Archives
Miscellaneous
Nodules and Cancer
Genetic Disorders
Disorders of Pregnancy and Childhood
Hypothyroidism and Hyperthyroidism
Thyroid Testing
Neonatal Hyperthyroidism
INTRODUCTION
Neonatal hyperthyroidism in most cases is transient and results from the transplacental passage of maternal stimulating TSH receptor antibodies (TRAb) known as neonatal Graves’ disease (GD). Permanent non autoimmune neonatal hyperthyroidism is rare and is due to activating mutations of TSH receptor or due to somatic activating mutations in the stimulatory alpha subunit of the guanine nucleotide-binding protein (GNAS gene) in McCune-Albright syndrome. Exposure to topical iodine has also been reported as a rare cause of hyperthyroidism in newborns.
TRANSIENT NEONATAL HYPERTHYROIDISM
Neonatal Graves’ disease (GD) is usually a self-limited disease, but it can be life threatening and permanently damage the brain. Neonatal GD is caused by transplacental passage of TSH receptor antibodies (TRAb) with stimulatory activity.
TRAb are Immunoglobulin of G class and freely cross the placenta. Different types of TRAb can be found: TRAb that bind to the TSH receptor and stimulates the production of thyroid hormones, (TSH receptor stimulating antibodies, TSI), TRAb that bind to the TSH receptor, do not stimulate the production of thyroid hormones and can block the binding of TSH (TSH receptor blocking antibodies TBI).
Hyperthyroidism develops only in babies born to mothers with the most potent stimulatory activity in serum. This corresponds to 1-2% of mothers with Graves’ disease, or 1 in 50,000 newborns, an incidence that is approximately four times higher than is that for transient neonatal hypothyroidism due to maternal TSH receptor blocking antibodies. Some mothers have mixtures of stimulating and blocking antibodies in their circulation, the relative proportion of which may change over time. Not surprisingly, the clinical picture in the fetus and neonate of these mothers is more complex and depends not only on the relative proportion of each activity in the maternal circulation at any one time but on the rate of their clearance from the neonatal circulation postpartum.
Occasionally, neonatal hyperthyroidism may even occur in infants born to hypothyroid mothers. A prospective study showed that 40% of patients treated for Graves’ disease with radioactive iodine had TRAb detectable after 5 years (13). In these situations, the maternal thyroid has been destroyed either by prior radioablation, surgery, or by coincident destructive autoimmune processes so that potent thyroid stimulating antibodies, present in the maternal circulation, are silent in contrast to the neonate whose thyroid gland is normal. Persistence of TRAb after thyroidectomy is higher in females with Graves’ ophthalmopathy or smokers. Fetal/neonatal thyrotoxicosis can occur also in newborn from hypothyroid mothers with chronic lymphocytic thyroiditis.
CLINICAL MANIFESTATIONS
|
TABLE 1. Situations That Should Prompt Consideration of Neonatal Hyperthyroidism |
|
· Unexplained tachycardia, goiter or stare |
|
Unexplained petechiae, hyperbilirubinemia, or hepatosplenomegaly |
|
· History of persistently high TSH receptor antibody titer in mother during pregnancy |
|
· History of persistently high requirement for antithyroid medication in mother during pregnancy |
|
· History of thyroid ablation for hyperthyroidism in mother |
|
· History of previously affected sibling |
Maternal TSH receptor antibody-mediated hyperthyroidism may present in utero. Fetal hyperthyroidism is suspected in the presence of fetal tachycardia (pulse greater than 160/min) especially if there is evidence of failure to thrive. Obstetric complications are common. Fetal goiter (fetal neck circumference >95%) can by monitored by ultrasound using nomograms for fetal thyroid growth. Fetal goiter can cause esophageal and/or tracheal obstructions and polyhydramnios. Fetal goiter can also be due to transplacental passage of antithyroid drugs that cause hypothyroidism in the fetus.
|
TABLE 2. Clinical Manifestations in the Fetus |
|
Unexplained tachycardia, Failure to thrive Intrauterine growth retardation Goiter Advanced bone age Prematurity Craniosynostosis, microcephaly Fetal death |
In the neonate infant typically, the onset is during the first one two weeks of life but can occur by 45 days. This is due both to the clearance of maternally administered antithyroid drug (propylthiouracil- PTU, methimazole- MMI, or carbimazole) from the infant ’s circulation and to the increased conversion of T4 to the more metabolically active T3 after birth. Rarely, as noted earlier, the onset of neonatal hyperthyroidism may be delayed until later if higher affinity blocking antibodies are also present.
