secretion during puberty, secondary sex characteristics fail to develop and closure of the epiphyseal plates of the long bones is delayed resulting in a eunuchoid habitus where the arm span is greater than the height.
In many cases individuals are started on birth control pills without the diagnosis being made, such that there is partial or complete development of secondary sexual characteristics. In the untreated patient, breast development is usually Tanner stage I or II while pubic hair development will be Tanner IV or V. Treatment of IGD will require estrogen therapy to induce progressive pubertal maturation (100 ng/kg/day of ethinyl estradiol). Patients should be monitored at 2 or 3 month intervals to determine the rate of skeletal growth and development. The addition of a progestin such as medroxyprogesterone acetate 10 mg/day for 12 days can then be used to shed the endometrium. Progesterone therapy should only be started after optimal breast development is achieved. Once sexual maturation is completed, the estrogen dose can be gradually increased and maintained on 2 mg of micronized estradiol or 0.625 mg to 1.25 mg of conjugated estrogens with 12 days of progestin each month. When fertility is desired, ovulation induction can be carried out with either human menopausal gonadotropins or pulsatile GnRH administration.
Postpartum Pituitary Necrosis (Sheehan Syndrome)
Postpartum pituitary necrosis is usually preceded by a history of severe obstetrical hemorrhage with hypotension, circulatory collapse, and shock. After fluid resuscitation of the patient, this condition may be manifested by clinical evidence of partial or panhypopituitarism. Simmonds was the first to describe this clinical syndrome although the most complete description has been attributed to Sheehan (56). This condition constitutes an endocrine emergency that can be life-threatening.
The pathophysiology of this process is not entirely clear. With pregnancy, there is an increase in blood supply to the pituitary bed and the pituitary gland enlarges. During the period of profound hypotension, Sheehan postulated that occlusive spasm of the arteries that supply the pituitary and stalk occurs. This leads to venous stasis and thrombosis of the pituitary portal vessels causing a variable degree of pituitary ischemia and cell death. Many patients initially present with a failure to have breast engorgement and lactation due to a deficiency in PRL secretion. These women may also have other anterior pituitary deficiencies. The posterior pituitary is usually spared because it is less dependent on the portal blood supply. In some patients, the absence of ACTH secretion leads to inadequate cortisol secretion resulting in postural hypotension, nausea, vomiting, and lethargy. Hypothyroidism may be noted later in this scenario. However, in the majority of women, there is a diagnostic delay due to the vague symptoms. Recovery of pituitary function has been reported in a few cases.
The extent of pituitary deficiencies can be characterized by provocative testing with a combined intravenous injection of the hypothalamic releasing factors GnRH, TRH, GHRH, and CRH (57). Appropriate replacement therapy can be instituted once the pituitary reserve is defined. For the patient who presents with hypotension, immediate administration of glucocorticoids is required (cortisone acetate 100 mg, IM). Once the patient stabilizes, a maintenance dose of cortisone acetate 20-25 mg/day or prednisone 5 mg/day can be given. With increased stressful conditions such as an infection, a doubling or tripling of daily doses should be used. For hypothyroid patients, thyroxine replacement should be replaced gradually beginning at 50 mg/day and increased at 50 mg increments at 1 week intervals until full replacement doses are reached (0.1 to 0.2 mg/day). Patients should be instructed to wear a medical alert bracelet. Estrogen replacement therapy will be required for persistent amenorrheic patients. If fertility is desired, ovulation induction with exogenous gonadotropins is required.
Post-traumatic Hypopituitarism
This condition can arise following severe head trauma as a result of a sudden deceleration of the head and occult damage to the pituitary stalk or hypothalamus during a traffic accident (58). Trauma may also be associated with a basal skull fracture or an episode of unconsciousness. The risk for post-traumatic hypopituitarism is generally higher after a severe brain injury requiring neurosurgical interventions than for a mild or moderate injury (59). These individuals will often manifest a delay from injury to presentation of up to 10 years post-injury, possibly due to ongoing atrophy of the sellar and perisellar structures. The symptoms range from partial to complete panhypopituitarism. These symptoms can include amenorrhea, galactorrhea, hypogonadism, loss of axillary and pubic hair, anorexia, and weight loss. For these patients, evaluation of the pituitary-adrenal axis is most important because hypocortisolism can be potentially life-threatening. The diagnostic evaluation and management is similar to that described for Sheehan syndrome.
Pituitary Apoplexy
This medical emergency is characterized by an acute infarction of the pituitary gland. Patients will complain of a sudden onset of a severe retro-orbital headache and visual disturbances which may be accompanied by lethargy or loss of consciousness (60). These symptoms may mimic other neurological emergencies such as hypertensive encephalopathy, cavernous sinus thrombosis, ruptured aneurysm, or basilar artery occlusion. CT or MRI imaging may indicate hemorrhagic changes in the pituitary sella region. Patients with pituitary tumors are at higher risk for this complication. For some patients, neurosurgical consultation and emergency decompression may become necessary. Provocative testing as describe for Sheehan syndrome should be performed to evaluate for multiple pituitary deficits. Appropriate replacement of target tissue hormones should be instituted based on testing.
Post radiation-Induced Hypopituitarism
Exposure to therapeutic radiation sources for treatment of midline CNS tumors can place patients at increased risk for delayed development of hypopituitarism (61). In general, sensitivity to radiation is greatest for somatrotropes and gonadotropes, followed by corticotropes and thyrotropes. The onset of pituitary deficiencies may be insidious but can occur within 1 year of radiotherapy. Periodic assessment of hypothalamic-pituitary function should be performed for an indefinite period of time and appropriate replacement hormone therapy should be instituted as these deficiencies develop.
SUMMARY
In this chapter, we have reviewed a variety of disorders that can disrupt the normal menstrual cycle. Whether the disorder is linked to central organic lesions, changes in life style, or functional, stress components, all appear to have a common pathway(s) resulting in alterations in the secretion of the GnRH pulse generator.
REFERENCES
- Berga S, Naftolin F. Neuroendocrine control of ovulation. Gynecol Endocrinol 2012; Mar;28 Suppl 1:9-13.
- Filicori M, Santoro N, Merriam GR, Crowley WF. Characterization of the physiological pattern of episodic gonadotropin secretion throughout the human menstrual cycle. J Clin Endocrinol Metabl 1986;62:1136-1144.
- Reiter EO, Kaplan SL, Conte FA, Grumbach MM. Responsivity of pituitary gonadotropes to luteinizing hormone-releasing factor in idiopathic precocious puberty, precocious thelarche, precocious adrenarche, and in patients treated with medroxyprogesterone acetate. Pediatr Res 1975;9:111–116.
- Boyar RM, Katz J, Finkelstein JW, Kapen S, Weiner H, Weitzman ED, Hellman L. Anorexia nervosa. Immaturity of the 24-hour luteinizing hormone secretory pattern. N Engl J Med 1974;291:861–865.
- Marshall JC and Kelch RP. Gonadotropin-releasing hormone: role of pulsatile secretion in the regulation of reproduction. N Engl J Med 1986;315:1459-1468.
- Naftolin F, Garcia-Segura LM, Horvath TL, Zsarnovszky A, Demir N, Fadiel A, Leranth C, et al. Estrogen-induced hypothalamic synaptic plasticity and pituitary sensitization in the control of the estrogen-induced gonadotrophin surge. Reprod Sci 2007;14:101–116.
- Liu JH, Yen SSC. Induction of a midcycle gonadotropin surge by ovarian steroids in women: a critical evaluation. 1983; J Clin Endocrinol Metab 57:797-802.
- Zsarnovszky A, Horvath TL, Garcia-Segura LM, Horvath B, Naftolin F. Oestrogen-induced changes in the synaptology of the monkey (Cercopithecus aethiops) arcuate nucleus during gonadotropin feedback. J Neuroendocrinol 2001;13:22–28.
- Kalló I, Vida B, Deli L, Molnár CS, Hrabovszky E, Caraty A, Ciofi P, Coen CW, Liposits Z. Co-localisation of kisspeptin with galanin or neurokinin B in afferents to mouse GnRH neurones. J Neuroendocrinol. Nov 2011. doi: 10.1111/j.1365–2826.2011.02262.x.
- Pau KY, Berria M, Hess DL, Spies HG. Preovulatory gonadotropinreleasing hormone surge in ovarian-intact rhesus macaques. Endocrinology 1993;133:1650–1656.
- Pineda R, Aguilar E, Pinilla L, Tena-Sempere M. Physiological roles of the kisspeptin/GPR54 system in the neuroendocrine control of reproduction. Prog Brain Res 2010;181:55-77.
- de RouxN,Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 2003;19:10972-10976.
13.Jayasena CN, Nijher GM, Abbara A, Murphy KG, Lim A, Patel D, Mehta A, Todd C,Donaldson M, Trew GH, et al. Twice-weekly administration of kisspeptin-54 for 8 weeks stimulates release of reproductive hormones in women with hypothalamic amenorrhea. Clin Pharmacol Ther 2010;6:840-847.
14.Abbara A, Jayasena CN, Nijher GK, Comninos AN, Christopoulos G, Ashby D,Ghatei MA, Bloom SR, Carby A, Trew G et al. Kisspeptin—a novel physiological trigger for oocyte maturation in IVF treatment. Hum Reprod 2013. Supplement 1; European Society of Human Reproduction and Embryology 29th Annual Meeting, London, Abstract O-107.
- Pineda R, Aguilar E, Pinilla L, Tena-Sempere M. Physiological roles of the kisspeptin/GPR54 system in the neuroendocrine control of reproduction. Prog Brain Res 2010;181:55-77.
- Wilson RC, Kesner JS, Kaufman JM, et al. Central electrophysiologic correlates of pulsatile luteinizing hormone secretion in the rhesus monkey. Nueroendocrinology 1984;39:256-260.
- Rasmussen DD. The interaction between mediobasohypothalamic dopaminergic and endorphinergic neuronal system.
- Berga S, Naftolin F. Neuroendocrine control of ovulation. Gynecol Endocrinol 2012; Mar;28 Suppl 1:9-13.
- Filicori M, Santoro N, Merriam GR, Crowley WF. Characterization of the physiological pattern of episodic gonadotropin secretion throughout the human menstrual cycle. J Clin Endocrinol Metabl 1986;62:1136-1144.
- Reiter EO, Kaplan SL, Conte FA, Grumbach MM. Responsivity of pituitary gonadotropes to luteinizing hormone-releasing factor in idiopathic precocious puberty, precocious thelarche, precocious adrenarche, and in patients treated s as a key regulator of reproduction: An hypothesis. J Endocrinol Invest. 1991;14:323-335.
- Rasmussen DD, Liu JH, Wolf PL, Yen SSC. GnRH neurosecretion in the human hypothalamus: in vitro regulation by dopamine. J Clin Endocrinol Metab 1986;62:479-483.
- Brann DW, Mahesh VB. Excitatory amino acids: Function and significance in reproduction and neuroendocrine regulation. Front Neuroendocrinol 1994;15:3.
23.Goodman RL, Lehman MN, Smith JT, Coolen LM, deOliveira CV, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, et al. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology2007;12:5752-5760.
- Skorupskaite K, George JT, Anderson RA. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum Reprod Update 2014;20(4):485-500.
- Ropert JF, Quigley ME, Yen SSC. Endogenous opiates modulate pulsatile luteinizing hormone release in humans. J Clin Endocrinol Metab 1981;52:583-585.
- Genazzani AD, Petraglia FP, Gastaldi M, Volpogni C, Gamba O, Genazzani AR. Naltrexone treatment restores menstural cycles in patients with weight loss-related amenorrhea. Fertil Steril 1995;64:951-956.
- Witchel SF, Tena-Sempere M. The Kiss1 system and polycystic ovary syndrome: lessons from physiology and putative pathophysiologic implications. Fertility and Sterility 2013:100;12-22.
- Signore AP, Zhang F, Weng Z, Gao Y, Chen J. Leptin neuroprotection in the CNS: mechanisms and therapeutic potentials. J Neurochemistry 2008;106:1977-1990.
- Laughlin GA, Yen SSC. Hypoleptinemia in women athletes: absence of a diurnal rhythm with amenorrhea. J Clin Endocrinol Metab 1997;82(1):318-21.
- Smith JT, Acohido BV, Clifton DK, Steiner RA. Kiss-1 neurons are direct targets for leptin in the ob/ob mouse. J of Neuroendocrinol 2006;18:298-303.
- Castellano JM, Nvarro VM, Fernandez R, Nogueiras R, Et al. Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology 2005;146:3917-3925.
- Liu JH. Hypothalamic amenorrhea: Clinical perspectives, pathophysiology, and management. Am J Obstet Gynecol 1990;163:1732-1736.
- Meczekalski B, Podfigurna-StopaA, Warenk-Szymankiewicz A, Genazzani AR. Functional hypothalamic amenorrhea: current view on neuroendocrine aberrations. Gynecol Endocrinol 2008;24:4-11.
- Brundu B, Loucks TL, Adler LJ, Cameron JL, Berga SL. Increased cortisol in the cerebrospinal fluid of women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab 2006;91:1561-1565.
- Berga SL, Marcus MD, Loucks TL, Hlastala S, Ringham R, Krohn MA. Recovery of ovarian activity in women with functional hypothalamic amenorrhea (FHA) treated with cognitive behavior therapy (CBT). Fertil Steril 2003;80:976-981.
- Michopoulos V, Mancini F, Loucks T, Berga S. Neuroendocrine recovery initiated by cognitive behavioral therapy in women with functional hypothalamic amenorrhea: a randomized controlled trial. Fertility and Sterility 2013:99(7):2084-91.
- Berga Sl, Mortola JF, Girton L, Suh B, Laughlin G, Pham P, Yen SSC. Neuroendocrine aberrations in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab 1989;68:301-308.
- Berga SL, Mortola JF, Yen SSC. Amplification of nocturnal melatonin secretion in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab 1988;66:242-244.
- Marcus MD, Loucks TL, Berga SL. Psychological correlates of functional hypothalamic amenorrhea. Fertil Steril 2001`;76:310-316.
- Adams Hillard PJ. Menstruation in adolescents: what’s normal, what’s not. Ann NY Acad Sci 2008;1135:29-35.
- Gordon C. Functional hypothalamic amenorrhea. N Engl J Med 2010;363:365-371.
- Scheiber M, Liu J. The use of gonadotropin-releasing hormone to induce ovulation. Global Library of Medicine 2011; DOI 10.3843/GLOWM.10340.
- J. Licinio, S. Caglayan, M. Ozata, B.O. Yildiz, P.B. de Miranda, F. O'Kirwan, et al.Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc Natl Acad Sci U S A, 101 (2004), pp. 4531–4536
- Pyle RL, Mitchell, JE, Eckert ED, Halvorson PA, Neuman PA Goff GM. The incidence of bulimia in freshman college students. Int J Eat Disord 1983;2:75-85.
- Vigersky RA, Loriaux D, Andersen AE, Lipsett MR. Anorexia nervosa: behavioral and hypothalamic aspects. Clin Endocrinol Metab 1976;5:517-535.
- Boyar RM, Hellman LD, Roffwarg H, Katz J, Zumoff B, O’Connor J. Bradlow HL, Fukushima DK. Cortisol secretion and metabolism in anorexia nervosa. N Engl J Med 1977;296:190-193.
- Kontula K, Anderson LC, Huttumen M, Pelkonen R. Reduced level of cellular glucocorticoid receptors in patients with anorexia nervosa. Horm Metab Res 1982;14:619-620.
- Kaye WH, Gwirtsman HE, George DT, Ebert MH, Jimerson DC, Tomai TP, Chrousos GP, Gold PW. Elevated cerebrospinal fluid levels of immunoreactive corticotropin-releasing hormone in anorexia nervosa: relation to state of nutrition, adrenal function, and intensity of depression. J Clin Endocrinol Metab 1987;64:203
- ChanJL& Mantzoros CS. 2005 Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet 366 74–85.
- Warren MP. Health issues for women athletes:exercise-induced amenorrhea. J Clin Endocrinol Metab 1999;84:1892-1896.
- Loucks AB, Verdun M, Heath EM. Low energy availability, not stress of exercise, alters LH pulsatility in exercising women. J Appl Physiol 1998;84:37-46.
- Rickenlund A, Eriksson MJ, Schenck-Gustafsson K, Hirschberg AL (2005) Amenorrhea in female athletes is associated with endothelial dysfunction and unfavorable lipid profile. J Clin Endocrinol Metab 90:1354–1359.
- Kallmann F, Schonfeld WA, Barrera SW. Genetic aspects of primary eunuchoidism. Am J Ment Defic. 1944;203-236.
- Layman LC. Clinical Genetic Testing for Kallmann Syndrome.J Clin Endocrinol Metab.2013;98:1860–2.
- Klingmuller D, Dewes W, Krahe T, Brecht G, Schweikert H. Magnetic resonance imaging of the brain in patients with anosmia and hypothalamic hypogonadism (Kallmanns syndrome). J Clin Endocrinol Metab 1987;65:581-584.
- Sheehan HL. Pituitary necrosis. Br Med Bull 1968;24:59-70.
- Sheldon WR, DeBold CR, Evans WS, DeCherney SG, Jackson RV, Island DP Thorner MD, Orth DN. Rapid sequential intravenous administration of four hypothalamic releasing hormones as a combined anterior pituitary function test in normal subjects. J Clin Endocrinol Metab 1985;60:623-630.
- Edwards OM, Clark JDA. Post-traumatic hypopituitarism. Medicine 1986;65:281-290.
- Nemes O, Kovacs N, Czeiter E, Kenyeres P, Tarjanyi Z, Bajnok L, Buki A, Doczi T, Mezosi E. Predictors of post-traumatic pituitary failure during long-term follow-up. Hormones 2015:14(3):383-391.
- Reid RL, Quigley ME, Yen SSC. Pituitary apoplexy: a review. Arch. Neurol. 1985;42:712-719.
- Lam KSL, Tse VKC, Wang C, Yeung RTT, Ma JTC, Ho, JHC. Early effects of cranial irradiation on hypothalamic-pituitary function. J Clin Endocrinol Metab 1987;64:418-424.
Clinical Management of Male Infertility
ABSTRACT
This chapter provides a comprehensive overview of the clinical, "hands-on" approach to the diagnosis and treatment of male infertility. It is a resource of evidence-based information for those who are practicing in this fascinating field as reproductive endocrinologists, fertility specialists, urologists, gynecologists and general practitioners. This chapter has been revised with a particular focus on current challenges in the management of severe male infertility. The chapter gives an overview of the genetic and chromosomal contribution to abnormal sperm production and covers the latest advances in the investigation of the topical DNA fragmentation and newest sperm selections strategies. Therapeutic interventions are presented in detail, mainly in the areas of sperm retrieval, varicocele repair and intra-cytoplasmic sperm injection, including potential complications and extreme challenges as complete failed fertilization. Various approaches to fertility preservation are described, including advances in prepubertal fertility preservation. The current chapter is a resource for those who wish to familiarize themselves with multiple aspects of donor sperm usage. For complete coverage of this and all related areas of Endocrinology, please visit our FREE web-book, www.endotext.org.
NATURE AND CAUSES OF MALE INFERTILITY
Definitions
Infertility is the inability to produce a pregnancy or failure to do so within a reasonable period of trying, usually 6 to 12 months. Sterility is a total inability to produce a pregnancy, and this may be reversible or irreversible. Subfertility is infertility without an absolute barrier to reproduction that would cause sterility, such as azoospermia. Hypogonadism is a nonspecific term for decreased testicular or ovarian function that could include a disorder of gamete production or function or a disorder of sex hormone production or action. Usually male hypogonadism indicates testicular failure associated with androgen deficiency. Primary hypogonadism results from disorders that affect the gonads directly, and secondary hypogonadism results from defective pituitary gonadotropin secretion.
Incidence and Distribution
Trends in Male Infertility
Controversial reports were published with regard to declining sperm count in some regions of the world, highlighting the potential adverse environment effect on infertility [7-10]. Other reports could not show this effect [11-13].
ETIOLOGY AND CLASSIFICATION OF INFERTILITY
At present, the precise cause cannot be determined in most men investigated for infertility.[6, 14] Relationships between testicular damage, semen quality, and fertility are not strong.[15-17] Even genetic disorders may have marked phenotypic variation. For example, with microdeletions in the long arm of the Y chromosome, testicular histology may show Sertoli-cell-only syndrome, germ cell arrest, or hypospermatogenesis.[18, 19] There are many suggestions for the causes of the common forms of male infertility, but new genetic causes are being found with the use of more efficient DNA sequencing technologies and large clinical databases (table 4). There are many other suggested etiological or contributory factors for which data is lacking or circumstantial including minor hormonal disturbances, dietary deficiencies of vitamins and minerals, disturbed scrotal thermoregulation with or without varicocele and accessory sex gland inflammation.[20] Currently the testicular dysgenesis syndrome and reactive oxygen species (ROS) damage are topical (see chapter 12 by Giwercman and Giwercman) [21, 22]. The concept of the testicular dysgenesis syndrome arose from concerns that toxins in the environment, acting in concert with genetic predisposition, are affecting testicular development. It encompasses hormonal inhibition (“endocrine disruption”) of the proliferation of Sertoli cell precursors, Leydig cells and germ cells during fetal life via adverse environmental, dietary, lifestyle or other influences affecting the mother and resulting in increased risks of cryptorchidism, hypospadias, primary spermatogenic defects, and testicular cancer.[21] Despite the attractively unifying nature of the TDS hypothesis for these disease association, the actual disease mechanism remain unknown and it remains unproven. ROS are believed to be causal in the relationship between abnormal spermatozoa in semen, DNA strand breaks in sperm heads and markers of apoptosis in sperm.[22] The importance of these pathogenetic mechanisms in male infertility remains to be determined.[23, 24]
A classification of causes of male infertility based on the effectiveness of treatment is shown in Table 1. In this classification, effective treatment means medical intervention known or proved by clinical trial to improve the chances of the man producing a conception by coitus or artificial insemination and does not include the use of in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI) to bypass the impairment.
| Table 1. Classification Of Male Infertility By Effectiveness Of Medical Intervention To Improve Natural Conception Rate | |
| TYPE OF INFERTILITY | FREQUENCY (%) |
| Untreatable sterility | 12% |
| Primary seminiferous tubule failure | 12% |
| Treatable conditions | 18% |
| Sperm autoimmunity | 7% |
| Obstructive azoospermia | 10% |
| Gonadotropin deficiency | 0.5% |
| Disorders of sexual function | 0.5% |
| Reversible toxin effects | 0.02% |
| Untreatable subfertility | 70% |
| Oligospermia | 35% |
| Asthenospermia and teratozoospermia | 30% |
| Normospermia with functional defects | 5% |
CLINICAL EVALUATION
Patients with irreversible sterility can be separated from those with potentially treatable conditions or subfertility usually by standard clinical evaluation (Table 2) and some simple investigations (Table 3).
Table 2 Clinical Evaluation of the Infertile Man
History:
- illness or injury affecting testes,
- pubertal development
- sexual performance
- fertility exposures
- occupation, habits
Physical examination:
- general
- virilization
- gynecomastia
- body proportions and BMI
- scrotal examination
testicular size
epididymides
vasa
varicocele
- scrotal examination
Table 3 Basic Investigation of the Infertile Man
Semen analysis:
Semen volume, sperm concentration, motility, morphology, and sperm antibodies
Hormone measurements:
Luteinizing hormone, follicle-stimulating hormone, prolactin, testosterone, sex hormone–binding globulin
Imaging:
Scrotal ultrasound, transrectal ultrasound of prostate and seminal vesicles, magnetic resonance imaging of pituitary
Testis biopsy:
Needle aspiration: cells or tissue, open
History
It cannot be overemphasized that both members of the couple need to be involved in the assessment and discussion of the results. The emotional reaction of the couple to the diagnosis of infertility may interfere with clinical evaluation and management. Intimate information may not be disclosed while the couple is embarrassed, hostile, or confused. Previous sexually transmissible infections or pregnancy may be concealed from the partner.
Nature and Duration of Infertility
Previous pregnancies and time taken to conceive each pregnancy and duration of infertility are important prognostic factors. The couple may be aware of an infertility-related problem, such as previous undescended testes or orchitis. Some who present with a short duration of infertility may be unaware of the normal human pregnancy rates. The plan for investigation depends on the possibility of finding remediable abnormalities and on the age of the female partner.
Family History
The family history should be considered but may not be known because infertility is often not discussed openly.[25, 26] Increasing numbers of chromosomal and genetic causes for male infertility are being discovered (Table 4).[27-29] Some of these cause sterility and are recessive disorders or de novo mutations. Others may only affect fertility slightly so that there may be no family history of infertility. The most important include Kallmann syndrome, myotonic dystrophy, androgen receptor defects, gonadotropin and gonadotropin receptor defects, cystic fibrosis and bilateral congenital absence of the vasa, and chromosomal rearrangements / aneuploides and Yq microdeletions.[18, 19, 27, 30-37] However, collectively they explain less than 10% of male infertility.
There are many pediatric syndromes that involve hypogonadism or undescended testes in association with ambiguous genitalia, multiple malformations, obesity, or mental retardation, but patients with these generally do not present for management of infertility. Other genetic diseases may be associated with infertility, for example, congenital adrenal hyperplasia, hemoglobinopathy, Huntington’s disease, polycystic kidneys, and mitochondrial disorders.[27, 38-41] Predispositions to some conditions may also have a genetic basis such as the anatomic variant of the tunica vaginalis which predisposes to testicular torsion, the association of Young’s syndrome with mercury poisoning in infancy and the familial aspects of sperm autoimmunity. Men with sperm autoimmunity have increased frequencies of both family histories of organ-specific autoimmune diseases and autoantibodies to thyroid and gastric parietal cells in their serum.[42] Furthermore, brothers of men with poor semen analysis results are more often infertile than expected.[25, 26] Thus, it is postulated that genetic causes or predispositions will be found for most male infertility. However, genetic factors remain unclear for the common types or associations of male infertility: idiopathic oligospermia, asthenospermia, teratospermia and undescended testes.
| Table 4. Genetic And Chromosomal Defects In Infertile Men: Known Or Suspected | ||
| Function | Defect | Phenotype (Approximate frequency) |
| Hormonal regulation | KALIG 1
Prokineticin-2 FGFR1 |
Kallmann syndrome, isolated gonadotropin |
| GnRH receptor | deficiency (1/10,000) | |
| DAX 1 | Adrenal hypoplasia congenita (rare) | |
| Steroidogenic enzymes | Congenital adrenal hyperplasia (rare) | |
| Haemochromatosis | iron deposition in gonadotroph: (1/1000) | |
| FSHβ
T allele of rs10835638 |
oligospermia (rare) | |
| FSH receptor | oligospermia (rare) | |
| Androgen receptor
CAG/CGG polymorphism |
oligospermia (1/20,000)
asthenozoospermia(?) |
|
| Spermatogenesis | XXY and variants | Klinefelter syndrome (1/800) |
| XYY | oligospermia (1/5000) | |
| Translocations | oligospermia (1/3000) | |
| Yq microdeletions (AZF regions, CDY) | Sertoli cell only, oligospermia (1/500) | |
| ETV5 gene variants | Sertoli cell-only syndrome | |
| TSPY gene (short arm of Y chromosome – Yp) | oligospermia | |
| DMPK CTG ext. | myotonic dystrophy (1/8000) | |
| INSL3 | undescended testes (?) | |
| Sex hormone binding globulin (SHBG) gene | Oligoasthenoteratozoospermia? | |
| Estrogen receptors (ESR1, ESR2) | undescended testes (?) | |
| MTHFR | oligospermia | |
| USP26 de-ubiquitinating enzyme family | Spermatogenic failure? | |
| TAF7L | Spermatogenic failure? | |
| PRM1-PRM2 (protamines responsible for chromatin compaction) | ||
| TNP1-TNP2 (transition nuclear protein) | ||
| Meiosis | Translocations | germ cell arrest (rare) |
| ?CREM | germ cell arrest (?) | |
| SYCP3 | Azoospermia | |
| DAZL (T54A) | germ cell arrest (?) | |
| Spermiogenesis | Fibrous sheath or | dysplasia (1/50,000) |
| DNAI1,DNAI2 DNAH5, DNAH11, DNAAF2 (dynein arm)
RSPH4A, RSPH9 (radial spoke) TXNDC3 (thioredoxin-nucleoside diphosphate kinase) |
immotile cilia (1/50,000) | |
| ? | absent acrosomes (rare) | |
| ? | decapitate sperm (rare) | |
| protamine II | teratozoospermia(?) | |
| LDH-x | asthenozoospermia(?) | |
| Genital tract | CFTR | BCAV (1/2000) |
| ? | Other obstructions (rare) | |
| ? | Necrozoospermia (rare) | |
| Sperm-oocyte interaction | ZPBP1 | Disordered zona pellucida induced acrosome reaction (1/4000) |
| ? | Defective sperm-zona binding with normal sperm morphology (rare) | |
Coital Adequacy and Timing
Information on impotence and ejaculatory disturbances is important because intravaginal deposition of semen near the time of ovulation is crucial for fertility. Infrequent coitus is common in couples seen for infertility. Low libido may result from androgen deficiency, general illness, or a psychological reaction to the infertility.
Childhood and Pubertal Development
Treatment in childhood for penile or scrotal disorders (e.g. hypospadias, epispadias, urethral valves, undescended testes, inguinal hernia, hydroceles) could be relevant (see chapter by Hutson). Sexual maturation may be delayed and incomplete with primary or secondary hypogonadism. There may have been associated growth problems that required treatment. Early puberty and growth resulting in short stature suggest congenital adrenal hyperplasia.[38, 39]General Health: Any illness, acute or chronic, can impair sperm production in a nonspecific manner.[43] Acute critical illness, such as severe trauma, surgery, myocardial infarction, burns, liver failure, intoxication, and starvation, is often accompanied by suppression of gonadotropin secretion and secondary hypogonadism. In contrast, a primary testicular disorder with elevated gonadotropin levels may occur with chronic illnesses. Increased peripheral conversion of androgens to estrogens may produce some features of feminization such as gynecomastia. The association of hypogonadism and feminization with chronic liver disease is well known. Similar primary, secondary or mixed hypogonadism may occur with other chronic illnesses such as chronic anemia, chronic renal failure, rheumatoid arthritis, chronic spinal cord injury, thyroid diseases, Cushing’s syndrome, obesity, human immunodeficiency virus (HIV) infection, and neoplasia. Sex hormone–binding globulin levels are increased with some conditions such as cirrhosis and thyrotoxicosis but suppressed with others such as obesity, hypercortisolism, and hypothyroidism.[43] Numerous drugs have side effects on the reproductive system.[43] Heroin addiction and intrathecal narcotic infusions to control chronic pain suppress luteinizing hormone secretion.[44] Fever can cause transient declines of a few months’ duration.[43, 45, 46] Diabetes mellitus may be associated with impotence in early uncontrolled stages, ejaculatory disorders with autonomic neuropathy, sperm autoimmunity and ROS damaged sperm.[47] Men with renal disease may have infertility of multifactorial origin, including testicular failure from chronic illness, cytotoxic drug exposure, zinc deficiency, and damage to the vasa or penile blood supply during kidney transplantation. However, as with cirrhosis, provided that metabolic decompensation is not severe, semen quality often is adequate for fertility.[43] Epididymal obstruction associated with chronic sinopulmonary disease (Young’s syndrome) was diagnosed frequently in Australia and the United Kingdom in the past yet is rare elsewhere.[48] Some cases of Young’s syndrome may have been caused by mercury poisoning in childhood from calomel-containing teething powders.[49] These were withdrawn from the market in the mid-1950s when it was found that they caused pink disease, and Young’s syndrome is seen rarely. Fetal rubella may result in defective development of the vasa. Bronchiectasis and sinusitis are common in men with immotile sperm from cilial defects.[50] Situs inversus may also be present.
Testicular Symptoms
Previously undescended testes are common in men being investigated for infertility.[14, 51, 52] Undescended testes may be associated with other congenital malformations and disorders of testicular hormone production or action during fetal development, such as Kallmann syndrome, insulin-like factor 3 receptor mutations, androgen receptor mutations or defects of androgen metabolism, and diethylstilbestrol exposure in utero. In Western countries, this condition is usually treated in early childhood, but whether early surgery reduces the severity of the subsequent spermatogenic disorder is unclear.
A randomized controlled trial of orchiopexy for unilateral palpable maldescended testis at 9 months versus 3 years of age showed that surgery at 9 months was followed by significant growth of the testis up to age 4 years but there was no change in testis size in those treated at age 3 [53]. This has led to clinical guidelines for treatment of maldescended testes that recommend orchiopexy for congenital forms between 6 and 12 months of age and as soon as possible for those discovered later and for acquired maldescent. Hopefully this will reduce the frequency of subsequent testicular tumours and spermatogenic defects. A testicular dystrophy may cause both the failure of descent and defective sperm production in adult life despite early surgery. It is difficult to explain otherwise how men with unilateral undescended testes are so frequent in the infertile population. Bilateral undescended testes carry a poorer outlook for fertility than unilateral undescended testes. Infertility after bilateral orchiopexy is approximately six times more common than in the general population and occurs in approximately half of the men, whereas after unilateral orchiopexy, infertility is increased by a factor of two and affects approximately 10%.[51] There may be associated malformation of the epididymides.[54] Rarely the testes may atrophy after surgery because of interference with the blood supply or coincidental torsion.
Episodes of severe testicular pain and swelling may result from torsion, orchitis, or epididymo-orchitis and may be followed by loss or atrophy of the testis. Post-inflammatory atrophy is particularly frequent with mumps orchitis but rare with other illnesses such as glandular fever and brucellosis.[55] Epididymo-orchitis of bacterial origin is commonly associated with urethritis or urinary tract infections and may follow straining with heavy lifting. Sexually transmitted diseases are important, particularly if there was associated epididymal pain or swelling. Some patients have post-gonococcal obstructions in the tails of the epididymides without clear or admitted histories of epididymitis.
Failure of development and a decrease in size of one or both testes are important symptoms of spermatogenic defects. Torsion of the testes may cause atrophy. The vasa may be damaged during hernia repairs and kidney transplantation. Testicular biopsy may inadvertently damage the epididymis, especially if retroversion of the testis is not recognized and the biopsy is performed without taking the testis out of the tunica. Similarly, surgery for torsion, hydroceles, or epididymal cysts may result in the obstruction of the epididymis. Hematomas in the scrotum and infarction of the testes may follow interference with the vascular supply of the testes. Rarely, autoimmune orchitis results from testicular injury or inflammation. Testicular tumors and carcinoma in situ occur with increased frequency in infertile men, even without a history of undescended testes.[21, 56]
Iatrogenic Infertility
Vasectomy and Sertoli-cell-only syndrome caused by cytotoxic chemotherapy and radiation therapy for malignant tumors of the testes, leukemia, lymphoma, and serious autoimmune diseases are the most common forms of medically induced infertility.[57, 58] Although some treatment regimens only suppress spermatogenesis temporarily, recovery of fertility is unpredictable. Alkylating agents, such as cyclophosphamide and busulfan, destroy spermatogonia.[58] Antimetabolites may be used to treat psoriasis, rheumatoid arthritis, or xenograft rejection and can have transient adverse effects on spermatogenesis.[43, 59] Treatment with sulfasalazine for inflammatory bowel disease or arthritis causes a reversible impairment of semen quality.[43] Cessation of sulfasalazine often results in a marked improvement in semen quality over several months. Many other drugs have real or potential adverse effects on spermatogenesis or sexual performance, including androgens, anabolic agents, estrogens, glucocorticoids, cimetidine, spironolactone, antibacterials (especially nitrofurantoin), antihypertensive drugs, and psychotropic agents. However, in practice these are not common causes of infertility.[43]
Anti-spermatogenic Factors
Occupational and environmental exposures may affect reproduction.[60, 61] Exposure to heat from frequent sauna baths, vehicle driving, furnaces, and perhaps working outdoors in summer may cause a decline in spermatogenesis. Impaired testicular heat exchange from obesity and varicoceles may accentuate the effect. Exposure to chemicals in the workplace or elsewhere, particularly nematocides; organophosphates; estrogens; benzene; and welding, zinc, lead, cadmium, and mercury fumes, may have anti-spermatogenic effects. Various social drugs, including tobacco, alcohol, marijuana, and narcotics, are potentially anti-spermatogenic, but these usually require heavy use for an adverse effect.[43, 62, 63] Some addicts have other organ damage, such as cirrhosis, which may further impair testicular function.[43]
Physical Examination
A general physical examination is performed (see Table 2) and specific abnormalities are sought in particular circumstances, for example, of the respiratory system with suspected genital tract obstructions or immotile sperm, the prostate for ejaculatory duct obstruction or prostatitis, the endocrine system for hypopituitarism or other defects associated with testicular failure, the nervous system for autonomic neuropathy with coital disorders, optic field defects with pituitary tumors, and hyposmia with Kallmann syndrome.
Virilization
Hair distribution varies markedly between men. The loss or reduced growth of facial, pubic, axillary, and body hair is an important feature of androgen deficiency but is often unrecognized by patients. Men may note a reduced frequency of the need to shave. The stages of genital and pubic hair development can be recorded according to the method of Tanner. Eunuchoidal proportions (arm span 6 cm greater than height or pubis-to-floor measurement greater than 6 cm longer than one half the height) result from delayed fusion of the epiphyses and are a sign of delayed or incomplete puberty in whites or Asians but can be found with normal testicular function.
Gynecomastia
Gynecomastia of mild degree is common in men with testicular failure of any cause.[43] Marked gynecomastia may be associated with Klinefelter’s syndrome, cirrhosis, androgen receptor defects, estrogen-producing tumors, or anabolic steroid and human chorionic gonadotropin abuse. Galactorrhea is rare in men and usually but not always associated with hyperprolactinemia.[64]
External Genitalia
Examination of the penis for the position of the meatus, phimosis, urethral strictures, and Peyronie’s disease is important because these may influence the adequacy or completeness of ejaculation. Inadequate penile size appears to be an exceptionally rare cause of infertility.[65]
Examination of the scrotal contents is critical in the evaluation of male infertility. A general approach to the examination is outlined in Figure 172-1. This examination should be performed with the man lying and standing.The body of the testes, the head, the body, and the tail of the epididymis and vas are palpated on both sides as shown. Sometimes it is difficult to examine the scrotum thoroughly because of ticklishness or because the scrotum is very tight. Testes may retract into the superficial inguinal region, especially if small. Testes not present in the scrotum may be palpable in the subcutaneous tissue in the groin or, occasionally, in the inguinal canal. Palpable remnants of the vas and epididymis in the scrotum suggest the testis has atrophied completely—the vanishing testis.[66]
Orchiometry
The volume of the testis is determined by comparison with an orchiometer (normal: 15–35 mL).[67] In the absence of varicoceles, the right and left testes are approximately equal in size. Testicular volume is related to body size and number of sperm per ejaculate. As seminiferous tubules occupy more than 90% of the volume of the testes, impairment of spermatogenesis is commonly associated with reduced testicular size. Testicular atrophy suggests severe impairment of spermatogenesis.

Figure 1. . Prader orchiometer for measuring testis volume.
Testicular Abnormalities
Pain on palpation or excessive tenderness suggests inflammation. Loss of normal testicular sensation may occur with chronic inflammations, neuropathy, or neoplasia. Reduced consistency or softness of the testes is a feature of reduced spermatogenesis. Abnormalities of shape and hard lumps suggest tumors or scars.
Epididymal Abnormalities
Palpable abnormalities include congenital absence of the vas or other failures of development, enlargements of the heads or nodules in the tails of the epididymides with obstruction, spermatoceles, and other cysts and tumors. In men with very small testes (<5 mL), small epididymides suggest severe androgen deficiency, and normal-size epididymides suggest postpubertal testicular atrophy or a severe seminiferous epithelial disorder, such as Klinefelter’s syndrome.
Vasal Abnormalities
Abnormalities of the vas include absence, nodules and gaps with vasectomy, and thickening or beading of the vas with severe post-inflammatory scarring as from tuberculosis.
Miscellaneous Abnormalities
Incidental scrotal findings include scars from surgery, scrotal dermatitis, and pubic fat pads around the genitals in extreme obesity. Inguinal hernias and lipomas and encysted hydroceles of the cord are palpated above and behind the epididymis. Cysts “hydatids” of the appendix testis or epididymis are typically anterior to the head of the epididymis. Spermatoceles and cysts of the paradidymis are in the head or body of the epididymis. Retroversion of the testes, where the vas and epididymis are anterior rather than posterior to the testes is common. Hydroceles of mild degree are common. A tense hydrocele may hide a testicular tumor. Unilateral absence of the vas may be associated with ipsilateral agenesis of the kidney and ureter on the same side. Many of these anomalies have little relationship with infertility.
Checking for Varicocele
With the man standing up, the scrotum can be inspected for swelling of the pampiniform plexus and a cough or Valsalva impulse seen or palpated by holding the spermatic cords between the thumb and index finger of each hand and elevating the testes toward the external inguinal ring. This maneuver reduces the risk of confusing contractions of the cremaster muscles with venous impulses. Varicocele size is graded: cough impulse without palpable enlargement of the spermatic cord (grade 1), palpable enlargement (grade 2), and visible enlargement (grade 3). Although predominantly a left-sided condition, varicoceles may occasionally be on the right side.
The accuracy and reproducibility of clinical examination, even for structures as accessible as those in the scrotum, may not be high. Varicoceles may vary in size from day to day. Even absence of the vasa may be overlooked. With practice, orchiometry can be repeated to within one orchiometer size.
SEMEN ANALYSIS AND OTHER INVESTIGATIONS
Investigations are outlined in Table 3.
Semen Analysis
The most important laboratory investigation in male infertility is semen analysis. The variables assessed and the methods are in the World Health Organization’s laboratory manual.[68] Automation of semen analysis is in progress and should be used in most specialized laboratories soon.[17]
It is crucial that the laboratory is experienced in the performance of semen analyses, and participates in quality assurance activities.[69] There should be a room nearby for the collection of semen. Semen may be obtained by masturbation or coitus using a special nontoxic condom. Ordinary latex contraceptive condoms are unsatisfactory because the rubber usually immobilizes the sperm.[70] If these methods of collection are not possible, postcoital examination of midcycle cervical mucus may give some information about the likelihood of adequate semen quality if many motile sperm are found. In contrast, a negative postcoital test on its own is of little diagnostic value because conception can occur in the same cycle.[71]
The man should be provided with a wide-mouth, sterile, and nontoxic collection jar and written instructions about collection and delivery to the laboratory. A period of abstinence from ejaculation from 2 to 5 (preferably 2) days, delivery of the sample to the laboratory within 1 hour of collection, and avoidance of exposure to lubricants or extremes of temperature are specified.
Because of the variability of results, several semen analyses at intervals of 2 or more weeks are necessary in a man with an abnormality in the first test. Even with complete collection of samples, there is variability caused by counting error, other technical errors, and differences in the ejaculate from day to day (Fig. 2).[68, 72] These large variations need to be remembered when interpreting results of semen analysis.
To check for retrograde ejaculation, urine collected immediately after ejaculation is centrifuged and the pellet examined for sperm.

Figure 2. Variability of semen analysis results in a fertile sperm donor. C, sperm concentration; V, semen volume; M, total motility; MI, motility index-product of grade and percentage of sperm with progressive motility graded 0 to 3. (Mallidis C et al: Variation of semen quality in normal men. Int J Androl 14:99-107, 1991. Used by permission Blackwell Scientific Publications.)
Assays of semen constituents from the accessory glands and testis are available: zinc and acid phosphatase from the prostate, fructose from the seminal vesicles, neutral α-glucosidase, glycerophosphocholine, and l-carnitine from the epididymis and inhibin B from the Sertoli cells. Prostatic fluid is acid (pH approximately 6.0), but the ejaculate is alkaline because of the admixture with seminal vesicle fluid. Semen biochemistry is of limited usefulness in clinical practice. Some examples are given in Table 5.
| Table 5. Common or Characteristic Patterns of Semen Abnormality | |||||
| Volume (mL) >1.5* | Concentration (106/mL) >15* | Motility (%) >40* | Normal Morphology (%) >4* | Comment | Cause |
| 0.4 | 0 | - | - | Fructose 1 nmol/L (low)
pH 6.5 (low) |
Congenital absence of vasa Ejaculatory duct obstruction Partial retrograde ejaculation Testicular failure with androgen deficiency (Spill or incomplete collection) |
| 4.0 | 0 | - | - | Fructose 15 nmol/L | Genital tract obstruction Primary seminiferous tubule failure Secondary seminiferous tubule failure with androgen treatment |
| 3.0 | 100 | 0 | 35 | Live 70% | (Contamination or condom collection) Immotile cilia |
| 3.0 | 100 | 5 | 35 | Live 20% | (Contamination or delayed examination) Necrospermia Sperm autoimmunity |
| 3.0 | 100 | 65 | 0 | Small round heads | Total teratospermia: absent acrosomes |
| 3.0 | 100 | 25 | 10 | Liquefaction delayed Sperm aggregation 2+Live 40% Polymorphs 1 x 106/mL |
Idiopathic asthenospermia Sperm autoimmunity Prostatitis (Delayed examination) |
| 3.0 | 4 | 30 | 3 | Mixed abnormal morphology | Oligospermia of specific or nonspecific causes |
| 3.0 | <1 | - | - | Motile sperm present | Severe oligospermia of specific or nonspecific causes Primary seminiferous tubule failure Partial genital tract obstruction |
| * WHO Reference Value (5%centile for fertile men) | |||||
Immunobead Test
Tests for sperm antibodies should be done routinely on all men being evaluated for infertility despite some clinical guidelines suggesting it is not necessary, because no semen analysis pattern is characteristic of sperm autoimmunity.[42, 68] The immunobead test (IBT) with beads binding to more than 50% of motile sperm is regarded as positive, but there is usually more than 70% to 80% immunoglobulin A (IgA) bead binding with clinically significant sperm autoimmunity. Tail tip–only IBT binding is not significant.[73] The mixed antiglobulin reaction test is an alternative to the IBT.[68] The indirect IBT, in which normal donor sperm are exposed to test serum or seminal plasma, can be used when there are too few motile sperm for the direct IBT. An alternative screening method for sperm autoimmunity in men with sperm in the semen would be to perform a sperm-mucus penetration test.[68]
Sperm-mucus penetration tests can be performed by postcoital examination of sperm in cervical mucus collected at mid-cycle or after estrogen treatment (ethinyl estradiol, 50 g twice daily for 4 days) to produce mucus of equivalent quality.[68] In vitro capillary mucus penetration (Kremer) tests are particularly important for evaluating the significance of sperm autoantibodies; failure of sperm to penetrate more than 2 cm in 1 hour indicates severe sperm autoimmunity with a poor prognosis and a high likelihood of failed fertilization with standard IVF.[42, 73]
Sperm Function Tests
A number of tests of sperm function are available to examine the human fertilization process (Fig. 3). These are only performed in specialist laboratories. If simpler approaches or active preparations of zona pellucida (ZP) or sperm receptor proteins become available, they will be widely used to improve the assessment of human sperm. IVF has permitted many conventional and new tests of sperm function to be examined. Groups of sperm variables that are independently significantly related to the proportion of oocytes that fertilize in vitro can be determined by regression analysis.[74] This approach has confirmed the importance of sperm morphology in the ability of sperm to interact with the coverings of the oocyte.

Figure 3. Stages of human fertilization. Spermatozoa swim through the surrounding medium and cumulus mass (not shown) and bind to the surface of the zona pellucida. The acrosome reaction is stimulated by zona proteins and the acrosome reacted sperm penetrates the zona, enters the perivitelline space and binds to the oolemma via the equatorial segment. Oocyte processes surround the sperm head and it enters the ooplasm and decondenses. Infertility could result from defects of any of these processes. For example, abnormal sperm particularly with defective head morphology bind poorly to the zona. (From Baker, H.W.G., Male Infertility. Chapter 141 In Endocrinology, 6th edition, Jameson J. L. and DeGroot L.J. (Chief Eds.), Saunders Elsevier Philadelphia PA. pp 2556-2579, 2010)
Human Sperm–Zona Pellucida Binding Ratio Test
Because the number of sperm bound to the ZP is strongly related to the fertilization rate, human sperm–ZP interaction tests have been developed using oocytes that failed to fertilize in vitro.[74] These oocytes can be used either fresh or after storage in concentrated salt solutions. Because the ZP binding capacity is variable, control (fertile donor) and test sperm are labeled with different fluorochromes (fluorescein and rhodamine). After incubation with equal numbers of control and test sperm, the oocytes are aspirated through a wide bore pipette to dislodge loosely adherent sperm and the numbers of sperm tightly bound to the ZP are counted with a fluorescence microscope. Results are expressed as a ratio of the number of test and control sperm bound to the ZP of four oocytes. An alternative method is to cut the zona and expose one half to test and the other to control sperm (Hemizona assay).[75]
Human Sperm–Zona Pellucida Penetration Test
It is difficult to determine the number of sperm penetrating the ZP when many sperm are bound to the surface. The sperm bound to the surface of the ZP can be sheared off by repeatedly aspirating the oocyte with a pipette with an internal diameter less than the diameter of the oocyte (120 m). The sperm penetrating the ZP or perivitelline space can then be counted easily, and the results of this test are the most predictive of fertilization rates with standard IVF.[74]
Zona Pellucida–Induced Acrosome Reaction Test
Sperm dislodged from the ZP can be stained with a fluorescein-labeled lectin such as pisum sativum agglutinin or an antibody specific for the acrosomal contents to determine the proportion that are acrosome reacted. This test is useful for diagnosis of disordered ZP-induced acrosome reaction.[74]
Human Sperm–Oolemma Binding Ratio Test
Sperm-oolemma binding has been studied in a similar way to the sperm-ZP binding test, using oocytes that have had the ZP removed. [74]
Interpretation of Semen Analysis Results
Table 5 shows various patterns of abnormality of semen quality and their common causes. It is always important to consider whether the result is spurious. Repeated tests are necessary to establish an average and to determine the variability within an individual man (see Fig. 2).
Variations in Semen Volume and Appearance
Low semen volume suggests incomplete collection, short duration of abstinence from ejaculation before the test, absence or obstruction of the seminal vesicles, or androgen deficiency. High semen volume (>8 mL) may be seen in association with oligospermia but is of little practical significance. Hemospermia is usually the result of minor bleeding from the urethra, but serious conditions, such as genital tract tumors, must be excluded. Other discoloration of the semen may indicate inflammation of accessory sex organs. The semen may be yellow with jaundice or salazopyrine administration. Defects of liquefaction and viscosity are relatively common and presumably result from malfunction of the accessory sex organs. Although these may cause problems with semen analysis and preparation of sperm for assisted reproductive technology (ART), they are probably of little relevance to fertility. Sperm agglutination is common with sperm autoimmunity but can also occur for other reasons.
Azoospermia
The total absence of sperm from the semen needs to be confirmed in repeated tests with vigorous centrifugation of the semen and careful examination of the pellet.[68] Cooper and others have shown that sperm maybe found in apparently azoospermic samples using more sensitive sperm-counting methods (e.g. fluorescence microscopy).[76] Rarely, an illness or difficulty with collection will cause transient azoospermia; however, this can also occur for unexplained reasons. With severe spermatogenic disorders and some obstructions, sperm may be present in the semen intermittently. If any live sperm can be found, these can be cryopreserved for intracytoplasmic sperm injection (ICSI).
Oligospermia
Sperm concentrations of less than 15 million/mL or preferably total number (concentration X volume) less than 39 million are classified as oligospermic.[68] This figure represents the 5th percentile derived from analysis of semen analyses preformed using WHO methods in about 1900 healthy volunteers whose partners had a time-to-pregnancy of ≤12 months and is used in the 5th edition of the WHO semen analysis manual .[77] There is a correlation between sperm concentration and other aspects of semen quality. Both motility and morphology are usually poor with oligospermia.
Asthenospermia
Asthenospermia is defined as less than 40% sperm motility or less than 32% with rapid progressive motility.[68] Spurious asthenospermia caused by exposure of sperm to rubber (particularly condoms), spermicides, extremes of temperature, or long delays between collection and examination, should be excluded. Low sperm motility is a frequent accompaniment of oligospermia, and is often also associated with a mixed picture of morphologic defects suggesting defective spermiogenesis.
Specific ultrastructural defects of the sperm can be evaluated by electron microscopy when there is zero sperm motility or extreme asthenospermia (less than 5% motile sperm).[50, 78] Absent dynein arms, other axonemal defects, mitochondrial abnormalities, disorganized fibrous sheath or outer dense fibers, or normal ultrastructure may be found. Standard semen analyses usually show normal sperm concentrations and morphology but there may be tail abnormalities: short, straight, or thick tails, or midpiece defects. Viability tests help to distinguish this group of patients from those with necrospermia.[79] Patients with structural defects in the sperm may be able to be treated by ICSI. Asthenospermia may also be associated with sperm autoimmunity. The causes of other motility defects of moderate degree are unidentified.
Absolute asthenospermia (the condition in which only immotile spermatozoa can be retrieved) is reported at a frequency of 1 in 5000 men and it implies a very poor fertility prognosis. Selection methods currently used to choose a viable sperm for ICSI in the case of absolute asthenospermia are based on chemical substances added to the sample before the procedure or biophysical approaches accomplished during the procedure. [80]
Immotile sperm treatment with pentoxifylline, a nonselective inhibitor of phosphodiesterase has been investigated and proved to be an efficient approach. [81, 82]
Theophylline, a nonselective inhibitor of phosphodiesterase with an increased half-life compared to pentoxifylline was also investigated, recently, [83] in a prospective study on thawed testicular sperm selected for ICSI. Theophylline-treated thawed sperm compared
with untreated sperm showed a significant improvement in searching time, increased fertilization and blastulation rates and higher implantation and clinical pregnancy rates. In a recent case report [84], theophylline has been demonstrated to be an efficient agent for stimulating immotile spermatozoa also in a patient with retrograde ejaculation and total sperm asthenospermia, and a healthy live birth was reported.
Other sperm selection methods as the sperm tail flexibility test (STFT) [85], the hypo-osmotic swelling test (HOS)[86, 87] and laser shot system [88] were reported to be useful.
Necrospermia
It is important to distinguish necrospermia from other types of severe asthenospermia because some patients with necrospermia produce pregnancies despite the low sperm motility.[40, 79, 89, 90] Necrospermia is characterized by usually less than 20% to 30% total motility, less than 5% progressive motility, and a viability test less than 30% to 40%, indicating a high proportion of dead sperm. Other causes of severe asthenospermia such as sperm autoimmunity and collection problems must be excluded. Necrospermia may fluctuate in severity, particularly with changes in coital frequency.[79, 90] Characteristic of necrospermia is an improvement of sperm motility with increased frequency of ejaculation. The condition may be caused by defective storage of sperm in the tails of the epididymides or stasis in the genital tract, and it also occurs with chronic spinal cord injury and with adult polycystic kidney disease associated with cysts in the region of the ejaculatory ducts.[40, 89] There are ultrastructural features of degeneration in the ejaculated sperm but normal structure of late spermatids in testicular biopsies.[79, 89] Treatment with antibiotics may have a beneficial effect, but this is not proved. The couple should have intercourse once or twice every day for 3 to 4 days up to the time of ovulation.
Teratospermia
Teratospermia is a reduced percentage (<4%) of sperm with normal morphology assessed by light microscopy.[68] It is important to distinguish mixed abnormalities of sperm morphology from those in which all or the majority of sperm show a single uniform defect, such as spherical heads with absence of the acrosomes (globospermia) and pinhead sperm. Pinhead sperm result when the centrioles from which the sperm tails develop are not correctly aligned opposite the developing acrosome. On spermiation, the sperm heads are disconnected from the tails and absorbed during epididymal transit so that there are only sperm tails in the ejaculate, the cytoplasmic droplet on the midpiece giving the pinhead appearance.[91] Both these conditions cause sterility but are extremely rare.
In general, human spermatozoa are very variable in appearance and the microscopic assessment of sperm morphology is highly subjective and difficult to standardize between laboratories. Only a small proportion (<25%) of the motile sperm from fertile men are capable of binding the ZP in vitro, and this zona binding capacity is closely related to the morphology of the sperm head.[92] The morphometric characteristics of the sperm that bind to the ZP may be useful as a standard for sperm morphology.[17, 93] Various histological assessments of morphology have been used. The simplest is to record as normal only those sperm that have no shape defects in head, midpiece or tail, regions.[68] In the strict morphology approach, although size measurements are set, the sperm are assessed by eye and those marginally abnormal are assigned abnormal. Automated methods involving image analysis by computer have been developed that could overcome the between-laboratory variability and greatly improve the predictive value of semen analysis for natural conception.[17, 93]
Before the introduction of ICSI, the percentage of sperm with normal morphology assessed by strict criteria after washing the sperm and adjusting the concentration to 80 million/mL, provided one of the most useful predictors of fertilization rates with standard IVF. There was a progressive reduction in oocytes fertilized from 60% to 20% as abnormal morphology increased from less than 70% to more than 95%.[94] Patients with high proportions of sperm with abnormal morphology are now treated by ICSI because of the risk of failure of fertilization with standard IVF. ICSI results are independent of sperm morphology.
Sperm chromatin and abnormal DNA assays
A variety of flow cytometric and other assays to measure sperm chromatin integrity, DNA fragmentation, sperm apoptosis or nuclear integrity have been developed. [95, 96] The usefulness of these tests for prediction of fertility remains controversial [97]
Hormone Assessment
It is not necessary to perform hormone measurements routinely. Follicle-stimulating hormone (FSH) levels in patients with azoospermia, normal testicular volume, and normal virilization may help distinguish genital tract obstruction from a spermatogenic disorder. The most useful FSH value for the upper limit in reproductively normal young men is ~8 IU/L.[98, 99] However, some men with primary seminiferous tubule failure have normal FSH levels. Normal FSH is common with germ cell arrest at the primary spermatocyte stage. Rarely, high FSH levels are seen with normal spermatogenesis.[100] Measurement of FSH, luteinizing hormone, and testosterone is useful in men with reduced testicular volume and signs of androgen deficiency, to distinguish primary from secondary hypogonadism. Inhibin B measurement may provide additional information about the state of spermatogenesis.[101] but is rarely used in routine practice.
Prolactin should be measured in men with galactorrhea or androgen deficiency and loss of libido.[102] Other hormone investigations are occasionally required, such as thyroid function tests with hyperprolactinemia, 17-hydroxyprogesterone measurements with congenital adrenal hyperplasia, estradiol with liver disease or tumors, iron studies to exclude hemochromatosis, human chorionic gonadotropin with tumors and estrogen excess, and pituitary function tests for panhypopituitarism.[43]
Chromosome and Genetic Studies
Chromosomal anomalies are 8-10X more common in infertile than fertile populations, and in many cases there are no other phenotypic changes. The prevalence is inversely correlated to the sperm density, being 14% of azoospermia.[103, 104] Accordingly a routine assessment is recommended in men with unexplained infertility and sperm densities < 5-10 million/ml. A karyotypes is performed in men with clinical evidence of primary testicular failure and small testes to confirm a clinical diagnosis of Klinefelter syndrome. Usually the karyotype is 47,XXY, but there may be higher numbers of X chromosomes or a sex-reversal 46,XX karyotype.[105-107] Although most men with Klinefelter syndrome produce no sperm in the semen, some are oligospermic and very rarely fertile.[105] Also, sperm for ICSI may be obtained by testicular biopsy in about 50% of patients.[106, 107] Defective spermatogenesis may occur with 47,XYY, but the clinical picture is much less uniform than it is for Klinefelter syndrome. The extra Y chromosome is deleted early in gametogenesis because the sperm, embryos, and children generally have normal karyotypes. However, an increased rate of sex chromosomal and autosomal aneuploidy has been noted in studies of sperm from XXY and XYY men.[107, 108] Some Y chromosome abnormalities, such as an isochromosome of two short arms, are associated with absences of spermatogenesis.
Infertile men have much higher rate of aneuploidies compared to fertile men and most of them have no other phenotypic features. An increased frequency of autosomal abnormalities is found with defective spermatogenesis, particularly balanced autosomal translocations (reciprocal and Robertsonian), which may be transmitted in unbalanced form to their offspring.[109] As part of their infertility investigation, it is imperative to screen severely oligospermic men as the result may affect treatment outcome.
Cystic fibrosis gene studies are important for evaluation of patients with congenital absence of the vas and their partners.[110] If the woman has a cystic fibrosis gene mutation, preimplantation genetic diagnosis of their embryos can be offered. Microdeletions in the long arm of the Y chromosome (AZF regions) have been found in 3% to 15% of men with severe primary spermatogenic disorders.[18, 19, 27, 36, 37] Sons of men with these microdeletions have the same microdeletions.[111]
Another gene involved in spermatogenesis (histones replacement) and different from the AZF regions Yq deletions is CDY. ([112] TSPY gene is located on the short arm of Y chromosome and may regulate the timing of spermatogenesis by signaling spermatogonia to enter meiosis. [29]
The sex hormone-binding globulin (SHBG) gene has been studied for possible role in spermatogenesis.[113] Other genes that have been investigated for a possible involvement in fertility are DAZL (sperm count) MTHFR and the estrogen receptors genes (ESR1 and ESR2). Polymorphisms of the promoter region of the estrogen receptor gene have been shown to be related to sperm production. Men with higher numbers of TA repeats have lower sperm counts. [29]
Androgen receptor defects have also been found in some men with unexplained primary spermatogenic disorders. Mutations in the gene impairing androgen receptor activity produce androgen insensitivity, which has a variable phenotypic expression from testicular feminization to otherwise normal males with gynecomastia or hypospermatogenesis and oligospermia.[33] Increases in the number of CAG repeats in exon 1 over approximately 40 are associated with Kennedy disease (progressive spinobulbar atrophy) and men with this condition may be infertile.
The field of epigenetic errors has also been studied for its possible contribution to male infertility. [114] (Table 4)
Other specific genetic tests and family studies may be indicated on clinical grounds (see Table 4).
At present, it is reasonable to screen all infertile men with otherwise unexplained primary spermatogenic defects with average sperm concentrations less than 5-15 million/mL by karyotype and Yq microdeletion testing.[115] All patients should be counseled about the possibility of transmitting known and unknown genetic defects.
Testicular Biopsy
Testicular biopsies are necessary to assess spermatogenesis in men with presumed genital tract obstruction. A significant proportion of men with azoospermia, normal testicular size, and normal FSH are found to have severe spermatogenic disorders.[14] Some severe spermatogenic defects may be incomplete, and because ICSI can be performed if sperm can be obtained from the testes, diagnostic testicular biopsies should be offered to men with severe primary spermatogenic tubule disorders with persistent azoospermia. If any elongated spermatids can be found, it should be possible to perform ICSI. However, if no elongated spermatids are seen in the diagnostic biopsies it still may be possible to find spermatids by more extensive sampling of testicular tissue with open biopsies (see later).
It is most important that tissue for histology is removed from the testes with minimal damage and placed in a suitable fixative, such as Bouin’s or Steive’s solution. Standard formalin fixatives destroy the cytoarchitecture.
Testis biopsies may be performed under local or general anesthesia. Needle biopsy many obtain only isolated cells but these may be sufficient for diagnosis based on cytology or for flow cytometry. The technique shown in Fig. 4 usually provides sufficient material for a histologic diagnosis of the state of the seminiferous epithelium despite some deformation artifacts.[116] It is also useful for obtaining testicular sperm for ICSI.[117] Complications are rare and include minor bleeding in the skin and testis, and rarely hematoma or reactions to the local anesthetic. Failure to obtain tissue occurs particularly with fibrosed or small (<5mL) testes
In the presence of azoospermia, an open testicular biopsy might not just be a therapeutic approach in the way of sperm retrieval, but also the only way of excluding early testicular germ cell neoplasia or even an overt testicular cancer.
Infertile men are more likely to develop testicular cancer compared to men with normal fertility.[118] Testicular intra-tubular germ cell neoplasia of the unclassified type (ITGCNU) also called carcinoma in situ (see below) is the precursor of testicular germ cell tumors in which the neoplastic cells are confined within the seminiferous tubules. This makes it a non-invasive stage of the disease. It can be found in testicular tissue adjacent to germ cell tumours in more than 90 percent of adult cases. [119, 120]
The incidence of ITGCNU in men undergoing fertility evaluation ranges between 0.4 - 1.1 percent.[121, 122] As ITGCNU is asymptomatic, patients remain undiagnosed until an overt tumour can be identified usually by palpation. A sample from a sperm retrieval open biopsy should be sent to histopathology routinely.

