ABSTRACT
While most patients with diabetes have Type 1 diabetes (T1D) or Type 2 diabetes (T2D) there are other etiologies of diabetes that occur less frequently. In this chapter we will discuss a number of these less common causes of diabetes. It is clinically very important to recognize these uncommon causes of diabetes as treatment directed towards the underlying etiology can at times result in the remission of diabetes (for example Cushing’s Syndrome) or be required to avoid other complications of the underlying disorder (for example hemochromatosis, which in addition to causing diabetes can lead to severe liver disease and congestive heart failure). In this chapter the following disorders that are associated with diabetes are discussed: 1) genetic disorders of insulin action (Type A insulin resistance, Donohue Syndrome/Leprechaunism, Rabson-Mendenhall syndrome); 2) maternally inherited diabetes mellitus and deafness syndrome; 3) disorders of the exocrine pancreas (pancreatitis, trauma/pancreatectomy, neoplasia, cystic fibrosis, hemochromatosis); 4) endocrinopathies (acromegaly, Cushing’s syndrome, glucagonoma, pheochromocytoma, hyperthyroidism, somatostatinoma, primary hyperaldosteronism); 5) drug induced; 6) infections; 7) immune mediated (stiff-man syndrome, anti-insulin receptor antibodies); 8) ketosis prone diabetes (Flatbush diabetes); and 9) genetic disorders sometimes associated with diabetes (Down syndrome, Klinefelter syndrome, Turner syndrome, Wilsons syndrome, Wolfram syndrome, Friedreich ataxia, Bardet-Biedl syndrome [Laurence-Moon-Biedl syndrome], myotonic dystrophy, Prader-Willi syndrome, Alström syndrome, and Werner syndrome). Gestational diabetes, monogenic diabetes (maturity onset diabetes of the young (MODY) and neonatal diabetes), lipodystrophy, fibrocalculous pancreatic disease, diabetes associated with HIV infection, diabetes due to the autoimmune polyglandular syndromes, and post-transplant diabetes are not discussed in this chapter as they are discussed in other Endotext chapters.
INTRODUCTION
While most patients with diabetes have Type 1 diabetes (T1D) or Type 2 diabetes (T2D) there are other etiologies of diabetes that occur less frequently. In this chapter we will discuss a number of these less common causes of diabetes (see table 1). Note that gestational diabetes, monogenic diabetes (maturity onset diabetes of the young (MODY) and neonatal diabetes), lipodystrophy, fibrocalculous pancreatic disease, malnutrition related diabetes (being written), diabetes associated with HIV infection, diabetes due to the autoimmune polyglandular syndromes, and post-transplant diabetes are discussed in separate Endotext chapters (1-7). It is clinically very important to recognize these uncommon causes of diabetes as treatment directed towards the underlying etiology can at times result in the remission of diabetes (for example Cushing’s Syndrome) or be required to avoid other complications of the underlying disorder (for example hemochromatosis, which in addition to causing diabetes can lead to severe liver disease and congestive heart failure). Additionally, recognizing the type of diabetes can allow for the appropriate treatment. For example, recognizing ketosis prone diabetes facilitates discontinuing insulin therapy.
|
Table 1. Non-Type 1 Non-T2D Classification
|
|
Genetic defects of beta-cell development and function
MODY (common causes- GCK, HNF1A, HNF4A, HNF1B)
Neonatal Diabetes (common causes- KCNJ11, ABCC8, INS, 6q24)
1. Mitochondrial DNA
|
|
Genetic defects in insulin action
1. Type A insulin resistance
2. Donohue Syndrome (Leprechaunism)
3. Rabson-Mendenhall syndrome
4. Lipoatrophic diabetes
|
|
Diseases of the exocrine pancreas
1. Pancreatitis
2. Fibrocalculous pancreatic disease
3. Trauma/pancreatectomy
4. Neoplasia
5. Cystic fibrosis
6. Hemochromatosis (iron overload)
Thalassemia (iron overload)
|
|
Endocrinopathies
1. Acromegaly
2. Cushing’s syndrome
3. Glucagonoma
4. Pheochromocytoma
5. Hyperthyroidism
6. Somatostatinoma
7. Primary hyperaldosteronism
|
|
Drug- or chemical-induced hyperglycemia
1. Vacor
2. Pentamidine
3. Nicotinic acid
4. Glucocorticoids
5. Diazoxide
6. Check point inhibitors
7. Phenytoin (Dilantin)
8. Interferon alpha
9. Immune suppressants
10. Others (statins, psychotropic drugs, b-Adrenergic agonists, thiazides, fluoroquinolones, beta-adrenergic drugs, teprotumumab, etc.)
|
|
Infections
1. Congenital rubella
2. Hepatitis C virus
3. HIV
COVID-19
|
|
Immune-mediated diabetes
1. Stiff-man syndrome
2. Anti-insulin receptor antibodies
3. Autoimmune polyglandular syndromes
|
|
Diabetes of unknown cause
1. Ketosis-prone diabetes (Flatbush diabetes)
|
|
Other genetic syndromes sometimes associated with diabetes
1. Down syndrome
2. Klinefelter syndrome
3. Turner syndrome
4. Wilsons syndrome
5. Wolfram syndrome
6. Friedreich ataxia
7. Bardet-Biedl syndrome (Laurence-Moon-Biedl syndrome)
8. Myotonic dystrophy
9. Prader-Willi syndrome
10. Alström syndrome
|
MATERNALLY INHERITED DIABETES MELLITUS AND DEAFNESS (MIDD)
Maternally inherited diabetes mellitus and deafness (MIDD) is a mitochondrial disorder characterized by diabetes and progressive sensorineural hearing loss (8-10). Mitochondrial DNA is only transmitted from the mother as the sperm lacks mitochondrial DNA (8). Therefore, over 50% of affected individuals with MIDD have a mother with diabetes. A mother with this disorder transmits the mutation to almost all of her offspring (11). However, the proportion of somatic cells with the mutation can vary considerably, a condition called heteroplasmy (9). The higher the number of somatic cells with a mutation the greater is the penetrance of symptoms and disease severity. Additionally, the proportion of somatic cells with a mutation can vary from tissue to tissue and may explain the variability in the manifestations of this disorder (9). The prevalence of mitochondrial diabetes in the diabetes population depends on ethnic background and ranges between 0.2% and 2%, with the highest prevalence in Japan (11).
MIDD is associated with a point mutation in a transfer ribonucleic acid (tRNA) gene at position 3243 with an A to G transition (8-10). In addition to diabetes and auditory impairment, the m.3243A>G mutation can cause other clinical manifestations including central neurological and psychiatric disorders, eye disease, myopathy, cardiac disorders, renal disease, endocrine disease, and gastrointestinal disease (8,9). Other point mutations in mitochondrial DNA can also result in diabetes and deafness but these mutations are rare in comparison to m.3243A>G (8,9,11).
It is thought that defects in mitochondrial function result in the decreased production of ATP following glucose uptake by beta cells resulting in decreased insulin secretion in response to elevated glucose levels (8,9). Additionally, mitochondrial dysfunction in the highly metabolically active pancreatic islets ultimately results in the loss of B‐cell mass further compromising insulin secretion (9). Insulin sensitivity is usually normal (11). Other tissues that are metabolically active may also be adversely effected by the inability of the mitochondria to produce ATP including the cells in the cochlea (9).
The clinical syndrome of MIDD can phenotypically resemble either T1D or T2D (9,11). The age of onset varies between childhood and mid-adulthood. Approximately 20% of patients present acutely with high glucose levels and even ketoacidosis (9). Most patients do not have islet cell antibodies but they are present in a small number of patients (9). This could be due to concomitant T1D or to the development of antibodies secondary to beta cell destruction due to mitochondria dysfunction. Patients tend to be thin rather than obese (9). This disorder can be distinguished from MODY by the presence of multi-organ involvement, particularly sensorineural hearing loss, and maternal rather than autosomal dominant transmission. Initially patients may be treated with diet and/or oral agents but overtime most patients with MIDD progress to requiring insulin therapy (8,9,11).
As the name implies, this disorder is recognized by the presence of diabetes and deafness and a family history of these conditions in maternal relatives (9,11). Hearing loss is present in approximately 75% of patients and typically precedes the development of diabetes (9). Hearing loss is more common and severe in males (9). Approximately 10-15% of patients, in addition to having diabetes and deafness, also have the syndrome of mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (9). The m.3243A>G mutation can cause a wide spectrum of abnormalities that include neurological abnormalities (strokes, dementia, seizures), psychiatric disorders including recurrent major depression, schizophrenia and a variety of phobias, macular retinal dystrophy with pigmentation, proximal myopathy, cardiomyopathy, renal failure, short stature, endocrine dysfunction, and gastrointestinal complaints (9). The finding of classical retinal dystrophy and hyperpigmentation on routine eye exam should suggest the diagnosis of maternally inherited diabetes mellitus and deafness. Once suspected the diagnosis of MIDD should be confirmed by genetic testing for the mitochondrial DNA point mutation at position 3243 (A>G). This is usually initially carried out on blood cells but if negative, urinary cells or skeletal muscle can be tested and if necessary one can test for other mutations that cause similar phenotypes (12). Once a diagnosis is confirmed first-degree family members at risk should be screened for the mutation and provided with genetic counseling. For those carrying the mutation without clinical manifestations, screening for diabetes and monitoring of kidney function, hearing, and cardiac function should be carried out.
GENETIC DEFECTS IN INSULIN ACTION
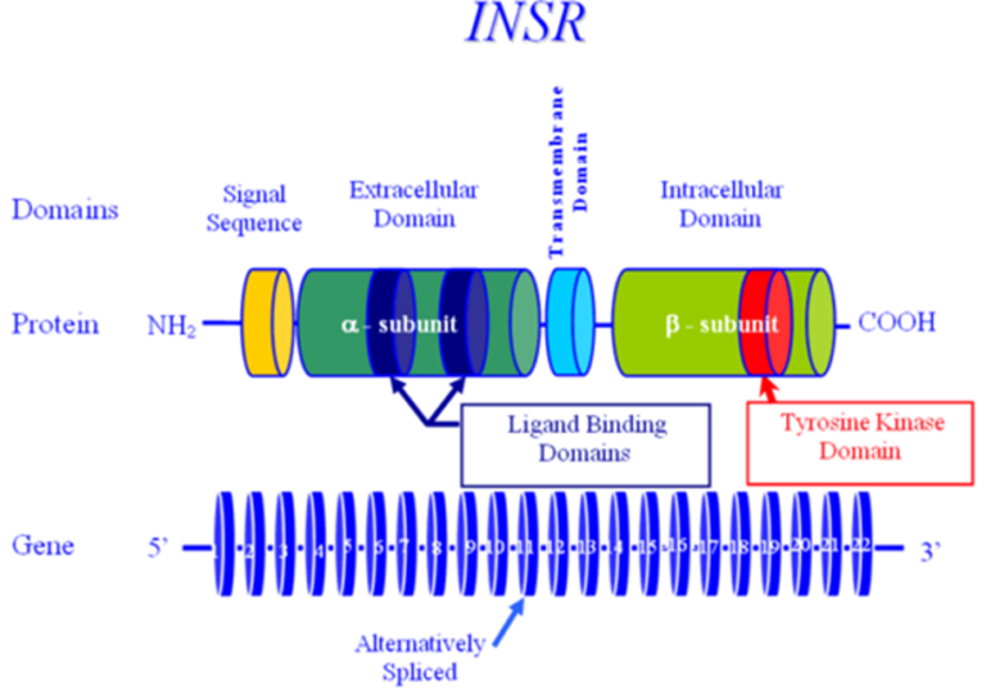
Overview of Insulin Receptor Defects
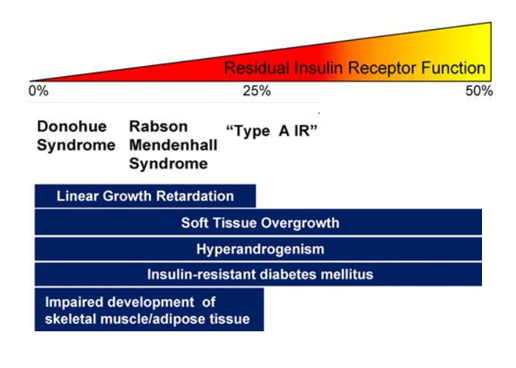
Mutations in the insulin receptor can cause different degrees of insulin resistance but do not need to be associated with diabetes per se (13). A large number of different mutations have been described and they can be classified as mutations that prevent synthesis of the receptor, inhibit transport of the receptor to the plasma membrane, decrease insulin binding to the receptor, impair transmembrane signaling, or increase receptor degradation (14). Pancreatic beta cell hyperplasia and hyperinsulinemia can compensate for the insulin resistance preventing hyperglycemia. Fasting hypoglycemia and postprandial hyperglycemia may be observed. Over time the beta cells’ ability to secrete insulin diminishes and frank diabetes usually develops. Treatment of the diabetes may require very high doses of insulin (15). Unfortunately, insulin sensitizers have not been very effective in patients with insulin receptor defects. In contrast to the typical patients with insulin resistance, obesity, dyslipidemia, hypertension, and fatty liver are not usually present (15,16). Acanthosis nigricans, pigmentation in the neck or axillae, is a visible sign of severe insulin resistance (13,15). In females, severe insulin resistance is usually associated with hyperandrogenism, oligomenorrhea or amenorrhea, anovulation, hirsutism, acne, and masculinization (13,15). It is hypothesized that ovarian dysfunction and acanthosis nigricans are due to high levels of insulin acting via the IGF1 receptors (16). The amount of residual insulin receptor function determines the specific syndrome in patients with insulin receptor mutations (Figure 1).
Type A Insulin Resistance
This autosomal dominant disorder includes patients with severe insulin resistance and acanthosis nigricans (13,15). Patients have normal growth and females show ovarian hyperandrogenism that typically presents in the peripubertal period (15). In females, hyperglycemia develops after ovarian hyperandrogenism and acanthosis nigricans. Males display only acanthosis nigricans and they often remain undiagnosed even after the development of symptomatic diabetes, which may not occur until the patients are adults. These patients have mutations in the insulin receptor gene that decreases the activity of the insulin receptor (14,15). In addition, mutations in transcription factors that stimulate the expression of insulin receptors can lead to a similar phenotype as mutations in the insulin receptor (13,16). Inherited defects in pathways downstream of the insulin receptor can also lead to clinical abnormalities similar to mutations in the insulin receptor (13,16).
Donohue Syndrome (Leprechaunism)
Donohue syndrome is a rare congenital (1:1,000,000), autosomal recessive syndrome characterized by very severe insulin resistance due to mutations in the insulin receptor gene, dysmorphic features such as protuberant and low-set ears, flaring nostrils and thick lips, growth retardation, failure to thrive, and early death (14). The name leprechaunism relates to the elfin features of those affected. Clinical features include in addition to acanthosis nigricans, hypertrichosis, hirsutism, dysmorphic facies, breast enlargement, abdominal distension, and lipoatrophy. Patients have extremely high levels of insulin and can develop impaired glucose tolerance or overt diabetes. The prognosis for infants with this condition is very poor and most will die in the first year of life. When parents, who are heterozygous for mutations in the insulin receptor are studied, many of these individuals are insulin resistant (14).
Rabson-Mendenhall Syndrome
The Rabson-Mendenhall syndrome represents another disorder of extreme insulin resistance (15). This autosomal recessive syndrome is associated with mutations in the insulin receptor gene (13). Initially fasting hypoglycemia, postprandial hyperglycemia, and marked hyperinsulinemia may be observed (13). When beta-cells decompensate, hyperglycemia may become very difficult to treat. Clinical features include in addition to acanthosis nigricans, phallic enlargement, precocious pseudopuberty, short stature, and abnormal teeth, hair, and nails (14,15). Hyperplasia of the pineal gland is an unusual feature (14). Prognosis is poor as diabetes is difficult to control even with high insulin doses (14). Hyperglycemia leads to microvascular disease and/or diabetic ketoacidosis resulting in death in the second and third decades of life (13). Leptin administration has resulted in an improvement in this syndrome (17,18).
DISEASES OF THE EXOCRINE PANCREAS
Diseases that destroy the pancreas can cause diabetes even in individuals who do not have risk factors for diabetes (19). In the medical literature this is often referred to as Type 3C diabetes. Acquired causes of damage to the pancreas include pancreatitis, trauma, infection, pancreatic carcinoma, and pancreatectomy. Inherited disorders that affect the endocrine pancreas, such as hemochromatosis, thalassemia, and cystic fibrosis, can also cause insulin deficiency and diabetes. The distribution of causes for diabetes secondary to pancreatic disorders in one study was chronic pancreatitis (79%), pancreatic ductal adenocarcinoma (8%), hemochromatosis (7%), cystic fibrosis (4%), and previous pancreatic surgery (2%) (20). The prevalence of diabetes secondary to pancreatic disease is estimated to range from 1% to 9% and likely will depend on the patient population studied (21). In a population study carried out in New Zealand the prevalence of diabetes secondary to pancreatic disorders was close to that of T1D (22).
Pancreatitis
Pancreatitis may lead to the destruction of the beta cells due to inflammation and irreversible fibrotic damage (23). In addition to destroying the beta cells, pancreatitis also leads to the destruction of glucagon secreting alpha-cells and pancreatic polypeptide secreting cells (23). The decrease in insulin secretion is the primary mechanism leading to hyperglycemia. In addition, the decrease in secretion of pancreatic polypeptide leads to a decrease in hepatic insulin sensitivity resulting in increased hepatic glucose production (21,23,24). Nutrient malabsorption that occurs secondary to pancreatitis leads to impaired incretin secretion that can result in diminished insulin release by the remaining beta-cells (25). Acute pancreatitis can induce transient hyperglycemia (stress hyperglycemia) that can last for several weeks or permanent hyperglycemia (21,26,27). The risk of developing diabetes after acute pancreatitis is increased after severe pancreatitis, hypertriglyceridemia or alcohol as the etiology of pancreatitis, and the occurrence of pancreatic necrosis (28). Other predictors of the development of diabetes include obesity, a family history of diabetes, exocrine pancreatic insufficiency, history of pancreatic surgery, pancreatic calcifications, and long duration of pancreatitis (29).
The prevalence of diabetes secondary to pancreatitis varies greatly with studies in North America estimating a prevalence of 0.5%-1.15% whereas in Southeast Asia, where tropical or fibrocalcific pancreatitis is endemic, a prevalence of approximately 15%-20% has been reported (23) (see chapter in Tropical Endocrinology Section of Endotext entitled “Fibrocalculous Pancreatic Diabetes” for an in depth discussion of this entity (7). Recently, data from the UK Royal College of General Practitioners Research and Surveillance Centre found 559 cases of diabetes following pancreatic disease in 31,789 cases of adults newly diagnosed with diabetes (1.8%) (30). Most cases of diabetes following pancreatic disease were classified as T2D (30). In another study approximately 50% of the patients with diabetes secondary to pancreatitis were not recognized and were incorrectly thought to have T2D. It is very likely that many cases of diabetes secondary to pancreatitis are not recognized to be due to pancreatic disease.
The prevalence of diabetes in patients with diagnosed pancreatitis has ranged between 26-80%, depending on the cohort and duration of follow up (21,23,31). The prevalence of diabetes increases with the duration of pancreatitis and early onset of calcific disease (23). Because of the high risk of diabetes in patients with pancreatitis these patients should be periodically screened for the presence of diabetes with measurement of fasting glucose and/or A1c levels.
At times it can be difficult to distinguish diabetes secondary to pancreatitis from T1D or T2D. The following diagnostic criteria have been proposed (Table 2) (23).
|
Table 2. Proposed Diagnostic Criteria for Diabetes Secondary to Pancreatitis
|
|
Major Criteria (must be present)
|
|
Presence of exocrine pancreatic insufficiency (monoclonal fecal elastase-1 test or direct function tests)
Pathological pancreatic imaging (endoscopic ultrasound, MRI, CT)
Absence of T1D associated autoimmune markers
|
|
Minor Criteria
|
|
Absent pancreatic polypeptide secretion
Impaired incretin secretion (e.g., GLP-1)
No excessive insulin resistance (e.g., HOMA-IR)
Impaired beta cell function (e.g., HOMA-B, C-Peptide/glucose-ratio)
Low serum levels of lipid soluble vitamins (A, D, E and K)
|
It should be recognized that these proposed criteria have not been rigorously tested nor are all criteria available in routine clinical practice. In addition, there are a number of key considerations. First, long-standing T1D and T2D are associated with exocrine pancreatic failure (32). It has been estimated that 26% to 74% of patients with T1D and 28% to 36% of patients with T2D have evidence of exocrine pancreatic insufficiency (19). Second, patients with diabetes are at a higher risk for developing acute and/or chronic pancreatitis (33). Lastly, patients with previous episodes of pancreatitis may also develop T1D or T2D independently of their exocrine pancreatic disease. When diabetes occurs in patients with a pre-existing diagnosis of chronic pancreatitis it is likely that pancreatitis is an important contributor to the development of diabetes.
Testing for T1D associated autoimmune markers can be helpful in separating T1D from diabetes secondary to pancreatic disease. The presence of islet cell antibodies supports the diagnosis of T1D. The pancreatic polypeptide response to insulin-induced hypoglycemia, secretin-infusion, or a mixed nutrient ingestion can be helpful in separating T2D from diabetes secondary to pancreatic disease. Patients with diabetes secondary to pancreatitis have an absent or reduced pancreatic polypeptide response while patients with T2D have an elevated pancreatic polypeptide response (23,31). Studies have shown that pancreatic polypeptide regulates hepatic insulin sensitivity and the absence of pancreatic polypeptide leads to hepatic insulin resistance and enhances hepatic glucose production, which could contribute to the abnormal glucose metabolism that occurs with pancreatic disease (21).
In patients with diabetes secondary to pancreatitis hyperglycemia can be mild to very severe depending upon the degree of pancreatic destruction leading to impaired insulin production and secretion (19,21,23). Glycemic control may be unstable due to the loss of glucagon secretion in response to hypoglycemia, carbohydrate malabsorption, and inconsistent food intake due to pain and/or nausea secondary to pancreatitis (i.e., “brittle diabetes”) (19,21,23). Whether glycemic control is worse in patients with diabetes secondary to pancreatitis is uncertain as older studies reported worse glycemic control and more recent studies have reported that glycemic control was similar to other patients with diabetes (19). The ability to obtain good glycemic control is likely to be related to the degree of pancreatic insufficiency with patients with a total absence of pancreatic function being more difficult to control.
In patients with relatively mild diabetes treatment with metformin is indicated. A nationwide cohort study in New Zealand and Denmark reported that metformin increased survival in patients with post pancreatitis diabetes (34,35). The GI side effects (nausea, abdominal complaints, diarrhea) of metformin may not be tolerable in some patients with pancreatitis. In observational studies metformin therapy has been associated with a reduction in the development of pancreatic cancer in patients with diabetes (36). Given the increased risk of pancreatic cancer in patients with diabetes and/or pancreatitis, a reduction in the development of pancreatic cancer would be a potential added benefit of metformin therapy (37-39). There are conflicting data on whether treatment with DPP4-inhibitors or GLP1-analogues can cause pancreatitis, but until this issue has been unequivocally settled, it is wise to refrain from using these drugs in patients who have had pancreatitis without a clear reversible etiology (for example, gallstone pancreatitis status post cholecystectomy). Note that two meta-analyses have demonstrated an 80% increased risk of acute pancreatitis in patients using DPP-4 inhibitors compared with those receiving standard care (40,41). In contrast, meta-analyses of large cardiovascular outcome studies have not demonstrated an increase in pancreatitis in patients treated with GLP-1 receptor agonists, but these studies typically excluded patients with a history of pancreatitis (42,43). Thiazolidinediones should probably be avoided as patients with pancreatitis and malabsorption are at increased risk for osteoporosis and thiazolidinediones may potentiate this problem.