In the newborn infant, characteristic signs and symptoms include tachycardia, irritability, poor weight gain, and prominent eyes. Goiter, when present, may be related to maternal antithyroid drug treatment as well as to the neonatal Graves’ disease itself.
Rarely, infants with neonatal Graves’ disease present with thrombocytopenia, jaundice, hepatosplenomegaly, and hypoprothrombinemia, a picture that may be confused with congenital infections such as toxoplasmosis, rubella, or cytomegalovirus. In addition, arrhythmias and cardiac failure may develop and may cause death, particularly if treatment is delayed or
inadequate. In addition to a significant mortality rate that approximates 20% in some older series, untreated fetal and neonatal hyperthyroidism is associated with deleterious long-term consequences, including premature closure of the cranial sutures (cranial synostosis), failure to
thrive, and developmental delay. The half-life of TSH receptor antibodies is 1 to 2 weeks. The duration of neonatal hyperthyroidism, a function of antibody potency and the rate of their metabolic clearance, is usually 2 to 3 months but may be longer.
|
TABLE 3. Clinical Manifestations in the Neonate |
|
Irritability, hyperexcitability, sleep disorders Tachycardia, hypertension, cardiac failure Flushing, sweating Respiratory distress, pulmonary hypertension Goiter, stare Feeding difficulties, increased appetite but no/poor weight gain Diarrhea Unexplained petechiae, hyperbilirubinemia, jaundice, or hepatosplenomegaly Craniosynostosis, microcephaly, Death |
LABORATORY EVALUATION
The recent guidelines for management of hyperthyroidism and the updated guidelines for the management of thyroid disease during pregnancy released from the American Thyroid Association ATA both suggest determining TRAb levels in pregnant women with Graves’ disease at 18-22 weeks instead of 20-24 weeks of gestation because a severe case of fetal Graves’ disease has occurred at 18 weeks of pregnancy.
Because of the importance of early diagnosis and treatment, infants at risk for neonatal hyperthyroidism should undergo both clinical and biochemical assessment as soon as possible.
All neonates born from a woman with TRAb positivity in pregnancy should undergo determination of TRAb from cord blood at delivery. If TRAb is negative, the risk to neonatal hyperthyroidism is negligible (Sensitivity is around 100%). FT3, FT4 and TSH determination from cord blood did not predict neonatal hyperthyroidism. Increases in FT4 on day 3 to 5 seems to better indicate the onset of hyperthyroidism. Situations that should prompt consideration of neonatal hyperthyroidism are listed in Table 1. A high index of suspicion is necessary in babies of women who have had thyroid ablation because in them a high titer of TSH receptor antibodies would not be evident clinically. Similarly, women with persistently elevated TSH receptor antibodies and with a high requirement for antithyroid medication are at an increased risk of having an affected child.
The diagnosis of hyperthyroidism is confirmed by the demonstration of an increased concentration of circulating T4 (and free T4, and T3, if possible) accompanied by a suppressed TSH level in neonatal or fetal blood. Results should be compared with normal values during gestation. Fetal ultrasonography may be helpful in detecting the presence of a fetal goiter and in monitoring fetal growth. Demonstration in the baby or mother of a high titer of TSH receptor antibodies will confirm the etiology of the hyperthyroidism and, in babies whose thyroid function testing is normal initially, indicate the degree to which the baby is at risk.
In general, babies likely to become hyperthyroid have the highest TSH receptor antibody titer but levels of maternal TRAb in the serum as low as 4.4 U/L has been associated with neonatal thyrotoxicosis. If TSH receptor antibodies are not detectable, the baby is very unlikely to become hyperthyroid. In the latter case, it can be anticipated that the baby will be euthyroid, have transient hypothalamic-pituitary suppression, or have a transiently elevated TSH, depending on the relative contribution of maternal hyperthyroidism versus the effects of maternal antithyroid medication, respectively. Close follow up of all babies with abnormal thyroid function tests or detectable TSH receptor antibodies is mandatory.