Figure 4. (A) Fine-needle tissue aspiration biopsy of the testis. Local anesthetic is injected around the vas to block testicular sensation. (B) Fine-needle tissue aspiration biopsy of the testis A 21 gauge butterfly needle is inserted into the testis. An assistant applies suction to the needle tubing via a 10mL syringe and the operator makes thrusting movements of the needle into the substance of the testis. (C) Fine-needle tissue aspiration biopsy of the testis while maintaining the suction the needle is removed carefully and any seminiferous tubules protruding from the needle are grasped with fine forceps to avoid them falling back into the puncture hole. With this technique seminiferous tubule sections are sucked into the needle and these are expelled into some culture medium. Portions can be sent for histology and the remainder used for extraction of sperm in the IVF laboratory by stripping the seminiferous tissue out of the connective tissue membrane of the seminiferous tubule.
For clinical purposes, testicular histology is classified as follows: normal or hypospermatogenesis (all the cellular elements of spermatogenesis are present but in reduced numbers), germ cell arrest (the earlier cellular elements of spermatogenesis are present but at a certain stage, the process stops, most often at the primary spermatocytes), Sertoli-cell-only syndrome or germ cell aplasia (the tubules contain Sertoli cells but no germ cells), hyalinization (the cellular elements have disappeared, leaving only thickened seminiferous tubule walls as in Klinefelter syndrome), and immature testis (no gonadotropin stimulation, prepubertal appearance).[123] Examples are shown in Figure 5. Other classifications such as partial or incomplete maturation arrest and partial germ cell aplasia cause confusion in the literature and should not be used.[124]

Figure 5A. Testicular histology from fine-needle aspiration samples. Normal.

Figure 5B. Testicular histology from fine-needle aspiration samples. Left mild hypospermatogenesis with elongated spermatids with poor head morphology: right normal.

Figure 5C. Testicular histology from fine-needle aspiration samples. Mild-moderate hypospermatogenesis.
Figure 5D. Testicular histology from fine-needle aspiration samples. Moderate-severe hypospermatogenesis.

Figure 5E. Testicular histology from fine-needle aspiration samples. Germ cell arrest at the primary spermatocyte stage.

Figure 5F. Testicular histology from fine-needle aspiration samples. Sertoli cell-only syndrome: low and high power.

Figure 5G. Testicular histology from fine-needle aspiration samples. Germ cell arrest at the spermatogonial stage from gonadotropin deficiency. Upper panel atrophic Leydig cell stained brown with anti-testosterone antibody.
Figure 5H. Testicular histology from fine-needle aspiration samples. Carcinoma in situ, only transformed spermatogonia and Sertoli cells present.
Other Investigations
Ultrasonography is useful to check for tumors in the testes, particularly when the testes are difficult to palpate because of a tense hydrocele.[125] Other abnormalities may also be found (Figs 6-8). It can also be used to measure testicular size and confirm the presence and nature of cysts or other abnormalities in the scrotum. Some argue scrotal ultrasound should be performed routinely in infertile men to measure testicular volume, assessing the texture and exclude impalpable malignant tumors in the testes.[126] However, some clinical guidelines do not support this. [127] Doppler blood flow assessment is valuable in assessing a painful swollen testis for torsion or inflammation and for evaluating varicoceles. Other tests of a varicocele, including thermography, technetium scans, and venography may be performed but, as pointed out later, the value of treating varicoceles to improve fertility is uncertain. Rectal ultrasound may demonstrate cysts in the prostate, enlarged seminal vesicles, or dilated ejaculatory ducts associated with distal genital tract obstructions (Figure 8).[128] Clinical suspicion of the presence of a pituitary tumor should be followed up by appropriate radiology. Abdominal imaging is necessary to check the position of impalpable testes.

FIGURE 6. Ultrasounds of testes with tumours. A Longitudinal, testis with seminoma in a man presenting with infertility, severe oligospermia and a large hard right testis; B Low power histological section of radical orchiectomy specimen with active tumour stained blue, and necrotic centre and seminiferous tubules stained red; C, Londitudinal and D transverse of impalpable 1cm diameter seminoma in the upper pole of the right testis in a man presenting with severe oligospermia and normal sized testes.

FIGURE 7. Miscellaneous findings on scrotal ultrasound. A,B multiple epididymal and intratesticular cysts in a man with von Hippel Lindau syndrome; C Simple hydrocele; D Multiloculated hydrocele; E Simple cysts in head of the epididymis; F Ectasia of the rete testis; G Hypoechoic area in periphery of testis of uncertain significance; H Microlithiasis in a severely atrophic testis; I Transverse blood vessel; J Intratesticular varicocele; K Blood flow in intratesticular varicocele.

FIGURE 8 Ultrasound of undescended testes. A - In neck of scrotum at external inguinal ring with small tumour; B - In inguinal canal; Incidental prostatic cyst in a healthy man: C - transverse, D - sagittal; Ejaculatory duct obstruction: E - prostate transverse - dilated ejaculatory duct; F - transverse, dilated seminal vesicle; G - sagittal, dilated duct and seminal vesicle.
MANAGEMENT OF SPECIFIC CONDITIONS
This section addresses the management of sperm autoimmunity, male genital tract obstructions, coital disorders, genital tract inflammation, and varicocele. Treatment of gonadotropin deficiency and androgen replacement therapy are covered in Chapters by Hayes and Pitteloud, and Handelsman.
Sperm Autoimmunity
Sperm autoimmunity is present in 6% to 10% of infertile men.[42] They have sperm coated with antibodies to the extent that sperm function is impaired, particularly sperm - mucus penetration and sperm zona-pellucida binding, resulting in severe infertility. Natural pregnancy rates without treatment are very low and fertilisation rates with standard IVF are low or zero. Other men have positive IBT, sometimes with only to tail tip binding and normal or only marginally impaired mucus penetration.[73] These low-level sperm autoantibodies are irrelevant to the infertility and other causes of the couple’s infertility should be sought. Sperm autoimmunity can be treated by glucocorticoids in immunosuppressive doses. Antibody levels fall and semen quality improves in about 50% of patients and about 25% produce natural conceptions during a 4-6 month course of treatment.[42] There are significant risks of severe side effects particularly aseptic necrosis of bone. The superior results of ICSI make this treatment obsolete and only useful in exceptional circumstances.
Genital Tract Obstruction
Clinical Characteristics
Most men with genital tract obstruction have azoospermia, normal testicular size, normal virilization, and normal serum FSH levels. However, some have combined obstruction and spermatogenic disorders or partial obstructions and severe oligospermia. There may be a history of an event that caused the obstruction, such as epididymitis with gonorrhea or associated respiratory disease. Because a few men with normal spermatogenesis have elevated FSH levels and some spermatogenesis may occur in association with a severe spermatogenic disorder, all patients should be offered further investigation. The presence of sperm antibodies in blood serum by indirect IBT indicates sperm are being formed but is an adverse prognostic factor for natural conception after surgery. With bilateral congenital absence of the vasa or ejaculatory duct obstruction, semen volume, pH, and fructose levels are low. The semen also does not have its characteristic smell and does not form a gel after ejaculation because it contains only prostatic and urethral fluid. Rectal ultrasound shows absent or atrophic seminal vesicles with bilateral congenital absence of the vasa but with ejaculatory duct obstruction the seminal vesicles and ejaculatory ducts are dilated and the cause of the obstruction may be obvious such as a cyst of the prostatic utricle.[128] Testicular biopsy is usually normal but there may be some reduction in spermatogenesis either as a coincidence or as a result of the obstruction, particularly after vasectomy.[129]
Pathophysiology
Degeneration of the Wolffian duct structures occurs with cystic fibrosis gene mutations but can be of variable extent. Although most often only the heads of the epididymides are palpable, some men with bilateral congenital absence of the vasa have parts or all of the epididymides and scrotal vasa present with absent or atrophic pelvic vasa and seminal vesicles. Other conditions may cause defective development of the vasa such as congenital rubella. Young’s syndrome which is now rare is not related to cystic fibrosis gene mutations. The pathology shows inspissated material in the head of the epididymis, and there are lipid inclusions in the epithelial cells.[48, 49] As some men with this syndrome have fathered children, the blockage may develop in adulthood.
Post-inflammatory obstructions after gonorrhea typically occur in the tail of the epididymis, whereas nonspecific bacterial inflammation produces more widespread destruction, and tuberculosis usually causes multiple obstructions in the epididymides and vasa or destruction of the prostate and seminal vesicles. Back pressure ‘blowout’ obstructions in the epididymis are frequent after vasectomy. Iatrogenic causes of genital tract obstruction include inadvertent epididymectomy during testicular biopsy, vasal damage during hernia repair or pelvic or lower abdominal surgery such as renal transplantation, and ejaculatory duct obstruction from prostatectomy or complicated bladder catheterization.
Differential Diagnosis
Men with persistent azoospermia, normal testicular size, normal virilization, and normal FSH levels can be assumed to have obstruction until proved otherwise. As many as one third of men with this clinical picture are found to have a serious spermatogenic disorder on testicular biopsy despite the normal serum FSH level.[14] There are rare instances of normal men who show azoospermia on single occasions or over a short period.[6, 20] This “spurious azoospermia” must be excluded before surgery is contemplated. Once diagnosis of obstruction is confirmed, it is necessary to determine the feasibility of surgery. Intratesticular and caput-epididymal obstructions have a poor prognosis, but cauda-epididymal and vasal obstructions can often be treated successfully with surgery.[130] Distal obstructions are important to diagnose because they may be reversed at transurethral endoscopy.[128] It is important to test for sperm in urine after ejaculation in patients with possible ejaculatory duct obstruction as partial retrograde ejaculation can produce the same the semen characteristics as ejaculatory duct obstruction (Table 5). Also large cysts causing ejaculatory duct obstruction can affect the bladder neck and cause retrograde ejaculation.
Treatment
General Management
Patients in whom one or both vasa deferentia are not palpable have congenital unilateral or bilateral absence of the vas deferens. Fifty to eighty-two percent of healthy men with CBAVD harbor detectable mutations in one or both alleles of the CFTR gene . These men should all be tested for CFTR abnormalities. CFTR testing is also indicated in azoospermic patients with normal testis volume, normal serum FSH, and palpable vasa deferentia, who have low semen volume (<1.5 ml). Female partners of carriers should be screened for cystic fibrosis gene mutations and the couple counseled accordingly. Although most consider genetic testing unnecessary, high proportion of men with idiopathic epididymal obstruction were shown to be carriers of CFTR mutations.[131] Preimplantation or prenatal genetic diagnosis may be performed if mutations are found in both partners. The woman should be investigated in detail to ensure her potential fertility before surgery is contemplated in the man. The prognosis of the procedure and the availability of other forms of treatment, including donor insemination, should be discussed with the couple. Sperm retrieval for ICSI, either from the testis or other parts of the genital tract, is an alternative to surgery.[117] ICSI is also used when reconstructive surgery is not possible, the female partner has an infertility problem, or the couple cannot wait 6 to 12 months to have a reasonable attempt at conceiving naturally after surgery. For ICSI, sperm may be obtained by testicular biopsy or percutaneous sperm aspiration from the epididymis under local anesthesia. If a spermatocele is present, usable sperm may be obtained by direct puncture through the scrotal skin. It may be possible to combine vasoepididymostomy with sperm aspiration for cryopreservation or ICSI.
Epididymal and Vasal Surgery
Treatment of male genital tract obstructions is best undertaken by specialist microsurgeons.[130] The testis is exposed and the most proximal (to the testis) level of obstruction determined. The patency of the vas is determined by syringing with saline or by vasography. The vas or epididymal tubule is opened proximal to the obstruction, and, if possible, the presence of motile sperm is demonstrated by microscopy. Microsurgical anastomosis between the ends of the vas or between the vas and the epididymal tubule is then undertaken.
Results
Vasovasostomy and vasoepididymostomy for caudal blocks produce relatively good results, with 50% to 80% of patients having sperm present in the semen. However, less than half of these produce a pregnancy within the first year.[130] There may be continuing obstruction, sperm autoimmunity, or coexisting spermatogenic disorders. The results of vasoepididymostomy for proximal blocks are poor. Although sperm may appear in the semen, pregnancies are extremely uncommon after vasoepididymostomy for caput epididymal blocks. In contrast, the results of ICSI with testicular or epididymal sperm, fresh or after cryopreservation, are similar to those obtained with sperm from semen.[117]
Gonadotrophin deficiency and suppression
Clinical Characteristics
Most men seeking treatment for infertility associated with gonadotropin deficiency have been treated with androgens, following presentation in adolescence with delayed puberty. The main diagnoses are Kallmann’s syndrome, other isolated gonadotropin deficiencies, combined gonadotropin and growth hormone deficiency and rarely pituitary tumours, trauma or craniopharyngiomas treated in childhood. [30] Occasionally men with previously undiagnosed prepubertal gonadotropin deficiency present with infertility. The clinical features are usually very small testes (<4mL) and severe androgen deficiency. There may be a child like appearance with lack of secondary sex hair development, failure of male pattern scalp hair recession and balding and eunuchoidal proportions. Gonadotropin deficiency may develop after puberty because of tumours, surgery or trauma of the pituitary or hemochromatosis. These men usually note loss of libido and may note reduced beard and body hair growth, low ejaculate volume and decreased testicular size. General lethargy, muscular weakness and hot flushes are also common but non-specific symptoms. Physical examination may show testicular atrophy, reduced secondary sex hair and dry finely wrinkled skin on the face. Gynaecomastia may be present. Features of underlying or associated conditions may be present for example: headache, visual disturbance and hormone excess or deficiency with pituitary tumours, or pigmentation, liver disease or diabetes with haemochromatosis.
Hyperprolactinemia is uncommon in men.[132, 133] It usually presents with loss of libido and impotence, low testosterone levels and variable semen analysis results from azoospermia to relatively normal. Galactorrhea may occur, sometimes with only minimal gynaecomastia. There is usually a pituitary tumour. Hyperprolactinemia associated with a pituitary macroadenoma is rare but important: as well as loss of libido there is usually progressively severe headache and visual field impairment. A number of paediatric syndromes include mental deficiency and gonadotropin deficiency but the patients rarely seek treatment for infertility as adults. Mutations of DAX1 cause adrenal hypoplasia and gonadotropin deficiency.
Gonadotropin suppression may occur in a variety of circumstances. The most common now appears to be the illicit use of anabolic and androgenic steroids or chorionic gonadotropin or opioid for chronic pain. [6, 14] [43, 134]Other hormones and drugs can cause gonadotropin suppression. Rarely men are seen with hormone producing tumours for example adrenal adenomas, Leydig cell tumours or hCG producing teratomas which will suppress gonadotropins, usually there are features of marked hyperestrogenization with progressive gynecomastia. Very rarely men are seen with congenital adrenal hyperplasia with gonadotropin suppression and azoospermia who can be treated successfully by glucocorticoid suppression of ACTH. [38, 39]
Spermatogenesis may occur despite severe androgen deficiency - the so-called fertile eunuch syndrome. This is believed to be due to predominant LH deficiency or partial gonadotropin deficiency. There may be normal sperm concentrations but usually there is low ejaculate volume and sperm motility. The fertile eunuch syndrome commonly occurs with hyperprolactinemia, hemochromatosis, starvation, illness or in athletes in negative energy balance. It is also seen with partial or mild Kallmann syndrome.
Pathophysiology
Commonly gonadotropin deficiency is caused by genetic disorders of gonadotropin releasing hormone production or the GnRH receptor, loss of function of gonadotrophs, or suppression of gonadotropin secretion by extraneous steroids, other drugs or illness. There is usually a combined defect of androgen and gamete production. If the underlying cause cannot be corrected life-long androgen replacement therapy is required. This is usually in the form of testosterone but when fertility is desired, treatment must be changed to gonadotropins. While experimental conditions may be found to indicate that either FSH or LH alone may be able to initiate spermatogenesis in humans, for practical clinical purposes treatment with LH alone (as hCG) is effective for fertile eunuch syndrome and may be effective where spermatogenesis has been stimulated before, either by natural puberty or previous gonadotropin therapy. In other situations both FSH and LH are required (see chapter by Hayes and Pitteloud).
Differential Diagnosis
In men with gonadotropin deficiency it is necessary to determine the cause of the disorder, or if this is not possible to exclude a serious underlying cause such as a pituitary tumour. With Kallmann syndrome there is hyposmia or anosmia from malformations of the rhinencephalon. Other abnormalities may also be present including colour blindness, cleft lip and cerebellar ataxia. [14] [30] Except where the diagnosis is obvious, detailed radiological examination of the pituitary and hypothalamic area is necessary, together with full pituitary function tests to determine if there are other hormone deficiencies.
Treatment
Gonadotropin suppression from administration of steroids or other agents is treated by withdrawal of the agents, and starvation induced gonadotropin suppression by refeeding. Hyperprolactinemia can be treated with bromocriptine or other dopamine agonist. [20] [102]
Gonadotropin deficiency caused by of gonadotrophe destruction or abnormalities of the GnRH receptor require treatment with gonadotropins. [20, 135] Some men with gonadotropin releasing hormone deficits can be treated successfully with pulsatile GnRH administration. For details of the gonadotropin and GnRH treatment, see chapter by Hayes and Pitteloud.
Coital Disorders
Male coital disorders impacting fertility include erectile dysfunction (impotence), failure of ejaculation, and retrograde ejaculation. Many men have problems with sexual performance after first learning about their infertility, but this usually ameliorates with time. Infrequent and poorly timed intercourse may result from incorrect advice, low libido, or the psychological reaction to infertility.[6]
Erectile dysfunction
Erectile dysfunction may be associated with low libido from androgen deficiency with primary or secondary hypogonadism. (This topic is considered in detail in Chapter 8 by Bochinski et al.) Erectile dysfunction related to vascular or neurologic abnormalities (diabetic autonomic neuropathy or pelvic nerve damage) is uncommon in men presenting with infertility.[14] Selective impotence at the time of ovulation may indicate psychological problems and ambivalence about having children.
Failure of Ejaculation
Failure of ejaculation is usual with chronic spinal cord injury and may also be caused by antihypertensive and psychotropic drugs but otherwise is an infrequent cause of infertility in most societies.[89, 136] Healthy men who cannot ejaculate with intercourse may be able to produce semen by masturbation, with a vibrator, or other stimulation.
Retrograde Ejaculation
Retrograde ejaculation occurs when the bladder neck fails to contract at the time of ejaculation so that all or most of the semen passes into the bladder. Usually, there is an obvious cause: prostatic surgery, diabetic neuropathy, pelvic nerve damage, or spinal cord injury. Retrograde ejaculation is diagnosed by the finding of sperm in urine passed after ejaculation.
Differential Diagnosis
Recognition of a coital disorder is crucial and thus all infertile patients must discuss their sexual history in detail. Once recognized, the contribution of organic and psychological factors needs to be evaluated.
General Treatment
An optimistic prognosis can be given provided that live sperm can be obtained. The couple is advised about the various techniques that might be used for collecting the sperm for artificial insemination or other ART. The woman’s potential fertility must be evaluated.
Specific Treatment
A drug, such as an antihypertensive or a tranquilizer, that may be contributing to the sexual disorder should be stopped temporarily or permanently.[43] Erectile dysfunction may respond to sex behavioral therapy, administration of phosphodiesterase 5 inhibitors, intrapenile injections of vasodilators and physical approaches with pumps and rubber occlusion devices to initiate and maintain erections or penile implants, but these are seldom needed in men with infertility. Some men with failure of ejaculation or retrograde ejaculation may be able to ejaculate during intercourse with a full bladder or after the administration of phosphodiesterase 5 inhibitors, imipramine, or cholinergic antihistamines, such as brompheniramine or ephedrine.[136] Others require more powerful stimulation with vibrators or electroejaculation.[89] If these are unsuccessful, sperm may be collected by needle biopsy of the testis.[137]
Use of Collected Semen
If semen can be obtained by masturbation or by wearing nontoxic condoms to collect nocturnal emissions, the couple can be taught to inseminate samples at home. The timing of ovulation can be determined by calendar and either symptoms of ovulation or luteinizing hormone surge detected with a urinary luteinizing hormone dipstick kit. Cryopreservation of samples for AIH or ICSI may also be possible.
Assisted Ejaculation
Ejaculation may be stimulated by applying a vibrator to the underside of the penis near the frenulum of the glans. Vibrators with a 2-mm pitch and frequency of 60 Hz or more are most effective. Men with complete spinal cord injuries below T10 are unlikely to respond and will require electroejaculation. Modern electroejaculation equipment is safe. The probe includes a thermal sensor and proctoscopy is performed before and after the procedure to ensure there are no burns or other damage to the rectum. A balloon catheter in the bladder is used to prevent retrograde ejaculation.[89]
Semen obtained by assisted ejaculation from able-bodied men or in the acute stages of spinal cord injuries is often normal.[89] In contrast, with chronic spinal cord injury, there is frequently low volume, high sperm concentration, and poor motility.[89] As with necrospermia, repeated ejaculation over several days can improve sperm motility. If the semen quality is too poor for AIH or the risks associated with electroejaculation are considered unacceptable, aspiration of sperm from the testis and ICSI produces good results. Assisted ejaculation may cause autonomic hyperreflexia with chronic spinal cord injuries above T6.[89] The resulting uncontrolled hypertension may cause cerebral hemorrhage. Careful monitoring of blood pressure and prophylactic nifedipine treatment usually prevents serious problems. Men without complete sensory deprivation require general anesthesia for electroejaculation.
Retrieval of Sperm with Retrograde Ejaculation
Motile sperm may be obtained from the urine after retrograde ejaculation.[138] Urinary pH is adjusted to above 7 and osmolality to between 200 and 400 mOsm/kg by administration of 80 g of sodium bicarbonate and 2.0 to 2.5 L of water daily for 3 days before the expected time of ovulation. On the day of ovulation, the man ejaculates and passes urine. Sperm are recovered from the urine by centrifugation, washed, and resuspended in an IVF culture medium. The final pellet is resuspended in approximately 0.5 mL of culture medium for insemination. It is also possible to cryopreserve the sample obtained. If this method fails, electroejaculation and catheterization of the bladder could be considered.
Effects of Systemic Illness and Reversible Exposures to Toxins or Drugs
A very large number of exposures to agents in the environment, drugs, and illnesses can adversely affect testicular function, but it is rare to find patients in whom such exposures can be confirmed as contributing to male infertility. However, this should always be considered during clinical evaluation. The most commonly encountered problems clinically are impairment of spermatogenesis by salazopyrine used for treatment of inflammatory bowel disease or arthritis, testosterone administration, anabolic steroid abuse, long-term high-dose opiate use, and febrile illnesses causing transient reduction of spermatogenesis.[43] Workplace exposures may be implicated in some patients, but the association is rarely clear-cut enough to advise change of occupation.[60, 61]
Acute Illnesses
Fever
The adverse effect of acute febrile illness on the semen quality is well known but only occasionally seen.[43, 45, 46] Frequent hot baths or saunas may also have a similar effect. There is a temporary suppression of spermatogenesis that recovers over 3 to 6 months. Whether increased scrotal temperature because of clothing, varicocele, obesity, or environmental temperature contributes to male infertility is controversial.
Critical Conditions
Suppression of gonadotropin secretion can occur with critical illness such as hepatic failure, myocardial infarction, head injury, stroke, respiratory failure, congestive cardiac failure, sepsis, burns, starvation, general anesthesia and severe stress, both psychological and physical.[43] Transient decreases occur after drug or alcohol intoxication, anesthesia, and surgery. The reduction in testosterone is proportional to the severity of some of the critical conditions and may predict the likelihood of recovery. There may also be direct effects on the testes and alterations in sex hormone–binding globulin levels. The shutdown of testicular function may be a useful adaptation to illness or starvation. During recovery from the critical condition, pulsatile secretion of gonadotropins increases in a manner reminiscent of the changes with puberty, and gynecomastia may develop.[43]
Nutritional Aspects
Starvation is associated with gonadotropin suppression. Specific deficiencies of vitamins and minerals such as B12, C, folate, and zinc may affect testicular function, but these are rare in Western countries.[139] Simple obesity may be associated with alterations in the hypothalamic-pituitary-testicular axis and impaired scrotal thermoregulation. The most common changes are increased conversion of androgens to estrogens in peripheral tissues and low sex hormone–binding globulin levels related to insulin resistance. Total testosterone, sex hormone–binding globulin levels and gonadotropin levels may be low and estrogen levels elevated. There are reports of hypogonadotrophic hypogonadism associated with gross obesity and indications that it can be treated with aromatase inhibitors.[140, 141] However, clinical androgen deficiency, estrogen excess, and abnormal semen analysis are not regularly seen in moderately obese men and the cause and effect relationship is not clear. It may be reduced testicular function predisposes to or aggravates obesity.
Obesity
Obesity in men is associated with low serum gonadotropin, total testosterone, and free testosterone concentrations. The low serum total testosterone concentrations is attributed to associated decrease in serum sex hormone binding globulin (SHBG)
Free testosterone concentrations appear to be inversely related to BMI, independent of changes in SHBG [142, 143] Other factors contributing to the hypogonadotropic hypogonadism seen with obesity include an increase in estrogens through aromatization in adipose tissue, insulin resistance, metabolic syndrome, diabetes mellitus, and sleep apnea [144-146]
Sperm quality has been reported to be inversely related to BMI. [147] U shaped relationship with lower sperm numbers was shown with both low and high BMI [148] Reversibility of obesity-associated male infertility with weight loss should be further investigated.[149]
Chronic Illnesses
Impairment of testicular function is common in uncontrolled or poorly controlled chronic diseases.[43] There are usually elevated gonadotropin levels, indicating a primary testicular defect, but impaired gonadotropin secretion, hyperprolactinemia, changes in sex hormone–binding globulin, and increased aromatization of androgens to estrogens may occur. Although this pattern of abnormal testicular function is a common nonspecific response to chronic illness, the mechanism is obscure. There may be symptoms and signs of androgen deficiency and estrogen excess. Hepatic cirrhosis is one of the classic conditions known to have a profound adverse effect on the male reproductive function. Testicular function may recover after liver transplantation. Similar primary hypogonadism may occur with non-cirrhotic liver disease, chronic alcoholism without liver disease, and a variety of chronic diseases without alcoholism: chronic anemias, chronic renal failure, thyroid hyper- or hypofunction, HIV infection, lymphoma, leukemia, advanced metastatic cancers, rheumatoid arthritis, severe cardiac disease, and chronic respiratory disease.
Effects of drugs
Drugs may contribute to male infertility by affecting gonadotropin (e.g., steroids, opiates) or prolactin secretion (psychotropic agents), or spermatogenesis (salazopyrine, alkylating agents) or by reducing sexual performance (psychotropic and antihypertensive drugs). Some drugs may also cause gynecomastia (antiandrogens, estrogens).[43, 134]
There is currently no place for testosterone treatment of infertile men, either continuously for low testosterone levels resulting from primary or secondary testicular failure, or as “testosterone rebound” therapy because testosterone suppresses gonadotropin secretion and reduces spermatogenesis. Abuse of androgens is widespread in people hoping to enhance athletic performance or bodybuilding. Some men are seen for infertility with azoospermia or oligospermia as a result. Others have sexual performance problems after stopping drug use. The patient may conceal the abuse. Normal virilization but low testosterone, low sex hormone–binding globulin, and low, normal, or transiently high gonadotropin levels may be seen. Recovery can take several months sometimes longer than 12 months, particularly after depot anabolic steroids.
Salazopyrine used for bowel disease and arthritis commonly causes spermatogenic defects. Usually, there is poor sperm motility and morphology or oligospermia. The semen may be stained yellow. The antispermatogenic effect is caused by the sulfapyridine in the drug. Stopping the drug results in a recovery of sperm output within a few months provided the patient’s health remains good and he does not have an underlying defect of spermatogenesis.
Other drugs and toxins are claimed to have adverse effects on spermatogenesis such as colchicine and anticonvulsants, and some antihypertensive drugs, calcium-channel blockers, and antiparasitic chemotherapeutic drugs may impair sperm motility, capacitation, or the acrosome reaction.[43, 134]
Smoking
A meta-analysis of 20 observational studies showed that men who smoked cigarettes were more likely to have low sperm counts.[150]
In utero exposure to smoking was studied in 1770 young, healthy, potential military recruits and results showed the possibility of a small effect. [151] Exposure to smoking in utero was associated with mean sperm concentrations which were 20 percent lower when compared with unexposed men. In another study, there were no significant differences in mean sperm concentrations in men whose mothers either smoked or did not smoke during pregnancy. [152] However, men whose mothers had smoked ≥10 cigarettes per day while pregnant were at higher risk of having oligospermia (sperm concentration <20 x 10(6)/mL)
Genital Tract Inflammation
Specific inflammations of the genital tract such as mumps orchitis or gonococcal epididymitis may cause sterility. Nonspecific inflammations in the accessory sex organs are more common in men with infertility than in fertile men.[6, 153-156] Also, male accessory sex organ inflammation and infertility may be more important in some countries than in others.[6] Symptoms include chronic low back pain, intermittent dysuria, discharges from the penis on straining, and discomfort in the pelvic region or testes after ejaculation or prolonged sexual abstinence. The prostate may be enlarged and tender. The semen may show discoloration, variations in volume, increased viscosity, delayed liquefaction, high pH, sperm agglutination, bacteriospermia, and pyospermia. The bacteria in semen are frequently not pathogens but the commensals of the urethra or skin.[153
To have more than 1 million polymorphs per milliliter in semen, as determined by peroxidase reaction or monoclonal antibodies to leukocyte antigens, is considered abnormal.[68] Although inflammatory cells could damage sperm by releasing oxygen free radicals or cytokines, bacteria could impair sperm motility, and inflammation could also cause partial genital tract obstruction, the actual contribution of nonspecific genital tract inflammation to male infertility is contentious.[155, 156] Routine cultures of semen are not warranted except for sperm donors.
General Management
Men with clinical evidence of prostatitis require full urologic assessment.[157] Specific infections with pathogenic agents are treated with appropriate agents. It remains unclear what should be done about the very common issue of asymptomatic pyospermia and nonspecific male accessory gland inflammation. Therapeutic trials generally show no benefit from antibacterial therapy on semen quality.[158, 159] Antibiotics or other agents may be used if it is thought that the pyospermia compromises semen quality or that bacteria might contaminate the IVF culture media. Because the organisms commonly implicated in nonspecific genital tract inflammation include chlamydia, mycoplasma, and various bacteria, broad-spectrum antimicrobial therapy is required if treatment it is to be given. Also, many of the standard drugs do not enter inflamed accessory sex organs. Trimethoprim, erythromycin, doxycycline, and norfloxacin are potentially effective.[158, 159] Increased frequency of ejaculation to facilitate drainage of the accessory glands and stress management may also help.
Varicocele
The mechanism by which varicoceles cause infertility and the effectiveness of treatment in improving semen quality and natural fertility are controversial.[6, 15, 160-162]
Varicoceles are found in approximately 25% of men being examined for infertility. An additional 15% may have a subclinical left varicocele indicated by a faint cough impulse in the spermatic cord or increase in diameter of the veins on ultrasound.[6, 15, 160] Varicoceles are also found in fertile men. Varicoceles are more common in tall men and in men with larger testes.[14] They are less frequent in men with severe testicular atrophy, for example, in Kallmann and Klinefelter syndromes. When there is a moderate to large left varicocele, the left testis is usually smaller than the right testis.
Pathophysiology
Men with varicoceles generally have poorer semen quality than those without varicoceles.[160, 163] Thus, varicoceles can have an adverse effect on testicular function. Various theories have been advanced for the effect, including vascular stasis, back pressure, interference with oxygenation, reflux of renal or adrenal products into the pampiniform plexus, ROS generation and interference with the heat exchange function of the pampiniform plexus.[160] Varicoceles are usually first noticed at puberty and thereafter may increase in size but remain relatively stable in size throughout the man’s lifetime. Symptoms, including swelling and a dragging sensation in the scrotum, are infrequent, and many men with a large varicocele are unaware of its presence. The sudden appearance of a varicocele in an adult should be taken seriously because it may be a feature of a renal carcinoma with extension into the left renal vein.
Differential Diagnosis
The semen quality in men with varicoceles varies from azoospermic to normal. There is no specific pattern of abnormality with varicocele. Testicular histology is also variable, the only common feature being that the defect in spermatogenesis is more severe on the left side than on the right. Varicocele may be an association rather than the cause of a couple’s infertility. Therefore, full evaluation of other aspects of male and of the female partner is necessary.
Treatment
The value of treatment of varicocele for infertility is particularly contentious.[160-162] One view is that treating varicoceles may not improve fertility; therefore, the varicoceles should only be treated for other reasons, such as symptoms.[15, 161] The other extreme is the belief that varicocele is the most important treatable cause of male infertility, therefore all varicoceles should be treated even if small.[162] In the middle are those who would select cases. When there is an absent, obstructed, or atrophic right testis and all sperm in the semen come from the left testis, treatment of the varicocele may produce a reasonable result.[164]
Treatment of the varicocele involves embolization of the incompetent veins or surgery to prevent venous back flow from the abdomen to the pampiniform plexus. Radiographic techniques involve placement of a sclerosant, glue, or coils that promotes clotting in the veins. They have lower morbidity than surgery. A variety of operations can be performed for varicocele. In the past, retroperitoneal ligation and division of the testicular veins with or without preservation of the testicular artery and lymphatics was performed. Inguinal and scrotal microsurgical approaches have lower failure, recurrence, and hydrocele rates. Successful venous occlusion will relieve pain and reduce the size of large varicoceles. Whether semen quality and fertility are improved is not certain.
Results
Because varicoceles are so frequent, treatment of varicocele for infertility became common and several large series were published with claims of high success rates for improving semen and fertility. Floating numerator pregnancy rates averaging 35% (range, 20%–60%) were commonly reported. Regression toward the mean in semen variables, the nature of subfertility, and the need to include time in the denominator of pregnancy rates were ignored.[165] Although there are reports of successful treatment of azoospermic men by varicocelectomy, transient azoospermia may follow a minor illness or occur for unknown reasons, and, thus, such examples do not prove the value of treatment. Most exponents of varicocele treatment regard azoospermia as a bad prognostic sign, especially if the FSH level is elevated.
Follow-Up Studies and Controlled Trials
Follow-up studies of groups of treated and untreated patients with varicoceles suggest pregnancies are as frequent without treatment as with treatment of the varicocele.[15, 161] Attempts have been made to conduct randomized, controlled clinical trials of varicocele treatment. Such trials are difficult because the ideal design with sham operations and blinding, which is so important in controlling for outcomes affected by psychological factors, is not possible. Large trials are also needed. For example, approximately 250 pregnancies are required to have a high chance of finding a 25% increase in pregnancy rate after treatment significant at the 5% level.[20]
So far, the trials have produced conflicting results and meta-analysis does not support varicocelectomy improves fertility.[161] But the trials are generally small and have problems. For example, the World Health Organization set up a multicenter controlled trial of Palomo ligation in men with infertility of more than 1-year duration, abnormal semen analyses, a moderate to large left varicocele, and a potentially fertile female partner. Volunteers were randomized to immediate operation or operation delayed for 12 months to provide an untreated control group. One of the participating centers reported their results separately.[166] There was a substantial effect on pregnancy rate. Two pregnancies occurred in 20 couples during the 1 year of observation without treatment compared with 15 pregnancies in 20 couples in the year after the operation. During the year after the operation in the remaining 18 control patients, there were 8 pregnancies. Semen analysis results also improved after the operation. There were another 248 couples in 12 countries in the trial, and there was a less marked but significant improvement, the life table pregnancy rates at 1 year being 35% for the operated group and 17% for the un-operated group (relative pregnancy rate, 2.7; 95% confidence interval, 1.6–4.4). Semen analysis results also improved over the first year in the operated group. In the control patients having the delayed operation, the life table pregnancy rate at 1 year after the operation was 21%. However, there were possible irregularities of randomization in some centers early in the trial and high dropout rates, and the results were not published in detail.[160] Also, the pregnancy rates in the control group are lower than expected for untreated subfertile men with varicoceles: approximately 30% produce a pregnancy in 12 months.[15, 167]
Thus, although some people remain convinced of the value of treating varicoceles for infertility, it is not easy to demonstrate this unequivocally and the apparent improvements in semen quality and fertility may result from random fluctuations and regression toward the mean. Although better trials are needed, meta-analyses do not support treatment of varicocele for infertility. It is clear that normal fertility is not achieved in a high proportion of patients treated for varicocele. ART is a realistic alternative for most couples who have not conceived after a reasonable time.
GENERAL MANAGEMENT
This section covers aspects of the management of couples with male infertility not amenable to specific treatment (Table 6). A number will conceive during investigation. Others will decide not to continue with medical intervention. Some patients with treatable conditions may choose ICSI instead of treatment or after a treatment has been unsuccessful. However, most couples with male infertility have conditions for which there is no clearly defined and certainly effective treatment. In these cases, it is important to discuss the prognosis for a natural pregnancy occurring, the ineffectiveness of treatments, and the availability of IVF and ICSI, donor insemination, and adoption. The investigation of the female partner should be reviewed and abnormalities treated when possible. Patients should be acquainted with the physiology of the menstrual cycle and symptoms of ovulation to help time sexual intercourse over the fertile phase of the cycle.[168] Good health practices should be promoted, particularly cessation of smoking because it reduces fertility in women. The psychological upheaval experienced by the couple should be discussed and additional help offered if necessary. Specialist infertility counselors and patient support groups are particularly valuable in this area.
Table 6 Current Management of Subfertility
- Estimate prognosis for natural conception
- Discuss doubtful value of “empirical therapies”
- Advise of alternatives: donor insemination, adoption, childlessness
- Review coital timing
- Review female partner’s potential fertility
- Consider artificial reproductive technology: in vitro fertilization/intracytoplasmic sperm injection
Prognosis for Natural Pregnancy
A number of factors in addition to semen quality affect the likelihood of natural pregnancies occurring. [1, 2, 4, 17, 169, 170] Some are obvious, such as female disorders and coital dysfunction. Female age is important because fertility declines after approximately 35 years of age. Duration of infertility is a major factor in most studies: The longer the infertility, the worse the outlook. The prognostic factors found in a study to determine the effect of varicocele surgery were duration of infertility (negative), mean sperm concentration (positive), untreated sperm autoimmunity (negative), ovulatory disorders (negative), occupational group (farmers doing better than other occupations), female age (negative), and previous fertility in the couple (positive).[15] Interestingly, varicocele presence and size were positive prognostic factors even though varicocele surgery was not significant. The pregnancy rate curves for different sperm concentration groups are shown in Figure 9. Subfertile patients seen in the late 1990s had similar natural conception rates.[17] Such factors can be used to advise patients about their chances of producing a natural pregnancy over time. The accuracy of prediction is low because the statistically significant factors only explain a small part of the variability of the pregnancy rates. New studies using automated methods for semen analysis reveal the percentage of sperm with characteristics conforming to morphometrics preferred for binding to the ZP, and the straight line velocity, may have better predictive value.[17] However, other factors currently not assessable, such as gamete transport, may have an important bearing on conception and may explain the occurrence of pregnancies in some couples despite severely abnormal semen analysis results. Patients should not be told natural conception is impossible unless there is an absolute barrier to fertility.

Figure 9. Pregnancy rate curves grouped according to average pretreatment sperm concentration. The number (n)of patients followed each year is shown. The numbers of men and pregnancies in each sperm concentration group are shown in the inset table (From Baker, H.W.G., Male Infertility. Chapter 141 In Endocrinology, 6th edition, Jameson J. L. and DeGroot L.J. (Chief Eds.), Saunders Elsevier Philadelphia PA. pp 2556-2579, 2010)
of the problem followed by a tendency to blame others and a period of depression before final acceptance of the infertility. The reaction may take years to resolve, and it can threaten the stability of the partnership, interfere with investigation and management of infertility, and lead to futile involvement in expensive “cures” offered by the unscrupulous. Participation in unsuccessful treatments during this phase is often particularly difficult emotionally for the patients. Stresses of ordinary existence are unlikely to influence semen quality. 162 An empathetic approach and involvement of independent counselors or self-help infertility groups may assist some couples. In most, the unpleasantness of the psychological reaction subsides with time.
TIMING OF COITUS
A practical approach is to advise intercourse each day when ovulation might occur. Ovulation can be predicted to occur approximately 14±2 days before a period is due. Knowing the range of menstrual cycle length allows calculation of the days when ovulation is most likely to occur. Symptoms of ovulation including mittelschmerz and mid-cycle mucus changes also help identify the fertile time. 159 Temperature charts may be used to indicate the end of the fertile time as the basal body temperature rises after ovulation. Ovulation timing by measurement of estrogen and progesterone metabolites in urine, urine or serum LH levels, or ovarian ultrasonography may also be used.
GENERAL HEALTH ASPECTS
Although correction of adverse lifestyle factors in the most men seen for infertility is unlikely to produce normal fertility, healthy living has positive long-term benefits. The following are advised: weight reduction for the obese, reduced alcohol intake for the moderate to heavy drinker, avoidance of social drugs including tobacco, avoidance of heat from frequent sauna and spa baths, and management of stress in the workplace, in their relationship, and that engendered by the infertility.
EMPIRCAL TREATMENTS: EVIDENCE-BASED VERSUS UNCONFIRMED TREATMENTS
Treatments of some causes of male infertility are available as discussed previously, but for the majority of patients with abnormal semen analyses, there are no methods of proved effectiveness. 20, 126 A medical or surgical treatment may become established because it is logical and obviously effective, for example, gonadotropin treatment for Kallmann syndrome or vasoepididymostomies for postinflammatory obstructions of the tails of the epididymides. However, in other situations in which semen quality is reduced and there is subfertility rather than absolute sterility, it is necessary to demonstrate that the treatment increases semen analysis results and pregnancy rates by a clinically meaningful amount. This evidence-based medicine approach generally requires controlled clinical trials of promising methods. These trials are usually designed to detect a certain magnitude of difference in the primary responses and thus a positive result supports the use of the method. However, if the trial is negative, it merely does not confirm the magnitude of benefit tested; it does not prove the method is of no value. In time, the results of several trials can be combined by meta-analysis to get better estimates of the overall effects of the method.
In the past, many treatments were used in an uncontrolled fashion for defects of sperm production. 20, 126 Androgens have been given to suppress spermatogenesis in the hope that there would be “rebound” improvement after the treatment is stopped. Low-dose testosterone or weak androgens, such as mesterolone, have been given in the hope of improving epididymal maturation of sperm. Human chorionic gonadotropin has been given for similar reasons. Antiestrogens have been used to increase gonadotropin secretion or gonadotropins (FSH and human chorionic gonadotropin) given to “stimulate” spermatogenesis. Antibiotics and anti-inflammatory drugs have been given for subtle infections or inflammations in the accessory sex organs. Antioxidants, amino acids, vitamins, herbs, and minerals such as zinc, cold baths, and testicular coolers have been used. There are difficulties with the interpretation of the results of these treatments. 20 Marked improvements in semen quality can occur spontaneously (Fig. 10). Semen analysis results also display the phenomenon of regression to the mean. That is, on average, repeated semen analyses improve in men with initially abnormal results. 156 Pregnancy rate data were not analyzed effectively in many early studies. Floating numerator pregnancy rates, in which a percentage of patients pregnant is given without regard for time of exposure, have caused confusion in the infertility literature. Statistical methods for life table analysis and regression analysis with censored data are especially useful for assessing the impact of groups of variables on pregnancy rates, for analysis of prognostic factors, and for testing results of therapeutic trials. 20

Figure 10. Sperm concentration and motility in a man with severe oligospermia and severe hypospermatogenesis included in a therapeutic trial of clomiphene. Semen quality improved and his wife conceived. He was given the placebo! (Baker HWG: Requirements for controlled therapeutic trials in male infertility. Clin Reprod Fertil 4:13-25, 1986.)
The empirical treatments either have not been submitted to adequately controlled clinical trials, or when they have, the trials have not shown consistently positive results. Meta-analyses have also produced conflicting results, probably because of the variable quality of the trials included in the analyses. Until there is sound evidence of the value of a drug or procedure from controlled therapeutic trials, patients should be advised that none of the empirical methods meet the requirements of evidence-based medicine.
AIH is widely practiced with dubious evidence of efficacy in patients who do not have coital difficulties. Ovulation induction with intrauterine artificial insemination probably does increase the pregnancy rates by increasing the number of oocytes exposed to the sperm.[172, 173]Results are lower with timed intercourse and multiple ovulation induction. Generally, the results are poor when the semen analysis is abnormal. Although this may be acceptable in countries where ART is expensive, the risk of multiple pregnancy is substantial. IVF or ICSI would be preferable because the number of embryos placed in the uterus can be controlled and high multiple pregnancies avoided.[172]
In Vitro Fertilization/Intracytoplasmic Sperm Injection for Male Infertility
ICSI has revolutionized the management of male infertility. It involves the injection of a single sperm into the ooplasm (Fig. 11).[174, 175] ICSI can be used with almost any live sperm with an expectation of results similar to those obtained with standard IVF using normal sperm. ICSI may not be needed with mild semen disorders. Provided that more than approximately 2 million motile sperm can be harvested from an ejaculate, IVF can be attempted with an expectation of success close to that of IVF for other indications. The outcome depends particularly on sperm morphology and the ability of the sperm to bind to and penetrate the ZP. ICSI should be offered if there is a chance of failure of fertilization with IVF: less than 2 million motile sperm per ejaculate, less than 4% of sperm with normal morphology, less than 5% of sperm with progressive motility, sperm autoimmunity, and defects of sperm-oocyte interaction.