Chronic pancreatitis is a progressive disease and therefore it is likely that glycemic control will worsen overtime and most patients will eventually require insulin therapy (23). Many patients will have severe insulin deficiency and will need to be treated with insulin therapy using regimens employed in patients with T1D. Because of the absence of glucagon secretion patients with diabetes secondary to pancreatitis are more susceptible to severe hypoglycemia with insulin therapy but diabetic ketoacidosis is not commonly observed due to the absence of glucagon.
Patients with diabetes secondary to pancreatitis are at risk for microvascular complications and lower extremity arterial disease and therefore routine testing for eye disease, kidney disease, foot ulcers, and neuropathy should be instituted (44-47).
Finally, it should be recognized that patients with diabetes secondary to pancreatitis will almost always have exocrine pancreatic insufficiency (31). Many patients with chronic pancreatitis manifest fat malabsorption without symptoms and therefore a thorough evaluation is required. Oral pancreatic enzyme replacement is beneficial for these patients. Of note, pancreatic enzyme supplementation can improve incretin secretion and thereby may benefit glycemic control (21,25,48). Fat soluble vitamin deficiency commonly occurs (Vitamin A, D, and K) and many patients require supplementation with fat-soluble vitamins.
Pancreatectomy
The metabolic abnormalities that occur after pancreatic surgery depend on the amount and area of the pancreas removed and whether the remaining pancreas is normal or diseased (24). The basis for this variability is due to the distribution of β and non-β islet cell types in the pancreas. Islet density is relatively low in the head of the pancreas and gradually increases through the body toward the tail region by greater than 2-fold and thus α- and β-cells predominate in the tail. In contrast, the cells that secrete pancreatic polypeptide are mainly localized in the head of the pancreas. Distal pancreatectomy usually causes little change in the metabolic status unless more than 50% of parenchyma is excised in patients with diffuse disease or more than 80% in patients with normal pancreatic function (24). The risk of a patient developing diabetes after a distal pancreatectomy varies greatly (24,49). The risk of new diabetes is reduced with central pancreatectomy compared to distal pancreatectomy (50). Resection of the head of the pancreas results in a decrease in pancreatic polypeptide, hepatic insulin resistance, and fasting hyperglycemia. Approximately 20% of patients develop diabetes after resection of the head of the pancreas (24). It should also be recognized that removal of pancreatic tissue can accelerate the development of T2D by decreasing insulin secretion in patients with impaired glucose metabolism (51).
Patients who have undergone total surgical pancreatectomy have a deficiency of insulin, glucagon, and pancreatic polypeptide and require insulin treatment. In general, there are several differences from typical T1D, including exocrine deficiency, low insulin requirements, and a higher risk of hypoglycemia due to the decrease in glucagon, which stimulates hepatic glucose production (glycogenolysis and gluconeogenesis). Pancreatectomized patients are prone to hypoglycemia and a delayed recovery from hypoglycemia. In an evaluation of 180 patients post total pancreatectomy 42% experienced one or more hypoglycemic events on a monthly basis (52). In addition to treatment with insulin, pancreatic enzyme supplements are always needed. SGLT2 inhibitors and GLP-1 receptor agonists have been shown to improve glycemic control in patients with diabetes post pancreatectomy (53,54). GLP-1 receptor agonists improve postprandial glycemia by decreasing gastric emptying and reducing postprandial responses of gut-derived glucagon (54). Intraportal islet auto transplantation has been used to prevent the development of diabetes with total pancreatectomy and/or reduce the risk of developing difficult to control diabetes (55-57).
Pancreatic Cancer
A high percentage of patients with pancreatic carcinoma have diabetes (21,58). In one study 68% of patients with pancreatic cancer also had diabetes (59). The prevalence of diabetes in patients with pancreatic cancer is much higher than in other common malignancies (58,59). In patients with pancreatic cancer who also have diabetes, the diagnosis of diabetes occurred less than 2 years prior to the diagnosis of pancreatic cancer in 74% of patients (60). In a population-based study less than 1% of patients over the age of 50 with newly diagnosed diabetes were diagnosed with pancreatic cancer within 3 years (61). In a study of 115 patients over 50 years of age who were hospitalized for new-onset diabetes 5.6% were found to have a pancreatic cancer (62). Many patients with pancreatic cancer lose weight and therefore deteriorating glycemic control in conjunction with weight loss and anorexia should raise the possibility of an occult pancreatic cancer (58,63). Other clues to the presence of pancreatic cancer in a patient with new onset diabetes are the lack of a family history of diabetes, a BMI < 25, recent thromboembolism, history of pancreatitis, GI symptoms, and the absence of features of the metabolic syndrome such as dyslipidemia and hypertension. Given the high incidence of diabetes relative to the incidence of pancreatic cancer the routine screening of all patients who develop diabetes is not cost effective. However, in selected patients with the features described above screening is appropriate.
Conversely, long standing T2D increases the risk of developing pancreatic cancer by approximately 1.5 to 2-fold indicating a bidirectional relationship (24,58,64). This risk may persist even after adjustment for obesity and smoking, risk factors for pancreatic cancer. Diabetes is both a risk factor for the development of pancreatic cancer and a complication of pancreatic cancer.
As discussed above diabetes may develop secondary to chronic pancreatitis. Chronic pancreatitis increases the risk of pancreatic cancer. Thus, patients with diabetes secondary to chronic pancreatitis are at a higher risk of developing pancreatic cancer (65).
The strongest evidence linking pancreatic cancer with incident diabetes is the beneficial effects of cancer resection on glycemic control (58). In a small study in 7 patients, Permert and colleagues reported an improvement in diabetes status and glucose metabolism after subtotal pancreatectomy (66). Similarly, Pannala and colleagues in a larger study reported that after pancreaticoduodenectomy, diabetes resolved in 17 of 30 patients (57%) with new-onset diabetes but was unaffected in patients with longstanding diabetes (60). Litwin and colleagues noted similar improvements in glucose metabolism after surgery in patients with pancreatic cancer but a deterioration in patients with chronic pancreatitis (67). Finally, studies have also shown that a good response to chemotherapy in patients with pancreatic cancer can also improve glucose levels (68). Taken together these results demonstrate a benefit from tumor removal and suggest that new-onset diabetes associated with pancreatic cancer may be a paraneoplastic phenomenon.
The mechanism accounting for the development of new onset diabetes by pancreatic cancers is unknown (58). In contrast to other pancreatic disorders the etiology of diabetes is not due to destruction of the pancreas as patients with new onset diabetes and pancreatic cancer have hyperinsulinemia rather than low insulin levels and as noted above the diabetes improves after resection (21). Additionally, pancreatic cancers may be very small and thus unlikely to cause pancreatic insufficiency. Pancreatic cancer is associated with insulin resistance but the factors leading to insulin resistance are unknown (21).
In patients with pancreatic cancer the main goal of the treatment of diabetes is to prevent the short- term metabolic complications and facilitate the ability of the patient to tolerate treatment of the pancreatic cancer (surgery and chemotherapy). Given the poor survival of patients with pancreatic cancer, prevention of the long-term sequelae of diabetes is not a major focus. Metformin is a preferred hypoglycemic agent because there are observational studies suggesting that metformin may improve survival in patients with pancreatic cancer (69-71). However, randomized trials have failed to demonstrate the benefit of metformin therapy in patients with pancreatic cancer (72,73).
Hemochromatosis
Hemochromatosis is an autosomal recessive disorder characterized by increased iron absorption by the GI tract and increased total body iron stores (74). The excess iron is sequestered in many different tissues including the liver, skin, heart, and pancreas. The classic triad of hemochromatosis is diabetes mellitus, hepatomegaly, and increased skin pigmentation (“bronze diabetes”), but clinical features also include gonadal failure, arthropathy, and cardiomyopathy (74).
In early studies diabetes was present in over 50% of patients with hemochromatosis (75,76). More recently the prevalence of diabetes in patients with hemochromatosis has decreased to approximately 20% of patients, presumably due to the early diagnosis and treatment of hemochromatosis due to genetic testing (75-78). In patients with hemochromatosis screening for the presence of diabetes should be periodically carried out.
Diabetes was typically observed in persons who also had severe iron overload and cirrhosis (76). It should be noted that iron overload from any cause can result in diabetes (79). For example, patients with thalassemia develop iron overload due to the need for frequent transfusions (80,81). The prevalence of diabetes in patients with thalassemia has been declining since the more aggressive and widespread use of iron chelation therapy (81).
There are two abnormalities that lead to abnormal glucose metabolism in patients with hemochromatosis and iron overload (75). First, iron overload leads to beta cell damage with decreased insulin production and secretion. Pathologic examination revealed hemosiderin deposition and iron-induced fibrosis of the islets (76). The decrease in insulin secretion is the primary defect leading to the development of diabetes (75,76,78). Of note glucagon secretion does not appear to be deficient in patients with diabetes and hemochromatosis suggesting that the iron overload has a preferential toxicity for beta cells compared to alpha cells (76,82,83). Similarly, basal and stimulated pancreatic polypeptide levels are also not decreased in diabetic patients with hemochromatosis (84). Thus, the hormonal abnormalities differ in patients with iron overload induced diabetes compared to patients with pancreatitis induced diabetes. The second abnormality is insulin resistance that occurs due to iron overload hepatic damage and/or secondary to obesity (75,76). In addition, a genetic predisposition to diabetes potentiates the development of metabolic dysfunction. Many patients with hemochromatosis and diabetes have a relative with diabetes (85).
The typical micro and macrovascular complications of diabetes occur in patients with hemochromatosis (76,85). In a study by Griffiths and colleagues, 11 of 49 patients with hemochromatosis and diabetes had diabetic retinopathy (86). Sixty percent of the patients with hemochromatosis who had diabetes for greater than 10 years had retinopathy. The incidence of retinopathy is similar to that observed in the general diabetes population (85,86). Similarly, Becker and Miller observed that 7 of 22 patients with diabetes and hemochromatosis had pathologic evidence of diabetic glomerulopathy (87).
The treatment of hemochromatosis by phlebotomy has a variable impact on glucose metabolism (75). In patients who do not yet have complications or organ damage an improvement of insulin secretory capacity and normalization of glucose tolerance has been observed (75,76). Glucose metabolism often improves in patients with impaired glucose tolerance (75,88). In patients with diabetes improvement in glucose metabolism by phlebotomy may occur but is not as common as in “pre-diabetics” (75,88,89). In one study 28% of patients with diabetes and hemochromatosis on insulin or oral agents showed improved glucose control following phlebotomy therapy (90).
Cystic Fibrosis
Cystic Fibrosis is an autosomal recessive disorder due to a defect in the chloride transport channel (91). Cystic fibrosis related diabetes is rare in children but is present in approximately 20% of adolescents and 40-50% of adults with cystic fibrosis (92,93). As patients with cystic fibrosis live longer it is likely that the number of patients with cystic fibrosis and diabetes will increase. The development of diabetes is associated with more severe cystic fibrosis gene mutations, increasing age, worse pulmonary function, undernutrition, liver dysfunction, pancreatic insufficiency, a family history of diabetes, female gender, and corticosteroid use (92,93).
The primary defect in patients with cystic fibrosis related diabetes is decreased insulin production and secretion due to fibrosis and atrophy of the pancreas with a reduction of islet mass (92). In addition, mutations in the cystic fibrosis transmembrane conductance regulator gene may have direct effects on the ability of beta cells to secrete insulin (93,94). Beta cell dysfunction is not complete with residual insulin secretion and thus patients with cystic fibrosis related diabetes do not typically develop ketosis (92). Reduced alpha cell mass also occurs so while fasting glucagon levels are normal, glucagon secretion in response to hypoglycemia is impaired (92). Peripheral and hepatic insulin resistance may also occur secondary to infections, inflammation, and cirrhosis (93).
Some of the clinical features of cystic fibrosis related diabetes are similar to T1D as patients are young, not obese, insulin deficiency is the primary defect, and features of the metabolic syndrome (hyperlipidemia, hypertension, visceral adiposity) are not usually present (92). However, cystic fibrosis related diabetes is not an autoimmune disease (islet cell antibodies are not present) and ketosis is rare because endogenous insulin is still produced (92). Fasting glucose levels are often normal initially with elevated postprandial glucose levels due to a reduced and delayed insulin response to carbohydrates while basal insulin is often adequate to maintain normal fasting glucose levels (95). Patients with cystic fibrosis related diabetes are not at high risk of developing atherosclerosis and heart disease is not a major issue (92-94). This is likely due to malabsorption leading to low life-long plasma cholesterol levels and the shortened length of life (92,93). As life expectancy increases and medical therapy with Cystic Fibrosis Transmembrane Regulator (CFTR) modulators improves, the risk of macrovascular disease may increase. Microvascular complications do occur in cystic fibrosis related diabetes and are related to the duration of diabetes and glycemic control (92,93,95). The American Diabetes Association recommends screening for complications of diabetes beginning 5 years after the diagnosis of cystic fibrosis related diabetes (96)
Lung disease is a major cause of morbidity and mortality in patients with cystic fibrosis and both insulin insufficiency and hyperglycemia negatively affect cystic fibrosis lung disease (97). Numerous studies have shown that the occurrence of diabetes in patients with cystic fibrosis is associated with more severe lung disease and increased mortality and this adverse effect disproportionately affects women (92,95,97). In patients with cystic fibrosis lung function is critically dependent on maintaining normal weight and lean body mass. Insulin deficiency leads to a catabolic state with the loss of protein and fat (92). Multiple studies have shown that insulin replacement therapy improves nutritional status and pulmonary function in patients with cystic fibrosis related diabetes (92). In addition, elevated blood glucose levels result in elevated blood glucose levels in the airways, which promotes the growth of pathogenic microorganisms and increases pulmonary infections (92,95). Of note recent studies have shown that the marked increase in mortality in patients with cystic fibrosis related diabetes compared to patients with cystic fibrosis only has decreased (98). It is likely that early diagnosis and aggressive treatment have improved survival in patients with cystic fibrosis related diabetes.
Because of the adverse effects of diabetes on lung function in patients with cystic fibrosis routine screening for diabetes is recommended (97). It is recommended that annual screening begin at age 10 (97). While fasting glucose and A1c levels are routine screening tests for diabetes, in patients with cystic fibrosis these tests are not sensitive enough (97). Fasting glucose and A1c testing will fail to diagnose approximately 50% of patients with cystic fibrosis related diabetes (94,97). However, recent studies have suggested that a screening A1c >5.5% would detect more than 90% of patients with diabetes and therefore with further confirming studies measuring A1c levels could become an initial screening approach (96). As noted above, abnormalities in postprandial glucose characterizes cystic fibrosis related diabetes and it is therefore recommended that an oral glucose tolerance test (OGTT) be utilized for the diagnosis of diabetes in patients with cystic fibrosis (97). Studies have shown that the diagnosis of diabetes by OGTT correlates with clinically important cystic fibrosis outcomes including the rate of lung function decline, the risk of microvascular complications, and the risk of early death (97). Moreover, the OGTT identified patients who benefited from insulin therapy (97). Additional screening recommendations are shown in Table 3 and the interpretation of these tests are shown in Table 4.
|
Table 3. ADA and Cystic Fibrosis Foundation Recommendations for Screening for Cystic Fibrosis Related Diabetes (CFRD) (97)
|
|
1) The use of A1C as a screening test for CFRD is not recommended.
2) Screening for CFRD should be performed using a 2-h 75-g OGTT.
3) Annual screening for CFRD should begin by age 10 years in all CF patients who do not have CFRD.
4) CF patients with acute pulmonary exacerbation requiring intravenous antibiotics and/or systemic glucocorticoids should be screened for CFRD by monitoring fasting and 2-h postprandial plasma glucose levels for the first 48 h. If elevated blood glucose levels are found by SMBG, the results must be confirmed by a certified laboratory.
5) Screening for CFRD by measuring mid- and immediate post-feeding plasma glucose levels is recommended for CF patients on continuous enteral feedings, at the time of gastrostomy feeding initiation, and then monthly by SMBG. Elevated glucose levels detected by SMBG must be confirmed by a certified laboratory.
6) Women with CF who are planning a pregnancy or confirmed pregnant should be screened for preexisting CFRD with a 2-h 75-g fasting OGTT if they have not had a normal CFRD screen in the last 6 months.
7) Screening for gestational diabetes mellitus is recommended at both 12–16 weeks’ and 24–28 weeks’ gestation in pregnant women with CF not known to have CFRD, using a 2-h 75-g OGTT with blood glucose measures at 0, 1, and 2 h.
8) Screening for CFRD using a 2-h 75-g fasting OGTT is recommended 6–12 weeks after the end of the pregnancy in women with gestational diabetes mellitus (diabetes first diagnosed during pregnancy).
9) CF patients not known to have diabetes who are undergoing any transplantation procedure should be screened preoperatively by OGTT if they have not had CFRD screening in the last 6 months. Plasma glucose levels should be monitored closely in the perioperative critical care period and until hospital discharge. Screening guidelines for patients who do not meet diagnostic criteria for CFRD at the time of hospital discharge are the same as for other CF patients.
|
CF= cystic fibrosis; CRFD= cystic fibrosis related diabetes; OGTT= oral glucose tolerance test, SMBG= self-monitored blood glucose
|
Table 4. Criteria for the Diagnosis of Cystic Fibrosis Related Diabetes (97)
|
|
1) During a period of stable baseline health the diagnosis of CFRD can be made in CF patients according to standard ADA criteria. Testing should be done on 2 separate days to rule out laboratory error unless there are unequivocal symptoms of hyperglycemia (polyuria and polydipsia); a positive FPG or A1C can be used as a confirmatory test, but if it is normal the OGTT should be performed or repeated. If the diagnosis of diabetes is not confirmed, the patient resumes routine annual testing.
· 2-h OGTT plasma glucose >200 mg/dl (11.1 mmol/l)
· FPG >126 mg/dl (7.0 mmol/l)
· A1C > 6.5% (A1C <6.5% does not rule out CFRD because this value is often spuriously low in CF.)
· Classical symptoms of diabetes (polyuria and polydipsia) in the presence of a casual glucose level >200 mg/dl (11.1 mmol/l)
2) The diagnosis of CFRD can be made in CF patients with acute illness (intravenous antibiotics in the hospital or at home, systemic glucocorticoid therapy) when FPG levels >126 mg/dl (7.0 mmol/l) or 2-h postprandial plasma glucose levels >200 mg/dl (11.1 mmol/ l) persist for more than 48 h.
3) The diagnosis of CFRD can be made in CF patients on enteral continuous drip feedings when mid- or post-feeding plasma glucose levels exceed 200 mg/dl (11.1 mmol/l) on 2 separate days.
4) Diagnosis of gestational diabetes mellitus is diagnosed based on 0-, 1-, and 2-h glucose levels with a 75-g OGTT if any one of the following is present:
· FPG >92 mg/dl (5.1 mmol/l)
· 1-h plasma glucose >180 mg/dl (10.0 mmol/l)
· 2-h plasma glucose >153 mg/dl (8.5 mmol/l)
CF patients with gestational diabetes mellitus are not considered to have CFRD, but require CFRD screening 6–12 weeks after the end of the pregnancy.
5) The onset of CFRD should be defined as the date a person with CF first meets diagnostic criteria, even if hyperglycemia subsequently abates.
|
CF= cystic fibrosis; CRFD= cystic fibrosis related diabetes; OGTT= oral glucose tolerance test
There is evidence that elevations in glucose below the levels typically used to diagnose diabetes result in adverse effects on the lungs (95). Thus, some experts recommend that treatment should be considered for individuals with abnormal glucose levels which do not meet the criteria for diabetes if there is evidence of declining lung function or weight loss (95).
A unique feature in the treatment of patients with cystic fibrosis related diabetes is that insulin is the treatment of choice in all patients (97). Studies have shown that cystic fibrosis patients on insulin therapy who achieve good glycemic control demonstrate improvement in weight, protein anabolism, pulmonary function, and survival (97). No specific insulin treatment regimen is recommended, and the regimen should be individualized for the patient. For example, a patient with elevated postprandial glucose levels will benefit from mealtime rapid acting insulin. It should be noted that patients with cystic fibrosis induced diabetes still have endogenous insulin production, which allows for the achievement of good glycemic control. Oral diabetes agents are not as effective as insulin in improving nutritional and metabolic outcomes and therefore are not recommended (97). However, in patients who do not tolerate insulin (for example frequent hypoglycemia), oral agents, such as DPP4 inhibitors, may be beneficial (99). For most patients with cystic fibrosis related diabetes an A1c < 7% is recommended but the A1c goal can be higher or lower for certain patients based on clinical judgement. Also of note is that cystic fibrosis patients require a high-calorie, high-salt, high-fat diet.
Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator modulator, is a relatively new agent to treat cystic fibrosis and has been shown to partially reverse the disease. Interestingly in case reports ivacaftor has been shown to markedly improve glycemic control (93,100). In a recent retrospective observation study approximately 1/3 of patients with CFRD had either a resolution of their diabetes or marked improvement with ivacaftor therapy (101). Additionally, the risk of developing CFRD is decreased in patients treated with ivacaftor (102). Studies using three Cystic Fibrosis Transmembrane Regulator (CFTR) modulators (elexacaftor /tezacaftor/ ivacaftor) improved glycemic control and reduced insulin requirements (103-105). These beneficial effects are likely to be due to an improvement in insulin secretion and/or insulin sensitivity (93,106,107). Note the response to these Cystic Fibrosis Transmembrane Regulator (CFTR) modulators depends on the specific mutation causing cystic fibrosis (107).
INFECTIONS
Viral Infections
Viral infections, particularly enterovirus and herpes virus infections, have been postulated to play a role in triggering the autoimmune reaction that leads to the development of T1D (108-111). This phenomenon is discussed in detail in the Endotext chapter on changing the course of the disease in T1D (112). A phase 2 study with the anti-viral agents, pleconaril and ribavirin, demonstrated preservation of residual insulin production in children and adolescents with new-onset T1D (113). In rare instances a viral infection has been associated with the fulminant development of diabetes due to the destruction of beta cells (114). For a review of the link of viral infections with the development of diabetes see a review by Jeremiah and colleagues (111).
Congenital Rubella
Congenital rubella infection has been shown to predispose to the development of T1D that usually presents before five years of age (115). It has been estimated that approximately 1-6% of individuals with the rubella syndrome will develop diabetes in childhood or adolescence (115,116). The mechanism for this association is unknown. In addition, studies have also shown that patients with congenital rubella also develop T2D (116). In one series 22% of individuals with congenital rubella developed diabetes later in life (116). Fortunately, with increased vaccinations, congenital rubella has become a disease of the past in developed countries.
Hepatitis C Virus (HCV)
Meta-analyses and large database studies have demonstrated that hepatitis V virus (HCV) infection is associated with an increased risk of T2D (117-122). In a meta-analysis of 34 studies the risk of diabetes in patients with HCV infection was increased by approximately 70% (117). Moreover, HCV infection is associated with an increased risk of T2D independent of the severity of the associated liver disease (i.e. occurs in patients without liver disease) but the risk of T2D was higher in HCV patients with cirrhosis (118). As expected, the risk of diabetes is increased in HCV patients if the BMI is increased, there is a family history of diabetes, older age, more severe liver disease, and male sex. Conversely, the prevalence of HCV infection in patients with T2D is higher than in non-diabetic controls (118,122,123). In a meta-analysis of 22 studies with 78,051 individuals it was found that patients with T2D were at a higher risk of HCV infection than non-diabetic patients (OR = 3.50; CI = 2.54-4.82) (123). Finally, diabetes is a significant risk factor for the development of liver cirrhosis and hepatocellular carcinoma in HCV infected patients (122,124-127).
Given the increased risk of diabetes in HCV infected patients it seems prudent to routinely screen HCV positive patients for diabetes. Conversely, screening patients with diabetes for HCV infection seems reasonable given the availability of drugs that can effectively treat HCV infections.
Patients with diabetes and HCV infection are insulin resistant in the liver and peripheral tissues (122,127,128). Insulin resistance is present in HCV infection in the absence of significant liver dysfunction and prior to the development of diabetes (128). Treatment that reduces viral load decreases insulin resistance and the risk of developing diabetes in HCV (122,128,129). The insulin resistance in individuals with HCV infections may be due to inflammation induced by cytokines such as TNF-alpha or monocyte chemoattractant protein-1, released from HCV-induced liver inflammation (122,127). Additionally, HCV may directly activate the mTOR/S6K1 signaling pathway, inhibiting IRS-1 protein function and thereby down-regulating GLUT-4 and up-regulating the gluconeogenic enzyme phosphoenolpyruvate carboxykinase-2 (122,127). Beta cell dysfunction may also contribute to the development of diabetes during HCV infection (111,130).