THERAPY
In the fetus, treatment is accomplished by maternal administration of antithyroid medication. Until recently PTU was the preferred drug for pregnant women in North America, but current recommendations suggest the use of MMI rather than PTU after the first trimester because of concerns about potential PTU-induced hepatotoxicity. The goals of therapy are to utilize the minimal dosage necessary to normalize the fetal heart rate and render the mother euthyroid or slightly hyperthyroid. In the neonate MMI (0.25 to 1.0 mg/kg/day) has been used initially in 3 divided doses. If the hyperthyroidism is severe, a strong iodine solution (Lugol’s solution or SSKI, 1 drop every 8 hours) is added to block the release of thyroid hormone immediately. Often the effect of MMI is not as delayed in infants as it is in older children or adults, a consequence of decreased intrathyroidal thyroid hormone storage. Therapy with both antithyroid drug and iodine is adjusted subsequently, depending on the response. Propranolol (2 mg/kg/day in 2 or 3 divided doses) is added if sympathetic overstimulation is severe, particularly in the presence of pronounced tachycardia. If cardiac failure develops, treatment with digoxin should be initiated, and propranolol should be discontinued. Rarely, prednisone (2 mg/kg/day) is added for immediate inhibition of thyroid hormone secretion. Measurement of TSH receptor antibodies in treated babies may be helpful in predicting when antithyroid medication can be safely discontinued. Lactating mothers on antithyroid medication can continue nursing as long as the dosage of PTU or MMI does not exceed 400 mg or 40 mg, respectively. The milk/serum ratio of PTU is 1/10 that of MMI, a consequence of pH differences and increased protein binding, so one might anticipate less transmission to the infant, but concerns about potential PTU toxicity need to be considered. At higher dosages of antithyroid medication, close supervision of the infant is advisable. A review about management of neonates born to mothers with Graves’ disease has been recently published.
PERMANENT NEONATAL HYPERTHYROIDISM
Rarely, neonatal hyperthyroidism is permanent and is due to a germline mutation in the TSH receptor (TSH-R) resulting in its constitutive activation. A gain of function mutation of the TSH-R should be suspected if persistent neonatal hyperthyroidism occurs in the absence of detectable TSH-R antibodies in the maternal circulation. Prematurity, low birth weight, and advanced bone age are common. Most cases result from a mutation in exon 10 which encodes the transmembrane domain and intracytoplasmic tail of the TSH-R, a member of the G protein coupled receptor superfamily. Less frequently, a mutation encoding the extracellular domain has been described. An autosomal dominant inheritance has been noted in many of these infants; other cases have been sporadic, arising from a de novo mutation.
Early recognition is important because the thyroid function of affected infants is frequently difficult to manage medically and, when diagnosis and therapy is delayed, irreversible sequelae, such as cranial synostosis and developmental delay may result. For this reason, early, aggressive therapy with either thyroidectomy or even radioablation has been recommended.
Two clinical forms were described: the first one is the “familial non-autoimmune autosomal dominant hyperthyroidism” (FNAH). High variable age of manifestation from neonatal period to 60 years, with variability also within the same family is reported. Goiter is present in children, with nodules in older age. The second one is “Persistent sporadic congenital non autoimmune hyperthyroidism” (PSNAH) includes forms with sporadic (de novo) germline mutations in the TSH-R. PSNAH is characterized by fetal-neonatal onset or within 11 months and more severe hyperthyroidism requiring early aggressive therapy.
ThyroidfunctioninbabieswithagainoffunctionmutationoftheTSH receptormaybe difficult tomanagemedically and,whendiagnosis andtherapyisdelayed,irreversible sequelae,suchas cranial synostosis anddevelopmental delaymayresult.Thyroidablationmayberequired.Thyroidsurgeryis thepreferredapproachif anexperienced pediatric surgeonisavailable.Thetimingatwhichthyroidectomy canbeperformedwilldependoninstitutionalpreference. Ifthis is notfeasible,thenradioablationmaybenecessary. Guidelines about this rare condition have recently been published.
MCCUNE ALBRIGHT SYNDROME
McCune Albright is a syndrome due to somatic activating mutations in Gsαgene and can rarely present with neonatal hyperthyroidism.
ACKNOWLEDGEMENTS
This chapter is, in part, based on the previous version written by Professor Rosalind Brown.
GUIDELINES
Ross DS, Burch HB, Cooper DS, Greenlee MC, Launberg P, Maia AL, Rivkees S, Samuels M, Sosa JA, Stan MN, Walter MA. 2016 American Thyroid Association Guidelines for Diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016; 2:1343-1421.