Figure 11. Intracytoplasmic sperm injection.
Preparation of the Patients
The couple needs to be counseled carefully about the procedures, predicted chance of a live birth, and the possible complications. Special arrangements for the collection of semen, or for its preparation, may be required. Trial-run sperm preparations help to identify those patients who have difficulty collecting semen. These patients should practice collections before attending for the IVF procedure. Men with many inflammatory cells in the semen could be treated with antibiotics. Those with low motility or sperm autoimmunity may have better sperm motility with short, 1- to 2-day durations of abstinence. Cryopreserved semen can be used as backup if the fresh semen is particularly poor on the day of ICSI in patients with fluctuating semen abnormalities. This is particularly useful when sperm are present in the semen only intermittently. Those patients who produce an unexpectedly poor sample on the day of IVF should provide a second sample later to supplement the first sample. Electroejaculation or needle biopsy of the testes can be used if the man is unexpectedly unable to collect semen. Patients with genital tract obstruction can have sperm retrieved from the testis or epididymis by needle aspiration.
ICSI also allows patients with severe primary spermatogenic disorders to be treated provided some live sperm or elongated spermatids can be recovered from the semen, testes, or genital tract (Table 172-7). If no sperm can be found in the semen, the likelihood of finding elongated spermatids in the testicular tissue can be estimated by the clinical situation and testicular histology. Sperm can be found by biopsies in approximately 50% of men with Klinefelter syndrome. Results are good if any tubules with complete spermatogenesis can be seen in diagnostic biopsies. However, if there are no elongated spermatids, the success rate with open biopsies is low: approximately 25% with Sertoli-cell-only syndrome and rarely with germ cell arrest at the primary spermatocyte stage.[117, 176] Yq microdeletions in the AZFa and b regions are also associated with a complete absence of spermatogenesis. Other factors such as hormone levels and testicular size do not seem to be predictive. The use of donor sperm should be discussed during preparation of the couple if the outlook is poor.
Testicular biopsy techniques should maximize the chance of finding sperm while minimizing damage. Microsurgery with examination of exposed testicular tissue under the operating microscope may allow selection of the larger diameter tubules, which are more likely to contain more advanced spermatogenesis.[176, 177] Alternatively, multiple sampling through small holes in the tunica may be used.[178] Large biopsies, particularly at multiple sites, have higher complication rates and will further impair testicular function (future androgen deficiency). Generally, repeat open biopsies for sperm collection should be performed only after the patient and testes have recovered from the previous surgery, and this may take 4-6 months.
Approaches
The standard approach is to stimulate multiple ovarian follicular development with FSH and collect the oocytes by ultrasound-guided transvaginal needle puncture of the ovaries after administration of human chorionic gonadotropin to mature the oocytes. ICSI or IVF is performed, and the resulting embryos are transferred into the uterine cavity, usually at the four- (day 2) to eight-cell (day 3) or blastocyst (day 5 or 6) stage with cryopreservation of remaining embryos. Although cryopreservation reduces the implantation potential frozen embryos produce good pregnancy outcomes.[179]
Biopsy of blastomeres for detection of chromosomal and genetic abnormalities is used to avoid transmission of serious hereditary diseases or to select embryos with higher cnances of implantation.[180]
Sperm Preparation
Various procedures have been developed for sperm preparation for IVF. Most popular is centrifugation on gradients of colloidal silica because this can be performed with high reproducibility.[181] Cryopreserved samples require especially gentle handling, particularly with dilution of the semen cryoprotectant medium with culture medium. Motility, even just an occasional slight twitch of the tail, is required in selecting sperm for ICSI. The morphology of the sperm cannot be assessed in detail at the magnification used, but obviously grossly abnormal sperm are excluded. If no motile sperm can be found, motility stimulation with pentoxifylline or hypoosmotic swelling can be used to show that the sperm are alive.[181]
Results
With IVF and ICSI, 60% of oocytes fertilize and cleave normally over the first 48 hours. For women younger than 35 years of age, the clinical pregnancy rate (fetal heart positive ultrasound at 6 weeks gestation) for transfer of one fresh 2-day old embryo is approximately 30% and for transfer of two 2 day old embryos 40%. Pregnancy rates are approximately 25% lower with cryopreserved embryos. The cumulative live birth rates with transfer of fresh and cryopreserved embryos from the first oocyte collection are approximately 50% and 80% by the third oocyte collection in women under 35 years of age. Approximately 25% of the births resulting from transfer of two 2-day old embryos are twins. Multiple pregnancy is more frequent if three or more cleave stage embryos or two or more blastocysts are transferred. Many clinics are transferring single embryos (elective single embryo transfer) in patients with good prognosis to minimize the multiple pregnancies. A number of other factors influence the results of ART, including embryo quality and particularly female age. Implantation and pregnancy rates decrease and pregnancy losses increase after age 35, mostly due to increasing abnormalities in the oocytes. For women aged 36-39 years the live birth rate is about 70% of that for women under 36 and for those 40 years and over using their own oocytes, about 30% that for women under 36.
Evaluation of Failed Fertilization
When most or all oocytes fail to fertilize in IVF, the cause is usually defective sperm. Oocytes may not fertilize because of immaturity or abnormality, but this is an unusual cause of total failure of fertilization of all the oocytes retrieved from a woman.[74] Unexpected failures of fertilization should be evaluated by examination of the number of sperm bound on the ZP and penetrating the ZP. Low numbers usually indicate sperm defects.[74] Low fertilization rates may also result from undiagnosed sperm autoimmunity, infected semen, or technical problems in the IVF laboratory. Careful evaluation of the semen quality and screening patients with idiopathic infertility for defects of sperm-oocyte interaction before IVF should allow most couples likely to have low fertilization with standard IVF to be directed to ICSI.[74, 182] ICSI of unfertilized oocytes with the man’s sperm 12 to 24 hours after standard IVF insemination may result in fertilization and pregnancy, but, overall, the results are poor and many clinics do not perform this “ICSI rescue” procedure. Re-insemination of failed fertilization oocytes with donor sperm is also possible for diagnostic or therapeutic purposes. This procedure is not permitted in some countries.
Individual oocytes may not fertilize with ICSI. In these, the sperm head is often only partially decondensed. Failure of fertilization of all oocytes with ICSI is rare. Globospermic, immotile and rarely sperm from patients with severe oligospermia may produce low or zero fertilization rates with ICSI. In these cases there may be a deficiency of an oocyte-activating factor from the sperm. Modified ICSI techniques (assisted activation) may be successful in artificially activating the oocytes in some of these cases.[91]
Complications of In Vitro Fertilization/Intracytoplasmic Sperm Injection
Potential adverse effects of ART include well-understood conditions in the woman and a variety of possible issues for the offspring. In general, the outcome and complications of ART are the same for standard IVF and ICSI.[183, 184] The implantation rate, pregnancy wastage, pregnancy complications, perinatal mortality, and risk of congenital abnormalities are no greater for ICSI than with IVF. Recent results for ART for male infertility are compared with those for infertility of other causes in Table 7.[185] The pregnancy, miscarriage and other outcomes are if anything better not worse for male infertility.
Table 7
Comparison of results of fresh and cryopreserved embryo transfers for male infertility versus other causes of infertility in Australia and New Zealand 2005.[185]
|
Type of infertility |
Male only | Other |
| Fresh embryo transfers
Clinical pregnancy (fetal heart(s)) Live birth |
7880
26.1% 21.0% |
20115
23.2% 18.4% |
| Cryopreserved embryo transfers
Clinical pregnancy (fetal heart(s)) Live birth |
4708
20.2% 15.8% |
12051
19.5% 14.6% |
| Clinical pregnancies (fetal heart(s))
Spontaneous abortion Induced abortion Ectopic pregnancy Stillbirth Preterm (<37 weeks gestation) |
3008
17.6% 0.5% 1.0% 0.8% 16.0% |
7013
19.4% 0.7% 1.7% 0.7% 18.3% |
In 2005 48% of embryo transfers were with single embryos and the multiple pregnancy rates were for fresh embryo transfers following IVF: 16.2%, ICSI 14.7% and thawed cryopreserved embryo transfers (FET) 10.9%. The perinatal mortality (stillbirth and neonatal deaths within 28 days of life) was for IVF 18.2, ICSI 15.3 and FET 11.8 per 1000 births. The perinatal mortality for ART singletons was 9.6 and for the general population in 2004: 10.2.
Risks for the Woman
The ovarian hyperstimulation syndrome is a major risk with gonadotropin stimulation of multiple follicular development.[186] Careful monitoring of the patients is necessary. If many follicles develop, embryo freezing rather than transfer avoids pregnancy and allows the ovaries to recover, reducing the risk of severe complications such as thromboembolism, renal failure, and death. Surgical complications including bleeding and infection from the oocyte collection procedure are rare. There is also a small risk of complications from anesthesia and sedation. Maternal complications of pregnancy increase in frequency with multiple pregnancy. Cesarean section is more frequent for singleton ART births than in the general community. There are also concerns about the ovarian stimulation drugs predisposing to breast or gynecologic cancers.[187]
Risks for the Child
Multiple Pregnancy
The risks of multiple pregnancy for the child, prematurity, low birth weight, increased perinatal mortality and morbidity, less parental attention during childhood, are well known and can be controlled by reducing the numbers of embryos transferred together.[183, 186, 188, 189]
Transmission of Genetic and Chromosomal Disorders
The known genetic risks were covered previously (see Table 4). For conditions such as cystic fibrosis and myotonic dystrophy, pre-implantation genetic diagnosis can be used so that only unaffected embryos are transferred. Balanced chromosomal translocations may become unbalanced in embryos and result in miscarriage or rarely in the birth of an abnormal child. Pre-implantation genetic diagnosis may also be used to detect unbalanced chromosomal constitution in the pre-implantation embryo.[180]
Defects Possibly Associated with Abnormal Spermatogenesis
There is a correlation between the production of abnormal sperm with poor morphology and motility and abnormal sperm DNA measured by a variety of techniques including acridine orange fluorescence, sperm chromatin structure assay, chromomycin staining, and comet assays. [22] Abnormal sperm produce reactive oxygen species that could damage sperm DNA and result in defects of implantation or pregnancy loss. [22] However, the results of ART do not reveal such problems (see Table 7).[24] A number of mechanisms reduce the likelihood such abnormal sperm would be involved in natural or assisted fertilization such as sperm aggregation caused by heavy coating with clusterin, poor motility, and limited ability to bind to the zona pellucida.[190, 191]
An increase in de novo sex chromosomal aneuploidy and structural autosomal defects reported with ICSI related to increased rates of chromosomal nondisjunction as a general association with abnormal spermatogenesis has not been confirmed by all studies.[109, 192-194]
Embryo Defects Possibly Caused by Laboratory Conditions
Laboratory conditions or procedures on gametes could affect embryo development and health of the child. In domestic animal IVF, there is a syndrome of large offspring that results from stress-induced changes in gene expression that may involve changes in DNA methylation and gene imprinting in embryos cultured to blastocysts.[195] Increased frequencies of rare conditions caused by disorders of imprinting such as Beckwith-Weidemann syndrome and Angelman’s syndrome have been reported in children born from ART procedures.[196] Increased frequencies of tumors in children born after ART have also been claimed, for example, retinoblastoma, but other studies do not support a general increase in childhood cancer rates.[197]
Surveillance Studies
In most studies the fertilization, implantation, and pregnancy failure rates and congenital malformation rates are no greater with ICSI than with standard IVF.[183, 184] However, overall results are different from those in the general population: preterm birth (6%), low birth weight (5%), major congenital malformations (2%), and perinatal mortality (1%)(see Table 7). The differences are partly explained by the high multiple pregnancy rate with IVF and ICSI, and perhaps female age and infertility factors. Closer surveillance and more accurate reporting may also contribute. The lower birth weight also affects singletons and there is a difference between the results of fresh and cryopreserved embryo transfers the low birth weight being more frequent in babies born after fresh embryo transfers.[179]
Studies of children born as a result of ART have not revealed any consistently associated congenital malformations, but there is data suggesting generally increased congenital malformation rates.[198-200] However, these studies may be biased. For example, if IVF and ICSI babies are examined more thoroughly and reporting to health registers is more complete, the malformation rates may appear higher than for naturally conceived children who are less carefully examined and reported. A higher uptake of prenatal screening for Down syndrome and other triploids in ART patients may also influence the reported rates of birth defects. Even with perfect reporting, differences in malformation rates could be caused by other factors, such as age, parity or health of the mothers, which have not been adequately allowed for in the statistical analysis.
USE OF DONOR SPERM
Donor insemination is a common method of managing male sterility.[201-204] Donor sperm may be involved in approximately 1 in 200 births in countries where it is permitted. The main indications for donor insemination are untreatable sterility in the man or when treatments and ICSI for severe or chronic subfertility have failed. The couple may choose donor insemination as the primary method of managing their infertility. Donor insemination is also used to avoid transmission of a severe genetic or infectious disease in the man. Women without a male partner may use donor insemination to have children. Donor sperm may also be used in IVF when there is a combination of female infertility and male sterility. Because of the higher pregnancy rates, IVF with donor sperm may be used if donor insemination fails.[203] Donor sperm may also be used in IVF procedures as a backup, for example, when there is a high risk of failure with of sperm extraction with a severe spermatogenic defect.
Cryopreservation of Semen
Donor insemination can be performed in the setting of a specialist infertility clinic with all donor and patient management available. Alternatively, the sperm bank may only supply semen for the patient and be separate from the clinics or physicians performing the artificial insemination. Because of the risk of transmission of infectious diseases, particularly HIV, and also for convenience, donor insemination services now use only cryopreserved semen.[201, 203] Semen cryopreservation with glycerol–egg yolk cryoprotectant and either vapor freezing or controlled-rate freezing in plastic straws or vials produces pregnancy rates equal to those with fresh semen. Importantly, cryopreservation allows the semen to be quarantined for 6 months for donors to be recalled and retested for infectious diseases before it is used.
Selection of Donors
Prospective donors have their medical and family histories evaluated and a physical examination to exclude the possibility of transmitting serious genetic diseases such as hemoglobinopathies or sexually transmissible infections. Donors sign a lifestyle declaration to indicate that they are not involved in any practices that might expose them to serious infections, such as HIV. There usually is an upper age limit of 40 to 45 years because of the increasing frequency of genetic abnormalities in sperm with age. Semen quality is selected to be in the upper part of the normal range, particularly for concentration and motility.[201, 202] The semen is cultured for bacteria and blood is tested for hepatitis and HIV antibodies. Genetic screening for hemoglobinopathies and cystic fibrosis or other conditions may be included depending on their prevalence in the community. The freezing of semen does not appear to cause any increase in the frequency of congenital abnormalities.[201, 203, 205]
It is usual to match the physical characteristics of the recipient’s husband and the donor including race, complexion, build, height, and hair and eye color. In addition, blood groups may be matched. In some programs, the recipient couple may be able to choose the donor on other information such as occupation and education. Known donors may also be used; these may be friends or relatives of the infertile couple. In this situation, special counseling of the donors and recipients is necessary. Also, there should be a full workup of the known donor as for an anonymous donor, including cryopreservation and quarantining of the semen.
Donor factors relevant to the success of donor insemination are mainly to do with the quality of the semen. Post-thaw motility has the strongest predictive value for high fertilization rates, but sperm morphology, motility, and concentration are also significant.[201, 203] Despite selection of high-quality semen, there remains considerable variability in the pregnancy rates between donors. A policy to discard semen from a donor who produces no pregnancy after a certain number (e.g., 20–40) of inseminations is necessary.[202]
Counseling
The special nature of the use of donor sperm is discussed in detail with the couple so that they are fully aware of the implications for the child and their family. Donor insemination is forbidden in some religions. There may be local legislation or regulations to control the use of donated gametes. In some countries, special laws have been enacted that may either allow or prevent the child from obtaining identifying information about the donor. The legal status of the child may also be specified in various ways. The couple needs to decide how and when to disclose the child’s donor sperm origin. What and how much they should tell their friends and relatives about their infertility treatment should also be discussed, as should their reaction to acquaintances questioning the paternity of the child. The possibility that in the future half-siblings may unwittingly find each other and attempt to have children is of concern to some prospective parents and donors. This needs to be discussed carefully and the risks explained in view of the number of pregnancies permitted per donor by the clinic. Studies of donor families in which there has been expert pretreatment counseling indicate no physical or emotional problems with the children, and greater marital stability than average.[205]
Procedures and Results
The prospective recipients are screened for HIV, hepatitis B and C, rubella immunity, blood group, and genetic conditions if necessary. Tests of tubal patency are performed if the history suggests pelvic pathology. The inseminations are timed to coincide with natural ovulation. Careful monitoring of ovulation and timing of intrauterine insemination of prepared motile sperm suspensions (as for IVF) appear to increase the pregnancy rate. Pregnancy rates are approximately 10% to 25% per month for the first 4 to 6 months and then 5% to 10% thereafter, so that approximately 50% of women are pregnant by 4 to 6 months.[201, 203, 204] Female age affects the pregnancy rates.[201, 204] Women with subfertile male partners have on average lower pregnancy rates than those with sterile male partners, indicating the presence of female factors contributing to the infertility when the male partner is subfertile.[201, 204] Cumulative pregnancy rates for women who have had more than one pregnancy by donor insemination indicate higher conception rates over the first few months for the second pregnancy, approximately 33% pregnant in the first cycle and 55% by the second cycle.[201]
Multiple ovulation induction and intrauterine insemination may increase the pregnancy rates but at the risk of multiple pregnancy.[201, 204] IVF may be used if no pregnancy has occurred after a reasonable number of inseminations (e.g., 4–6).[203] Live birth pregnancy rates with IVF in such patients are high, and women in good health can be advised that they have an 80% chance of having a child within 2 years.
PREVENTION OF INFERTILITY
Prevention is difficult because of the lack of understanding of the causes of most types of male infertility. Mumps orchitis was an uncommon cause of infertility, and childhood immunization for this disease should make it very rare. It is important to recognize that subfertility often is a couple problem, with both partners contributing. Therefore, general factors that would change a society’s attitude to child bearing could have an important impact on the frequency of infertility, for example, a trend toward having children at earlier ages. On the other hand, toxins and environmental factors known to cause defects of sperm production, such as heat, dibromochlorobenzine, lead, benzene, ionizing radiation, and microwaves, are probably well controlled by environmental health measures.
Preventable Diseases and Conditions
Sexually Transmissible Infections
Post-gonococcal epididymal obstructions appear to be the most important cause of infertility from sexually transmitted diseases. In countries where gonorrhea is treated promptly, post-gonococcal epididymal obstruction is rare. On the other hand, it remains a common preventable cause of infertility in other countries.
Undescended Testes
Although undescended testes have been sought and treated aggressively over the past 50 years, previously undescended testes remain a common association of male infertility, affecting approximately 7% of the men seen. It is therefore uncertain whether early surgery for undescended testes has any impact on subsequent fertility. It is possible that the failure of normal descent is a feature of testicular dystrophy and that the sperm production will be poor whether or not the testes are placed in the scrotum.
A randomized controlled trial of orchiopexy for unilateral palpable maldescended testis at 9 months versus 3 years of age showed that surgery at 9 months was followed by significant growth of the testis up to age 4 years but there was no change in testis size in those treated at age 3 [50]. This has led to clinical guidelines for treatment of maldescended testes that recommend orchiopexy for congenital forms between 6 and 12 months of age and as soon as possible for those discovered later and for acquired maldescent. Hopefully this will reduce the frequency of subsequent testicular tumours and spermatogenic defects.
Varicocele
The effectiveness of varicocelectomy for sperm defects is controversial. Varicoceles are common and usually appear about the time of puberty. Although some groups believe that varicoceles should be sought actively and treated in adolescence to prevent infertility, this approach could pose a major burden on the health resources because at least 15% of men have varicoceles. Long-term prospective trials are needed.
Vasectomy
Vasectomy reversal and treatment for continuing infertility after attempted vasectomy reversal are now common. Better counseling about the limited effectiveness of vasectomy reversal is needed and cryopreservation of semen before vasectomy in men who are uncertain about their need for future fertility should be promoted.
Androgen deficiency
In patients with symptomatic androgen deficiency, testosterone therapy should be deferred until fertility issues have been addressed. Alternatively, Human chorionic gonadotropin (hCG) preparation can be given in order to stimulate testosterone production without inhibition of spermatogenesis.
If fertility is not immediately sought, sperm cryopreservation allows testosterone replacement to be commenced.
Options for fertility preservation in males
Advances in the treatment of cancer in young patients have led to great improvements in life expectancy which approaches 80% 5-year survival rate. As a result, fertility preservation and desire for paternity have become a significant issue in this group. A major concern is the negative impact of chemo and radiotherapy on fertility. Up to two thirds of patients are azoospermic following chemotherapy. Recovery of spermatogenesis is strongly dependent upon the chemotherapy and radiation regimen and the patient’s baseline reproductive function. Alkylating agents seem to have the most profound reproductive effects. Thus men about to have treatment for malignant conditions may have sperm cryopreserved before commencing chemotherapy or radiotherapy.[206] Although pretreatment semen quality may be too poor for AIH, ICSI now has improved the outlook for successful pregnancies. Semen collected during chemotherapy or radiotherapy must not be used because of the likelihood of induced mutations.[207]
Infertile men with conditions such as orchitis or severe primary spermatogenic disorders that might involve progressively declining semen quality such as Klinefelter syndrome, when sperm can be recovered, should also store any live sperm that can be obtained as insurance for the future. A similar approach could be extended to adolescents with risk factors for infertility such as undescended testes in childhood, testicular torsion, and possibly a family history of infertility or a father with a Yq microdeletion. Semen may also be stored after treatment of gonadotropin deficiency or surgery for genital tract obstruction in case of re-stenosis. Storage of sperm before premature death or after a sudden unexpected death is also possible. Although the use of gametes from a dead person is surrounded by complex ethical and legal issues it is permissible in some countries. Semen cryopreservation can also be offered to adolescents. Sperm may be obtainable from the semen or testis after mid puberty. Although only a small proportion of men who store semen may use the frozen sperm, the service provides insurance for future fertility.
Methods of Collection
Sperm for fertility preservation may be collected in a variety of ways. Ejaculated sperm cryopreservation is the most common technique used as enough sperm is usually found for freezing in majority of men. Unfortunately, some men with testicular and other malignancies may present initially with oligospermia or even azoospermia. This may be related to a basic infertility condition which puts them under higher risk to develop testicular cancer or the stress effect of the disease on spermatogenesis. Some patients will not be able to produce a sample. Some patients are unable to ejaculate for social, religious or medical reasons or may be unfamiliar with masturbation for example peri-pubertal boys. For them, sperm can be retrieved by penile vibratory stimulation, electroejaculation or surgically from the epididymis or the testis as discussed above.
Fertility Preservation in Prepubertal Boys
Preserving fertility in postpubertal boys facing chemotherapy can be achieve similar success rates to those in adults. However fertility preservation in prepubertal boys presents a great challenge as sperm banking is not possible. Alternative strategies have been developed but all are currently experimental. These strategies are based on immature spermatogenic cell cryopreservation as cell suspensions or whole testicular tissue for future fertility restoration using autografting, xenografting or in-vitro spermatogenesis. [208] More research is needed to establish the best approach to generate spermatozoa from immature stem cells via in-vivo or in-vitro maturation. In the meantime, prepubertal tissue preservation should be discussed with the boys and their parents and samples should be banked only after careful counseling emphasizing the experimental nature of this approach.
Post Chemotherapy Sperm Recovery
Various barriers to sperm banking such as prepubertal age, under-referral or inadequate understanding of the sterilizing effect of chemotherapy and defective spermatogenesis before the treatment may have prevented sperm banking.[209, 210] In these patients an approach similar to that used for men with severe primary spermatogenic disorders can be used as recovery of spermatogenesis after gonadotoxic treatments is highly variable. Men with persistent azoospermia following chemotherapy or radiotherapy can be offered microsurgical testicular sperm extraction (TESE) and ICSI as there is some chance of success. Schlegel et al. reported 84 microdissection TESE procedures performed in 73 patients with 43% having sperm retrieved and these produced an average 57% fertilization rate with ICSI and 42% had live births.[211]
DNA FRAGMENTATION
The study of sperm DNA fragmentation has become topical in recent years. It has been proposed that DNA damage may contribute to infertility in a way that is not revealed by simple morphological evaluation of spermatozoa. Sperm showing a high proportion of fragmented DNA has been blamed for a lower fertilization rate [212], poorer embryo development and reduced implantation rates. [213-215]. It is now assumed that a significant proportion of infertile men have clinically important levels of DNA damage in their spermatozoa. [216]
Basically, once a sperm nucleus has been introduced into the oocyte, a rapid de condensation should occur to allow the DNA formation of the paternal pronucleus. Abnormalities in the structural DNA organization can cause delays and errors in the paternal DNA delivery. Animal studies demonstrated that sperm DNA damage can cause embryo development compromise.[217, 218].
Apoptosis plays a key role and is considered to be the main pathway leading to DNA breakage in sperm.[219] The known main external inducers of sperm DNA fragmentation are chemotherapy [220], advanced age [216], environmental factors such as cigarette smoking [221], genital tract inflammation and varicoceles [222-224] and the presence of leukocytes in the semen [225]
Three main mechanisms have been proposed as contributors to the generation of DNA fragmentation; 1) DNA nicks occurring to promote the remodeling of sperm chromatin are not completely repaired due to an impairment of the sperm maturation process, [226]; 2) DNA cleavage produced by a process of apoptosis first triggered and later interrupted [227, 228] and; 3) reactive oxygen species (ROS) , acting both in testis and in posttesticular sites [229]
Based on the above understanding, strategies to identify fragmented sperm gained popularity.[230] Novel sperm selection techniques like annexin V–magnetic activated cell sorting (annexin V–MACS), zeta potential selection, electrophoretic systems for the rapid isolation of sperm exhibiting high levels of DNA integrity and hyaluronic acid binding techniques, have been recently described.[231] Currently, the evidence is insufficient to recommend one specific method of sperm selection in the case of high sperm DNA fragmentation.
REFERENCES
- Leridon, H., Human Fertility: The Basic Components. 1977, Chicago: University of Chicago Press.
- Greenhall, E. and M. Vessey, The prevalence of subfertility: a review of the current confusion and a report of two new studies. Fertil Steril, 1990. 54(6): p. 978-83.
- Joffe, M., Time trends in biological fertility in Britain. Lancet, 2000. 355(9219): p. 1961-5.
- Gnoth, C., et al., Definition and prevalence of subfertility and infertility. Hum Reprod, 2005. 20(5): p. 1144-7.
- Holden, C.A., et al., Sexual activity, fertility and contraceptive use in middle-aged and older men: Men in Australia, Telephone Survey (MATeS). Hum Reprod, 2005. 20(12): p. 3429-34.
- Comhaire, F.H., et al., Towards more objectivity in diagnosis and management of male infertility. International Journal of Andrology, 1987. Suppl 7: p. 1-53.
- Carlsen, E., et al., Evidence for decreasing quality of semen during past 50 years. BMJ, 1992. 305(6854): p. 609-13.
- Irvine, D.S., Falling sperm quality. BMJ, 1994. 309(6952): p. 476.
- Auger, J., et al., Decline in semen quality among fertile men in Paris during the past 20 years. N Engl J Med, 1995. 332(5): p. 281-5.
- Irvine, S., et al., Evidence of deteriorating semen quality in the United Kingdom: birth cohort study in 577 men in Scotland over 11 years. BMJ, 1996. 312(7029): p. 467-71.
- Povey, A.C. and S.J. Stocks, Epidemiology and trends in male subfertility. Hum Fertil (Camb), 2010. 13(4): p. 182-8.
- Itoh, N., et al., Have sperm counts deteriorated over the past 20 years in healthy, young Japanese men? Results from the Sapporo area. J Androl, 2001. 22(1): p. 40-4.
- Fisch, H., Declining worldwide sperm counts: disproving a myth. Urol Clin North Am, 2008. 35(2): p. 137-46, vii.
- Baker, H.W.G., et al., Relative incidence of etiologic disorders in male infertility., in Male Reproductive Dysfunction: Diagnosis and Management of Hypogonadism, Infertility and Impotence., R.J. Santen, Swerdloff, R. S., Editor. 1986, Marcel Dekker Inc.: New York. p. 341-372.
- Baker, H.W.G., et al., Testicular vein ligation and fertility in men with varicoceles. British Medical Journal (Clin Res Ed), 1985. 291(6510): p. 1678-80.
- Guzick, D.S., et al., Sperm morphology, motility, and concentration in fertile and infertile men. N Engl J Med, 2001. 345(19): p. 1388-93.
- Garrett, C., et al., Automated semen analysis: 'zona pellucida preferred' sperm morphometry and straight-line velocity are related to pregnancy rate in subfertile couples. Hum Reprod, 2003. 18(8): p. 1643-9.
- Krausz, C. and S. Degl'Innocenti, Y chromosome and male infertility: update, 2006. Front Biosci, 2006. 11: p. 3049-61.
- Vogt, P.H., et al., The AZF proteins. Int J Androl, 2008. 31(4): p. 383-94.
- Baker, H.W.G., Medical treatment for idiopathic male infertility: is it curative or palliative? Baillieres Clin Obstet Gynaecol, 1997. 11(4): p. 673-89.
- Olesen, I.A., et al., Environment, testicular dysgenesis and carcinoma in situ testis. Best Pract Res Clin Endocrinol Metab, 2007. 21(3): p. 462-78.
- Aitken, R.J. and G.N. De Iuliis, Origins and consequences of DNA damage in male germ cells. Reprod Biomed Online, 2007. 14(6): p. 727-33.
- Phillips, K.P. and N. Tanphaichitr, Human exposure to endocrine disrupters and semen quality. J Toxicol Environ Health B Crit Rev, 2008. 11(3-4): p. 188-220.
- Collins, J.A., K.T. Barnhart, and P.N. Schlegel, Do sperm DNA integrity tests predict pregnancy with in vitro fertilization? Fertil Steril, 2008. 89(4): p. 823-31.
- Lilford, R., A. Jones, and D. Bishop, Case-control study of whether subfertility in men is familial. BMJ, 1994. 309: p. 570-573.
- Van der Avoort, I.A., et al., Underestimation of subfertility among relatives when using a family history: taboo bias. J Androl, 2003. 24(2): p. 285-8.
- Baker, H.W.G., Clinical approach to molecular genetic diagnosis in andrology., in Andrology in the 21st century, B. Robaire, Chemes, H., Morales, CR., Editor. 2001, Medimond: Englewood NJ. p. 373-382.
- O'Bryan, M.K. and D. de Kretser, Mouse models for genes involved in impaired spermatogenesis. Int J Androl, 2006. 29(1): p. 76-89; discussion 105-8.
- Nuti, F. and C. Krausz, Gene polymorphisms/mutations relevant to abnormal spermatogenesis. Reprod Biomed Online, 2008. 16(4): p. 504-13.
- Fechner, A., S. Fong, and P. McGovern, A review of Kallmann syndrome: genetics, pathophysiology, and clinical management. Obstet Gynecol Surv, 2008. 63(3): p. 189-94.
- Huhtaniemi, I. and M. Alevizaki, Mutations along the hypothalamic-pituitary-gonadal axis affecting male reproduction. Reprod Biomed Online, 2007. 15(6): p. 622-32.
- Tuttelmann, F., et al., Gene polymorphisms and male infertility--a meta-analysis and literature review. Reprod Biomed Online, 2007. 15(6): p. 643-58.
- Rajender, S., L. Singh, and K. Thangaraj, Phenotypic heterogeneity of mutations in androgen receptor gene. Asian J Androl, 2007. 9(2): p. 147-79.
- Kaplan, E., et al., Reproductive failure in males with cystic fibrosis. N Engl J Med, 1968. 279(2): p. 65-9.
- Anguiano A, O.R., Amos JA, Dean M, Gerrard B, Stewart C, et al., Congenital bilateral absence of the vas deferens: a primary genital form of cystic fibrosis. JAMA, 1992(267): p. 1794-7.
- Reijo, R., et al., Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet, 1995. 10(4): p. 383-93.
- Najmabadi, H., et al., Substantial prevalence of microdeletions of the Y-chromosome in infertile men with idiopathic azoospermia and oligozoospermia detected using a sequence-tagged site-based mapping strategy. J Clin Endocrinol Metab, 1996. 81(4): p. 1347-52.
- Wischusen, J., H.W.G. Baker, and B. Hudson, Reversible male infertility due to congenital adrenal hyperplasia. Clin Endocrinol (Oxf), 1981. 14(6): p. 571-7.
- New, M.I., Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab, 2006. 91(11): p. 4205-14.
- Fang, S. and H.W.G. Baker, Male infertility and adult polycystic kidney disease are associated with necrospermia. Fertil Steril, 2003. 79(3): p. 643-4.
- St John, J.C., R.P. Jokhi, and C.L. Barratt, The impact of mitochondrial genetics on male infertility. Int J Androl, 2005. 28(2): p. 65-73.
- Baker, H. and G. Clarke, Management of immunological factors in male infertility, in Male reproductive dysfunction pathophysiology and treatemnt, K. FR, Editor. 2007, Informa: New York. p. 419-430.
- Baker, H.W.G., Reproductive effects of nontesticular illness. Endocrinology & Metabolism Clinics of North America, 1998. 27(4): p. 831-50.
- Pedrazzoni, M., et al., Effects of chronic heroin abuse on bone and mineral metabolism. Acta Endocrinol (Copenh), 1993. 129(1): p. 42-5.
- MacLeod, J. and R. Hotchkiss, The effect of hyperpyrexia on spermatozoa counts in men. Endocrinology, 1941. 28: p. 780.
- Carlsen, E., et al., History of febrile illness and variation in semen quality. Hum Reprod, 2003. 18(10): p. 2089-92.
- Mallidis, C., et al., Advanced glycation end products accumulate in the reproductive tract of men with diabetes. Int J Androl, 2008.
- Handelsman, D.J., et al., Young's syndrome. Obstructive azoospermia and chronic sinopulmonary infections. N Engl J Med, 1984. 310(1): p. 3-9.
- Hendry, W.F., R.P. A'Hern, and P.J. Cole, Was Young's syndrome caused by exposure to mercury in childhood? British Medical Journal, 1993. 307(6919): p. 1579-82.
- Chemes, E.H. and Y.V. Rawe, Sperm pathology: a step beyond descriptive morphology. Origin, characterization and fertility potential of abnormal sperm phenotypes in infertile men. Hum Reprod Update, 2003. 9(5): p. 405-28.
- Lee, P.A., M.F. Bellinger, and M.T. Coughlin, Correlations among hormone levels, sperm parameters and paternity in formerly unilaterally cryptorchid men. Journal of Urology, 1998. 160(3 Pt 2): p. 1155-7; discussion 1178.
- Hutson, J.M. and M.C. Clarke, Current management of the undescended testicle. Semin Pediatr Surg, 2007. 16(1): p. 64-70.
- Kollin, C., et al., Surgical treatment of unilaterally undescended testes: testicular growth after randomization to orchiopexy at age 9 months or 3 years. J Urol, 2007. 178(4 Pt 2): p. 1589-93; discussion 1593.
- Barthold, J.S. and J.F. Redman, Association of epididymal anomalies with patent processus vaginalis in hernia, hydrocele and cryptorchidism. Journal of Urology, 1996. 156(6): p. 2054-6.
- Masarani, M., H. Wazait, and M. Dinneen, Mumps orchitis. J R Soc Med, 2006. 99(11): p. 573-5.
- Pasqualotto, F.F., et al., Detection of testicular cancer in men presenting with infertility. Rev Hosp Clin Fac Med Sao Paulo, 2003. 58(2): p. 75-80.
- Sandeman, T., The effects of X-irradiation on male human fertility. Br J Radiol, 1966. 39: p. 901-907.
- Fairley, K.F., J.U. Barrie, and W. Johnson, Sterility and testicular atrophy related to cyclophosphamide therapy. Lancet, 1972. 1(7750): p. 568-9.
- Janssen, N.M. and M.S. Genta, The effects of immunosuppressive and anti-inflammatory medications on fertility, pregnancy, and lactation. Arch Intern Med, 2000. 160(5): p. 610-9.
- Henderson, J., H.W.G. Baker, and P.J. Hanna, Occupation-related male infertility: a review. Clinical Reproduction and Fertility, 1986. 4(2): p. 87-106.
- Sheiner, E.K., et al., Effect of occupational exposures on male fertility: literature review. Ind Health, 2003. 41(2): p. 55-62.
- Ramlau-Hansen, C.H., et al., Is smoking a risk factor for decreased semen quality? A cross-sectional analysis. Hum Reprod, 2007. 22(1): p. 188-96.
- Dorfman, S.F., Tobacco and fertility: our responsibilities. Fertil Steril, 2008. 89(3): p. 502-4.
- Baker, H.W.G. and R.J. Pepperell, Lack of effect of bromocriptine on semen quality in men with normal or slightly elevated prolactin levels. Aust N Z J Obstet Gynaecol, 1980. 20(3): p. 158-61.
- Gebhard, P. and A. Johnson, The Kinsey data: Marginal tabulations of the 1938-1963. 1979, interviews conducted by the Institute for Sex Research: Philadelphia. p. 116-120.
- Smith, N.M., R.W. Byard, and A.J. Bourne, Testicular regression syndrome--a pathological study of 77 cases. Histopathology, 1991. 19(3): p. 269-72.
- Prader, A., Testicular size: Assessment and clinical importance. Triangle, 1966. 7: p. 240-243.
- WHO, World Health Organization laboratory manual for the exmination of human semen and sperm-cervical mucus interaction. 1999, Cambridge: Cambridge University Press.
- McLachlan, R.I., et al., Semen analysis: its place in modern reproductive medical practice. Pathology, 2003. 35(1): p. 25-33.
- Jones, D.M., et al., Immobilization of sperm by condoms and their components. Clinical Reproduction and Fertility, 1986. 4(6): p. 367-72.
- Kovacs, G.T., G.B. Newman, and G.L. Henson, The postcoital test: what is normal? British Medical Journal, 1978. 1(6116): p. 818.
- Mallidis, C., E.J. Howard, and H.W.G. Baker, Variation of semen quality in normal men. International Journal of Andrology, 1991. 14(2): p. 99-107.
- Clarke, G. and H. Baker, Immunological evaluation of male infertility, in Male reproductive function pathophysiology and treatment, F. Kandeel, Editor. 2007, Informa: New York. p. 293-300.
- Liu, D.Y., C. Garrett, and H.W. Baker, Clinical application of sperm-oocyte interaction tests in in vitro fertilization--embryo transfer and intracytoplasmic sperm injection programs. Fertil Steril, 2004. 82(5): p. 1251-63.
- Burkman, L.J., et al., The hemizona assay (HZA): development of a diagnostic test for the binding of human spermatozoa to the human hemizona pellucida to predict fertilization potential. Fertil Steril, 1988. 49(4): p. 688-97.
- Cooper, T.G., et al., Azoospermia: virtual reality or possible to quantify? J Androl, 2006. 27(4): p. 483-90.
- Cooper, T.G., et al., World Health Organization reference values for human semen characteristics. Human Reproduction Update, 2010. 16(3): p. 231-45.
- Zariwala, M.A., M.R. Knowles, and H. Omran, Genetic defects in ciliary structure and function. Annu Rev Physiol, 2007. 69: p. 423-50.
- Wilton, L.J., et al., Human male infertility caused by degeneration and death of sperm in the epididymis. Fertility and Sterility, 1988. 49(6): p. 1052-8.
- Ortega, C., et al., Absolute asthenozoospermia and ICSI: what are the options? Hum Reprod Update, 2011. 17(5): p. 684-92.
- Kovacic, B., V. Vlaisavljevic, and M. Reljic, Clinical use of pentoxifylline for activation of immotile testicular sperm before ICSI in patients with azoospermia. J Androl, 2006. 27(1): p. 45-52.
- Mangoli, V., et al., Selection of viable spermatozoa from testicular biopsies: a comparative study between pentoxifylline and hypoosmotic swelling test. Fertil Steril, 2011. 95(2): p. 631-4.
- Ebner, T., et al., Pharmacological stimulation of sperm motility in frozen and thawed testicular sperm using the dimethylxanthine theophylline. Fertil Steril, 2011. 96(6): p. 1331-6.
- Ebner, T., et al., Healthy live birth using theophylline in a case of retrograde ejaculation and absolute asthenozoospermia. Fertil Steril, 2014. 101(2): p. 340-3.
- Nordhoff, V., How to select immotile but viable spermatozoa on the day of intracytoplasmic sperm injection? An embryologist's view. Andrology, 2015. 3(2): p. 156-62.
- Check, J.H., A. Tubman, and C. Wilson, Intracytoplasmic sperm injection allows normal pregnancy rates for males 40 with low hypoosmotic swelling test scores even when complicated by very low motility percentage. Clin Exp Obstet Gynecol, 2013. 40(1): p. 18-9.
- Check, J.H., et al., Low hypoosmotic swelling test scores correlate better with lower percent motility than any other abnormal semen parameters. Clin Exp Obstet Gynecol, 2012. 39(2): p. 137-8.
- Nordhoff, V., et al., Optimizing TESE-ICSI by laser-assisted selection of immotile spermatozoa and polarization microscopy for selection of oocytes. Andrology, 2013. 1(1): p. 67-74.
- Brown, D.J., S.T. Hill, and H.W. Baker, Male fertility and sexual function after spinal cord injury. Prog Brain Res, 2006. 152: p. 427-39.
- Correa-Perez, J.R., et al., Clinical management of men producing ejaculates characterized by high levels of dead sperm and altered seminal plasma factors consistent with epididymal necrospermia. Fertil Steril, 2004. 81(4): p. 1148-50.
- Francavilla, S., et al., Isolated teratozoospermia: a cause of male sterility in the era of ICSI? Front Biosci, 2007. 12: p. 69-88.
- Liu, D.Y., C. Garrett, and H.W.G. Baker, Low proportions of sperm can bind to the zona pellucida of human oocytes. Hum Reprod, 2003. 18(11): p. 2382-9.
- Garrett, C. and H.W.G. Baker, New Fully Automated System For the Morphometric Analysis of Human Sperm Heads. Fertility & Sterility, 1995. 63(6): p. 1306-1317.
- Liu, D.Y. and H.W.G. Baker, Tests of human sperm function and fertilization in vitro. Fertil.Steril., 1992. 58: p. 465-83.
- Larson, K.L., et al., Sperm chromatin structure assay parameters as predictors of failed pregnancy following assisted reproductive techniques. Human Reproduction, 2000. 15(8): p. 1717-22.
- Chohan, K.R., et al., Comparison of chromatin assays for DNA fragmentation evaluation in human sperm. J Androl, 2006. 27(1): p. 53-9.
- Collins, J.A., K.T. Barnhart, and P.N. Schlegel, Do sperm DNA integrity tests predict pregnancy with in vitro fertilization? Fertility and Sterility, 2008. 89(4): p. 823-31.
- Sikaris, K., et al., Reproductive hormone reference intervals for healthy fertile young men: evaluation of automated platform assays. J Clin Endocrinol Metab, 2005. 90(11): p. 5928-36.
- Schoor, R.A., et al., The role of testicular biopsy in the modern management of male infertility. J Urol, 2002. 167(1): p. 197-200.
- Karpas, A.E., et al., Elevated serum follicle-stimulating hormone levels in men with normal seminal fluid analyses. Fertility and Sterility, 1983. 39(3): p. 333-6.
- de Kretser, D.M., et al., Inhibins, activins and follistatin in reproduction. Hum Reprod Update, 2002. 8(6): p. 529-41.
- Strachan, M.W., et al., Clinical and radiological features of patients with macroprolactinaemia. Clin Endocrinol (Oxf), 2003. 59(3): p. 339-46.
- McLachlan, R.I. and M.K. O'Bryan, Clinical Review#: State of the art for genetic testing of infertile men. J Clin Endocrinol Metab, 2010. 95(3): p. 1013-24.
- Johnson, M.D., Genetic risks of intracytoplasmic sperm injection in the treatment of male infertility: recommendations for genetic counseling and screening. Fertility and Sterility, 1998. 70(3): p. 397-411.
- Bourne, H., et al., Delivery of Normal Twins Following the Intracytoplasmic Injection of Spermatozoa From a Patient With 47,XXY Klinefelters-Syndrome. Human Reproduction, 1997. 12(11): p. 2447-2450.
- Tournaye, H., et al., Testicular sperm recovery in nine 47,XXY Klinefelter patients. Human Reproduction, 1996. 11(8): p. 1644-9.
- Wikstrom, A.M. and L. Dunkel, Testicular function in Klinefelter syndrome. Horm Res, 2008. 69(6): p. 317-26.
- Staessen, C., et al., PGD in 47,XXY Klinefelter's syndrome patients. Hum Reprod Update, 2003. 9(4): p. 319-30.
- Martin, R.H., Cytogenetic determinants of male fertility. Hum Reprod Update, 2008. 14(4): p. 379-90.
- Claustres, M., Molecular pathology of the CFTR locus in male infertility. Reprod Biomed Online, 2005. 10(1): p. 14-41.
- Silber, S.J. and S. Repping, Transmission of male infertility to future generations: lessons from the Y chromosome. Hum Reprod Update, 2002. 8(3): p. 217-29.
- Vogt, P.H., Azoospermia factor (AZF) in Yq11: towards a molecular understanding of its function for human male fertility and spermatogenesis. Reprod Biomed Online, 2005. 10(1): p. 81-93.
- Lazaros, L., et al., Evidence for association of sex hormone-binding globulin and androgen receptor genes with semen quality. Andrologia, 2008. 40(3): p. 186-91.
- O'Flynn O'Brien, K.L., A.C. Varghese, and A. Agarwal, The genetic causes of male factor infertility: a review. Fertility and Sterility, 2010. 93(1): p. 1-12.
- Sigman M, Kolettis PN, Lipshultz LR, McClure RD , Nangia AK NC, Prins GS, Sandlow J 2010 The Optimal Evaluation of the Infertile Male: Best Practice Statement Revised (2010). American Urological Association Education and Research, Inc
- Mallidis, C. and H.W.G. Baker, Fine Needle Tissue Aspiration Biopsy of the Testis. Fertility & Sterility, 1994. 61(2): p. 367-375.
- Watkins, W., et al., Testicular and Epididymal Sperm in a Microinjection Program - Methods of Retrieval and Results. Fertility & Sterility, 1997. 67(3): p. 527-535.
- Bettocchi, C., et al., A review of testicular intratubular germ cell neoplasia in infertile men. J Androl, 1994. 15 Suppl: p. 14S-16S.
- Jacobsen, G.K., O.B. Henriksen, and H. von der Maase, Carcinoma in situ of testicular tissue adjacent to malignant germ-cell tumors: a study of 105 cases. Cancer, 1981. 47(11): p. 2660-2.
- Huyghe, E., et al., Conservative management of small testicular tumors relative to carcinoma in situ prevalence. J Urol, 2005. 173(3): p. 820-3.
- Pryor, J.P., et al., Carcinoma in situ in testicular biopsies from men presenting with infertility. Br J Urol, 1983. 55(6): p. 780-4.
- Giwercman, A., et al., Prevalence of carcinoma in situ of the testis in 207 oligozoospermic men from infertile couples: prospective study of testicular biopsies. BMJ, 1997. 315(7114): p. 989-91.
- de Kretser, D.M., H.G. Burger, and B. Hudson, The relationship between germinal cells and serum FSH levels in males with infertility. J Clin Endocrinol Metab, 1974. 38(5): p. 787-93.
- McLachlan, R.I., et al., Histological evaluation of the human testis--approaches to optimizing the clinical value of the assessment: mini review. Hum Reprod, 2007. 22(1): p. 2-16.
- Ragheb, D. and J.L. Higgins, Jr., Ultrasonography of the scrotum: technique, anatomy, and pathologic entities. J Ultrasound Med, 2002. 21(2): p. 171-85.
- Pierik, F.H., et al., Is routine scrotal ultrasound advantageous in infertile men? J Urol, 1999. 162(5): p. 1618-20.
- The Optimal Evaluation of the Infertile Male: AUA Best Practice Statement.Revised,2010.http://www.auanet.org/content/media/optimalevaluation2010.pdf
- Heshmat, S. and K.C. Lo, Evaluation and treatment of ejaculatory duct obstruction in infertile men. Can J Urol, 2006. 13 Suppl 1: p. 18-21.
- Hirsch, I.H., et al., Objective assessment of spermatogenesis in men with functional and anatomic obstruction of the genital tract. International Journal of Andrology, 1994. 17(1): p. 29-34.
- Heidenreich, A., P. Altmann, and U.H. Engelmann, Microsurgical vasovasostomy versus microsurgical epididymal sperm aspiration/testicular extraction of sperm combined with intracytoplasmic sperm injection. A cost-benefit analysis. Eur Urol, 2000. 37(5): p. 609-14.
- Mak, V., et al., Proportion of cystic fibrosis gene mutations not detected by routine testing in men with obstructive azoospermia. JAMA, 1999. 281(23): p. 2217-24.
- De Rosa, M., et al., Hyperprolactinemia in men: clinical and biochemical features and response to treatment. Endocrine, 2003. 20(1-2): p. 75-82.
- Buvat, J., Hyperprolactinemia and sexual function in men: a short review. Int J Impot Res, 2003. 15(5): p. 373-7.
- Nudell, D.M., M.M. Monoski, and L.I. Lipshultz, Common medications and drugs: how they affect male fertility. Urol Clin North Am, 2002. 29(4): p. 965-73.
- Liu, P.Y. and D.J. Handelsman, The present and future state of hormonal treatment for male infertility. Hum Reprod Update, 2003. 9(1): p. 9-23.
- Kamischke, A. and E. Nieschlag, Update on medical treatment of ejaculatory disorders. Int J Androl, 2002. 25(6): p. 333-44.
- Watkins, W., et al., Testicular Aspiration of Sperm For Intracytoplasmic Sperm Injection - an Alternative Treatment to Electro-Emission - Case Report. Spinal Cord, 1996. 34(11): p. 696-698.
- Mahadevan, M., J.F. Leeton, and A.O. Trounson, Noninvasive method of semen collection for successful artificial insemination in a case of retrograde ejaculation. Fertil Steril, 1981. 36(2): p. 243-7.
- Wong, W.Y., et al., Male factor subfertility: possible causes and the impact of nutritional factors. Fertil Steril, 2000. 73(3): p. 435-42.
- Roth, M.Y., J.K. Amory, and S.T. Page, Treatment of male infertility secondary to morbid obesity. Nat Clin Pract Endocrinol Metab, 2008. 4(7): p. 415-9.
- Pauli, E.M., et al., Diminished paternity and gonadal function with increasing obesity in men. Fertil Steril, 2008. 90(2): p. 346-51.
- Jensen, T.K., et al., Body mass index in relation to semen quality and reproductive hormones among 1,558 Danish men. Fertility and Sterility, 2004. 82(4): p. 863-70.
- Gapstur, S.M., et al., Serum androgen concentrations in young men: a longitudinal analysis of associations with age, obesity, and race. The CARDIA male hormone study. Cancer Epidemiol Biomarkers Prev, 2002. 11(10 Pt 1): p. 1041-7.
- Hammoud, A.O., et al., Obesity and male reproductive potential. J Androl, 2006. 27(5): p. 619-26.
- Isidori, A.M., et al., Leptin and androgens in male obesity: evidence for leptin contribution to reduced androgen levels. J Clin Endocrinol Metab, 1999. 84(10): p. 3673-80.
- Tsai, E.C., et al., Association of bioavailable, free, and total testosterone with insulin resistance: influence of sex hormone-binding globulin and body fat. Diabetes Care, 2004. 27(4): p. 861-8.
- Kort, H.I., et al., Impact of body mass index values on sperm quantity and quality. J Androl, 2006. 27(3): p. 450-2.
- Stewart, T.M., et al., Associations between andrological measures, hormones and semen quality in fertile Australian men: inverse relationship between obesity and sperm output. Human Reproduction, 2009. 24(7): p. 1561-8.
- Hammoud, A.O., et al., Impact of male obesity on infertility: a critical review of the current literature. Fertility and Sterility, 2008. 90(4): p. 897-904.
- Vine, M.F., et al., Cigarette smoking and sperm density: a meta-analysis. Fertility and Sterility, 1994. 61(1): p. 35-43.
- Jensen, T.K., et al., Association of in utero exposure to maternal smoking with reduced semen quality and testis size in adulthood: a cross-sectional study of 1,770 young men from the general population in five European countries. Am J Epidemiol, 2004. 159(1): p. 49-58.
- Jensen, M.S., et al., Lower sperm counts following prenatal tobacco exposure. Human Reproduction, 2005. 20(9): p. 2559-66.
- McGowan, M.P., et al., The incidence of non-specific infection in the semen in fertile and sub- fertile males. International Journal of Andrology, 1981. 4(6): p. 657-62.
- Jennings, M.G., M.P. McGowan, and H.W.G. Baker, Is conventional bacteriology useful in the management of male infertility? Clinical Reproduction and Fertility, 1986. 4(6): p. 359-66.
- Aitken, R.J. and H.W.G. Baker, Seminal Leukocytes - Passengers, Terrorists or Good Samaritans. Human Reproduction, 1995. 10(7): p. 1736-1739.
- Alvarez, J.G., et al., Increased DNA damage in sperm from leukocytospermic semen samples as determined by the sperm chromatin structure assay. Fertil Steril, 2002. 78(2): p. 319-29.
- Meares, E. and T. Stamey, Bacteriologic localisation patterns in bacterial prostatis and urethritis. Invest Urol, 1968. 5: p. 492-518.
- Baker, H.W.G., et al., A controlled trial of the use of erythromycin for men with asthenospermia. International Journal of Andrology, 1984. 7(5): p. 383-8.
- Comhaire, F.H., P.J. Rowe, and T.M. Farley, The effect of doxycycline in infertile couples with male accessory gland infection: a double blind prospective study. Int J Androl, 1986. 9(2): p. 91-8.
- Hargreave, T.B., Varicocele: overview and commentary on the results of the World Health Organisation varicocele trial., in Current advances in andrology, G.M.H. Waites, J. Frick, and H.W.G. Baker, Editors. 1997, Monduzzi Editore: Bologna. p. 31-44.
- Evers, J.L. and J.A. Collins, Surgery or embolisation for varicocele in subfertile men. Cochrane Database Syst Rev, 2004(3): p. CD000479.
- Ficarra, V., et al., Treatment of varicocele in subfertile men: The Cochrane Review--a contrary opinion. Eur Urol, 2006. 49(2): p. 258-63.
- WHO, The influence of varicocele on parameters of fertility in a large group of men presenting to infertility clinics. World Health Organization. Fertility and Sterility, 1992. 57(6): p. 1289-93.
- Asci, R., et al., The outcome of varicocelectomy in subfertile men with an absent or atrophic right testis. Br Journal of Urology, 1998. 81(5): p. 750-2.
- Baker, H.W.G. and G.T. Kovacs, Spontaneous improvement in semen quality: regression towards the mean. International Journal of Andrology, 1985. 8(6): p. 421-6.
- Madgar, I., et al., Controlled trial of high spermatic vein ligation for varicocele in infertile men. Fertility and Sterility, 1995. 63(1): p. 120-4.
- Bostofte, E., et al., Fertility prognosis for infertile men: results of follow-up study of semen analysis in infertile men from two different populations evaluated by the Cox regression model. Fertil Steril, 1990. 54(6): p. 1100-6.
- Billings, E.L., et al., Symptoms and hormonal changes accompanying ovulation. Lancet, 1972. 1(7745): p. 282-4.
- Zinaman, M.J., et al., Semen quality and human fertility: a prospective study with healthy couples. J Androl, 2000. 21(1): p. 145-53.
- Rowe, T., Fertility and a woman's age. J Reprod Med, 2006. 51(3): p. 157-63.
- Hjollund, N.H., et al., Reproductive effects of male psychologic stress. Epidemiology, 2004. 15(1): p. 21-7.
- Duran, H.E., et al., Intrauterine insemination: a systematic review on determinants of success. Hum Reprod Update, 2002. 8(4): p. 373-84.
- Akanji Tijani, H. and S. Bhattacharya, The role of intrauterine insemination in male infertility. Hum Fertil (Camb), 2010. 13(4): p. 226-32.
- Palermo, G., et al., Pregnancies after intracytoplasmic injection of single spermatozoon into an oocyte. Lancet, 1992. 340(8810): p. 17-8.
- Harari, O., et al., Intracytoplasmic Sperm Injection - a Major Advance in the Management of Severe Male Subfertility. Fertility & Sterility, 1995. 64(2): p. 360-368.
- Goldstein, M. and C. Tanrikut, Microsurgical management of male infertility. Nat Clin Pract Urol, 2006. 3(7): p. 381-91.
- Donoso, P., H. Tournaye, and P. Devroey, Which is the best sperm retrieval technique for non-obstructive azoospermia? A systematic review. Hum Reprod Update, 2007. 13(6): p. 539-49.
- Shah, R.S., Surgical and non-surgical methods of sperm retrieval., in Advanced Infertility Management., M. Hansotia, Desai, S and Parihar, M, Editor. 2002, Jaypee Brothers: New Delhi. p. 253-8.
- Shih, W., et al., Factors affecting low birthweight after assisted reproduction technology: difference between transfer of fresh and cryopreserved embryos suggests an adverse effect of oocyte collection. Hum Reprod, 2008. 23: p. 1644-53.
- Ogilvie, C.M., P.R. Braude, and P.N. Scriven, Preimplantation genetic diagnosis--an overview. J Histochem Cytochem, 2005. 53(3): p. 255-60.
- Bourne, H., D.H. Edgar, and H.W.G. Baker, Sperm preparation techniques., in Textbook of Assisted Reproductive Techniques Laboratory and Clinical Perspectives., G. D.K., et al., Editors. 2004, Taylor and Francis: London. p. 79-92.
- Liu, D.Y. and H.W.G. Baker, Disordered acrosome reaction of sperm bound to the zona pellucida: a newly discovered sperm defect with reduced sperm-zona pellucida penetration and reduced fertilization in vitro. Hum Reprod, 1994. 9: p. 1694-700.
- Hurst, T. and P. Lancaster, Assisted conception Australia and New Zealand 1999 and 2000. Assisted Conception Series no. 6. Vol. 6. 2001, Sydney: Australian Institute of Health and Welfare National Perinatal Statistics Unit. 73.
- Verpoest, W. and H. Tournaye, ICSI: hype or hazard? Hum Fertil (Camb), 2006. 9(2): p. 81-92.
- Wang, Y.A., J.H. Dean, and E.A.Sullivan, Assisted reproduction technology in Australia and New Zealand 2005., in Assisted reproduction technology series no. 11. Cat. No. PER 36. 2007, AIHW National Perinatal Statistics Unit.: Sydney.
- Van Voorhis, B.J., Outcomes from assisted reproductive technology. Obstet Gynecol, 2006. 107(1): p. 183-200.
- Venn, A., et al., Risk of cancer after use of fertility drugs with in-vitro fertilisation [see comments]. Lancet, 1999. 354(9190): p. 1586-90.
- Niederberger, C., Cryptorchidism, fertility, and cancer. J Androl, 2003. 24(1): p. 19-20.
- Min, J.K., et al., What is the most relevant standard of success in assisted reproduction? The singleton, term gestation, live birth rate per cycle initiated: the BESST endpoint for assisted reproduction. Hum Reprod, 2004. 19(1): p. 3-7.
- O'Bryan, M.K., et al., Human seminal clusterin (SP-40,40). Isolation and characterization. J Clin Invest, 1990. 85(5): p. 1477-86.
- Liu, D.Y. and H.W. Baker, Human sperm bound to the zona pellucida have normal nuclear chromatin as assessed by acridine orange fluorescence. Hum Reprod, 2007. 22(6): p. 1597-602.
- Bonduelle, M., et al., Incidence of Chromosomal Aberrations in Children Born After Assisted Reproduction Through Intracytoplasmic Sperm Injection. Human Reproduction, 1998. 13(4): p. 781-782.
- Egozcue, J., et al., Meiotic abnormalities in infertile males. Cytogenet Genome Res, 2005. 111(3-4): p. 337-42.
- Halliday, J., Outcomes for offspring of men having ICSI for male factor infertility. Asian J Androl, 2012. 14(1): p. 116-20.
- Sinclair, K.D., et al., In-utero overgrowth in ruminants following embryo culture: lessons from mice and a warning to men. Hum Reprod, 2000. 15 Suppl 5: p. 68-86.
- Paoloni-Giacobino, A., Epigenetics in reproductive medicine. Pediatr Res, 2007. 61(5 Pt 2): p. 51R-57R.
- Bruinsma, F., et al., Incidence of cancer in children born after in-vitro fertilization. Hum Reprod, 2000. 15(3): p. 604-7.
- Hansen, M., et al., Assisted reproductive technologies and the risk of birth defects--a systematic review. Hum Reprod, 2005. 20(2): p. 328-38.
- Halliday, J., Outcomes of IVF conceptions: are they different? Best practive and research clinical obstertics and gynaecology, 2007. 21: p. 67-81.
- Davies, M.J., et al., Reproductive technologies and the risk of birth defects. N Engl J Med, 2012. 366(19): p. 1803-13.
- Barratt, C. and I.D. Cooke, Donor Insemination. 1993, Cambridge: Cambridge University Press.
- McGowan, M.P., et al., Selection of high fertility donors for artificial insemination programmes. Clinical Reproduction and Fertility, 1983. 2(4): p. 269-74.
- Clarke, G.N., et al., Artificial Insemination and in-Vitro Fertilization Using Donor Spermatozoa - a Report On 15 Years of Experience. Human Reproduction, 1997. 12(4): p. 722-726.
- Ferrara, I., R. Balet, and J.G. Grudzinskas, Intrauterine insemination with frozen donor sperm. Pregnancy outcome in relation to age and ovarian stimulation regime. Hum Reprod, 2002. 17(9): p. 2320-4.
- Lansac, J. and D. Royere, Follow-up studies of children born after frozen sperm donation. Hum Reprod Update, 2001. 7(1): p. 33-7.
- Tournaye, H., et al., Preserving the reproductive potential of men and boys with cancer: current concepts and future prospects. Hum Reprod Update, 2004. 10(6): p. 525-32.
- Arnon, J., et al., Genetic and teratogenic effects of cancer treatments on gametes and embryos. Hum Reprod Update, 2001. 7(4): p. 394-403.
- Wyns, C., et al., Options for fertility preservation in prepubertal boys. Human Reproduction Update, 2010. 16(3): p. 312-28.
- Holoch, P. and M. Wald, Current options for preservation of fertility in the male. Fertility and Sterility, 2011. 96(2): p. 286-90.
- Stahl, P.J., et al., Indications and strategies for fertility preservation in men. Clin Obstet Gynecol, 2010. 53(4): p. 815-27.
- Hsiao, W., et al., Successful treatment of postchemotherapy azoospermia with microsurgical testicular sperm extraction: the Weill Cornell experience. J Clin Oncol, 2011. 29(12): p. 1607-11.
- Simon, L., et al., Sperm DNA damage has a negative association with live-birth rates after IVF. Reprod Biomed Online, 2013. 26(1): p. 68-78.
- Zini, A., et al., Sperm DNA damage is associated with an increased risk of pregnancy loss after IVF and ICSI: systematic review and meta-analysis. Hum Reprod, 2008. 23(12): p. 2663-8.
- Zini, A., O. Albert, and B. Robaire, Assessing sperm chromatin and DNA damage: clinical importance and development of standards. Andrology, 2014. 2(3): p. 322-5.
- Aitken, R.J., G.N. De Iuliis, and R.I. McLachlan, Biological and clinical significance of DNA damage in the male germ line. Int J Androl, 2009. 32(1): p. 46-56.
- Palermo, G.D., et al., Perspectives on the assessment of human sperm chromatin integrity. Fertil Steril, 2014. 102(6): p. 1508-17.
- Gawecka, J.E., et al., Mouse zygotes respond to severe sperm DNA damage by delaying paternal DNA replication and embryonic development. PLoS One, 2013. 8(2): p. e56385.
- Gawecka, J.E., et al., A model for the control of DNA integrity by the sperm nuclear matrix. Asian J Androl, 2015. 17(4): p. 610-5.
- Muratori, M., et al., Investigation on the Origin of Sperm DNA Fragmentation: Role of Apoptosis, Immaturity and Oxidative Stress. Mol Med, 2015. 21: p. 109-22.
- Morris, I.D., Sperm DNA damage and cancer treatment. Int J Androl, 2002. 25(5): p. 255-61.
- Kunzle, R., et al., Semen quality of male smokers and nonsmokers in infertile couples. Fertil Steril, 2003. 79(2): p. 287-91.
- Saleh, R.A., et al., Evaluation of nuclear DNA damage in spermatozoa from infertile men with varicocele. Fertil Steril, 2003. 80(6): p. 1431-6.
- Esteves, S.C., M. Roque, and A. Agarwal, Outcome of assisted reproductive technology in men with treated and untreated varicocele: systematic review and meta-analysis. Asian J Androl, 2015.
- Moazzam, A., R. Sharma, and A. Agarwal, Relationship of spermatozoal DNA fragmentation with semen quality in varicocele-positive men. Andrologia, 2015. 47(8): p. 935-44.
- Aitken, R.J. and M.A. Baker, Oxidative stress, spermatozoa and leukocytic infiltration: relationships forged by the opposing forces of microbial invasion and the search for perfection. J Reprod Immunol, 2013. 100(1): p. 11-9.
- Sakkas, D., et al., Relationship between the presence of endogenous nicks and sperm chromatin packaging in maturing and fertilizing mouse spermatozoa. Biol Reprod, 1995. 52(5): p. 1149-55.
- Sakkas, D., et al., The presence of abnormal spermatozoa in the ejaculate: did apoptosis fail? Hum Fertil (Camb), 2004. 7(2): p. 99-103.
- Sakkas, D., et al., Abnormal spermatozoa in the ejaculate: abortive apoptosis and faulty nuclear remodelling during spermatogenesis. Reprod Biomed Online, 2003. 7(4): p. 428-32.
- Wright, C., S. Milne, and H. Leeson, Sperm DNA damage caused by oxidative stress: modifiable clinical, lifestyle and nutritional factors in male infertility. Reprod Biomed Online, 2014. 28(6): p. 684-703.
- Sakkas, D., Novel technologies for selecting the best sperm for in vitro fertilization and intracytoplasmic sperm injection. Fertil Steril, 2013. 99(4): p. 1023-9.
- Rubino, P., et al., The ICSI procedure from past to future: a systematic review of the more controversial aspects. Hum Reprod Update, 2015.
Surgical Treatment of Obesity
ABSTRACT
Obesity is one of the most prevalent pathogens in the developed world, causing numerous common and lethal diseases. Non-surgical treatments to date have failed to provide an effective, durable solution. Bariatric surgery includes the procedures of gastric bypass, sleeve gastrectomy, gastric banding and biliopancreatic bypass. These procedures have been shown to produce substantial and durable weight loss yet are provided annually to less than 0.2% of eligible obese people, making current bariatric surgery largely irrelevant to public health.
The principal mechanisms of effect vary between procedures and include control of hunger, change of appetite, restriction of intake, diversion of food from the proximal small intestine, malabsorption of macronutrients, increased energy expenditure, food aversion and possibly changes to the gut microflora and changes to serum bile acid levels.
The mortality risk reflects the type of surgery and varies between 0.1% for gastric banding to 1-2% for other procedures.
The criteria for consideration of bariatric surgery include the presence of obesity (BMI > 30), a history of multiple attempts at weight reduction by non-surgical means, an awareness of the potential risks and a commitment to attend the follow up program. The decision on which procedure should be used is based on patient or surgeon preference, availability of appropriate aftercare and the patient’s tolerance of risk and permanent anatomical change. For complete coverage of this and related areas of endocrinology, plese see our on-line free web-book, www.endotext.org.
BACKGROUND
Obesity is one of the most significant pathogens in the developed world. It causes or exacerbates numerous common and lethal diseases. It can markedly reduce the quality of life and it competes with smoking as the commonest cause of premature death. The prevalence of obesity has increased markedly in the last thirty years. A systematic review of 199 countries in 2008 estimated 502 million people worldwide were obese1. According to the World Health Organization, obesity has more than doubled worldwide since 1980 and more than 600 million or 13% of the adult population were obese in 2014.
The epidemic is quite recent. In the United States, between 1960 and 1980, there was a relatively modest rise in the number of adults with obesity, from 12% to 14% only. This number has more than doubled since 19802. Currently, approximately 80 million people in the USA, 34% of the adult population, are obese3 and this number is estimated to increase to more than 140 million by 20304. The direct health care costs were estimated to be $75 billion per year in 2003. These would rise to $140 billion per year and it would be an associated loss of productivity (indirect costs) of $580 billion4 Based on the Health and Wellness Survey in 2008 there is estimated to be loss of 1.7 - 3.0 million productive person-years in the USA, representing a cost of $390 - $580 billion due to the current state of obesity4 .
The morbidity caused by obesity makes it our greatest current health challenge because of its direct contribution to many chronic, debilitating and life-threatening diseases. These include type 2 diabetes, cardiovascular diseases such as ischemic heart disease, stroke, hypertension and dyslipidemia and several common cancers. The health care costs of obesity are now a major component of health budgets across the developed world. Currently there is no non-surgical method for predictably achieving major weight loss in the obese and maintaining that weight loss for an extended period. Current programs involving diet, behavioral modification, exercise and activity, with or without drug supplementation, are able to achieve a modest weight loss which is generally sustained only for the duration of the program.
In this chapter I will review the surgical options, their strengths and weaknesses, to provide a framework of evidence that enables a logical treatment approach to this major healthcare challenge. In doing so, I will seek an alignment of the views of health professionals and the general community so that they recognize obesity as a disease and not a sign of personal failing, they acknowledge the severity of this disease and they recognize the spread of invasiveness and risk and effectiveness associated with options for surgical treatments.
Surgical methods have been known to achieve substantial and durable weight loss for more than half a century and yet they have not achieved a significant impact on community health. Currently, less 1 in a 1000 world-wide and less than 1 in 250 in the USA and Australia of those who would benefit by treatment are having bariatric surgery. In terms of the public health, bariatric surgery is used so infrequently as to be largely irrelevant. In its early days the risks were high, undoubtedly discouraging many. But these risks have been reduced markedly5. The risk to benefit ratio has now become much more favorable and the cost to benefit ratio is also favorable. It is time for a change and the data are there to justify a change.
AN HISTORICAL PERSPECTIVE OF BARIATRIC SURGERY
Although the weight loss that accompanied surgery, particularly if it involved the stomach or small intestine, had been noted for as long as the procedures have existed, a direct use of surgery for the specific purpose of weight loss - bariatric surgery - really began in 1954 with the small bowel bypass procedure. It can be seen to pass through three phases so far. The small bowel bypass phase was replaced by the gastric stapling phase in the late 1960s and then, in the early 1990s, the introduction of laparoscopic surgery and gastric banding led to the third phase.
The Initial Phase (1950 – 1970) – Small Bowel Bypass.
Surgical management of obesity began with the introduction of the jejunoileal bypass (JIB) in the 19546. In this procedure the proximal jejunum was diverted to distal part of the gut, leaving a long segment of excluded small intestine and a marked reduction in absorptive capacity. Many variations existed. In a typical procedure, the proximal 35 cm of proximal jejunum was joined end-to-side to the last 10 cm of ileum. The JIB procedures represented the best and the worst of bariatric surgery. Major and sustained weight loss was achieved and there were impressive health benefits, particularly in relation to lipid metabolism. However it was associated with serious side-effects including copious offensive diarrhea, electrolyte imbalances, oxalate calculi in the kidneys and progressive hepatic fibrosis with eventual liver failure7-11. For these reasons this group of procedures was generally abandoned by the 1970s in favor of stomach stapling procedures
The Middle Phase (1970 – 1990) – Stomach Stapling.
The Roux-en-Y gastric bypass (RYGB) operation was introduced by Edward Mason in 196012. In this procedure the stomach was completely partitioned into a small upper gastric pouch, draining into a Roux–en-Y limb of proximal jejunum of variable length from 40 to 150 cm, and a distal excluded stomach. This procedure provided a hybrid between the malabsorptive approach of JIB and later, more purely restrictive, operations. It has undergone various modifications over the subsequent 40 years and still serves us well as an effective anti-obesity operation. However its drawbacks of perioperative death and significant perioperative and late morbidity, although markedly less threatening than those of JIB, have been nevertheless sufficient to cause most of the obese to stay away.
Dr Mason and his colleague, Dr Printen, then introduced a purely restrictive operation of gastroplasty in 197313. The procedure involves partitioning the stomach into a small upper pouch draining through a narrow stoma into the remainder of the stomach. Numerous variations of this procedure have followed, the most significant variant being the vertical banded gastroplasty (VBG) which was first described by Dr Mason in 198214 . It was hoped that this group of operations would provide greater short and long term safety and yet retain the power of gastric bypass. Unfortunately both randomized controlled trials and observational studies have consistently shown that it has failed in both aspirations15-18.
In the meantime there was a resurgence of malabsorptive surgery with Italian surgeon, Nicola Scopinaro, introducing the biliopancreatic diversion procedure (BPD) in 197619. It too has undergone change with time and experience. The basic procedure involves distal gastrectomy leaving a proximal gastric pouch of 200 – 500 ml, a 200 cm length of terminal ileum anastomosed to the gastric pouch and the biliopancreatic limb entering at 50 cm from the ileocecal valve20. The most notable remodeling of the procedure has been the so-called duodenal switch variant (BPD-DS) proposed by Picard Marceau’s group in 199321, 22 in which a longitudinal gastrectomy (sleeve gastrectomy) enabled retention of the gastric antrum for controlled gastric emptying, and the ileal limb was anastomosed to the proximal duodenum. The benefit of this variation remains controversial.
The Current Phase (1990 - Present) – Laparoscopic Procedures.
This phase is characterized by the advent of the laparoscopic approach to bariatric surgery particularly gastric bypass23 and biliopancreatic diversion24, introduction of the laparoscopic adjustable gastric band (LAGB) with the particular features of minimal anatomical disturbance, adjustability and potential reversibility25, 26 and growth of the sleeve gastrectomy as a stand-alone procedure27. The reduced invasiveness and increased safety of these approaches has led to a major rise in the use of bariatric surgery for obesity across the world. In the United States the estimated total bariatric procedures in 1990 was 30,000. The total for 2008 was estimated to be 220,000 cases28
Adjustable gastric banding had first been proposed by two Austrian surgical researchers, Szinicz and Schnapka, in 198229. The idea was brought into clinical practice as an open operation by Lubomyr Kusmak in 198630. It did not attract major interest until the advent of the technology that enabled the performance of complex laparoscopic surgical procedures became widespread in the early 1990s. The BioEnterics® Lap-Band ® system (LAGB) was specifically designed for laparoscopic placement and was introduced into clinical practice by Mitiku Belachew from Huy, Belgium in September 1993. Because of the dual attractions of a controlled level of effect through adjustability and of laparoscopic placement without resection of gut or anastomoses, through the 1990s this procedure rapidly became the dominant bariatric procedure in all regions of the developed world except the USA, where its introduction was delayed until regulatory requirements were completed in June 2001. More recently, the challenges of maintaining good aftercare for the banded patient and the relative simplicity of the sleeve gastrectomy has seen this latter procedure take a dominant position.
In the meantime, laparoscopic approach to the RYGB was introduced by Wittgrove and Clark in 199431. The technical challenge of completing the gastrojejunostomy laparoscopically has been variously managed by transoral passage of a circular stapler, end-to-end or side-to-side stapling transabdominally or a simple hand-sewn anastomosis. As the technical challenges of the laparoscopic approach were overcome, this procedure became the dominant approach in the USA until it was overtaken by gastric banding around 2008 and then by sleeve gastrectomy which, in 2015, is the most used procedure world-wide.
Laparoscopic BPD or its DS variant has remained clinically challenging. The mortality can be high24 and few are now performed28. Most procedures are still performed by open technique and as they represents less than 2% of bariatric surgery worldwide, it is unlikely they will grow beyond that.
CURRENT METHODS IN BARIATRIC SURGERY
Sleeve Gastrectomy
The sleeve gastrectomy involves excision of approximately 80% of the stomach by using multiple firings of a linear stapler/cutter to separate a narrow tube or sleeve of the lesser curve of the stomach from the greater curve aspect. The antrum is preserved to maintain gastric emptying. A bougie is placed in the lesser curve segment during the resection to maintain adequate lumen yet achieve a standardised gastric remnant. The optimal size of this bougie is not yet agreed upon. A 36 French is the most common size but sizes from 30F to 50F are reported. The procedure is non-adjustable and non-reversible.
The procedure is a laparoscopic procedure, taking between 30 and 90 minutes. Some patients are treated on an outpatient basis but a one to two day stay is more common.

Figure 1. Sleeve gastrectomy
Gastric imbrication is a non-resectional variant of the sleeve gastrectomy. The greater curve vascular pedicles are ligated and then the gastric wall is imbricated using two rows of sutures to create a narrow lumen, similar in size to the sleeve gastrectomy. The costs are reduced by avoiding the use of multiple firings of stapling devices and the risks are expected to be lower by the absence of the stapled closure of divided stomach.

Figure 2. Gastric imbrication. The upper panel shows the placement of the initial continuous suture after division of the greater curve vascular pedicles. The lower panel shows completion of the second row of sutures.
Laparoscopic Adjustable Gastric Banding (LAGB).
The LAGB is the safest of the surgical options and therefore has tended to be considered ahead of other bariatric procedures on the hierarchy of risk because of its effectiveness in the setting of a better safety profile, minimal invasiveness and complete and easy reversibility. Its requirement for skilled follow up and adjustments has proved challenging for many.
The procedure was introduced into clinical practice by Dr Mitiku Belachew with the laparoscopic placement of a Lap-Band in a patient in Huy, Belgium on the 3rd September, 1993. Others quickly followed and soon it was being embraced as an important new procedure. However initially there were significant gaps in the knowledge of the process of care the LAGB required and skills in LAGB placement, optimal aftercare and management of late adverse events. It was not known how the band worked or even if it would work. There were no data on optimal placement and fixation. Protocols for the aftercare process, the adjustment protocols and the education of the patient into the specific requirements for eating and activity after the band were not present.
There has been important growth in knowledge since that time with more than 1,000 peer-reviewed papers defining the LAGB process and outcomes. It is now more studied than any bariatric procedure with better knowledge of its mechanisms and a higher quality evidence base than all the other bariatric options.
There are a number of adjustable gastric bands available. The principal two, which have approval of the US Food and Drugs Administration for use in the USA, are the Lap-Band ™ (Apollo Endosurgery, Austin, Tx) and the Realize Band™ (Ethicon Endosurgery, Cincinnati, OH). Others include the Mid-Band, the Heliogast band, the Minimizer band and the AMI band. These are in use principally in Europe. They lack adequate published data attesting to their effectiveness, they are not approved for use in the USA and so their uptake has been modest. The discussion below will focus primarily on the data from the Lap-Band.
The current version of the Lap-Band, known as the Lap-Band AP (for Advanced Platform), is shown in figure 3.

Figure 3. The band consists of a ring of silicone with an inner balloon. The balloon is connected to an access port.
The LAGB is placed laparoscopically. Commonly 3 x 5mm, 1 x 10mm and 1 x 15 mm ports are required. A path is developed from the top of the lesser curve of the stomach to the angle of His, the band is placed around this path, closed and then stabilized at this site with some sutures. The tubing passes to an access port, placed through a 3 cm incision over the left rectus abdominis muscle. The procedure takes 30 -50 minutes to complete and the patient is able to go home at a mean of 2 hours after completion of the procedure32.

Figure 4. The LAGB is placed over the cardia of the stomach within 1cm of the esophago-gastric junction.
Roux en Y Gastric Bypass (RYGB)
Gastric bypass combines a marked reduction in the size of stomach available for food with a narrow stoma passing from the gastric pouch to a Roux-en-Y loop of jejunum diverting food from the duodenum and proximal jejunum. In a typical current laparoscopic version (figure 5), the stomach is divided completely by multiple firings of a device which places two rows of staples and cuts the gastric wall in between. This creates a small proximal gastric pouch of volume of 50ml or less and a large residual stomach, now excluded from the food. A Roux limb of jejunum is formed by dividing the proximal jejunum completely at about 50cm from the duodeno-jejunal flexure, taking the distal aspect of this point of division up to form a small anastomosis with the small gastric pouch and anastomosing the proximal aspect to the more distal jejunum at 50 cm below the gastro-jejunal anastomosis. The procedure takes 90 -150 minutes and the patient stays in hospital for 2 - 4 days.