Studies have shown that direct-acting antiviral agents that eradicate HCV infection are associated with improved glycemic control in patients with diabetes indicated by decreased A1c levels and decreased insulin use (127,131). Additionally, in a database study of anti-viral treatment for HCV infection, a decrease in end-stage renal disease, ischemic stroke, and acute coronary syndrome was reported in patients with diabetes (132). These beneficial results on key outcomes need to be confirmed in randomized trials (this may be impossible as withholding treatment of HCV is not appropriate). Treatment of diabetes with metformin or thiazolidinediones is preferred as studies have suggested that these drugs may lower the risk of hepatocellular carcinoma, liver-related death, or liver transplantation in patients infected with HCV (133,134).
COVID-19
There is a bidirectional relationship between diabetes and COVID-19. Both T1D and T2D are important risk factors for morbidity and mortality with COVID-19 infections, which is discussed in the Endotext chapter entitled “Diabetes Mellitus and Infections” (135). Studies have also shown that COVID-19 infections are associated with hyperglycemia and new onset diabetes (136,137). In a large meta-analysis of 20 studies the risk of new incident type 1 diabetes was increased (HR1.44; 95% CI: 1.13-1.82) and the risk of new incident type 2 diabetes was also increased (HR 1.47; 95% CI: 1.36-1.59) (138). Other meta-analyses have reported similar results (139-142). A meta-analysis observed that the risk of diabetes increased 1.17-fold (1.02-1.34) after COVID-19 infection compared to patients with general upper respiratory tract infections (140). The risk of new diabetes in patients with COVID-19 was highest for patients in intensive care (HR 2.88) and hospitalized patients (HR 2.15) (138). For non-hospitalized patients the risk of developing new diabetes was much lower (HR=1.16; 95% CI: 1.07-1.26; p = 0.002) (138).
A very large data-based study with millions of patients found that the risk of developing T2D prior to the availability of COVID-19 vaccination was increased and that this increased risk was still elevated by approximately 30% 1 year after the COVID-19 infection (143). The risk of developing T2D is highest soon after COVID-19 infection (four times higher during the first 4 weeks) and 60% of those diagnosed with T2D after COVID-19 still had evidence of diabetes 4 months after infection (i.e. persistent T2D) (143). The risk of developing T1D was elevated but this increase was no longer seen one year after the COVID-19 infection (143). The increased risk of developing both T1D and T2D was greater in people who were hospitalized with COVID-19 and therefore the risk of developing diabetes was reduced, but not entirely ameliorated, in vaccinated individuals compared with unvaccinated people (143). The absolute risk of developing diabetes was greatest in patients at increased risk for diabetes (obesity, certain ethnic groups, individuals with “prediabetes”, etc. (143).
The mechanisms that account for an increased risk of diabetes following COVID-19 infections are unresolved. There are a number of suggested mechanisms.
- Diabetes could be secondary to acute illness and stress induced hyperglycemia. Stress induced hyperglycemia has been observed after other acute conditions including other infections (137,144).
- Pre-existing diabetes could first be recognized during a COVID-19 infection or during follow-up. This could account for some of the patients that appear to develop new T2D as many patients with T2D are unaware that they have diabetes (145).
- COVID-19 infection could trigger beta cell autoimmunity. This is particularly relevant to the development of T1D as viral infections have been hypothesized to initiate beta cell autoimmunity (112,137).
- The SARS-CoV-2 virus could directly damage the beta cells leading to decreased insulin secretion and hyperglycemia (137).
- SARS-CoV-2 virus could lead to pancreatitis indirectly affecting beta cell function.
- The strong immune response that is seen in COVID-19 infections (cytokine storm) could indirectly lead to beta cell dysfunction and insulin resistance. Additionally, elevated cytokines could persist for an extended period leading to insulin resistance and abnormal glucose metabolism (146)
- SARS-CoV-2 virus infects adipose tissue and may cause adipose dysfunction (decreased adiponectin and increased insulin resistance) (147,148). Persistent adipose tissue infection could result in inflammation and alteration of adipokines and cytokines leading to diabetes.
- The use of high dose glucocorticoids in patients with severe COVID-19 could lead to hyperglycemia and diabetes.
- Changes in environment that occurred during the COVID-19 pandemic, such as decreased exercise, increased food intake, increased weight gain, etc., could enhance the risk of developing diabetes (149).
Hopefully, future studies will better characterize the mechanisms leading to new onset diabetes in patients with COVID-19 infections and determine whether there are unique mechanisms for this association.
ENDOCRINOPATHIES
A number of endocrine disorders are associated with an increased occurrence of diabetes (Table 1). Increased levels of growth hormone, glucocorticoids, catecholamines, and glucagon cause insulin resistance while increased levels of catecholamines, somatostatin, and aldosterone (by producing hypokalemia) decrease insulin secretion and hence can adversely affect glucose homeostasis. The disturbance in glucose metabolism occurring secondary to endocrine disorders may vary from a moderate degree of glucose intolerance to overt diabetes with symptomatic hyperglycemia. Additionally, endocrine disorders can worsen glycemic control in patients with pre-existing diabetes.
Acromegaly
This condition is caused by excessive production of growth hormone (GH) from the pituitary (150). The prevalence of DM in patients with acromegaly is between 10-40%; the prevalence of diabetes and glucose intolerance effects more than 50% of patients (150-153). As expected, there is an increased prevalence of diabetes with age, elevated BMI, a family history of diabetes, and longer duration of acromegaly (151). Diabetes may be present at the time of the diagnosis of acromegaly (154). Higher plasma IGF-1 concentrations correlate with an increased risk of diabetes, suggesting that the biochemical severity of acromegaly influences the risk of developing abnormalities of glucose metabolism (155). Patients with acromegaly should be screened for abnormalities in glucose metabolism (152). The prevalence of acromegaly in patients with diabetes is unknown but is likely to be very low given that acromegaly is an uncommon disorder (60 per million) and diabetes is very common (150).
GH is a counter regulatory hormone to insulin and is secreted during hypoglycemia (156,157). In patients with acromegaly insulin resistance is the major abnormality leading to disturbances in glucose metabolism (151,153,154,158). The insulin resistance is driven primarily through GH induced lipolysis, which results in glucose-fatty acid substrate competition leading to decreased glucose utilization in muscle (151,154,158). Additionally, inhibition of post receptor signaling pathway of the insulin receptor also likely plays a role in the insulin resistance (158). Increased hepatic gluconeogenesis also contributes to the hyperglycemia (151,158). Lipolysis increases the delivery of glycerol and fatty acids to the liver, which serves as a substrate and energy source for gluconeogenesis. In some patients with acromegaly increased insulin secretion compensates for the insulin resistance and glucose metabolism remains normal (151). If insulin secretion cannot increase sufficiently to compensate for the insulin resistance glucose intolerance or diabetes develops (151).
Treatment is directed at the cause of the acromegaly (150). Successful surgical removal of the pituitary adenoma improves hyperglycemia and glucose metabolism has been reported to normalize in 23–58% of people with pre-operative diabetes after surgical cure of acromegaly (151,152,154). Lower IGF-1 and growth hormone levels post operatively correlate with remission of diabetes (159). A meta-analysis of 31 studies with 619 patients treated with somatostatin analogues for acromegaly reported a decrease in insulin levels and glucose levels during a glucose tolerance test but no change in fasting glucose or A1c levels (160). Another meta-analysis of 47 studies with 1297 participants reported that somatostatin analogues also did not affect fasting plasma glucose but worsened 2 hour oral glucose tolerance test and resulted in a mild but significant increase in HbA1c (161) The absence of greater benefit in glucose homeostasis with somatostatin analogues could be secondary to somatostatin analogues inhibiting insulin secretion (150). Of note, while first generation somatostatin analogues appear to have mild or neutral effects on glucose metabolism in patients with acromegaly, treatment with pasireotide, a second-generation somatostatin analogue, aggravated glucose metabolism leading to the development of diabetes in some instances (162-164). The adverse effect of pasireotide is due to inhibiting insulin secretion and decreasing the incretin effect. There is little data on the impact of cabergoline on glucose homeostasis in patients with acromegaly, but the available studies suggest that it modestly improves glucose metabolism or has no effect (152,165). Studies have shown that bromocriptine can improve glucose homeostasis (151,152,166). Finally, treatment with the growth hormone receptor antagonist, pegvisomant, has beneficial effects on glucose homeostasis (164,167,168).
The treatment of diabetes in patients with acromegaly is similar to the treatment in other patients with diabetes (151,153). Patients with acromegaly are often lean with low body fat and therefore dietary recommendations may need to be modified. Additionally, since insulin resistance is the primary defect in patients with acromegaly the use of insulin sensitizers may be especially effective but there are no studies comparing the efficacy of various hypoglycemic agents in patients with acromegaly (151). Data suggests that active acromegaly with elevated GH levels enhances the development of microvascular disease (154). The effect of acromegaly on the development of macrovascular disease is unclear (154). Ketoacidosis is uncommon in patients with diabetes and acromegaly.
Cushing’s Syndrome
Cushing’s syndrome is due to elevated glucocorticoids that can be caused by the overproduction of ACTH by pituitary adenomas or ectopic ACTH producing tumors, overproduction of glucocorticoids by the adrenal glands due to adenomas or hyperplasia, or the exogenous administration of glucocorticoids (169). In patients with Cushing’s syndrome diabetes is present in 20-47% of the patients, while impaired glucose tolerance (IGT) is present in 21-64% of cases (170). Risk factors for the development of diabetes in patients with Cushing syndrome include age, obesity, and a family history of diabetes (170). The prevalence of diabetes varies depending on the etiology of Cushing’s syndrome (pituitary 33%, ectopic 74%, adrenal 34%) (171). In patients with endogenous Cushing’s syndrome the relationship of the degree of hypercortisolism and abnormalities in glucose metabolism has been inconsistent with some studies showing a correlation and other studies no relationship (172). For example, in one study, in patients with endogenous Cushing’s syndrome the prevalence of abnormalities in glucose metabolism and diabetes did not differ in patients with slightly elevated (not greater than 2x the upper limit of normal), moderately elevated (2-5X the ULN), and severely elevated (>5x the ULN) levels of urinary free cortisol (173). In patients with exogenous Cushing’s syndrome high doses of glucocorticoids and longer duration of treatment are more likely to cause diabetes (152,172). Elevated glucocorticoids are more likely to cause high glucose levels in the afternoon or evening and in the postprandial state (152). Hyperglycemia resulting from exogenous steroids occurs in concert with the time-action profile of the steroid regimen employed, such that once daily morning administration of an intermediate acting steroid (prednisone or methylprednisone) causes peak hyperglycemia within 12 hours (post-prandial) while long-acting or frequently administered steroids cause both fasting and postprandial hyperglycemia.
Patients with Cushing’s syndrome should be screened for the presence of abnormalities in glucose metabolism (172). It should be noted that fasting glucose levels are often normal with abnormalities present during an oral glucose tolerance test (170,172). Screening with A1c levels or with an oral glucose tolerance test are therefore preferred. The abnormalities in glucose metabolism may contribute to the increased risk of atherosclerosis in patients with Cushing’s syndrome.
The prevalence of Cushing’s syndrome in patients with diabetes is uncertain with studies reporting very different results ranging from 0 to 9% (172). The selection process used, and the criteria used to determine the presence of Cushing’s syndrome likely greatly influences the results with studies that select patients with marked obesity, poor glycemic control, and poorly controlled hypertension finding a higher percentage of patients with diabetes having Cushing’s syndrome. A recent meta-analysis of 14 studies with a total of 2827 patients with T2D reported that 1.4% had Cushing’s syndrome based on biochemical analysis (174). In a multicenter study carried out in Italy between 2006 and 2008, 813 patients with known T2D without clinically overt hypercortisolism were evaluated for Cushing’s syndrome (175). After extensive evaluation 6 patients (0.7%) were diagnosed with Cushing’s syndrome. Four patients had an adrenal adenoma and their diabetes was markedly improved with the disappearance of diabetes in three patients and discontinuation of insulin therapy in the remaining patient. One patient had bilateral macronodular adrenal hyperplasia and one patient had ACTH dependent Cushing’s syndrome with a normal pituitary MRI.
In approximately 15-35% of patients with an incidental adrenal nodule mild autonomous cortisol secretion with T2D is present (176,177). After surgical removal of the adenoma in patients with autonomous cortisol secretion diabetes normalized or improved in 62.5% of patients (5 of 8) (178). However, not all studies have seen such dramatic improvements in diabetes after adenoma removal (179). Clearly additional studies (preferably large, randomized trials) are required to better define the prevalence of mild subclinical Cushing’s syndrome in patients with diabetes and whether treating the subclinical Cushing’s syndrome in these patients will improve their glycemic control. For a detailed discussion of autonomous cortisol secretion see the chapter on Adrenal Incidentalomas in the Adrenal section of Endotext (180).
Currently, routinely screening patients with T2D for Cushing’s syndrome is not recommended (172). Nevertheless, clinicians should be aware of the possibility of Cushing’s syndrome and screen appropriate patients with T2D (young patients, negative family history of diabetes, patients with physical findings suggestive of Cushing’s syndrome, patients with difficult to control diabetes or hypertension) (172).
Glucocorticoids function as a counter regulatory hormone to insulin and increase in response to hypoglycemia (181). Glucocorticoids disrupt glucose metabolism primarily by inducing insulin resistance in liver and muscle and by stimulating hepatic gluconeogenesis (170,172). The increase in hepatic gluconeogenesis is mediated by several mechanisms including a) directly stimulating the expression of gluconeogenic enzymes b) stimulating proteolysis and lipolysis leading to an increase delivery of amino acids, glycerol, and fatty acids to the liver that provides substrates and energy sources for gluconeogenesis c) inducing insulin resistance and d) enhancing the action of glucagon (170,172). The glucocorticoid induced increase in insulin resistance is due to inhibition of the post-receptor signaling pathway of the insulin receptor, which will result in a decrease in the uptake of glucose by skeletal muscle and adipose tissue (170). In addition to the above glucocorticoids can accelerate the degradation of Glut4 in beta cells, which impairs the ability of beta cells to secrete insulin in response to glucose (182).
Treatment of Cushing’s syndrome by removal of a pituitary tumor, an ectopic ACTH producing tumor, or an adrenal lesion result in a marked improvement in glucose metabolism and in many patients a remission of the diabetes (170,172). In patients with persistent Cushing’s syndrome drug therapy may be needed to normalize cortisol levels. Studies have shown that ketoconazole (200–1200 mg/day), levoketoconazole (150-600 mg twice daily), metyrapone (250–4500 mg/day), mifepristone (300–2000 mg/day) (approved to treat diabetes in patients with Cushing’s syndrome),osilodrostat (1-30 mg twice daily), or cabergoline (1-7mg/day) improves glucose metabolism in patients with Cushing’s syndrome (152,172,183). In contrast, pasireotide has been shown to significantly worsen glucose tolerance, despite control of hypercortisolism, in patients with Cushing’s syndrome (152,172,183). In patients with exogenous Cushing’s syndrome it is important to use as low a dose as possible of glucocorticoids for the shortest period of time to avoid complications of therapy including disrupting glucose homeostasis (184).
The management of diabetes in patients with Cushing’s syndrome is similar to the treatment of other patients with diabetes (152,183). Since insulin resistance is a key defect in patients with Cushing’s syndrome the use of insulin sensitizers may be especially effective but there are no studies comparing the efficacy of various hypoglycemic agents in patients with Cushing’s syndrome (152). Pioglitazone and rosiglitazone can increase the risk of osteoporosis, and it should be noted that patients with Cushing’s syndrome also have a high risk of osteoporosis. As noted above, postprandial glucose levels are preferentially increased in Cushing’s syndrome and therefore drugs that lower postprandial glucose levels, such as DPP4 inhibitors, GLP1 receptor agonists, alpha glucosidase inhibitors, and rapid acting insulin may be very useful (152). In glucocorticoid-treated patients requiring a basal-bolus insulin regimen, a higher requirement of short-acting insulin than basal insulin is frequently required (usually approximately 70% of total insulin dose as prandial and 30% as basal) (152). Because of the insulin resistance in patients with Cushing’s syndrome higher doses of insulin are often required to achieve glycemic control. Patients with Cushing’s syndrome are at higher risk for developing macrovascular disease and therefore aggressive treatment of dyslipidemia and hypertension is required (185,186).
Pheochromocytoma
Pheochromocytomas are rare neuroendocrine tumors that secrete norepinephrine, epinephrine, and dopamine (187). In pheochromocytomas the prevalence of diabetes has been estimated to be between 15-40% and impaired glucose tolerance as high as 50% (188-191). Patients with diabetes were older, had a longer known duration of hypertension, higher plasma epinephrine and norepinephrine levels, increased urinary metanephrine excretion, and larger tumors (189-191). Surprisingly the BMI did not differ between patients with and without diabetes perhaps because more active tumors with higher catecholamine levels lead to weight loss (189,190). In most instances the diabetes is relatively mild but in rare instances can be severe with ketoacidosis (192). The association of hypertension with diabetes in a young patient who is not overweight is a clue to the presence of a pheochromocytoma (189).
Catecholamines, acting primarily by the beta-adrenergic receptors, stimulate glucose production in the liver by increasing glycogenolysis and increase insulin resistance leading to a decrease in tissue disposal of glucose, which together result in elevations in glucose levels (193,194). In addition, catecholamines acting via the alpha-adrenergic receptors, inhibit insulin secretion by the beta cells and acting via the beta-adrenergic receptors, increase glucagon secretion by the alpha cells (195). A decrease in insulin secretion and an increase in glucagon secretion would facilitate the development of hyperglycemia.
With tumor resection diabetes resolves or markedly improves in the majority of patients (>50-90%) with a pheochromocytoma (189,190,196). A duration of diabetes of less than 3 years is associated with a remission of diabetes (197). It should be noted that post-surgical removal of a pheochromocytoma, hypoglycemia can occur in approximately 5% of patients (198). Most of these hypoglycemic episodes occur in the first 24 hours and are more likely to occur in patients with large tumors and high urinary metanephrine levels (198). If surgery is unsuccessful the use of alpha and beta blockers may improve insulin resistance and glucose homeostasis (199).
Hyperthyroidism
Hyperthyroidism induces insulin resistance and hyperglycemia, by increasing intestinal glucose absorption and hepatic glucose production (200,201). Thyroid hormones increase hepatic glucose production by stimulating endogenous glucose production by increasing gluconeogenesis and glycogenolysis (201). Hyperthyroidism in patients without diabetes may increase glucose intolerance (202). Whether hyperthyroidism causes frank diabetes is unclear because much of the older literature that purports that hyperthyroidism causes diabetes used criteria for diabetes that differs greatly from current guidelines. For example, the study of Kreines and colleagues reported that 57% of patients with hyperthyroidism had diabetes but the criteria for diabetes was a 1 hour glucose >160mg/dL plus a 2 hour value >120 mg/dL during an oral glucose tolerance test (203). A study from China using oral glucose tolerance tests did not find a major difference in the prevalence of diabetes in patients with Grave’s disease (11.3%) vs controls (10.0%) (204). In a careful review of the literature Roa Dueñas et al found that hyperthyroidism was not related to type 2 diabetes except for one study in which hyperthyroidism had a positive association with the risk of T2D (5 studies, with a total of 148,684 participants and 11,154 incident cases of type 2 diabetes) (200). In a meta-analysis of these studies the results showed a non-significant association with the risk of T2D (HR 1.16; 95% CI 0.90-1.49). A meta-analysis of 12 cohorts with 32,747 participants also failed to demonstrate that subclinical hyperthyroidism was associated with the development of T2D (205). In contrast, a large retrospective cohort study found that after 10 years of follow-up T2D was increased (HR 1.30; 95% CI 1.21-1.39) (206). Thus, the effect of hyperthyroidism on the development of T2D if present is likely to be modest with most studies demonstrating no relationship. The duration of hyperthyroidism may be a key variable and health care systems where hyperthyroidism is promptly treated may fail to demonstrate that hyperthyroidism leads to incident T2D.
It should be noted that hyperthyroidism may worsen glycemic control in patients with diabetes by increasing intestinal glucose absorption, decreasing insulin sensitivity, and increasing glucose production (207). Teprotumumab, which is used to treat thyroid eye disease, may worsen glycemic control in patients with diabetes and induce hyperglycemia in patients without diabetes (208,209) (discussed in drug induced diabetes section below).
Additionally, T1D and Grave’s disease can occur together as part of the autoimmune polyglandular syndrome (210).
Glucagonoma
Glucagonomas are extremely rare and are associated with a characteristic rash termed necrolytic migratory erythema (82% of patients), painful glossitis, cheilitis, angular stomatitis, normochromic normocytic anemia (50-60%), weight loss (60-90%), mild diabetes mellitus (68-80%), hypoaminoacidemia, low zinc levels, deep vein thrombosis (50%), and depression (50%) (211,212). Glucagon stimulates hepatic glucose production by increasing gluconeogenesis and glycogenolysis leading to an increase in plasma glucose levels (213). Removal of the tumor results in the remission of diabetes.
Somatostatinoma
Somatostatinomas are extremely rare tumors that may present with a triad of diabetes mellitus, diarrhea/steatorrhea, and gallstones, but weight loss and hypochlorhydria also occur (214). Approximately seventy-five percent of patients with pancreatic somatostatinomas have diabetes while diabetes occurs in only approximately 10% of patients with intestinal tumors. Typically, the diabetes is relatively mild and can be controlled with diet, oral hypoglycemic agents, or small doses of insulin (214). Somatostatin inhibits insulin secretion which can result in elevations in plasma glucose levels (214). Increased secretion of somatostatin by cells in the pancreas may be in closer proximity to beta cells and more effective in inhibiting insulin secretion than somatostatin secreted by intestinal cells. Somatostatin also inhibits glucagon secretion and therefore diabetic ketoacidosis is very unusual but has been reported (214,215). Additionally, replacement of functional islet cell tissue by the pancreatic tumor may also contribute to the development of diabetes in patients with a pancreatic somatostatinoma (214). Removal of the tumor results in remission of diabetes.
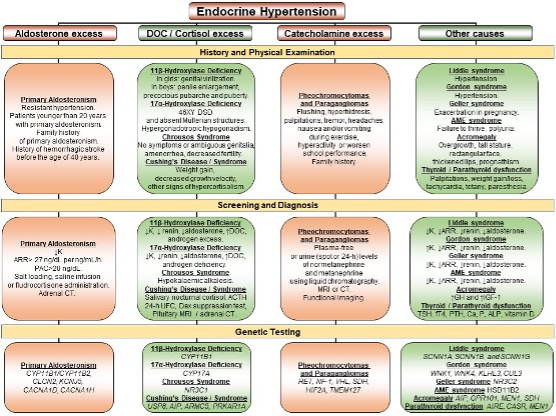
Primary Hyperaldosteronism
Hypokalemia secondary to hyperaldosteronism can impair insulin secretion and result in diabetes. Potassium replacement will improve glucose homeostasis. Additionally, aldosterone induces insulin resistance in adipocytes, skeletal muscle, and liver and decreases insulin secretion independent of potassium levels (216).
In a large meta-analysis the risk of diabetes (OR 1.33, 95% CI 1.01–1.74) and the metabolic syndrome (OR 1.53, 95% CI 1.22–1.91) was modestly increased in patients with primary hyperaldosteronism (217). Hyperaldosteronism increases the risk of cardiovascular disease and renal disease (216,218,219).