Alexander EK, Pearce EN, Brent GA, Brown RS, Chen H, Dosiou C, Grobman WA, Launberg P, Lazarus JH, Mandel SJ, Peeters RP, Sullivan S. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid: 27:315.
REFERENCES
Samuels SL, Namoc SM, Bauer AJ. Neonatal thyrotoxicosis Clin Perinatol 45:31-40 2018.
Barbesino G, Tomer Y. Clinical utility of TSH receptor antibodies. J Clin Endocrinol Metab.
2013; 98:2247-2255.
van Dijk MM, Smits IH, Fliers E, Bisschop PH. Maternal thyrotropin receptor antibody concentration and the risk of fetal and neonatal thyrotoxicosis: a systematic review. Thyroid 2018: 28:257-
Rivkees S, Pediatric Graves’ disease: management in the post -propylthiouracil Era. Int J Pediatr Endocrinol. 2014 10
Van der Kaay D, Wasserman JD, Palmert MR. Management of neonates born to mothers with Graves’ disease. Pediatrics. 2016;137:e20151878
Paschke R, Niedzela M, Vaidya B, Persani L, Rapaport B, Leclere J. The management of
familial and persistent sporadic non-autoimmune hyperthyroidism caused by thyroid stimulating
hormone receptor germline mutations. Eur Thyroid J. 2012; 1:142-147.
Segni M. Disorders of the Thyroid Gland in Infancy, Childhood and Adolescence. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2017 Mar 18.
Kleinau G, Vassart G. TSH Receptor Mutations and Diseases. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2017 Jul 24.
Congenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment
CLINICAL RECOGNITION
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders that arise from defective steroidogenesis. The production of cortisol in the zona fasciculata of the adrenal cortex occurs in five major enzyme-mediated steps. CAH results from deficiency in any one of these enzymes. Impaired cortisol synthesis leads to chronic elevations of ACTH via the negative feedback system, causing overstimulation of the adrenal cortex and resulting in hyperplasia and over-secretion of the precursors to the enzymatic defect. The forms of CAH are summarized in Table 1. Impaired enzyme function at each step of adrenal cortisol biosynthesis leads to a unique combination of elevated precursors and deficient products. The most common enzyme deficiency that accounts for more than 90% of all CAH cases is 21-hydroxylase deficiency (21OHD).
|
|
||
|
Condition Onset Abnormality |
Genitalia Mineralocorticoid Effect |
Gene Chromosomal Location Typical Features |
|
Lipoid CAH |
Female, with no sexual development |
StAR 8p11.2 |
|
Lipoid CAH Congenital P450scc |
Female, with no sexual development |
CYP11A 15q23-24 |
|
3β-HSD deficiency Congenital |
Females virilized, males hypovirilized |
HSD3B2 1p13.1 |
|
17α-OH deficiency Congenital P450c17 |
Males hypovirilized, Hyperkalemic low-renin hypertension |
CYP17 CYP17 10q24.3 |
|
Classic 21-OH deficiency, salt wasting P450c21 |
Females prenatally virilized, males unchanged |
CYP21 6p21.3 |
|
Classic 21-OH deficiency, simple virilizing |
Females prenatally virilized, males unchanged |
CYP21 6p21.3 |
|
Non-classic 21-OH deficiency Postnatal |
All with normal genitalia at birth, hyperandrogenism postnatally |
CYP21 6p21.3 |
|
11β-OH deficiency Congenital P450c11B1 |
Females virilized, males unchanged |
CYP11B1 8q24.3 |
|
P450 Oxidoreductase deficiency (POR), Congenital P450 oxidoreductase
|
Males undervirilized, females unchanged Variable degree of mineralocorticoid deficiency |
P450 Oxidoreductase gene (POR) 7q11.2 Combined and variable enzymatic defects of P450c21, P450c17 and P450aro Wide range of phenotypes: normal to genital ambiguity +/- skeletal abnormalities (Antley Bixler type)
|
Classical CAH occurs in 1:13,000 to 1:15,000 live births. It is estimated that 75% of patients have the salt-wasting (SW) phenotype and the rest have simple-virilizing (SV) phenotype. Non-classical 21-OHD CAH (NC-CAH) is more common, and is one of the most common disorders in the Ashkenazi Jewish population with 1 in 27 Jews affected. CAH owing to 11β-hydroxylase deficiency (11β-OHD) is the second most common cause of CAH, accounting for 5-8% of all cases. The other forms of CAH are considered rare diseases and the incidence is unknown in the general population.