Figure 5. RYGB showing a small gastric pouch, a narrow gastrojejunostomy and exclusion of foods from the duodenum and proximal jejunum.
Single anastomosis Gastric Bypass (SAGB)
This form of gastric bypass is increasingly popular as an alternative to the RYGB. It is variously known as the SAGB, OAGB (one anastomosis gastric bypass), OLGB (Omega loop gastric bypass) or MGB (Mini gastric bypass). It differs from the RYGB by having a loop of small bowel rather than a Roux limb, a long and narrow lesser curve gastric pouch and a longer bypass of the duodenum and proximal jejunum (typically, 150 cm rather than 40 cm) (figure 6).

Figure 6. SAGB showing the gastric pouch as a sleeve of lesser curve of stomach and the loop gastrojejunostomy.
There are now extensive published data on outcomes which include large patient groups, long-term follow up and systematic literature reviews33-36. Early concerns regarding bile reflux leading to intractable gastritis and esophagitis have not proven to be valid.
In a prospective randomized controlled clinical trial performed on 80 patients with a two year follow up37, the SAGB proved to be simpler and safer than RYGB, yet achieved similar outcomes for effects on the metabolic syndrome and quality of life and demonstrated no disadvantages over RYGB.
Biliopancreatic Diversion / Duodenal Switch (BPD/DS)
The BPD contains a "restrictive" component and a "malabsorptive" component. The restrictive component is a partial gastrectomy leaving a large proximal gastric segment of between 200 and 500ml. The actual size is only loosely defined. The originator of BPD, Dr Nicola Scopinaro, moved to the "ad hoc gastrectomy" with its wide variability of gastric pouch size after he had treated nearly 1000 patients. He recommended tailoring the gastrectomy to the patient's weight and some physical characteristics. He also recommended tailoring the intestinal lengths38. The validity and effect of these variations has not been reported.
The malabsorptive component consists of division of the small intestine, usually at 250cm proximal to the ileocecal valve, anastomosis of the distal side of this division to the gastric pouch and end-to-side anastomosis of the proximal side of the division to the terminal ileum, usually at 50cm proximal to the junction with the cecum.
In the duodenal switch variant of the BPD (figure 7) a sleeve gastrectomy is performed, preserving the pylorus and proximal duodenum. The distal end of the transected small bowel is anastomosed to the duodenum. This structure is designed to avoid dumping syndrome but any particular benefit of one version over the other is unclear.

Figure 7. The DS variant of BPD with a sleeve gastrectomy, retention of the gastric antrum, diversion of food into the mid small gut and diversion of pancreatic and biliary secretions to the distal small gut .Note both limbs are passing behind the transverse colon and a colour difference is added to help follow the respective pathways. The common channel is the normal ileum terminating at the ileo-caecal junction.
It is the most metabolically severe of the current options and therefore hasn’t proved to be popular with patients or surgeons in spite of favorable published outcomes. BPD has been available for 30 years19 and yet remains a very minor part of bariatric surgery. Worldwide, it constitutes less than 2% of bariatric surgery28. However, it does generate good weight loss and should be considered on occasions as a second line bariatric surgical option.
MECHANISMS OF ACTION IN BARIATRIC SURGERY
Historically, bariatric surgical procedures were all subdivided into two groups. Some were classified as restrictive, with the creation of a small pouch that limited the size of a meal and a small opening from that pouch into the rest of the gut so that the transit of food was abnormally delayed. The best model of a restrictive procedure was the vertical banded gastroplasty used during the 1970s and 80s. Others were described as malabsorptive, in which the changed anatomy reduced normal macronutrient absorption. The best example of this was the original jejunoileal bypass. Some procedures are considered to be a hybrid containing both elements, the BPD being an example. Other possible mechanisms were not considered. This narrow dichotomous concept has happily faded as better research and more careful consideration has led to a much broader understanding of the mechanistic options39. Table 1 provides a list of current known mechanisms. It is undoubtedly incomplete but provides a more up-to-date view of how bariatric surgical procedures can achieve weight loss.
Table 1. Possible mechanisms of bariatric surgical effect.
| Induce satiety, reduce appetite, control hunger |
| Change of taste preference - less sweet foods; lower fat content; |
| Restrict Intake |
| Diversion from upper GI tract |
| Malabsorption of macronutrients |
| Increased energy expenditure; Increased diet-induced thermogenesis |
| Aversion to food through side-effects |
| Inhibition of the metabolic adaptation to weight loss |
| Changes to the gut microflora |
| Changes to plasma bile acid levels |
| Changes in gut hormones: candidates include the incretins (GLP-1; GIP), ghrelin, CCK, Peptide YY, |
| Central mechanisms: Modify hedonics; central appetite control; altered liking and wanting; |
Mechanisms in Sleeve Gastrectomy
Although primarily seen as a gastric restrictive procedure, it is likely that satiety induction is centrally important and occurs because of removal of most of the source for ghrelin. Further, with rapid gastric emptying the increase of nutrients into the small intestine may lead to change in distal gut hormonal responses. The sleeve gastrectomy is the first element of the duodenal switch variant of the BPD. It has lately become popular as a single procedure because of ease of surgery, early effectiveness and perceived lack of need for close follow up ("sleeve and leave").
Mechanisms of Gastric Bypass (RYGB and SAGB)
RYGB is a complex procedure both anatomically and physiologically. There are several mechanisms of action involved. First, there is early satiation after eating a small amount of food due to the small volume and slow emptying of the gastric pouch. The small stoma provides a restrictive component through delayed gastric emptying. The diversion of food away from the distal stomach, duodenum and proximal jejunum reduces the digestion and possibly the absorption of food by this area of the gut. However, as most of the small bowel remains in the absorptive pathway, it is unlikely the absorption of macronutrients is affected and so the standard RYGB should not be regarded as having a malabsorptive component. The long-limb version of RYGB where food is diverted from the digestive enzymes of the pancreas for 150cm or more40 is more likely to contain a malabsorptive component. Micronutrients which are normally absorbed in the upper gut, such as calcium, are changed. Finally it can have an aversion effect with the symptoms of dumping syndrome occurring if there is ingestion of simple sugars or small osmotically-active molecules, leading to reduce sweet-eating. The diversion of food from the duodenum and proximal jejunum may mediate gut hormonal effects with increased release of GLP-1 and GIP from the distal gut. These hormones act as incretins, increasing release of insulin from pancreatic β cells.
Mechanisms in Laparoscopic adjustable gastric banding (LAGB)
The band lies at the very top of the stomach, around the cardia and within one cm of the esophago -gastric junction. The access port is place in the subcutaneous layer of the anterior abdominal wall and is accessed by a percutaneous injection. The primary mechanism of action of the gastric band is by the induction of a sense of satiety, a lack of appetite or hunger41. There are two components to this - satiety and satiation.
Satiety is the state of not being hungry. It is achieved for the LAGB patient by adding or removing of fluid from the system to change the degree of compression of the band on the gastric wall. When this compression is optimal, it induces a sense of satiety which is present throughout the day. Although some hunger may develop at times during the day, there is a general reduction of appetite, less interest in food and less concern about not eating.
Satiation is the resolution of hunger with eating. For the LAGB patient, it is induced by each bite of food as it passes across the band. When the band is optimally adjusted each bite is squeezed across by esophageal peristalsis, generating increased pressure on that segment of the gastric wall. This reduces any appetite that may have been present and induces a feeling of not being hungry after eating a small amount. The combination of these effects allows the person to eat three or less small meals per day. The mean energy intake of the banded patient should be between 1,000 and 1,200 kcals per day42.

Figure 8. The Lap-Band AP with and without added fluid.
Figure 8 shows two views of the Lap-Band AP. On the left, it contains only the basal volume 3ml of saline and on the right it is well inflated band containing 7 ml of saline. The space within which is occupied by the cardia of the stomach. With 3ml added the internal space has an area of 357cm2. This is reduced to an area of 139cm2 with 7.0ml is present. These two areas represent the limits within which the LAGB is set for most patients. This ability to titrate the level of adjustment against the level of satiety is central to the effectiveness of the band.The optimally adjusted band modifies the normal transit of a food bolus into the stomach. With normal swallowing, a food bolus is swallowed and carried by oesophageal peristalsis down the esophagus. The lower esophageal sphincter (LES) relaxes and the bolus passes intact smoothly into the stomach. The LES facilitates the final transfer with an aftercontraction. With the band in its correct place with only 1-2 cm of cardia above the upper edge of the band and with the band optimally adjusted (exerting a pressure of between 25 and 35 mm Hg on the gastric lumen43), the esophagus must generate stronger peristalsis and the aftercontraction of the LES become more important. The bolus is squeezed through by these forces. It takes between two and six squeezes to achieve complete transit of a single small bite. This may take up to one minute.
Figure 9. A small bite of food is being squeezed across the band, thereby compressing the vagal afferents and generating a feeling of satiety.
The aftercontraction of the LES is evident. Just part of each bite will transit on each peristaltic sequence. The remainder will reflux into the body of the distal esophagus, generate a secondary peristalsis wave and a further squeeze will occur. After several squeezes the bite will have passed. Importantly, each squeeze generates signals to the satiety centre of the hypothalamus. A second swallow should not commence until all of the previous bite has passed totally into the stomach below the band.
The signalling of both satiety and satiation to the arcuate nucleus of the hypothalamus does not appear to be mediated by any of the hormones known to arise from the cardia as none has been shown to be increased in a basal state after band placement and none increases post-prandially44. Vagal afferents are the more probable mediators and, among these, the intraganglionic laminar endings (IGLEs) demonstrate the characteristics needed to subserve this role45, 46
Figure 10 shows the components of the lower esophageal contractile segment (LECS), an entity described by Dr Paul Burton from extensive study of the "physiology" of the gastric band47. It brings together the key elements that together generate early onset of satiation after eating. The distal esophagus squeezes each bite of food to the stomach proximal to the band. The lower esophageal sphincter relaxes to allow passage and then contracts to maintain the forward pressure. The proximal segment of stomach maintains tonic contraction and detects the pressure increase. The band maintains an optimal compression to provide sufficient resistance to stimulate afferent signals but not sufficient to stop transit. There should be no restrictive component for normal functioning of the LAGB.
Figure 10. The four components of the Lower Esophageal Contractile Segment (LECS)
Mechanisms in Biliopancreatic bypass (BPD)
The initial element of the BPD is a partial gastrectomy either as a standard Bilroth II procedure for the standard BPD or as a sleeve gastrectomy in the DS variant of the BPD which generates a restrictive element and also through reduction of the ghrelin sources, and sense of satiety is induced. The gastric volume is reduced to 200 -500ml thus limiting food intake and this is considered to be the initial mechanism of effect48. Subsequently the separation of ingested food from the digestive enzymes until the last 50 cm of terminal ileum leads to malabsorption of all macronutrients. A number of positive and negative secondary effects occur. Through interruption of the enterohepatic circulation of bile and the bypassing of the proximal small intestine, there is a marked reduction of total cholesterol, triglycerides and LDL-cholesterol. Insulin sensitivity, as measured by HOMA, is improved49. Malabsorption of amino acids can lead to hypoproteinemia. Malabsorption of micronutrients and minerals can lead to osteoporosis and anaemia.
OUTCOME FROM BARIATRIC SURGERY
Mortality And Adverse Events
Perioperative mortality. There is a mortality risk with any surgery and this risk was strongly evident for bariatric surgery prior to the general use of laparoscopic approach. Pories et al50 reported a perioperative mortality of 1.9% of the 605 patients treated by open gastric bypass. In a major series from Richmond, Virginia there were 31 perioperative deaths (1.5%) in 2011 patients having RYGB between 1992 and 2004. The mortality occurring at the level of community surgery is probably higher than from the major academic centers. Flum and Dellinger51 used the Washington State Comprehensive Hospital Abstract Reporting System database and the Vital Statistics database to evaluated 30 day mortality of all people having a RYGB procedure in that state during the period 1987 to 2001. Of 3,328 procedures there were 64 deaths, a mortality rate of 1.9%. This period included both laparoscopic and open surgery and could be seen to reflect community practice.
The overall mortality has decreased in more recent years, particularly with the widespread use of a laparoscopic surgical approach as reported in the systematic reviews of the published data. Death after LAGB is rare and in most reports is 10-15 times less likely than after RYGB52, 53. At the Centre for Bariatric Surgery in Melbourne, we have performed more than 9000 primary LAGB procedures and have performed revisional LAGB surgery on more than 1500 of these or other patients without any 30-day mortality or any later death related to the LAGB procedure.
The most definitive evaluation of mortality currently available is derived from the Longitudinal Assessment of Bariatric Surgery (LABS) study report in 20095. This NIH-sponsored study of bariatric surgery involved 10 sites, carefully selected for their expertise and experience. The 30-day rate of death was monitored closely. Of the 4776 patients studied, 3412 had RYGB, 1198 had LAGB and 166 had other procedures unspecified in the report. There were 15 deaths in the RYGB group (0.44%), 6 after a laparoscopic approach and 9 after an open approach. There were no deaths in the LAGB group of patients. The difference was highly significant.
Sleeve gastrectomy was not included in the LABS study. In a recent systematic review54 there were 26 deaths in 8922 patients (0.3%) after sleeve gastrectomy, an outcome similar to RYGB in the LABS study.
Early adverse events. The LABS study serves also to inform on early adverse events for the two major bariatric procedures of RYGB and LAGB. Not surprisingly, the incidence of adverse events mirrored the perioperative mortality rates. Using a composite end-point of death, DVT or pulmonary embolism, reintervention or failure to be discharged by 30 days, they identified 189 who were positive to that end-point, 177 in the RYGB group (5.2%) and 12 in the LAGB group (1.0%), a difference that was highly significant. The complication rate for sleeve gastrectomy is at least comparable to RYGB. In the systematic review of all reports available on the sleeve gastrectomy reported by Brethauer in 200955, the quality of the data prevented careful analysis but those papers that included complications declared rates from 0% to 24%. Postoperative leaks from the staple line after sleeve gastrectomy are relatively frequent (2-3%) and appear to be independent of surgeon experience56. In contrast to leaks after RYGB, they tend to be slow to close thereby generating mortality, greater morbidity, long-term patient suffering and high hospital costs.
Late adverse events. There is, and always will be, a maintenance requirement with any bariatric procedure as we are treating a chronic disease. The procedure needs to remain effective over decades rather than years. It is inevitable that there will be the need to correct or repair. Whilst reversal of a bariatric procedure should be counted as failure, revision to correct or repair should not. It is a part of the process of care.
All bariatric procedures have been shown to have a maintenance requirement. The revisional surgery rate of the studies which report 10 or more years of follow-up, as shown in table 3, was a median value of 24% and it was not different between procedures. Sleeve gastrectomy is not included as there are no 10 year follow-up studies available to date.
The median rate for the six RYGB reports that provided data was 22% with a range of 8% - 38%. All of the LAGB sets provided data on revisional surgery. The median value was 26% with a range of 8% - 60%. As almost no details of the type of revisional procedures were provided and, as there were too few reports at ten years on other procedures, a more detailed analysis is not possible.
Weight Loss Outcomes
Weight loss can be described in different ways, each of which has its advantages and drawbacks. In bariatric surgery the percent of excess weight lost (%EWL) is the preferred method. The excess weight approximates the excess fat within the body. That is what we are trying to reduce and therefore a measure of that effect has more relevance in this clinical setting than measures of total weight change as are commonly used in the non-surgical weight loss literature. It could be argued that percent of excess BMI lost (%EBL) is the most relevant to health status. However as it has a fixed linear relationship to %EWL and it currently lacks broad usage, it has not yet been embraced. Most reports in the bariatric surgical literature provide %EWL, and thus allow comparison between studies.
As obesity is a chronic disease, for treatments to be effective they must also be effective in the long term. Short-term studies (1 - 3 years) are plentiful but simply suggest a potential effectiveness. Medium term studies (3 -10 years) are far fewer but are more assuring of real effectiveness. Long-term studies (greater than 10 years) are very few and yet are the only ones that truly enable rational decision making on effectiveness. There have been a number of systematic reviews bringing these data together. These provide perhaps the best understanding available of the reasonable weight loss expectations for bariatric surgery in general and for specific bariatric procedures. I will deal with these in order of the duration of follow up provided.
One year outcomes: The most quoted of all the systematic reviews is the report by Buchwald et al57 published in JAMA in 2004. There was 47.5%EWL at one year after LAGB, 61.6%EWL for RYGB, 68.2%EWL for gastroplasty, principally vertical banded gastroplasty, and 70.1%EWL for biliopancreatic diversion. In dealing with the outcome data with one year follow up only, the study gave advantage to the stapling procedures as the weight loss generally peaks at this time for these procedures. However, for the LAGB procedures, weight loss continues for 2 -3 years and therefore assessment at one year was premature. Note that in this review gastroplasty, a procedure now largely discontinued, appeared to provide better weight loss than RYGB and was equivalent to BPD in this study.
Short-term (1 - 3 yr)outcomes: Chapman et al52 performed a systematic review of the literature available up to mid-2001 and compared the published reports on LAGB, RYGB and VBG. They found that, although LAGB showed less weight loss at 1 and 2 years, all three groups of procedures produced comparable weight loss at 3-4 years, that being the longest follow up available on LAGB at that time. They noted the lower mortality associated with LAGB.
Maggard et al53 reviewed the literature up to mid-2003 and identified 89 reports that provided weight loss data after bariatric surgery. Weight loss data were available at or beyond three years in 57 of these reports. Table 2 shows a summary of their weight loss findings for 3 or more years of follow up.
Table 2. Summary of published data providing three or more years of follow up (adapted from Maggard, 200553). Note that they identified only one report for laparoscopic RYGB and for BPD at that time.
| Procedure | Weight Loss (kg) | 95% CI | No. of Studies | No. of Patients |
| Open RYGB | 41.6 | 37.4 - 45.8 | 20 | 1266 |
| Laparoscopic RYGB | 38.2 | 28.0 - 48.6 | 1 | 15 |
| VBG | 32.0 | 27.7 - 36.4 | 18 | 1877 |
| LAGB | 34.8 | 29.5 - 40.1 | 17 | 3076 |
| BPD | 53.1 | 47.4 - 58.8 | 1 | 50 |
Based on the confidence intervals, only BPD appears to offer better weight loss than the other procedures.
Medium-term (3 - 10 year)outcomes: A single systematic review has focused on the medium term-term outcomes and thus included only reports that provided at least three year data58. A total of 43 reports were included, 18 related to LAGB, 18 related to RYGB and 7 on BPD or its DS variant. Figure 11 shows the %EWL for these three procedures.

Figure 11. The pattern of weight loss over time after RYGB, LAGB and BPD ( from reference 58, with permission).
The most significant single finding was that each of these procedures was effective in achieving substantial weight loss over the medium term. RYGB was significantly more effective than LAGB at years 1 and 2 but not beyond that point. BPD appeared to be more effective than the other procedures but the difference was shown to be statistically significant at 5 years follow up alone.
Long-Term ( >10 Year) Outcomes:
It is not hard to achieve substantial weight loss in the short term. Most lifestyle programs can do it. Showing weight loss after bariatric surgery in the short term cannot justify its costs or risks. The key weakness of the lifestyle programs is not their impotence but their lack of durability. For bariatric surgery to be worthwhile the effects must last. Yet most of the published studies deal with the short-term outcomes only.
Data published up to the end of 2011 on weight loss at 10 or more years after bariatric surgery have been subjected to systematic review59. At that time there were 9 reports of RYGB, 7 reports of LAGB, and 4 reports of BPD/DS with data beyond 10 years. No long-term outcome data for sleeve gastrectomy are yet available.
Table 3 summarizes the findings. Notably, all three procedures have generated substantial long-term weight loss. BPD/DS appears to be the most effective with 72% EWL. RYGB (54.0% EWL) and LAGB (54.2% EWL) are identical.
Table 3. Bariatric surgical procedures: systematic review of long-term outcomes (Adapted from ref 59).
| Procedure | RYGB | LAGB | BPD / DS |
| No. of Reports | 9 | 7 | 4 |
| No. of patients – initial | 3194 | 6369 | 3408 |
| Perioperative mortality | 21/2102 (1.01%) | 1/6369 (0.002%) | 27/3066 (1%) |
| % Follow up achieved | 64% | 82% | 83% |
| % EWL at 10+ yr: Weighted mean % EWL (range) |
54.0 (28-68) |
54.2 (33-64) |
71.7 (69-75) |
|
Revisional procedures: Range as % of total followed up |
8-38 |
8-60 |
NR |
NR = not reported. As perioperative mortality was not reported in all studies, the denominator for mortality rates may be lower than the initial number of patients.
The results emphasize the laudable effectiveness of all of these procedures, each of which has been able to maintain more than 50% EWL beyond 10 years. Bariatric surgery does work and RYGB and LAGB appear to be equal in the long-term.
Sleeve Gastrectomy Outcomes.
Adequate weight loss reporting for sleeve gastrectomy remains a problem with no long term data, poor medium term data and major loss to follow up in all series providing this information. Brethauer et al provided a literature review of the 36 published reports involving 2570 patients that were available in 200955. Only one report gave data beyond 3 years. There was a mean weight loss for the full group of 55% EWL. The only study exceeding three years showed a weight regain of approximately 40% from 3 - 5 years60.
In 2014, Diamantis et al61 reported a systematic review of the literature which included 16 studies, involving a total of 492 patients, which reported more than 5 year follow-up. There was 62% EWL at 5 years, 54% at 6 years, 43% at 7 years and 54% at 8 years. Not included in this review were the data from Weiner from Frankfurt who has a large series with longer follow up62. He has reported the outcomes of 746 patients with up to 8 year follow up. The %EWL was 59% at 2 yr, 45% at 5 yr and 36% at 8 yr.
Gastric imbrication is too new to know much of the benefits or risks. Very few reports of weight loss outcomes are available. Brethauer et al report a 53%EWL in 6 patients at one year63. The anticipated freedom from perioperative complications needs to be shown. Just as with sleeve gastrectomy, there is a high likelihood of failure of initial weight loss or weight regain in the medium term. It remains an experimental procedure at this time.
Type 2 Diabetes and Bariatric Surgery
Type 2 diabetes mellitus is the paradigm of an obesity-related illness and improved control for diabetes and other metabolic disorders represents the greatest potential strength for bariatric surgery. The metabolic effects of weight loss following bariatric surgery have been well documented and provide clinicians with an obvious path for the prevention and treatment64-66. The effects of bariatric surgery on type 2 diabetes have been subject of 2 systematic reviews64, 66. Buchwald reported 86% of 1835 patients from multiple case studies showed remission or improved control64. Maggard et al66 reviewed 21 case series and reported a range of 64% - 100% showing remission or improvement.
Sadly, few patients with diabetes have been offered this benefit to date. Arguments against the broad application of bariatric surgery have included: Safety concerns, lack of evidence from RCTs, insufficient understanding of mechanism, lack of proven durability of effect and lack of proven reduction of the complications of diabetes in association with long-term remission. All of these arguments are losing substance as new data continue to appear. However, not all have yet been answered fully. The question regarding long-term outcomes, in particular, needs to be better supported by data.
Safety.
The safety of bariatric surgery has improved greatly in recent years with almost no perioperative mortality risk associated with LAGB and, in expert centres, a mortality rate of 0.44% after RYGB5. Compared to the mortality risk of diabetes, bariatric surgery is a safe path.
Remission rates in RCTs.
Until recently, the lack of high levels of evidence has been the most valid criticism of reports of bariatric surgery and diabetes. The medical literature had been littered with numerous observational studies. These studies often carried major deficiencies including poor definition of both diabetes and its remission and extensive or unreported loss to follow up. Systematic reviews have sought to resolve these deficiencies through the pooling of data64, 66.
We have now been provided with at least five high-quality randomised controlled trials67-71. Their findings should over-ride the past observational studies. All studies have compared one or more bariatric procedures with a group having non-surgical treatment (NST). However the studies are not directly comparable due to extensive heterogeneity, including different criteria for patient selection, various treatment durations and various definitions of remission of diabetes, particularly the cut-off values for HbA1c. Nevertheless, they serve to provide key comparisons with NST, the current offering to more than 99% of people with diabetes.
The key outcome of remission of diabetes from each of these studies is shown in figure 12. The first of the studies was performed by our group at the Centre for Obesity Research and Education (CORE) in Melbourne67. 60 patients were randomised to LAGB or non-surgical therapy (NST). They were required to have a BMI between 30 and 40 and to have been known to have type 2 diabetes for less than 2 years. Inter alia, remission was defined as a HbA1c of less than 6.2%. The results reported were after 2 yr of follow up.
The study by Schauer et al68 from the Cleveland Clinic randomised 150 patients and included NST, RYGB and sleeve gastrectomy. They defined remission, in part, as a HbA1c of less than 6.0%. They reported their results at one year and then at 3 years72.
Mingrone et al69 randomised 60 patients to three groups – NST, RYGB or BPD by open laparotomy and reported their outcomes after a two year follow up. They used a HbA1c cut-off of 6.5% to define remission. They have recently reported the five year follow up of these groups of patients73.
Ikamuddin and coworkers70, from Minnesota and other sites, performed a similar study to the Cleveland Clinic study except for restricting it to a comparison of NST with RYGB. It involved 120 patients and was multicentered, including a group from Taiwan. They defined remission as HbA1c of less than 6.0% and to date have reported the one70 and two year74 follow up data of a planned 5 year study.
Courcoulas et al from Pittsburgh randomised 69 patients into 3 groups – NST, RYGB and LAGB. They defined partial and complete remission, in part, as HbA1c values of less than 6.5% and 5.7% respectively. They have published the one year71 and three year75 outcomes to date. The data shown in figure 11 below are for partial remission.

Figure 12. Comparison of remission of type 2 diabetes in five RCTs of bariatric surgical procedures versus non-surgical treatment.
Three of the studies included a RYGB arm and report remissions rates of 44% at one year in 2 studies68, 70 and 75% at two years in one study69. The remission rates for LAGB were 73% at two years67, for biliopancreatic diversion (BPD) 95% at two years69 and for VSG 37% at one year68. The studies contained sufficient heterogeneity to not permit head-to-head comparison between procedures but have established the significantly greater effectiveness of all bariatric surgical options over non-surgical treatment.
Mechanisms of effect.
This remains an area of contention and learning and is linked to the general study of mechanisms reviewed above. Optimal selection of the patients and the procedure requires more knowledge of what drives better outcomes. The two primary effects of bariatric procedures is an immediate and sustained reduction of nutritional intake and a reduction of insulin resistance associated with loss of weight. Additional factors include improvement on β cell function, changes in appetite and changes in the gut-associated incretins.
The relative effectiveness of different bariatric procedures has been subject to considerable debate. RYGB is associated with the incretin effects with increased release of GLP-1 and GIP from the distal gut, increasing β cell function, at least in the short term after the procedure76. However comparison with non-surgical patients taking a diet equivalent to the early post-operative RYGB patient suggests that an improvement in insulin resistance immediately after RYGB is primarily due to caloric restriction, and the enhanced incretin response after RYGB does not improve postprandial glucose homeostasis during this time77.
The improvement in diabetes following weight loss is, in part, related to the dual effects of improvement in insulin sensitivity and pancreatic beta-cell function78. As beta-cell function deteriorates progressively over time in those with type 2 diabetes, early weight loss intervention should therefore be a central part of initial therapy in severely obese subjects who develop type 2 diabetes. For obese patients with type 2 diabetes, weight loss provides benefit not equalled by any other therapy, and may prove to be the only therapy that substantially changes the natural history of the disease50, 79.
Durability of remission. Recent reports derived from several of the RCTs suggest a possible lack of durability of effect. For RYGB there has been reasonable stability for three of the four studies. In the Cleveland clinic study, at three years the remission rate for RYGB did not change significantly (43% to 38%). Partial remission for the RYGB patients in the Pittsburgh study changed from 50% at one year to 40% at three years. The multi-centered study based in Minnesota also showed stability of the RYGB effect between one and two years74. In contrast, for RYGB, the Rome study, found the remission rate fell from 75% at 2 years to 37% at 5 years, a relapse rate of 53%.
Sleeve gastrectomy was measured only in the Cleveland study and between one and three years it showed a marked fall from 37% to 24%, a 50% reduction72. The remission rate for the BPD group of the Rome study fell from 95% to 63% a 37% relapse rate73 between the two and five year study points. LAGB partial remission rate in the Pittsburgh study was unchanged at three years (27% at year 1 and 29% at year 3).In the Pittsburgh study, comparing the relative effect of RYGB and LAGB, complete remission of diabetes had occurred in 17% of RYGB patients and 23% of LAGB patients at year 3.
Macrovascular and microvascular complication rates.
The durability of remission and the reduction of complications has been demonstrated at a fifteen year follow up in the Swedish Obese Subjects (SOS) study, a prospective matched cohort study80. They reported that the remission rate for the surgical group, predominantly gastroplasty, at two years was 72% and at 15 years was 31%. This remission rate, though reduced with time, was significantly better than their control group and indicates an important long-term benefit. Furthermore, and arguably more important than the remission rate, they found the macrovascular and microvascular complications of diabetes were fewer at 15 years in the surgical group than in the controls.
Earlier surgery improves outcome.
The duration of diabetes prior to bariatric surgery is a consistent and strong predictor of remission72, 80. A commitment to substantial weight loss must be a component of the early management of the obese person with diabetes. In a recent report from the SOS study, the patient who had bariatric surgery within the 1st year of diagnosis had more than twice the likelihood of remission of diabetes at two years and approximately six times the likelihood of remission at 15 years when compared to the patients who was diagnosed more than 4 years before surgery. If an obese patient with diabetes cannot achieve substantial weight loss by lifestyle change, early referral for consideration of bariatric surgery is an essential responsibility of the health care provider.
OTHER HEALTH OUTCOMES AFTER BARIATRIC SURGERY
Dyslipidemia Of Obesity
Increased fasting triglyceride and decreased high-density lipoprotein (HDL)-cholesterol concentrations characterize the dyslipidemia of obesity and insulin resistance81. This dyslipidemic pattern is highly atherogenic and the most common pattern associated with coronary artery disease 82. Weight loss surgery produces substantial decreases in fasting triglyceride levels, an elevation of HDL-cholesterol levels to normal, and improved total cholesterol–to–HDL-cholesterol ratio 83-85. Although elevation of total cholesterol is not obesity-driven, hypercholesterolemia can be controlled by malabsorptive procedures such as BPD86 and, to a lesser extent, RYGB87.
Hypertension
There is evidence of a reduction in both systolic and diastolic blood pressure (BP) following weight loss in association with a bariatric procedure88. We have studied the outcome of 147 consecutive hypertensive patients at 12 months after LAGB. Preoperatively, only 17 of these patients had BP within the normal range, all on therapy. Hypertension was present in 130 patients preoperatively; 101 of these were taking antihypertensive medications and the remaining 29 were not on therapy. Mean BP for these patients was 156/97 mm Hg prior to surgery. At 12 months after LAGB, 105 patients had normal BP, 42 remained hypertensive, and only 42 were taking any antihypertensive medication at that time. Mean BP was 127/76 mm Hg. From these data, we found that 80 patients (55%) had resolution of the problem (i.e., normal BP and taking no antihypertensive therapy), 45 patients (31%) were improved (less therapy and easier control), and 22 patients (15%) were unchanged89. We have demonstrated that the fall in blood pressure is sustained to at least 4-years after surgery but durability of blood pressure reduction over a longer period is uncertain90.
Gastro Esophageal Reflux Disease (GERD)
GERD is much more common in the obese. The community adult prevalence is estimated to be 8-26% whereas 39 - 53% of an obese population have been shown to suffer the disease91. The reasons for this increase are still unclear but suggestions include reduced basal pressure at the lower esophageal sphincter, increased transient lower esophageal sphincter relaxations, hiatal hernia and abnormal motility patterns in the esophagus or stomach. Multiple observational studies of LAGB and RYGB have reported strong benefit with improvement in LES function, reduced acid reflux and a level of symptom relief comparable to standard anti-reflux surgery91, 92. However it appears that to date no RCTs have been performed directly comparing the two approaches in a well-constructed study. We studied 87 patients who had moderate or severe GERD and have been followed for at least 12 months after LAGB. 73 (89%) have had total resolution of the problem, as defined by the absence of symptoms without treatment for the previous month. Preoperative and postoperative pH study and manometry have been performed on 12 of these patients who had severe symptoms preoperatively. The mean DeMeester score was 38 +/- 15 preoperatively and 7.9 +/- 8 at follow-up (p<0.001). In all but one of these patients symptoms had resolved completely93.
Asthma
There is a positive relationship between asthma and obesity with a possible dose-response effect in evidence94, 95. The Nurses' Health study identified a five-fold increase in the relative risk of asthma with a weight gain of 25kg from age 18 when compared to a weight stable group96. In the setting of obesity, asthma is more difficult to control97, 98. Weight loss by non-surgical means has shown to improve asthma. In an RCT from Finland99 the weight loss group (14% of total body weight lost) showed improved lung function, symptoms, morbidity, and health status. However lack of durability of the weight loss programs limits the clinical application of the non-surgical approach.
Several observational studies have report major improvement in asthma after bariatric surgery. A study of 32 consecutive patient with asthma treated by LAGB reported improvement in all measured aspects of the disease, including symptoms, severity, need for asthma medications (including corticosteroids), and hospital admissions100. The asthma severity score fell from 44.5 before operation to 14.3 at 12 months after operation. Other studies have also reported benefit after LAGB101 and community studies indicate an important reduction of asthma medications after bariatric surgery in the state of Michigan at a time when RYGB was the dominant procedure 102, 103. No RCTs of bariatric surgery versus best medical care appear to have been performed to date.
Obstructive Sleep Apnea
A range of sleep disorders is associated with obesity. The most serious of these is obstructive sleep apnea (OSA). Severe obesity is the greatest risk factor for the development of sleep apnea, with a 10-fold increase in prevalence. Excessive daytime sleepiness, a disabling and potentially dangerous condition, is very common in the obese population and is not necessarily related to OSA104. There are major improvements in sleep quality, excessive daytime sleepiness, snoring, nocturnal choking, and observed OSA with weight loss following LAGB surgery105.
Obstructive sleep apnea and other sleep disturbances has been studied in 313 patients prior to LAGB and repeated at one year after operation in 123 of the patients105. There was a high prevalence of significantly disturbed sleep in both men (59%) and women (45%). Observed sleep apnea was decreased from 33% to 2%, habitual snoring from 82% to 14%, abnormal daytime sleepiness from 39% to 4% and poor sleep quality from 39% to 2%.
Depression
Depression is common in the morbidly obese. Does the presence of obesity cause the person to be depressed or does depression cause the person to eat too much? We have investigated the effect of weight loss induced by LAGB on depression as measured by the Beck Depression Inventory (BDI)106. Preoperative BDI on 487 consecutive patients was a mean of 17.7+/- 9.5, a level within the range for moderate depression. Weight loss was associated with a significant and sustained fall in BDI scores with a mean score of 7.8 +/- 6.5 (N=373) at one year after surgery. By four years after surgery, the 134 patients studied had lost 54% of excess weight and had a BDI of 9.6 +/- 7.7. Although a small number remained in the major depressive illness category, the shift of the majority to normal values for BDI strongly indicates that most of the depression of obesity is reactive to the problem of obesity rather than a cause of obesity and is resolved by weight loss.
IMPROVEMENT IN QUALITY OF LIFE
Improvement in QOL is one of the most gratifying outcomes of bariatric surgery. A number of studies clearly demonstrate major QOL improvements following bariatric procedures 107-111. A large prospective study of QOL after weight loss surgery employed the Medical Outcomes Trust Short Form-36 (SF-36). The SF-36 is a reliable, broadly used instrument that has been validated in obese people. In this study, 459 severely obese subjects had lower scores compared with community normal values for all 8 aspects of QOL measured, especially the physical health scores. Weight loss provided a dramatic and sustained improvement in all measures of the SF-36. Improvement was greater in those with more preoperative disability, and the extent of weight loss was not a good predictor of improved QOL. Mean scores returned to those of community normal values by 1 year after surgery, and remained in the normal range throughout the 4 years of the study. It is significant that patients who required revisional surgery during the follow-up period achieved the same improvement in measures of QOL. Similar improvements in QOL have been demonstrated in patients having LAGB for previously failed gastric stapling112.
Studies of appearance orientation and appearance evaluation indicate that the severely obese usually have quite normal pride and investment in their appearance and presentation but they evaluate their appearance as being very poor113. Weight loss following LAGB has been shown to produce major improvements in self-evaluation of appearance, although it does not return to community normal levels. The extent of the improvement in appearance is related to the percent of excess weight loss. The discrepancy between one’s pride and investment in appearance and presentation and one’s self evaluation of appearance is lower with weight loss, reducing psychological stress113.
IMPROVEMENT IN SURVIVAL
The ultimate test of effectiveness of a treatment is the reduction of mortality. A comparison of the long-term mortality of bariatric surgical patients with obese controls shows improved survival. A systematic review by Pontiroli and Morabito114 identified 6 relevant trials, shown in table 4. They reported a reduction of risk of global mortality with an odds ratio of 0.55 (95% CI = 0.49 - 0.63) . The overall survival advantage was not different between LAGB (odds ratio 0.57) and RYGB (odds ratio 0.55).
Table 4. The relative risk of mortality compared to controls after weight loss following bariatric surgery.
| Study | Operation | Relative Risk | Source of controls |
| Christou (2004)115 | RYGB | 0.11 | Medical |
| Flum (2004)51 | RYGB | 0.67 | Medical |
| SOS study (2007)116 | Various | 0.71 | Medical |
| Busetto (2007)117 | LAGB | 0.38 | Medical |
| Adams (2007)118 | RYGB | 0.40 | Community |
| Peeters (2007)119 | LAGB | 0.28 | Community |
The source of the control patients should be known and considered in the interpretation of the trials as sourcing a control group from a medical setting can preselect those who already have an existing disease that will shorten their life expectancy. As an example, the study by Christou115 used patients admitted to the Quebec provincial health insurance database with a diagnostic profile which included obesity as their control group. The reasons for admission were not known. This group had a mean age of 47 yr and had a mortality rate of 12.6 deaths per 1000 patient.years. By comparison, the Peeters' study (mean age of 55 yr) and Adams study (mean age of 39 yr) used true community controls and had mortality rates for the controls of 8.6 and 5.7 deaths per 1000 patient.years respectively. Adjusting for age, the mortality rate or the Christou patients was approximately twice those using community control, suggesting they included a number with late-stage illness
The study by Adams118 had sufficient numbers and details to provide some sub-group analysis. They found that mortality was significantly reduced for deaths from diabetes, heart disease and cancer whereas the rates of death from accidents and suicide were higher in the surgical group.
In spite of some weaknesses in the data and the inability to do a matched controlled study, the consistent pattern of the various reports indicates that global mortality is reduced in association with weight loss after RYGB and LAGB
THE HIERARCHY OF OBESITY THERAPIES
Resolution of the disease of obesity requires substantial and durable weight loss. The therapeutic options available are listed in table 5 in order of their risk, side effects, invasiveness and costs. We should always begin with the simplest and safest and work down the list. Only when a simpler and safer option has failed should we seek a more complex or risky option.
Table 5. Hierarchy of weight loss techniques in order of rating for invasiveness /risk / complexity /potential problems.
| Therapy | Rating |
| Lifestyle - Eat less and do more | 1.0 |
|
Cognitive behavioural therapy Very low calorie diets Drug therapy |
2.0 |
| Endoscopic - gastric balloons et al | 4.0 |
| Laparoscopic adjustable gastric banding (LAGB) | 5.0 |
| Sleeve gastrectomy | 6.0 |
| Laparoscopic gastric bypass (RYGB and SAGB) | 7.0 |
| Open RYGB | 8.0 |
| Open Biliopancreatic diversion (BPD) | 9.0 |
| Laparoscopic BPD | 10.0 |
Lifestyle therapies (diet, exercise, behavioral change) should always be the first line of management. Multiple RCTs have shown that a modest weight loss of between 2 and 5 kg can be achieved at 12 months120-123. This level of weight loss is associated with a clinically valuable reduction of metabolic risk124-126 but generally will not solve the problems of obesity. Lifestyle therapies should be applied optimally and sought to be maintained permanently. If however they fail to resolve the obese patient’s problems, the next level of therapy should be considered. Current drug therapies add little further benefit127. Very low energy diets can be effective if taken correctly but are inevitably short term. The recent versions of the intragastric balloon have yet to show effectiveness by RCT and remain short term options. In spite of vigorous research effort, no additional endoscopic approaches are yet available which can provide even medium term benefit.
In general, this table provides a path along which the obese patient seeking treatment should travel. The procedure of gastric imbrication has not been placed on the list as its safety and effectiveness are not yet known.
Comparison of bariatric surgery with non-surgical therapies
We have performed three randomised controlled trials (RCT) in which we have compared gastric banding with optimal non-surgical programs. The initial RCT was of mild to moderately obese adults (BMI 30-35). We compared optimal non-surgical therapy, including lifestyle measures, drug therapy and very low energy diets with the gastric band and showed significantly better weight loss, health and quality of life for the banding group128. Adverse events were similar between groups. The gastric band patients had lost 86% of their excess weight (%EWL) compared to 21% EWL in the non-surgical group. The effects are durable as the substantial weight loss, health benefits and improved quality of life have remained at 10 yr follow up129.