DRUG-INDUCED DIABETES
A large number of different drugs have been shown to adversely affect glucose homeostasis (Table 1). Most of these drug’s act in conjunction with other risk factors for T2D and are usually not the sole cause of diabetes. Drug-induced hyperglycemia is often mild and may be clinically asymptomatic, but in some instances can result in the development of severe hyperglycemia manifesting as diabetic ketoacidosis. There are a number of mechanisms by which drugs induce alterations in glucose metabolism including inducing insulin resistance or inhibiting insulin secretion. In most cases diabetes remits when the drug is stopped but in some instances diabetes can be permanent. Use of a rodenticide (N-3 pyridylmethyl-N’4 nitrophenylurea, VacorÒ), structurally related to streptozotocin, was removed from the market in the 1980s because the ingestion of this compound resulted in insulin-dependent diabetes due to beta cell destruction (220). In this section we will focus on drugs that cause major changes in glucose homeostasis or drugs that are commonly used in clinical practice.
Antihypertensive Drugs
In a meta-analysis the risk of developing diabetes varied between different classes of antihypertensive drugs (221). The odds ratios were: angiotensin receptor blocker (ARB) 0.57; angiotensin converting enzyme (ACE) inhibitor 0.67; calcium channel blocker (CCB); 0.75; placebo; 0.77; beta blocker; 0.90 with the thiazide group set at 1.00. Similarly, in the ALLHAT study the risk of developing diabetes was greater in the thiazide group than in patients treated with an ACE inhibitor or a CCB (222). In a meta-analysis of 10 studies of beta-blockers and 12 studies of diuretics in patients without diabetes it was found that beta-blockers increased fasting blood glucose concentrations by 11.5 mg/dL and diuretics by 13.9 mg/dl (223). As one would expect, lower doses of thiazides (hydrochlorothiazide or chlorthalidone ≤25 mg daily) had less effect on glucose levels (224). In a meta-analysis of twelve studies with 94,492 patients beta-blocker therapy resulted in a 22% increased risk for new-onset diabetes compared with nondiuretic antihypertensive agents (225). Thus, both thiazide diuretics and beta blockers increase the risk of developing diabetes while ARBs and ACE inhibitors reduce the risk (226). CCB, alpha receptor blockers (prazosin and doxazosin), and clonidine do not increase the risk of developing of diabetes (227).
The hyperglycemia secondary to thiazide diuretics may in some instances be due to decreased insulin secretion secondary to potassium loss, which can be improved with potassium replacement (228). Studies have suggested that combining thiazides with potassium-sparing diuretics reduces the development of hyperglycemia (229). In addition, thiazides may directly affect insulin secretion similar to diazoxide (see below). Finally, thiazides also increase insulin resistance and enhance hepatic glucose production (228).
The effect on glucose metabolism differs between different beta-blockers and carvedilol, a third-generation beta-blocker, has beneficial effects on glucose metabolism (226,228). A greater inhibition of insulin secretion occurs with non-selective beta-blocking agents (228). Beta blockers decrease insulin secretion and increase insulin resistance (226,228). In addition, beta-blockers increase weight, which also could adversely affect glucose homeostasis (230). Finally, beta-blockers increase the risk of severe hypoglycemia by decreasing the recovery from hypoglycemia and masking the symptoms of hypoglycemia (231).
Diazoxide
Diazoxide is a non-diuretic benzothiadiazine derivative, which increases plasma glucose levels by inhibiting insulin secretion through opening the potassium/ATP channels in beta cells (232). Diazoxide is used to control hypoglycemia in patients with insulinomas (233).
Statins
In a meta-analysis of 13 trials with over 90,000 subjects, there was a 9% increase in the incidence of diabetes during follow-up among subjects receiving statin therapy (234). All statins appear to increase the risk of developing diabetes. A more recent meta-analysis comparing low or moderate intensity statin therapy vs. placebo found that there was a 10% increase in the incidence of new diabetes whereas with high intensity statin therapy there was a 36% increase (235). In patients on intensive vs. moderate statin therapy, Preiss et al observed that patients treated with intensive statin therapy had a 12% greater risk of developing diabetes compared to subjects treated with moderate dose statin therapy (236). Older subjects, obese subjects, and subjects with high glucose levels were at a higher risk of developing diabetes while on statin therapy (235,237). Thus, statins may be unmasking and accelerating the development of diabetes that would have occurred naturally in these subjects at some point in time. In patients without risk factors for developing diabetes, treatment with statins does not appear to increase the risk of developing diabetes.
The mechanism by which statins increase the risk of developing diabetes is unknown (237). Studies suggest that the inhibition of HMG-CoA reductase per se may be leading to the statin induced increased risk of diabetes via weight gain (237). However, a large number of studies have now shown that polymorphisms in a variety of different genes that lead to a decrease in LDL cholesterol levels are also associated with an increase in diabetes suggesting that decreases in LDL cholesterol levels per se alter glucose metabolism and increase the risk of diabetes (237). How decreased LDL cholesterol levels effect glucose metabolism is unknown.
In some studies statins have been shown to increase insulin resistance (238) and in some studies to decrease insulin secretion (239,240). Clearly further studies are required to understand the mechanisms by which statins increase the risk of developing diabetes.
Niacin
A meta-analysis examined the effect of niacin therapy on the development of new onset diabetes (241). In 11 trials with 26,340 non-diabetic participants, niacin therapy was associated with a 34% increased risk of developing diabetes. This increased risk results in one additional case of diabetes per 43 initially non-diabetic individuals who are treated with niacin for 5 years. Results were similar in patients who were receiving niacin therapy in combination with statin therapy. It has been recognized for many years that niacin induces insulin resistance (242). The mechanisms by which niacin induces insulin resistance are unknown but possible mechanisms include a rebound increase in free fatty acids with niacin therapy or the accumulation of diacylglycerol (242).
Pentamidine
Pentamidine is an antiprotozoal agent known to cause hypoglycemia and hyperglycemia (228). Pentamidine induces a direct cytolytic effect on pancreatic beta cells leading to insulin release and hypoglycemia, which is then followed by beta cell destruction and insulin deficiency resulting in diabetes (243,244).
Phenytoin (Dilantin)
Phenytoin can cause hyperglycemia and there have been cases of diabetic ketoacidosis (245,246). The adverse effect of phenytoin on glucose metabolism is mediated primarily by an inhibition of insulin secretion (228).
Alpha Interferon
Treatment with alpha interferon in rare instances can cause T1D. Of 987 patients treated with alpha interferon for chronic hepatitis C, 5 patients developed T1D (247). The clinical course is characterized by the abrupt development of severe hyperglycemia at times with ketoacidosis (248). High titers of anti-islet autoantibodies are present and almost all patients require permanent insulin therapy (248). Treatment with interferon alpha facilitates the development of autoimmune disorders including T1D (249). Other autoimmune disorders frequently occur, particularly thyroid dysfunction.
Checkpoint Inhibitors
There are several checkpoint inhibitors; ipilimumab a cytotoxic T-lymphocyte-associated protein 4 inhibitor (CTLA-4 inhibitor); nivolumab and pembrolizumab, programmed cell death protein 1 inhibitors (PD-1 inhibitors); atezolizumab, avelumab, and durvalumab. programmed cell death 1 ligand inhibitors (PD-L1 inhibitors) (250). Both CTLA-4 and PD-1 play a key role in the maintenance of immunological tolerance to self-antigens thereby preventing autoimmune disorders (250). Immune mediated hypothyroidism, hyperthyroidism, hypophysitis, primary adrenal insufficiency, hypoparathyroidism, and insulin-deficient diabetes have been reported as a complication of the use of these drugs (250,251). In a meta-analysis of 38 randomized clinical trials with 7551 patients, autoimmune diabetes occurred in only 0.2% of the patients and was primarily seen with the use of PD-1 inhibitors (250). In another meta-analysis of 101 studies with 19,922 patients the incidence of autoimmune diabetes was 2.0% (95% CI, 0.7–5.8) for nivolumab and 0.4% (95% CI, 0.2–1.3) for pembrolizumab (251). The occurrence of autoimmune diabetes with other checkpoint inhibitors was rare (251).
The onset of diabetes ranges from a few weeks up to one year after initiating therapy and typically presents with polyuria, polydipsia, weight loss, and dehydration (251,252). Severe hyperglycemia and ketoacidosis are commonly observed (252). Because of the acute occurrence, A1c levels may not be elevated. C-peptide levels are very low and approximately 50% of patients have islet cell antibodies (GAD, ICA, IAA or IA-2; GAD antibodies are the most commonly observed) (251,252). Insulin treatment is required, and it is likely that the diabetes will be irreversible (251,252).
For additional information on the checkpoint inhibitor associated diabetes see the Endotext chapter “Immune Checkpoint Inhibitors Related Endocrine Adverse Events” in the Disorders that Affect Multiple Organs section (253).
Antipsychotic Drugs
Many studies have linked second generation antipsychotic medications with the development of T2D (Table 5) (254,255). In a meta-analysis of a large number of studies it was reported that olanzapine and clozapine treatment resulted in a greater increase in glucose abnormalities than aripiprazole, quetiapine, risperidone and ziprasidone (256). Another meta-analysis has further shown that aripiprazole has a reduced risk of T2D compared to other antipsychotic agents (257). With regards to first generation antipsychotic drugs, chlorpromazine has a high risk of disrupting glucose metabolism while haloperidol, fluphenazine, and perphenazine have a low risk (254). It is thought that antipsychotic drugs induce diabetes by multiple mechanisms: (1) they inhibit insulin signalling in muscle cells, hepatocytes, and adipocytes thereby causing insulin resistance; (2) they induce obesity, which can also cause insulin resistance; and (3) they can cause direct damage to β-cells, leading to dysfunction and apoptosis of β-cells (255,258).
|
Table 5. Risk of Diabetes of Selected First- and Second-Generation Antipsychotics
|
|
|
Risk of diabetes*
|
|
First-generation antipsychotic
Chlorpromazine
Fluphenazine
Perphenazine
Haloperidol
|
+++
+
+
+
|
|
Second-generation antipsychotic
Clozapine
Olanzapine
Quetiapine
Risperidone
Ziprasidone
Aripiprazole
Paliperidone
Lurasidone
|
+++
+++
++
++
+
+
+
+
|
*Relative to other antipsychotics. Not all the risk of diabetes or weight gain are related to the antipsychotics. Table modified from (255)
Androgen Deprivation Therapy
A number of studies have shown that androgen deprivation therapy increases the risk of developing diabetes (259). For example, a study by Tsai reported that androgen deprivation therapy was associated with a 1.61-fold increased diabetes risk and the number needed to harm was 29 (260). The androgen deprivation induced diabetes typically develops after a year of treatment (259). Androgen deprivation therapy induces insulin resistance (259). The increase in insulin resistance may be due to an increase in visceral fat mass and/or an increase in pro-inflammatory adipokines such as TNF-a, IL-6, and resistin (259).
Immunosuppressive Drugs
Immunosuppressive drugs used after organ transplantations increase the risk of diabetes (261). In general, tacrolimus has been associated with a greater risk of developing diabetes compared to cyclosporine (261,262). The calcineurin inhibitors, tacrolimus and cyclosporine, decrease insulin secretion and synthesis (261,262). Additionally, tacrolimus and cyclosporine inhibit glucose uptake in human subcutaneous and omental adipocytes (262).
Mechanistic Target of Rapamycin Inhibitors (mTOR inhibitors)
mTOR inhibitors, sirolimus and everolimus, can induce diabetes (263). The adverse effect of mTOR inhibitors on glucose metabolism is due to insulin resistance secondary to a reduction of the post receptor insulin signalling pathway and a reduction of insulin secretion via a direct effect on the pancreatic beta cells (261,263).
Asparaginase
Hyperglycemia is common with the use of asparaginase treatment ranging from 2.5% to 23% in the pediatric population and as high as 76% in adults with PEG-asparaginase use (264,265). Hyperglycemia usually resolves within 12 days after the last dose (264). Risk factors predisposing to hyperglycemia with asparaginase treatment include a history of impaired glucose tolerance, age >10 years, obesity, family history of diabetes mellitus, and history of Down syndrome (264,265). Diabetic ketoacidosis has been described with asparaginase treatment but is not a common occurrence (264). Decreased insulin secretion, increased insulin resistance, and increased glucagon secretion may contribute to the hyperglycemia observed with asparaginase. Additionally, asparaginase can induce pancreatitis, which can also lead to hyperglycemia (264).
Antibiotics
Fluoroquinolones have been associated with an increased risk of hyperglycemia, particularly in the elderly (228). The risk of developing hyperglycemia is greatest with gatifloxacin (228).
Beta-Adrenergic Drugs
High doses of beta-adrenergic drugs can lead to hyperglycemia likely due to the stimulation of hepatic gluconeogenesis (228).
Teprotumumab
Teprotumumab blocks the activation of the insulin-like growth factor-1 receptor (IGF-1 receptor). Initial studies in patients with thyroid eye disease found that approximately 10% of patients had hyperglycemia and one-third of these individuals with hyperglycemia did not have pre-existing diabetes or impaired glucose tolerance but the study by Amarikwa et al found a higher risk of hyperglycemia (209,266,267). Older age, prediabetes/diabetes, and Asian and Hispanic ethnicity may increase the risk of hyperglycemia. Case reports of diabetic ketoacidosis and hyperglycemic hyperosmolar state have been reported (268,269). In some but not all patients glycemia returns towards normal when teprotumumab treatment is completed. Blocking the activation of the IGF-1 receptor may lead to an increase in growth hormone secretion leading to hyperglycemia (270). Additionally, adverse effects on insulin receptors that interact with the IGF-1 receptor may lead to insulin resistance (270).
Glucocorticoids, Somatostatin, Growth Hormone, and Glucagon
The effects of these hormones on glucose metabolism were discussed in the section on Endocrinopathies.
HIV Antiretroviral Therapy
The effect of the drugs used to treat patients living with HIV on the development of diabetes is discussed in the Endotext chapter “Diabetes in People Living with HIV” in the Diabetes section (6).
Latent Autoimmune Diabetes in Adults (LADA)
LADA is an autoimmune disorder that resembles T1D but shows a later onset and slower progression towards requiring insulin therapy (271-273). The ADA includes LADA as T1D whereas WHO classifies LADA as a hybrid form of diabetes (T1D and T2D) (96) (https://www.who.int/publications/i/item/classification-of-diabetes-mellitus). Epidemiological studies suggest that LADA may account for 2–12% of all cases of diabetes in the adult population (271,272,274). To differentiate LADA from T1D and T2D, the Immunology of Diabetes Society has proposed three criteria: (a) adult age of onset (> 30 years of age); (b) presence of at least one circulating autoantibody (GAD, ICA, IAA or IA-2) and; (c) insulin independence for the first 6 months after the time of diagnosis (271,272). Of the various antibodies associated with autoimmune diabetes, GAD antibodies are present in most patients with LADA (271,272,274). Patients with high titers of GAD antibodies progress to requiring insulin more rapidly (273). LADA subjects appear to have a faster decline in C-peptide levels compared to autoantibody negative patients with T2D (274). It should be noted that classic T1D can occur in adults and this is defined as those adult patients with antibodies (GAD, ICA, IAA or IA-2) that require insulin therapy at diagnosis or soon after diagnosis (272,274). In contrast, patients with LADA can often go many years before requiring insulin therapy (272). Whether LADA is just a slowly progressing form of T1D or a hybrid T1D and T2D is unclear (Table 6).
|
Table 6. Comparison of T1D, LADA, and T2D
|
|
|
T1D
|
LADA
|
T2D
|
|
Age of onset
|
Tend to be young
|
>age 25
|
Tend to be adult
|
|
Family history
|
Occasional
|
Occasional
|
Usually
|
|
C-peptide
|
Low, often undetectable
|
Varies
|
Normal or high
|
|
Auto-ab
|
+
|
+
|
-
|
|
Weight
|
Tend to be lean
|
Tend to be lean
|
Usually overweight
|
|
Metabolic syndrome
|
No
|
Varies
|
Usually
|
|
Insulin requirement
|
Yes
|
Varies
|
Varies
|
|
Genetic risk
|
HLA
PTPN22
INS
SH283
PFKFB3
|
Intermediate between T1D & T2D
|
TCF7L2
FTO
SLC30A8
|
In a retrospective study, Fourlanos and colleagues pointed out several features that increase the likelihood of a patient with “T2D” having LADA (275). These features include age of onset <50 years of age, acute symptoms (polyuria, polydipsia, weight loss), BMI <25 kg/m2, personal history of autoimmune disease, and family history of autoimmune disease (275). The presence of at least two of these clinical features indicated a 90% sensitivity and 71% specificity for identifying a patient with LADA (275). As compared to patients with T2D, LADA patients have a lower rate of hypertension, lower total cholesterol levels, higher HDL cholesterol levels, and a decreased frequency of the metabolic syndrome (272,274). HLA-DQB1 risk genotypes have been consistently positively associated and protective genotypes have been negatively associated with LADA (273). However, in addition to genotypes that are associated with T1D, patients with LADA also have an increased frequency of genotypes that are associated with T2D (TCF7L2, FTO, and SLC30A8) (273). Having a healthy lifestyle and a BMI<25 is associated with a reduced risk of LADA including in individuals with a genetic susceptibility (276). Individuals with other autoimmune diseases are more likely to develop LADA (277).
Some have proposed GAD antibody testing all patients with T2D (278) to diagnose LADA but given the given the increased costs and the relatively frequent occurrence of false positive tests compared to true positives in a low-risk population this strategy is not widely accepted (279). The ADA suggests selective testing in adults without traditional risk factors for T2D and/or younger age (96).
In LADA patients glycemic control can initially be achieved with hypoglycemic agents other than insulin but overtime patients progress to requiring insulin therapy. Sulfonylureas seem to accelerate the progress to requiring insulin therapy and therefore should be avoided (280). Because of the progressive loss of beta cell function there is an increased risk of diabetic ketoacidosis with SGLT2 inhibitors and therefore these drugs should be used with caution. Monitoring ketone levels in patients with LADA treated with SGLT2 inhibitors would be prudent. Novel therapies to preserve beta cell function would be ideal for patients with LADA but at this time there are no proven strategies to preserve beta cell function. However, there are several studies from China that suggest that vitamin D may slow the loss of beta cell function (281-283)
In a long- term follow-up (median 17.3 years) comparing microvascular outcomes in patients with LADA or T2D it was observed that the risk of renal failure/death, blindness, vitreous hemorrhage, or retinal photocoagulation was decreased in the patients with LADA during the first 9 years (adjusted HR 0.45; p<0.0001), whereas in subsequent years their risk was higher (HR 1·25; p=0.047) (284). This difference was attributed to higher A1c levels in the LADA patients. The prevalence of coronary heart disease and cardiovascular mortality is similar in patients with LADA and T2D (285,286). Mortality is increased in patients with LADA (HR 1.44), compared to controls (287).
Autoimmune Polyglandular Syndromes
T1D can occur as part of the autoimmune polyglandular syndromes. These disorders are discussed in the Endotext chapter “Autoimmune Polyglandular Syndromes” in the Disorders that Affect Multiple Organs section (4).
Stiff-Person syndrome
Stiff-person syndrome is a rare autoimmune disorder of the nervous system with fluctuating stiffness and spasm of the skeletal muscles that occurs more frequently in females than males (approximately 2/3 women) (288). Rigidity is caused by simultaneous contracture of agonist and antagonist muscles. Muscle involvement is symmetrical, and the lower extremities are affected more commonly than the upper extremities and proximal limb and axial muscles are affected more severely than distal muscles (288). Most patients have very high levels of anti-glutamic acid decarboxylase (GAD) antibodies (288). 30-65% of these individuals also develop beta cell destruction and T1D (289). Diabetes may occur several years prior to the development of the stiff-person syndrome (60%) or after the development of the stiff-person syndrome (288,289). The stiff person syndrome without GAD antibodies is not associated with diabetes (289). The GAD antibodies in patients with T1D and stiff-person syndrome recognize a different set of epitopes and have distinct biological effects (290). Other autoimmune manifestations are also common, particularly thyroid disorders and pernicious anemia (288,289). It should be noted that a variety of neurological disorders (cerebellar ataxia, limbic and extra-limbic encephalitis, nystagmus/oculomotor dysfunction, drug-resistant epilepsy, etc.) are associated with GAD antibodies (290).
Autoimmune Insulin Resistance Type B Syndrome
Insulin resistance can result from autoantibodies directed against the insulin receptor, which either inhibit insulin from binding to the receptor or stimulate the receptor (291). Thus, they can cause either hyperglycemia or hypoglycemia, even alternating in the same patient. Low titers of insulin receptor antibodies typically lead to hypoglycemia while high titers result in hyperglycemia but the possibility of epitope switching may also determine if hyperglycemia or hypoglycemia is manifest (292,293). The patients usually present with very high glucose levels with high fasting insulin levels and significant weight loss (291,293). Serum triglyceride levels are typically low and HDL cholesterol levels normal, which contrasts with typical patients with insulin resistance who usually have high triglyceride levels and low HDL cholesterol levels. This difference is explained by post receptor insulin resistance stimulating lipogenesis whereas insulin resistance localized to the receptor does not (294). The diagnosis can be confirmed by demonstrating the presence of autoantibodies to the insulin receptor. The prevalence of type B insulin resistance syndrome is unknown but is quite rare (291). Middle-aged women are most often affected and often have other manifestations of autoimmune disease such as SLE or Sjogren’s. However, this disorder can also affect males and younger patients. In some instances, the type B insulin resistance syndrome occurs as a paraneoplastic manifestation of lymphoma or multiple myeloma. Patients may have signs of insulin resistance including acanthosis nigricans and ovarian hyperandrogenism. Of note the acanthosis nigricans may involve the lips and the periocular region resulted in a typical facial appearance (291). Serum testosterone levels are often elevated in females (291). Patients often need excessive amounts of insulin (1,000 U or more per day). One can add insulin sensitizers such as metformin and/or thiazolidinediones to try to reduce the insulin dose, which can in some patients be greater than 10,000U per day (291). Treatment includes immunosuppression and/or plasmapheresis to halt the autoantibody production and decrease antibody levels (295). Treatment with rituximab, high-dose steroids, and cyclophosphamide until remission, followed by maintenance therapy with azathioprine is very effective in inducing and maintaining remissions. Approximately 1/3 of patients will undergo a spontaneous remission with reversal of the hyperglycemia/hypoglycemia and the clinical manifestations (291).
DIABETES OF UNKNOWN CAUSE
Ketosis-Prone Diabetes in Adults (Flatbush Diabetes)
This syndrome is characterized by the acute onset of severe hyperglycemia with or without ketoacidosis, which after several weeks to months no longer requires insulin therapy and can be treated with diet or oral hypoglycemic agents (296,297). These patients typically have a history of polyuria, polydipsia, and weight loss for less than 4 to 6 weeks indicating an abrupt onset of the disorder in glucose metabolism and no history of an event that could have precipitated the hyperglycemia (297). The initial presentation is suggestive of T1D. While in most patients insulin therapy can be stopped there are some patients who continue to require insulin treatment (296). This syndrome occurs in black populations (African American, African-Caribbean, sub-Saharan African), Hispanic populations, and Asian (Chinese, Indian, and Japanese) populations but is not typically seen in Caucasians (296,297). The typical patient is male, middle-aged, overweight or modestly obese with a strong family history of diabetes (296,297). Patients are negative when tested for islet cell antibodies (GAD, ICA, IAA or IA-2) (296). Recurrent episodes of ketoacidosis can occur, but the clinical course is typical of patients with T2D (296,297). Treatment with hypoglycemic agents reduces the risk of recurrence (297,298). SGLT2 inhibitors should be used with caution given the risk of recurrent ketoacidosis. With long-term follow-up many patients eventually require insulin therapy similar to what is observed in patients with T2D (298).
During the episode of severe hyperglycemia patients with ketosis-prone diabetes have lost the ability of glucose to stimulate beta cell insulin secretion but nonglycemic pharmacologic agents (glucagon and arginine) can stimulate insulin secretion (296). After restoration of normal glycemia the ability of glucose to stimulate insulin secretion returns towards normal and by 8-12 weeks has maximally improved (296). Usually patients with this syndrome have a modest reduction in stimulated insulin secretion (296). Why these patients temporarily lose the ability for glucose to stimulate insulin secretion is unknown. Additionally, during the acute episode of hyperglycemia the patients are severely insulin resistant, which improves during a period of euglycemia (297,298).