PATHOPHYSIOLOGY
Adrenal steroidogenesis occurs in three major pathways: glucocorticoids, mineralocorticoids, and sex steroids as shown in Figure 1. Glucocorticoids (particularly cortisol), androgens, and estrogens are synthesized in the zona fasciculata and reticularis; and aldosterone in the zona glomerulosa. The HPA feedback system is mediated through the circulating level of plasma cortisol by negative feedback of cortisol on CRF and ACTH secretion. Therefore, a decrease in cortisol secretion leads to increased ACTH production, which in turn stimulates (1) excessive synthesis of adrenal products in those pathways unimpaired by the enzyme deficiency and (2) an increase of precursor molecules in pathways blocked by the enzyme deficiency.
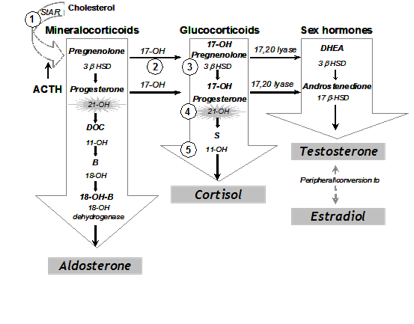
Figure 1. Pathways of Adrenal Steroidogenesis: Five enzymatic steps necessary for cortisol production are shown in numbers. 1= 20, 22 desmolase, 2= 17 hydroxylase (17-OH), 3=3ß-hydroxysteroid dehydrogenase (3ß HSD), 4=21 hydroxylase (21-OHD), 5=11ß hydroxylase (11-OH) In the first step of adrenal steroidogenesis, cholesterol enters mitochondria via a carrier protein called StAR. ACTH stimulates cholesterol cleavage, the rate limiting step of adrenal steroidogenesis.
The clinical symptoms of the five different forms of CAH result from the particular hormones that are deficient and those that are produced in excess as outlined in Table 1. In 21 OHD-CAH, there is an accumulation of 17-hydroxyprogesterone (17-OHP), a precursor to the 21-hydroxylation step, which is then shunted into the intact androgen pathway, where the 17,20-lyase enzyme converts the 17-OHP to D4-androstenedione, which is converted into androgens. Mineralocorticoid deficiency is a feature of SW-CAH, the most severe form of CAH. The enzyme defect in NC-CAH is only partial and salt wasting in this mild form of the disease does not occur. The analogy of all other enzyme deficiencies in terms of precursor retention and product deficiencies are shown in Table 1.
CLINICAL FEATURES
Genitalia
Females with Classical 21-OHD and 11β-hydroxylase deficiency CAH present at birth with virilization of their genitalia. Adrenocortical function begins around the 7th week of gestation; thus, a female fetus with classical CAH is exposed to adrenal androgens at the critical time of sexual differentiation (approximately 9 to 15 weeks gestational age). This leads to clitoral enlargement, fusion and scrotalization of the labial folds, and rostral migration of the urethral/vaginal perineal orifice, placing the phallus in the male position. Degrees of genital virilization are classified into five Prader stages (see Figure 2).
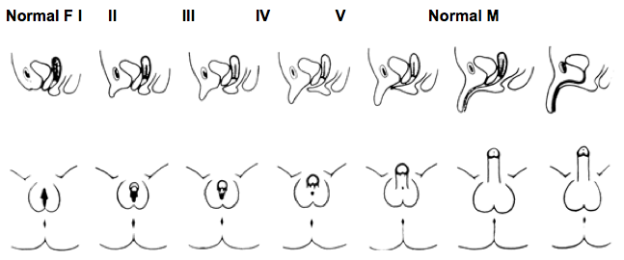
Figure 2. Different degrees of virilization according to the scale developed by Prader
Stage I: clitoromegaly without labial fusion
Stage II: clitoromegaly and posterior labial fusion
Stage III: greater degree of clitoromegaly, single perineal urogenital orifice, and almost complete labial fusion
Stage IV: increasingly phallic clitoris, urethra-like urogenital sinus at base of clitoris, and complete labial fusion
Stage V: penile clitoris, urethral meatus at tip of phallus, and scrotum-like labia (appear like males without palpable gonads)
Prader, A. Helv Paediatr Acta, 1954. 9:230-248.