Figure 13. RCT outcomes at ten years (adapted from reference 129). Weight loss outcomes for the surgical group for the ten year period are shown by the continuous line. After completion of the two year randomised study, 17 of 40 patients in the non-surgical arm elected to cross-over to the LAGB. Their weight loss following cross-over is shown by the dotted line.
The second study was of obese adults (BMI 30 – 40) with type 2 diabetes. There was 73% remission rate of diabetes in the gastric band group and 13% in the lifestyle group130. Again, the metabolic syndrome was significantly improved in the banding group alone. The third RCT was of obese adolescents (BMI > 35; age 14 -18yr)131. The gastric band group lost 79% of their excess weight and showed a significant improvement on the metabolic syndrome which reduced from 36% to zero. There was also an improved quality of life.
Comparison between bariatric surgical procedures.
Table 6 lists a range of comparators and the position of each current option against these comparators.
Table 6. Comparison of attributes of the principal bariatric procedures
| Attribute | LAGB | RYGB | Sleeve Gastrectomy | BPD +/- DS |
| Safe | +++ | ++ | ++ | + |
| Weight loss: 1. Short term 2. Medium term3. Long term |
++ ++ ++ |
+++ ++ ++ |
+++++ Not known |
+++ |
| Durable | ++ | ++ | ? | ++ |
| Side effects | ++ | ++ | + | ++ |
| Reversible easily | Yes | No | No | No |
| Minimally invasive | +++ | ++ | ++ | + |
| Controllable/adjustable | Yes | No | No | No |
| Low revision rate | + | ++ | ? | ++ |
| Requires follow up | +++ | ++ | ++ | ++ |
The key outcome comparators between procedures are weight loss and safety. Short term data (<3 yr) are largely irrelevant as durability is essential. Systematic review of the medium term weight loss outcomes have shown no difference between RYGB and LAGB and suggest there is a better weight loss to be achieved with biliopancreatic diversion. Figure 10 shows the relative % EWL for the three principal procedures. Long term (> 10 years) weight loss as shown in table 3 also appears to be similar for RYGB and LAGB but several studies have a major potential for bias, particularly those with low numbers of patients.
WHO SHOULD BE CONSIDERED AND WHO SHOULD NOT?
BMI Criteria
There is level 1 evidence supporting a better outcome for using LAGB in the mild to moderately obese (BMI 30-35) when compared with lifestyle therapy128, 130. This approach is cost-effective132, 133. When the two treatment paths are modelled over time, the LAGB approach is dominant i.e. it provides increased number of quality-adjusted life years at a lower cost than the existing option of non-surgical therapies. Any person who is obese (BMI > 30), is suffering from the medical, physical or psychosocial consequences of the obesity and has diligently sought a solution through a range of lifestyle options over time, should be considered for LAGB.
Because the stapling group of surgical options lack level 1 data, are of greater risk, and are not controllable or reversible, maintenance of existing cut-off of BMI >40 or BMI > 35 with major comorbidities should remain for these procedures.
Age criteria.
We now have the results of a RCT showing the clear benefit for LAGB for the obese teenager134 and we offer this option to severely obese teens from age 14 years. We do not consider offering an irreversible procedure to teens when a safe, effective and reversible option is available.
We are generally hesitant to offer bariatric surgery above the age of 65 but do at least consider people between age 65 - 70 who do not have chronic cardiovascular or pulmonary disease.
Contraindications.
Bariatric surgery is contraindicated in those with portal hypertension. Sleeve gastrectomy is considered by many to be contraindicated in those with severe gastro-esophageal reflux disease. LAGB is unsuitable for those who are mentally defective or otherwise unable to engage in the “partnership” needed for optimal outcome. Because of its particular aftercare needs, LAGB is also contraindicated in those who live remote from a follow-up center.
NEEDS AND CHALLENGES
Bariatric surgery is never a quick fix. It is a process of care that begins with a careful initial clinical evaluation and detailed patient education and it continues beyond the operative procedure through a permanent follow up. All procedures have the potential for perioperative complications and death. Revisional surgery is relatively common as maintenance of the correct anatomy is intrinsic to effectiveness. But bariatric surgery does provide a solution to the problem of obesity. It achieves substantial weight loss, improved health and quality of life and a longer life. We need to optimize these benefits and minimize the risks and the costs. The following are some of the areas for further research and development:
- A better understanding of the mechanisms of action of each procedure is required to enable optimum surgery and follow up.
- Accurate and comprehensive data management for all patients. Bariatric surgical procedures should be incorporated into national clinical registries to enable objective assessment of the risks and benefits across the community.
- More randomized controlled trials to define the benefits of weight loss on various comorbidities of obesity. More study is needed in particular for the patients with metabolic diseases – type 2 diabetes, metabolic syndrome, non-alcoholic steatohepatitis, the dyslipidaemias, polycystic ovary syndrome and obstructive sleep apnoea.
- Better definition of who is suitable and who is not suitable for bariatric surgery.
- Definition of safe and efficient pathways for assessment, surgery and aftercare.
- Cost-effectiveness evaluation of the bariatric surgical approach to disease management in comparison with existing options.
Bariatric surgery has the potential to be one of the most important and powerful treatment approaches in medicine. High quality of clinical care, good science and comprehensive data management will allow optimal application of this approach to be realized.
REFERENCES
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, Farzadfar F, Riley LM, Ezzati M. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557-567
- Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the united states: Prevalence and trends, 1960-1994. International Journal of Obesity and Related Metabolic Disorders. 1998;22:39-47
- Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among us adults, 1999-2008. Journal of the American Medical Association. 2010;303:235-241
- Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the uk. Lancet. 2011;378:815-825
- Flum DR, Belle SH, King WC, Wahed AS, Berk P, Chapman W, Pories W, Courcoulas A, McCloskey C, Mitchell J, Patterson E, Pomp A, Staten MA, Yanovski SZ, Thirlby R, Wolfe B. Perioperative safety in the longitudinal assessment of bariatric surgery. New England Journal of Medicine. 2009;361:445-454
- Kremen A, Linner JH, Nelson CH. An experimental evaluation of the nutritional importance of proximal and distal small intestine. Annals of Surgery. 1954;140:439-444
- Jorizzo JL, Apisarnthanarax P, Subrt P, Hebert AA, Henry JC, Raimer SS, Dinehart SM, Reinarz JA. Bowel-bypass syndrome without bowel bypass. Bowel-associated dermatosis-arthritis syndrome. Archives of Internal Medicine. 1983;143:457-461
- O'Leary JP. Hepatic complications of jejunoileal bypass. Seminars in Liver Disease. 1983;3:203-215
- Corrodi P. Jejunoileal bypass: Change in the flora of the small intestine and its clinical impact. Reviews of Infectious Diseases. 1984;6 S80-84
- Parfitt AM, Podenphant J, Villanueva AR, Frame B. Metabolic bone disease with and without osteomalacia after intestinal bypass surgery: A bone histomorphometric study. Bone. 1985;6:211-220
- DeWind LT, Payne JH. Intestinal bypass surgery for morbid obesity. Long-term results. Journal of the American Medical Association. 1976;236:2298-2301
- Mason EE, Ito C. Gastric bypass in obesity. Surgical Clinics of North America. 1967;47:1345-1351
- Printen KJ, Mason EE. Gastric surgery for relief of morbid obesity. Archices of Surgery. 1973;106:428-431
- Mason EE. Vertical banded gastroplasty for obesity. Archices of Surgery. 1982;117:701-706
- Hall JC, Watts JM, O'Brien PE, Dunstan RE, Walsh JF, Slavotinek AH, Elmslie RG. Gastric surgery for morbid obesity. The adelaide study. Annals of Surgery. 1990;211:419-427
- Pories WJ, Flickinger EG, Meelheim D, Van Rij AM, Thomas FT. The effectiveness of gastric bypass over gastric partition in morbid obesity: Consequence of distal gastric and duodenal exclusion. Annals of Surgery. 1982;196:389-399.
- Sugerman HJ, Starkey JV, Birkenhauer R. A randomized prospective trial of gastric bypass versus vertical banded gastroplasty for morbid obesity and their effects on sweets versus non- sweets eaters. Annals of Surgery. 1987;205:613-624
- Sugerman HJ, Londrey GL, Kellum JM, Wolf L, Liszka T, Engle KM, Birkenhauer R, Starkey JV. Weight loss with vertical banded gastroplasty and roux-y gastric bypass for morbid obesity with selective versus random assignment. American Journal of Surgery. 1989;157:93-102
- Scopinaro N, Gianetta E, Civalleri D, Bonalumi U, Bachi V. Bilio-pancreatic bypass for obesity: Ii. Initial experience in man. British Journal of Surgery. 1979;66:618-620
- Scopinaro N, Gianetta E, Adami GF, Friedman D, Traverso E, Marinari GM, Cuneo S, Vitale B, Ballari F, Colombini M, Baschieri G, Bachi V. Biliopancreatic diversion for obesity at eighteen years. Surgery. 1996;119:261-268
- Marceau P, Biron S, Bourque RA, Potvin M, Hould FS, Simard S. Biliopancreatic diversion with a new type of gastrectomy. Obesity Surgery. 1993;3:29-35
- Marceau P, Hould FS, Simard S, Lebel S, Bourque RA, Potvin M, Biron S. Biliopancreatic diversion with duodenal switch. World Journal of Surgery. 1998;22:947-954
- Wittgrove AC, Clark GW, Schubert KR. Laparoscopic gastric bypass, roux-en-y: Technique and results in 75 patients with 3-30 months follow-up. Obesity Surgery. 1996;6:500-504
- Ren CJ, Patterson E, Gagner M. Early results of laparoscopic biliopancreatic diversion with duodenal switch: A case series of 40 consecutive patients. Obesity Surgery. 2000;10:514-523; discussion 524
- Belachew M, Legrand MJ, Defechereux TH, Burtheret MP, Jacquet N. Laparoscopic adjustable silicone gastric banding in the treatment of morbid obesity. A preliminary report. Surgical Endoscopy. 1994;8:1354-1356
- Belachew M, Legrand MJ, Vincent V. History of lap-band: From dream to reality. Obesity Surgery. 2001;11:297-302.
- Almogy G, Crookes PF, Anthone GJ. Longitudinal gastrectomy as a treatment for the high-risk super-obese patient. Obes Surg. 2004;14:492-497
- Buchwald H, Oien DM. Metabolic/bariatric surgery worldwide 2008. Obesity Surgery. 2009;19:1605-1611
- Szinicz G, Schnapka G. A new method in the surgical treatment of disease. Acta Chirirgica Austrica. 1982;Supp 43
- Kuzmak LI. A review of seven years' experience with silicone gastric banding. Obesity Surgery. 1991;1:403-408
- Wittgrove A, Clark GW, Tremblay LJ. Laparoscopic gastric bypass, roux-en-y: Preliminary report of five cases. Obesity Surgery. 1994;4:353-357
- Cobourn C, Mumford D, Chapman MA, Wells L. Laparoscopic gastric banding is safe in outpatient surgical centers. Obesity Surgery. 2010;20:415-422
- Chevallier JM, Arman GA, Guenzi M, Rau C, Bruzzi M, Beaupel N, Zinzindohoue F, Berger A. One thousand single anastomosis (omega loop) gastric bypasses to treat morbid obesity in a 7-year period: Outcomes show few complications and good efficacy. Obes Surg. 2015;25:951-958
- Lee WJ, Lin YH. Single-anastomosis gastric bypass (sagb): Appraisal of clinical evidence. Obes Surg. 2014;24:1749-1756
- Lee WJ, Ser KH, Lee YC, Tsou JJ, Chen SC, Chen JC. Laparoscopic roux-en-y vs. Mini-gastric bypass for the treatment of morbid obesity: A 10-year experience. Obes Surg. 2012;22:1827-1834
- Rutledge R, Walsh TR. Continued excellent results with the mini-gastric bypass: Six-year study in 2,410 patients. Obesity Surgery. 2005;15:1304-1308
- Lee WJ, Yu PJ, Wang W, Chen TC, Wei PL, Huang MT. Laparoscopic roux-en-y versus mini-gastric bypass for the treatment of morbid obesity: A prospective randomized controlled clinical trial. Ann Surg. 2005;242:20-28
- Scopinaro N, Adami GF, Marinari GM, Gianetta E, Traverso E, Friedman D, Camerini G, Baschieri G, Simonelli A. Biliopancreatic diversion. World Journal of Surgery. 1998;22:936-946
- Miras AD, le Roux CW. Mechanisms underlying weight loss after bariatric surgery. Nat Rev Gastroenterol Hepatol. 2013;10:575-584
- Brolin RE, Kenler HA, Gorman JH, Cody RP. Long-limb gastric bypass in the superobese. A prospective randomized study. Annals of Surgery. 1992;215:387-395
- Dixon AF, Dixon JB, O'Brien P E. Laparoscopic adjustable gastric banding induces prolonged satiety: A randomised blind crossover study. J Clin Endocrinol Metab. 2004
- Colles SL, Dixon JB, O'Brien PE. Hunger control and regular physical activity facilitate weight loss after laparoscopic adjustable gastric banding. Obesity Surgery. 2008;18:833-840
- Burton PR, Brown WA, Laurie C, Richards M, Hebbard G, O'Brien PE. Effects of gastric band adjustments on intraluminal pressure. Obesity Surgery. 2009;19:1508-1514
- Pournaras DJ, Le Roux CW. The effect of bariatric surgery on gut hormones that alter appetite. Diabetes & Metabolism. 2009;35:508-512
- Berthoud HR. Vagal and hormonal gut-brain communication: From satiation to satisfaction. Neurogastroenterol Motil. 2008;20 Suppl 1:64-72
- Zagorodnyuk VP, Chen BN, Brookes SJ. Intraganglionic laminar endings are mechano-transduction sites of vagal tension receptors in the guinea-pig stomach. Journal of Physiology. 2001;534:255-268
- Burton PR, Brown WA, Laurie C, Hebbard G, O'Brien PE. Criteria for assessing esophageal motility in laparoscopic adjustable gastric band patients: The importance of the lower esophageal contractile segment. Obes Surg. 2010;20:316-325
- Scopinaro N. Biliopancreatic diversion: Mechanisms of action and long-term results. Obesity Surgery. 2006;16:683-689
- Adami GF, Cordera R, Camerini G, Marinari GM, Scopinaro N. Long-term normalization of insulin sensitivity following biliopancreatic diversion for obesity. Int J Obes Relat Metab Disord. 2004;28:671-673
- Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, Barakat HA, deRamon RA, Israel G, Dolezal JM, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Annals of Surgery. 1995;222:339-350; discussion 350-332
- Flum DR, Dellinger EP. Impact of gastric bypass operation on survival: A population-based analysis. Journal of the American College of Surgeons. 2004;199:543-551
- Chapman A, Kiroff G, Game P, Foster B, O'Brien P, Ham J, Maddern G. Laparoscopic adjustable gastric banding in the treatment of obesity: A systematic review. Surgery. 2004;135:326-351
- Maggard MA, Shugarman LR, Suttorp M, Maglione M, Sugerman HJ, Livingston EH, Nguyen NT, Li Z, Mojica WA, Hilton L, Rhodes S, Morton SC, Shekelle PG. Meta-analysis: Surgical treatment of obesity. Annals of Internal Medicine. 2005;142:547-559
- Parikh M, Issa R, McCrillis A, Saunders JK, Ude-Welcome A, Gagner M. Surgical strategies that may decrease leak after laparoscopic sleeve gastrectomy: A systematic review and meta-analysis of 9991 cases. Ann Surg. 2013;257:231-237
- Brethauer SA, Hammel JP, Schauer PR. Systematic review of sleeve gastrectomy as staging and primary bariatric procedure. Surg Obes Relat Dis. 2009;5:469-475
- Aurora AR, Khaitan L, Saber AA. Sleeve gastrectomy and the risk of leak: A systematic analysis of 4,888 patients. Surg Endosc. 2011
- Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K. Bariatric surgery: A systematic review and meta-analysis. Journal of the American Medical Association. 2004;292:1724-1737
- O'Brien PE, McPhail T, Chaston TB, Dixon JB. Systematic review of medium-term weight loss after bariatric operations. Obesity Surgery. 2006;16:1032-1040
- O'Brien P, McDonald L, Anderson M, Brennan L, Brown WA. Long term outcomes after bariatric surgery: Fifteen year follow up after gastric banding and a systematic review of the literature. Annals of Surgery. 2013;257:87-94
- Weiner RA, Weiner S, Pomhoff I, Jacobi C, Makarewicz W, Weigand G. Laparoscopic sleeve gastrectomy--influence of sleeve size and resected gastric volume. Obes Surg. 2007;17:1297-1305
- Diamantis T, Apostolou KG, Alexandrou A, Griniatsos J, Felekouras E, Tsigris C. Review of long-term weight loss results after laparoscopic sleeve gastrectomy. Surg Obes Relat Dis. 2014;10:177-183
- Weiner R. Sleeve gatrectomy - late results. Obesity Surgery. 2010;20:O-031
- Brethauer SA, Harris JL, Kroh M, Schauer PR. Laparoscopic gastric plication for treatment of severe obesity. Surgery for Obesity and Related Diseases. 2011;7:15-22
- Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, Bantle JP, Sledge I. Weight and type 2 diabetes after bariatric surgery: Systematic review and meta-analysis. American Journal of Medicine. 2009;122:248-256 e245
- Dixon JB, le Roux CW, Rubino F, Zimmet P. Bariatric surgery for type 2 diabetes. Lancet. 2012;379:2300-2311
- Maggard-Gibbons M, Maglione M, Livhits M, Ewing B, Maher AR, Hu J, Li Z, Shekelle PG. Bariatric surgery for weight loss and glycemic control in nonmorbidly obese adults with diabetes: A systematic review. JAMA. 2013;309:2250-2261
- Dixon JB, O'Brien PE, Playfair J, Chapman L, Schachter LM, Skinner S, Proietto J, Bailey M, Anderson M. Adjustable gastric banding and conventional therapy for type 2 diabetes: A randomized controlled trial. Journal of the American Medical Association. 2008;299:316-323
- Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, Thomas S, Abood B, Nissen SE, Bhatt DL. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med. 2012;366:1567-1576
- Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L, Nanni G, Pomp A, Castagneto M, Ghirlanda G, Rubino F. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med. 2012;366:1577-1585
- Ikramuddin S, Korner J, Lee WJ, Connett JE, Inabnet WB, Billington CJ, Thomas AJ, Leslie DB, Chong K, Jeffery RW, Ahmed L, Vella A, Chuang LM, Bessler M, Sarr MG, Swain JM, Laqua P, Jensen MD, Bantle JP. Roux-en-y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: The diabetes surgery study randomized clinical trial. JAMA. 2013;309:2240-2249
- Courcoulas AP, Goodpaster BH, Eagleton JK, Belle SH, Kalarchian MA, Lang W, Toledo FG, Jakicic JM. Surgical vs medical treatments for type 2 diabetes mellitus: A randomized clinical trial. JAMA surgery. 2014;149:707-715
- Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Brethauer SA, Navaneethan SD, Aminian A, Pothier CE, Kim ES, Nissen SE, Kashyap SR. Bariatric surgery versus intensive medical therapy for diabetes--3-year outcomes. N Engl J Med. 2014;370:2002-2013
- Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Nanni G, Castagneto M, Bornstein S, Rubino F. Bariatric-metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. Lancet. 2015;386:964-973
- Ikramuddin S, Billington CJ, Lee WJ, Bantle JP, Thomas AJ, Connett JE, Leslie DB, Inabnet WB, 3rd, Jeffery RW, Chong K, Chuang LM, Sarr MG, Jensen MD, Vella A, Ahmed L, Belani K, Schone JL, Olofson AE, Bainbridge HA, Laqua PS, Wang Q, Korner J. Roux-en-y gastric bypass for diabetes (the diabetes surgery study): 2-year outcomes of a 5-year, randomised, controlled trial. The lancet. Diabetes & endocrinology. 2015;3:413-422
- Courcoulas AP, Belle SH, Neiberg RH, Pierson SK, Eagleton JK, Kalarchian MA, DeLany JP, Lang W, Jakicic JM. Three-year outcomes of bariatric surgery vs lifestyle intervention for type 2 diabetes mellitus treatment: A randomized clinical trial. JAMA surgery. 2015;150:931-940
- Laferrere B. Effect of gastric bypass surgery on the incretins. Diabetes & Metabolism. 2009;35:513-517
- Isbell JM, Tamboli RA, Hansen EN, Saliba J, Dunn JP, Phillips SE, Marks-Shulman PA, Abumrad NN. The importance of caloric restriction in the early improvements in insulin sensitivity after roux-en-y gastric bypass surgery. Diabetes Care. 2010;33:1438-1442
- Dixon JB, Dixon AF, O'Brien PE. Improvements in insulin sensitivity and beta-cell function (homa) with weight loss in the severely obese. Diabetic Medicine. 2003;20:127-134
- Pinkney JH, Sjostrom CD, Gale EA. Should surgeons treat diabetes in severely obese people? Lancet. 2001;357:1357-1359.
- Sjostrom L, Peltonen M, Jacobson P, Ahlin S, Andersson-Assarsson J, Anveden A, Bouchard C, Carlsson B, Karason K, Lonroth H, Naslund I, Sjostrom E, Taube M, Wedel H, Svensson PA, Sjoholm K, Carlsson LM. Association of bariatric surgery with long-term remission of type 2 diabetes and with microvascular and macrovascular complications. JAMA. 2014;311:2297-2304
- Despres J. The insulin resistance-dyslipidemia syndrome: The most prevalent cause of coronary artery disease. Canadian Medical Association Journal. 1993;148:1339-1340
- Koba S, Hirano T, Sakaue T, Sakai K, Kondo T, Yorozuya M, Suzuki H, Murakami M, Katagiri T. Role of small dense low-density lipoprotein in coronary artery disease patients with normal plasma cholesterol levels. Journal of Cardiology. 2000;36:371-378.
- Busetto L, Pisent C, Rinaldi D, Longhin PL, Segato G, De Marchi F, Foletto M, Favretti F, Lise M, Enzi G. Variation in lipid levels in morbidly obese patients operated with the lap-band adjustable gastric banding system: Effects of different levels of weight loss. Obesity Surgery. 2000;10:569-577.
- Bacci V, Basso MS, Greco F, Lamberti R, Elmore U, Restuccia A, Perrotta N, Silecchia G, Bucci A. Modifications of metabolic and cardiovascular risk factors after weight loss induced by laparoscopic gastric banding. Obesity Surgery. 2002;12:77-82.
- Dixon J, O'Brien P. Ovarian dysfunction, androgen excess and neck circumference in obese women: Changes with weight loss (abstract). Obesity Surgery. 2002;12:193
- Scopinaro N, Adami GF, Marinari GM, Gianetta E, Traverso E, Friedman D, Camerini G, Baschieri G, Simonelli A. Biliopancreatic diversion. World J Surg. 1998;22:936-946.
- Brolin RE, Bradley LJ, Wilson AC, Cody RP. Lipid risk profile and weight stability after gastric restrictive operations for morbid obesity. Journal of Gastrointestinal Surgery. 2000;4:464-469.
- Sjostrom CD, Lissner L, Wedel H, Sjostrom L. Reduction in incidence of diabetes, hypertension and lipid disturbances after intentional weight loss induced by bariatric surgery: The sos intervention study. Obesity Research. 1999;7:477-484
- Dixon JB, O'Brien PE. Changes in comorbidities and improvements in quality of life after lap-band placement. American Journal of Surgery. 2002;184:S51-54
- Sjostrom CD, Peltonen M, Wedel H, Sjostrom L. Differentiated long-term effects of intentional weight loss on diabetes and hypertension. Hypertension. 2000;36:20-25
- Sise A, Friedenberg FK. A comprehensive review of gastroesophageal reflux disease and obesity. Obesity Reviews. 2008;9:194-203
- Ikramuddin S. Surgical management of gastroesophageal reflux disease in obesity. Digestive Diseases and Sciences. 2008;53:2318-2329
- Dixon JB, O'Brien PE. Gastroesophageal reflux in obesity: The effect of lap-band placement. Obes Surg. 1999;9:527-531
- Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: A meta-analysis of prospective epidemiologic studies. American Journal of Respiratory and Critical Care Medicine. 2007;175:661-666
- Young SY, Gunzenhauser JD, Malone KE, McTiernan A. Body mass index and asthma in the military population of the northwestern united states. Archives of Internal Medicine. 2001;161:1605-1611.
- Camargo C, Weiss S, Zhang S, Willett W, Speizer F. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch Intern Med. 1999;159:2582-2588
- Lavoie KL, Bacon SL, Labrecque M, Cartier A, Ditto B. Higher bmi is associated with worse asthma control and quality of life but not asthma severity. Respiratory Medicine. 2006;100:648-657
- Saint-Pierre P, Bourdin A, Chanez P, Daures JP, Godard P. Are overweight asthmatics more difficult to control? Allergy. 2006;61:79-84
- Stenius-Aarniala B, Poussa T, Kvarnstrom J, Gronlund EL, Ylikahri M, Mustajoki P. Immediate and long term effects of weight reduction in obese people with asthma: Randomised controlled study. British Medical Journal. 2000;320:827-832
- Dixon JB, Chapman L, O'Brien P. Marked improvement in asthma after lap-band surgery for morbid obesity. Obes Surg. 1999;9:385-389
- Maniscalco M, Zedda A, Faraone S, Cerbone MR, Cristiano S, Giardiello C, Sofia M. Weight loss and asthma control in severely obese asthmatic females. Respiratory Medicine. 2008;102:102-108
- Sikka N, Wegienka G, Havstad S, Genaw J, Carlin AM, Zoratti E. Respiratory medication prescriptions before and after bariatric surgery. Annals of Allergy, Asthma, & Immunology. 2010;104:326-330
- Reddy RC, Baptist AP, Fan Z, Carlin AM, Birkmeyer NJO. The effects of bariatric surgery on asthma severity. Obesity Surgery. 2011;21:200-206
- Vgontzas AN, Papanicolaou DA, Bixler EO, Hopper K, Lotsikas A, Lin HM, Kales A, Chrousos GP. Sleep apnea and daytime sleepiness and fatigue: Relation to visceral obesity, insulin resistance, and hypercytokinemia. Journal of Clinical Endocrinology & Metabolism. 2000;85:1151-1158.
- Dixon JB, Schachter LM, O'Brien PE. Sleep disturbance and obesity: Changes following surgically induced weight loss. Archives of Internal Medicine. 2001;161:102-106
- Dixon JB, Dixon ME, O'Brien PE. Depression in association with severe obesity: Changes with weight loss. Archives of Internal Medicine. 2003;163:2058-2065
- Dixon JB. Elevated homocysteine with weight loss. Obesity Surgery. 2001;11:537-538.
- Weiner R, Datz M, Wagner D, Bockhorn H. Quality-of-life outcome after laparoscopic adjustable gastric banding for morbid obesity. Obesity Surgery. 1999;9:539-545.
- Schok M, Geenen R, van Antwerpen T, de Wit P, Brand N, van Ramshorst B. Quality of life after laparoscopic adjustable gastric banding for severe obesity: Postoperative and retrospective preoperative evaluations. Obesity Surgery. 2000;10:502-508.
- Balsiger BM, Kennedy FP, Abu-Lebdeh HS, Collazo-Clavell M, Jensen MD, O'Brien T, Hensrud DD, Dinneen SF, Thompson GB, Que FG, Williams DE, Clark MM, Grant JE, Frick MS, Mueller RA, Mai JL, Sarr MG. Prospective evaluation of roux-en-y gastric bypass as primary operation for medically complicated obesity [see comments]. Mayo Clinic Proceedings. 2000;75:673-680
- Horchner R, Tuinebreijer MW, Kelder PH. Quality-of-life assessment of morbidly obese patients who have undergone a lap-band operation: 2-year follow-up study. Is the mos sf- 36 a useful instrument to measure quality of life in morbidly obese patients? Obesity Surgery. 2001;11:212-218; discussion 219.
- O'Brien P, Brown W, Dixon J. Revisional surgery for morbid obesity- conversion to the lap-band system. Obesity Surgery. 2000;10:557-563.
- Dixon JB, Dixon ME, O'Brien PE. Body image: Appearance orientation and evaluation in the severely obese. Changes with weight loss. Obesity Surgery. 2002;12:65-71.
- Pontiroli AE, Morabito A. Long-term prevention of mortality in morbid obesity through bariatric surgery. A systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann Surg. 2011;253:484-487
- Christou NV, Sampalis JS, Liberman M, Look D, Auger S, McLean AP, MacLean LD. Surgery decreases long-term mortality, morbidity, and health care use in morbidly obese patients. Annals of Surgery. 2004;240:416-423;
- Sjostrom L, Narbro K, Sjostrom CD, Karason K, Larsson B, Wedel H, Lystig T, Sullivan M, Bouchard C, Carlsson B, Bengtsson C, Dahlgren S, Gummesson A, Jacobson P, Karlsson J, Lindroos AK, Lonroth H, Naslund I, Olbers T, Stenlof K, Torgerson J, Agren G, Carlsson LM. Effects of bariatric surgery on mortality in swedish obese subjects. New England Journal of Medicine. 2007;357:741-752
- Busetto L, Mirabelli D, Petroni ML, Mazza M, Favretti F, Segato G, Chiusolo M, Merletti F, Balzola F, Enzi G. Comparative long-term mortality after laparoscopic adjustable gastric banding versus nonsurgical controls. Surg Obes Relat Dis. 2007;3:496-502; discussion 502
- Adams TD, Gress RE, Smith SC, Halverson RC, Simper SC, Rosamond WD, Lamonte MJ, Stroup AM, Hunt SC. Long-term mortality after gastric bypass surgery. New England Journal of Medicine. 2007;357:753-761
- Peeters A, O'Brien PE, Laurie C, Anderson M, Wolfe R, Flum D, MacInnis RJ, English DR, Dixon J. Substantial intentional weight loss and mortality in the severely obese. Annals of Surgery. 2007;246:1028-1033
- Bravata DM, Sanders L, Huang J, Krumholz HM, Olkin I, Gardner CD. Efficacy and safety of low-carbohydrate diets: A systematic review. Journal of the American Medical Association. 2003;289:1837-1850
- Dansinger ML, Gleason JA, Griffith JL, Selker HP, Schaefer EJ. Comparison of the atkins, ornish, weight watchers, and zone diets for weight loss and heart disease risk reduction: A randomized trial. Journal of the American Medical Association. 2005;293:43-53
- Sacks FM, Bray GA, Carey VJ, Smith SR, Ryan DH, Anton SD, McManus K, Champagne CM, Bishop LM, Laranjo N, Leboff MS, Rood JC, de Jonge L, Greenway FL, Loria CM, Obarzanek E, Williamson DA. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. The New England journal of medicine. 2009;360:859-873
- Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, Greenberg I, Golan R, Fraser D, Bolotin A, Vardi H, Tangi-Rozental O, Zuk-Ramot R, Sarusi B, Brickner D, Schwartz Z, Sheiner E, Marko R, Katorza E, Thiery J, Fiedler GM, Bluher M, Stumvoll M, Stampfer MJ. Weight loss with a low-carbohydrate, mediterranean, or low-fat diet. The New England journal of medicine. 2008;359:229-241
- Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa M. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. New England Journal of Medicine. 2001;344:1343-1350.
- Aucott L, Poobalan A, Smith WC, Avenell A, Jung R, Broom J, Grant AM. Weight loss in obese diabetic and non-diabetic individuals and long-term diabetes outcomes - a systematic review. Diabetes, Obesity and Metabolism. 2004;6:85-94
- Sjostrom L LA, Peltonen M, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. New England Journal of Medicine. 2004;351:2683-2693
- Padwal R, Li SK, Lau DC. Long-term pharmacotherapy for overweight and obesity: A systematic review and meta-analysis of randomized controlled trials. Int J Obes Relat Metab Disord. 2003;27:1437-1446
- O'Brien PE, Dixon JB, Laurie C, Skinner S, Proietto J, McNeil J, Strauss B, Marks S, Schachter L, Chapman L, Anderson M. Treatment of mild to moderate obesity with laparoscopic adjustable gastric banding or an intensive medical program: A randomized trial. Annals of internal medicine. 2006;144:625-633
- O'Brien PE, Brennan L, Laurie C, Brown W. Intensive medical weight loss or laparoscopic adjustable gastric banding in the treatment of mild to moderate obesity: Long-term follow-up of a prospective randomised trial. Obes Surg. 2013;23:1345-1353
- Dixon J, O'Brien PE, Playfair J, Chapman L, Schachter LM, Skinner S, Proietto J, Bailey M, Anderson M. Adjustable gastric banding and conventional therapy for type 2 diabetes: A randomized controlled trial. JAMA. 2008;299:316 - 323
- O'Brien PE, Sawyer SM, Laurie C, Brown WA, Skinner S, Veit F, Paul E, Burton PR, McGrice M, Anderson M, Dixon JB. Laparoscopic adjustable gastric banding in severely obese adolescents: A randomized trial. JAMA. 2010;303:519-526
- Keating CL, Dixon JB, Moodie ML, Peeters A, Playfair J, O'Brien PE. Cost-efficacy of surgically induced weight loss for the management of type 2 diabetes: A randomized controlled trial. Diabetes Care. 2009;32:580-584
- Keating CL, Dixon JB, Moodie ML, Peeters A, Bulfone L, Maglianno DJ, O'Brien PE. Cost-effectiveness of surgically induced weight loss for the management of type 2 diabetes: Modeled lifetime analysis. Diabetes Care. 2009;32:567-574
- O'Brien PE, Sawyer SM, Laurie C, Brown WA, Skinner S, Veit F, Paul E, Burton PR, McGrice M, Anderson M, Dixon JB. Laparoscopic adjustable gastric banding in severely obese adolescents: A randomized trial. Journal of the American Medical Association. 2010;303:519-526
Autoimmunity to the Thyroid Gland
ABSTRACT
This discussion stresses the normal occurrence of immune self-reactivity, the genetic and environmental forces that may amplify such responses, the role of the antigen-driven immune attack, secondary disease-enhancing factors, and the important contributory role of antigen-independent immune reactivity. Research on thyroid autoimmunity has benefited greatly by knowledge of the specific target antigens and easy access to blood cells and involved target tissue. As research moves apace in realm of molecular genetics and investigation of environmental factors that cause disease, we may look for rapid progress in understanding and controlling these common illnesses. For complete coverage of this and all related ares of Endocrinology, please see our FREE web-book www.endotext.org.
A Brief Review of Immunologic Reactions
T lymphocytes develop from precursor stem cells in fetal liver and bone marrow and differentiate into mature cell types during residence in the thymus. Mature T lymphocytes are present in thymus, spleen, lymph nodes, throughout skin and other lymphatic organs, and in the bloodstream. B lymphocytes (immunoglobulin producing cells) develop from precursor cells in fetal liver and bone marrow and are found in all lymphoid organs and in the bloodstream. The ontogeny and functions of these cells have been identified in a variety of ways, including morphologic and functional criteria, and by antibodies identifying surface proteins which correlate to a varying extent with specific functions. Lymphocytes develop through stages leading to pools of cells which can be operationally defined, and be recognized by acquisition of specific antigenic determinants (1) (Fig. 7-1, Table 7-1). Human B and T cells normally express class I (HLA-A, B, C) major histocompatibility complex (MHC) antigens on their surface, and B cells express class II antigens (HLA‑DR, DP, DQ). Activated T cells also express class II antigens on their surface, and are then described as DR+.
TABLE 7-1
KEY DIFFERENTIATION ANTIGENS WHICH CHARACTERIZE SPECIFIC LYMPHOCYTE SUBSETS
| Primary | |||
|---|---|---|---|
| Antigen | Synonyms | Distribution | Comment |
| CD2 | LFA-2 Cells | T cells | Cytoadhesion molecule; NK Cells cognate to LFA-3 |
| CD3 | T3, Leu 4 | All peripheral T Cells | T Cell reseptor complex cells |
| CD4 | T4, Leu 3 (L3T4 in mice) |
Class II restricted T Cells | CD4 binkds to MHC clas II (55-70% of peripheral T cells) |
| CD8 | T8, Leu 2 Lyt 2 | Class I restricted T Cells | CD8 binds to MHC class I (25-40% of peripheral T cells) |
| CD11a | LFA-1 chain | Leukocytes | LFA-1 chain adhesion molecule, binds to ICAM-1 |
| CD14 | LPS Receptor | Monocytes | Marker for monocytes |
| CD16 | Fc R111 | NK cells, Granulocytes |
Low affinity Fc receptor |
| CD20 | B1 | B cells | Marker for B cells |
| CD25 | TAC, IL2 | Activated T and B cells and monocytes | Complexes with chain; T cell growth |
| CD28 | Tp44 | Most T cells | T cell receptor for B7-1 |
| CD29 | - | 40-45% of CD4+ and CD8+ cells | 1 chain of VLA protein, an "integrin" type of adhesion molecule |
| CD40 | - | B Cells | B cell activation |
| CD45RO | - | 25-40% of peripheral T cell subsets | Expressed on naive T cells |
| CD54 | ICAM-1 | T and B Cells | Cognate to LFA-1 |
| CD56 | NKH1 | NK Cells, some T cells | Neural cell adhesion molecule; NK marker |
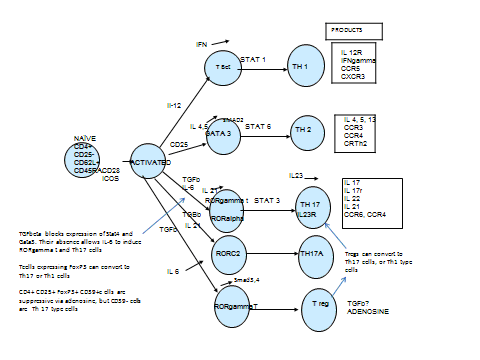
Figure 7-1: Development of T Cell Subsets. In the thymus, undifferentiated precursors give rise to CD4+ and CD8+ cells. In the peripheral lymphoid tissues CD4+ cells (CHO) differentiate following activation by exposure to cognate antigen into two subsets (TH1 and TH2), which are well characterized in the mouse, less so in man. Development of these cells is to some extent reciprocally controlled by cytokines, and the cytokines secreted are also distinct. CD8+ cells similarly mature after antigenic stimulation into less well defined subsets. or = effect on subset proliferation. = cytokines produced.
Lymphocyte Surface Molecules
T cells have on their surface T cell antigen receptors (TCR) which recognize an antigen/HLA complex, accessory molecules which recognize HLA determinants, and adhesion molecules which recognize their counterpart ligands on antigen presenting cells (APCs). After activation, T cells also have new receptors for cytokines, the hormone products mainly produced by macrophages, T cells and B cells, which control other T or B cells (2) (Table 7-2). The T cell antigen recognition complex consists of disulfide-linked TCR heterodimers, usually the TCR-a and TCR-b chains, plus five or more associated peptides making up the CD3 complex (3). A small proportion of T cells have TCRg and TCRd chain instead of a and b chains. TCR-a and b peptides and gd peptides are derived from rearranged genes coding for proteins which are unique in each cell clone. The germline TCR genes are very large, containing 40 - 100 different V (variable) segments, D (diversity) segments (in genes), many J (junctional) segments, and one or two C (constant) segments (Fig. 7-2).
TABLE 7-2
CYTOKINES
| Cytokine | Cell Source | Targets | Primary Effects On Targets |
| Type 1 IFN (IFN-α, Β) | Mononuclear phagocyte, fibroblast | All | Antiviral, antiproliferative, increased class I MHC expression |
| Tumor necrosis factor | Mononuclear phagocyte, T cell |
Neutrophil Liver Muscle Hypothalamus |
Inflammation, Acute phase reactants, Catabolism, Fever |
| Interleukin-1 | Mononuclear phagocyte |
Thymocyte Endothelial cell Hypothalamus Liver Muscle fat |
Costimulator Inflammation Fever Acute phase reactants Catabolism (cachexia) |
| Interleukin-6 | Mononuclear phagocyte, endothelial cell, T cell |
Thymocyte Mature B cell Liver |
Costimulator, Growth, Acute phase reactants |
| Interleukin-2 | T cells |
T cell NK cell B cell |
Growth; cytokine production, Growth, activation, Growth, antibody synthesis |
| Interleukin-4 |
CD4+ T cell, mast cell |
B cell Mononuclear phagocyte T cell |
Isotype switching, Inhibit activation, Growth |
| Transforming growth factor- β | T cells, mononuclear phagocyte, other |
T cell Mononuclear phagocyte Other cell types |
Inhibit activation, Inhibit activation Growth regulation |
| Interferon-γ | T cell, NK cell |
Mononuclear phagocyte Endothelial cell All cells |
Activation Activation Activation. Increased class I and class II MHC |
| Cytokine | Cell Source | Primary Effects On Targets | |
| Lymphotoxin | T cell |
Neutrophil Endothelial cell NK cell |
Activation Activation Activation |
| Interleukin- 10 | T cell |
Mononuclear phagocyte B cell |
Inhibition Activation |
| Interleukin-5 | T cell |
Eosinophil B cell |
Activation Growth and activation |
| Interleukin- 12 | Macrophages |
NK cells T cells |
Activation Activation |
Adapted from tables in Cellular and Molecular Immunology, Edition II by AK Abbas, AH Lichtman, and JS Pober, WB Saunders Company, Philadelphia
Figure 7-2: Cartoon of the human T cell receptor and its subunits. Part A shows subunit composition of the human T cell receptor. The TCR subunits are held together by S-S bonds and are closely associated with either the CD4 or CD8 molecule and chains of the CD3 complex. The subunits are anchored in the cell membrane. The CD3 complex consists of three subunits referred to as gamma, delta, and epsilon. Associated in the TCR complex is another pair of 16 kD homodimer (32 kD nonreduced), subunits existing as homodimers of zeta or heterodimers of zeta and eta. Part B shows the structure of the Ti subunits. The predicted primary structure of the -chain subunit after translation from the cDNA sequence is depicted, as are the variable region leader (L), V, D, and J segments, a hydrophobic transmembrane segment (TM) and cytoplasmic part (Cyt) in the C region, potential intrachain sulfhydryl bonds (S-S), and the single SH group (S) that can form a sulfhydryl bondwith the subunit. Part C shows a scheme of the genomic organization of human -and -chain genes. In the locus, V indicates the V gene pool located 5', at an unknown distance from the D 1 element, the J 1 cluster, and the C 1 constant-region gene. Further downstream, a second D 2 element, J 2 cluster, and C 2 constant-region gene are indicated. A similar nomenclature is used for the Ti locus, in which only a single constant region is found. ?D indicates the uncertainly about the existence of a putative Ti -diversity element. (From Reference 1).
During development of each T cell, segments of the germline gene are rearranged so that one TCR gene V segment becomes associated with one D (in the case of TCR-b), one J, and one C segment to produce a unique gene sequence. This random combination of different V, D, and J and C segments, and additional variations in DNA sequence introduced in the J and D region during recombination, provides the enormous diversity of specific TCRs required to recognize the entire universe of T cell antigens. This process also means that all individuals have (before clonal deletion) preformed TCRs able to recognize thyroid autoantigens as well as thousands of other autoantigens.
Each TCR recognizes one specific antigenic peptide sequence termed an epitope (5), which consists of 8 - 9 amino acids for class I restricted T cells, and 13 - 17 amino acids for class II restricted T cells. However, T cells respond to several portions epitopes of any one antigen; these may represent overlapping peptide segments of the epitope. Thus the response of each individual T (and B) cell is extremely specific, but the combined effect of many T (and B) cells acting together is observed in the typical final polyclonal response.
T cells recognize antigen presented by an MHC-molecule; CD4+ T cells (often functioning as helper cells) recognize MHC class II molecules plus antigenic epitope, and CD8+ T cells (often functioning as cytotoxic cells) recognize MHC class I molecules plus antigenic epitope. The epitope fits within a cleft in the HLA-DR molecule and the TCR functions to recognize this complex (Fig. 7-3). The five associated peptides of the CD3 complex are believed to be signal-transducers and to initiate intracellular events following antigen recognition. The normal response proceeds via TCR antigen recognition, then activation of the T cell through the combined effect of antigen recognition and costimulatory signals (see below) leading to T cell IL-2 secretion and IL-2 receptor expression, followed by proliferation of the T cell into an active clone.
Figure 7-3: In this diagram the antigen is depicted in a cleft of the HLA-DR molecule on an APC, being recognized by the T cell TCR. "Adhesive" peptide segments may augment close contact. A CD4 molecule is associated with the TCR. Presumably the APC surface is normally covered with many DR molecules, each studded with an antigen. T cells must somehow scan these complexes in order to find the one that best fits their TCR.
Lymphocyte development is controlled by cytokines released by macrophages, dendritic cells, lymphocytes, and many other cells. Both T and B cells release a large array of cytokines which carry out their effector functions and alter the function of other cells (Fig. 7-1, Table 7-2). As lymphocytes mature in the thymus, and become activated on exposure to antigen, the types of cytokines to which they respond -- and produce -- become altered. In animals, and to a lesser extent in man, types of lymphocytes can be operationally defined by the cytokines produced. For example, Th1 T cells produce IL-2, IFN-g and TNF and are predominant in delayed hypersensitivity type reactions, whereas Th2 T cells produce IL-4 and IL-5, stimulate B cells, and are involved especially in antibody-mediated reactions. Cytokines produced by Th1 cells enhance the activity of this subset but inhibit Th2 cells, and vice versa. This type of regulation may be critical in determining an immune response and in suppressor phenomena. Additional Th subsets are now recognized, including Th17 cells which secrete IL-17 see below), as well as Th9 and Th22 cells which also have discrete pathological roles.
As well as cytokines and their receptors, T cells express a number of receptors for chemokines, integrins and selectins which are involved in the sequential stages of cell adhesion which leads to T cell homing to tissues (7). A word of caution is necessary however in terms of translating these findings into the human situation where boundaries between the subsets are less clear. It is also increasingly recognized that the simple dichotomy of T cells into two types is over-simple, with cytokines such as IL-12 being assigned to the Th1 subset although not being secreted by T cells, and production of this cytokine is stimulated by the Th2 cytokines IL-4 and IL-13, which will drive the immune response from Th2 towards Th1. The blurring of pattern that is seen in many autoimmune diseases challenges the dogma of an easy divide in the type of immune response.
Each B cell produces a unique immunoglobulin (Ig) programmed by an Ig gene which has also been rearranged from the germline V, D, J, and C segments (as for the TCR) (8). The TCR and Ig genes are, not surprisingly, members of one gene superfamily. Further diversity is provided by antigen-driven somatic mutations which occur during amplification of the progeny of a stimulated B cell, causing the production of a family of antibodies with slightly different sequences. B cells secrete their unique antibodies into surrounding fluids, and also express surface Ig which is therefore a B cell receptor for antigen (Fig. 7-4). The recognition process by antibodies involves the shape of the epitope - i.e. it is conformational and for B cells normally involves unprocessed antigen. Thus B cell and T cell epitopes for the same antigen are usually different segments or forms of the molecule.
Figure 7-4: The B cell surface is studded with specific Ig molecules which function as high affinity receptors for specific antigen epitopes which match the shape of the Ig recognition idiotype.
Antigen Presentation On Mhc Molecules
The genes for the HLA‑A, B, C and HLA‑DR, DP, DQ molecules are on chromosome 6, and comprise some of the genes in a large immune response control complex (Fig. 7-5). Each cell surface HLA molecule is made up of 2 peptide chains; an a chain and b2 microglobulin for class I molecules, and a and b chains for class II. Each individual inherits from each parent one HLA-A, B, and C, one DRa and 3 DRb genes, a pair each of DP and DQa and b genes, and other related genes which are not expressed, including DX and DO (Fig. 7-5). The genes are expressed in a co-dominant manner, and (in contrast to TCR and Ig molecules) are invariant in individuals. However, the genes are all highly polymorphic, that is, many alleles may exist for each gene. The actual evolutionary drive for this diversity is unknown. While TCR gene rearrangement provides the T cell repertoire to respond to individual antigens, HLA diversity guarantees that different individuals will have different T cell repertoires, which confers evolutionary advantage to the species in terms of responding to new pathogens.
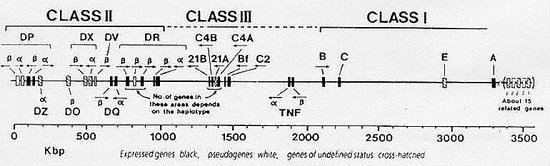
Figure 7-5: Partial map of the short arm of human chromosome 6 showing the molecular organization of the area containing the MHC loci, with details of the HLA Class I, II, and III genes. Map distances in kilobases were determined by pulsed-field gel electrophoresis. Genes are not drawn to scale. Expressed genes are designated by filled boxes _ (|_|). (From Trowsdale, J. and Campbell, R.D. Physical map of the human HLA region. Immunology Today, 9:34, 1988.)
The HLA molecules play a central role in T cell clonal selection during fetal development, in normal immune responses, and in presentation of self-antigens. In many instances -- including autoimmune thyroid disease (AITD) as detailed below -- inheritance of a specific HLA gene correlates with increased susceptibility to disease. In some cases this can be related to a gene coding for a specific amino acid in the HLA molecule which is believed to control epitope selection (often called determinant selection) and thus to be associated with disease susceptibility.
Antigen can be presented to CD4+ T cells by conventional (or "professional") APCs, particularly dendritic cells (9), and also by B cells and activated T cells, and less effectively by a variety of other cells (fibroblasts, glial cells, thyrocytes), when these normally HLA-DR-negative cells are altered and express HLA class II molecules on their surface. This is because non-classical APCs cannot provide the necessary costimulatory signals, including the B7-1 (CD80) and B7-2 (CD86) molecules, which bind to CD28 on the T cell and are necessary for activation of certain T cells. If B7 molecules bind instead to CD152 (CTLA-4) on the T cell, the immune response is terminated. The individual roles of CD80 and CD86 are not clearly established, although some functions appear to be distinct (e.g. CD80 appears to stimulate CD152) and some overlapping (e.g. both stimulate CD28), and the tempo of their involvement at different times of the immune response is likely to be critical to the type of response produced. The maturation state of the dendritic cell is another determinant of immune homeostasis. Semi-maturation, induced by proinflammatory cytokines like TNF-a, allows the development of a tolerogenic stage for these cells. Full maturation, induced by signaling through toll-like receptors, complement receptors or antibody Fc receptors, induces proinflammatory cytokine production by the dendritic cell and allows them to generate T cell immunity.
T And B Cell Responses
Antigen presentation to T cells leads to a variety of responses which include proliferative or suppressive functions, development of cell cytotoxic responses, control of Ig secretion, and many more. In addition, under specific circumstances, antigen presentation may cause the T cell to become non-responsive or anergic (10). Presentation of antigen and the accompanying second signal are required to activate a naive T cell and initiate an immune response; previously activated T cells are much less dependent on B7-mediated costimulation. Antigen recognition and APC-produced cytokines (Table 7-2) together cause T cell stimulation. This activates the T cell to express IL-2 receptors and to secrete IL-2 itself. Increased T-cell secreted IL-2 induces the responding T cell, and nearby ("bystander") T cells to proliferate. T-cell secreted IL-2, IL-6 and other cytokines and IL-4 cause B cells to be stimulated and proliferate and cell surface receptors such as CD40 on B cells and its ligand on T cells are also involved in B cell activation (11).
B cells themselves secrete distinct profiles of cytokines, in response to the engagement on CD40, and these cytokines can upregulate or downgregulate an immune response in a manner which depends on whether the B cell is simultaneously stimulated by antigen (12). Intimate T-cell to B-cell contact may account for antigen-specific help for T cell and B cell responses, whereas the effect of T cell-secreted cytokines on bystander T or B cells may account for stimulation of non-antigen-specific responses by these lymphocytes. The beneficial effects of rituximab, a CD20 specific, B depleting monoclonal antibody, in autoimmune conditions including Graves’ disease (13) is related to its effects on inhibiting this interaction between T and B cells.
Th1 cells function as inflammatory cells, typical of a delayed hypersensitivity type reaction, while Th2 cells are more specifically helper cells for B cell immunoglobulin synthesis. A number of factors including TCR affinity and ligand density, and non-T cell-derived cytokines such as IL-4 and IL-12, determine whether the outcome of an immune response is predominantly by Th1 or Th2 cells. A third population of T helper cells has been defined recently, based on their secretion of the pleiotropic proinflammatory cytokine IL-17, and are so called Th17 cells. The differentiation and expansion of these cells depends on the coordinate effects of IL-6, transforming growth factor beta (TGFβ) and IL-23 (14). These Th17 cells are responsible for defense against certain micro-organisms such as Klebsiella, Borrelia and fungi. Of relevance to this discussion, they also have important roles in tissue inflammation and organ-specific autoimmunity.
Although the concept of suppressor cells fell into disrepute during the late 1980s, there has been resurgence in interest with the recognition that CD4+ cells expressing high levels of the IL-2 receptor, CD25, act in a way entirely in keeping with the previously defined suppressor population. These CD4+, CD25+ T cells have been termed regulatory or Treg cells. Such cells can prevent autoimmunity when transferred from healthy, naïve animals and their depletion results in autoimmune disease. Such cells express Foxp3 which encodes a critical transcription factor for their function: mutation of this gene in man results in the lethal immunological disorder IPEX syndrome that includes autoimmune hypothyroidism amongst its manifestations (15).
APCs have a central role in controlling Treg cells, with resting APCs (including thymic epithelial cells) promoting their development through the induction of the transcription factor Foxp3 (16). Activation of APCs, for instance through their T cell-like receptors, has the opposite effect, and at least one component responsible for the suppression of Tregs then is the cytokine IL-6; this pathway allows effector T cells to predominate over Tregs, thereby shifting the dynamic equilibrium in favor of an immune (or autoimmune) response. Another critical molecule in the Treg cell pathway is the costimulatory signal receptor, CD28, which is required for both development and maintenance of Treg function. TGFβ exposure induces Tregs, but when combined with IL-6, Th17 effector cells are generated. The absence or presence of IL-6 is thus critical to determining whether there is a regulatory milieu or a proinflammatory response mounted by Th17 cells. Both Th1 and Th17 cells are potent inducers of organ-specific autoimmunity, but their relative roles in each type of disease remain to be clarified.
It is increasingly clear that Treg are more complex a group of cells than originally clear. T regulatory cells can be classified as those which arise within the thymus and express Foxp3, and a Th3-like population which probably does not express this molecule and which develops in the periphery. More recently described regulatory cells have been phenotyped as CD4+CD69+ and CD4+NKG2D+ T cells. The glucocorticoid inducible tumor necrosis factor receptor (GITR) is expressed by both populations but CD25bright expression is not a requirement for regulatory T cell function.
The reciprocal relationship between Th1 and Th2 cells, exerted through secretion of cytokines, serves as another model of suppressor function. This paradigm is conceptually useful but is almost certainly too simplistic, not least because there may exist within the Th2 compartment different types of T cells, some with pathological effector function and others which act as physiological regulators of Th1 responses. Endeavors to manipulate the entire Th2 population, to deviate an immune response away from Th1 cells, may therefore lead to exacerbation of the immune response, and may explain the reciprocal relation between the prevalence of infectious disease and autoimmunity (18).
Killer (K) And Natural Killer (Nk) Cells
In addition to the standard T cell function described above, other cells participate in immune responses. Macrophages may destroy cells having immune complexes on their surface through recognition of the Fc portion of bound Ig. Other cells which do not bear the CD3 marker of T cell lineage exist (K and NK cells) and have the ability to spontaneously kill other cells (especially those expressing HLA antigens). NK cells can be detected by specific monoclonal antibodies such as anti-CD16, and are recognized phenotypically as large granular lymphocytes. Like T cells, NK cells can have a type 1 or 2 pattern of cytokine release. Macrophages, T, K, NK, or other cells also kill cells coated with immune complexes in the process of antibody-dependent-cell-cytotoxicity (ADCC) (Fig. 7-6).
Figure 7-6: Some of the proposed mechanisms which could produce thyroid damage in AITD. Emperipolesis is the movement of lymphocytes and macrophages between epithelial cells and occurs in many organs such as gut, bronchus, and thyroid. The existence of interepithelial cells with immunoreactive potential is obviously relevant to an understanding of how autoantigens at the luminal surface of the thyroid cells may be exposed to allow recognition. (From Weetman AP, McGregor AM. Endocrine Rev 5: 309-355, 1984 ).
Self-Non-Self Discrimination
The immune system, which evolved to defend us from invading foreign proteins, normally tolerates (i.e. does not develop recognizable responses to) self‑antigens. The level of this control is variable. For example, self‑reactivity to serum albumin is not seen. However, antibodies to thyroid antigens exist in up to 20% of adult women, and their presence must be considered effectively normal. The development of tolerance is closely associated with the restriction of TCRs to recognizing an antigen only when presented by an HLA molecule. The process, which for T cells occurs in the fetal thymus, leads to elimination of some T cells, and retention of others with TCRs having desirable features. Self-antigens are believed to be presented on HLA molecules to T cells developing in the thymus. This implies that antigen must be in the thymus or in the circulation for tolerance to develop and indeed we now know that specialized cells in the thymus can express a panoply of autoantigens during development. T cells bearing autoreative TCRs are largely inactivated or destroyed. T cells which have the capacity to react with foreign antigens presented by self MHC molecules are allowed and retained (Fig. 7-7). This system is imperfect however and some T cells which react with MHC molecules plus self-antigen are not deleted, which is the fundamental explanation for autoimmunity.
Figure 7-7: Left: Fetal Thymus; T cells strongly activated by DR alone, or strongly reactive to self-antigen presented by HLA molecules, are selectively destroyed. T cells, with a weak or absent response to DR alone, or to DR+ self-antigen, survive. Center: Normal Adult Immune Reaction; T cell TCR and APC-DR interaction is normally a weak or neutral signal. The presence of allo-antigen serves to switch the signal to positive. Right: Allo-MLR; Allogeneic DR is sufficiently different from autologous DR to act as a positive signal with or without antigen present.
The best evidence that thymic T cell deletion prevents autoimmunity in man comes from autoimmune polyglandular syndrome (APS) type 1, which is the result of an autosomal recessive mutation in the AIRE (AutoImmune REgulator) gene. Such patients have multiple autoimmune disorders, principally Addison’s disease and hypoparathyroidism but including thyroid autoimmunity. The AIRE protein is expressed in the thymus by medullary epithelial cells and regulates the surprising expression of an array of self proteins (normally confined to extrathymic tissues) by these cells during fetal development. When through the AIRE mutation such self-antigens cannot be expressed to allow clonal deletion, autoimmunity ensues and this accounts for the early onset multiple autoimmunity found in this syndrome (reviewed in 19). Recently, dominant mutations in AIRE have been identified and such patients have later-onset, milder phenotypes (19b). During maturation in the thymus, probably 95% or more of the lymphocytes produced are negatively selected, and die through a process described as programmed cell death or apoptosis. This process involves several genes including those required for apoptosis, such as Fas. A similar process is thought to ensue whenever a T cell is stimulated by its cognate antigen but does not receive a "second signal", and during induction of anergy by other mechanisms. Defects in Fas lead to preservation of autoreactive T cells in some models of animal autoimmune disease.
This trade-off between perfection in clonal deletion and repertoire maintenance allows a limited number of autoreactive T cells to survive, and thus sets the stage for autoimmune disease. The main mechanism to prevent autoimmunity by these escaped cells and also to induce tolerance to autoantigens not present in the fetal thymus or circulation is termed peripheral (i.e. non-thymic or central) tolerance, and is mainly effected by the Tregs described above.
B cells undergo a similar selection process in fetal bone marrow or liver, except for the participation of MHC molecules. If exposed to antigen during this early stage of development, B cells are permanently inactivated. As for T cells, the selection process is not perfect, and leaves some B cells having the ability to make antibodies directed to self‑antigens in the adult. However, B cells require T cell help in order to proliferate and differentiate into mature Ig secreting cells. In the absence of self‑reactive T helper cells, these B cells remain dormant and expanding clones do not develop. Although such clonal ignorance may be an important pathway in preventing B cell autoreactivity, it is not the only mechanism, and physiological concentrations of autoantigen may induce anergy of B cells, even when their affinity for autoantigen is low.
Tolerance to self‑antigen can be overcome ("broken") in animals by injecting the antigen in an unusual site on the body, especially in the presence of adjuvant compounds such as mycobacterial fragments and oil, or alum, or by slightly altering the antigen structure, or by altering the responding immune system (for example, by whole body irradiation, or depletion of suppressor T cells). An additional mechanism for the inflammatory component of many autoimmune disorders has recently been proposed based on the evolutionary origins of mitochondria from bacteria. Given that the prime function of the immune system is to defend the organism from microbes, it is possible that the immune system may mistake mitochondria released from damaged tissue through pattern-recognition receptors and thereby induce a ‘mistaken’ inflammatory response (20).
The Syndromes of Thyroid Autoimmunity
The three syndromes classically comprising autoimmune thyroid disease are (1) Graves' disease with goiter, hyperthyroidism and, in many patients, associated ophthalmopathy (2) Hashimoto's thyroiditis with goiter and euthyroidism or hypothyroidism; and (3) primary thyroid failure or myxedema. Many variations of these syndromes are also recognized, including transient thyroid dysfunction occurring independently of pregnancy and in 5 - 6% of postpartum women, neonatal hyperthyroidism, and neonatal hypothyroidism. The syndromes are bound together by their similar thyroid pathology, similar immune mechanisms, co-occurrence in family groups, and transition from one clinical picture to the other within the same individual over time. The immunological mechanisms involved in these three diseases must be closely related, while the phenotypes probably differ because of the specific type of immunological response that occurs. For example, if immunity against the TSH receptor leads to production of thyroid stimulating antibodies, Graves' disease is produced, whereas if TSH blocking antibodies are formed or a cell destructive process occurs, the result is Hashimoto's thyroiditis or primary myxedema.
Associated with autoimmune thyroid disease in some patients are other organ specific autoimmune syndromes including pernicious anemia, vitiligo, myasthenia gravis, primary adrenal autoimmune disease, ovarian insufficiency, rarely pituitary insufficiency, alopecia, and sometimes Sjögren's syndrome or rheumatoid arthritis or lupus, as manifestations of non-organ specific autoimmunity. There has also been a description of pituitary antibodies and growth hormone deficiency in around a third of patients with autoimmune hypothyroidism, implying the existence of a substantial reservoir of pituitary autoimmunity in these patients but further work is needed to confirm these findings and to understand the basis for the autoimmune response against the pituitary (21).
The Antigens in AUTOIMMUNE THYROID DISEASE
Thyroglobulin
The three most important antigens involved in thyroid autoimmunity are clearly defined. First to be recognized was thyroglobulin (TG), the 670 kD protein synthesized in thyroid cells and in which T3 and T4 are produced. Four to six B cell epitopes of TG are known to be involved in the human autoimmune responses and epitope recognition is similar in both Graves’ disease and Hashimoto’s thyroiditis (22). Animal studies suggest that antigenicity of the molecule is related to iodine content, but studies on human antisera do not consistently bear this out: these species differences and the role of measuring TG antibodies in thyroid disease are reviewed elsewhere (23).
Mouse experiments suggest that, to induce autoimmunity to TG, initial tolerance to dominant epitopes must be overcome, and the immune response then spreads to cryptic epitopes that are the major inducers of thyroidal T cell infiltration (24). One particular TG T cell epitope, Tg.2098, has been identified which is a strong and specific binder to the MHC class II disease susceptibility HLA-DRβ1-Arg74 molecule, and stimulates T cells from both mice and humans that develop AITD (25). This could be a major T cell epitope which might be involved in pathogenesis through initiating an immune response that then spreads to involve other autoantigens. Furthermore, screening a diverse library of small molecules has identified one, cepharanthine, which blocked Tg.2098 peptide binding and presentation to T cells in mice with experimental autoimmune thyroiditis; such an approach has obvious therapeutic potential (25a).
Tsh Receptor
The second antigen to be identified was the TSH receptor (TSH-R), a 764 aa glycoprotein. Antibodies to TSH‑R mimic the function of TSH, and cause disease by binding to the TSH‑R and stimulating (or inhibiting) thyroid cells, as described later. The human TSH-R is a member of a family of cell surface hormone receptors which are characterized by an extra-membranous portion, seven transmembrane loops, and an intracellular domain which binds the GS subunit of adenyl cyclase (26, 27). Uniquely among G-protein-coupled receptors TSH-R undergoes post-translational cleavage to comprise a 53kD extracellular A subunit (53 kDa) and transmembrane and intracellular B subunit coupled by disulfide bridges. The A subunit may be shed provoking speculation on the role of this in stimulating autoimmunity. Recent evidence indicates that in Graves' disease TSHR antibody affinity maturation is driven by A-subunit multimers rather than monomers (27a). Human TSH-R B cell epitopes are conformational and composed of several segments of the protein.
The initial description of mouse and hamster monoclonal TSH-R antibodies was significant for several reasons (28-30). Firstly, these antibodies confirmed that a single antibody was sufficient to activate the receptor, rather than two or more simultaneously. Secondly, they have permitted epitope mapping. One antibody preferentially recognized the free A subunit, not the holoreceptor, suggesting that free A subunit, shed from thyroid cells, may initiate or amplify the autoimmune response. Another antibody, in contrast to TSH, did not enhance post-translational TSH-R cleavage, which may extend the receptor half-life and thus account for the prolonged thyroid stimulation seen following antibody binding. Finally, these antibodies paved the way for the development of human monoclonal antibodies which have allowed a greatly improved understanding of the mechanisms involved in Graves’ disease.
The first human monoclonal TSH-R stimulating antibody bound to the TSH-R with high affinity, either as IgG or as Fab fragment, and the monoclonal had similar features in all respects to known TSAb (thyroid stimulating antibodies) (31). This observation indicated that only a single species of antibody is needed to stimulate the receptor. More conventional approaches based on different methods of expressing the TSH-R have shown that TSAb preferentially recognize the free A subunit rather than the holoreceptor, either because of steric hindrance from the plasma membrane or membrane spanning region of the receptor or because of TSH-R dimerization (32). The epitopes for TBAb overlap with those for TSAb but are more focused on the C terminus and are able to recognize holoreceptor more efficiently. These observations have provided support from the hypothesis that shedding of free TSH-R A subunits may be critical in initiating or amplifying the autoimmune response in Graves’ disease. Further evidence comes from immunization of mice with adenoviruses expressing different structural forms of the TSH-R: goiter and hyperthyroidism occur more frequently when mice are given virus that expresses the free A subunit rather than a receptor with minimal cleavage into subunits (33).