Clinically, it is important to recognize this syndrome as some patients presenting with diabetic ketoacidosis, particularly if they are non-Caucasians, may not have T1D but rather have ketosis-prone diabetes. It is estimated that between 20% and 50% of African-American and Hispanic patients with a new diagnosis of diabetic ketoacidosis have ketosis-prone diabetes (297). After restoration of euglycemia the management of these patients is similar to the management of patients with T2D, and they frequently do not require permanent insulin treatment.
OTHER GENETIC SYNDROMES SOMETIMES ASSOCIATED WITH DIABETES
There are a number of inherited monogenic disorders that secondarily can be associated with diabetes. The mechanisms linking these disorders with diabetes are often not clear.
Chromosomal Abnormalities
DOWN SYNDROME
Down syndrome is due to trisomy of chromosome 21 and occurs in 1 in every 787 liveborn babies (299). Down syndrome is often associated with autoimmune disorders like T1D and thyroiditis (299,300). The prevalence rate of T1D in patients with Down syndrome has been estimated to be between 1.4 and 10.6%, which is higher than in the general population (301). In another study there was a 4-fold increased prevalence of diabetes in patients with Down syndrome (302). Diabetes in patients with Down syndrome often presents earlier in life with 22% of participants developing diabetes by 2 years of age (303). The presence of diabetes is often associated with other autoimmune disorders, particularly hypothyroidism and celiac disease (300). Anti-glutamic acid decarboxylase antibodies (GAD antibodies) are very frequently present in Down syndrome subjects who develop diabetes (300). Downs syndrome patients with diabetes have similar HLA genotypes as non-Down syndrome patients with T1D (300). Interestingly, while patients with Down syndrome and diabetes are typically treated with simpler regimens their glycemic control tends to be as good or better than the usual patient with T1D, perhaps related to a simpler lifestyle and acceptance of routine (300). The cause of the increased autoimmunity in patients with Down syndrome may be due to the abnormal expression of the AIRE gene, which regulates T-cell function and self-recognition and is located on chromosome 21 (21q22.3 region) (299,300).
While the incidence of T2D is similar between patients with Down syndrome and controls, the onset of T2D occurs at a much earlier age (304). The incidence for T2D is >10 times higher in patients aged 5–14 years with Down syndrome compared to controls. In individuals under the age of 45, Down syndrome is associated with an increased incidence of diabetes while over the age of 45, the incidence of diabetes is increased in controls. Notable the BMI in increased in younger patients with Down syndrome compared to controls and could contribute to the increase in T2D.
KLINEFELTER SYNDROME
Klinefelter syndrome is due to an extra X chromosome in men (XXY) resulting in hypergonadotropic hypogonadism, low testosterone levels, gynecomastia, and reduced intelligence (305,306). The prevalence of Klinefelter syndrome is approximately 1 in 500 to 1 in 1000 males (305,306). Patients with Klinefelter syndrome are frequently obese, insulin resistant, and at increased risk to develop T2D (50% have the metabolic syndrome) (307,308). The decreased muscle mass and increased fat mass that often occur in patients with Klinefelter syndrome contribute to the high prevalence of insulin resistance and metabolic syndrome. The prevalence of overt diabetes in Klinefelter syndrome is estimated to be between 10-39% (307,309). Additionally, the prevalence of diabetes is even higher (up to 57%) in patients with more severe karyotypes (48, or 49 chromosomes) (307). Klinefelter syndrome patients develop diabetes earlier in life (onset around 30 years) and their BMI is lower than what is usually observed in patients with T2D (307). Hypogonadism is associated with insulin resistance and an increased risk of diabetes and whether testosterone therapy will be of benefit in preventing or treating diabetes in patients with Klinefelter syndrome is uncertain (309). Given the increased risk of developing T2D, patients with Klinefelter syndrome should be periodically screened for diabetes.
Interestingly, one study reported an increased prevalence of T1D in patients with Klinefelter syndrome (310). Furthermore, a study reported that 8.2% of patients with Klinefelter syndrome had autoantibodies specific to T1D (Insulin Abs, GAD Abs, IA-2 Abs, Znt8 Abs) (311). Additional studies are required to better elucidate whether Klinefelter syndrome increases the risk of developing T1D.
TURNER SYNDROME
Turner syndrome is the most common chromosomal abnormality in girls, affecting approximately 1:2,500 female live births (312). The condition is caused by complete or partial deletion of an X chromosome (312). The incidence of both T1D and T2D has been reported to be increased in patients with Turner syndrome (313). However, the link between T1D and Turner syndrome is not well characterized while the link with T2D is clearly established (314,315). For example, in a study of 224 patients with Turner syndrome 56 (25%) had T2D whereas only 1 patient (<0.5%) had T1D (316). Patients with Turner syndrome have an increased risk of autoimmune disorders, particularly hypothyroidism and celiac disease, but the prevalence of autoimmune T1D is much less (315,317). Four percent of patients with Turner syndrome have been shown to have GAD antibodies, which is greater than the 1% prevalence seen in the general population (317).
The prevalence of glucose intolerance is estimated to be from 15-50% while the prevalence of T2D is estimated to be approximately 10-25% (315,316). T2D occurs at a relatively young age in patients with Turner syndrome. A 25%-70% lifetime risk for diabetes has been described (318). Decreased beta cell function and decreased insulin sensitivity was observed in teenagers with Turner syndrome and was accompanied by an increased prevalence of impaired fasting glucose and impaired glucose tolerance compared to controls (319). Increased obesity is common in patients with Turner syndrome, which likely contributes to the abnormalities in glucose metabolism (314). Both insulin resistance and decreased insulin secretion are present in patients with Turner syndrome but the development of hyperglycemia in patients with T2D appears to be driven by decreased insulin secretion (314,315). Because of the high prevalence of diabetes, it is recommended to screen A1c with or without fasting glucose levels annually beginning at 10-12 years of age or sooner with symptoms of diabetes and then every 1-2 years (315,318). When diabetes is diagnosed,measuring diabetes auto-antibodies is helpful in differentiating T1D vs, T2D in patients with Turner syndrome (318). Growth hormone therapy does not appear to increase the risk or worsen diabetes (314,315). Growth hormone therapy may lead to a decrease in adiposity and impaired glucose tolerance, which suggests it may actually improve glucose homeostasis (315). Similarly, sex steroid hormone replacement therapy also does not appear to have major adverse effects on glucose metabolism in patients with Turner syndrome (314).
WILLIAMS SYNDROME
Williams syndrome (Williams-Beuren syndrome) is a multisystem disorder characterized by transient infantile hypercalcemia, distinctive facial dysmorphism, and supravalvular aortic stenosis (320,321). In addition, gastrointestinal problems, dental anomalies, developmental delay/intellectual disability, anxiety disorders, and attention deficit disorder may occur as well as a variety of endocrine abnormalities including reduced statural growth, obesity, dyslipidemia, early pubertal development, hypothyroidism, and decreased bone density (320,322). Williams syndrome is due to a deletion on chromosome 7q, leading to the loss of 25–27 contiguous genes and thus individuals with Williams syndrome have only a single copy of these genes (321). This deletion almost always arises de novo in the affected individual. The estimated prevalence of Williams syndrome is ~1/7,500 and effects both males and females (321).
Numerous studies have shown a high prevalence of T2D and impaired glucose tolerance in patients with Williams syndrome (320). The abnormalities in glucose metabolism occur during adolescence and are not necessarily associated with obesity (320). Of note insulin resistance is observed initially followed by a loss of insulin secretion (320). Markers of islet autoimmunity are not observed (320). In a review of 7 studies with 154 participants with Williams syndrome and an average age ranging from 13 to 35 years of age it was observed that 18% had diabetes and 42% impaired glucose tolerance (320). Because of this high risk for diabetes, it is recommended that patients with Williams syndrome be screened for diabetes beginning in adolescence (320). Note-worthy is that A1c was frequently not abnormal and therefore screening should be with fasting glucose levels or an oral glucose tolerance test (320).
Diseases of the Endoplasmic Reticulum
The endoplasmic reticulum folds and modifies newly formed proteins to make them function properly. Therefore, diseases affecting the endoplasmic reticulum usually affect many organs. Wolfram syndrome is the best known but there are other genetic syndromes that affect the endoplasmic reticulum and cause diabetes (323).
WOLFRAM SYNDROME
Wolfram syndrome is a rare autosomal recessive genetic disorder characterized by T1D, diabetes insipidus, optic nerve atrophy, hearing loss, and neurodegeneration (324,325). There are also rare autosomal dominant forms of this disorder (325). This syndrome is sometimes called DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness). The prevalence is approximately one per 770,000 but varies depending upon the specific population (324,325). The onset of the clinical picture is highly variable in both severity and clinical manifestations (325). This disorder typically has a very poor prognosis with the median age at death being 30 years (324). Diabetes mellitus is usually the first manifestation, typically diagnosed around age 6 (324). The diabetes is not immune mediated but is characterized by insulin deficiency (325). Almost all patients require insulin therapy (325). Residual beta cell function persists and therefore good glycemic control tends to be easier to achieve in Wolfram syndrome than immune mediated T1D (325). However, over time C-peptide levels decrease (326). The development of optic atrophy and hearing loss in children with diabetes are clues to the presence of this syndrome. Until the onset of optic atrophy and hearing loss these patients are usually thought to have typical T1D with an absence of antibodies (325). Confirmation of the diagnosis can be made by identifying mutations in the WFS1 gene (Wolfram syndrome type 1) (324). The WFS1 gene encodes a transmembrane protein (wolframin) localized to the ER (endoplasmic reticulum) and mutations result in ER stress leading to beta cell dysfunction and death (324).
Mutations in CISD2 gene cause a similar recessive type of Wolfram syndrome (Wolfram syndrome type 2) with patients exhibiting bleeding from upper intestinal ulcers and defective platelet aggregation without diabetes insipidus and psychiatric disorders (325). CISD2 encodes for a protein that moves between the ER and mitochondrial outer membrane (325).
Unfortunately, there are currently no specific treatments to restore ER function and prevent the complications of this disorder.
Base Pair Repeat Syndromes
FRIEDRICHS ATAXIA
Friedreich ataxia is a rare recessive disorder caused by triplet repeats (GAA) in the mitochondrial frataxin gene characterized by slowly progressive ataxia associated with dysarthria, muscle weakness, spasticity particularly in the lower limbs, scoliosis, bladder dysfunction, absent lower limb reflexes, and loss of position and vibration sense (327). The onset usually occurs before 25 years of age (327). Cardiomyopathy occurs in 2/3 of patients and up to 30% of patients have diabetes (327). The disorder affects approximately 1 in 30,000 Caucasians.
Diabetes occurs in 8-32% of patients with Friedrichs ataxia, and an even higher percentage have impaired glucose tolerance (328,329). Hyperglycemia commonly develops approximately 15 years after the manifestation of neurological symptoms often presenting acutely with patients requiring insulin therapy (328,329). In a number of instances patients present with ketoacidosis (329). Both insulin deficiency and insulin resistance have been reported in patients with Friedreich ataxia (329). It is hypothesized that mutations in frataxin result in alterations in mitochondria function that impair the ability of beta cells to secrete insulin in response to glucose and increase the risk of beta cell death (329). Diabetes is an independent predictor of reduced survival in Friedrichs ataxia (330).
There are no controlled studies comparing different diabetes therapies in patients with Friedreich ataxia. Metformin and thiazolidinediones inhibit the mitochondrial respiratory chain and therefore they should probably be used with caution in patients with mitochondrial disease (329). Additionally, thiazolidinediones increase the risk of congestive heart failure and patients with Friedreich ataxia have a high risk of cardiomyopathies and therefore should be avoided. Insulin is often required to achieve glycemic control.
HUNTINGTON’S DISEASE
Huntington’s disease is an autosomal dominant disorder that begins in adulthood (usually 30-50 years of age) and has distinctive motor defects (chorea, dystonia, and dyskinesia), psychiatric symptoms (depression and anxiety), and cognitive decline (331). This disorder is due to an unstable expansion of CAG repeats in the first exon of the gene that encodes the protein huntingtin (331). The prevalence of this disorder is approximately 5-12 per 100,000 (331,332). While an early study reported that approximately 10% of patients with Huntington’s disease have diabetes a careful review of recent studies reached the conclusion that the prevalence of diabetes in patients with Huntington’s disease is not increased and might actually be decreased (332,333).
MYOTONIC DYSTROPHY
Myotonic dystrophy type 1 is an autosomal-dominantly inherited disease characterized by myotonia, distal muscular dystrophy, cataracts, hypogonadism, and frontal hair loss that occurs in middle age (334). The disease is due to a CTG triplet repeat expansion in the myotonic dystrophy protein kinase gene (334). In genetic new-born screening the estimated prevalence is approximately 1:2100 individuals (335). Diabetes is not a characteristic finding in myotonic dystrophy type 1, but the prevalence is increased 2-4-fold in patients with this disorder compared to the general population (334,336,337). A large study in Korea with 387 patients with myotonic dystrophy type 1 found that 27% had diabetes (338). Patients with myotonic dystrophy type 1 and diabetes have elevated insulin levels suggesting insulin resistance (334,336). Pioglitazone alone and in combination with metformin has been reported to improve glycemic control in patients with myotonic dystrophy and diabetes (339,340).
Obesity Syndromes
BARDET-BIEDLE SYNDROME (BBS)
Bardet-Biedl syndrome (BBS), also earlier referred to as Laurence Moon Biedl syndrome, is a rare autosomal recessive disease with a prevalence of about 1/125,000 (341,342). BBS belongs to the group of ciliopathies characterized by obesity, retinal degeneration, finger anomalies, hypogonadism, renal abnormalities, and intellectual impairment (341,342). It can result from autosomal recessive mutations in at least 26 genes (BBS), which play a key role in the structure and function of cilia (341-343). In a study of 152 patients with BBS it was reported that approximately 75% were obese and the average BMI was 35.7kg/m2 (342). Twenty-five of these patients with BBS had diabetes (16.4%) with 24 having T2D and 1 having T1D (342). Of the 24 patients with T2D six patients were diet controlled, eight were taking metformin, and 10 were on insulin therapy. The mean A1c of subjects with T2D was 7.8 (342). The risk of developing diabetes increases with age. In the BBS patients without diabetes fasting glucose, insulin levels, and HOMA-IR were significantly increased in the BBS group compared with an age and BMI matched control group (342). The metabolic syndrome was present in 54% of the patients with BBS (342). There is evidence that BBS genes affect cilia that alter leptin trafficking and signaling thereby impacting the melanocortin 4 receptor (MC4R) pathway (344). In a small trial setmelanotide reduced body weight (16.3% decrease at 12 months) and hunger in individuals with BBS (345). Other trials also observed a decrease in body weight and hunger in individuals with BBS treated with setmelanotide (346,347). One would anticipate that weight loss will reduce the risk of diabetes on patients with BBS. Setmelanotide is approved by the FDA for the treatment of BBS.
PRADER WILLI SYNDROME (PWS)
PWS is a rare autosomal dominant disorder, affecting 1 in 15,000 newborns, due to a mutation or deletion of several genes in an imprinting region on chromosome 15 (344,348). PWS in children is associated with excessive eating and morbid obesity, hypogonadism, low muscle tone, growth hormone deficiency, and short stature (344,348). The hyperphagia that occurs in PWS is believed to be due to a hypothalamic abnormality resulting in lack of satiety. This leads to excessive obesity in children, which is often associated with T2D due to severe insulin resistance (348). Approximately 20- 25% of adults with PWS have T2D with a mean age of onset of 20 years (348,349). Individuals with PWS who develop early diabetes have severe obesity, a high prevalence of psychiatric and metabolic disorders, and a family history of overweight and T2D (349). Screening for diabetes is recommended annually if obese or beginning in adolescence or with rapid significant weight gain or other symptoms. In recent years the earlier diagnosis and education of parents, use of growth hormone therapy, and the frequency of group homes specific for PWS have led to a reduction in the development of morbid obesity resulting in a decrease in the development of T2D among individuals with PWS (348). Metformin has been shown to be effective in the treatment of PWS patients with diabetes (350). Studies of GLP1 receptor agonists demonstrated lowering of A1c levels and weight loss is some patients (351,352).
ALSTROM SYNDROME
Alstrom syndrome is a rare autosomal recessive disorder with a prevalence of less than one per million characterized by retinal dystrophy, hearing loss, childhood truncal obesity, insulin resistance and hyperinsulinemia, T2D, hypertriglyceridemia, short stature in adulthood, cardiomyopathy, and progressive pulmonary, hepatic, and renal dysfunction (353-355). Symptoms appear in infancy and multi-organ pathology lead to a decreased life expectancy (353,354). The syndrome is caused by mutations in ALMS1, which is a ciliary protein and hence many of the features of Alstrom syndrome resemble those seen in the Bardet-Biedl syndrome (353-355). Diagnosis is confirmed by finding biallelic pathogenic variants in ALMS1 gene.
Severe insulin resistance secondary to abnormalities in GLUT4 trafficking, hyperinsulinemia, and impaired glucose tolerance frequently present in early childhood and are often accompanied by acanthosis nigricans (353,355). T2D develops early in life with a mean age of onset at 16 years (353). In one study 82% of patients with Alstrom syndrome older than 16 years of age had T2D (356). Weight loss with diet, exercise, and medications is indicated (357). Therapy with oral agents, particularly insulin sensitizing agents, may be effective but insulin therapy may be required (353,357). GLP-1 receptor agonists induced weight loss and improved glycemic control (358).
Miscellaneous
WERNER SYNDROME (WS)
Werner syndrome (WS) is an autosomal recessive progeroid syndrome due to biallelic WRN pathogenic variants (359). Clinical features include accelerated aging, short stature, skin atrophy, decreased skeletal muscle mass, hair graying and baldness, partial loss of subcutaneous fat, and cataracts (359). Clinical problems include hypogonadism, osteoporosis, dyslipidemia, atherosclerosis, and cancer (359). Diabetes occurs in 50-75% of patients with WS (360). It is recommended that patients with WS be screened yearly for diabetes (359). The mean age of onset of diabetes is 30-40 years (360). The BMI of most patients with WS is low to normal but patients with diabetes have a higher BMI that is still in the normal range and increased visceral fat (360). It is thought that a decrease in subcutaneous fat leads to visceral fat and insulin resistance ultimately resulting in diabetes. Treatment with thiazolidines (TZDs) or metformin is felt to be beneficial but one needs to balance the beneficial effects of TZDs on glucose metabolism with the risk of osteoporosis associated with TZDs in WS patients who are increased risk for osteoporosis (360). In a case report metreleptin improved glycemia and reduced triglyceride and cholesterol levels (361).
PORPHYRIA
Porphyria cutanea tarda has been associated with diabetes (362), but given that many patients with this disorder also have iron overload, genes for hemochromatosis, and HCV and HIV infection it is very difficult to tell if porphyria cutanea tarda per se is responsible for the association with diabetes (363,364).
ACKNOWLEDGEMENTS
This work was supported by grants from the Northern California Institute for Research and Education.
REFERENCES
- Akinci B, Sahinoz M, Oral E. Lipodystrophy Syndromes: Presentation and Treatment. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Naylor RN, Philipson LH. Diagnosis and Clinical Management of Monogenic Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2020.
- Pham PT, Sidhu HS, Pham PM, Pham PC. Diabetes Mellitus After Solid Organ Transplantation. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Sperling MA, Angelousi A, Yau M. Autoimmune Polyglandular Syndromes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Wang XY, Cleary EM, Thung SF, KK V, Buschur EO. Gestational Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Sarkar S, Brown TT. Lipid Disorders in People with HIV. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2023.