Internal female genitalia, such as the uterus, fallopian tubes and ovaries, develop normally. Females with classical CAH maintain the internal genitalia potential for fertility.
Postnatal Effects, Growth and Puberty
Lack of appropriate postnatal treatment in boys and girls results in continued exposure to excessive androgens, causing progressive penile or clitoral enlargement, the development of premature pubic hair, axillary hair and acne. Advanced somatic and epiphyseal development occurs with exaggerated growth and is usually accompanied by premature epiphyseal maturation and closure, resulting in a final adult height that is typically significantly below that expected from parental heights. Excess glucocorticoid treatment can also lead to poor growth. The mean age at onset of puberty in both males and females is slightly younger than the general population. In those who are inadequately treated, central precocious puberty can occur. Following the onset of puberty, in a majority of successfully treated patients, the milestones of further development of secondary sex characteristics in general appear to be normal. In female adolescents and adults, signs of hyperandrogenism may include male-pattern alopecia (temporal balding), acne, hirsutism, menstrual irregularities, secondary PCOS and impaired fertility. Although the expected age of menarche may be delayed in females with classical CAH, when adequately treated many have regular menses after menarche. In males, short stature and impaired fertility are observed.
Gender Role Behavior and Cognition
Prenatal androgen exposure in females affected with classical forms of CAH not only has a masculinizing effect on the development of the external genitalia, but also on childhood behavior. Both physical and behavioral masculinization are related to each other and to genotype, indicating that behavioral masculinization in childhood is a consequence of prenatal androgen exposure. The majority of genetic females with CAH retain the female gender identity even in the setting of prenatal androgen exposure and postnatal hyperandrogenism.
Fertility
Difficulty with fertility in females with CAH may be due to anovulation, secondary polycystic ovarian syndrome, irregular menses, non-suppressible serum progesterone levels, or an inadequate introitus. Fertility is reduced in SW-CAH with rare reports of pregnancy. Non-classical CAH is an important and frequently unrecognized form of infertility. Males with CAH, particularly if poorly treated, may have reduced sperm counts and low testosterone as a result of high androstenedione concentrations which suppress gonadotropins and testicular adrenal rest tumors. Testicular adrenal rest tumors (TART) are thought to arise from aberrant adrenal cells in the testes; TARTs are always benign and mostly bilateral. Microscopic examination shows that adrenal rest cells are present in the testicles of all male patients with CAH and often detected radiographically in those with longstanding poorly controlled disease. Regular testicular examination and periodic testicular ultrasonography are recommended for early detection of adrenal rest tumors of the testes. However, MRI studies have been increasingly used to diagnose TARTs.
Salt-Wasting 21-Hydroxylase Deficiency
When the loss of 21-hydroxylase function is severe, adrenal aldosterone secretion is not sufficient for sodium reabsorption by the distal renal tubules, and individuals suffer from salt wasting as well as cortisol deficiency and androgen excess. Infants with renal salt wasting have poor feeding, weight loss, failure to thrive, vomiting, dehydration, hypotension, hyponatremia, and hyperkalemic metabolic acidosis progressing to adrenal crisis (azotemia, vascular collapse, shock, and death). Adrenal crisis can occur as early as age one to four weeks. Affected males who are not detected in a newborn screening program are at high risk for a salt-wasting adrenal crisis because their normal male genitalia do not alert medical professionals to their condition. It is important to recognize that the extent of genital virilization may not differ among SV-CAH and SW-CAH.
Simple-Virilizing 21-Hydroxylase Deficiency
The salient features of classical SV-CAH are prenatal virilization and progressive postnatal masculinization with rapid somatic growth and advanced epiphyseal maturation leading to early epiphyseal closure and likely short stature. There is no evidence of mineralocorticoid deficiency in this disorder and serum electrolyte concentrations are normal. Diagnosis at birth of a female with SV-CAH is usually made immediately because of the apparent genital ambiguity. Since the external genitalia are not affected in newborn males, hyperpigmentation may be the only clue suggesting increased ACTH secretion and cortisol deficiency. Diagnosis at birth in males thus rests on prenatal or newborn screening.