Patients with autoimmune thyroid disease may have both stimulating and blocking antibodies in their sera, the clinical picture being the result of the relative potency of each species. Switching between one type of antibody and another in unusual patients, involving changes in concentration, potency and affinity, may be caused by a number of factors including levothyroxine treatment, antithyroid drug treatment and pregnancy, and can lead to difficulties in clinical care (35). TSH-R neutral antibodies have also been identified which do not block TSH binding and are unable to stimulate cAMP production; these antibodies are capable of inducing thyroid cell apoptosis in vitro and therefore could conceivably play a role in pathogenesis by inducing release of thyroid autoantigens (36).
Identification of the critical T cell epitopes has proved elusive although peptides 132-150 do appear to constitute one key epitope; there is poor correlation between binding affinity and T cell immunogenicity in experiments to attempt such localization (37). In animal studies, however, there is clear evidence of epitope spreading when mice are immunized with TSH-R peptide epitopes or TSH-R cDNA, indicating that dominant TSH-R epitopes are, at best, elusive (38). TSH-R mRNA transcripts and protein have been identified in retrobulbar ocular tissue, particularly the preadipocyte fibroblast, suggesting that TSH-R expression in the orbit could well be involved in the development of autoimmunity and ophthalmopathy, and similar TSH-R-expressing fibroblasts have also been found in the thyroid gland itself (39). Further support for involvement of the TSH-R comes from experiments showing that activation of the TSH-R stimulates early differentiation of preadipocytes, but terminal differentiation is not induced (40). An animal model with some features of similarity to human ophthalmopathy has been induced in mice by immunization with TSH-R A subunit plasmid given by a specific electroporation protocol (41). Oddly there was no thyroid lymphocytic infiltrate to accompany these orbital changes, which were very heterogeneous between immunized animals. It should also be noted parenthetically that an alternative pathway for fibroblast involvement in ophthalmopathy has been proposed which depends on the production of insulin-like growth factor antibodies in these patients but it is difficult to reconcile these findings with the orbital specificity of the autoimmune process in thyroid eye disease (42). Most recently, TSH-R has been identified in immature thymocytes, which can be stimulated by TSAb. This could in turn explain why thymic hyperplasia is seen in occasional cases of Graves’ disease (42a).
Thyroid Peroxidase
The third thyroid antigen was described as "microsomal antigen" was identified as thyroid peroxidase (TPO) in 1985 (43) (Fig. 7-8). DeGroot’s laboratory demonstrated that human antisera reacting to "microsomal antigen" precipitated human thyroid peroxidase (TPO) prepared from Graves' disease thyroid tissue () (Fig. 7-8) and at the same time Czarnocka et al. purified human TPO and confirmed identity with the microsomal antigen (44). The cDNA was cloned and sequenced in several laboratories (45-48). The interaction of human anti-TPO antisera and monoclonal antibodies also indicate the presence of several B cell epitopes which map to two main domains, A and B (reviewed in 49). Further experiments with monoclonal antibodies have defined individual amino acid residues that are critical for the two immunodominant regions (50). The epitopes recognized by antibodies are stable within a patient and may be genetically determined (51). Investigation of TPO epitopes recognized by T cells from patients with AITD has produced conflicting results but certain sequences are beginning to emerge which are shared between reports on various patients (52, 53). There is also debate as to whether patients with autoimmune hypothyroidism differ in their pattern of epitope recognition from healthy controls who are TPO antibody positive, and further work is required to analyze this in detail, as it might allow better prediction of those antibody positive individuals who will progress to overt hypothyroidism (54)
TPO is expressed on the thyroid cell surface as well as in the cytoplasm, and likely represents the cell-surface antigen involved in complement-mediated cytotoxicity as well as antibody-dependent cell mediated cytotoxicity (55). Intracytoplasmic binding of antibodies to TPO indicates that there is access
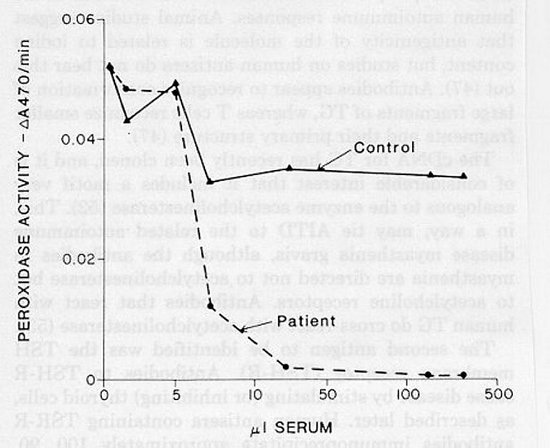
Figure 7-8: Precipitation of peroxidase activity by sera from a patient with autoimmune thyroid disease and positive "microsomal" antibodies, and from a control subject without circulating antibodies. TPO was precipitated by primary incubation with human sera, and removal of TPO-Ig complexes was achieved by addition of Protein H-Sepharose CL-4B. Residual hTPO activity in the supernatant was assayed in a guaiacol assay.
Other Antigens
Antibodies against the sodium/iodide symporter (NIS) were first shown functionally in cultured dog thyroid cells (56). Up to a third of Graves’ disease sera contain antibodies capable of blocking NIS-mediated iodide uptake in cells transfected with the human NIS but the relevance of this for thyroid function is unclear (57). The same antibodies have also been detected using an immunoprecipitation assay (58). Others have found no such blocking activity using assays with cell lines displaying much higher 131I uptake, in turn suggesting that any NIS blocking activity only occurs at limiting conditions (59). This implies that NIS autoantibodies probably have no effect in vivo. NIS expression on TECs is upregulated by TSH and downregulated by cytokines and the latter could impair thyroid function in the setting of AITD when such cytokines are synthesized in the thyroid (60). Pendrin, an apical protein responsible for mediating iodide efflux from thyroid cells into the follicular lumen, has also been identified as an autoantigen. Autoantibodies were initially found in 81% of patients with AITD by immunoblotting (more frequently and at higher titer in Hashimoto’s than Graves’ patients) and also in 9% of controls (61), but the frequency of these autoantibodies detected using a radioligand binding assay is rather low at around 10% of patients and no controls (62).
Antibodies to a variety of other thyroid cell components are also occasionally present in AITD, including antibodies that react with thyroxine or triiodothyronine (63). The insulin-like growth factor receptor has also emerged as a possible autoantigen involved in ophthalmopathy, with antibodies being detected in patients with this complication, and this receptor co-localizes with the TSH-R on both fibroblasts and thyrocytes (64).
Immune Reactions in Autoimmune thyroid disease
Humoral Immunity
The principal autoantibodies identified in AITD and the methods for detecting them are listed in Table 7-3. Antibodies to the TSH receptor are discussed in detail in Chapter 10, but, in brief, observation of a factor in serum of patients with Graves' disease causing long acting stimulation of thyroid hormone release from an animal's thyroid, in contrast to the short acting stimulation produced by TSH, led directly to our knowledge of TSH-R antibodies. We summarize here a huge amount of clinical and laboratory research. The antibodies directed to the TSH-R are currently separated into three types. Some antibodies bind to an important epitope in TSH-R and activate the receptor, producing the same effects as TSH, in particular causing generation of cyclic AMP. These antibodies may be referred to as TSI or TSAb -- thyroid stimulating immunoglobulins or thyroid stimulating antibodies. Other antibodies bind to different, or the same epitopes and interfere with radiolabelled TSH binding in certain assays -- thus they are known as thyrotropin binding inhibitory immunoglobulins or TBII. Still others bind and prevent the action of TSH -- thus blocking antibodies. These may either interfere directly with TSH binding or have less well characterized
TABLE 7-3
ANTIBODIES REACTING WITH THYROID AUTOANTIGENS IN AITD AND TECHNIQUES FOR DETECTION
| Antigen | Test Used To Identify Antibody |
|---|---|
| TG | Precipitin Hemagglutination assay Immunofluorescence on fixed sections of thyroid tissue: colloid localization |
| localization | Solid-phase RIA Immunoradiometric assay Hemolytic plaque assay |
| Colloid component other than TG | Immunofluorescence on fixed sections: colloid localization |
| Microsomal antigen/ TPO |
Complement fixation Immunofluorescence on unfixed sections; thyroid tissue cell localization Cytotoxic effect on cultured thyroid cells Hemagglutination assay ELISA Solid-phase RIA TPO activity inhibition |
| TSH-R | Bioassay in mice cAMP production by thyroid cells, TSH- R transfected cells or membranes Iodide uptake by thyroid cells Thymidine incorporation by thyroid cells Inhibition of TSH action on thyroid cells Inhibition of TSH binding to cells or membranes Immunoprecipitation |
| Sodium/iodide symporter | Western blotting Immunoprecipitation Bioassay using cultured thyroid cells or cells transfected with the symporter |
TSI cause non-TSH dependent stimulation of thyroid function, which, if of sufficient intensity, is hyperthyroidism. TBII comprise the mixture of TSI and TSH blocking antibodies, and therefore function cannot be predicted from the TBII level. Predominance of TSI characterizes Graves' disease, and TSH blocking antibodies are present in a small proportion of patients with Hashimoto's disease and primary myxedema. Probably a combination is present in most patients with AITD. Recent work indicates that both types of TSH-R antibody are present in Graves’ sera at low concentration with high affinity and similar (but nonetheless subtly distinct) binding epitopes (65). TSI directly cause thyroid overactivity, their level correlates roughly with disease intensity, and a drop in levels correlates loosely with disease remission. Unlike TG and TPO antibodies which are polyclonal and not restricted by immunoglobulin subclass (reviewed in 66), there is evidence that some TSH-R are restricted to particular heavy and light chain subclasses, which may indicate an oligoclonal origin (67), and TSH-R stimulating antibodies are present at much lower concentration than TG and TPO antibodies.
Normal subjects can have TSH-R antibodies that bind to but do not activate the TSH-R and that generally have low affinity. These natural autoantibodies may be the precursors of the TSI that cause Graves’ disease and it is possible that affinity maturation, with class switching of immunoglobulin isotype, is critical in determining the clinical consequences of TSH-R antibody production. Conversely, using the most sensitive binding assays, there are still a very small number of patients with Graves’ disease who are apparently negative for these antibodies when their serum is tested; it is likely that the explanation lies in either assay sensitivity or exclusively intrathyroidal production of these antibodies (68).
Precipitating antibodies to TG were first detected by mixing antibody and antigen in equivalent concentrations, or by agar gel diffusion, as in the Ouchterlony plate technique. Subsequently, much more sensitive methods were developed, such as solid phase ELISA (69) and RIA (70), although for many years the tanned red cell hemagglutination test remained the assay of choice (71). Immunoradiometric assays (IRMA) used currently involve binding of serum antibodies to solid phase antigen, and secondary quantitation of antibody by binding labelled monoclonal anti-human Ig antibody. These tests are very sensitive but lack specificity as so many healthy individuals are positive, albeit with a future risk of developing AITD.
Antibodies directed against TG are rarely present in children without evidence of thyroid disease. The prevalence in healthy persons increases with age, and low levels are frequently present in normal adults (72). The greatest frequency occurs in women aged 40‑60 years. The frequency of antibodies in well persons correlates with the incidence of focal lymphocytic infiltration found on microscopic examination of thyroid tissue form healthy individuals (73). Over 90% of patients with Hashimoto's thyroiditis and primary myxedema have these antibodies. Low to moderate titers are found in half of patients with Graves' disease. TG antibodies are either absent or low in patients with subacute (De Quervain's) thyroiditis, who may present clinically like patients with Hashimoto's thyroiditis. In general human TG and its autoantibody bind complement weakly due to the widely scattered epitopes which are unable to allow antibody cross-linking.
The second important antigen-antibody system was originally recognized by antibodies which, by immunofluorescence, were observed to bind to non-denatured thyroid cytoplasm, to fix complement in the presence of human thyroid membranes (microsomes), or to bind to microsome‑coated red cells (the MCHA assay). We now know this antigen is TPO (see previous Section 3) (Fig. 7-8). Almost all patients with Hashimoto's thyroiditis have TPO antibodies. They also occur in the normal population in the absence of clinically significant thyroid disease: in a recent survey of a population followed for 20 years, 26% of adult women and 9% of adult men had TPO and/or TG antibodies (74). However, the presence of such antibodies was shown to be associated with an increased risk of future hypothyroidism, especially if the TSH was also raised (subclinical hypothyroidism). Few sera from AITD contain TG antibodies in the absence of TPO antibodies, but the converse is not always true, so it has been proposed that screening for AITD could be undertaken initially with assays for TPO antibodies (75). This is particularly the case if the hemagglutination assay is used for TG antibodies; sensitive RIAs may detect a very high frequency of TG antibodies in individuals with autoimmune thyroid disease, even more than TPO antibodies (76). Using modern types of assay, TG antibodies occurring in isolation from TPO antibodies are more commonly found, and thus measurement of both antibodies might have clinical utility in certain situations, for instance in diagnosing possible causes for impaired fertility in women (77).
Antibodies detected by these techniques are believed to be similar to antibodies first described in the 1950s that fix complement in the presence of extracts from a thyrotoxic gland (78) and that have cytotoxic effects on thyroid cells (79). Sera from patients with Hashimoto's thyroiditis usually have high cytotoxic activity (80). Complement-mediated sublethal injury probably occurs in vivo since complement containing complexes have been identified in thyroid tissue of patients with Graves’ disease and Hashimoto’s thyroiditis (81). Thyroid cell expression of membrane proteins, especially CD59, helps prevent complement-mediated lysis (82), and this protein is upregulated by IL-1 and IFN-g.
The cytotoxicity of circulating antibodies has also been explored using systems to detect antibody-dependent cell-mediated cytotoxicity (ADCC) in which nonimmunized lymphocytes (NK cells) or macrophages act as effector cells and kill antigen-coated target cells, following incubation of the targets with antibody (83, 84). This reaction does not require complement, instead depending on the interaction of antibody on the target cell with Fc receptors on the effector cells. The exact role of ADCC in the pathogenesis of autoimmune thyroid disease is unclear, as it has been investigated only as an in vitro phenomenon. Antibodies capable of mediating ADCC on target cells include those against TG and TPO, but other antigens may also be targets, and sera from patients with Hashimoto’s thyroiditis, primary myxedema and Graves’ disease cause ADCC, although the frequency is lower in Graves’ disease (85). A further possible role for TPO antibodies has been suggested by the finding that these bind to cultured astrocytes and it is therefore possible that the controversial entity of Hashimoto’s encephalopathy is the result of some autoimmune cross-reaction between thyroid and central nervous system (86).
Titers for all types of thyroid autoantibody obviously increase during the process of development of AITD. It is possible that one critical step in the production of TG autoimmune responsiveness is the generation of immunoreactive C-terminal fragments during hormone synthesis (which results in oxidative stress); these fragments may also lead to preferential presentation of TG epitopes by thyroid cells (87). Natural autoantibodies against TG may be more important in the initiation of the response than previously thought. These low affinity, mainly IgM antibodies, which are frequent in healthy individuals, can complex TG with complement and such opsonized complexes can be taken up by B cells and presented to CD4+ T cells (88). After first observation, antibody levels tend to be stable over months.
Radioactive iodine therapy in Graves' disease leads to a rise in thyroid antibody levels during the first few months after treatment (89), and exposure to high levels of IFN-a in those with pre-existing autoantibodies also does this (90, 91). With treatment of Graves' disease, or replacement therapy in Hashimoto's thyroiditis or myxedema, there is characteristically a gradual reduction in antibody levels over months or years, and some patients with total destruction of thyroid tissue eventually lose detectable antibody titers.
There are two major conformational epitopes on the TG molecule that are recognized differentially by sera from healthy subjects and those with AITD; linear epitopes are recognized by polyclonal antibodies from healthy individuals (92-94). Similar studies on TPO have indicated at least eight major domains for human autoantibodies which are probably conformational epitopes. Using recombinant proteins and synthetic peptides, human anti-TPO antibodies are found to recognize apparently linear epitopes in the area of amino acids 590-622 and 710-722 (95) but, again, the important B cell epitopes are conformational.
Peripheral blood mononuclear cells (PBMC) and thyroid lymphocytes from patients with AITD have among them activated cells that spontaneously secrete TG and TPO antibodies (96). B cell production of antibodies to TPO and TG is most easily shown using cells incubated with mitogens (97). Specific antibody secretion in response to PBMC stimulation by TG or purified TPO is more difficult to demonstrate (98). In patients with AITD, approximately 50 B cells secreting anti-TG antibodies are found per 106 PBMC (~2% of total Ig secreting cells) by using plaque-forming assays after stimulation of PBMC with pokeweed mitogen. B cells from AITD patients synthesize antibodies in response to insolubilized TG bound to Sepharose (98), which appears to function as an especially good antigen. There are reports of production of anti-TSH-R antibodies in vitro, but in general this response has been difficult to observe.
In fully developed AITD, the thyroid is clearly an important source of autoantibody and spontaneous autoantibody secretion by B cells is easily demonstrable (99). This is also supported by the histopathological features, including the demonstration of thyroid antigen-specific B cells and the occurrence of secondary immunoglobulin gene rearrangement in intrathyroidal lymphoid follicles, together with a congruent pattern of adhesion molecule and chemokine expression (100). However, lymph nodes, bone marrow and possibly other organs also contribute to autoantibody production (101) and this explains why patients with apparently destroyed thyroid tissue, or those with resected thyroids, continue to have circulating thyroid auto-antibodies
Cell-Mediated Immunity In Autoimmune Thyroid Disease
Techniques for identification of T lymphocyte reactivity to foreign or autologous antigens depend on culturing mixed peripheral leukocytes or semi-purified thyroid or blood lymphocytes with an antigen to which the cells may have been pre-sensitized. Upon re-exposure to antigen, the sensitized cells change to a blast-like immature form, synthesize new protein, RNA, and DNA, and directly or through liberated effector molecules alter the function of target cells. Different endpoints characterize the various assays, including measurement of [3H]-thymidine uptake, assay of migration inhibition factor (MIF), or leukocyte migration inhibition (LMI) (102), assessment of the mobility of lymphocytes, and cytokine assay, all after stimulation with antigen in culture.
Numerous reports have shown that T cell immunity can be detected in Graves' disease, Hashimoto’s thyroiditis, and primary myxedema, although responsivity of T cells to thyroid antigens is much less than to exogenous antigens such as tetanus toxoid or tuberculin. Peripheral blood T cells respond to incubation with TG or TPO in the form of a microsomal preparation by thymidine incorporation, the so-called proliferation assay (103, 104). Responses by separated lymphocytes are generally weak; better responses are seen by adding IL-2 to thyroid antigen-stimulated cultures of diluted whole blood (105). Thyroid T cells responding to TG are of the CD4+ T helper type (106), or occasionally CD8+ cells (107). T cells also respond to crude thyroid antigen extracts in LMI assays (102). T cell lines and short term T cell clones (CD4+) are stimulated during co-culture with TECs to incorporate [3H]-thymidine; DR+ TECs are especially effective stimulators (108 - 110). The identity of the antigen recognized on TECs is unknown but may well be TPO and/or TG.
The specific peptide epitope fragments of TPO recognized by lymphocytes of patients with HT were noted previously. T cell epitopes present within the extracellular domain of the TSH-R are also heterogeneous with peptides bearing sequences of aa 158-176, 237-252, and 248-263 and 343-362 being especially important (111) but other epitopes (aa 57-71, 142-161, 202-221, 247-266) have been identified by others using different assay parameters (112). HLA-DR3 molecules bind TSH-R peptides with high affinity, which may explain the genetic association of this HLA specificity with Graves’ disease (113).
T cell responses to an antigenic stimulus may use a wide variety of variable (V) TCR gene segments, or the response may be restricted to a few V segments. Restriction of autoreactive T cells to use of one or more V gene segments has been found in some experimental autoimmune models (4). Restricted Va and Vb usage in the whole intrathyroidal lymphocyte population has been reported (114, 115) but not confirmed by others (116, 117). However, intrathyroidal CD8+ T cells do display a degree of restriction although their autoreactive potential is at present not known (118). Presumably at an early stage of disease, the T cell response is clonally restricted, but as it advances, spreading of the immune response occurs, involving many more epitopes, leading to an unrestricted response as demonstrated in an animal model of AITD (119). Evidence has emerged of a combined TG and TPO epitope-specific cellular immunity, with CD8+ T cells reacting against these epitopes rising to 9% in the peripheral blood of patients with long-standing Hashimoto's thyroiditis (120).
While T cell immunization is conventionally recognized by a stimulatory effect of antigen, direct T cell cytotoxicity of thyroid cells has been recognized in a few studies. For example, Davies and co‑workers developed a CD8+ T cell clone which was cytotoxic to autologous TEC and was appropriately class I restricted (121). Another potential consequence of T lymphocytic adherence to thyroid cells is the stimulation of thyroid cell proliferation via ICAM-1/LFA-3 interaction, rather than their destruction, which could lead to goiter formation (122).
Immune Complexes
In addition to the antibody and T cell responses, circulating immune complexes are found in patients with autoimmune thyroid disease as would be anticipated], although their pathogenic importance appears minimal. In a certain sense this is most fortuitous. Since many individuals have circulating TG antibodies and antigen, if the immune complexes caused serious disease, it would be a catastrophe. Fortunately the immune complexes of TG and its antibody do not bind complement and do not cause serious illness such as immune-complex nephritis, except in rare instances (123, 124). Immune complexes, including complement terminal components, can however be recognized around the basement membrane of thyroid follicular cells (81) and may cause sublethal effects including release of proinflammatory mediators by TECs (125).
K And Nk Cell Responses
Many studies have been reported on natural killer (NK) cell activity and antibody dependent cell-mediated cytotoxicity (ADCC); their conclusions vary. Endo et al (126) found NK cells were decreased in Graves' disease and Hashimoto's thyroiditis, and presented evidence that this was due to saturation of their Fc receptors by immune complexes. Normal NK effector function was found in Hashimoto's thyroiditis PBMC (127) in one study, although by phenotyping, decreased NK cells in Graves' disease, and increased NK cells in Hashimoto's thyroiditis were reported in another (128). ADCC of thyroid cells, mediated by normal PBMC, was induced by TPO antibody positive sera (129) but other, unknown antibody-antigen systems may also contribute (85). Effector cell activity in ADCC was increased in Hashimoto's thyroiditis and in post-partum thyroiditis, and thought to be related to thyroid cell destruction (130). Other data have indicted that ADCC may be more important in primary myxedema than Hashimoto’s thyroiditis explaining the difference in clinical presentation (131), but this has not been confirmed in studies showing equal ADCC activity in sera from both diseases (132).
Cytokines
Cytokines lie at the heart of the autoimmune response and can have a number of direct and indirect effects (Fig. 7-9). For example, IFN-g is produced in the thyroid by infiltrating lymphocytes and causes HLA class I expression on the surface of TECs to increase and initiates class II expression. It also has a direct inhibitory function on TEC iodination and TG synthesis (133, 134). Caveolin-1 and TPO form part of the apical thyroxisome, responsible for thyroid hormone synthesis. Recent studies have shown that Th1 cytokines down-regulate caveolin-1, leading to intracytoplasmic thyroxine synthesis and mislocalization of the thyroxisome. Disruption of the thyroxisome in this manner may then lead to damage by reactive oxygen metabolites and apoptosis in Hashimoto’s thyroiditis (135).
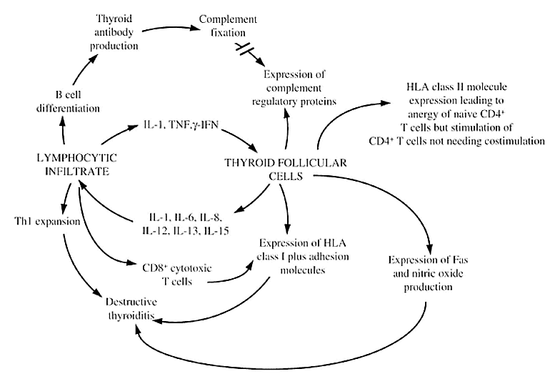
Figure 7-9: Interactions between thyroid follicular cells and the immune system in autoimmune thyroid disease. Reproduced from Weetman AP, Ajjan R, Watson PF. Bailliere’s Clin Endocrinol Metab 11: 481-497, 1997 with permission.
IFN-g is not essential for the development of AITD in mice but exacerbates disease activity (136). IL-2 can activate lymphocytes to produce IFN-g, and activate NK cells. TNF is produced by infiltrating macrophages and is potentially cytotoxic to TEC. TEC can produce several cytokines, including IL-1, which may activate T cells, IL-6, which stimulates T and B cells and IL-8, a chemokine which attracts inflammatory cells (reviewed in 134). More recently IL-14 (taxilin) and IL-16 production by TECs has been described: the former regulates B cell growth and the latter is a chemoattractant for CD4+ cell (136a). Dendritic cells are important sources of IL-1b and IL-6 in the thyroid and can inhibit thyroid follicular cell growth (137). As an aside, plasmacytoid dendritic cell numbers are decreased in the blood in AITD, together with an alteration in their phenotype, but these cells increase in the thyroid gland, also suggesting that this cell type may be important in pathogenesis (138)
IL-1a causes dissociation of junctional complexes between thyroid cells which could expose hidden autoantigens (139). An ever wider array of factors besides the classical cytokines has been implicated in the pathogenesis of AITD, including the finding that thyroid cells can release angiopoietin-1 and -2 (140). These ligands serve as a chemoattractant for monocytes and the angiopoietin receptor, Tie-2, is increased in monocytes form AITD patients, suggesting a role for monocytes in thyroid damage. Vascular endothelial growth factor expression is increased in AITD and is important in angiogenesis in autoimmune goiters (141). Cytokines also seem to play a major role in the pathogenesis of thyroid-associated ophthalmopathy through their stimulatory actions on orbital fibroblasts (142). Exogenous cytokines given therapeutically can also precipitate autoimmune thyroid disease, probably in predisposed individuals. The best described such reaction is α interferon used in hepatitis C and cancer therapy (90). Destructive thyroiditis accounts for the majority of thyroid dysfunction after treatment with this cytokine, and risks are highest in white women, whereas smoking is protective (91).
SUMMARY
To summarize, augmented pools of activated and resting T and B cells reactive to thyroid antigen exist in patients with AITD. The time course of development of these reactive cells, before clinical disease is apparent, has not been established. The cells respond to biochemically normal antigen, and some reactive cells exist in otherwise healthy individuals. Immune complex formation appears to be of limited importance in the disease process. K and NK activity may be reduced in Graves' disease and increased in Hashimoto's thyroiditis and may contribute to the course of the disease: proliferative in Graves' disease and destructive in Hashimoto's thyroiditis. Cytokines have multiple actions in the thyroid in AITD and are likely to determine clinical manifestations such as ophthalmopathy. The role of the TEC in the autoimmune response is not simply passive and, as discussed below, the interaction between TECs and cells of the classical immune system may be critical in determining the outcome of an initially mild thyroiditis.
EXPERIMENTAL THYROIDITIS IN ANIMALS
Chronic thyroiditis histologically identical to that in Hashimoto's thyroiditis occurs spontaneously in Obese strain (OS) chickens (143), beagles (144), mice, and rats. It can be induced in dogs (145), mice, rats, hamsters, guinea pigs, rabbits, monkeys (146), and baboons (147) by immunization with autologous or allogenic thyroid homogenate mixed with adjuvants, or by using heterologous TG , or TG that has been arsenylated or otherwise chemically modified. The need for modification of TG or adjuvant to break tolerance can also be overcome by immunization with cDNA (148). An important thyroiditogenic epitope includes a thyroxine residue (aa 2553) in human TG (149, 150) but the role of iodination at this site is unclear and may depend on the type of T cell assay system used, as well as other parameters (151). Mice have been the most frequently used model and have provided key insights into genetic susceptibility, pathogenesis and the development of Treg and autoreactive T cell repertoires (152).
Induced thyroiditis leads to formation of humoral antibodies and T cell- mediated immunity. Usually the histologic pattern conforms to that of T cell-mediated immunity (153). The role of TG antibodies is unclear but likely to be minor. An idiotype-anti-idiotype network exists for TG antibodies in mice but the induction of those antibodies does not lead to thyroiditis (154). Furthermore, the intensity of the thyroiditis correlates better with T cell-mediated immunity than with antibody levels, and can be transferred by T cells but not antibodies, and both CD4+ and CD8+ T cells are usually needed for transfer (155). In normal mice, thyroiditis can be produced by immunization with mouse TG in adjuvant, and transferred to isogenic animals by sensitized Ly-1+ T cells. The same cells, given before immunization, vaccinate against the development of thyroiditis during subsequent immunization (156).
However, a subpopulation of CD4+ T cells has an important regulatory role in tolerance to murine TG, keeping in check those TG-reactive T cells which escape thymic deletion and peripheral anergy-inducing mechanisms (157). Amelioration of thyroiditis by oral administration of TG (158) operates through enhancing the activity of these regulatory T cells although other mechanisms are possible. More recent studies have emphasized the importance of regulatory T cells in suppression of thyroiditis in animals immunized with TG. In particular, semi-mature dendritic cells, which can be induced with granulocyte-macrophage colony stimulating factor, can induce the function of TG-specific CD4+, CD25+ T cells which can suppress thyroiditis through the production of IL-10 (159, 160).
Another model has used homologous (murine) TPO in an immunization protocol and this method established thyroiditis and TPO antibody production although none of the immunized mice developed hypothyroidism (161). HLA-DRB1*0301 (DR3) transgenic mice have been created which are susceptible to thyroiditis induced by TG immunization, unlike DR2 transgenics, thus confirming that HLA-DRB1 polymorphism determines susceptibility to autoimmune thyroiditis, and his model has been extended to study of the immune response to TSH-R, with results again showing the importance of the DR3 specificity (162). However, when modeling has attempted to reproduce Graves’ disease by immunization of mice with adenovirus expressing the TSH-R, it is non-MHC genes which play a major role in controlling the development of hyperthyroidism (163). This concurs with the polygenic susceptibility and rather weak effect of HLA-DR3 in Graves’ disease. The DR3-transgenic model has also been used to show that dietary iodide enhances the development of thyroid disease and depletion of CD4+, CD25+ Tregs exacerbates this iodide-induced thyroiditis (193a)
Spontaneous thyroiditis in OS chickens more closely resembles Hashimoto’s thyroiditis than the immunization models just discussed, particularly as the birds develop hypothyroidism as a consequence of the autoimmune process. Some evidence suggests that the thyroid of the newly hatched chick is intrinsically abnormal, since its function is partially non-suppressible by thyroid hormone and this constitutes an important element of the genetic susceptibility of these birds, together with genes controlling T cell responses and possibly glucocorticoid tonus. The MHC conversely has only a limited effect. Iodine plays a critical role in the induction of thyroid injury in OS chickens, most likely through the generation of reactive oxygen metabolites, and this injury is an early event, preceding lymphocytic infiltration (165). Iodination of TG is a second path by which iodine influences disease in OS chickens, as autoreactive T cells respond to the antigen only if it is iodinated (166).
Lymphocytic thyroiditis occurs spontaneously in the Buffalo and BB/W rat strains and the NOD (non-obese diabetic) mouse, especially the NOD.H-2h4 line (167). In both species, there are associated abnormalities in the animals' immune system. As in the OS chicken, administration of excess iodine augments the incidence of rat thyroiditis and iodine depletion reduces it (168). Iodine also enhances the susceptibility of NOD mice to thyroiditis, and further exploration of this model has demonstrated a key role for Th17 cells which accumulate within the thyroid (169). IL-17-deficient mice have a markedly reduced frequency of TG autoantibodies and thyroid lesions. Furthermore, selenium supplementation lowers serum TG antibody levels and decreases the prevalence of thyroiditis and the degree of infiltration of lymphocytes in iodine-treated NOD mice (169a). The susceptibility of NOD mice has also been exploited in a model in which the CCR7 gene was knocked out in this strain: such mice do not develop diabetes but do develop severe inflammation elsewhere including a severe thyroiditis with TG autoantibody formation and hypothyroidism (170). CCR7 is a chemokine receptor which is expressed by Tregs; the CC7-deficient mice had lower numbers of these cells. As well as this effect, it is possible that CCR7 deficiency impaired negative selection of thyroid reactive T cells.
Another intriguing aspect of this model comes from long-term observations in NOD.H-2h4 mice which have shown that TG antibodies occur initially and much later TPO antibodies appear, suggesting that tolerance at the B cell and presumably T cell level is broken first for TG and then by spreading (see above) for TPO (171). These results suggest a more important role for TG as an autoantigen in AITD than it is currently assigned. When engineered through CD28 knockout to have a deficiency of Treg cells, NOD mice develop more severe thyroiditis than control animals, with thyroid fibrosis and hypothyroidism. Transferring healthy Treg cells reduces thyroiditis without increasing the total number of Treg cells, suggesting that endogenous Tregs in these mice are functionally defective (172).
The iodine-accelerated thyroid autoimmunity which occurs in NOD.H2(h4) mice is associated with TG and TPO but not TSH-R autoantibodies However transgenic animals expressing the human TSH-R A-subunit develop pathogenic TSH-R antibodies which can be detected in standard bioassays, and this is especially the case in female animals (172a). These antibodies only weakly cross-react with the murine TSH-R and so do not cause hyperthyroidism.
A third kind of model is produced by manipulation of T cells. The original description of thyroiditis in genetically susceptible rats by sublethal irradiation and thymectomy (173) has been followed by a number of more refined models in which T cell subsets can be perturbed more or less specifically to induce disease. For instance CD7/CD28 double-deficient mice have impaired Treg function and such animals develop spontaneous thyroiditis after 1 year of age (174). These experiments clearly demonstrate the recurrence of autoreactive T and B cells in normal animals and show that any of a number of factors which can perturb the regulation of these could result in autoimmune thyroiditis (Fig. 7-10). The most elegant model resulting from T cell manipulation is the generation of transgenic mice expressing a human T cell receptor specific for a TPO epitope, which resulted in a spontaneous destructive hypothyroidism and hypothyroidism (175). The CD8 T cells recognizing the epitope in these animals unconventionally were MHC class II rather than class I restricted and it is unclear whether this atypical behavior is significant to the creation of the model, nor is it yet clear what the mechanism is for thyroid cell destruction.
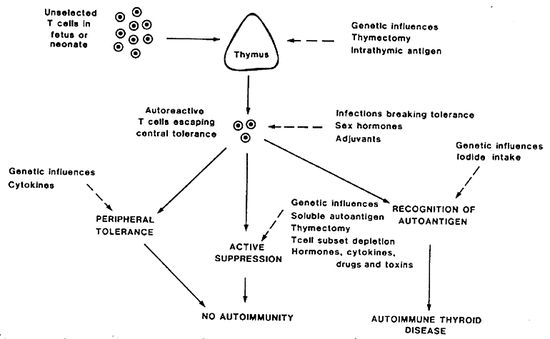
Figure 7-10: Control of thyroid antigen-specific T cells in experimental autoimmune thyroiditis. Development of disease depends on the balance of these factors, and their sites of operation are shown as dotted lines. Reproduced from (255) with permission.
Another intriguing model is one in which necrotic thyroid cells can induce maturation of dendritic cells in vitro, and when injected back into autologous mice EAT is induced, with a lymphocytic thyroiditis and TG-specific IgG (176). It is not clear whether this protocol yields cryptic TG epitopes which can break tolerance. It is possible that such work could be reversed therapeutically to allow attenuation of EAT by pulsing tolerogenic dendritic cells.
Establishing an animal model of Graves’ disease has been surprisingly difficult despite the cloning of the TSH-R. Spontaneous models are not obvious, suggesting that critical differences in the TSH-R receptor between man and other mammals (such as glycosylation) may be necessary to break tolerance (177). However, immunization of AKR/N mice (but not other strains sharing the same MHC haplotype) with murine fibroblasts doubly transfected with the human TSH-R and haploidentical MHC class II genes results in a syndrome similar to Graves’ disease except that thyroid lymphocytic infiltration was not induced (178), whereas thyroiditis is a feature of immunization with the TSH-R (179). This is a promising model although its exact physiological parallel remains unclear, particularly as fibroblasts may behave differently to TECs in terms of antigen presentation. This is because the fibroblasts used express the critical costimulatory molecule B7-1 and also because the procedure causes generalized in vivo immune activation. This model is therefore not evidence that thyroid follicular cells (which do not normally express B7) could initiate thyroid autoimmunity.
More recent models include the use of transgenic mice expressing the A-subunit of the TSH-R, which develop lymphocytic infiltration of the thyroid, hypothyroidism and autoantibodies against TG and TPO as well as TSH-R following immunization with the TSH-R expressed in adenovirus and regulatory T cell depletion (180). Although obviously a contrived system, this model does clearly show that spreading of the immune response can occur to include the normal array of antibodies found in patients, and that this can result in a severe thyroiditis. Some of the difficulties in producing reliable animal models of Graves’ disease are seen in the disparity between hyperthyroidism in the animal and the presence of TSH-R antibodies detected by bioassays using human TSH-R. This may be the result of loci in the immunoglobulin heavy chain variable region contributing in a strain-specific manner to the development of antibodies specific for the human or the mouse TSH-R (181). This novel finding of a role for immunoglobulin heavy chain variable region genes in TSI specificity indicates a possible role for them genetic susceptibility to human Graves' disease.
One unexpected finding has been the observation that mice with a TSH-R knockout do not differ in their response to immunization with TSH-R when compared to healthy animals, whereas the expectation was that such animals would have no tolerance to this autoantigen (as it had been absent throughout development) and therefore a greater immune response would be predicted (182). This suggests that thymic (central) tolerance is not a critical step in self tolerance to this autoantigen. The same conclusions have been drawn from the finding of similar intrathymic transcript levels of thyroid autoantigens (TPO and TSH-R) in mice which are genetically susceptible or resistant to the development of EAT (183). However the situation may be more complex than originally imagined, as the same group has identified a role of the Aire gene in the response to TSH-R and in Aire-deficient mice, intrathymic transcripts of TSH-R and TG are reduced while the expression of TPO is nearly abolished (184). These results are compatible with the finding of an increase in AITD in autoimmune polyglandular syndrome type 1, but at a much lower frequency than the classical disorders of Addison’s disease and hypoparathyroidism. It is also intriguing that TPO transcripts are so much more affected in the Aire-deficient murine thymus, perhaps explaining (via more rigorous tolerance) the rather weak response to this autoantigen, compared to TG, in the mouse.
Balb/c strain mice appeared to develop orbital changes suggestive of ophthalmopathy when given TSH-R primed T cells derived from donor mice immunized with TSH-R protein or cDNA but this model has not proved reproducible by the original authors, for reasons which are not yet clear, although complex histological artefacts may be part of the answer (185). A somewhat more convincing model of ophthalmopathy has been described recently in which deep injection of plasmid containing the TSH-R A subunit into the leg muscles of BALB/c mice followed by electroporation resulted in a wide variety of histological orbital changes and obvious eye signs (41). However the animals developed TSH-R blocking rather than stimulating antibodies and thyroiditis was absent. Nonetheless these finding support a pathogenic role for the TSH-R in the pathogenesis of thyroid eye disease.
The clear general concept to be derived from all of these studies is that a genetically controlled balance of helper and suppressor T cell function is needed to prevent autoimmunity, and that a variety of perturbations can lead to onset of the disease.
Relation of the Immune Response to THE Thyroid Cell: Stimulation and Destruction
For certain we know that the autoantibodies can stimulate the thyroid and cause overactivity in Graves' disease, and can in select circumstances inhibit thyroid function and cause hypothyroidism in neonates and some adults. Whether thyroid antibodies are primary cytotoxic agents in AITD remains an unsettled issue. TG antibodies are probably not normally cytotoxic, but TPO antibodies can certainly mediate complement-dependent thyroid cell cytotoxicity and ADCC. However, the frequently reproduced natural experiment of transplacental antibody passage from a mother with AITD to her fetus, without evidence of thyroid damage, clearly shows that antibodies alone are not destructive to the thyroid.
Cell-mediated immunity is thought to be important in thyroid cell destruction, and T cells have been shown to be reactive to TECs. T cell lines or clones have been shown to react to TECs (108-110), but the nature of the antigen recognized is unknown. One CD8+ T cell clone in man has been shown to be cytotoxic specifically to autologous TECs (121), suggesting that cell-mediated TEC destruction is an important process, and similar activity has been reported in CD8+ T cell lines and clones derived from mice with experimental autoimmune thyroiditis (186). A second type of T cell-mediated cytotoxicity is that mediated by gd TCR-bearing T cells and specific recognition of TECs by such cells has been reported in Graves’ disease, but the exact autoantigen involved is unknown (187). In animals it is clearly shown that there can be a marked dissociation between the extent of histologic thyroiditis and the levels of antibodies, again suggesting that T cells rather than antibodies mediate cell destruction. However, it must be admitted that the hard evidence for direct T cell-mediated cytotoxicity in thyroid autoimmunity in man is meagre at present.
There are 3 mechanisms by which T cells might mediate TEC destruction and evidence for all 3 operating in AITD has accrued. Firstly, cell lysis might be effected via T cell-derived perforin, which leads to pore formation in target cell surfaces, and certainly the thyroid lymphocytic infiltrate contains perforin-expressing T cells in AITD (188). Secondly, T cells expressing Fas ligand, especially the CD8+ subset, can induce apoptosis in TECs expressing Fas (189). Fas is induced by IL-1b on TECs, whereas TSH-R stimulation inhibits Fas expression (190) and this may lead to the involvement of this pathway in Hashimoto’s thyroiditis but not Graves’ disease, as TSI would act like TSH in the latter to diminish Fas expression (and other regulatory molecules). It has been suggested that T cells may not be necessary, as Hashimoto TEC may express Fas ligand, and autocrine/paracrine interaction with Fas may lead to TEC death (191). The mechanisms for this are unclear and as yet there is no consensus on the role this may have in AITD. The picture is complicated by the upregulation of molecules which protect against apoptosis such as Bcl-2. The pattern of expression of this molecule is different in Graves’ and Hashimoto’s diseases, suggesting that TECs are protected in the former and more sensitive to destruction in the latter (192). Whether these differences depend on cytokines, genetics or other factors is unknown (193). Finally, T cell-derived cytokines can injure the TECs directly, leading to functional impairment (133-135), and by triggering other phlogistic pathways such as nitric oxide synthesis (194).
Possible Explanations for Autoimmunity
Many reasons for the development of autoimmunity have been advanced, and these are briefly catalogued below. Cross-reacting epitopes, aberrant T or B cell regulatory mechanisms, inheritance of specific immune response-related genes, and aberrant HLA-DR expression on TECs have all at some time been considered important for development and progression of thyroid autoimmunity.
- Abnormal presentation of antigen could occur due to cell destruction, or viral invasion, so that large amounts of antigen or cell fragments are liberated locally into the lymphatics. Excessive levels of antigen are produced, thereby overwhelming the usual low dose tolerance mechanism.
- Abnormal antigen could be produced by a malignancy, or damage to the cell by viral attack, or other means. This antigen could be a partially degraded or denatured normal antigen, for example.
- Cross-reacting bacterial or viral epitopes e.g. Yersinia enterocolitica (195) could induce immune responses that happen to cross-react with a self-antigen having identical conformation. An extension of this concept is that the normal anti-idiotypic control response happens to produce an Ig or T cell that cross-reacts with self-antigen. For example, experimentally produced anti-idiotypic monoclonal antibodies directed to TSH antibodies bind to and stimulate the TSH-R (196).
- Somatic mutation of a TCR gene could lead to a clone of self-reactive cells. However, somatic mutation of TCR genes is believed to occur very rarely if at all, and such monoclonal or oligoclonal activation has not been documented in autoimmune disease. Somatic mutation of B cell Ig genes is, as described above, a normal phenomenon during an antigen‑driven proliferative response. Such an event could occur by chance during response to any antigen and this does not effectively introduce any new variable, since B cells capable of producing Igs that can react with self-antigens are already normally present. However, TSI seem to be clonally restricted and, until the V gene usage of these antibodies is documented, it remains possible that Graves’ disease is due to the inheritance of a unique, etiologically critical V gene encoding TSI.
- Inheritance of specific HLA, TCR, or other genes that code for proteins having especially effective ability to process or present antigen.
- T cell or B cell feedback control mechanisms could be aberrant due to hereditary or environmental factors.
- Failure of clonal deletion could leave self-reactive T cells present in the adult. In fact this is clearly normal, as described above.
- Failure of normal maturation of immune system could allow fetal T and B cells that are autoreactive and of wide specificity to persist.
- Polyclonal activation of T or B cells, by some unknown stimulus, could lead to B cells producing self-reactive Ig, in the apparent absence of antigenic stimulus. This theory is in a sense impossible to disprove but would need to co-exist with other abnormalities to explain disease remission, genetic associations, associated diseases, etc. Polyclonal activation is not typical of peripheral lymphocytes of patients with AITD (197).
- TECs could express MHC class II molecules as a primary event and then could function as APCs, including antigens on their cell surface.
- Environmental factors could distort normal control. For example, stress or steroids may alter immunoregulation, and the potential role of dietary iodine has been mentioned above.
Abnormal Exposure To Thyroid Antigens And The Effects Of Pregnancy
Damage to the thyroid might release normally sequestered antigens, inducing an immune response. Damage to thyroid cells does indeed occur in viral thyroiditis, such as in association with mumps or in subacute thyroiditis of unknown cause, but autoantibodies appear only transiently at low titer, and progressive lesions of the thyroid do not usually occur (reviewed in 198, 199). External irradiation to the thyroid, including that from nuclear fallout, can also lead to an increase in Graves' disease or thyroid antibody production (200, 201), but it is unclear if this is caused by autoantigen release or an effect on the lymphocytes which are radio-sensitive. Even occupational exposure to ionizing radiation appears to be a risk factor for the development of autoimmune thyroiditis (202). Another possible example where exposure to thyroid antigens released by gland injury leads to autoimmunity is the rare case of precipitation of Graves’ disease and ophthalmopathy after ethanol injection of thyroid nodules (203).
A powerful argument against the hidden antigen hypothesis is that TG is a normal component of circulating plasma (204). One might turn the first argument around and suggest that thyroiditis results from a lack of exposure to TG at some period, an exposure that is necessary to depress continuously an otherwise inevitable immune response. This suggestion has no clinical or experimental support, and the available evidence indicates that TG is present in the plasma of patients with active immunity. It remains to be seen how sequestered TPO and TSH-R are, but the appearance of T cells capable of proliferating in response to these antigens, in apparently healthy individuals, also argues against any sequestration (205). What is clear is that availability of the thyroid autoantigen is essential to maintain the autoimmune response: complete removal of thyroid antigens following thyroidectomy and remnant ablation with radioiodine leads to disappearance of antibodies to TG, TPO and TSH-R (206). Although this is not surprising, it does suggest that extrathyroidal sources TSH-R are insufficient normally to maintain an autoimmune response.
A variant on this theme is that of microchimerism, the persistence of fetal cells in maternal tissues. Studies have found evidence of microchimerism in thyroid tissue from patients with and without AITD (207, 208). Could such sequestered fetal material make the thyroid prone to an alloimmune response, and be responsible for the exacerbation of AITD seen in the postpartum period? If so, this phenomenon would help to explain the high frequency of AITD in women. Twins from opposite sex pairs should have an increased risk of thyroid autoimmunity compared to monozygotic twins if microchimerism has a role, and indeed such twins have been found to have more frequent thyroid autoantibodies (209). However, although parity is associated with an 11% increase in the risk of all female-associated autoimmune disorders, there is no increase with multiple pregnancies, which rather argues against a microchimerism mechanism (210). During and after pregnancy, major changes in Treg function occur and direct effects on the cytokines produced by T cells can also be demonstrated (211). It is these alterations that are most probably the ultimate cause of the increase in autoimmunity after pregnancy.
It seems likely that sex steroids play a role in determining the autoimmune response. For instance, in a recent study of an animal model of Graves’ disease, 5α-dihydrotestosterone was given to mice a week before immunization with TSH-R, and this reduced both the severity of the hyperthyroidism that developed and downregulated the Th1 response (211a). Another hypothetical reason for the unequal sex ratio is that skewed X chromosome inactivation could contribute through the failure of some autoantigens expressed on one X chromosome to be expressed at a critical point in the disease pathway. A recent survey of 309 patients with Graves’ disease and 490 with Hashimoto’s thyroiditis found skewed inactivation of the X chromosome in Graves’ disease (odds ratio 2.2) but not Hashimoto’s thyroiditis; when combined with 4 other studies in a meta-analysis, the results remained significant for Graves’ disease and reached significance for Hashimoto’s thyroiditis (odds ratio 2.4) (212).
Abnormal Antigens
An abnormal antigen might also serve to produce an immune reaction. The protein abnormality could be either congenital or acquired by an injury such as a virus infection. To date there is no evidence which indicates that TG, TPO, or other proteins of the thyroid of a patient with autoimmunity are abnormal. Minor allelic differences apparently do occur but attempts to associate thyroid disease with polymorphisms of the TPO and TSH-R genes have been unsuccessful.
Cross-Reacting Antigens
The theory that immune reactivity to an environmental antigen could lead to antibodies that cross‑react with thyroid antigens has been bolstered by studies which show a relationship between Graves' disease and antibodies to the common enteropathogen Yersinia enterocolitica. An increased incidence of antibodies to Yersinia is found by some, but not all authors, in patients who have Graves' disease, and there are saturable binding sites for TSH on Yersinia proteins (213). After infection by Yersinia, human sera contain Igs that bind to TEC cytoplasm (195), and IgGs which appear to compete with TSH for binding to thyroid membrane TSH receptors (214). The antigens involved may in fact include proteins encoded by plasmids present in the Yersinia, rather than intrinsic Yersinia proteins, but that does not alter the general concept (215). Arguing strongly against a role for Yersinia is the fact that there is no unique pattern of serological immunoreactivity to Yersinia antigens in patients with AITD (216), and most patients with this infection do not develop Graves’ disease. Moreover, there was no association between Yersinia infection and autoimmune thyroid disease in a large prospective study of individuals developing AITD (217).
In theory an initial response to one antigen might proceed by reacting to the other antigen, and thereby spread and augment the autoimmune process. In the context of T cell autoreactivity there is much greater scope for molecular mimicry whereby a response to an exogenous epitope leads to a cross-reactive response to an endogenous autoantigenic epitope. Simple sequence homology is insufficient to predict this, as shown elegantly by the cross-reactivity of two TPO epitopes showing a similar surface but not amino acid sequence (218). This makes the prediction and study of molecular mimicry much more difficult than is generally appreciated (219). For these reasons, it may be naïve to believe that the putative orbital antigen responsible for ophthalmopathy has to be an identical protein to that expressed in the thyroid.
Virus Infection
Virus infection has for years been speculated to be an etiological factor in most autoimmune diseases, by causing cell destruction and liberating antigens, by forming altered antigens or causing molecular mimicry, by inducing DR expression, or by inducing CD8+ T cell responses to viral antigens expressed on the cell surface. Thyroid autoantibodies are elevated transiently after subacute thyroiditis, which is thought to be a virus-associated syndrome, but no clear evidence of virus-induced autoimmune thyroiditis in humans has been presented. In this regard it is of interest that persistent, apparently benign virus infection of the thyroid can be induced in mice (220), and that infection of neonatal mice with reo virus induces a polyendocrine autoimmunity (Fig. 7-11). These agents could work by liberating thyroid antigens. Virus infection might also augment autoimmunity by causing non-specific secretion of IL-2, or by inducing MHC class II expression on TEC. Despite many attempts to implicate retroviruses in AITD, results to date remain inconclusive (221). Human T lymphotrophic virus-1 has been repeatedly associated with various autoimmune disorders, including Hashimoto’s thyroiditis; presumably the virus alters immunoregulatory pathways allowing autoimmunity to emerge (222).
Figure 7-11: Autoantibodies to thyroid in sera of reovirus-infected mice detected by indirect immunofluorescence. (a) Frozen section of normal mouse thyroid incubated with sera obtained from mice 21 days after infection, showing staining of colloid characteristic of antithyroglobulin antibody (original magnification, X200). (b) Section of normal mouse thyroid (fixed in Bouin's solution) incubated with sera obtained from mice 21 days after infection, showing staining of thyroid acinar cells (original magnification, X 200). Reproduced with permission from I. Okayasu and S. Hatokeyama, Clin. Immunol. Immunopath., 31:334, 1984.
Lymphocyte Mutation And Oligoclonality
Apart from the evidence that some TSI may have an oligoclonal origin (67, 223), there is no evidence to support a clonal B cell abnormality in AITD. V gene usage by TSI will need to be analyzed to determine whether Graves’ disease has a unique pathogenesis determined by germ-line immunoglobulin genes. Thyroid-reactive T cells are present in healthy animals and man, as noted above, and therefore a defect at the clonal T cell level is less likely as a primary event in etiology than previously thought. A few autoreactive T cells can be expected to escape tolerance normally, particularly if the autoantigen in question is not available to delete T cells in thymus during fetal development. Stochastic events later in life affecting such undeleted T cells could readily explain the lack of complete concordance for AITD in genetically identical twins (224), and this lack of such concordance argues against an inherited pathogenic TCR as a primary event in AITD.
Genetic Predisposition
A role of heredity in AITD is clearly demonstrated by family studies (225, 226). The role of heredity in AITD is clear, since there is an increased frequency of AITD among family members, first degree relatives, and twins of patients with the illness (227). Indeed a detailed analysis of concordance in Danish twins with Graves’ disease came up with the estimate that 79% of the liability for this disorder was attributable to genetic factors (228). Another strand of evidence is the variation of disease with race, although of course this is complicated by environmental influences too. Analyzing military personnel in the USA, it has been shown that HT is more frequent in white individuals, and lowest in black and Asian/Pacific Islander individuals (229). Despite some shared genetic susceptibility factors (see below), in Graves’ disease the opposite is true. It is unknown why these ethnic differences occur and this is clearly an area that could be fruitfully explored further.
In an investigation of the relatives of a group of propositi with high circulating antibody levels and clinical thyroid disease, approximately half of the siblings and parents (first‑order relatives) were found to have significant titers of thyroid antibodies, many being without clinical thyroid disease (230) but the transmission of thyroid autoantibodies is a more complex trait than the dominant inheritance originally thought (231, 232).
Together, such observations suggest that these diseases have a common genetic defect, although other genes are likely to be disease-specific in their effects, as reviewed extensively elsewhere (233). The most important susceptibility factor so far recognized is the inheritance of certain MHC class II genes. Inheritance of HLA-DR3 causes a 2 to 6-fold increased risk for the occurrence of Graves' disease or autoimmune thyroiditis in Caucasians, and inheritance of HLA-DR4 and DR5 has been found in some studies to increase the incidence of goitrous hypothyroidism (234). In post-partum painless thyroiditis an association with HLA-DR5 has been reported (235). HLA-DQA1*0501, which is often linked to DR3, may have an even more pronounced predisposing effect in Caucasians with Graves’ disease (236), whereas HLA-DRB1*07 may be protective (227). A large series of 991 Japanese patients with AITD has been studied and the HLA susceptibility to Graves’ disease differentiated from that to Hashimoto’s thyroiditis, while 3 common haplotypes were identified which conferred protection against Graves’ disease; one of these acted epistatically with the HLA-DP5 susceptibility molecule and another also conferred protection to Hashimoto’s thyroiditis (238). It is noteworthy also that the relative risks conferred by HLA alleles is rather modest, borne out by the relatively low concordance for Graves’ disease in HLA-identical siblings of patients with Graves’ disease (239). This suggests the operation of other genetic susceptibility loci, also emphasized by the weak lod scores for linkage with the HLA region in family studies of AITD (240, 241).
The nature of these other loci is unclear and their identification is likely to require an extensive analysis involving thousands of families in studies using modern molecular techniques. Association studies have been the method of choice until recently, investigating various candidate genes, but with mixed success. Inconclusive results have been reported for associations of AITD with TCR polymorphisms, immunoglobulin allotype and TSH-R polymorphisms. The most consistent non-HLA association is between polymorphisms in the CTLA-4 gene and both Graves’ disease and Hashimoto’s thyroiditis (242, 243). Despite claims to the contrary, there appears to be no additional risk conferred by CTLA-4 (or HLA) polymorphisms in Graves’ patients with clinical evidence of ophthalmopathy (244), but these CTLA-4 polymorphisms may partially determine outcome after antithyroid drug (245, 246). Given the most important role of the interaction between CTLA-4 on T cells and the B7 family of molecules on APCs, it is possible that this association represents a genetic effect on immunoregulation, although, as with HLA-DR3, this is not specific for thyroid autoimmunity; the same polymorphism is also associated with type I diabetes mellitus and several other autoimmune disorders. Fine mapping of the CTLA-4 region has confirmed that it is indeed this gene, rather than those in linkage disequilibrium, which is responsible for the associations, and the polymorphisms may exert their effects by causing variation in levels of soluble CTLA-4, which in turn may after T cell activation, especially in Treg cells (247).
Polymorphism of the vitamin D receptor has been linked with Graves’ disease, an association which has some biological plausibility as vitamin D has immunological effects (248). However a large survey comprising 768 patients with Graves’ disease from the UK, compared to 864 controls, found no evidence of an association (249) and there is not yet any prospective evidence yet for vitamin D deficiency being associated with AITD (250). Polymorphisms in genes encoding molecules involved the NFkB inhibitor pathway modulating B cell function (FCRL3 and MAP3K7IP2) are more likely to be involved in susceptibility to Graves’ disease (251, 252).
Another genetic susceptibility locus in Graves’ disease is polymorphism in the lymphoid tyrosine phosphatase LYP/PTPN22 gene, which has been associated with functional changes in T cell receptor signaling. A study of 549 patients and 429 controls found that a codon 620 tryptophan allele conferred an odds ratio of 1.88 (253), although it should be noted that similar effects have been seen in many other autoimmune diseases. This result has recently been confirmed (254) and another likely locus is the IL-2 receptor alpha (CD25) gene region, which is also associated with other autoimmune diseases like type diabetes (255).
As well as genes controlling the immune response, genes that control the target organ susceptibility to autoimmunity are logical candidates for investigation. There is to be conclusive proof from both linkage disequilibrium and association studies, that polymorphisms in the TSH-R gene confer susceptibility to Graves’ disease but not autoimmune hypothyroidism (256, 257). This is one of the few susceptibility factors that segregates with one rather than both types of thyroid autoimmunity, although polymorphisms in the PDE10A and MAF genes (which have many actions, including immune regulation) may also influence whether patients develop Graves' disease or Hashimoto's disease (257a). Although not thyroid-specific in tissue location, selenoproteins (SEP) are central to thyroid hormone deiodination and a significant association of HT with SNP in SEPS1 (odds ratio 2.2) has been reported in a series of 481 Portuguese HT patients (258).
A different approach to chasing candidate genes has been genome scanning, although huge effort is required to undertake such studies. Based largely on this approach, other loci which may be important have been identified on chromosomes 14q31, 20q11 and Xq21 (241, 259), and the importance of a gene on the X chromosome is supported by the increased frequency of AITD in women with Turners syndrome, especially those with an isochromosome-X karyotype (260). However in a genome scan involving 1119 relative pairs, there was no replication of these findings (261). A more impressive genome wide scan of thousands of individuals with Graves’ disease confirmed susceptibility loci in the major histocompatibility complex, TSHR, CTLA4 and FCRL3 and identified two new loci; the RNASET2-FGFR1OP-CCR6 region at 6q27 and an intergenic region at 4p14 (262). Seven new loci for AITD, including MMEL1, LPP, BACH2, FGFR1OP and PRICKLE1, have been uncovered by using a custom made SNP array across 186 susceptibility loci known for immune-mediated diseases (263). In another study of almost 10000 Chinese patients with Graves’ disease, five additional novel loci were identified and polymorphism in the TG gene was also confirmed to be associated with Graves’ disease (264). Thus the genetic factors involved in AITD are increasingly more complex and their interactions with each other and with environmental factors in disease pathogenesis will be a major task to uncover.