- Unnikrishnan AG, R.C. B, Kumaran S, Kalra S. Fibrocalculous Pancreatic Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Li HZ, Li RY, Li M. A review of maternally inherited diabetes and deafness. Front Biosci (Landmark Ed) 2014; 19:777-782
- Murphy R, Turnbull DM, Walker M, Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med2008; 25:383-399
- Chanoine JP, Thompson DM, Lehman A. Diabetes Mellitus Associated with Maternally Inherited Diabetes and Deafness (MIDD): From Pathogenic Variant to Phenotype. Diabetes 2025; 74:153-163
- Maassen JA, Jahangir Tafrechi RS, Janssen GM, Raap AK, Lemkes HH, t Hart LM. New insights in the molecular pathogenesis of the maternally inherited diabetes and deafness syndrome. Endocrinol Metab Clin North Am2006; 35:385-396, x-xi
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, Gropman AL, Haas R, Hirano M, Morgan P, Sims K, Tarnopolsky M, Van Hove JL, Wolfe L, DiMauro S. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 2015; 17:689-701
- Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev 2011; 32:498-514
- Taylor SI. Lilly Lecture: molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes 1992; 41:1473-1490
- Musso C, Cochran E, Moran SA, Skarulis MC, Oral EA, Taylor S, Gorden P. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore)2004; 83:209-222
- Stears A, O'Rahilly S, Semple RK, Savage DB. Metabolic insights from extreme human insulin resistance phenotypes. Best Pract Res Clin Endocrinol Metab 2012; 26:145-157
- Cochran E, Young JR, Sebring N, DePaoli A, Oral EA, Gorden P. Efficacy of recombinant methionyl human leptin therapy for the extreme insulin resistance of the Rabson-Mendenhall syndrome. J Clin Endocrinol Metab 2004; 89:1548-1554
- Okawa MC, Cochran E, Lightbourne M, Brown RJ. Long-term effects of metreleptin in Rabson-Mendenhall Syndrome on glycemia, growth, and kidney function. J Clin Endocrinol Metab 2021;
- Ewald N, Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur J Intern Med 2013; 24:203-206
- Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, Hardt PD. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev 2012; 28:338-342
- Hart PA, Bellin MD, Andersen DK, Bradley D, Cruz-Monserrate Z, Forsmark CE, Goodarzi MO, Habtezion A, Korc M, Kudva YC, Pandol SJ, Yadav D, Chari ST, Consortium for the Study of Chronic Pancreatitis D, Pancreatic C. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol Hepatol 2016; 1:226-237
- Pendharkar SA, Mathew J, Petrov MS. Age- and sex-specific prevalence of diabetes associated with diseases of the exocrine pancreas: A population-based study. Dig Liver Dis 2017; 49:540-544
- Ewald N, Hardt PD. Diagnosis and treatment of diabetes mellitus in chronic pancreatitis. World J Gastroenterol2013; 19:7276-7281
- Salvatore T, Marfella R, Rizzo MR, Sasso FC. Pancreatic cancer and diabetes: A two-way relationship in the perspective of diabetologist. Int J Surg 2015; 21 Suppl 1:S72-77
- Ebert R, Creutzfeldt W. Reversal of impaired GIP and insulin secretion in patients with pancreatogenic steatorrhea following enzyme substitution. Diabetologia 1980; 19:198-204
- Petrov MS. DIAGNOSIS OF ENDOCRINE DISEASE: Post-pancreatitis diabetes mellitus: prime time for secondary disease. Eur J Endocrinol 2021; 184:R137-R149
- Hart PA, Bradley D, Conwell DL, Dungan K, Krishna SG, Wyne K, Bellin MD, Yadav D, Andersen DK, Serrano J, Papachristou GI. Diabetes following acute pancreatitis. Lancet Gastroenterol Hepatol 2021; 6:668-675
- Zahariev OJ, Bunduc S, Kovacs A, Demeter D, Havelda L, Budai BC, Veres DS, Hosszufalusi N, Eross BM, Teutsch B, Juhasz MF, Hegyi P. Risk factors for diabetes mellitus after acute pancreatitis: a systematic review and meta-analysis. Front Med (Lausanne) 2023; 10:1257222
- Goodarzi MO, Petrov MS, Andersen DK, Hart PA. Diabetes in chronic pancreatitis: risk factors and natural history. Curr Opin Gastroenterol 2021; 37:526-531
- Woodmansey C, McGovern AP, McCullough KA, Whyte MB, Munro NM, Correa AC, Gatenby PAC, Jones SA, de Lusignan S. Incidence, Demographics, and Clinical Characteristics of Diabetes of the Exocrine Pancreas (Type 3c): A Retrospective Cohort Study. Diabetes Care 2017; 40:1486-1493
- Rickels MR, Bellin M, Toledo FG, Robertson RP, Andersen DK, Chari ST, Brand R, Frulloni L, Anderson MA, Whitcomb DC. Detection, evaluation and treatment of diabetes mellitus in chronic pancreatitis: recommendations from PancreasFest 2012. Pancreatology 2013; 13:336-342
- Hardt PD, Krauss A, Bretz L, Porsch-Ozcurumez M, Schnell-Kretschmer H, Maser E, Bretzel RG, Zekhorn T, Klor HU. Pancreatic exocrine function in patients with type 1 and type 2 diabetes mellitus. Acta Diabetol 2000; 37:105-110
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834-838
- Cho J, Scragg R, Pandol SJ, Goodarzi MO, Petrov MS. Antidiabetic Medications and Mortality Risk in Individuals With Pancreatic Cancer-Related Diabetes and Postpancreatitis Diabetes: A Nationwide Cohort Study. Diabetes Care 2019; 42:1675-1683
- Davidsen L, Jensen MH, Cook ME, Vestergaard P, Knop FK, Drewes AM, Olesen SS. Metformin treatment is associated with reduced risk of hypoglycaemia, major adverse cardiovascular events, and all-cause mortality in patients with post-pancreatitis diabetes mellitus: a nationwide cohort study. Eur J Endocrinol 2024; 190:44-53
- Wang Z, Lai ST, Xie L, Zhao JD, Ma NY, Zhu J, Ren ZG, Jiang GL. Metformin is associated with reduced risk of pancreatic cancer in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract 2014; 106:19-26
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993; 328:1433-1437
- Magruder JT, Elahi D, Andersen DK. Diabetes and pancreatic cancer: chicken or egg? Pancreas 2011; 40:339-351
- Cho J, Scragg R, Petrov MS. Postpancreatitis Diabetes Confers Higher Risk for Pancreatic Cancer Than Type 2 Diabetes: Results From a Nationwide Cancer Registry. Diabetes Care 2020; 43:2106-2112
- Roshanov PS, Dennis BB. Incretin-based therapies are associated with acute pancreatitis: Meta-analysis of large randomized controlled trials. Diabetes Res Clin Pract 2015; 110:e13-17
- Tkac I, Raz I. Combined Analysis of Three Large Interventional Trials With Gliptins Indicates Increased Incidence of Acute Pancreatitis in Patients With Type 2 Diabetes. Diabetes Care 2017; 40:284-286
- Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, Pagidipati NJ, Chan JC, Gustavson SM, Iqbal N, Maggioni AP, Ohman P, Poulter NR, Ramachandran A, Zinman B, Hernandez AF, Holman RR. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol 2018; 6:105-113
- Liu Y, Tian Q, Yang J, Wang H, Hong T. No pancreatic safety concern following glucagon-like peptide-1 receptor agonist therapies: A pooled analysis of cardiovascular outcome trials. Diabetes Metab Res Rev 2018; 34:e3061
- Couet C, Genton P, Pointel JP, Louis J, Gross P, Saudax E, Debry G, Drouin P. The prevalence of retinopathy is similar in diabetes mellitus secondary to chronic pancreatitis with or without pancreatectomy and in idiopathic diabetes mellitus. Diabetes Care 1985; 8:323-328
- Tiengo A, Segato T, Briani G, Setti A, Del Prato S, Devide A, Padovan D, Virgili F, Crepaldi G. The presence of retinopathy in patients with secondary diabetes following pancreatectomy or chronic pancreatitis. Diabetes Care1983; 6:570-574
- Briani G, Riva F, Midena E, Trevisan R, Sgnaolin E, Jori E, Munari R, Bruttomesso D, Segato T, Tiengo A. Prevalence of microangiopathic complications in hyperglycemia secondary to pancreatic disease. J Diabet Complications 1988; 2:50-52
- Ziegler O, Candiloros H, Guerci B, Got I, Crea T, Drouin P. Lower-extremity arterial disease in diabetes mellitus due to chronic pancreatitis. Diabete Metab 1994; 20:540-545
- Kuo P, Stevens JE, Russo A, Maddox A, Wishart JM, Jones KL, Greville H, Hetzel D, Chapman I, Horowitz M, Rayner CK. Gastric emptying, incretin hormone secretion, and postprandial glycemia in cystic fibrosis--effects of pancreatic enzyme supplementation. J Clin Endocrinol Metab 2011; 96:E851-855
- Imamura S, Niwano F, Babaya N, Hiromine Y, Matsumoto I, Kamei K, Yoshida Y, Taketomo Y, Yoshida S, Takeyama Y, Noso S, Maeda N, Ikegami H. High Incidence of Diabetes Mellitus After Distal Pancreatectomy and Its Predictors: A Long-term Follow-up Study. J Clin Endocrinol Metab 2024; 109:619-630
- Klotz R, Schilling C, Kuner C, Hinz U, Klaiber U, Holze M, Tjaden C, Loos M, Buchler MW, Hackert T. Central pancreatectomy prevents postoperative diabetes. J Hepatobiliary Pancreat Sci 2023; 30:951-961
- Mezza T, Cefalo CMA, Cinti F, Quero G, Pontecorvi A, Alfieri S, Holst JJ, Giaccari A. Endocrine and Metabolic Insights from Pancreatic Surgery. Trends Endocrinol Metab 2020; 31:760-772
- Suto H, Kamei K, Kato H, Misawa T, Unno M, Nitta H, Satoi S, Kawabata Y, Ohtsuka M, Rikiyama T, Sudo T, Matsumoto I, Hirao T, Okano K, Suzuki Y, Sata N, Isaji S, Sugiyama M, Takeyama Y. Diabetic control and nutritional status up to 1 year after total pancreatectomy: a nationwide multicentre prospective study. Br J Surg2021; 108:e237-e238
- Baekdal M, Nielsen SW, Hansen CP, Storkholm JH, van Hall G, Hartmann B, Holst JJ, Vilsboll T, Lund A, Knop FK. Empagliflozin Normalizes Fasting Hyperglycemia and Improves Postprandial Glucose Tolerance in Totally Pancreatectomized Patients: A Randomized, Double-Blind, Placebo-Controlled Crossover Study. Diabetes Care2024; 47:71-80
- Juel CTB, Lund A, Andersen MM, Hansen CP, Storkholm JH, Rehfeld JF, van Hall G, Hartmann B, Wewer Albrechtsen NJ, Holst JJ, Vilsboll T, Knop FK. The GLP-1 receptor agonist lixisenatide reduces postprandial glucose in patients with diabetes secondary to total pancreatectomy: a randomised, placebo-controlled, double-blinded crossover trial. Diabetologia 2020; 63:1285-1298
- Chaouch MA, Leon P, Cassese G, Aguilhon C, Khayat S, Panaro F. Total pancreatectomy with intraportal islet autotransplantation for pancreatic malignancies: a literature overview. Expert Opin Biol Ther 2021:1-7
- Robertson GS, Dennison AR, Johnson PR, London NJ. A review of pancreatic islet autotransplantation. Hepatogastroenterology 1998; 45:226-235
- Jablonska B, Mrowiec S. Total Pancreatectomy with Autologous Islet Cell Transplantation-The Current Indications. J Clin Med 2021; 10
- Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, Chari ST, Abbruzzese JL. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017; 66:1103-1110
- Aggarwal G, Kamada P, Chari ST. Prevalence of diabetes mellitus in pancreatic cancer compared to common cancers. Pancreas 2013; 42:198-201
- Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008; 134:981-987
- Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M, Petersen GM. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005; 129:504-511
- Damiano J, Bordier L, Le Berre JP, Margery J, Dupuy O, Mayaudon H, Bauduceau B. Should pancreas imaging be recommanded in patients over 50 years when diabetes is discovered because of acute symptoms? Diabetes Metab 2004; 30:203-207
- Hart PA, Kamada P, Rabe KG, Srinivasan S, Basu A, Aggarwal G, Chari ST. Weight loss precedes cancer-specific symptoms in pancreatic cancer-associated diabetes mellitus. Pancreas 2011; 40:768-772
- Song S, Wang B, Zhang X, Hao L, Hu X, Li Z, Sun S. Long-Term Diabetes Mellitus Is Associated with an Increased Risk of Pancreatic Cancer: A Meta-Analysis. PLoS One 2015; 10:e0134321
- Roy A, Sahoo J, Kamalanathan S, Naik D, Mohan P, Kalayarasan R. Diabetes and pancreatic cancer: Exploring the two-way traffic. World J Gastroenterol 2021; 27:4939-4962
- Permert J, Ihse I, Jorfeldt L, von Schenck H, Arnquist HJ, Larsson J. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg 1993; 80:1047-1050
- Litwin J, Dobrowolski S, Orlowska-Kunikowska E, Sledzinski Z. Changes in glucose metabolism after Kausch-Whipple pancreatectomy in pancreatic cancer and chronic pancreatitis patients. Pancreas 2008; 36:26-30
- Gardner TB, Hessami N, Smith KD, Ripple GH, Barth RJ, Klibansky DA, Colacchio TA, Zaki B, Tsapakos MJ, Suriawinata AA, Putra J, Tsongalis GJ, Mody K, Gordon SR, Pipas JM. The effect of neoadjuvant chemoradiation on pancreatic cancer-associated diabetes mellitus. Pancreas 2014; 43:1018-1021
- Zhou PT, Li B, Liu FR, Zhang MC, Wang Q, Li YY, Xu C, Liu YH, Yao Y, Li D. Metformin is associated with survival benefit in pancreatic cancer patients with diabetes: a systematic review and meta-analysis. Oncotarget2017; 8:25242-25250
- Jian-Yu E, Graber JM, Lu SE, Lin Y, Lu-Yao G, Tan XL. Effect of Metformin and Statin Use on Survival in Pancreatic Cancer Patients: a Systematic Literature Review and Meta-analysis. Curr Med Chem 2018; 25:2595-2607
- Wan G, Sun X, Li F, Wang X, Li C, Li H, Yu X, Cao F. Survival Benefit of Metformin Adjuvant Treatment For Pancreatic Cancer Patients: a Systematic Review and Meta-Analysis. Cell Physiol Biochem 2018; 49:837-847
- Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R, Liberati D, Pasquale V, Scavini M, Maggiora P, Sordi V, Lampasona V, Ceraulo D, Di Terlizzi G, Doglioni C, Falconi M, Piemonti L. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin Cancer Res 2016; 22:1076-1085
- Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A, Punt CJ, Richel DJ, Wilmink JW. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol 2015; 16:839-847
- Kawabata H. The mechanisms of systemic iron homeostasis and etiology, diagnosis, and treatment of hereditary hemochromatosis. Int J Hematol 2018; 107:31-43
- Pelusi C, Gasparini DI, Bianchi N, Pasquali R. Endocrine dysfunction in hereditary hemochromatosis. J Endocrinol Invest 2016; 39:837-847
- Barton JC, Acton RT. Diabetes in HFE Hemochromatosis. J Diabetes Res 2017; 2017:9826930
- O'Sullivan EP, McDermott JH, Murphy MS, Sen S, Walsh CH. Declining prevalence of diabetes mellitus in hereditary haemochromatosis--the result of earlier diagnosis. Diabetes Res Clin Pract 2008; 81:316-320
- McClain DA, Abraham D, Rogers J, Brady R, Gault P, Ajioka R, Kushner JP. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 2006; 49:1661-1669
- Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab 2013; 17:329-341
- Toumba M, Sergis A, Kanaris C, Skordis N. Endocrine complications in patients with Thalassaemia Major. Pediatr Endocrinol Rev 2007; 5:642-648
- Gamberini MR, De Sanctis V, Gilli G. Hypogonadism, diabetes mellitus, hypothyroidism, hypoparathyroidism: incidence and prevalence related to iron overload and chelation therapy in patients with thalassaemia major followed from 1980 to 2007 in the Ferrara Centre. Pediatr Endocrinol Rev 2008; 6 Suppl 1:158-169
- Nelson RL, Baldus WP, Rubenstein AH, Go VL, Service FJ. Pancreatic alpha-cell function in diabetic hemochromatotic subjects. J Clin Endocrinol Metab 1979; 49:412-416
- Passa P, Luyckx AS, Carpentier JL, Lefebvre PJ, Canivet J. Glucagon secretion in diabetic patients with idiopathic haemochromatosis. Diabetologia 1977; 13:509-513
- Lassmann V, Passa P, Vague P, Vialettes B. Pancreatic polypeptide secretion in diabetic patients with idiopathic haemochromatosis. Diabete Metab 1987; 13:106-109
- Dymock IW, Cassar J, Pyke DA, Oakley WG, Williams R. Observations on the pathogenesis, complications and treatment of diabetes in 115 cases of haemochromatosis. Am J Med 1972; 52:203-210
- Griffiths JD, Dymock IW, Davies EW, Hill DW, Williams R. Occurrence and prevalence of diabetic retinopathy in hemochromatosis. Diabetes 1971; 20:766-770
- Becker D, Miller M. Presence of diabetic glomerulosclerosis in patients with hemochromatosis. N Engl J Med1960; 263:367-373
- Hatunic M, Finucane FM, Norris S, Pacini G, Nolan JJ. Glucose metabolism after normalization of markers of iron overload by venesection in subjects with hereditary hemochromatosis. Metabolism 2010; 59:1811-1815
- Buysschaert M, Paris I, Selvais P, Hermans MP. Clinical aspects of diabetes secondary to idiopathic haemochromatosis in French-speaking Belgium. Diabetes Metab 1997; 23:308-313
- Bomford A, Williams R. Long term results of venesection therapy in idiopathic haemochromatosis. Q J Med 1976; 45:611-623
- Sferra TJ, Collins FS. The molecular biology of cystic fibrosis. Annu Rev Med 1993; 44:133-144
- Moran A, Becker D, Casella SJ, Gottlieb PA, Kirkman MS, Marshall BC, Slovis B, Committee CCC. Epidemiology, pathophysiology, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabetes Care2010; 33:2677-2683
- Yoon JC. Evolving Mechanistic Views and Emerging Therapeutic Strategies for Cystic Fibrosis-Related Diabetes. J Endocr Soc 2017; 1:1386-1400
- Kayani K, Mohammed R, Mohiaddin H. Cystic Fibrosis-Related Diabetes. Front Endocrinol (Lausanne) 2018; 9:20
- Bridges N, Rowe R, Holt RIG. Unique challenges of cystic fibrosis-related diabetes. Diabet Med 2018;
- Draznin B, Aroda VR, Bakris G, Benson G, Brown FM, Freeman R, Green J, Huang E, Isaacs D, Kahan S, Leon J, Lyons SK, Peters AL, Prahalad P, Reusch JEB, Young-Hyman D, Das S, Kosiborod M. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022; 45:S17-S38
- Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, Robinson KA, Sabadosa KA, Stecenko A, Slovis B. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 2010; 33:2697-2708
- Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care 2009; 32:1626-1631
- Santhakumar A, Lewis F, Pickles J, Winterbottom H, Punt S, Beynon J, Tofeec K, Barry P, Brennan A. Role for DPP4 inhibitor therapy in cystic fibrosis related diabetes: A single centre experience. J Cyst Fibros 2024; 23:853-856
- Hayes D, Jr., McCoy KS, Sheikh SI. Resolution of cystic fibrosis-related diabetes with ivacaftor therapy. Am J Respir Crit Care Med 2014; 190:590-591
- Gaines H, Jones KR, Lim J, Medhi NF, Chen S, Scofield RH. Effect of CFTR modulator therapy on cystic fibrosis-related diabetes. J Diabetes Complications 2021; 35:107845
- Volkova N, Moy K, Evans J, Campbell D, Tian S, Simard C, Higgins M, Konstan MW, Sawicki GS, Elbert A, Charman SC, Marshall BC, Bilton D. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J Cyst Fibros 2020; 19:68-79
- Aguilera Garcia I, Garcia Moreno RM, Lopez Plaza B, Barquiel Alcala B, Vazquez Perez P, Barreda Bonis AC, Zamarron de Lucas E, Palma Milla S, Prados Sanchez MC, Gonzalez Perez de Villar N. Impact of triple transmembrane regulator therapy on glucose metabolism in cystic fibrosis related diabetes during clinical practice. Diabetes Res Clin Pract 2024; 216:111839
- Taelman V, Declercq D, Van Biervliet S, Weygaerde YV, Lapauw B, Van Braeckel E. Effect of 18 months elexacaftor-tezacaftor-ivacaftor on body mass index and glycemic control in adults with cystic fibrosis. Clin Nutr ESPEN 2023; 58:73-78
- Nielsen BU, Olsen MF, Mabuza Mathiesen IH, Pressler T, Ritz C, Katzenstein TL, Olesen HV, Skov M, Jensen-Fangel S, Almdal TP, Faurholt-Jepsen D. Decline in HbA1c during the first year of elexacaftor/tezacaftor/ivacaftor treatment in the Danish cystic fibrosis cohort: Short title: Decline in HbA1c after elexacaftor/tezacaftor/ivacaftor treatment. J Cyst Fibros 2024; 23:103-108
- Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, Moran A. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatr Diabetes 2013; 14:417-421
- Lurquin F, Buysschaert M, Preumont V. Advances in cystic fibrosis-related diabetes: Current status and future directions. Diabetes Metab Syndr 2023; 17:102899
- Richardson SJ, Morgan NG. Enteroviral infections in the pathogenesis of type 1 diabetes: new insights for therapeutic intervention. Curr Opin Pharmacol 2018; 43:11-19
- Principi N, Berioli MG, Bianchini S, Esposito S. Type 1 diabetes and viral infections: What is the relationship? J Clin Virol 2017; 96:26-31
- Rodriguez-Calvo T, Sabouri S, Anquetil F, von Herrath MG. The viral paradigm in type 1 diabetes: Who are the main suspects? Autoimmun Rev 2016; 15:964-969
- Jeremiah SS, Moin ASM, Butler AE. Virus-induced diabetes mellitus: revisiting infection etiology in light of SARS-CoV-2. Metabolism 2024; 156:155917
- VanBuecken D, Lord S, Greenbaum CJ. Changing the Course of Disease in Type 1 Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Krogvold L, Mynarek IM, Ponzi E, Mork FB, Hessel TW, Roald T, Lindblom N, Westman J, Barker P, Hyoty H, Ludvigsson J, Hanssen KF, Johannesen J, Dahl-Jorgensen K. Pleconaril and ribavirin in new-onset type 1 diabetes: a phase 2 randomized trial. Nat Med 2023; 29:2902-2908
- Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med 1979; 300:1173-1179
- Gale EA. Congenital rubella: citation virus or viral cause of type 1 diabetes? Diabetologia 2008; 51:1559-1566
- Burgess MA, Forrest JM. Congenital rubella and diabetes mellitus. Diabetologia 2009; 52:369-370; author reply 373
- White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol 2008; 49:831-844
- Fabiani S, Fallahi P, Ferrari SM, Miccoli M, Antonelli A. Hepatitis C virus infection and development of type 2 diabetes mellitus: Systematic review and meta-analysis of the literature. Rev Endocr Metab Disord 2018; 19:405-420
- Choi HY, Kim Y, Cho H, Kim BH, Ki M. Risk of diabetes in viral hepatitis B or C patients compared to that in noninfected individuals in Korea, 2002-2013: A population-based cohort study. J Viral Hepat 2018; 25:272-280
- Naing C, Mak JW, Ahmed SI, Maung M. Relationship between hepatitis C virus infection and type 2 diabetes mellitus: meta-analysis. World J Gastroenterol 2012; 18:1642-1651
- Lin YJ, Shaw TG, Yang HI, Lu SN, Jen CL, Wang LY, Wong KH, Chan SY, Yuan Y, L'Italien G, Chen CJ, Lee MH, Group REVEALHS. Chronic hepatitis C virus infection and the risk for diabetes: a community-based prospective study. Liver Int 2017; 37:179-186
- Gastaldi G, Goossens N, Clement S, Negro F. Current level of evidence on causal association between hepatitis C virus and type 2 diabetes: A review. J Adv Res 2017; 8:149-159
- Guo X, Jin M, Yang M, Liu K, Li JW. Type 2 diabetes mellitus and the risk of hepatitis C virus infection: a systematic review. Sci Rep 2013; 3:2981
- Li X, Gao Y, Xu H, Hou J, Gao P. Diabetes mellitus is a significant risk factor for the development of liver cirrhosis in chronic hepatitis C patients. Sci Rep 2017; 7:9087
- Li X, Xu H, Gao Y, Pan M, Wang L, Gao P. Diabetes mellitus increases the risk of hepatocellular carcinoma in treatment-naive chronic hepatitis C patients in China. Medicine (Baltimore) 2017; 96:e6508
- Cao LH, Lu FM, Lu XJ, Zhu LY. Study on the relationship between insulin growth factor 1 and liver fibrosis in patients with chronic hepatitis C with type 2 diabetes mellitus. J Cell Biochem 2018; 119:9513-9518
- Colaci M, Malatino L, Antonelli A, Fallahi P, Giuggioli D, Ferri C. Endocrine disorders associated with hepatitis C virus chronic infection. Rev Endocr Metab Disord 2018; 19:397-403
- Vanni E, Bugianesi E, Saracco G. Treatment of type 2 diabetes mellitus by viral eradication in chronic hepatitis C: Myth or reality? Dig Liver Dis 2016; 48:105-111
- Delgado-Borrego A, Jordan SH, Negre B, Healey D, Lin W, Kamegaya Y, Christofi M, Ludwig DA, Lok AS, Chung RT. Reduction of insulin resistance with effective clearance of hepatitis C infection: results from the HALT-C trial. Clin Gastroenterol Hepatol 2010; 8:458-462
- Narita R, Abe S, Kihara Y, Akiyama T, Tabaru A, Otsuki M. Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J Hepatol 2004; 41:132-138
- Hum J, Jou JH, Green PK, Berry K, Lundblad J, Hettinger BD, Chang M, Ioannou GN. Improvement in Glycemic Control of Type 2 Diabetes After Successful Treatment of Hepatitis C Virus. Diabetes Care 2017; 40:1173-1180
- Hsu YC, Lin JT, Ho HJ, Kao YH, Huang YT, Hsiao NW, Wu MS, Liu YY, Wu CY. Antiviral treatment for hepatitis C virus infection is associated with improved renal and cardiovascular outcomes in diabetic patients. Hepatology2014; 59:1293-1302
- Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol 2012; 107:46-52
- Nkontchou G, Cosson E, Aout M, Mahmoudi A, Bourcier V, Charif I, Ganne-Carrie N, Grando-Lemaire V, Vicaut E, Trinchet JC, Beaugrand M. Impact of metformin on the prognosis of cirrhosis induced by viral hepatitis C in diabetic patients. J Clin Endocrinol Metab 2011; 96:2601-2608
- Zhou K, Lansang MC. Diabetes Mellitus and Infection. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Khunti K, Del Prato S, Mathieu C, Kahn SE, Gabbay RA, Buse JB. COVID-19, Hyperglycemia, and New-Onset Diabetes. Diabetes Care 2021; 44:2645-2655
- Hirani D, Salem V, Khunti K, Misra S. Newly detected diabetes during the COVID-19 pandemic: What have we learnt? Best Pract Res Clin Endocrinol Metab 2023; 37:101793
- Zhou J, Wang Y, Xu R. Association of COVID-19 infection and the risk of new incident diabetes: a systematic review and meta-analysis. Front Endocrinol (Lausanne) 2024; 15:1429848
- Sathish T, Kapoor N, Cao Y, Tapp RJ, Zimmet P. Proportion of newly diagnosed diabetes in COVID-19 patients: A systematic review and meta-analysis. Diabetes Obes Metab 2021; 23:870-874
- Zhang T, Mei Q, Zhang Z, Walline JH, Liu Y, Zhu H, Zhang S. Risk for newly diagnosed diabetes after COVID-19: a systematic review and meta-analysis. BMC Med 2022; 20:444
- Lai H, Yang M, Sun M, Pan B, Wang Q, Wang J, Tian J, Ding G, Yang K, Song X, Ge L. Risk of incident diabetes after COVID-19 infection: A systematic review and meta-analysis. Metabolism 2022; 137:155330
- Wong R, Lam E, Bramante CT, Johnson SG, Reusch J, Wilkins KJ, Yeh HC. Does COVID-19 Infection Increase the Risk of Diabetes? Current Evidence. Curr Diab Rep 2023; 23:207-216
- Taylor K, Eastwood S, Walker V, Cezard G, Knight R, Al Arab M, Wei Y, Horne EMF, Teece L, Forbes H, Walker A, Fisher L, Massey J, Hopcroft LEM, Palmer T, Cuitun Coronado J, Ip S, Davy S, Dillingham I, Morton C, Greaves F, Macleod J, Goldacre B, Wood A, Chaturvedi N, Sterne JAC, Denholm R. Incidence of diabetes after SARS-CoV-2 infection in England and the implications of COVID-19 vaccination: a retrospective cohort study of 16 million people. Lancet Diabetes Endocrinol 2024; 12:558-568
- Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet 2009; 373:1798-1807
- Harris MI. Undiagnosed NIDDM: clinical and public health issues. Diabetes Care 1993; 16:642-652
- Montefusco L, Ben Nasr M, D'Addio F, Loretelli C, Rossi A, Pastore I, Daniele G, Abdelsalam A, Maestroni A, Dell'Acqua M, Ippolito E, Assi E, Usuelli V, Seelam AJ, Fiorina RM, Chebat E, Morpurgo P, Lunati ME, Bolla AM, Finzi G, Abdi R, Bonventre JV, Rusconi S, Riva A, Corradi D, Santus P, Nebuloni M, Folli F, Zuccotti GV, Galli M, Fiorina P. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat Metab 2021; 3:774-785
- Martinez-Colon GJ, Ratnasiri K, Chen H, Jiang S, Zanley E, Rustagi A, Verma R, Chen H, Andrews JR, Mertz KD, Tzankov A, Azagury D, Boyd J, Nolan GP, Schurch CM, Matter MS, Blish CA, McLaughlin TL. SARS-CoV-2 infection drives an inflammatory response in human adipose tissue through infection of adipocytes and macrophages. Sci Transl Med 2022; 14:eabm9151
- Reiterer M, Rajan M, Gomez-Banoy N, Lau JD, Gomez-Escobar LG, Ma L, Gilani A, Alvarez-Mulett S, Sholle ET, Chandar V, Bram Y, Hoffman K, Bhardwaj P, Piloco P, Rubio-Navarro A, Uhl S, Carrau L, Houhgton S, Redmond D, Shukla AP, Goyal P, Brown KA, tenOever BR, Alonso LC, Schwartz RE, Schenck EJ, Safford MM, Lo JC. Hyperglycemia in acute COVID-19 is characterized by insulin resistance and adipose tissue infectivity by SARS-CoV-2. Cell Metab 2021; 33:2174-2188 e2175
- Valabhji J, Barron E, Bradley D, Bakhai C, Khunti K, Jebb S. Effect of the COVID-19 pandemic on body weight in people at high risk of type 2 diabetes referred to the English NHS Diabetes Prevention Programme. Lancet Diabetes Endocrinol 2021; 9:649-651
- Carroll PV, MN J. Acromegaly. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Hannon AM, Thompson CJ, Sherlock M. Diabetes in Patients With Acromegaly. Curr Diab Rep 2017; 17:8
- Baroni MG, Giorgino F, Pezzino V, Scaroni C, Avogaro A. Italian Society for the Study of Diabetes (SID)/Italian Endocrinological Society (SIE) guidelines on the treatment of hyperglycemia in Cushing's syndrome and acromegaly. J Endocrinol Invest 2016; 39:235-255
- Esposito D, Boguszewski CL, Colao A, Fleseriu M, Gatto F, Jorgensen JOL, Ragnarsson O, Ferone D, Johannsson G. Diabetes mellitus in patients with acromegaly: pathophysiology, clinical challenges and management. Nat Rev Endocrinol 2024; 20:541-552
- Frara S, Maffezzoni F, Mazziotti G, Giustina A. Current and Emerging Aspects of Diabetes Mellitus in Acromegaly. Trends Endocrinol Metab 2016; 27:470-483
- Alexopoulou O, Bex M, Kamenicky P, Mvoula AB, Chanson P, Maiter D. Prevalence and risk factors of impaired glucose tolerance and diabetes mellitus at diagnosis of acromegaly: a study in 148 patients. Pituitary 2014; 17:81-89
- Lim CT, Khoo B. Normal Physiology of ACTH and GH Release in the Hypothalamus and Anterior Pituitary in Man. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2025.