Further developments in genetic analysis will no doubt bring even greater complexity to this area, albeit with the prospect of better defining patient subsets (265). It is now clear that to detect common, low-risk variants with reliability, huge sample sizes are essential facilitated by the haplotypic data available from the HapMap project, which means that genome wide variability can be detected using half a million single nucleotide polymorphisms (266). These studies present considerable logistical challenges, and many older studies of genetic associations in AITD have produced conflicting results as because of lack of power or population stratification issues. However a good example of the utility of such studies is a massive genome-wide association study in which a new set of SNPs, which includes polymorphism in MAGI3, has been associated with an increased risk of progression from TPO antibody positivity without hypothyroidism to the development of hypothyroidism (267).
As an aside, it should be noted that low birth weight, a known risk factor for several chronic disorders, has not associated with clinically overt thyroid disease or with the production of thyroid autoantibodies in one study (268) but others have come to an opposite conclusion, with prematurity irrespective of birth size being another risk factor (269, 270).
Co-Occurrence Of Autoimmune Diseases
The co-existence of AITD and other diseases possibly of autoimmune cause has often been reported, and suggests some intrinsic abnormality in immune regulation. An extensive review of these associations has been published (271) and extensive population data bases have clarified the strength of the various associations (272). A striking association is with pernicious anemia. Perhaps 45% of patients with autoimmune thyroiditis have circulating gastric parietal cell antibodies (273), and the reverse association is almost as strong (274). Up to 14% of patients with pernicious anemia have primary myxedema, and pernicious anemia is increased in prevalence in patients with hypothyroidism (275).
Another strong association is with celiac disease, which is found 3 times more commonly in patients with AITD. Intriguingly the autoantibodies which are the hallmark of celiac disease, directed against transglutaminase, can bind to thyroid cells and thus could be implicated directly in thyroid disease pathogenesis (276). The association of Sjögren's syndrome and thyroiditis is not uncommon and both systemic lupus erythematosus (SLE) and rheumatoid arthritis are also significantly associated with AITD (277, 278). A high frequency of antibodies to nucleus, smooth muscle, and single-stranded DNA (26-36%) is found in AITD (279). Although multiple sclerosis has stood out as a putative autoimmune disease which is not obviously associated with AITD, meta-analysis has revealed there is an odds ratio of 1.7 for AITD in these patients (280).
Autoimmune Addison's disease and/or type I diabetes mellitus and AITD occasionally co-exist and this forms the autoimmune polyglandular syndrome (APS) type 2 (281). This is an autosomal dominant disorder with incomplete penetrance and is often associated with other disorders, such as vitiligo, celiac disease, myasthenia gravis, premature ovarian failure and chronic active hepatitis (282, 283). AITD is an infrequent feature of the much rarer APS type I (284) and there is no association between mutations in the AIRE gene, which causes APS type I, and sporadic AITD (285).
Together these data provide convincing proof of an association of other autoimmune phenomena with AITD. Most typically, this immunity is organ specific, but in one subset of patients, thyroid autoimmunity develops in association with the non-organic-specific collagen diseases. A syndrome of running together, of course, does not prove a causal association. Nevertheless, the plethora of associations and their familial occurrence indicates that a defect in the immune system may be more likely than primary defects in each organ. This in turn suggests a shared immunoregulatory defect, which is at least partly genetically determined, as these diseases often share similar genetic associations, including HLA, CTLA-4, PTPN22 and CD25 gene polymorphisms. Recently, analysis of HLA molecules has shown a pocket amino acid signature, DRβ-Tyr-26, DRβ-Leu-67, DRβ-Lys-71, and DRβ-Arg-74, that was strongly associated with type 1 diabetes and AITD (286). This could confer joint susceptibility to these diseases in the same individual by causing significant structural changes in the MHC II peptide binding pocket and influencing peptide binding and presentation. It is also clear however that there is a difference in the kind of clustering of other autoimmune disease in Hashimoto’s thyroiditis and Graves’ disease, presumably related to differences between these two types of thyroid disease in genetic predisposition (287).
Immunoregulation: Phenomena And Mechanisms
Possible abnormalities in immunoregulation have been addressed in hundreds of studies. The basic hypothesis of this work is that a deficiency of functional T suppressor cells, now termed regulatory cells, may allow uncontrolled T and B cell immune responses to thyroid (or other) antigens. As noted above, this concept is a major theme in experimentally induced or naturally occurring thyroiditis in animal models. Most of the studies to define immunoregulatory responses in AITD have relied on phenotyping (which may relate poorly to effector function in vivo) or in vitro assays done in unique conditions; as we have previously noted, T cell antigen expression and function can vary depending on source of cells, stage of disease, the use of any stimulating agent in vitro, culture conditions, etc.
Sridama and DeGroot found decreased suppressor cells, defined as CD8+ peripheral blood T cells in patients with Graves' disease (288, 289). These results have been challenged, and some investigators have reported depression of CD4+ cells in AITD (290). However, overall, there is now agreement that, in thyrotoxic patients with Graves' disease, a decrease in CD8+ T cell number (291, 292) is characteristically present, and that a similar abnormality exists in the thyroid. CD8+ cells return gradually toward normal during therapy, and are usually but not always normal at the end of therapy (292) (Fig. 7-12). The phenomenon is present but less evident in Hashimoto's thyroiditis patients. It has been attributed by some workers to increased thyroid hormone levels (293), although this issue is clouded, since there are reports disproving the idea that hyperthyroidism per se induces suppressor cell abnormalities in humans, and reduced suppressor T cells (Ts) are found present long after thyrotoxicosis is cured (294). Our interpretation is that the abnormality is not due specifically to excess T4 in blood, but is a manifestation of ongoing active autoimmunity, for reasons which are unclear. Reduced nonspecific "suppressor" T cell function may be in part an inherited abnormality, and is probably also a manifestation of the augmented immune reactivity ongoing in AITD patients. It may be largely a secondary phenomenon, but one which augments and continues the immunological disease. The mechanism causing such reduced Ts number and function is unclear.
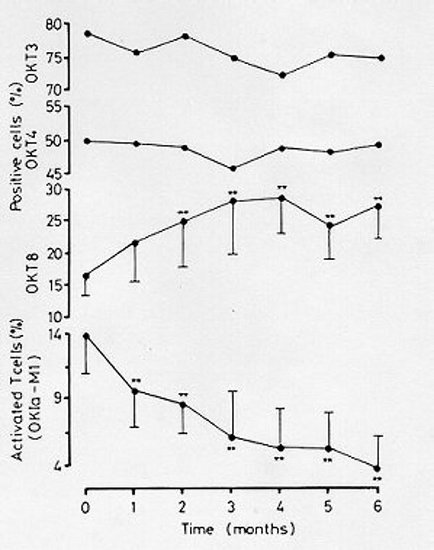
Figure 7-12: Influence of a 6 month course of carbimazole on peripheral blood T cell subsets of 29 patients with hyperthyroid Graves' disease (Mean SD). OKT4 = CD4+ OKT3 = CD3+ OKT8 = CD8+ ** = p < .001 vs. zero time value (From Reference 265)
These older findings need to be related to recent developments in understanding Treg function. One study has found that despite increased numbers of CD4+ T cells bearing the T regulatory cell markers CD25, Foxp3, GITR and CD69, in both thyroid and PBMC of patients with AITD, there is a non-specific defect in regulatory function in vitro, which in turn must explain somehow why the increased number of regulatory T cells are so patently ineffective (295). For example there is an increase in circulating CD69+ regulatory lymphocytes in AITD, and numbers are even higher in the thyroid glands of these patients and yet they are functionally deficient in vitro (295a). The existence of a functional rather than numerical deficiency in regulatory T cells has also been suggested in a study of AITD patients, in which the defect was found to be detectable only when optimal in vitro conditions were achieved (296). Analysis in the earliest phases of disease may of course yield different results and unlocking how T regulatory cells can be activated seems an obvious but at present unrealizable therapeutic strategy. The finding that many thyroid infiltrating lymphocytes, early on in the disease process, are in fact recent thymic emigrants does suggest that there is a problem with central tolerance that allows autoreactive T cells to accumulate in the gland where the strength of local immunoregulation could be critical in determining whether disease progresses (297).
Thyrotoxic Graves' disease patients and those with active Hashimoto's thyroiditis have a high proportion of DR+ T cells in their peripheral circulation (291, 298), which indicates the presence of activated T cells. It is unlikely that these cells (> 20% of circulating T cells) are all responsive related to thyroid antigens, so they must include DR+ T cells with TCRs for many other antigens. There is also a marked increase in circulating soluble IL-2 receptors in thyrotoxic Graves' disease, but this appears to be typical of thyrotoxicosis per se, and not specifically Graves' disease (299). Nevertheless, there is no evidence for a generalized ongoing immune hyper-responsiveness in thyrotoxic patients. Perhaps these T cells (for many different specificities) are stimulated by IL-2, but in the absence of the required second signal provided by antigen exposure, do not induce B cell proliferation or cytotoxic responses.
Diminished, non-specific suppressor cell function is also observed in many autoimmune diseases including lupus, and multiple sclerosis and the results in AITD are equally non-specific. The most likely explanation for many “suppressor” phenomena is the reciprocal inhibition of Th1 and Th2 cells by their cytokine products, and powerful evidence shows how important this regulatory mechanism is in exacerbating or inhibiting autoimmune disease, at least in animal models. However regulatory phenomena utilizing cytokines are much more complex, and include both Th17 cells and invariant NKT (iNKT) cells. The latter share receptors with T and NK cells, with the α chain of the T cell receptor being invariant gene segment-encoded, and are notable for releasing cytokines when stimulated by antigen, thus endowing them with regulatory properties which may be either stimulatory or inhibitory. Recently iNKT cell lines have been identified that can be stimulated with TG to induce EAT (300).
In keeping with the importance of the Th17 subset in inflammatory autoimmune diseases discussed earlier, there is an increased differentiation of circulating Th17 lymphocytes and an enhanced synthesis of Th17 cytokines in AITD, mainly in those patients with Hashimoto thyroiditis (301). Nonetheless a recent study has found an increase in both Th22 and Th17 cells and the levels of plasma IL-22 and IL-17 in patients with Graves’ disease; the magnitude of these increases correlated TSH-R antibody levels (302). Circulating platelet-derived microvesicles are significantly raised in AITD patients and these can inhibit the differentiation of Foxp3+ Treg cells and induce differentiation of Th17 cells (302a). Another newly recognized T cell subset involved in the regulation of antibody production, comprising follicular helper T cells, is increased in the circulation of patients with AITD and correlates with autoantibody levels (303).
Many studies have examined T cell subsets in thyroid tissue of patients with active AITD. For example, Margolick et al (304) found increased CD8+ cytotoxic/suppressor cells and also increased CD4+ T helper cells, and a normal Th/Ts ratio. Canonica et al (305) found increased proportions of cytotoxic/suppressor T cells in thyroids of Hashimoto's thyroiditis patients. Infiltrating cytotoxic/suppressor cells in Hashimoto's thyroiditis were found usually to be activated and to express DR antigen, whereas this response was not so obvious in Graves' disease (306). Canonica et al (305) reported an increased proportion of activated T helper/inducer cells in both Graves' disease and Hashimoto's thyroiditis, and increased cells thought to represent cytotoxic T cells in Hashimoto's thyroiditis. Chemokine expression within the thyroid is likely to be an important determinant of this infiltration (307).
Increased CD8+CD11B- cells, presumed to be cytotoxic cells, were found in Graves' disease thyroids (in comparison to PBMC of Graves' disease or normal subjects), whereas "dull" CD8+CD11B+ natural killer cells were diminished (308). Other studies have suggested a reduction in NK cells in Graves' disease and an increase in Hashimoto's thyroiditis. Tezuka et al found decreased NK cells in Graves' disease thyroid tissue, no differences in the NK activity of PBMC between Graves' and normal patients, and that the NK cells in Graves' disease did not kill autologous thyroid epithelial cells (309). We have already indicated other reports of normal NK and ADCC in Hashimoto's PBMC, and of increased ADCC in Hashimoto's thyroiditis. Most studies that have looked at Graves' disease tissues also indicate an increased proportion of B cells compared to peripheral blood subsets.
Cell cloning has also been used to examine thyroid and peripheral blood lymphocyte subsets. Bagnasco et al (310) found a predominance of cytolytic clones, releasing IFN-g, in Hashimoto's thyroiditis but not in Graves' disease. Del Prete et al (311) found a high proportion of cytolytic cells with the CD8+ phenotype in clones from thyroid tissue, and felt these results may relate to autoimmune destruction of TEC but the non-specific methods used to derive such cytotoxic T cells raises questions about any pathophysiological relevance.
There is no clear predominance of Th1 or Th2 cytokines in the thyroid of patients with Graves’ disease or Hashimoto’s thyroiditis (312), although Th1 clones seem to predominate in the retrobulbar tissues in ophthalmopathy (313). It might simplistically be thought that Graves’ disease represents a Th2 response, but the fact that some patients end up with hypothyroidism itself indicates the likely presence of a Th1 response too. This is supported by evidence from an animal model of Graves’ disease: immune deviation away from a Th1 response, in g-IFN knockout mice, did not enhance the response to TSH-R cDNA vaccination (314).
One situation in which it is likely that perturbation the cytokine milieu is responsible for the emergence of Graves’ disease is during reconstitution of the immune system following lymphopenia induced by alemtuzumab treatment for multiple sclerosis, bone marrow or stem cell transplantation or after highly active antiretroviral therapy for HIV infection (315, 316). In these situations there is an initial increase in the Th1 response flowed by a Th2 response at the time when Graves’ disease becomes apparent. Defects in T regulatory cells may also contribute.
A general summary of these data is difficult. The results probably at least indicate there are increased B cells, increased DR+ T cells, increased CD4+DR+ T helper cells, decreased CD8+DR+ T suppressor/cytotoxic cells, and possibly lower NK cells in Graves' disease AITD tissue and in blood than among normal subjects' PBMCs. The intrathyroidal T cells are a mix of Th1 and Th2. Such studies have been performed primarily on patients with well developed and often treated disease, and do not bear directly on early stages of the disease, or on whether the changes represent primary or secondary phenomena. To date there has been no certain indication that a non-specific or specific suppressor cell defect exists in patients who are genetically predisposed to have AITD, or in most patients who have recovered from the illness, although observations on Treg and other recently defined T cell subsets appear to indicate defects that are likely to be causal.
Anti-Idiotype Antibodies
Whereas anti-idiotypic antibodies are thought to play a physiological role in immunoregulation, there is little evidence for participation in, or abnormality of, this function in AITD. Immunoglobulins from some patients with Graves' disease bind TSH (317). This suggests that anti-idiotypes to TSH antibodies are present and might theoretically function as thyroid stimulating immunoglobulins; or conversely that anti-idiotypes to thyroid stimulating antibody exist and can bind TSH. Either possibility remains to be confirmed. Sikorska (318) demonstrated the presence of antibodies in sera of AITD patients which inhibit binding of TG to monoclonal TG antibodies, and interpreted these as anti-idiotypes. We have looked for anti-TG anti-idiotypes in patients with autoimmune thyroid disease and failed to find them (319). On the other hand, weak anti-idiotypes of the IgM class have been found which bind to TPO antibodies and are present in pooled normal immunoglobulins as well as certain patient sera (320). Although one could postulate that a failure to produce anti-idiotype antibodies could be a feature of AITD, a more likely hypothesis is that anti-idiotypic antibodies are simply rarely produced at a detectable level. Since anti-idiotype antibodies raised in animals will suppress in vitro TG antibody production, the theory that lack of anti-idiotype control is causal in AITD remains attractive, but data to support it are scant.
De Novo Expression Of Class Ii Antigens On Thyroid Cells
De novo expression of HLA-DR on thyroid epithelial cells, from patients with Graves' disease, was first reported by Hanafusa et al (321) and was proposed as the cause of autoimmunity by Bottazzo et al. (322) who suggested that de novo expression of MHC class II molecules on these cells, which are normally negative, allows them to function as APCs. Lymphocyte-produced IFN-g augments the expression of HLA-DR (also DP and DQ) on thyroid epithelial cells, and that TNF-a further increases the induction caused by IFN-g (323, 324). HLA-DR+ TECs definitely can stimulate T cells (325, 326) but this is critically dependent on the requirements of the T cell for a costimulatory signal, as Graves’ TECs do not express B7-1 or B7-2 (327, 328). In contrast B7.1 expression on Hashimoto TEC has been recorded, but how this is differentially regulated, compared to Graves’ disease, is unknown (329). We have shown that TECs can present antigen to T cell clones which no longer require costimulation through B7, yet not only fail to stimulate B7-dependent T cells but also induce anergy in these cells by at least two mechanisms, one of which is Fas-dependent (330, 331). Perhaps the most conclusive proof that class II expression by thyroid cells cannot induce thyroiditis comes from the creation of transgenic mice expressing such molecules on TECs – such animals have no thyroiditis and have normal thyroid functionm (332). For reasons which remain unclear, thyroid follicular and papillary cancers may express B7.1 and B7.2, and B7.2 expression is associated with an unfavourable prognosis (333).
HLA-DR is also expressed on TECs in multinodular goiter and in many benign and malignant thyroid tumors, and this does not appear to induce thyroid autoimmunity (334). Aberrant DR expression has not been shown to develop before autoimmunity. Normal animal thyroids not expressing class II molecules can become the focus of induced thyroiditis, and then express class II molecules (335). Furthermore, HLA-DR expression on Graves' disease thyroid tissue is lost when tissue is transplanted to nude mice (336). Thus a consensus position is that class II expression could be important, but is a secondary phenomenon in AITD, dependent on the T cell-derived cytokine, g-IFN, and only allowing TECs to become APCs for T cells which have already received B7-dependent costimulation elsewhere. This could clearly exacerbate AITD once initiated, but teleologically the role of class II expression seems to be as a peripheral tolerance mechanism, allowing the induction of anergy in potentially autoreactive but still naive (ie. B7-dependent) T cells (Fig. 7-13). The recent description of hyperinducibility of HLA class II expression by TECs from Graves’ disease suggests that such patients may be genetically predisposed to display a more vigorous local class II response and this would increase the likelihood of disease progression (337). The genes controlling this response are therefore worthwhile candidates for future studies of genetic susceptibility.
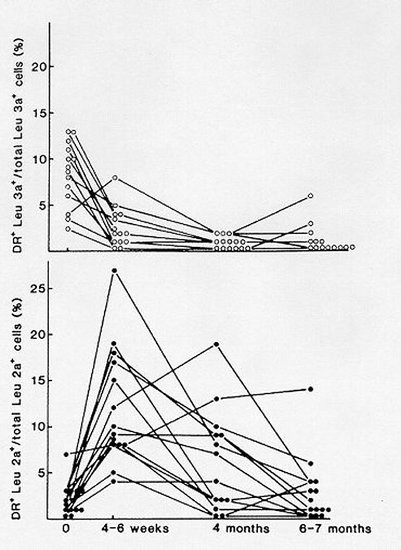
Figure 7-13: Alternative outcomes of MHC class II expression by thyroid follicular cells. Reproduced from Weetman (1997) New Aspects of Thyroid Autoimmunity. Hormone Research 48 (Suppl 4), 5154 with permission.
ENVIRONMENTAL FACTORS
Environmental factors include viral and other infections, discussed above. Strong evidence for an important role for environmental factors is provided by the incomplete concordance seen in the monozygotic twins or other siblings of individuals with AITD. Also, there are temporal changes in disease incidence that can only be the result of environmental influences, such as the rise in Graves’ disease in children in Hong Kong, the steady rise in autoimmune thyroid disease in Calabria, Italy, the more than two-fold increase in lymphocytic thyroiditis over 31 years in Austria, and the changes in the rates of histologically diagnosed Hashimoto’s thyroiditis over a 124 year period (338, 339, 340, 341).
Such studies also show that environmental factors may change rapidly, making their ascertainment difficult and challenging. Epidemiological studies have also shown that there is a higher prevalence of thyroid autoimmunity in children raised in environments that have higher prosperity and standards of hygiene (342). This falls in line with the so-called hygiene hypothesis, that is, the idea that early exposure to infections may skew the immune system away from Th2 responses like allergy and also away from autoimmunity. IL-2 administration for treatment of cancer leads to the production of antithyroid antibodies, and hypothyroidism (and possibly a better tumor response) (343). IFN-a administration and other cytokines (91), as well as highly active antiretroviral therapy for HIV infection (344), have a similar effect, although interferon-b1b treatment has no significant adverse effect on AITD (345). However long-term follow up studies have shown that around a quarter of multiple sclerosis patients treated with this latter cytokine may develop autoimmune thyroid disease within the first year of treatment (346). It remains unclear how relevant any lessons from these observations are for AITD pathogenesis, as of course the doses of cytokines and drugs used therapeutically are vast. However, it has been reported that thyrotoxicosis tends to recur following attacks of allergic rhinitis (347). Possibly this is due to a rise in endogenous cytokines and the recent association of raised IgE levels with newly diagnosed Graves’ disease indicates that this may be mediated by preferential Th2 activation (348).
Cigarette smoking is associated with Graves' disease, and with ophthalmopathy (reviewed in 349) although it seems to be that smoking is associated with a lower risk of autoimmune hypothyroidism (350). The mechanisms behind these complex changes uncertain and is doubtless more complex than a local irritative effect. Environmental tobacco smoke induces allergic sensitization in mice, associated with increased production of Th2 cytokines, but a reduction in Th1 cytokines, by the respiratory tract (351). It is therefore possible that modulation of cytokines contributes to the worsening of ophthalmopathy with smoking. On the other hand, as noted above, the opposite effect prevails in hypothyroidism and smoking exposure was associated with a lower prevalence of thyroid autoantibodies in a large population survey of over 15000 US citizens (352) and smoking cessation is known to induce a transient rise in AITD (353). To explain this, investigations have been undertaken on anatabine, an alkaloid found in tobacco; this compound ameliorates EAT and reduces TG antibody levels in human subjects with Hashimoto’s thyroiditis (354).
More general environmental pollutants have not been thoroughly explored for their possible effects (although there is some evidence from older experiments that methylcholanthracene can induce thyroiditis) but a recent study has demonstrated that polychlorinated biphenyls can induce the formation of TPO antibodies and lymphocytic thyroiditis in rats (355). A cross-sectional survey in Brazil has fond that Hashimoto’s thyroiditis and thyroid autoantibodies are more frequent in individuals living near to a petrochemical complex than in controls (356). In addition pesticide use, especially of the fungicides benomyl and maneb/mancozeb, has been associated with an increased odds of developing thyroid dysfunction although the mechanism of action is unclear (357). it clear that this aspect of the environment warrants further study in human thyroid disease.
The role of dietary iodine is clearly established in animal models of AITD and circumstantial evidence exists for a similar role in man (358-360). The response is complex and recently it has been shown that iodide may exacerbate thyroiditis in NOD mice but not affect the production of TSH-R antibodies in the same strain (361). Such findings are intriguing as they raise the possibility that the thyroiditis which accompanies Graves’ disease may not be due to the immune response to the TSH-R. Iodine may affect several aspects of the autoimmune response, as detailed in the section on experimental thyroiditis above. In addition, iodide stimulates thyroid follicular cells to produce the chemokines CCL2, CXCL8, and CXCL14 (362). These observations suggest that iodide at high concentrations could induce AITD through chemokine upregulation thus attracting lymphocytes into thyroid gland.
Dietary selenium has also been proposed as a contributor. A recent large epidemiological survey of two counties of Shaanxi Province, China, one with adequate and the other with low selenium intake, showed that higher serum selenium was associated with lower odds ratio of autoimmune thyroiditis (0.47) and hypothyroidism (0.75) (362a). However a recent Cochrane Systematic Review of trials of selenium supplementation has shown no clear beneficial clinical effect in HT, although TPO autoantibody levels do fall over a 3 month period of supplementation (363). Vitamin D may be important in autoimmunity and many other disorders, as it is now recognized that individuals living in northerly latitudes may have suboptimal levels based on a fresh understanding of what normal levels of this vitamin should be. A significant inverse correlation has been observed between 25(OH)D levels and TPO antibody levels in Indian subjects, although the overall impact of this effect in terms of causality was low (364), and another prospective study has found no evidence for a role of vitamin D (250). A more recent large scale survey has found that for every 5 nmol/L increase in serum 25(OH)D there was an associated 1.5 to 1.6-fold reduction in the risk for developing Graves’ disease, Hashimoto’s thyroiditis or postpartum thyroiditis, but vitamin D was not strong associated with the level of thyroid autoantibodies (364a). These new results are also supported by a meta-analysis of all studies prior to this, indicating that low vitamin D levels, as well as frank deficiency, are indeed risk factors for AITD (364b).
A variety of lifestyle factors that are difficult to investigate may also be involved. It is otherwise difficult to account for the increase in AITD seen in same-sex marriages (365). Stress is likely to be important in the etiology of Graves’ disease, although studies to date have had to rely on retrospective measures of this (reviewed in 366). Moreover stress does not appear to be asociated with the development of TPO antibodies in euthyroid women (369). Presumably stress acts on the immune system via pertubations in the neuroendocrine network, including alterations in glucocorticoids, but the complex interaction between the nervous, endocrine and immune systems includes the actions of neurotransmitters, CRF, leptin and melanocyte stimulating hormone as well and so unravelling the pathways whereby stress may alter the course of autoimmunity is difficult in the extreme (370). Indirect support for such a mechanism, mediated through norepinephrine, comes from experiments showing dramatic enhancement of delayed-type hypersensitivity by acute stress, the result of sympathetic nervous system activation on the migration of dendritic cells and subsequent enhanced T cell stimulation (371). Moderate consumption of alcohol appears to have a protective effect with regard to AIITD (371, 372). Given the diversity of these environmental factors, presumably operating on different genetic backgrounds, it will be difficult (if not impossible with current tools) to establish the relative importance of each in AITD.
NORMAL AUTOIMMUNITY
"Normal" people express antithyroid immunity, as previously described, and this must be important in understanding the overall mechanism of AITD. Antibodies to TG and TPO are present in both Graves’ disease and Hashimoto’s thyroiditis up to 7 years prior to diagnosis, increasing over time in the former and consistently elevated in the alter (374). Many people with low levels of antibodies but without clinical disease can be shown to have lymphocyte infiltrates in the thyroid at autopsy. B cells from normal individuals can be induced to secrete anti‑TG antibody in vitro. These observations clearly show that incomplete deletion of clonal self‑reactive T cells is indeed the normal (and indeed perhaps necessary) circumstance, and provide strong support for the idea that disordered control of this low level immunity may be important in the etiology of AITD.
Effect of Antithyroid Drugs on the Immune Response
Antithyroid drugs are used in Graves' disease to decrease production of thyroid hormone, and also lead to diminution in TSI and other antibody levels. Clinical studies show that antithyroid drug administration also leads to a diminution in antibody production in thyroxine replaced Hashimoto's thyroiditis patients (375), proving that their effect is not simply due to control of hyperthyroidism in Graves' disease. Somewhat surprisingly (376), administration of KClO4 to patients with Graves' disease leads to diminished serum antibodies, suggesting that the effect of treatment is not specific for thionamide drugs, but could be mimicked by this compound. Antithyroid drugs inhibit macrophage function, interfering with oxygen metabolite production (377).
Following antithyroid drug treatment of active Graves' disease, there is a prompt short‑term increase of DR+CD8+ T cells in the bloodstream as described above. Antithyroid drugs inhibit the production of cytokines, reactive oxygen metabolites and prostaglandin E2 by TECs and the reduction in these inflammatory mediators may explain the site-specificity of the immunomodulation produced by antithyroid drugs (125). Another pathway for an immunomodulatory action of these drugs is via the upregulation of Fas ligand expression, which may then attenuate the autoimmune response of Fas-expressing T cells (378). Only approximately 50% of patients enter remission after treatment with antithyroid drugs, a fact which must be accommodated in any hypothesis concerning an immunomodulatory action of these agents. Those patients with Graves’ disease who have the highest IgE and IL-13 levels in the circulation are the most likely to relapse (379). In turn, this suggests that antithyroid drugs only effect remission in individuals who do not have a strong Th2 response; those with the strongest such responses seem unlikely to be affected by the relatively weak action of such drugs.
AITD AS A CONSEQUENCE OF A MULTIFACTORAL PROCESS
(TABLE 4) (FIG. 7-14)
TABLE 4
DEVELOPMENT OF AUTOIMMUNE THYROID DISEASE
| Stage 1 --Basal State |
|---|
| Normal exposure to antigen such as TG and normal low levels of antibody response Inherited susceptibility via HLA-DR, DQ, or other genes |
| Stage 2a --Initial Thyroid Damage and Low Level Immune Response |
| Viral or other damage with release of normal or altered TG, TPO, or TSH-R Increased antibody levels in genetically susceptible host with high efficiency HLA-DR, DQ, TCR molecules Infection induced elevation of IL-2 or IFN-γIL-2 stimulation of antigen specific or nonspecific ThIFN-γ stimulation of DR expression and NK activation Glucocorticoid-induced alterations in lymphocyte function during stress |
| Stage 2b --Spontaneous Regression of Immune Response |
| Diminished antigen exposure Anti-idiotype feedback Antigen specific Ts induction |
| Stage 3 --Antigen Driven Thyroid Cell Damage (or Stimulation) |
| Complement dependent antibody mediated cytotoxicity Fc receptor+ cell ADCC by T, NK, or macrophage cells NK cell attack Direct CD4+ or CD8+ T cell cytotoxicity Antibody-mediated thyroid cell stimulation |
| Stage 4 --Secondary Disease Augmenting Factors |
| Thyroid cell DR, DQ expression --APC function Other molecules (cytokines, CD40, adhesion molecules) expressed by thyroid cell Immune complex binding and removal of Ts |
| Stage 5 --Antigen Independent Disease Progression |
| Recruitment of nonspecific Th or autoreactive Th Autoreactive Th bind DR+ TEC or B cells IL-2 activation of bystander Th |
| Stage 6 --Clonal Expansion with Development of Associated Diseases |
| Antigen release and new Th and B recruitment Cross reactivity with orbital antigen IL-2, IFN-γ augmentation of normal immune response to intrinsic factor, acetylcholine receptor, DNA, melanocytes, hair follicles, etc. |
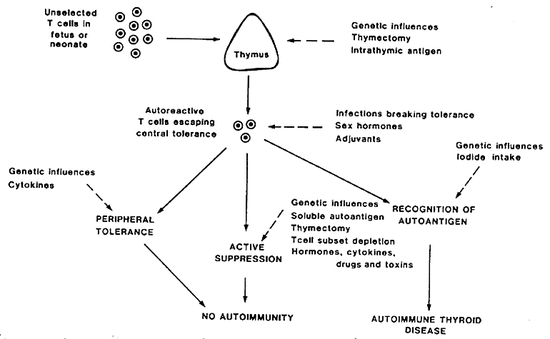
Figure 7-14: Theoretical Sequence of Development of AITD.
Thus one is led to the uncomfortable position that AITD is probably not caused by a single factor, but rather due to very many factors which interact. In terms of genetic and environmental factors, as well as factors that may be termed existential (such as age, being female and parity), these may all have to coincide in a favorable way for AITD to occur, in keeping with the Swiss-cheese model for accidents (Fig 7-15). We have divided the roles of these potential disease activity factors into a series of stages, emphasizing the predisposing events, antigen driven responses, and then the secondary and nonspecific amplification which ensues.
Figure 7-15: A Swiss cheese model for the causation of autoimmune thyroid disease, showing the effect of cumulative environmental, genetic and existential weaknesses lining up to allow AITD to occur, like the holes in the slices of cheese. In reality each of the slices depicted is composed of many individual components. The Swiss cheese model for accident causation, for instance an airplane crashing, incorporates active failures (e.g. pilot error) and latent failures (e.g. maintenance deficiency). Some factors contributing to the initiation of AITD are latent (e.g. ageing, growing up in a hygienic environment) and others are active (e.g. possession of an HLA allele which permits presentation of a thyroid autoantigen). Reproduced with permission from Weetman AP, Europ Thy J 2013 in press.
Stage 1 -- In the basal state, Stage 1, immune reactivity to autologous antigen occurs as a normal process. This probably exists at a physiologically insignificant level, since not all T or B cells reacting with TSH-R, TPO or TG are clonally deleted, and Ag is normally present in the circulation. If assays become sensitive enough, we probably will find some level of antibodies to TSH-R, TPO and TG present in most or even all healthy persons, increasing in prevalence and concentration with age, and especially in women, since being female somehow augments antithyroid immunity many-fold. Patients who have inherited certain susceptibility genes will be especially prone to develop AITD because their T and B cell repertoire includes cells recognizing self-antigen, or their immunocytes are especially good at collecting, presenting, and responding to antigen.
Stage 2 -- Possibly viral infection, or other causes of cell damage, or cross‑reacting antibodies present after Yersinia (or other) infection, leads to release of increased amounts of (possibly modified) thyroid antigens which, in genetically prone individuals, leads to an increased but still a low level immune response. Nonspecific production of TNF-a and IFN-g, in response to any infection or immune response, may augment MHC class expression on TECs, allow these cells to function as APCs, and increase production of the already established, normally occurring low levels of antibodies. The process may be affected by stress, although the mechanism remains quite uncertain. The process may go on over years, and wax and wane, as it has been shown that thyroiditis can be clinically apparent and then disappear. Factors involved in temporary or permanent suppression of the autoimmune response may include diminished thyroidal release of antigen, peripheral tolerance induction by DR-positive TECs which cannot provide a costimulatory signal, B cell anti-idiotype feedback, or the induction of T cells with a regulatory function, including those engendered by the mutual regulation of Th1 and Th2 subsets. In some individuals, thyroid cells may be less able to express DR, or may secrete TGF-b and suppress immune responses. Glucocorticoid administration and other immunosuppressives can also temporarily prevent the expression of nascent autoimmunity.
Stage 3 -- If suppressive factors do not control the developing immune response, the disease progresses to a new intensity, now driven by specific antigens, inducing cell hyperfunction (TSI), or hypofunction (TSH blocking or NIS antibodies), or cell destruction. Direct T cell cytolysis and apoptosis, ADCC, and K or NK cell attack play an important role at this stage, and now the disease becomes clinically evident.
Stage 4 -- As the disease develops, a variety of secondary factors come into play, and augments antithyroid immunoreactivity. Any stimulus which causes increased DR expression on thyroid cells, such as T cell release of IFN-g, combined with increased TSH stimulation, may allow TECs to function as APCs. Although perhaps poor in this function, they are large in number and localized in one area. The TECs may also participate in the autoimmune process by several other pathways, including the expression of adhesion molecules, Fas, Fas ligand, CD40 and complement regulatory proteins, and the production of a number of inflammatory mediators such as cytokines, reactive oxygen metabolites, nitric oxide and prostaglandins. These events are, like class II expression, dependent on cytokines and other signals generated by the intrathyroidal lymphocytic infiltrate. Some patients may inherit diminished T regulatory cell function. The ongoing immune reaction itself, may lead to nonspecific suppressor dysfunction, further augmenting immunoreactivity.
Stage 5 -- T cell derived cytokines may non-specifically induce bystander antigen specific T and B cells to be activated and produce antibody. Autoreactive cells will now accumulate in thyroid tissue because of the many strongly DR+ positive lymphocytes and TECs, and augment the developing response by lymphokine secretion or cytolysis, in a manner independent of thyroid antigens. At this stage in the disease, non-specific autoreactive immune processes may dominate a disease process which no longer depends upon antigen for its continuation.
Stage 6 -- As the concentration of activated T and B cells builds in thyroid tissue, and autoreactive and antigen nonspecific T cells become progressively involved, cell destruction may lead to release of new antigens. Cross-reacting epitopes, and nonspecific stimulation of T cells in genetically prone individuals, may cause the addition of new immunologic syndromes (exophthalmos, pretibial myxedema, atrophic gastritis) typical of older patients with more long standing and florid disease.
THYROIDITIS, MYXEDEMA, AND GRAVES' DISEASE AS AUTOIMMUNE DISEASES
HASHIMOTO'S THYROIDITIS (FIG. 7-16)
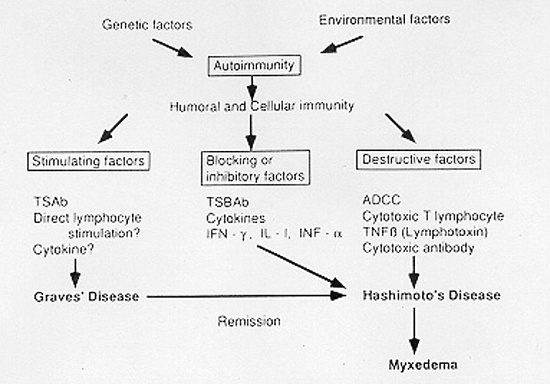
Figure 7-16: Balance of Immune Reactions Favoring Graves' or Hashimoto's Disease.
How well do the changes of Hashimoto's thyroiditis fulfill the criteria of an immunologic reaction? Neither the presence of autoantibodies in the serum of patients with Hashimoto's thyroiditis nor the demonstration in vitro of cytotoxicity of the serum constitutes definitive evidence that autoimmunity is the cause of the disease. Rarely, if ever, is there a well-defined initial immunizing event, and accordingly a shortened latent period after a secondary stimulus has not been observed. Further, experimental passive transfer of the immune state in normal recipients has not yet been attempted and has failed when human sera have been transfused into monkeys and other animals. This experiment is conducted by nature during pregnancy, since maternal antibodies cross the placenta. Transplacental passage of thyroid stimulating antibodies can produce neonatal thyrotoxicosis, and TSH blocking antibodies can produce transient neonatal hypothyroidism. Passage of TG antibody or TPO antibody has no detectable cytotoxic effect.
Assays for T cell reactivity in man, supplemented by data from animal models, provide compelling evidence of the autoimmune basis for Hashimoto’s thyroiditis, but this does not exclude an amplifying role for TG and TPO antibodies via ADCC, and, for TPO antibodies, via complement fixation. It may well be that T cell-mediated damage is required initially for all of these antibody-mediated events to take place, as this could be necessary for such access. Another striking feature of Hashimoto’s thyroiditis is the development of Hürthle cells, with granular eosinophilic cytoplasm. This appears to be the result of a chronic inflammatory milieu, resulting in overexpression of immunoproteasomes (3680
The evidence is now overwhelming that an immune reaction mediated by T lymphocytes is involved in the development of experimental thyroiditis in animals and several mechanisms may operate singly or together in man to injure TECs. Lymphocytes presensitized to antigens of the thyroid are present in the circulation of most if not all patients and are believed to localize to the thyroid itself. Since T cell mediated immunity is frequently lethal to cells, it is logical to assume that the T cell mediated immune response in thyroiditis could cause first a goiter, with lymphocyte infiltration and compensatory thyroid cell hyperplasia, and then gradual cell death and gland atrophy. The circulating antibodies may also be a functional part of this reaction. We can accept the idea that T cell-mediated immunity is the major pathogenic factor in thyroiditis.
Idiopathic Myxedema
Even before the present era of immunologic study, the basic unity of Hashimoto's thyroiditis and myxedema was realized. To quote from Crile, writing in 1954 (381): "Struma lymphomatosa is responsible not only for large lymphadenoid goiters, but also for fibrosis and atrophy of the thyroid. The clinical spectrum of struma lymphomatosa extends from spontaneous myxedema with no palpable thyroid tissue to a rapidly growing goiter associated with no clinical evidence of thyroid failure."
Hubble (382) also drew attention to the occurrence of syndromes intermediate between those of myxedema and Hashimoto’s thyroiditis, in which a small, firm thyroid gland can be felt on careful palpation. The histologic studies of Bastenie (383) and Douglass and Jacobson (384) revealed a close similarity in appearance of the thyroid remnant in myxedema and the Hashimoto gland. The immunologic studies of Owen and Smart (385), and the experience in most thyroid laboratories, indicate a similar incidence and titer of antibodies in myxedema and Hashimoto's thyroiditis. The familial association of myxedema and thyroiditis was described earlier and so far no clear genetic susceptibility difference has been reported in the two diseases. Attempts to ascribe atrophy of the thyroid gland in myxedema to particular antibodies, such as those inhibiting growth or TSH (386), or which mediate ADCC have not been confirmed by other studies (reviewed in 234).
Thus, idiopathic myxedema is the end result of Hashimoto's thyroiditis, in which the phase of thyroid enlargement was minimal or was overlooked. We may assume that in idiopathic myxedema the cell‑destructive T cell-mediated immune response is an important pathogenic factor in the illness, and that cytotoxic antibodies and TSH blocking antibodies contribute to the development of hypothyroidism, but perhaps in only a proportion.
Graves' Disease
Graves' disease is associated with a similar type of thyroid autoimmunity, since most hyperthyroid patients have circulating TG and TPO antibodies. High antibody levels are found in a small group of hyperthyroid patients and histologic examination of their glands show changes of both cell stimulation and focal thyroiditis (387). Some patients with clinical Graves' disease have tissue changes in the thyroid that are typical of thyroiditis (388). This type of patient with Graves' disease most often becomes hypothyroid after operation (389), or after 131I therapy (390). It is also well known that some patients fluctuate from hyper- to hypothyroidism over a period of months and others behave in the converse fashion, and of course the familial association of Graves’ disease with autoimmune hypothyroidism is well established.
The humoral response in Graves' disease leads to production of TG and microsomal TPO antibodies, but most importantly, as described in Chapter 10, B cells produce TSI, TBII and, in some, TSH blocking antibodies (35). TSI stimulate thyroid release of hormone primarily via cyclic AMP, although other pathways may also be activated by TSI in a proportion of patients. TSI are true cell stimulators and can even induce experimental goiter. However, the clinical picture in Graves’ disease will be a balance between the stimulation produced by TSI and the opposing effects of any TSH blocking antibodies which may be present.
Evidence also supports a role for T cell mediated immunity to thyroid antigens in Graves' disease, and against orbital antigens in patients with associated ophthalmopathy. We speculate that Graves' disease may be a condition representing a semistable balance between stimulatory, blocking, and cell‑lethal immune responses. Thus, TSI could cause thyroid hyperplasia and produce hyperthyroidism. Other antibodies might block the action of TSI either directly or, as in the case of NIS antibodies, indirectly, and prevent this hyperplastic response in some patients. Cytotoxic T cells will also gradually destroy cells and produce hypothyroidism either spontaneously or after therapy. It must be admitted that the etiology of ophthalmopathy still remains rather obscure, although the key role of cytokines in pathogenesis, causing fibroblast activation, seems firmly established.
RELATION TO OTHER DISEASES
Thyroid Cancer
Thyroid antibodies are present in increased prevalence (up to 32%) in patients with carcinoma of the thyroid, and usually are at low titer. Histologic evidence of thyroiditis is found in up to 26% of tumors. Histologic changes range from diffuse thyroiditis to focal collections of lymphocytes around the tumor or reactive lymphoid hyperplasia. Possibly release of antigens leads to increased thyroid autoimmunity. Some evidence suggests that patients who have thyroid antibodies have a better prognosis than antibody negative patients. Lymphoma and lymphosarcoma of the thyroid are associated with Hashimoto's thyroiditis (391), and there is compelling evidence that thyroiditis precedes development of the tumor. An increased frequency of carcinoma, especially of the papillary type, has been suggested in Hashimoto's thyroiditis but this relationship remains to be fully established (392).
Adolescent Goiter Enlargement of the thyroid during the second decade, accompanied by normal results of function tests, usually is labeled adolescent goiter. If the examination includes needle biopsy, an appreciable incidence of Hashimoto's thyroiditis is found (393) - up to 65%. Eighty percent of these children with thyroiditis have a positive thyroid antibody test result. The parents of many of them have either overt thyroid disease or circulating thyroid antibodies. Hyperplasia, in response to an increased demand for thyroid hormone, and colloid involution are at the root of some of these goiters, but Hashimoto's thyroiditis is the most frequent explanation of adolescent goiter in iodine sufficient areas.
Transient Thyrotoxicosis, Painless Thyroiditis, Postpartum Thyroiditis, And Related Syndromes
These illnesses, all similar, involve an acute exacerbation of thyroid autoimmunity occurring independent of, or following pregnancy in women, and in men. They are characterized by sequential inflammation-induced T4 and TG release, transient hypothyroidism, usually return to euthyroidism, and are discussed in Chapters 8 and 14. They are considered subtypes of Hashimoto's thyroiditis, and in the postpartum period, appear to result from release of the immunoregulatory effects of normal pregnancy (211).
Focal Thyroiditis
Focal lymphocytic infiltrations are frequently seen in Graves' disease, nodular goiter, nontoxic or colloid goiter, and thyroid carcinoma. The significance of these changes is not precisely known, but they correlate with positive antibody titers and may represent variations that do not differ qualitatively from thyroiditis.
Riedel’s Thyroiditis And Ig4 Disease
This rare thyroid disorder is associated with both Hashimoto’s thyroiditis and Graves’ disease and in addition many patients have evidence of fibrosis elsewhere, such as the retroperitoneum, lung, biliary tract and orbit. In one large series, 12 of 15 patients had positive TPO antibodies (389) It is now recognized that some of these patients with multifocal fibrosclerosis have IgG4-related sclerosing disease in which lymphocytes and IgG4-positive plasma cells infiltrate the affected tissues, especially the lacrimal gland, biliary tree and pancreas but the exact relationship of this entity to Riedels’ thyroiditis is unclear at present. There is a predominance of IgA rather than IgG4 in Riedel’s thyroiditis, but the effects of steroids may obscure the few analyses that have been undertaken (395, 396). However it does appear that Hashimoto’s thyroiditis can be divided into two discrete entities based on whether IgG4 plasma cells predominate in the thyroid infiltrate: in those individuals with IgG4 predominance, there is a greater male frequency, more rapid progression to hypothyroidism and more intense gland fibrosis (397). These studies have been predominantly undertaken in Japanese subjects. In a recent survey from Europe, only 12.5% of Hashimoto thyroid glands showed this feature: there was an association with younger age and male sex, and fibrosis was identified in 96 % of the IgG4-related cases but also in 18 % of the non-IgG4-related cases (397a). The authors note that unlike other forms of IgG4-related disease, the fibrosis is not accompanied by intense eosinophilia or obliterative phlebitis. IgG4 subclass thyroid autoantibodies display heritability in individuals with high levels of both TPO and TG antibodies; it is possible that more sophisticated analysis of autoantibody subtypes could lead to new methods to predict the natural history of disease (398).
Other Problems
An association between the occurrence of maternal antithyroid antibodies and recurrent abortion has been reported (399) and although this association has been disputed, a recent study showed clear evidence that the presence of TPO antibodies was associated with a 3-4-fold increased risk of miscarriage in women having in vitro fertilization (400). There is also an association between breast cancer and thyroid autoimmunity (401, 402) and between depression in middle-aged women and the presence of TPO antibodies (403). The nature of these associations is unclear; does thyroid autoimmunity predispose to such adverse events, or is the presence of thyroid autoimmunity simply a marker of a non-specific disturbance in the immune system due to whatever has caused miscarriage, cancer or depression? Having thyroid autoimmunity is not all bad news. Community-dwelling older women who have TG and TPO antibodies are less likely to be frail than those who are antibody-negative (404). Again the reason for this unexpected finding is unclear but it certainly warrants follow-up.
REFERENCES
- Janeway CA, Carding S, Jones B, Murray J, Portoles P, Rasmussen R, Rojo J, Saizawa K, West J, Bottomly K. CD4+ T cells: specificity and function. Immunol Rev 101:39-80, 1988.
- Kronenberg M, Siu G, Hood LE, Shastri N. The molecular genetics of the T-cell antigen receptor and T-cell antigen recognition. Ann Rev Immunol 4:529, 1986.
- Isakov N. Cell activation and signal initiation. Immunol Today 9:251-253, 1988.
- Panzara MA, Oksenberg JR, Steinman L. The polymerase chain reaction for detection of T-cell antigen receptor expression. Current Opinion in Immunol 4:205-210, 1992.
- Germain RN. MHC-dependent antigen processing and peptide presentation: Providing ligands for T lymphocyte activation. Cell 76:287-299, 1994.
- Gor DO, Rose NR, Greenspan NS. Th1-Th2: a Procrustean paradigm. Nature Immunol 4:503-505, 2004.
- Von Andrian UH, Mackay CR. T-cell function and migration. New Engl J Med 343:1020-1034, 2000.
- Matsuda F, Honjo T. Organization of the human immunoglobulin heavy chain locus. Adv Immunol 62:1-29, 1996.
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 392:245-252, 1998.
- Goodnow CC. Balancing immunity and tolerance: Deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci USA 93:2264-2271, 1996.
- Pérez-Melgosa M, Hollenbaugh D, Wilson CB. CD40 ligand is a limiting factor in the humoral response to T cell-dependent antigens. J Immunol 163:1123-1127, 1999.
- Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol 172:3422-3427, 2004.
- El Fassi D, Nielsen CH, Bonnema SJ, Hasselbalch HC, Hegedüs L. B lymphocyte depletion with the monoclonal antibody rituximab in Graves' disease: a controlled pilot study J Clin Endocrinol Metab. 92:1769-72, 2007.
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 8:345-50, 2007.
- Sakaguchi S. Naturally arising Foxp3-expressing CD25+ CD4+ regulatory T cells in immunological tolerance to self and non-self. Nature Immunol 6: 345-352, 2005.
- Powrie F, Maloy KJ. Regulating the regulators. Science 299:1030-1031, 2003.
- Jiang H, Chess L. Regulation of immune responses by T cells. New Engl J Med 354: 1166-1176, 2006.
- Bach J-F. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347:911-920, 2002.
- Cheng MH, Anderson MS. Monogenic autoimmunity. Annu Rev Immunol 30:393-427, 2012.
19a. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, Kemp EH, Fiskerstrand T, Viken MK, Weetman AP, Fleishman SJ, Banka S, Newman WG, Sewell WA, Sozaeva LS, Zayats T, Haugarvoll K, Orlova EM, Haavik J, Johansson S, Knappskog PM, Løvås K, Wolff AS, Abramson J, Husebye ES. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity 42:1185-96, 2015.
- Manfredi AA, Rovere-Querini P. The mitochondrion--a Trojan horse that kicks off inflammation? N Engl J Med. 362:2132-4, 2010.
Prevalence and functional significance of antipituitary antibodies in patients with autoimmune and non-autoimmune thyroid diseases.
J Clin Endocrinol Metab.92:2176-81, 2007.
- Latrofa F, Ricci D, Grasso L, Vitti P, Masserini L, Basolo F, Ugolini C, Mascia G, Lucacchini A, Pinchera A. Characterization of thyroglobulin epitopes in patients with autoimmune and non-autoimmune thyroid diseases using recombinant human monoclonal thyroglobulin autoantibodies. J Clin Endocrinol Metab 93:591-6, 2008.
- McLachlan SM, Rapoport B. Why measure thyroglobulin autoantibodies rather than thyroid peroxidase autoantibodies? Thyroid 14:510-520, 2004.
- Vali M, Rose NR, Caturegli P. Thyroglobulin as autoantigen: Structure-function relationships. Rev Endocr Met Dis 1:69-77, 2000.
- Menconi F, Huber A, Osman R, Concepcion E, Jacobson EM, Stefan M, David CS, Tomer Y. Tg.2098 is a major human thyroglobulin T-cell epitope. J Autoimmun. 35:45-51, 2010.
25a. Li CW, Menconi F, Osman R, Mezei M, Jacobson EM, Concepcion E, David CS, Kastrinsky DB, Ohlmeyer M, Tomer Y. Identifying a small molecule blocking antigen presentation in autoimmune thyroiditis. J Biol Chem. jbc.M115.694687. [Epub ahead of print], 2015
- Libert F, Lefort A, Gerard C, Parmentier M, Perret J, Ludgate M, Dumont JE, Vassart G: Cloning, sequencing, and expression of the human thyrotropin (TSH) receptor: Evidence for binding of autoantibodies. Biochem Biophys Res Commun 165:1250‑1255, 1989.
- Nagayama Y, Kaufman KD, Seto P, Rapoport B: Molecular cloning, sequence and functional expression of the cDNA for the human thyrotropin receptor. Biochem Biophys Res Commun 165:1184‑1190, 1989.
27a. Rapoport B, Aliesky HA, Chen CR, McLachlan S. Evidence that TSH receptor A-subunit multimers, not monomers, drive antibody affinity maturation in Graves' disease. J Clin Endocrinol Metab. 100:E871-5, 2015.
- Sanders J, Jeffreys J, Depraetere H, Richards T, Evans M, Kiddie A, Brereton K, Groenen M, Oda Y, Furmaniak J, Rees Smith B. Thyroid-stimulating monoclonal antibodies. Thyroid 12:1043-1050, 2002.
- Costagliola S, Franssen JDF, Bonomi M, Urizar E, Willnich M, Bergmann A, Vassart G. Generation of a mouse monoclonal TSH receptor antibody with stimulating activity. BBSRC 299:891-896, 2002.
- Ando T, Latif R, Pritsker A, Moran T, Nagayama Y, Davies TF. A monoclonal thyroid-stimulating antibody. J Clin Invest 110:1667-1674, 2002.
- Sanders J, Evans M, Premawardhana LDKE, Depraetere H, Jeffreys J, Richards T, Furmaniak J, Rees Smith B. Human monoclonal thyroid stimulating antibody. Lancet 362:126-28, 2003.
- Chazenbalk GD, Pichurin P, Chen C-R, Latrofa F, Johnstone AP, McLachlan SM, Rapoport B. Thyroid-stimulating autoantibodies in Graves’ disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J Clin Invest 110:209-217, 2002.
- Chen C-R, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves’ disease is the culprit as well as the victim. J Clin Invest 111:1897-1904, 2003.
- McLachlan SM, Rapoport B. Thyrotropin-blocking autoantibodies and thyroid-stimulating autoantibodies: potential mechanisms involved in the pendulum swinging from hypothyroidism to hyperthyroidism or vice versa. Thyroid 23:14-24, 2013
- Morshed SA, Ando T, Latif R, Davies TF. Neutral antibodies to the TSH receptor are present in Graves' disease and regulate selective signaling cascades. Endocrinology. 151:5537-49, 2010.
- Inaba H, Martin W, De Groot AS, Qin S, De Groot LJ. Thyrotropin receptor epitopes and their relation to histocompatibility leukocyte antigen-DR molecules in Graves' disease. J Clin Endocrinol Metab 91:2286-94, 2006.
- Inaba H, Pan D, Shin YH, Martin W, Buchman G, De Groot LJ. Immune response of mice transgenic for human histocompatibility leukocyte Antigen-DR to human thyrotropin receptor-extracellular domain. Thyroid 19:1271-80, 2009.
- Smith TJ, Padovani-Claudio DA, Lu Y, Raychaudhuri N, Fernando R, Atkins S, Gillespie EF, Gianoukakis AG, Miller BS, Gauger PG, Doherty GM, Douglas RS. Fibroblasts expressing the thyrotropin receptor overarch thyroid and orbit in Graves' disease. J Clin Endocrinol Metab 96:3827-37, 2011.
- Zhang L, Baker G, Janus D, Paddon CA, Fuhrer D, Ludgate M. Biological effects of thyrotropin receptor activation on human orbital preadipocytes. Invest Ophthalmol Vis Sci.47:5197-203, 2006.
- Moshkelgosha S, So PW, Deasy N, Diaz-Cano S, Banga JP. Cutting edge: retrobulbar inflammation, adipogenesis, and acute orbital congestion in a preclinical female mouse model of Graves' orbitopathy induced by thyrotropin receptor plasmid-in vivo electroporation. Endocrinology 154:3008-15, 2013.
- Douglas RS, Gianoukakis AG, Kamat S, Smith TJ. Aberrant expression of the insulin-like growth factor-1 receptor by T cells from patients with Graves' disease may carry functional consequences for disease pathogenesis. J Immunol 178:3281-7, 2007.
42a. Giménez-Barcons M, Colobran R, Gómez-Pau A, Marín-Sánchez A, Casteràs A, Obiols G, Abella R, Fernández-Doblas J, Tonacchera M, Lucas-Martín A, Pujol-Borrell R. Graves' disease TSHR-stimulating antibodies (TSAbs) induce the activation of immature thymocytes: a clue to the riddle of TSAbs generation? J Immunol. 194:4199-206, 2015.
- McLachlan SM, Rapoport B. The molecular biology of thyroid peroxidase: Cloning, expression, and role as autoantigen in autoimmune thyroid disease. Endocrine Rev 13:192-206, 1992.
- Portmann L, Hamada N, Heinrich G, DeGroot LJ. Antithyroid peroxidase antibody in patients with autoimmune thyroid disease: possible identity with anti‑microsomal antibody. J Clin Endocrinol Metab 61:1001‑1003, 1985.
- Czarnocka B, Ruf J, Ferrand M, Carayon P, Lissitzky S. Purification of the human thyroid peroxidase and its identification as the microsomal antigen involved in autoimmune thyroid diseases. FEBS Letters 190:147‑152, 1985.
- Libert F, Ruel J, Ludgate M, Swillens S, Alexander N, Vassart G, Dinsart C: Thyroperoxidase, an autoantigen with a mosaic structure made of nuclear and mitochondrial gene modules. EMBO J, 6:4193‑4196, 1987.
- Kimura S, Kotani T, McBride OW, Umeki K, Hirai K, Nakayama T, Ohtaki S: Human thyroid peroxidase: Complete cDNA and protein sequence, chromosome mapping, and identification of two alternately spliced mRNAs. Proc Natl Acad Sci USA 84:5555‑5559, 1987.
- Magnusson RP, Chazenbalk GD, Gestautas J, Seto P, Filetti S, DeGroot, LJ, Rapoport B. Molecular cloning of the complementary deoxyribonucleic acid for human thyroid peroxidase. Mol Endocrinol 1:856‑861, 1987.
- McIntosh RS, Watson P MS, Weetman AP. Somatic hypermutation in autoimmune thyroid disease. Immunol Rev 162:219-231, 1998.
- Dubska M, Banga JP, Plochocka D, Hoser G, Kemp EH, Sutton BJ, Gardas A, Gora M. Structural insights into autoreactive determinants in thyroid peroxidase composed of discontinuous and multiple key contact amino acid residues contributing to epitopes recognized by patients' autoantibodies. Endocrinology 147: 5995-6003, 2006.
- Jaume JC, Guo J, Pauls DL, Zakarija M, McKenzie JM, Egeland JA, Burek CL, Rose NR, Hoffman WH, Rapoport B, McLachlan SM. Evidence for genetic transmission of thyroid peroxidase autoantibody epitopic “fingerprints”. J Clin Endocrinol Metab 84: 1424-1431, 1999.
- Tandon N, Freeman M, Weetman AP: T cell responses to synthetic thyroid peroxidase peptides in autoimmune thyroid disease. Clin Exp Immunol 86:56‑60, 1991.
- Fisfalen M-E, Soliman M, Okamoto Y, Soltani K, DeGroot LJ. Proliferative responses of T-cells to thyroid antigens and synthetic thyroid peroxidase peptides in autoimmune thyroid disease. J Clin Endocrinol Metab 80:1597-1604, 1995.
- Nielsen CH, Brix TH, Gardas A, Banga JP, Hegedüs L. Epitope recognition patterns of thyroid peroxidase autoantibodies in healthy individuals and patients with Hashimoto's thyroiditis. Clin Endocrinol (Oxf) 69:664-8, 2008.
- Rebuffat SA, Nguyen B, Robert B, Castex F, Peraldi-Roux S. Antithyroperoxidase antibody-dependent cytotoxicity in autoimmune thyroid disease. J Clin Endocrinol Metab 93:929-34, 2008.
- Raspé E, Costagliola S, Ruf J, Mariotti S, Dumont JE, Ludgate M. Identification of the thyroid Na+/I- cotransporter as a potential autoantigen in thyroid autoimmune disease. Europ J Endocrinol 132:399-405, 1995.
- Ajjan RA, Findlay C, Metcalfe RA, Watson PF, Crisp M, Ludgate M, Weetman AP. The modulation of the human sodium iodide symporter activity by Graves’ disease sera. J Clin Endocrinol Metab 83: 1217, 1998.
- Ajjan RA, Kemp EH, Waterman EA, Watson PF, Endo T, Onaya T, Weetman AP. Detection of binding and blocking autoantibodies to the human sodium-iodide symporter in patients with autoimmune thyroid disease. J Clin Endocrinol Metab 85:2020-2027, 2000.
- Chin HS, Chin DKH, Morgenthaler NG, Vassart G, Costagliola S. Rarity of anti-Na+/I- transporter (NIS) antibody with iodide uptake inhibiting activity in autoimmune thyroid diseases (AITD). J Clin Endocrin Metab 85:3937-3940, 2000.
- Ajjan RA, Kamaruddin NA, Crisp M, Watson PF, Ludgate M, Weetman AP. Regulation and tissue distribution of the human sodium iodide symporter gene. Clin Endocrinol 49: 517, 1998.
- Yoshida A, Hisatome I, Taniguchi S, Shirayoshi Y, Yamamoto Y, Miake J, Ohkura T, Akama T, Igawa O, Shigemasa C, Kamijo K, Ikuyama S, Caturegli P, Suzuki K. Pendrin is a novel autoantigen recognized by patients with autoimmune thyroid diseases. J Clin Endocrinol Metab. 94:442-8, 2009
- Kemp EH, Sandhu HK, Watson PF, Weetman AP. Low frequency of pendrin autoantibodies detected using a radioligand binding assay in patients with autoimmune thyroid disease. J Clin Endocrinol Metab 98:E309-313, 2013.
- Benvenga S, Trimarchi F, Robbins J. Circulating thyroid hormone autoantibodies. J Endocrinol Invest 10:605, 1987.
- Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Sinha Hikim A, Gianoukakis AG, Douglas RS, Smith TJ. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves' disease. J Immunol 181:4397-405, 2008.
- Morgenthaler NG, Ho SC, Minich WB. Stimulating and blocking thyroid-stimulating hormone (TSH) receptor autoantibodies from patients with Graves' disease and autoimmune hypothyroidism have very similar concentration, TSH receptor affinity, and binding sites. J Clin Endocrinol Metab. 92:1058-65, 2007.
- Weetman AP, Black CM, Cohen SB, Tomlinson R, Banga JP, Reimer CB. Affinity purification of IgG subclasses and the distribution of thyroid auto-antibody reactivity in Hashimoto’s thyroiditis. Scand J Immunol 30:73-82, 1989
- Weetman AP, Yateman ME, Ealey PA, Black CM, Reimer CB, Williams RC Jr, Shine B, Marshall NJ. Thyroid‑stimulating antibody activity between different immunoglobulin G subclasses. J Clin Invest 86:723‑727, 1990.
- Vos XG, Smit N, Endert E, Tijssen JG, Wiersinga WM. Frequency and characteristics of TBII-seronegative patients in a population with untreated Graves' hyperthyroidism: a prospective study. Clin Endocrinol (Oxf) 69:311-7, 2008.
- Ewins DI, Wilkin TJ. A clinical comparison of the enzyme‑linked immunosorbent assay (ELISA) and hemagglutination (TRC) in the routine detection of antithyroglobulin antibodies. Acta Endocrinol 103:216‑222, 1983.
- Takeda Y, Kriss JP. Radiometric measurement of thyroglobulin‑antithyroglobulin immune complex in human serum. J Clin Endocrinol Metab 44:46, 1977.
- Boyden SV. The adsorption of proteins on erythrocytes treated with tannic acid and subsequent hemagglutination by antiprotein sera. J Exp Med 93:107, 1951.
- Vanderpump MPJ, Tunbridge WMG, French JM, Appleton D, Bates D, Clark F, Grimley Evans J, Hasan DM, Rodgers H, Tunbridge F, Young ET. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol 43:55-68, 1995.
- Williams ED, Doniach I. Post‑mortem incidence of focal thyroiditis. J Pathol Bact 83:255, 1962.
- Vanderpump MPJ, Tunbridge WMG, French JM, Appleton D, Bates D, Clark F, Grimley Evans J, Hasan DM, Rodgers H, Tunbridge F, Young ET. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol 43:55-68, 1995.
- Nordyke RA, Gilbert JR, FI, Miyamoto LA, Fleury KA. The superiority of antimicrosomal over antithyroglobulin antibodies for detecting Hashimoto’s thyroiditis. Arch Int Med 153:862-865, 1993.
- Lindberg G, Svensson J, Ericsson U-B, Nilsson P, Svenonius E, Ivarsson S-A. Comparison of some different methods for analysis of thyroid autoantibodies: Importance of thyroglobulin autoantibodies. Thyroid 11:265-269, 2001.
- Unuane D, Velkeniers B, Anckaert E, Schiettecatte J, Tournaye H, Haentjens P, Poppe K. Thyroglobulin autoantibodies: is there any added value in the detection of thyroid autoimmunity in women consulting for fertility treatment? Thyroid 23:1022-1028, 2013.
- Trotter WR, Belvayin G, Waddams A. Precipitating and complement‑fixing antibodies in Hashimoto's disease. Proc R Soc Med 50:961, 1957.
- Pulvertaft RJV, Doniach D, Roitt IM, Hudson RV. Cytotoxic effects of Hashimoto serum on human thyroid cells in tissue culture. Lancet 2:214, 1959.
- Chiovato L, Bassi P, Santini F, Mammoli C, Lapi P, Carayon P, Pinchera A. Antibodies producing complement-mediated thyroid cytotoxicity in patients with atrophic or goitrous autoimmune thyroiditis. J Clin Endocrinol Metab 77:1700-1705, 1993.
- Weetman AP, Cohen SB, Oleesky DA, Morgan BP. Terminal complement complexes and C1/C1 inhibitor complexes in autoimmune thyroid disease. Clin Exp Immunol 77:25-30, 1989.
- Weetman AP, Freeman MA, Morgan BP. Thyroid follicular cell function after non-lethal complement membrane attack. Clin Exp Immunol 82:69-74, 1990.
- Calder EA, Penhale WJ, McCleman D, Barnes EW, Irvine WJ. Lymphocyte dependent antibody‑mediated cytotoxicity in Hashimoto’s thyroiditis. Clin Exp Immunol 14:153, 1973.
- Suzuki S, Mitsunaga M, Miyoshi M, Hirakawa S, Nakagawa O, Miura H, Ofuji T. Cytophilic antithyroglobulin antibody and antibody‑dependent monocyte‑mediated cytotoxicity in Hashimoto's thyroiditis. J Clin Endocrinol Metab 51:446, 1980.
- Metcalfe RA, Oh YS, Stroud C, Arnold K, Weetman AP. Analysis of antibody-dependent cell-mediated cytotoxicity in autoimmune thyroid disease. Autoimmunity 25:65-72, 1997.
- Blanchin S, Coffin C, Viader F, Ruf J, Carayon P, Potier F, Portier E, Comby E, Allouche S, Ollivier Y, Reznik Y, Ballet JJ Anti-thyroperoxidase antibodies from patients with Hashimoto's encephalopathy bind to cerebellar astrocytes. J Neuroimmunol 192:13-20, 2007.
- Duthoit C, Estienne V, Delom F, Durand-Gorde J-M, Mallet B, Carayon P, Ruf J. Production of immunoreactive thyroglobulin C-terminal fragments during thyroid hormone synthesis. Endocrinology 141:2518-2525, 2000.
- Nielson CH, Leslie RGQ, Jepsen BS, Kazatchkine MD, Kaveri SV, Fischer E. Natural autoantibodies and complement promote the uptake of a self antigen, human thyroglobulin, by B cells and the proliferation of thyroglobulin-reactive CD4+ T cells in healthy individuals. Eur J Immunol 31:2660-2668, 2001.
- Fenzi G, Hashizume K, Roudebush CP, DeGroot LJ. Changes in thyroid‑stimulating immunoglobulins during antithyroid therapy. J Clin Endocrinol Metab 48:572, 1979.
- Carella C, Mazziotti G, Amato G, Braverman LE, Roti E. Interferon-α-related thyroid disease: pathophysiological, epidemiological and clinical aspects. J Clin Endocrinol Metab 89:3656-3661, 2004.
- Mammen JS, Ghazarian SR, Rosen A, Ladenson PW. Patterns of interferon-alpha-induced thyroid dysfunction vary with ethnicity, sex, smoking status, and pretreatment thyrotropin in an international cohort of patients treated for hepatitis C. Thyroid 23:1151-1158, 2013.
- Henry M, Zanelli E, Piechaczyk M, et al. A major human thyroglobulin epitope defined with monoclonal antibodies is mainly recognized by human autoantibodies. Eur J Immunol 22:315-319, 1992.
- Prentice L, Kiso Y, Fukuma N, Horimoto M, Petersen V, Grennan F, Pegg C, Furmaniak J, Rees Smith B. Monoclonal thyroglobulin autoantibodies: Variable region analysis and epitope recognition. J Clin Endocrinol Metab 80:977-986, 1995.
- Tomer Y. Anti-thyroglobulin autoantibodies in autoimmune thyroid diseases: Cross-reactive or pathogenic? Clinl Immunol Immunopathol 82:3-11, 1997.
- Libert F, Ludgate M, Dinsart C, Vassart G. Thyroperoxidase, but not the thyrotropin receptor, contains sequential epitopes recognized by autoantibodies in recombinant peptides expressed in the pUEX vector. J Clin Endocrinol Metab 73:857‑860, 1991.
- McLachlan SM, Pegg CAS, Atherton MC, Middleton SL, Dickinson A, Clark F, Proctor SJ, Proud G, Smith BR. Subpopulations of thyroid autoantibody secreting lymphocytes in Graves' and Hashimoto thyroid glands. Clin Exp Immunol 65:319‑328, 1986.
- McLachlan SM, Dickinson AM, Malcolm A, Farndon JR, Young E, Proctor SJ, Smith BR. Thyroid autoantibody synthesis by cultures of thyroid and peripheral blood lymphocytes. I. Lymphocyte markers and response to pokeweed mitogen. Clin Exp Immunol 52:45‑53, 1983.
- Logtenberg T, Kroon A, Gmelig‑Meyling FHJ, Ballieux RE. Production of anti‑thyroglobulin antibody by blood lymphocytes from patients with autoimmune thyroiditis, induced by the insolubilized autoantigen. J Immunol 136:1236‑1240. 1986.
- Weetman AP, McGregor AM, Lazarus JH, Hall R. Thyroid antibodies are produced by thyroid‑derived lymphocytes. Clin. Exp. Immunol. 48:196‑200, 1982.
- Armengol MP, Juan M, Lucas-Martin A, Fernández-Figueras MT, Jaraquemada D, Gallart T, Pujol-Borrell R. Demonstration of thyroid antigen-specific B cells and recombination-activating gene expression in chemokine-containing intrathyroidal germinal centers. Am J Pathol 159, 861-873, 2001.
- Weetman AP, McGregor AM, Wheeler MH, Hall R. Extrathyroidal sites of autoantibody synthesis in Graves’ disease. Clin Exp Immunol 56:330-336, 1984.
- Okita N, Kidd A, Row VV, Volpe R. Sensitization of T‑lymphocytes in Graves' and Hashimoto's diseases. J Clin Endocrinol Metab 51:316, 1980.
- Aoki N, DeGroot LJ. Lymphocyte blastogenic response to human thyroglobulin in Graves' disease, Hashimoto's thyroiditis, and metastatic thyroid cancer. Clin Exp Immunol 38:523‑530, 1979.
- Weetman AP. Autologous CD8-positive cells suppress T cell proliferation in response to thyroid antigens in Hashimoto’s thyroiditis. Clin Immunol Immunopathol 51:303-310, 1989.
- Butscher WG, Ladenson PW, Burek CL. Whole-blood proliferation assay for autoimmune thyroid disease: Comparison to density-gradient sepaated-peripheral blood lymphocytes. Thyroid 11, 531-537, 2001.
- Canonica GW, Cosulich ME, Croci R, Ferrini S, Bagnasco M, Dirienzo W, Ferrini O, Bargellesi A, Giordano G. Thyroglobulin‑induced T‑cell in vitro proliferation in Hashimoto's thyroiditis: identification of the responsive subset and effect of monoclonal antibodies directed to Ia antigens. Clin Immunol Immunopathol 32:132‑141, 1984.
- Canonica GW, Caria M, Bagnasco M, Cosulich ME, Giordano G, Moretta L. Proliferation of T8‑positive cytolytic T lymphocytes in response to thyroglobulin in human autoimmune thyroiditis: analysis of cell interactions and culture requirements. Clin Immunol Immunopathol 36:40‑48, 1985.
- Davies TF. Cocultures of human thyroid monolayer cells and autologous T cells: impact of HLA Class II antigen expression. J Clin Endocrinol Metab 61:418‑422, 1985.
- Weetman AP, Volkman DJ, Burman KD, Margolick JB, Petrick P, Weintraub BD, Fauci AS. The production and characterization of thyroid‑derived T‑cell lines in Graves’ disease and Hashimoto's thyroiditis. Clin Immunol Immunopathol 39:139‑150, 1986.
- Londei M, Bottazzo GF, Feldmann M. Human T‑cell clones from autoimmune thyroid glands: specific recognition of autologous thyroid cells. Science 228:85‑88, 1985.
- Soliman M, Kaplan E, Yanagawa T, Hidaka Y, Fisfalen M-E, DeGroot LJ. T-cells recognize multiple epitopes in the human thyrotropin receptor extracellular domain. J Clin Endocrinol Metab 80:905-914, 1995.
- Martin A, Nakashima M, Zhou A, Aronson D, Werner AJ, Davies TF. Detection of major T cell epitopes on human thyroid stimulating hormone receptor by overriding immune heterogeneity in patients with Graves’ disease. J Clin Endocrinol Metab 82:3361-3366, 1997.
- Sawai Y, DeGroot LJ. Binding of human thyrotropin receptor peptides to a Graves’ disease predisposing human leukocyte antigen class II molecule. J Clin Endocrinol Metab 85:1176-1179, 2000.
- Davies TF, Martin A, Concepcion ES, Graves P, Lahat N, Cohen WL, Ben-Nun A. Evidence for selective accumulation of intrathyroidal T lymphocytes in human autoimmune thyroid disease based on T cell receptor V gene usage. J Clin Invest 89:157-162, 1992.
- Davies TF, Concepcion ES, Ben-Nun A, Graves P, Tarjan G. T-cell receptor V gene use in autoimmune thyroid disease: Direct assessment by thyroid aspiration. J Clin Endocrinol Metab 76:660-666, 1993.
- McIntosh RS, Tandon N, Pickerill AP, Davies R, Barnett D, Weetman AP. IL-2 receptor positive intrathyroidal lymphocytes in Graves’ disease: Analysis of Va transcript microheterogeneity. J Immunol 91:3884-3893, 1993.
- Caso-Peláez E, McGregor AM, Banga JP. A polyclonal T cell repertoire of V-alpha and V-beta T cell receptor gene families in intrathyroidal T lymphocytes of Graves’ disease patients. Scand J Immunol 41:141-147, 1995.
- McIntosh RS, Watson PA, Weetman AP. analysis of T cell receptor Va repertoire in Hashimoto’s thyroiditis: Evidence for the restricted accumulation of CD8+ T cells in the absence of CD4+ T cell restriction. J Clin Endocrinol Metab 82:1140-1146, 1997.
- McMurray RW, Hoffman RW, Tang H, Braley-Mullen H. T cell repertoire Vb usage in murine experimental autoimmune thyroiditis. Cell Immunol 172:1-9, 1996.
- Ehlers M, Thiel A, Bernecker C, Porwol D, Papewalis C, Willenberg HS, Schinner S, Hautzel H, Scherbaum WA, Schott M. Evidence of a combined cytotoxic thyroglobulin and thyroperoxidase epitope-specific cellular immunity in Hashimoto's thyroiditis. J Clin Endocrinol Metab 97:1347-54, 2012.
- MacKenzie WA, Davies TF. An intrathyroidal T‑cell clone speci-fically cytotoxic for human thyroid cells. Immunology 61:101‑103, 1987.
- Arao T, Morimoto I, Kakinuma A, Ishida O, Zeki K, Tanaka Y, Ishikawa N, Ito K, Ito K, Eto S. Thyrocyte proliferation by cellular adhesion to infiltrating lymphocytes through the intercellular adhesion molecule-1/lymphocyte function-associated antigen-1 pathway in Graves’ disease. J Clin Endocrinol Metab 85:382-389, 2000.
- Ploth DW, Fitz A, Schnetzler D, Seidenfeld J, Wilson CB. Thyroglobulin‑anti‑thyroglobulin immune complex glomerulonephritis complicating radioiodine therapy. Clin Immunol Immunopathol 9:327‑334, 1978.
- O'Regan S, Fong JSC, Kaplan BS, de Chadarevian J‑P, Lapointe N, Drummond KN. Thyroid antigen‑antibody nephritis. Clin Immunol Immunopathol 6:341‑346, 1976.
- Weetman AP, Tandon N, Morgan BP. Antithyroid drugs and release of inflammatory mediators by complement-attacked thyroid cells. Lancet 340:633-636, 1992.
- Endo Y, Aratake Y, Yamamoto I, Nakagawa H, Kuribayashi T, Ohtaki S. Peripheral K cells in Graves' disease and Hashimoto's thyroiditis in relation to circulating immune complexes. Clin Endocrinol 18:187‑194, 1983.
- Sack J, Baker JR Jr, Weetman AP, Wartofsky L, Burman KD. Killer cell activity and antibody‑dependent cell‑mediated cytotoxicity are normal in Hashimoto's disease. J Clin Endocrinol Metab 62:1059‑1064, 1986.
- Amino N, Mori H, Iwatani Y, Asari S, Izumiguchi Y, Miyai K. Peripheral K lymphocytes in autoimmune thyroid disease: decrease in Graves' disease and increase in Hashimoto's disease. J Clin Endocrinol Metab 54:587‑591, 1982.
- Bogner U, Schleusener H, Wall JR. Antibody‑dependent cell mediated cytotoxicity against human thyroid cells in Hashimoto’s thyroiditis but not Graves' disease. J Clin Endocrinol Metab 59:734‑738, 1984.
- Hidaka Y, Amino N, Iwatani Y, et al. Increase in peripheral natural killer cell activity in patients with autoimmune thyroid disease. Autoimmunity 11:239-246, 1992.
- Bogner U, Hegedüs L, Hansen JM, Finke R, Schleusener H. Thyroid cytotoxic antibodies in atrophic and goitrous autoimmune thyroiditis. Europ J Endocrinol 132:69-74, 1995.
- Bornet H, Orgiazzi J. Assessment of antibody dependent cell cytotoxicity in autoimmune thyroid disease using porcine thyroid cells. Autoimmunity 13:177-185, 1992.
- Kung AWC, Lau LMA, Lau KS. The role of interferon-g in lymphocytic thyroiditis: Its functional and pathological effect on human thyrocytes in culture. Clin Exp Immunol 87:261-265, 1992.
- Weetman AP, Ajjan RA. Cytokines and autoimmune thyroid disease. (www.hotthyroidology.com) June, No 1, 2002.
- Marique L, Van Regemorter V, Gérard AC, Craps J, Senou M, Marbaix E, Rahier J, Daumerie C, Mourad M, Lengelé B, Colin IM, Many MC. The expression of dual oxidase, thyroid peroxidase, and caveolin-1 differs according to the type of immune response (TH1/TH2) involved in thyroid autoimmune disorders. J Clin Endocrinol Metab 99:1722-32, 2014.
- Alimi E, Huang S, Brazillet M-P, Charreire J. Experimental autoimmune thyroiditis (EAT) in mice lacking the IFN-g receptor gene. Eur J Immunol 28:201-208, 1998.
136a. Kemp EH, Ajjan RA, Metcalfe RA, Watson PF, Weetman AP. IL-14 and IL-16 are expressed in the thyroid of patients with either Graves' disease or Hashimoto's thyroiditis. Clin Endocrinol (Oxf). 83:726-32, 2015.
- Simons PJ, 27
elemarre FGA, Drexhage HA. Antigen-presenting dendritic cells as regulators of the growth of thyrocytes: a role of interleukin-1b and interleukin-6. Endocrinology 139:3148-3156, 1998.
- Leskela S, Rodríguez-Muñoz A, de la Fuente H, Figueroa-Vega N, Bonay P, Martín P, Serrano A, Sánchez-Madrid F, González-Amaro R, Marazuela M. Plasmacytoid dendritic cells in patients with autoimmune thyroid disease. J Clin Endocrinol Metab. 98:2822-2833, 2013.
- Nilsson M, Husmark J, Björkman U, Ericson LE. Cytokines and thyroid epithelial integrity: interleukin-1a induces dissociation of the junctional complex and paracellular leakage in filter-cultured human thyrocytes. J Clin Endocrinol Metab 83:945-952, 1998.
- Figueroa-Vega N, Alfonso-Pérez M, Cuesta-Mateos C, Sánchez-Madrid F, Moreno-Otero R, González-Amaro R, Marazuela M. Tie-2 is overexpressed by monocytes in autoimmune thyroid disorders and participates in their recruitment to the thyroid gland. J Clin Endocrinol Metab. 94:2626-33, 2009.
- Itaka M, Miura S, Yamanaka K, Kawasaki S, Kitahama S, Kawakami Y, Kakinuma S, Oosuga I, Wada S, Katayama S. Increased serum vascular endothelial growth factor levels and intrathyroidal vascular area in patients with Graves’ disease and Hashimoto’s thyroiditis. J Clin Endocrinol Metab 83:3908-3912, 1998.
- Weetman AP. Determinants of autoimmune thyroid disease. Nature Immunol 2:405, 2001.
- Kite JH, Witebsky E. Hereditary autoimmune thyroiditis in the fowl. Science 160:1357, 1968.
- Beierwaltes WH, Nishyama RH. Dog thyroiditis. Occurrence and similarity to Hashimoto's struma. Endocrinology 83:501, 1968.
- Evans TC, Beierwaltes WH, Nishiyama RH. Experimental canine Hashimoto's thyroiditis. Endocrinology 84:641, 1969.
- Kite JH, Argue H, Rose NR. Experimental thyroiditis in the rhesus monkey. I. Cytotoxic, mixed‑agglutinating and complement‑fixing antibodies. Clin Exp Immunol 1:139, 1966.
- Beall GN, Daniel PM, Pratt OE, Solomon DH. Effects of immunization of baboons with human thyroid tissue. J Clin Endocrinol Metab 29:1460, 1969.
- Jacobson EM, Concepcion E, Ho K, Kopp P, Vono Toniolo J, Tomer Y. cDNA immunization of mice with human thyroglobulin generates both humoral and T cell responses: a novel model of thyroid autoimmunity. PLoS One;6:e19200, 2011.
- Dawe KI, Hutchings PR, Geysen M, Champion BR, Cooke A, Roitt IM. Unique role of thyroxine in T cell recognition of a pathogenic peptide in experimental autoimmune thyroiditis. Europ J Immunol 26:768-772, 1996.
- Kong Y-C, McCormick DJ, Wan Q, Motte RW, Fuller BE, Giraldo AA, David CS. Primary hormonogenic sites as conserved autoepitopes on thyroglobulin in murine autoimmune thyroiditis. J Immunol 155:5847-5854, 1995.
- Carayanniotis G, Rao VP. Searching for pathogenic epitopes in thyroglobulin: parameters and caveats. Immunol Today 18:83-88, 1997.
- Kong YC, Morris GP, Brown NK, Yan Y, Flynn JC, David CS. Autoimmune thyroiditis: a model uniquely suited to probe regulatory T cell function. J Autoimmun. 33:239-46, 2009.
- Sugihara S, Fujiwara H, Shearer GM. Autoimmune thyroiditis induced in mice depleted of particular T cell subsets. J Immunol 150:683-694, 1993.
- Tomer Y, Gilburd B, Sack J, Davies TF, Meshorer A, Burek CL, Rose NR, Shoenfeld Y. Induction of thyriod autoantibodies in naive mice by idiotypic manipulation. Clin Immunol Immunopathol 78:180-187, 1996.
- Flynn JC, Conaway DH, Cobbold S, Waldmann H, Kong Y-CM. Depletion of L3T4+ and Lyt-2+ cells by rat monoclonal antibodies alters the development of adoptively transferred experimental autoimmune thyroiditis. Cell Immunol 122:377-390, 1989.
- Maron R, Zerubavel R, Friedman A, Cohen IR. T lymphocyte line specific for thyroglobulin produces or vaccinates against autoimmune thyroiditis in mice. J Immunol 131:2316‑2322, 1983.
- Taguchi O, Takahashi T. Mouse models of autoimmune disease suggest that self-tolerance is maintained by unresponsive autoreactive T cells. Immunology 89:13-19, 1996.
- Guimaraes VC, Quintans J, Fisfalen M-E, Straus FH, Wilhelm K, Medeiros-Neto GA, DeGroot LJ. Suppression of development of experimental autoimmune thyroiditis by oral administration of thyroglobulin. Endocrinology 136:3353-3359, 1995.
- Gangi E, Vau C, Cheatam D, Prabhakar B. IL-10-producing CD4+, CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J Immunol 174: 7006-7013, 2005.
- Verginis P, Li HS, Carayanniotis G. Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4+, CD25+ T cells. J Immunol 174, 7433-7439, 2005.
- Ng HP, Banga JP, Kung AWC. Development of a murine model of autoimmune thyroiditis induced with homologous mouse thyroid peroxidase. Endocrinolgy 145: 809-816, 2004.
- Pichurin P, Chen C-R, Pichurina O, David C, Rapoport B, McLachlan SM. Throtropin receptor-DNA vaccination of transgenic mice expressing HLA-DR3 or HLA-DQ6b. Thyroid 13:911-917, 2003.
- Nagayama Y, McLachlan SM, Rapoport B, Niwa M. A major role for non-major histocompatibility complex genes but not for microorganisms in a novel murine model of Graves’ hyperthyroidism. Thyroid 13:233-238, 2003.
- Flynn JC, Meroueh C, Snower DP, David CS, Kong YM. Depletion of CD4+CD25+ regulatory T cells exacerbates sodium iodide-induced experimental autoimmune thyroiditis in human leucocyte antigen DR3 (DRB1*0301) transgenic class II-knock-out non-obese diabetic mice. Clin Exp Immunol.147:547-54, 2007.
- Bagchi N, Brown TR, Sundick RS. Thyroid cell injury is an initial event in the induction of autoimmune thyroiditis by iodine in obese strain chickens. Endocrinology 136:5054-5060, 1995.
- Bagchi N, Sundick RS, Hu LH, Cummings GD, Brown TR. Distinct regions of thyroglobulin control the proliferation and suppression of thyroid-specific lymphocytes in obese strain chickens. Endocrinology 137:-3286-3390, 1996.
- Damotte D, Colomb E, Cailleau C, Brousse N, Charreire J, Carnaud C. Analysis of susceptibility of NOD mice to spontaneous and experimentally induced thyroiditis. Europ J Immunol 27:2854-2862, 1997.
- Allen EM, Appel MC, Braverman LE. Iodine‑induced thyroiditis and hypothyroidism in the hemithyroidectomized BB/W rat. Endocrinology 121:481‑485, 1987.
- Horie I, Abiru N, Nagayama Y, Kuriya G, Saitoh O, Ichikawa T, Iwakura Y, Eguchi K. T helper type 17 immune response plays an indispensable role for development of iodine-induced autoimmune thyroiditis in nonobese diabetic-H2h4 mice. Endocrinology. 150:5135-42, 2009.
169a. Wang W, Xue H, Li Y, Hou X, Fan C, Wang H, Zhang H, Shan Z, Teng W. Effects of selenium supplementation on spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Clin Endocrinol Metab. 100:4037-47, 2015.
- Martin AP, Marinkovic T, Canasto-Chibuque C, Latif R, Unkeless JC, Davies TF, Takahama Y, Furtado GC, Lira SA. CCR7 deficiency in NOD mice leads to thyroiditis and primary hypothyroidism.
J Immunol. 183:3073-80, 2009.
- Chen CR, Hamidi S, Braley-Mullen H, Nagayama Y, Bresee C, Aliesky HA, Rapoport B, McLachlan SM. Antibodies to thyroid peroxidase arise spontaneously with age in NOD.H-2h4 mice and appear after thyroglobulin antibodies Endocrinology. 151:4583-93, 2010.
- Ellis JS, Hong SH, Zaghouani H, Braley-Mullen H. Reduced effectiveness of CD4+Foxp3+ regulatory T cells in CD28-deficient NOD.H-2h4 mice leads to increased severity of spontaneous autoimmune thyroiditis. J Immunol.191:4940-4949, 2013.
172a. Rapoport B, Aliesky HA, Banuelos B, Chen CR, McLachlan SM. A unique mouse strain that develops spontaneous, iodine-accelerated, pathogenic antibodies to the human thyrotrophin receptor. J Immunol. 194:4154-61, 2015.
- Penhale WJ, Farmer A, McKenna RP, Irvine WJ. Spontaneous thyroiditis in thymectomized and irradiated Wistar rats. Clin Exp Immunol 15:225-236, 1973.
- Sempowski GD, Cross SJ, Heinly CS, Scearce RM, Haynes BF. CD7 and CD28 are required for murine CD4+,CD25+ regulatory T cell homeostasis and prevention of thyroiditis. J Immunol 172, 787-794, 2004.
- Quarantino S, Badami E, Pang YY, Bartok I, Dyson J, Kioussis D, Londei M, Maiuri L. Degenerate self-reactive human T cell receptor causes spontaneous autoimmune disease in mice. Nature Med 10:920-926, 2004.
- Li HS, Verginis P, Carayanniotis G. Maturation of dendritic cells by necrotic thyrocytes facilitates induction of experimental autoimmune thyroiditis.Clin Exp Immunol. 144:467-74, 2006
- McLachlan SM, Alpi K, Rapoport B. Review and hypothesis: does Graves' disease develop in non-human great apes? Thyroid 21:1359-66, 2011.
- Kohn LD, Shimojo N, Kohno Y, Suzuki K. An animal model of Graves’ disease: Understanding the cause of autoimmune hyperthyroidism. Rev Endocr Met Dis 1:59-67, 2000
- McLachlan SM, Nagayama Y, Rapoport B. Insight into Graves’ hyperthyroidism from animal models. Endocrine Rev 26: 800-832, 2005.
- Mizutori Y, Nagayama Y, Flower D, Misharin A, Aliesky HA, Rapoport B, McLachlan SM. Role of the transgenic human thyrotropin receptor A-subunit in thyroiditis induced by A-subunit immunization and regulatory T cell depletion. Clin Exp Immunol 154: 305-15, 2008.
- Rapoport B, Williams RW, Chen CR, McLachlan SM. Immunoglobulin heavy chain variable region genes contribute to the induction of thyroid-stimulating antibodies in recombinant inbred mice. Genes Immun. 11:254-63, 2010.
- Pichurin PN, Pichurina O, Marians RC, Chen CR, Davies TF, Rapop0rt B, McLachlan SM. Thyrotropin receptor knockout mice: studies on immunological tolerance to a major thyroid autoantigen. Endocrinology 145:1294-1301,2004.
- Misharin AV, Rapoport B, McLachlan SM. Thyroid antigens, not central tolerance, control responses to immunization in BALB/c versus C57BL/6 mice. Thyroid. 19:503-9, 2009.
- Misharin AV, Nagayama Y, Aliesky HA, Rapoport B, McLachlan SM. Studies in mice deficient for the autoimmune regulator (Aire) and transgenic for the thyrotropin receptor reveal a role for Aire in tolerance for thyroid autoantigens. Endocrinology. 150:2948-56, 2009.
- Baker G, Mazziotti G, von Ruhland C, Ludgate M. Re-evaluating thyrotropin receptor-induced mouse models of Graves’ disease. Endocrinol 146:835-844, 2005.
- Sugihara S, Fujiwara H, Niimi H, Shearer GM. Self-thyroid epithelial cell (TEC)-reactive CD8+ T cell lines/clones derived from autoimmune thyroiditis lesions. J Immunol 155:1619-1628, 1995.
- Catálfamo M, Roura-Mir C, Sospedra M, Aparicio P, Costagliola S, Ludgate M, Pujol-Borrell R, Jaraquemada D. Self-reactive cytotoxic gd T lymphocytes in Graves’ disease specifically recognize thyroid epithelial cells. J Immunol 156:804-811, 1996.
- Wu Z, Podack ER, McKenzie JM, Olsen KJ, Zakarija M. Perforin expression by thyroid-infiltrating T cells in autoimmune thyroid disease. Clin Exp Immunol 98:470-477, 1994.
- Giordano C, Stassi G, De Maria R, Todaro M, Richiusa P, Papoff G, Ruberti G, Bagnasco M, Testi R, Galluzzo A. Potential involvement of Fas and its ligand on the pathogenesis of Hashimoto’s thyroiditis. Science 275:960-963, 1997.
- Kawakami A, Eguchi K, Matsuoka N, Tsuboi M, Yrayama S, Kawabe Y, Tahara K, Ishikawa N, Ito K, Nagataki S. Modulation of Fas-mediated apoptosis of human thyroid epithelial cells by IgG from patients with Graves’ disease (GD) and idiopathic myxoedema. Clin Exp Immunol 110:434-439, 1997.
- Stassi G, Todaro M, Bucchieri F, Stoppacciaro A, Farina F, Zummo G, Testi R, De Maria R. Fas/Fas ligand-driven T cell apoptosis as a consequence of ineffective thyroid immunoprivilege in Hashimoto’s thyroiditis. J Immunol 162: 263-267, 1999.
- Giordano C, Richiusa P, Bagnasco M, Pizzolanti G, Di Blasi F, Sbriglia MS, Mattina A, Pesce G, Montagna P, Capone F, Misiano G, Scorsone A, Publiese A, Galluzzo A. Differential regulation of Fas-mediated apoptosis in both thyrocyte and lymphocyte cellular compartments correlates with opposite phenotypic manifestations of autoimmune thyroid disease. Thyroid 11:233-244, 2001.
- Baker Jr JR. Guest editorial. Thyroid 11:245-247, 2001.
- Kasai K, Hattori Y, Nakanishi N, Manaka K, Banba N, Motohashi S, Shimoda S-I. Regulation of inducible nitric oxide production by cytokines in human thyrocytes in culture. Endocrinology 136:4261-4270, 1995.
- Lidman K, Eriksson U, Norberg R, Fagraeus A. Indirect immunofluorescence staining of human thyroid by antibodies occurring in Yersinia enterocolitica infections. Clin Exp Immunol 23:429‑435, 1976.
- Costagliola S, Ruf J, Durand-Gorde M-J, Carayon P. Monoclonal anti-idiotypic antibodies interact with the 93 kilodalton thyrotropin receptor and exhibit heterogeneous biological activities. Endocrinology 128:1555-1562, 1991.
- Dziarski R. Autoimmunity: polyclonal activation or antigen induction? Immunol Today, 9:340‑342, 1988.
- Tomer Y, Davies TF. Infection, thyroid disease, and autoimmunity. Endocrine Rev 14:107-120, 1993.
- Bartalena L, Bogazzi F, Pecori F, Martino E. Graves’ disease occurring after subacute thyroiditis: Report of a case and review of the literature. Thyroid 6:345-348, 1996.
- Vykhovanets EV, Chernyshov VP, Slukvin II, Antipkin YG, Vasyuk AN, Klimenko HF, Strauss KW. 131I dose-dependent thyroid autoimmune disorders in children living around Chernobyl. Clin Immunol Immunopathol 84:251-259, 1997.
- Vermiglio F, Castagna MG, Volnova E, Lo Presti VP, Moleti M, Violi MA, Artemisia A, Trimarchi F. Post-Chernobyl increased prevalence of humoral thyroid autoimmunity in children and adolescents from a moderately iodine-deficient area in Russia. Thyroid 9: 781-786, 1999.
- Volzke H, Werner A, Wallaschofski H et al. Occupational exposure to ionizing radiation is associated with autoimmune thyroid disease. J Clin Endocrinol Metab 90: 4587-4592, 2005.
- Regalbuto C, Le Moli R, Muscia V, Russo M, Vigneri R, Pezzino V. Severe Graves' ophthalmopathy after percutaneous ethanol injection in a nontoxic thyroid nodule. Thyroid 22:210-3, 2012.
- Van Herle AJ, Uller RP, Mathews NL, Brown J. Radioimmunoassay for measurement of thyroglobulin in human serum. J Clin Invest 52:1320,1973.
- Tandon N, Freeman MA, Weetman AP. T cell responses to synthetic TSH receptor peptides in Graves’ disease. Clin Exp Immunol 89:468-473, 1992.
- Chiovato L, Latrofa F, Braverman LE, Pacini F, Capezzone M, Masserini L, Grasso L, Pinchera A. Disappearance of humoral thyroid autoimmunity after complete removal of thyroid antigens. Ann Intern Med 139:346-351, 2003.
- Klintschar M, Schwaiger P, Mannweiler S, Regauer S, Kleiber M. Evidence of fetal microchimerism in Hashimoto’s thyroiditis. J Clin Endocrinol Metab 86:2494-2498, 2001.
- Imaizumi M, Pritsker A, Unger P, Davies TF. Intrathyroidal fetal microchimerism in pregnancy and postpartum. Endocrinology 143:247-253, 2002.
- Brix TH, Hansen PS, Kyvik KO, Hegedüs L. Aggregation of thyroid autoantibodies in twins from opposite-sex pairs suggests that microchimerism may play a role in the early stages of thyroid autoimmunity. J Clin Endocrinol Metab. 94:4439-43, 2009.
- Jørgensen KT, Pedersen BV, Nielsen NM, Jacobsen S, Frisch M.
Childbirths and risk of female predominant and other autoimmune diseases in a population-based Danish cohort. J Autoimmun. 38:J81-7, 2012.
- Weetman AP. Immunity, thyroid function and pregnancy: molecular mechanisms. Nat Rev Endocrinol 6:311-8, 2010.
211a. Liu L, Wu L, Gao A, Zhang Q, Lv H, Xu L, Xie C, Wu Q, Hou P, Shi B. The influence of dihydrotestosterone on the development of Graves' disease in female BALB/c mice. Thyroid. 2016 Jan 5. [Epub ahead of print]
- Simmonds MJ, Kavvoura FK, Brand OJ, Newby PR, Jackson LE, Hargreaves CE, Franklyn JA, Gough SC. Skewed X chromosome inactivation and female preponderance in autoimmune thyroid disease: an association study and meta-analysis. J Clin Endocrinol Metab 99:E127-31, 2014.
- Weiss M, Ingbar SH, Winblad S, Kasper DL. Demonstration of a saturable binding site for thyrotropin in Yersinia enterocolitica. Science 219:1331‑1333, 1983.
- Wolf MW, Misaki T, Bech K, Tvede M, Silva JE, Ingbar SH: Immunoglobulins of patients recovering from Yersinia enterocolitica infections exhibit Graves' disease‑like activity in human thyroid membranes. Thyroid 1:315, 1991.
- Wenzel BE, Heesemann J, Wenzel KW, Scriba PC. Patients with autoimmune thyroid diseases have antibodies to plasmid encoded proteins of enteropathogenic Yersinia. J Endocrinol Invest 11:139‑140, 1988.
- Arscott P, Rosen ED, Koenig RJ, Kaplan MM, Ellis T, Thompson N, Baker JR. Immunoreactivity to Yersinia enterocolitica antigens in patients with autoimmune thyroid disease. J Clin Endocrinol Metab 75:295-320, 1992.
- Effraimidis G, Tijssen JG, Strieder TG, Wiersinga WM. No causal relationship between Yersinia enterocolitica infection and autoimmune thyroid disease: evidence from a prospective study. Clin Exp Immunol. 165:38-43, 2011.
- Quaratino S, Thorpe CJ, Travers PJ, Londei M. Similar antigenic surfaces, rather than sequence homology, dictate T-cell epitope molecular mimicry. Proc Natl Acad Sci USA 92:10398-10402, 1995.
- Epstein FH. Molecular mimicry and autoimmunity. New Engl J Med 341:2068-2074, 1999.
- Klavinskis LS, Notkins AL, Oldstone MBA. Persistent viral infection of the thyroid gland: alteration of thyroid function in the absence of tissue injury. Endocrinology 122:567‑575, 1988.
- Jaspan JB, Sullivan K, Garry RF, Lopez M, Wolfe M, Clejan S, Yan C, Tenenbaum S, Sander DM, Ahmed B, Bryer-ash M. The interaction of a type A retroviral particle and class II human leukocyte antigen susceptibility genes in the pathogenesis of Graves’ disease. J Clin Endocrinol Metab 81:2271-2279, 1996.
- Tomoyose T, Komiya I, Takara M, Yabiku K, Kinjo Y, Shimajiri Y, Yogi H, Kouki T, Masuda M, Takasu N. Cytotoxic T-lymphocyte antigen-4 gene polymorphisms and human T-cell lymphotrophic virus-1 infection: their associations with Hashimoto’s thyroiditis in Japanese patients. Thyroid 12:673-677, 2002.
- Knight J, Laing P, Knight A, Adams D, Ling N. Thyroid‑stimulating autoantibodies usually contain only lambda‑light chains: evidence for the "forbidden clone" theory. J Clin Endocrinol Metab 62:342‑347, 1986.
- Brix TH, Christensen K, Holm NV, Harvald B, Hegedüs L. A population-based study of Graves’ disease in Danish twins. Clin Endocrinol 48:397-400, 1998.
- Bartels ED. Heredity in Graves' disease. Copenhagen, Enjnar Munksgaards Forlag, 1941.
- Brix TH, Kyvik KO, Hegedüs L. What is the evidence of genetic factors in the etiology of Graves’ disease? A brief review. Thyorid 8:727-734, 1998.
- Brix T, Kyvik KO, Hegedüs L. A population-based study of chronic autoimmune hypothyroidism in Danish twins. J Clin Endocrinol Metab 85:536-539, 2000.
- Brix TH, Kyvik KO, Christensen K, Hegedüs L. Evidence for a major role of heterdity in Graves’ disease: A population-based study of two Danish twin cohorts. J Clin Endocrinol Metab 86:930-934, 2001.
- McLeod DS, Caturegli P, Cooper DS, Matos PG, Hutfless S. Variation in rates of autoimmune thyroid disease by race/ethnicity in US military personnel. JAMA. 2014 311:1563-5.
- Hall R, Owen SG, Smart GA. Evidence for genetic predisposition to formation of thyroid auto‑antibodies. Lancet 2:187, 1960.
- Phillips D, Prentice L, Upadhyaya M, Lunt P, Chamberlain S, Roberts DF, McLachlan S, Smith BR. Autosomal dominant inheritance of autoantibodies to thyroid peroxidase and thyroglobulin--studies in families not selected for autoimmune thyroid disease. J Clin Endocrinol Metab 72:973-975, 1991.
- Phillips DIW, Shields DC, Dougoujon JM, Prentice L, McGuffin P, Rees Smith B. Complex segregation analysis of thyroid autoantibodies: Are they inherited as an autosomal dominant trait? Hum Hered 43:141-146, 1993.
- Wiebolt J, Koeleman BP, van Haeften TW.Endocrine autoimmune disease: genetics become complex. Eur J Clin Invest. 40:1144-55, 2010.
- Weetman AP, McGregor AM. Autoimmune thyroid disease: Further developments in our understanding. Endocrine Rev 15:788-830, 1994.
- Parkes AB, Darke C, Othman S, Thomas M, Young N, Richards CJ, Hall R, Lazarus JH. Major histocompatibility complex class II and complement polymorphisms in postpartum thyroiditis. Europ J Endocrinol 134:449-453, 1996.
- Yanagawa T, Mangklabruks A, Chang Y-B, Okamoto Y, Fisfalen M-E, Curran PG, DeGroot LJ. Human histocompatibility leukocyte antigen-DQA1*0501 allele associated with genetic susceptibility to Graves' disease in a Caucasian population. J Clin Endocrinol Metab 76:1569-1574, 1993.
- Chen Q-Y, Huang W, She J-X, Baxter F, Volpe R, Maclaren NK. HLA-DRB1*08, DRB1*03/DRB3*0101, and DRB3*0202 are susceptibility genes for Graves’ disease in North American Caucasians, whereas DRB1*07 is protective. J Clin Endocrinol Metab 84: 3182-3186, 1999.
- Ueda S, Oryoji D, Yamamoto K, Noh JY, Okamura K, Noda M, Kashiwase K, Kosuga Y, Sekiya K, Inoue K, Yamada H, Oyamada A, Nishimura Y, Yoshikai Y, Ito K, Sasazuki T. Identification of independent susceptible and protective HLA alleles in Japanese autoimmune thyroid disease and their epistasis. J Clin Endocrinol Metab 99:E379-83, 2014.
- Stenszky V, Kozma L, Balázs C, Rochlitz S, Bear JC, Farid NR. The genetics of Graves’ disease: HLA and disease susceptibility. J Clin Endocrinol Metab 61:735-740, 1985.
- Shields DC, Ratanachaiyavong S, McGregor AM, Collins A, Morton NE. Combined segregation and linkage analysis of Graves’ disease with a thyroid autoantibody diathesis. Amer J Hum Genet 55:540-554, 1994.
- Tomer Y, Barbesino G, Kedache M, Greenberg DA, Davies TF. Mapping of a major susceptibility locus for Graves’ disease (GD-1) to chromosome 14q31. J Clin Endocrinol Metab 82:1645-1648, 1997.
- Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ. CTLA-4 gene polymorphism associated with Graves’ disease in Caucasian population. J Clin Endocrinol Metab 80:41-45, 1995.
- Kotsa K, Watson PF, Weetman AP. A CTLA-4 gene polymorphism is associated with both Graves’ disease and autoimmune hypothyroidism. Clin Endocrinol 46:551-554, 1997.
- Villanueva R, Inzerillo AM, Tomer Y, Barbesino G, Meltzer M, Concepcion ES, Greenberg DA, MacLaren N, Sun ZS, Zhang DM, Tucci S, Davies TF. Limited genetic susceptibility to severe Graves’ ophthalmopathy: No role for CTLA-4 but evidence for an environmental etiology. Thyroid 10:791-798, 2000.
- Kinjo Y, Takasu N, Komiya I, Tomoyose T, Takara M, Kouki T, Shimajiri Y, Yabiku K, Yoshimura H. Remission of Graves’ hyperthyroidism and A/G polymorphism at position 49 in exon 1 of cytotoxic T lymphocyte-associated molecular-4 gene. J Clin Endocrinol Metab 87:2593-2596, 2002.
- Wang PW, Chen IY, Liu RT, Hsieh CJ, Hsi E, Juo SH. Cytotoxic T lymphocyte-associated molecule-4 gene polymorphism and hyperthyroid Graves' disease relapse after antithyroid drug withdrawal: a follow-up study. J Clin Endocrinol Metab. 92:2513-8, 2007.
- Ueda H, Howson JMM, Esposito L, Heward J, Snool Hywel, Chamberlain G, Rainbow DB, Huner KMD, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RCJ, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia Am Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehot J, Bingley P, Gillespie KM, Undlein DE, RØnningen KS, Guja C, Ionescu-Tirgovişte C, Savage DA, Maxwell AP. Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SCL. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506-511, 2003.
- Ban Y, Ban Y, Taniyama M, Katagiri T. Vitamin D receptor initiation codon polymorphism in Japanese patients with Graves’ disease. Thyroid 16:475-480, 2000.
- Collins JE, Heward JM, Nithiyananthan R, Nejentsev S, Todd JA, Franklyn JA, Gough SCL. Lack of an association of the vitamin D receptor gene with Graves’ disease in UK Caucasians. Clin Endocrinol 60:618-624,2004.
- Effraimidis G, Badenhoop K, Tijssen JG, Wiersinga WM. Vitamin D deficiency is not associated with early stages of thyroid autoimmunity. Eur J Endocrinol 167:43-8, 2012.
- Simmonds MJ, Heward JM, Carr-Smith J, Foxall H, Franklyn JA, Gough SC. Contribution of single nucleotide polymorphisms within FCRL3 and MAP3K7IP2 to the pathogenesis of Graves' disease. J Clin Endocrinol Metab. 91:1056-61, 2006.
- Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC). Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 39:1329-37, 2007.
- Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P, Pearce SHS. The codon 620 tryptophan allele of the lymphoid tyrosine phosphates (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 89: 5862-5865, 2004.
- Heward JM, Brand OJ, Barrett JC, Carr-Smith JD, Franklyn JA, Gough SC. Association of PTPN22 haplotypes with Graves' disease. J Clin Endocrinol Metab. 92:685-90, 2007.
- Brand OJ, Lowe CE, Heward JM, Franklyn JA, Cooper JD, Todd JA, Gough SC. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves' disease using a multilocus test and tag SNPs. Clin Endocrinol (Oxf). 66:508-12, 2007.
- Dechairo BM, Zabaneh D, Collins J, Brand O, Dawson GJ, Green AP, Mackay I, Franklyn JA, Connell JM, Wass JA, Wiersinga WM, Hegedus L, Brix T, Robinson BG, Hunt PJ, Weetman AP, Carey AH, Gough SC. Association of the TSHR gene with Graves' disease: the first disease specific locus. Eur J Hum Genet. 131: 223-30, 2005.
- 257. Brand OJ, Barrett JC, Simmonds MJ, Newby PR, McCabe CJ, Bruce CK, Kysela B, Carr-Smith JD, Brix T, Hunt PJ, Wiersinga WM, Hegedüs L, Connell J, Wass JA, Franklyn JA, Weetman AP, Heward JM, Gough SC. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves' disease. Hum Mol Genet.18:1704-13, 2009.
257a. Campbell P, Brix TH, Wilson SG, Ward LC, Hui J, Beilby JP, Hegedüs L, Walsh JP. Common genetic variants associated with thyroid function may be risk alleles for Hashimoto's disease and Graves' disease.Clin Endocrinol (Oxf). [Epub ahead of print], 2015.
- Santos LR, Durães C, Mendes A, Prazeres H, Alvelos MI, Moreira CS, Canedo P, Esteves C, Neves C, Carvalho D, Sobrinho-Simões M, Soares P. A polymorphism in the promoter region of the selenoprotein S gene (SEPS1) contributes to Hashimoto's thyroiditis susceptibility. J Clin Endocrinol Metab 99:E719-23, 2014.
- Barbesino G, Tomer Y, Concepcion ES, Davies TF, Greenberg DA and the International Consortium for the Genetics of Autoimmune Thyroid Disease. Linkage analysis of candidate genes in autoimmune thyroid disease. II. Selected gender-related genes and the X-chromosome. J Clin Endocrinol Metab 83: 3290-3295, 1998.
- Elsheikh M, Wass JAH, Conway GS. Autoimmune thyroid disease in women with Turner’s syndrome - the association with karyotype. Clin Endocrinol 55:223-226, 2001.
- Taylor JC, Gough SC, Hunt PJ, Brix TH, Chatterjee K, Connell JM, Franklyn JA, Hegedus L, Robinson BG, Wiersinga WM, Wass JA, Zabaneh D, Mackay I, Weetman AP. A genome-wide screen in 1119 relative pairs with autoimmune thyroid disease. J Clin Endocrinol Metab 91:646-53, 2005.
- Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, Zhang XM, Yuan GY, Li CG, Xue LQ, Shen M, Liu W, Xie F, Yang SY, Wang HF, Shi JY, Sun WW, Du WH, Zuo CL, Shi JX, Liu BL, Guo CC, Zhan M, Gu ZH, Zhang XN, Sun F, Wang ZQ, Song ZY, Zou CY, Sun WH, Guo T, Cao HM, Ma JH, Han B, Li P, Jiang H, Huang QH, Liang L, Liu LB, Chen G, Su Q, Peng YD, Zhao JJ, Ning G, Chen Z, Chen JL, Chen SJ, Huang W, Song HD; China Consortium for Genetics of Autoimmune Thyroid Disease. A genome-wide association study identifies two new risk loci for Graves' disease. Nat Genet. 43:897-901, 2011.
- Cooper JD, Simmonds MJ, Walker NM, Burren O, Brand OJ, Guo H, Wallace C, Stevens H, Coleman G; Wellcome Trust Case Control Consortium, Franklyn JA, Todd JA, Gough SC. Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet 21:5202-8, 2012.
2764 Zhao SX, Xue LQ, Liu W, Gu ZH, Pan CM, Yang SY, Zhan M, Wang HN, Liang J, Gao GQ, Zhang XM, Yuan GY, Li CG, Du WH, Liu BL, Liu LB, Chen G, Su Q,Peng YD, Zhao JJ, Ning G, Huang W, Liang L, Qi L, Chen SJ, Chen Z, Chen JL, Song HD; China Consortium for the Genetics of Autoimmune Thyroid Disease. Robust evidence for five new Graves' disease risk loci from a staged genome-wide association analysis. Hum Mol Genet 22:3347-3362, 2013.
- Simmonds MJ. GWAS in autoimmune thyroid disease: redefining our understanding of pathogenesis. Nat Rev Endocrinol. 9:277-287, 2013.
- Hardy J, Singleton A. Genomewide association studies and human
disease. N Engl J Med. 360:1759-68, 2009.
- Medici M, Porcu E, Pistis G, Teumer A, Brown SJ, Jensen RA, Rawal R, Roef GL, Plantinga TS, Vermeulen SH, Lahti J, Simmonds MJ, Husemoen LL, Freathy RM, Shields BM, Pietzner D, Nagy R, Broer L, Chaker L, Korevaar TI, Plia MG, Sala C, Völker U, Richards JB, Sweep FC, Gieger C, Corre T, Kajantie E, Thuesen B, Taes YE, Visser WE, Hattersley AT, Kratzsch J, Hamilton A, Li W, Homuth G, Lobina M, Mariotti S, Soranzo N, Cocca M, Nauck M, Spielhagen C, Ross A, Arnold A, van de Bunt M, Liyanarachchi S, Heier M, Grabe HJ, Masciullo C, Galesloot TE, Lim EM, Reischl E, Leedman PJ, Lai S, Delitala A, Bremner AP, Philips DI, Beilby JP, Mulas A, Vocale M, Abecasis G, Forsen T, James A, Widen E, Hui J, Prokisch H, Rietzschel EE, Palotie A, Feddema P, Fletcher SJ, Schramm K, Rotter JI, Kluttig A, Radke D, Traglia M, Surdulescu GL, He H, Franklyn JA, Tiller D, Vaidya B, de Meyer T, Jørgensen T, Eriksson JG, O'Leary PC, Wichmann E, Hermus AR, Psaty BM, Ittermann T, Hofman A, Bosi E, Schlessinger D, Wallaschofski H, Pirastu N, Aulchenko YS, de la Chapelle A, Netea-Maier RT, Gough SC, Meyer Zu Schwabedissen H, Frayling TM, Kaufman JM, Linneberg A, Räikkönen K, Smit JW, Kiemeney LA, Rivadeneira F, Uitterlinden AG, Walsh JP, Meisinger C, den Heijer M, Visser TJ, Spector TD, Wilson SG, Völzke H, Cappola A, Toniolo D, Sanna S, Naitza S, Peeters RP. Identification of novel genetic loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLoS Genet. 10:e1004123, 2014.
- Brix TH, Hansen PS, Rudbeck AB, Hansen JB, Skytthe A, Kyvik KO, Hegedus L. Low birth weight is not associated with thyroid autoimmunity: a population-based twin study. J Clin Endocrinol Metab. 91:3499-502, 2006.
- Kajantie E, Phillips DI, Osmond C, Barker DJ, Forsén T, Eriksson JG. Spontaneous hypothyroidism in adult women is predicted by small body size at birth and during childhood. J Clin Endocrinol Metab. 91:4953-6, 2006.
- Radetti G, Fanolla A, Pappalardo L, Gottardi E. Prematurity may be a risk factor for thyroid dysfunction in childhood. J Clin Endocrinol Metab. 92:155-9, 2007.
- Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid 12:975-986, 2002.
- Boelaert K, Newby PR, Simmonds MJ, Holder RL, Carr-Smith JD, Heward JM, Manji N, Allahabadia A, Armitage M, Chatterjee KV, Lazarus JH, Pearce SH, Vaidya B, Gough SC, Franklyn JA. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. 123:183.e1-9, 2010.
- Irvine WJ, Davies SH, Teitelbaum S, Delamore IW, Williams AW. The clinical and pathological significance of gastric parietal cell antibody. Ann NY Acad Sci 124:657, 1965.
- Doniach D, Roitt IM, Taylor KB. Autoimmune phenomena in pernicious anemia. Serological overlap with thyroiditis, thyrotoxicosis, and systemic lupus erythematosus. Br Med J 1:1374, 1963.
- Tudhope GR, Wilson GM. Deficiency of vitamin B12 in hypothyroidism. Lancet 1:703, 1962.
- Naiyer AJ, Shah J, Hernandez L, Kim SY, Ciaccio EJ, Cheng J, Manavalan S, Bhagat G, Green PH. Tissue transglutaminase antibodies in individuals with celiac disease bind to thyroid follicles and extracellular matrix and may contribute to thyroid dysfunction. Thyroid. 18:1171-8, 2008.
- Buchanan WW, Crooks J, Alexander WD, Koutras DA, Wayne EJ, Gray KG. Association of Hashimoto's thyroiditis and rheumatoid arthritis. Lancet 1:245, 1961.
- Mulhern LM, Masi AT, Shulman LE. Hashimoto's disease. A search for associated disorders in 170 clinically detected cases. Lancet 2:508, 1966.
- Morita H, Arima T, Matsuda M. Prevalence of nonthyroid specific autoantibodies in autoimmune thyroid diseases. Journal of Clinical Endocrinology and Metabolism 80:1203-1206, 1995.
- Dobson R, Giovannoni G. Autoimmune disease in people with multiple sclerosis and their relatives: a systematic review and meta-analysis. J Neurol 260:1272-85, 2013
281 Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine 60:355-362, 1981.
- Weetman AP. Autoimmunity to steroid-producing cells and familial polyendocrine autoimmunity. Balliere’s Clin Endocrinol Metab 9:157-174, 1995.
- Falorni A, Laureti S, Santeusanio F. Autoantibodies in autoimmune polyendocrine syndrome type II. Endocrinol Metab Clin N Am 31:369-389, 2002.
- Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy - candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 322:1829-1836, 1990.
- Nithiyamanthan R, Heward JM, Allahabadia A, Barnett AJ, Franklyn JA, Gough SCL. A heterozygous deletion of the autoimmune receptor (AIRE1) gene, autoimmune thyroid disease, and type 1 diabetes: No evidence for association. J Clin Endocrinol Metab 85:1320-1322, 2000.
- Menconi F, Osman R, Monti MC, Greenberg DA, Concepcion ES, Tomer Y. Shared molecular amino acid signature in the HLA-DR peptide binding pocket predisposes to both autoimmune diabetes and thyroiditis. Proc Natl Acad Sci U S A. 107:16899-903, 2010.
- Wiebolt J, Achterbergh R, den Boer A, van der Leij S, Marsch E, Suelmann B, de Vries R, van Haeften TW. Clustering of additional autoimmunity behaves differently in Hashimoto's patients compared with Graves' patients. Eur J Endocrinol 164:789-94, 2011.
- Sridama V, Pacini F, DeGroot LJ. Decreased suppressor T lymphocytes in autoimmune thyroid diseases detected by monoclonal antibodies. J Clin Endocrinol Metab 54:316-319, 1982.
- Pacini F, DeGroot LJ. Studies of immunoglobulin synthesis in cultures of peripheral T and B lymphocytes: Reduced T-suppressor cell activity in Graves’ disease. Clin Endocrinol 18L219-232, 1983.
- Fournier C, Chen H, Leger A, Charreire J. Immunological studies of autoimmune thyroid disorders: abnormalities in the inducer T cell subset and proliferative responses to autologous and allogeneic stimulation. Clin Exp Immunol 54:539-546, 1983.
- Ludgate ME, McGregor AM, Weetman AP, Ratanachaiyavong S, Lazarus JH, Hall R, Middleton GW. Analysis of T cell subsets in Graves' disease: alterations associated with carbimazole. Br Med J 288:526-530, 1984.
- Misaki T, Konishi J, Iida Y, Endo K, Torizuka K. Altered balance of immunoregulatory T lymphocyte subsets in autoimmune thyroid diseases. Acta Endocrinol 105:200-204, 1984.
- Volpe R, Karlsson A, Jansson R, Dahlberg PA. Evidence that antithyroid drugs induce remissions in Graves' disease by modulating thyroid cellular activity. Clin Endocrinol 25:453‑462, 1986.
- How J, Topliss DJ, Strakosch C, Lewis M, Row VV, Volpe R.T lymphocyte sensitization and suppressor T lymphocyte defect in patients long after treatment for Graves' disease. Clin Endocrinol 18:61‑71, 1983.
- Marazuela M, Garcia-Lopez MA, Figueroa-Vega N, de la Fuente H, Alvarado-Sanchez B, Monsivais-Urenda A, Sanchez-Madrid F, Gonzalez-Amaro R. Regulatory T cells in human autoimmune thyroid disease.J Clin Endocrinol Metab. 2006;91:3639-46.
295a. Rodríguez-Muñoz A, Vitales-Noyola M, Ramos-Levi A, Serrano-Somavilla A, González-Amaro R, Marazuela M. Levels of regulatory T cells CD69+NKG2D+IL-10+ are increased in patients with autoimmune thyroid disorders. Endocrine. Jun 23. [Epub ahead of print], 2015.
- Glick AB, Wodzinski A, Fu P, Levine AD, Wald DN. Impairment of regulatory T-cell function in autoimmune thyroid disease. Thyroid 23:871-878, 2013.
- Armengol MP, Sabater L, Fernández M, Ruíz M, Alonso N, Otero MJ, Martínez-Cáceres E, Jaraquemada D, Pujol-Borrell R. Influx of recent thymic emigrants into autoimmune thyroid disease glands in humans. Clin Exp Immunol 153:338-50, 2008.
- Jackson RA, Haynes BF, Burch WM, Shimizu K, Bowring MA, Eisenbarth GS. Ia+ T cells in new onset Graves' disease. J Clin Endocrinol Metab 59:187‑190, 1984.
- Koukkou E, Panayiotidis P, Alevizou‑Terzaki V, Thalassinos N: High levels of serum soluble interleukin‑2 receptors in hyperthyroid patients: Correlation with serum thyroid hormones and independence from the etiology of the hyperthyroidism. J Clin Endocrinol Metab 73:771‑776, 1991.
- Sharma RB, Fan X, Caturegli P, Rose NR, Burek CL. Invariant NKT cell lines derived from the NOD·H2 mouse enhance autoimmune thyroiditis. J Thyroid Res 1:895923, 2011.
- Figueroa-Vega N, Alfonso-Pérez M, Benedicto I, Sánchez-Madrid F, González-Amaro R, Marazuela M. Increased circulating pro-inflammatory cytokines and Th17 lymphocytes in Hashimoto's thyroiditis. J Clin Endocrinol Metab. 95:953-62, 2010.
- Peng D, Xu B, Wang Y, Guo H, Jiang Y. A high frequency of circulating th22 and th17 cells in patients with new onset graves' disease. PLoS One 8:e68446, 2013.
302a. Rodríguez-Muñoz A, Martínez-Hernández R, Ramos-Leví AM, Serrano-Somavilla A, González-Amaro R, Sánchez-Madrid F, de la Fuente H, Marazuela M. Circulating microvesicles regulate Treg and Th17 differentiation in human autoimmune thyroid disorders. J Clin Endocrinol Metab. 100:E1531-9, 2015.
- Zhu C, Ma J, Liu Y, Tong J, Tian J, Chen J, Tang X, Xu H, Lu L, Wang S. Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J Clin Endocrinol Metab 97:943-50, 2012.
- Margolick JB, Hsu S‑M, Volkman DJ, Burman KD, Fauci AS. Immunohistochemical characterization of intrathyroid lymphocytes in Graves' disease. Interstitial and intraepithelial populations. Amer J Med 76:815‑821, 1984.
- Canonica GW, Caria M, Torre G, Risso A, Cosulich ME, Bagnasco M. Autoimmune thyroid disease: purification and phenotypic analysis of intrathyroid T cells. J Endocrinol Invest 7:641, 1984.
- Misaki T, Konishi J, Arai K, Iida Y, Kasagi K, Kuma K, Torizuka K. HLA‑DR antigen expression on intrathyroidal lymphocytes and thyrocytes in Hashimoto's thyroiditis and Graves' disease: an immunohistological study. Endocrinol Japon 34:257‑262, 1987.
- Ashhab Y, Dominguez O, Sospedra M, Roura-Mir C, Lucas-Martin A, Pujol-Borrell R. A one-tube polymerase chain reaction protocol demonstrates CC chemokine overexpression in Graves’ disease glands. J Clin Endocrinol Metab 84: 2873-2882, 1999.
- Ueki Y, Eguchi K, Otsubo T, Kawabe Y, Shimomura C, Matsunaga M, Tezuka H, Nakao H, Kawakami A, Izumi M, Ishikawa N, Ito K, Nagataki S. Phenotypic analyses and Concanavalin‑A‑ induced suppressor cell dysfunction of intrathyroidal lymphocytes from patients with Graves' disease. J Clin Endocrinol Metab 67:1018‑1024, 1988.
- Tezuka H, Eguchi K, Fukuda T, Otsubo T, Kawabe Y, Ueki Y, Matsunaga M, Shimomura C, Nakao H, Ishikawa N, Ito K, Nagataki S. Natural killer and natural killer‑like cell activity of peripheral blood and intrathyroidal mononuclear cells from patients with Graves' disease. J Clin Endocrinol Metab 66:702, 1988.
310 Bagnasco M, Venuti D, Prigione I, Torre GC, Ferrini S, Canonica GW. Graves' disease: phenotypic and functional analysis at the clonal level of the T‑cell repertoire in peripheral blood and in thyroid. Clin Immunol Immunopathol 47:230‑239, 1988.
- Del Prete GF, Maggi E, Mariotti S, Tiri A, Vercelli D, Parronchi P, Macchia E, Pinchera A, Ricci M, Romagnani S. Cytolytic T lymphocytes with natural killer activity in thyroid infiltrate of patients with Hashimoto's thyroiditis: analysis at clonal level. J Clin Endocrinol Metab 62:52, 1986.
- Ajjan RA, Watson PF, McIntosh RS, Weetman AP. Intrathyroidal cytokine gene expression in Hashimoto’s thyroiditis. Clin Exp Immunol 105:523-528, 1996.
- Yang D, Hiromatsu Y, Hoshino T, Inoue Y, Itoh K, Nonaka K. Dominant infiltration of TH1-type CD4+ T cells at the retrobulbar space of patients with thyroid-associated ophthalmopathy. Thyroid 9: 305-310, 1999.
- Pichurin P, Pichurina O, Chazenbalk GD, Paras C, Chen C-R, Rapoport B, McLachlan SM. Immune deviation away from Th1 in interferon-g knockout mice does not enhance TSH receptor antibody production after naked DNA vaccination. Endocrinology 143:1182-1189, 2002.
- WeetmanA. Immune reconstitution syndrome and the thyroid. Best Pract Res Clin Endocrinol Metab 23:693-702, 2009.
- SinhaA, Abinun M, Gennery AR, Barge D, Slatter M, Cheetham T. Graves' immune reconstitution inflammatory syndrome in childhood. Thyroid 23:1010-1014, 2013.
- Raines KB, Baker JR, Lukes YG, Wartofsky L, Burman KD. Antithyrotropin antibodies in the sera of Graves' disease patients. J Clin Endocrinol Metab 61:217‑222, 1985.
- Sikorska HM. Anti‑thyroglobulin anti‑idiotypic antibodies in sera of patients with Hashimoto's thyroiditis and Graves' disease. J Immunol 137:3786‑3795, 1986.
- Hara Y, Sridama V, DeGroot LJ. Auto‑anti‑idiotype antibody against antithyroglobulin auto‑antibody in humans. J Clin Lab Immunol 26:13‑20, 1988.
- Tandon N, Jayne DRW, McGregor AM, Weetman AP. Analysis of anti-idiotypic antibodies against anti-microsomal antibodies in patients with thyroid autoimmunity. J Autoimmunity 5:557-570, 1992.
- Hanafusa T, Chiovato L, Doniach D, Pujol‑Borrell R, Russell RCG, Bottazzo GF. Aberrant expression of HLA‑DR antigen on thyrocytes in Graves' disease: relevance for autoimmunity. Lancet 2:1111‑1115, 1983.
- Bottazzo GF, Pujol‑Borrell R, Hanafusa T, Feldmann F. Role of aberrant HLA‑DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet 2:1115‑1119, 1983.
- Buscema M, Todd I, Deuss U, Hammond L, Mirakian R, Pujol‑Borrell R, Bottazzo GF: Influence of tumor necrosis factor‑ on the modulation by interferon‑a of HLA class II molecules in human thyroid cells and its effect on interferon‑g binding. J Clin Endocrinol Metab 69:433, 1989.
- Weetman AP, Volkman DJ, Burman KD, Gerrard TL, Fauci AS. The in vitro regulation of human thyrocyte HLA‑DR antigen expression. J Clin Endocrinol Metab 61:817‑824, 1985.
- Davies TF, Bermas B, Platzer M, Roman SH. T‑cell sensitization to autologous thyroid cells and normal nonspecific suppressor T‑cell function in Graves' disease. Clin Endocrinol 22:155‑167, 1985.
- Eguchi K, Otsubo T, Kawabe Y, Shimomura C, Ueki Y, Nakao H, Tezuka H, Matsunaga M, Fukuda T, Ishikawa N, Ito K, Nagataki S. Synergy in antigen presentation by thyroid epithelial cells and monocytes from patients with Graves' disease. Clin Exp Immunol 72:84‑90, 1988.
- Tandon N, Metcalfe RA, Barnett D, Weetman AP. Expression of the costimulatory molecule B7/BB1 in autoimmune thyroid disease. Quart J Med 87:231-236, 1994.
- Matsuoka N, Eguchi K, Kawakami A, Tsuboi M, Nakamura H, Kimura H, Ishikawa N, Ito K, Nagataki S. Lack of B7-1/BB1 and B7-2/B70 expression on thyrocytes of patients with Graves’ disease. Delivery of costimulatory signals from bystander professional antigen-presenting cells. J Clin Endocrinol Metab 81:4137-4143, 1996.
- Battifora M, Pesce G, Paolieri F, Fiorino N, Giordano C, Riccio M, Torre G, Olive D, Bagnasco M. B7.1 costimulatory molecule is expressed on thyroid follicular cells in Hashimoto’s thyroiditis, but not in Graves’ disease. J Clin Endocrinol Metab 83:4130-4139, 1998.
- Lombardi G, Arnold K, Uren J, Marelli-Berg F, Hargreaves F, Inami N, Weetman AP, Lechler R. Antigen presentation by IFN-g treated thyroid follicular cells inhibits IL-2 and supports IL-4 production by B7-dependent human T cells. Europ J Immunol 27:62-71, 1997.
- Marelli-Berg F, Weetman AP, Frasca L, Deacock SJ, Inami N, Lombardi G, Lechler RI. Antigen presentation by epithelial cells induces anergic immunoregulatory CD45RO+ T cells and deletion of CD45RA+ T cells. J Immunol 159:5853-5861, 1997.
- Li YS, Kanamoto N, Hataya Y, Moriyama K, Hiratani H, Nakao K, Akamizu T. Transgenic mice producing major histocompatibility complex class II molecules on thyroid cells do not develop apparent thyroid autoimmunity. J Clin Endocrinol Metab 145: 2524- 2530, 2004.
- Shah R, Banks K, Patel A, Dogra S, Terrell R, Powers PA, Fenton C, Dinauer CA, Tuttle RM, Francis GL. Intense expression of the B7-2 antigen presentation coactivator is an unfavorable prognostic indicator for differentiated thyroid carcinoma of children and adolescents. J Clin Endocrinol Metab 87:4391-4397, 2002.
- Fisfalen M-E, Franklin WA, DeGroot LJ, Cajulis RS, Soltani K, Ryan M, Jones N. Expression of HLA-ABC and -DR antigens in thyroid neoplasia and correlation with mononuclear leukocyte infiltration. J Endocrinol Invest 13:41-48, 1990.
- Cohen SB, Dijkstra CD, Weetman AP. Sequential analysis of experimental autoimmune thyroiditis induced by neonatal thymectomy in the Buffalo strain rat. Cell Immunol 114:126-136, 1988.
- Leclere J, Bene MC, Duprez A, Faure G, Thomas JL, Vignaud JM, Burlet C. Behavior of thyroid tissue from patients with Graves' disease in nude mice. J Clin Endocrinol Metab 59:175, 1984.
- Sospedra M, Obiols G, Babi LFS, Tolosa E, Vargas F, Roura-Mir C, Lucas-Martin A, Ercilla G, Pujol-Borrell R. Hyperinducibility of HLA class II expression of thyroid follicular cells from Graves’ disease. J Immunol 154:4213-4222, 1995.
- Wong GWK, Cheng PS. Increasing incidence of childhood Graves’ disease in Hong Kong: A follow-up study. Clin Endocrinol 54:547-550, 2001.
- Benvenga S, Trimarchi F. Changed presentation of Hashimoto's thyroiditis in North-Eastern Sicily and Calabria (Southern Italy) based on a 31-year experience. Thyroid 18:429-41, 2008.
- Ott J, Meusel M, Schultheis A, Promberger R, Pallikunnel SJ, Neuhold N, Hermann M. The incidence of lymphocytic thyroid infiltration and Hashimoto's thyroiditis increased in patients operated for benign goiter over a 31-year period. Virchows Arch 459:277-81, 2011.
- Caturegli P, De Remigis A, Chuang K, Dembele M, Iwama A, Iwama S. Hashimoto's thyroiditis: celebrating the centennial through the lens of the Johns Hopkins hospital surgical pathology records. Thyroid 23:142-150, 2013.
- Kondrashova A, Viskari H, Haapala AM, Seiskari T, Kulmala P, Ilonen J, Knip M, Hyöty H. Serological evidence of thyroid autoimmunity among schoolchildren in two different socioeconomic environments. J Clin Endocrinol Metab 93:729-34, 2008.
- Kaplan MM. Hypothyroidism after treatment with interleukin‑2 and lymphokine‑activated killer cells. N Engl J Med 318:1557‑1563, 1988.
- Gilquin J, Viard J-P, Jubault , Sert C, Kazatchkine MD. Delayed occurrence of Graves’ disease after immune restoration with HAART. Lancet 352:1907-1908, 1998.
- Durelli L, Ferrero B, Oggero A, Verdun E, Ghezzi A, Montanari E, Zaffaroni M and the Betaferon Safety Trial Study Group. Thyroid function and autoimmunity during interferon b-1b treatment: A multicenter prospective study. J Clin Endocrinol Metab 86:3525-3532, 2001.
- Craccio N, Dradano A, Manfredonia F et al. Long-term follow-up of 106 multiple sclerosis patients undergoing interferon-β 1a or 1b therapy : predictive factors of thyroid disease development and duration. J Clin Endocrinol Metab 90:4133-4137, 2005.
- Hidaka Y, Amino N, Iwatani Y, Itoh E, Matsunaga M, Tamaki H. Recurrence of thyrotoxicosis after attack of allergic rhinitis in patients with Graves' disease. J Clin Endocrinol Metab 77:1667-1670, 1993.
- Yamada T, Sato A, Komiya I, Nishimori T, Ito Y, Terao A, Eto S, Tanaka Y. An elevation of serum immunoglobulin E provides a new aspect of hyperthyroid Graves’ disease. J Clin Endocrinol Metab 85:2775-2778, 2000.
- Bartalena L, Bogazzi F, Tanda ML, Manetti L, Dell’Unto E, Martino E. Cigarette smoking and the thyroid. Europ J Endocrinol 133:507-512, 1995.
- Andersen SL, Olsen J, Wu CS, Laurberg P. Smoking reduces the risk of hypothyroidism and increases the risk of hyperthyroidism: evidence from 450 842 mothers giving birth in Denmark. Clin Endocrinol (Oxf) 80:307-14, 2014.
- Rumold R, Jyrala M, Diaz-Sanchez D. Secondhand smoke induces allergic sensitization in mice. J Immunol 167:4765-4770, 2001.
- Belin RM, Astor BC, Powe NR, Ladenson PW. Smoke exposure is associated with a lower prevalence of serum thyroid autoantibodies and thyrotropin concentration elevation and a higher prevalence of mild thyrotropin concentration suppression in the third National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 89: 6077-6086, 2004.
- Carlé A, Bülow Pedersen I, Knudsen N, Perrild H, Ovesen L, Banke Rasmussen L, Jørgensen T, Laurberg P. Smoking cessation is followed by a sharp but transient rise in the incidence of overt autoimmune hypothyroidism - a population-based, case-control study. Clin Endocrinol (Oxf) 77:764-72, 2012.
- Schmeltz LR, Blevins TC, Aronoff SL, Ozer K, Leffert JD, Goldberg MA, Horowitz BS, Bertenshaw RH, Troya P, Cohen AE, Lanier RK, Wright C 4th. Anatabine supplementation decreases thyroglobulin antibodies in patients with chronic lymphocytic autoimmune(Hashimoto's) thyroiditis: A randomized controlled clinical trial. J Clin Endocrinol Metab 99:E137-42, 2014.
- Gu JY, Qian CH, Tang W, Wu XH, Xu KF, Scherbaum WA, Schott M, Liu C. Polychlorinated biphenyls affect thyroid function and induce autoimmunity in Sprague-Dawley rats. Horm Metab Res. 41:471-4, 2009.
- de Freitas CU, Grimaldi Campos RA, Rodrigues Silva MA, Panachão MR, de Moraes JC, Waissmann W, Roberto Chacra A, Maeda MY, Minazzi Rodrigues RS, Gonçalves Belchor J, Oliveira Barbosa S, Santos RT. Can living in the surroundings of a petrochemical complex be a risk factor for autoimmune thyroid disease? Environ Res. 110:112-7, 2010.
- Goldner WS, Sandler DP, Yu F, Hoppin JA, Kamel F, Levan TD Pesticide use and thyroid disease among women in the Agricultural Health Study. Am J Epidemiol.171:455-64, 2010.
- Papanastasiou L, Alevizaki M, Piperingos G, Mantzos E, Tseleni-Balafouta S, Koutras DA. The effect of iodine administration on the development of thyroid autoimmunity in patients with nontoxic goiter. Thyroid 10:493-497, 2000.
- Zois C, Stavrou I, Svarna E, Seferiadis K, Tsatsoulis A. Natural course of autoimmune thyroiditis after elimination of iodine deficiency in northwestern Greece. Thyroid. 16: 289-93, 2006.
- Teng X, Shan Z, Chen Y, Lai Y, Yu J, Shan L, Bai X, Li Y, Li N, Li Z, Wang S, Xing Q, Xue H, Zhu L, Hou X, Fan C, Teng W. More than adequate iodine intake may increase subclinical hypothyroidism and autoimmune thyroiditis: a cross-sectional study based on two Chinese communities with different iodine intake levels. Eur J Endocrinol 164:943-50, 2011.
- McLachlan SM, Braley-Mullen H, Chen CR, Aliesky H, Pichurin PN, Rapoport B. Dissociation between iodide-induced thyroiditis and antibody-mediated hyperthyroidism in NOD.H-2h4 mice. Endocrinol 146: 294-300, 2005.
- Yamazaki K, Tanigawa K, Suzuki K, Yamada E, Yamada T, Takano K, Obara T, Sato K. Iodide-induced chemokines and genes related to immunological function in cultured human thyroid follicles in the presence of thyrotropin. Thyroid. 20:67-76, 2010.
362a. Wu Q, Rayman MP, Lv H, Schomburg L, Cui B, Gao C, Chen P, Zhuang G, Zhang Z, Peng X, Li H, Zhao Y, He X, Zeng G, Qin F, Hou P, Shi B. Low population selenium status is associated with increased prevalence of thyroid disease. J Clin Endocrinol Metab. 100:4037-47, 2015.
- Van Zuuren EJ, Albusta AY, Fedorowicz Z, Carter B, Pijl H. Selenium supplementation for Hashimoto's thyroiditis: Summary of a Cochrane Systematic Review. Eur Thyroid J. 2014 3:25-31.
- Goswami R, Marwaha RK, Gupta N, Tandon N, Sreenivas V, Tomar N, Ray D, Kanwar R, Agarwal R. Prevalence of vitamin D deficiency and its relationship with thyroid autoimmunity in Asian Indians: a community-based survey. Br J Nutr. 102:382-6, 2009.
364a. Ma J, Wu D, Li C, Fan C, Chao N, Liu J, Li Y, Wang R, Miao W, Guan H, Shan Z, Teng W. Lower serum 25-hydroxyvitamin D level is associated with 3 types of autoimmune thyroid diseases. Medicine (Baltimore). 94:e1639, 2015.
364b. Wang J, Lv S, Chen G, Gao C, He J, Zhong H, Xu Y. Meta-analysis of the association between vitamin D and autoimmune thyroid disease. Nutrients. 7:2485-98, 2015.
- Frisch M, Nielsen NM, Pedersen BV. Same-sex marriage, autoimmune thyroid gland dysfunction and other autoimmune diseases in Denmark 1989-2008. Eur J Epidemiol 29:63-71, 2014.
- Chiovato L, Pinchera A. Stressful life events and Graves’ disease. Europ J Endocrinol 134:680-682, 1996.
- Streider TGA, Prummel MF, Tijssen JCP, Brosschot JF, Wiersinga W. Stress is not associated wirthh thyroid peroxidase autoantibodies in euthyroid women. Brain Behav Immun 19: 203-206, 2004.
- Steinman L. Elaborate interactions between the immune and nervous systems. Nature Immunol 5:575-581, 2004.
- Saint-Mezard P, Chavagnac C, Bosset S, Ionescu M, Peyron E, Kaiserlian D, Nicolas J-F, Bérard F. Psychological stress exerts an adjuvant effect on skin dendritic cell functions in vivo. J Immunol 171:4073-4080, 2003.
- Carlé A, Pedersen IB, Knudsen N, Perrild H, Ovesen L, Rasmussen LB, Jørgensen T, LaurbergP. Moderate alcohol consumption may protect against overt autoimmune hypothyroidism: a population-based case-control study. Eur J Endocrinol 167:483-490, 2012.
- Effraimidis G, Tijssen JGP, Wiersinga WM. Alcohol consumption as a risk factor for autoimmune thyroid disease: a prospective study. Eur Thyroid J 1:99–104, 2012.
- Hutfless S, Matos P, Talor MV, Caturegli P, Rose NR. Significance of prediagnostic thyroid antibodies in women with autoimmune thyroid disease. J Clin Endocrinol Metab 96:E1466-71, 2011
- McGregor AM, Ibbertson HK, Smith BR, Hall R. Carbimazole and autoantibody synthesis in Hashimoto's thyroiditis. Brit Med J 281:968‑969, 1980.
- Wenzel KW, Lente JR. Similar effects of thionamide drugs and perchlorate on thyroid‑stimulating immunoglobulins in Graves' disease: evidence against an immunosuppressive action of thionamide drugs. J Clin Endocrinol Metab 58:62‑ 69, 1984.
- Weetman AP, Holt ME, Campbell AK, Hall R, McGregor AM. Methimazole and generation of oxygen radicals by monocytes: potential role in immunosuppression. Brit Med J 288:518‑520, 1984.
- Mitsiades N, Poulaki V, Tseleni-Balafouta S, Chrousos GP, Koutras DA. Fas ligand expression in thyroid follicular cells from patients with thionamide-treated Graves’ disease. Thyroid 10:527-532, 2000.
- Komiya I, Yamada T, Sato A, Kouki T, Nishimori T, Takasu N. Remission and recurrence of hyperthyroid Graves’ disease during and after methimazole treatment when assessed by IgE and interleukin 13. J Clin Endocrinol Metab 86:3540-3544, 2001.
- Kimura HJ, Chen CY, Tzou SC, Rocchi R, Landek-Salgado MA, Suzuki K, Kimura M, Rose NR, Caturegli P. Immunoproteasome overexpression underlies the pathogenesis of thyroid oncocytes and primary hypothyroidism: studies in humans and mice. PLoS One. 4:e7857, 2009.
- Crile G Jr. The treatment of diseases of the thyroid gland. Ann R Coll Surg Engl 14:158, 1954.
- Hubble D. The diagnosis and treatment of auto‑immunizing thyroiditis. Scott Med J 4:55, 1959.
- Bastenie PA. Contribution a l'etiologie du myxodeme spontane de l'adulte. Bull Acad Med Belg 9:179, 1944.
- Douglass RC, Jacobson SD. Pathologic changes in adult myxedema. Survey of necropsies. J Clin Endocrinol Metab 17:1354, 1957.
- Owen SG, Smart GA. Thyroid antibodies in myxedema. Lancet 2:1034, 1958.
- Drexhage HA. Autoimmunity and thyroid growth. Where do we stand? European Journal of Immunology 135:39-45, 1996.
- Buchanan WW, Alexander WD, Crooks J, Koutras DA, Wayne EJ, Anderson JR, Goudie RB. Association of thyrotoxicosis and autoimmune thyroiditis. Br Med J 1:843, 1961.
- Fatourechi V, McConahey WM, Woolner LB. Hyperthyroidism associated with histologic Hashimoto's thyroiditis. Mayo Clin Proc 46:682, 1971.
- Whitesell FB, Black BM. Statistical study of clinical significance of lymphocytic and fibrocytic replacements in hyperplastic thyroid gland. J Clin Endocrinol 9:1202, 1949.
- Blagg CR. Antibodies to thyroglobulin in patients with thyrotoxicosis treated with radioactive iodine. Lancet 2:1364, 1960.
- Matsuzuka F, Miyauchi A, Katayama S, Narabayashi I, Ikeda H, Kuma K, Sugawara M. Clinical aspects of primary thyroid lymphoma: Diagnosis and treatment based on our experience of 119 cases. Thyroid 3:93-99, 1993.
- Ehlers M, Schott M. Hashimoto's thyroiditis and papillary thyroid cancer: are they immunologically linked? Trends Endocrinol Metab 25:656-664, 2014.
- Gribetz D, Talbot NB, Crawford JD. Goiter due to lymphocytic thyroiditis (Hashimoto's struma): Its occurrence in preadolescent and adolescent girls. N Engl J Med 250:555, 1954.
- Fatourechi MM, Hay ID, McIver B, Sebo TJ, Fatourechi V. Invasive fibrous thyroiditis (Riedel thyroiditis): the Mayo Clinic experience, 1976-2008. Thyroid 21:765-72, 2011.
- Jakobiec FA, Stacy RC, Hatton P. Clinical characterization and immunopathologic features of sclerosing dacryoadenitis and Riedel thyroiditis. Arch Ophthalmol 128:1626-1628, 2010.
- Li Y, Nishihara E, Hirokawa M, Taniguchi E, Miyauchi A, Kakudo K. Distinct clinical, serological, and sonographic characteristics of Hashimoto's thyroiditis based with and without IgG4-positive plasma cells. J Clin Endocrinol Metab. 95:1309-17, 2010.
- Kakudo K, Li Y, Hirokawa M, Ozaki T. Diagnosis of Hashimoto's thyroiditis and IgG4-related sclerosing disease. Pathol Int 61:175-83, 2011.
397a. Jokisch F1, Kleinlein I, Haller B, Seehaus T, Fuerst H, Kremer M. A small subgroup of Hashimoto's thyroiditis is associated with IgG4-related disease. Virchows Arch. Dec 15. [Epub ahead of print], 2015
- Outschoorn IM, Talor MV, Burek CL, Hoffman WH, Rose NR. Heritability analysis of IgG4 antibodies in autoimmune thyroid disease. Autoimmunity 47:320-6, 2014.
- Pratt DE, Kaberlein G, Dudkiewicz A, Karande V, Gleicher N. The association of antithyroid antibodies in euthyroid nonpregnant women with recurrent first trimester abortions in the next pregnancy. Fertil Steril, 60:1001-1005, 1993.
- Poppe K, Glinoer D, Tournaye H, Devroey P, van Steirteghem A, Kaufman L, Velkeniers B. Assisted reproduction and thyroid autoimmunity: an unfortunate combination? J Clin Endocrinol Metab 88:4149-4152, 2003.
- Giani C, Fierabracci P, Bonacci R, Gigliotti A, Campani D, De Negri F, Cecchetti D, Martino E, Pinchera A. Relationship between breast cancer and thyroid disease: Relevance of autoimmune thyroid disorders in breast malignancy. J Clin Endocrinol Metab 81:990-994, 1996.
- Smyth PPA, Shering SG, Kilbane MT, Murray MJ, McDermott EWM, Smith DF, O’Higgins NJ. Serum thyroid peroxidase autoantibodies, thyroid volume, and outcome in breast carcinoma. J Clin Endocrinol Metab 83:2711-2716, 1998.
- Pop VJ, Maartens LH, Leusink G, Van Son MJ, Knottnerus AA, Ward AM, Metcalfe R, Weetman AP. Are autoimmune thyroid dysfunction and depression related? J Clin Endocrinol Metab 83:3194-3197, 1998.
- Wang GC, Talor MV, Rose NR, Cappola AR, Chiou RB, Weiss C, Walston JD, Fried LP, Caturegli P. Thyroid autoantibodies are associated with a reduced prevalence of frailty in community-dwelling older women. J Clin Endocrinol Metab. 95:1161-8, 2010.
.