- Yuen KCJ. Growth hormone Stimulation Tests in Assessing Adult Growth Hormone Deficiency. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2019.
- Olarescu N, Gunawardane K, Hansen T, Møller N, Jørgensen J. Normal Physiology of Growth Hormone in Adults. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2019.
- Dutta P, Hajela A, Gupta P, Rai A, Sachdeva N, Mukherjee KK, Pathak A, Dhandapani S. The predictors of recovery from diabetes mellitus following neurosurgical treatment of acromegaly: A prospective study over a decade. Neurol India 2019; 67:757-762
- Mazziotti G, Floriani I, Bonadonna S, Torri V, Chanson P, Giustina A. Effects of somatostatin analogs on glucose homeostasis: a metaanalysis of acromegaly studies. J Clin Endocrinol Metab 2009; 94:1500-1508
- Cozzolino A, Feola T, Simonelli I, Puliani G, Pozza C, Giannetta E, Gianfrilli D, Pasqualetti P, Lenzi A, Isidori AM. Somatostatin Analogs and Glucose Metabolism in Acromegaly: A Meta-analysis of Prospective Interventional Studies. J Clin Endocrinol Metab 2018;
- Gadelha MR, Bronstein MD, Brue T, Coculescu M, Fleseriu M, Guitelman M, Pronin V, Raverot G, Shimon I, Lievre KK, Fleck J, Aout M, Pedroncelli AM, Colao A. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol 2014; 2:875-884
- Schmid HA, Brue T, Colao A, Gadelha MR, Shimon I, Kapur K, Pedroncelli AM, Fleseriu M. Effect of pasireotide on glucose- and growth hormone-related biomarkers in patients with inadequately controlled acromegaly. Endocrine 2016; 53:210-219
- Gatto F, Arecco A, Amaru J, Arvigo M, Campana C, Milioto A, Esposito D, Johannsson G, Cocchiara F, Maggi DC, Ferone D, Puddu A. Differential Impact of Medical Therapies for Acromegaly on Glucose Metabolism. Int J Mol Sci 2025; 26
- Varaldo E, Prencipe N, Bona C, Cuboni D, Aversa LS, Sibilla M, Bioletto F, Berton AM, Gramaglia C, Gasco V, Ghigo E, Grottoli S. Effect of Cabergoline on weight and glucose metabolism in patients with acromegaly. J Endocrinol Invest 2024; 47:3019-3028
- Wass JA, Cudworth AG, Bottazzo GF, Woodrow JC, Besser GM. An assessment of glucose intolerance in acromegaly and its response to medical treatment. Clin Endocrinol (Oxf) 1980; 12:53-59
- Urbani C, Sardella C, Calevro A, Rossi G, Scattina I, Lombardi M, Lupi I, Manetti L, Martino E, Bogazzi F. Effects of medical therapies for acromegaly on glucose metabolism. Eur J Endocrinol 2013; 169:99-108
- Brue T, Lindberg A, Jan van der Lely A, Akerblad AC, Koltowska-Haggstrom M, Gomez R, Droste M, Hey-Hadavi J, Strasburger CJ, Camacho-Hubner C. Diabetes in patients with acromegaly treated with pegvisomant: observations from acrostudy. Endocrine 2019; 63:563-572
- Juszczak A, Morris D, Grossman A. Cushing's Syndrome. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Ferrau F, Korbonits M. Metabolic Syndrome in Cushing's Syndrome Patients. Front Horm Res 2018; 49:85-103
- Valassi E, Santos A, Yaneva M, Toth M, Strasburger CJ, Chanson P, Wass JA, Chabre O, Pfeifer M, Feelders RA, Tsagarakis S, Trainer PJ, Franz H, Zopf K, Zacharieva S, Lamberts SW, Tabarin A, Webb SM. The European Registry on Cushing's syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol 2011; 165:383-392
- Scaroni C, Zilio M, Foti M, Boscaro M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr Rev 2017; 38:189-219
- Guarnotta V, Amato MC, Pivonello R, Arnaldi G, Ciresi A, Trementino L, Citarrella R, Iacuaniello D, Michetti G, Simeoli C, Colao A, Giordano C. The degree of urinary hypercortisolism is not correlated with the severity of cushing's syndrome. Endocrine 2017; 55:564-572
- Steffensen C, Pereira AM, Dekkers OM, Jorgensen JO. DIAGNOSIS OF ENDOCRINE DISEASE: Prevalence of hypercortisolism in type 2 diabetes patients: a systematic review and meta-analysis. Eur J Endocrinol 2016; 175:R247-R253
- Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, Limone P, Crivellaro C, Martinelli I, Montini M, Disoteo O, Ambrosi B, Lanzi R, Arosio M, Senni S, Balestrieri A, Solaroli E, Madeo B, De Giovanni R, Strollo F, Battista R, Scorsone A, Giagulli VA, Collura D, Scillitani A, Cozzi R, Faustini-Fustini M, Pia A, Rinaldi R, Allasino B, Peraga G, Tassone F, Garofalo P, Papini E, Borretta G. Screening of Cushing's syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab 2012; 97:3467-3475
- Aresta C, Favero V, Morelli V, Giovanelli L, Parazzoli C, Falchetti A, Pugliese F, Gennari L, Vescini F, Salcuni A, Scillitani A, Persani L, Chiodini I. Cardiovascular complications of mild autonomous cortisol secretion. Best Pract Res Clin Endocrinol Metab 2021; 35:101494
- Elhassan YS, Alahdab F, Prete A, Delivanis DA, Khanna A, Prokop L, Murad MH, O'Reilly MW, Arlt W, Bancos I. Natural History of Adrenal Incidentalomas With and Without Mild Autonomous Cortisol Excess: A Systematic Review and Meta-analysis. Ann Intern Med 2019; 171:107-116
- Toniato A, Merante-Boschin I, Opocher G, Pelizzo MR, Schiavi F, Ballotta E. Surgical versus conservative management for subclinical Cushing syndrome in adrenal incidentalomas: a prospective randomized study. Ann Surg 2009; 249:388-391
- Sereg M, Szappanos A, Toke J, Karlinger K, Feldman K, Kaszper E, Varga I, Glaz E, Racz K, Toth M. Atherosclerotic risk factors and complications in patients with non-functioning adrenal adenomas treated with or without adrenalectomy: a long-term follow-up study. Eur J Endocrinol 2009; 160:647-655
- Chatzellis E, Kaltsas G. Adrenal Incidentalomas. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Babic N, Yeo K, Hannoush Z, Weiss R. Endocrine Testing Protocols: Hypothalamic Pituitary Adrenal Axis. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2023.
- Gremlich S, Roduit R, Thorens B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells. Comparison with the effects of fatty acids. J Biol Chem 1997; 272:3216-3222
- Mehlich A, Bolanowski M, Mehlich D, Witek P. Medical treatment of Cushing's disease with concurrent diabetes mellitus. Front Endocrinol (Lausanne) 2023; 14:1174119
- Nicolaides NC, Pavlaki AN, Maria Alexandra MA, Chrousos GP. Glucocorticoid Therapy and Adrenal Suppression. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2018.
- Juszczak A, Sulentic P, Grossman A. Cushing's Syndrome. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2004.
- Feingold K. The Effect of Endocrine Disorders on Lipids and Lipoproteins. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Pacak K, Tella SH. Pheochromocytoma and Paraganglioma. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2018.
- Adlan MA, Bondugulapati LN, Premawardhana LD. Glucose intolerance and diabetes mellitus in endocrine disorders - two case reports and a review. Curr Diabetes Rev 2010; 6:266-273
- La Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens 2003; 21:1703-1707
- Beninato T, Kluijfhout WP, Drake FT, Lim J, Kwon JS, Xiong M, Shen WT, Gosnell JE, Liu C, Suh I, Duh QY. Resection of Pheochromocytoma Improves Diabetes Mellitus in the Majority of Patients. Ann Surg Oncol 2017; 24:1208-1213
- Zhang W, Yu J, Chen Y, Zhou Y, Cui Y, Li T, Wang Y, Ren W, Tong A, Li Y. Glucose Disorders in Patients With Pheochromocytoma/Paraganglioma: Profile and Influence Effects in a Large Cohort With 705 Patients. Endocr Pract 2024;
- Douma S, Petidis K, Kartali N, Mahera K, Sabanis C, Zamboulis C. Pheochromocytoma presenting as diabetic ketoacidosis. J Diabetes Complications 2008; 22:295-296
- Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest 1980; 65:717-721
- Rizza RA, Cryer PE, Haymond MW, Gerich JE. Adrenergic mechanisms for the effects of epinephrine on glucose production and clearance in man. J Clin Invest 1980; 65:682-689
- Halter JB, Beard JC, Porte D, Jr. Islet function and stress hyperglycemia: plasma glucose and epinephrine interaction. Am J Physiol 1984; 247:E47-52
- Araujo-Castro M, Minguez Ojeda C, Garcia Centeno R, Lopez-Garcia MC, Lamas C, Hanzu FA, Mora M, Del Castillo Tous M, Rodriguez de Vera Gomez P, Parra Ramirez P, Alvarez-Escola C, Blanco Carrera C, Barahona San Millan R, Recasens M, Valdes N, Gracia Gimeno P, de Miguel Novoa P, Vicente A, Manjon L, Garcia Sanz I, Michalopoulou T, Calatayud M. Glycemic disorders in patients with pheochromocytomas and sympathetic paragangliomas. Endocr Relat Cancer 2022; 29:645-655
- Khatiwada S, Agarwal S, Kandasamy D, Jyotsna VP, Kumar R, Kumar Bansal V, Pandey RM, Gupta N, Tandon N. Diabetes mellitus in pheochromocytoma and paraganglioma: Prevalence, dynamics of insulin secretion / sensitivity and predictors of remission. Diabetes Metab Syndr 2020; 14:2169-2175
- Chen Y, Hodin RA, Pandolfi C, Ruan DT, McKenzie TJ. Hypoglycemia after resection of pheochromocytoma. Surgery 2014; 156:1404-1408; discussion 1408-1409
- Diamanti-Kandarakis E, Zapanti E, Peridis MH, Ntavos P, Mastorakos G. Insulin resistance in pheochromocytoma improves more by surgical rather than by medical treatment. Hormones (Athens) 2003; 2:61-66
- Roa Duenas OH, Van der Burgh AC, Ittermann T, Ligthart S, Ikram MA, Peeters R, Chaker L. Thyroid Function and the Risk of Prediabetes and Type 2 Diabetes. J Clin Endocrinol Metab 2022; 107:1789-1798
- Nishi M. Diabetes mellitus and thyroid diseases. Diabetol Int 2018; 9:108-112
- Andersen OO, Friis T, Ottesen B. Glucose tolerance and insulin secretion in hyperthyroidism. Acta Endocrinol (Copenh) 1977; 84:576-587
- Kreines K, Jett M, Knowles HC, Jr. Observations in hyperthyroidism of abnormal glucose tolerance and other traits related to diabetes mellitus. Diabetes 1965; 14:740-744
- Yang L, Shen X, Yan S, Yuan X, Lu J, Wei W. HbA1c in the diagnosis of diabetes and abnormal glucose tolerance in patients with Graves' hyperthyroidism. Diabetes Res Clin Pract 2013; 101:28-34
- Alwan H, Villoz F, Feller M, Dullaart RPF, Bakker SJL, Peeters RP, Kavousi M, Bauer DC, Cappola AR, Yeap BB, Walsh JP, Brown SJ, Ceresini G, Ferrucci L, Gussekloo J, Trompet S, Iacoviello M, Moon JH, Razvi S, Bensenor IM, Azizi F, Amouzegar A, Valdes S, Colomo N, Wareham NJ, Jukema JW, Westendorp RGJ, Kim KW, Rodondi N, Del Giovane C. Subclinical thyroid dysfunction and incident diabetes: a systematic review and an individual participant data analysis of prospective cohort studies. Eur J Endocrinol 2022; 187:S35-S46
- Sarabhai T, Kostev K. Thyroid disorders and the incidence of type 2 diabetes: insights from a 10-year cohort study in Germany. Endocr Connect 2025; 14
- Mouradian M, Abourizk N. Diabetes mellitus and thyroid disease. Diabetes Care 1983; 6:512-520
- Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, Harris GJ, Antonelli A, Salvi M, Goldberg RA, Gigantelli JW, Couch SM, Shriver EM, Hayek BR, Hink EM, Woodward RM, Gabriel K, Magni G, Douglas RS. Teprotumumab for Thyroid-Associated Ophthalmopathy. N Engl J Med 2017; 376:1748-1761
- Douglas RS, Kahaly GJ, Patel A, Sile S, Thompson EHZ, Perdok R, Fleming JC, Fowler BT, Marcocci C, Marino M, Antonelli A, Dailey R, Harris GJ, Eckstein A, Schiffman J, Tang R, Nelson C, Salvi M, Wester S, Sherman JW, Vescio T, Holt RJ, Smith TJ. Teprotumumab for the Treatment of Active Thyroid Eye Disease. N Engl J Med 2020; 382:341-352
- Sperling M, A. A, Yau M. Autoimmune Polyglandular Syndromes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- de Herder WW, Hofland J. Glucagon & Glucagonoma Syndrome. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2023.
- Song X, Zheng S, Yang G, Xiong G, Cao Z, Feng M, Zhang T, Zhao Y. Glucagonoma and the glucagonoma syndrome. Oncol Lett 2018; 15:2749-2755
- Wewer Albrechtsen NJ, Kuhre RE, Pedersen J, Knop FK, Holst JJ. The biology of glucagon and the consequences of hyperglucagonemia. Biomark Med 2016; 10:1141-1151
- de Herder WW, Hofland J. Somatostatinoma. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2023.
- Theodoraki A, Khoo B, Hamda A, Grillo F, Meyer T, Bouloux PM. Malignant somatostatinoma presenting with diabetic ketoacidosis and inhibitory syndrome: pathophysiologic considerations. Endocr Pract 2010; 16:835-837
- Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 2009; 150:776-783
- Monticone S, D'Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, Mulatero P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 2018; 6:41-50
- Katsuragawa S, Tsurutani Y, Takiguchi T, Saito J, Nishikawa T. Impact of primary aldosteronism on renal function in patients with type 2 diabetes. J Diabetes Investig 2021; 12:217-225
- Papadopoulou-Marketou N, Vaidya A, Dluhy R, Chrousos GP. Hyperaldosteronism. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2020.
- Feingold KR, Lee TH, Chung MY, Siperstein MD. Muscle capillary basement membrane width in patients with vacor-induced diabetes mellitus. J Clin Invest 1986; 78:102-107
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201-207
- Allhat Officers Coordinators for the Allhat Collaborative Research Group The Antihypertensive Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981-2997
- Hirst JA, Farmer AJ, Feakins BG, Aronson JK, Stevens RJ. Quantifying the effects of diuretics and beta-adrenoceptor blockers on glycaemic control in diabetes mellitus - a systematic review and meta-analysis. Br J Clin Pharmacol 2015; 79:733-743
- Zhang X, Zhao Q. Association of Thiazide-Type Diuretics With Glycemic Changes in Hypertensive Patients: A Systematic Review and Meta-Analysis of Randomized Controlled Clinical Trials. J Clin Hypertens (Greenwich)2016; 18:342-351
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254-1262
- Rizos CV, Elisaf MS. Antihypertensive drugs and glucose metabolism. World J Cardiol 2014; 6:517-530
- Li Z, Wei H, Li R, Wu B, Xu M, Yang X, Zhang Y, Liu Y. The effects of antihypertensive drugs on glucose metabolism. Diabetes Obes Metab 2024; 26:4820-4829
- Fathallah N, Slim R, Larif S, Hmouda H, Ben Salem C. Drug-Induced Hyperglycaemia and Diabetes. Drug Saf2015; 38:1153-1168
- Martins VM, Ziegelmann PK, Ferrari F, Bottino LG, Lucca MB, Correa HLR, Blum GB, Helal L, Fuchs SC, Fuchs FD. Thiazide diuretics alone or combined with potassium-sparing diuretics to treat hypertension: a systematic review and network meta-analysis of randomized controlled trials. J Hypertens 2023; 41:1108-1116
- Verhaegen AA, Van Gaal LF. Drugs That Affect Body Weight, Body Fat Distribution, and Metabolism. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2019.
- Davis H, Spanakis E, Cryer P, Siamashvili M, Davis S. Hypoglycemia During Therapy of Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Koch-Weser J. Diazoxide. N Engl J Med 1976; 294:1271-1273
- de Herder WW, Hofland J. Insulinomas. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2023.
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735-742
- Cholesterol Treatment Trialists Collaboration. Effects of statin therapy on diagnoses of new-onset diabetes and worsening glycaemia in large-scale randomised blinded statin trials: an individual participant data meta-analysis. Lancet Diabetes Endocrinol 2024; 12:306-319
- Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N, Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 2011; 305:2556-2564
- Feingold KR. Cholesterol Lowering Drugs. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Abbasi F, Lamendola C, Harris CS, Harris V, Tsai MS, Tripathi P, Abbas F, Reaven GM, Reaven PD, Snyder MP, Kim SH, Knowles JW. Statins Are Associated With Increased Insulin Resistance and Secretion. Arterioscler Thromb Vasc Biol 2021; 41:2786-2797
- Cederberg H, Stancakova A, Yaluri N, Modi S, Kuusisto J, Laakso M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia 2015; 58:1109-1117
- Carmena R, Betteridge DJ. Diabetogenic Action of Statins: Mechanisms. Curr Atheroscler Rep 2019; 21:23
- Goldie C, Taylor AJ, Nguyen P, McCoy C, Zhao XQ, Preiss D. Niacin therapy and the risk of new-onset diabetes: a meta-analysis of randomised controlled trials. Heart 2016; 102:198-203
- Feingold KR, Grunfeld C. Triglyceride Lowering Drugs. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2024.
- Shen M, Orwoll ES, Conte JE, Jr., Prince MJ. Pentamidine-induced pancreatic beta-cell dysfunction. Am J Med1989; 86:726-728
- Bouchard P, Sai P, Reach G, Caubarrere I, Ganeval D, Assan R. Diabetes mellitus following pentamidine-induced hypoglycemia in humans. Diabetes 1982; 31:40-45
- Carter BL, Small RE, Mandel MD, Starkman MT. Phenytoin-induced hyperglycemia. Am J Hosp Pharm 1981; 38:1508-1512
- Gharib H, Munoz JM. Endocrine manifestations of diphenylhydantoin therapy. Metabolism 1974; 23:515-524
- Okanoue T, Sakamoto S, Itoh Y, Minami M, Yasui K, Sakamoto M, Nishioji K, Katagishi T, Nakagawa Y, Tada H, Sawa Y, Mizuno M, Kagawa K, Kashima K. Side effects of high-dose interferon therapy for chronic hepatitis C. J Hepatol 1996; 25:283-291
- Zornitzki T, Malnick S, Lysyy L, Knobler H. Interferon therapy in hepatitis C leading to chronic type 1 diabetes. World J Gastroenterol 2015; 21:233-239
- Lombardi A, Tsomos E, Hammerstad SS, Tomer Y. Interferon alpha: The key trigger of type 1 diabetes. J Autoimmun 2018; 94:7-15
- Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, Tolaney SM. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-analysis. JAMA Oncol 2018; 4:173-182
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm Metab Res 2019; 51:145-156
- Venetsanaki V, Boutis A, Chrisoulidou A, Papakotoulas P. Diabetes mellitus secondary to treatment with immune checkpoint inhibitors. Curr Oncol 2019; 26:e111-e114
- Elshimy G, Raj R, Akturk HK, Schriber A, Sisterna N, Ahmad I, Jacob A, Michels AW, Correa R. Immune Checkpoint Inhibitors Related Endocrine Adverse Events. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Whicher CA, Price HC, Holt RIG. Mechanisms in endocrinology: Antipsychotic medication and type 2 diabetes and impaired glucose regulation. Eur J Endocrinol 2018; 178:R245-R258
- Holt RIG. Association Between Antipsychotic Medication Use and Diabetes. Curr Diab Rep 2019; 19:96
- Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, Schmid F, Lobos CA, Kissling W, Davis JM, Leucht S. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res 2010; 123:225-233
- Citrome L, Kalsekar I, Baker RA, Hebden T. A review of real-world data on the effects of aripiprazole on weight and metabolic outcomes in adults. Curr Med Res Opin 2014; 30:1629-1641
- Chen J, Huang XF, Shao R, Chen C, Deng C. Molecular Mechanisms of Antipsychotic Drug-Induced Diabetes. Front Neurosci 2017; 11:643
- Tzortzis V, Samarinas M, Zachos I, Oeconomou A, Pisters LL, Bargiota A. Adverse effects of androgen deprivation therapy in patients with prostate cancer: focus on metabolic complications. Hormones (Athens) 2017; 16:115-123
- Tsai HT, Keating NL, Van Den Eeden SK, Haque R, Cassidy-Bushrow AE, Ulcickas Yood M, Smith MR, Potosky AL. Risk of diabetes among patients receiving primary androgen deprivation therapy for clinically localized prostate cancer. J Urol 2015; 193:1956-1962
- Pham P, Sarkar M, Pham P, Pham P. Diabetes Mellitus After Solid Organ Transplantation. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Chakkera HA, Kudva Y, Kaplan B. Calcineurin Inhibitors: Pharmacologic Mechanisms Impacting Both Insulin Resistance and Insulin Secretion Leading to Glucose Dysregulation and Diabetes Mellitus. Clin Pharmacol Ther2017; 101:114-120
- Verges B. mTOR and Cardiovascular Diseases: Diabetes Mellitus. Transplantation 2018; 102:S47-S49
- Ahmad MH, Shafiq I. Diabetic ketoacidosis following PEG-asparaginase therapy. Endocrinol Diabetes Metab Case Rep 2018; 2018:18-0064
- Pui CH, Burghen GA, Bowman WP, Aur RJ. Risk factors for hyperglycemia in children with leukemia receiving L-asparaginase and prednisone. J Pediatr 1981; 99:46-50
- Kahaly GJ, Douglas RS, Holt RJ, Sile S, Smith TJ. Teprotumumab for patients with active thyroid eye disease: a pooled data analysis, subgroup analyses, and off-treatment follow-up results from two randomised, double-masked, placebo-controlled, multicentre trials. Lancet Diabetes Endocrinol 2021; 9:360-372
- Amarikwa L, Mohamed A, Kim SH, Kossler AL, Dosiou C. Teprotumumab-Related Hyperglycemia. J Clin Endocrinol Metab 2023; 108:858-864
- Carter C, Marks M, Bundeff AW, Adewodu T, Alderman L. A case of rapidly declining glycemic control and diabetic ketoacidosis in a newly diagnosed diabetes patient after starting teprotumumab for thyroid eye disease. Endocrine 2024; 83:65-68
- Shah K, Charitou M. A Novel Case of Hyperglycemic Hyperosmolar State After the Use of Teprotumumab in a Patient With Thyroid Eye Disease. AACE Clin Case Rep 2022; 8:148-149
- Stan MN, Krieger CC. The Adverse Effects Profile of Teprotumumab. J Clin Endocrinol Metab 2023; 108:e654-e662
- Guglielmi C, Palermo A, Pozzilli P. Latent autoimmune diabetes in the adults (LADA) in Asia: from pathogenesis and epidemiology to therapy. Diabetes Metab Res Rev 2012; 28 Suppl 2:40-46
- Kumar A, de Leiva A. Latent autoimmune diabetes in adults (LADA) in Asian and European populations. Diabetes Metab Res Rev 2017; 33
- Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L. The many faces of diabetes: a disease with increasing heterogeneity. Lancet 2014; 383:1084-1094
- Naik RG, Brooks-Worrell BM, Palmer JP. Latent autoimmune diabetes in adults. J Clin Endocrinol Metab 2009; 94:4635-4644
- Fourlanos S, Perry C, Stein MS, Stankovich J, Harrison LC, Colman PG. A clinical screening tool identifies autoimmune diabetes in adults. Diabetes Care 2006; 29:970-975
- Herzog K, Ahlqvist E, Alfredsson L, Groop L, Hjort R, Lofvenborg JE, Tuomi T, Carlsson S. Combined lifestyle factors and the risk of LADA and type 2 diabetes - Results from a Swedish population-based case-control study. Diabetes Res Clin Pract 2021; 174:108760
- Santoso C, Wei Y, Ahlqvist E, Tuomi T, Carlsson S. Autoimmune diseases and the risk and prognosis of latent autoimmune diabetes in adults. Diabetologia 2025; 68:331-341
- Buzzetti R, Tuomi T, Mauricio D, Pietropaolo M, Zhou Z, Pozzilli P, Leslie RD. Management of Latent Autoimmune Diabetes in Adults: A Consensus Statement From an International Expert Panel. Diabetes 2020; 69:2037-2047
- Jones AG, McDonald TJ, Shields BM, Hagopian W, Hattersley AT. Latent Autoimmune Diabetes of Adults (LADA) Is Likely to Represent a Mixed Population of Autoimmune (Type 1) and Nonautoimmune (Type 2) Diabetes. Diabetes Care 2021; 44:1243-1251
- Brophy S, Davies H, Mannan S, Brunt H, Williams R. Interventions for latent autoimmune diabetes (LADA) in adults. Cochrane Database Syst Rev 2011:CD006165
- Li X, Liao L, Yan X, Huang G, Lin J, Lei M, Wang X, Zhou Z. Protective effects of 1-alpha-hydroxyvitamin D3 on residual beta-cell function in patients with adult-onset latent autoimmune diabetes (LADA). Diabetes Metab Res Rev 2009; 25:411-416
- Zhang Z, Yan X, Wu C, Pei X, Li X, Wang X, Niu X, Jiang H, Zeng X, Zhou Z. Adding vitamin D3 to the dipeptidyl peptidase-4 inhibitor saxagliptin has the potential to protect beta-cell function in LADA patients: A 1-year pilot study. Diabetes Metab Res Rev 2020; 36:e3298
- Yan X, Li X, Liu B, Huang J, Xiang Y, Hu Y, Tang X, Zhang Z, Huang G, Xie Z, Zhou H, Liu Z, Wang X, Leslie RD, Zhou Z. Combination therapy with saxagliptin and vitamin D for the preservation of beta-cell function in adult-onset type 1 diabetes: a multi-center, randomized, controlled trial. Signal Transduct Target Ther 2023; 8:158
- Maddaloni E, Coleman RL, Agbaje O, Buzzetti R, Holman RR. Time-varying risk of microvascular complications in latent autoimmune diabetes of adulthood compared with type 2 diabetes in adults: a post-hoc analysis of the UK Prospective Diabetes Study 30-year follow-up data (UKPDS 86). Lancet Diabetes Endocrinol 2020; 8:206-215
- Isomaa B, Almgren P, Henricsson M, Taskinen MR, Tuomi T, Groop L, Sarelin L. Chronic complications in patients with slowly progressing autoimmune type 1 diabetes (LADA). Diabetes Care 1999; 22:1347-1353
- Maddaloni E, Coleman RL, Pozzilli P, Holman RR. Long-term risk of cardiovascular disease in individuals with latent autoimmune diabetes in adults (UKPDS 85). Diabetes Obes Metab 2019; 21:2115-2122
- Wei Y, Herzog K, Ahlqvist E, Andersson T, Nystrom T, Zhan Y, Tuomi T, Carlsson S. All-Cause Mortality and Cardiovascular and Microvascular Diseases in Latent Autoimmune Diabetes in Adults. Diabetes Care 2023; 46:1857-1865
- McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, Pittock SJ. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol 2012; 69:230-238
- Rathbun JT, Imber J. Stiff Person Syndrome and Type 1 Diabetes Mellitus: a Case of the Chicken or the Egg? J Gen Intern Med 2019;
- Baizabal-Carvallo JF. The neurological syndromes associated with glutamic acid decarboxylase antibodies. J Autoimmun 2019; 101:35-47
- Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore) 2002; 81:87-100
- Taylor SI, Shahidzadeh Yazdi Z, Oral EA. Letter to the Editor From Taylor et al "Type B Insulin Resistance Syndrome: A Rare Cause of Hypoglycemia". JCEM Case Rep 2024; 2:luae001
- Brown N, Elston MS. Systematic Review-Type B Insulin Resistance With Isolated Hypoglycemia and Suppressed Insulin. J Clin Endocrinol Metab 2024; 109:936-943
- Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, Soos MA, Carpenter TA, Lomas DJ, Cochran EK, Gorden P, O'Rahilly S, Savage DB. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest 2009; 119:315-322
- Klubo-Gwiezdzinska J, Lange M, Cochran E, Semple RK, Gewert C, Brown RJ, Gorden P. Combined Immunosuppressive Therapy Induces Remission in Patients With Severe Type B Insulin Resistance: A Prospective Cohort Study. Diabetes Care 2018; 41:2353-2360
- Lebovitz HE, Banerji MA. Ketosis-Prone Diabetes (Flatbush Diabetes): an Emerging Worldwide Clinically Important Entity. Curr Diab Rep 2018; 18:120
- Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann Intern Med2006; 144:350-357
- Smiley D, Chandra P, Umpierrez GE. Update on diagnosis, pathogenesis and management of ketosis-prone Type 2 diabetes mellitus. Diabetes Manag (Lond) 2011; 1:589-600
- Whooten R, Schmitt J, Schwartz A. Endocrine manifestations of Down syndrome. Curr Opin Endocrinol Diabetes Obes 2018; 25:61-66
- Guaraldi F, Rossetto Giaccherino R, Lanfranco F, Motta G, Gori D, Arvat E, Ghigo E, Giordano R. Endocrine Autoimmunity in Down's Syndrome. Front Horm Res 2017; 48:133-146
- Anwar AJ, Walker JD, Frier BM. Type 1 diabetes mellitus and Down's syndrome: prevalence, management and diabetic complications. Diabet Med 1998; 15:160-163
- Bergholdt R, Eising S, Nerup J, Pociot F. Increased prevalence of Down's syndrome in individuals with type 1 diabetes in Denmark: A nationwide population-based study. Diabetologia 2006; 49:1179-1182
- Aitken RJ, Mehers KL, Williams AJ, Brown J, Bingley PJ, Holl RW, Rohrer TR, Schober E, Abdul-Rasoul MM, Shield JP, Gillespie KM. Early-onset, coexisting autoimmunity and decreased HLA-mediated susceptibility are the characteristics of diabetes in Down syndrome. Diabetes Care 2013; 36:1181-1185
- Aslam AA, Baksh RA, Pape SE, Strydom A, Gulliford MC, Chan LF, Consortium G-D. Diabetes and Obesity in Down Syndrome Across the Lifespan: A Retrospective Cohort Study Using U.K. Electronic Health Records. Diabetes Care 2022; 45:2892-2899
- Behre HM, Bergmann M, Simoni M, Tuttelmann F. Primary Testicular Failure. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2015.
- Blackburn J, Ramakrishnan A, Graham C, Bambang K, Sriranglingam U, Senniappan S. Klinefelter Syndrome: A Review. Clin Endocrinol (Oxf) 2025;
- Salzano A, D'Assante R, Heaney LM, Monaco F, Rengo G, Valente P, Pasquali D, Bossone E, Gianfrilli D, Lenzi A, Cittadini A, Marra AM, Napoli R. Klinefelter syndrome, insulin resistance, metabolic syndrome, and diabetes: review of literature and clinical perspectives. Endocrine 2018; 61:194-203
- Groth KA, Skakkebaek A, Host C, Gravholt CH, Bojesen A. Clinical review: Klinefelter syndrome--a clinical update. J Clin Endocrinol Metab 2013; 98:20-30
- Gravholt CH, Chang S, Wallentin M, Fedder J, Moore P, Skakkebaek A. Klinefelter Syndrome: Integrating Genetics, Neuropsychology, and Endocrinology. Endocr Rev 2018; 39:389-423
- Bojesen A, Juul S, Birkebaek NH, Gravholt CH. Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocrinol Metab 2006; 91:1254-1260
- Panimolle F, Tiberti C, Granato S, Semeraro A, Gianfrilli D, Anzuini A, Lenzi A, Radicioni A. Screening of endocrine organ-specific humoral autoimmunity in 47,XXY Klinefelter's syndrome reveals a significant increase in diabetes-specific immunoreactivity in comparison with healthy control men. Endocrine 2016; 52:157-164
- Sybert VP, McCauley E. Turner's syndrome. N Engl J Med 2004; 351:1227-1238
- Gravholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in Turner syndrome. J Clin Epidemiol 1998; 51:147-158
- Sun L, Wang Y, Zhou T, Zhao X, Wang Y, Wang G, Gang X. Glucose Metabolism in Turner Syndrome. Front Endocrinol (Lausanne) 2019; 10:49
- Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, Lin AE, Mauras N, Quigley CA, Rubin K, Sandberg DE, Sas TCJ, Silberbach M, Soderstrom-Anttila V, Stochholm K, van Alfen-van derVelden JA, Woelfle J, Backeljauw PF, International Turner Syndrome Consensus G. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017; 177:G1-G70
- Bakalov VK, Cheng C, Zhou J, Bondy CA. X-chromosome gene dosage and the risk of diabetes in Turner syndrome. J Clin Endocrinol Metab 2009; 94:3289-3296
- Mortensen KH, Cleemann L, Hjerrild BE, Nexo E, Locht H, Jeppesen EM, Gravholt CH. Increased prevalence of autoimmunity in Turner syndrome--influence of age. Clin Exp Immunol 2009; 156:205-210
- Gravholt CH, Andersen NH, Christin-Maitre S, Davis SM, Duijnhouwer A, Gawlik A, Maciel-Guerra AT, Gutmark-Little I, Fleischer K, Hong D, Klein KO, Prakash SK, Shankar RK, Sandberg DE, Sas TCJ, Skakkebaek A, Stochholm K, van der Velden JA, International Turner Syndrome Consensus G, Backeljauw PF. Clinical practice guidelines for the care of girls and women with Turner syndrome. Eur J Endocrinol 2024; 190:G53-G151
- Sheanon N, Elder D, Khoury J, Casnellie L, Gutmark-Little I, Cernich J, Backeljauw PF. Increased Prevalence of Beta-Cell Dysfunction despite Normal HbA1c in Youth and Young Adults with Turner Syndrome. Horm Res Paediatr 2021; 94:297-306
- Stanley TL, Leong A, Pober BR. Growth, body composition, and endocrine issues in Williams syndrome. Curr Opin Endocrinol Diabetes Obes 2021; 28:64-74
- Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, Pober BR. Williams syndrome. Nat Rev Dis Primers 2021; 7:42
- Morris CA. Williams Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, eds. GeneReviews((R)). Seattle (WA)2017.
- Stone SI, Abreu D, McGill JB, Urano F. Monogenic and syndromic diabetes due to endoplasmic reticulum stress. J Diabetes Complications 2021; 35:107618
- Urano F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr Diab Rep 2016; 16:6
- Rigoli L, Bramanti P, Di Bella C, De Luca F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res 2018; 83:921-929
- Ray MK, Chen L, White NH, Ni R, Hershey T, Marshall BA. Longitudinal progression of diabetes mellitus in Wolfram syndrome: The Washington University Wolfram Research Clinic experience. Pediatr Diabetes 2022; 23:212-218
- Bidichandani SI, Delatycki MB. Friedreich Ataxia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993.
- Pappa A, Hausler MG, Veigel A, Tzamouranis K, Pfeifer MW, Schmidt A, Bokamp M, Haberland H, Wagner S, Bruckel J, de Sousa G, Hackl L, Bollow E, Holl RW. Diabetes mellitus in Friedreich Ataxia: A case series of 19 patients from the German-Austrian diabetes mellitus registry. Diabetes Res Clin Pract 2018; 141:229-236
- Cnop M, Mulder H, Igoillo-Esteve M. Diabetes in Friedreich ataxia. J Neurochem 2013; 126 Suppl 1:94-102
- Bidichandani SI, Delatycki MB, Napierala M, Duquette A. Friedreich Ataxia. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993.
- Pandey M, Rajamma U. Huntington's disease: the coming of age. J Genet 2018; 97:649-664
- Montojo MT, Aganzo M, Gonzalez N. Huntington's Disease and Diabetes: Chronological Sequence of its Association. J Huntingtons Dis 2017; 6:179-188
- Farrer LA. Diabetes mellitus in Huntington disease. Clin Genet 1985; 27:62-67
- Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl) 2004; 82:510-529
- Hartman J, Patki T, Johnson NE. Diagnosis and Management of Myotonic Dystrophy Type 1. JAMA 2024; 331:1227-1228
- Winters SJ. Endocrine Dysfunction in Patients With Myotonic Dystrophy. J Clin Endocrinol Metab 2021; 106:2819-2827
- Alsaggaf R, Pfeiffer RM, Wang Y, St George DMM, Zhan M, Wagner KR, Amr S, Greene MH, Gadalla SM. Diabetes, metformin and cancer risk in myotonic dystrophy type I. Int J Cancer 2020; 147:785-792
- Lee JY, Jeong HN. Nationwide incidence of myotonic dystrophy type 1 and the status of multi-organ involvement. J Neurol 2021;
- Abe H, Mita T, Kudo K, Funayama T, Tokoro M, Kaga H, Ikeda F, Kanazawa A, Hirose T, Kawamori R, Watada H. Dramatic improvement of blood glucose control after pioglitazone treatment in poorly controlled over-weight diabetic patients with myotonic dystrophy. Endocr J 2009; 56:911-913
- Takeshima K, Ariyasu H, Ishibashi T, Kawai S, Uraki S, Koh J, Ito H, Akamizu T. Myotonic dystrophy type 1 with diabetes mellitus, mixed hypogonadism and adrenal insufficiency. Endocrinol Diabetes Metab Case Rep 2018; 2018
- Tsang SH, Aycinena ARP, Sharma T. Ciliopathy: Bardet-Biedl Syndrome. Adv Exp Med Biol 2018; 1085:171-174
- Mujahid S, Hunt KF, Cheah YS, Forsythe E, Hazlehurst JM, Sparks K, Mohammed S, Tomlinson JW, Amiel SA, Carroll PV, Beales PL, Huda MSB, McGowan BM. The Endocrine and Metabolic Characteristics of a Large Bardet-Biedl Syndrome Clinic Population. J Clin Endocrinol Metab 2018; 103:1834-1841
- Dollfus H, Lilien MR, Maffei P, Verloes A, Muller J, Bacci GM, Cetiner M, van den Akker ELT, Grudzinska Pechhacker M, Testa F, Lacombe D, Stokman MF, Simonelli F, Gouronc A, Gavard A, van Haelst MM, Koenig J, Rossignol S, Bergmann C, Zacchia M, Leroy BP, Mosbah H, Van Eerde AM, Mekahli D, Servais A, Poitou C, Valverde D. Bardet-Biedl syndrome improved diagnosis criteria and management: Inter European Reference Networks consensus statement and recommendations. Eur J Hum Genet 2024; 32:1347-1360
- Esbati R, Yazdani O, Simonetti J. Management of Obesity-Related Genetic Disorders. Endocrinol Metab Clin North Am 2025; 54:17-38
- Haws R, Brady S, Davis E, Fletty K, Yuan G, Gordon G, Stewart M, Yanovski J. Effect of setmelanotide, a melanocortin-4 receptor agonist, on obesity in Bardet-Biedl syndrome. Diabetes Obes Metab 2020; 22:2133-2140
- Haqq AM, Chung WK, Dollfus H, Haws RM, Martos-Moreno GA, Poitou C, Yanovski JA, Mittleman RS, Yuan G, Forsythe E, Clement K, Argente J. Efficacy and safety of setmelanotide, a melanocortin-4 receptor agonist, in patients with Bardet-Biedl syndrome and Alstrom syndrome: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial with an open-label period. Lancet Diabetes Endocrinol 2022; 10:859-868
- Argente J, Verge CF, Okorie U, Fennoy I, Kelsey MM, Cokkinias C, Scimia C, Lee HM, Farooqi IS. Setmelanotide in patients aged 2-5 years with rare MC4R pathway-associated obesity (VENTURE): a 1 year, open-label, multicenter, phase 3 trial. Lancet Diabetes Endocrinol 2025; 13:29-37
- Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993.
- Clerc A, Coupaye M, Mosbah H, Pinto G, Laurier V, Mourre F, Merrien C, Diene G, Poitou C, Tauber M. Diabetes Mellitus in Prader-Willi Syndrome: Natural History during the Transition from Childhood to Adulthood in a Cohort of 39 Patients. J Clin Med 2021; 10
- Chan NN, Feher MD, Bridges NA. Metformin therapy for diabetes in Prader-Willi syndrome. J R Soc Med 1998; 91:598
- Ng NBH, Low YW, Rajgor DD, Low JM, Lim YY, Loke KY, Lee YS. The effects of glucagon-like peptide (GLP)-1 receptor agonists on weight and glycaemic control in Prader-Willi syndrome: A systematic review. Clin Endocrinol (Oxf) 2022; 96:144-154
- Gimenez-Palop O, Romero A, Casamitjana L, Pareja R, Rigla M, Caixas A. Effect of semaglutide on weight loss and glycaemic control in patients with Prader-Willi Syndrome and type 2 diabetes. Endocrinol Diabetes Nutr (Engl Ed) 2024; 71:83-87
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics2011; 12:225-235
- Hearn T. ALMS1 and Alstrom syndrome: a recessive form of metabolic, neurosensory and cardiac deficits. J Mol Med (Berl) 2019; 97:1-17
- Dassie F, Favaretto F, Bettini S, Parolin M, Valenti M, Reschke F, Danne T, Vettor R, Milan G, Maffei P. Alstrom syndrome: an ultra-rare monogenic disorder as a model for insulin resistance, type 2 diabetes mellitus and obesity. Endocrine 2021; 71:618-625
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 2005; 165:675-683
- Tahani N, Maffei P, Dollfus H, Paisey R, Valverde D, Milan G, Han JC, Favaretto F, Madathil SC, Dawson C, Armstrong MJ, Warfield AT, Duzenli S, Francomano CA, Gunay-Aygun M, Dassie F, Marion V, Valenti M, Leeson-Beevers K, Chivers A, Steeds R, Barrett T, Geberhiwot T. Consensus clinical management guidelines for Alstrom syndrome. Orphanet J Rare Dis 2020; 15:253
- Ali S, Baig S, Wanninayake S, da Silva Xavier G, Dawson C, Paisey R, Geberhiwot T. Glucagon-like peptide-1 analogues in monogenic syndromic obesity: Real-world data from a large cohort of Alstrom syndrome patients. Diabetes Obes Metab 2024; 26:989-996
- Oshima J, Martin GM, Hisama FM. Werner Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993.
- Takemoto M, Kubota Y, Taniguchi T, Motegi SI, Taniguchi A, Nakagami H, Maezawa Y, Koshizaka M, Kato H, Tsukamoto K, Mori S, Kuzuya M, Yokote K. Management guideline for Werner syndrome 2020. 3. Diabetes associated with Werner syndrome. Geriatr Gerontol Int 2021; 21:142-145
- Atallah I, McCormick D, Good JM, Barigou M, Fraga M, Sempoux C, Superti-Furga A, Semple RK, Tran C. Partial lipodystrophy, severe dyslipidaemia and insulin resistant diabetes as early signs of Werner syndrome. J Clin Lipidol 2022; 16:583-590
- Christiansen AL, Bygum A, Hother-Nielsen O, Rasmussen LM. Diagnosing diabetes mellitus in patients with porphyria cutanea tarda. Int J Dermatol 2018; 57:763-769
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med 2017; 377:862-872
- Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab 2019;