Non-Classical 21-Hydroxylase Deficiency
Individuals with the non-classical (NC) form of 21-OHD have only mild to moderate enzyme deficiency and present postnatally, eventually developing signs of hyperandrogenism. Females with NC-CAH do not have virilized genitalia at birth. NC-CAH may present at any age after birth with a variety of hyperandrogenic symptoms. While serum cortisol concentration is typically low in patients with the classic form of the disease, it is usually normal in patients with NC 21-OHD. Similar to classical CAH, NC-CAH may cause premature development of pubic hair, acne, secondary PCOS, advanced bone age with accelerated linear growth velocity, and short stature. In adult males, early balding, acne, infertility or short stature may prompt the diagnosis of NC-CAH.
DIAGNOSIS
Diagnosis of CAH must be suspected in infants born with ambiguous genitalia. The physician is obliged to make the diagnosis as quickly as possible to initiate therapy. The diagnosis and rational decision of sex assignment must rely on the determination of genetic sex, the hormonal determination of the specific deficient enzyme, genotype, and an assessment of the patient's potential for future sexual activity and fertility. As indicated in Table 1, each form of CAH has its own unique hormonal profile, consisting of elevated levels of precursors and elevated or diminished levels of adrenal steroid products. Diagnosis of the 21-OHD CAH can also be confirmed biochemically by a hormonal evaluation. In a randomly timed blood sample, a very high concentration of 17-hydroxyprogesterone (17-OHP), the precursor of the defective enzyme, is diagnostic of classical 21-OHD. Such testing is the basis of the newborn-screening program developed to identify classically affected patients who are at risk for salt wasting crisis. False-positive results are, however, common with premature infants. Appropriate references based on weight and gestational age are therefore in place in many screening programs. False negative results may occur if samples are drawn late in the afternoon as adrenal hormones exhibit diurnal variation. The gold standard for hormonal diagnosis is the corticotropin stimulation test (250 μg cosyntropin intravenously), measuring levels of 17-OHP and Δ4 androstenedione at baseline and 60 min. These values can then be plotted in the published nomogram (Figure 4) to ascertain disease severity. The corticotropin stimulation test should not be performed during the initial 24 hours of life as samples from this period are typically elevated in all infants and may yield false-positive results. Establishing a genetic diagnosis is not only important for the genotype-phenotype correlation, but also for genetic counseling for future pregnancies and for genetic counseling for the patient and his/her reproductive future.
For 21-OHD CAH, genetic analysis of the CYP21A2 gene may provide more clues to predict phenotypic severity. In about 50% of the causative genotypes, genotype-phenotype correlation can be found, although certain mutations can lead to variable phenotypes in different population groups especially in the simple virilizer group. Sequencing of the entire gene should be performed to detect rare mutations when genotype–phenotype non-concordance is observed in patients with CAH.
Newborn screening for CAH, which utilizes 17 hydroxyprogesterone levels, is a useful tool for early detection of CAH prior to the development of adrenal crisis in the affected neonate. However, screening is associated with a high rate of false positive results as levels are affected by prematurity and birth weight. Molecular genetics, especially genotyping of the CYP21A2 gene should be considered as a second-tier screening test in the new born screening program.
Prenatal testing for CAH in utero has historically utilized invasive techniques like amniocentesis and chorionic villus sampling which cannot be done prior to 14 weeks of gestation. Prenatal dexamethasone treatment must begin prior to genital formation occurring at approximately 9 weeks, in order to avoid genital ambiguity in the affected female fetus. Massive parallel sequencing using hybridization probes on cell-free fetal DNA in maternal plasma indicated that the fetal CAH status was correctly deduced as early as 5 weeks 6 days of gestation. This is a noninvasive technique that accurately diagnoses CAH before the ninth week of gestation.
TREATMENT
Routine Treatment
br />
The goal of therapy in CAH is to both correct the deficiency in cortisol secretion and to suppress ACTH overproduction. Proper treatment with glucocorticoid reduces stimulation of the androgen pathway, thus preventing further virilization and allowing normal growth and development. The usual requirement of hydrocortisone (or its equivalent) for the treatment of classical 21-OHD form of CAH is about 10-15 mg/m2/day divided into 2 or 3 doses per day and for non-classical 21-OHD 5-8 mg/m2/day divided into 2 or 3 doses per day. Hydrocortisone is the glucocorticoid of choice in the pediatric age group. Prednisolone and dexamethasone are not used in growing children given growth suppressive effects. A small dose of dexamethasone at bedtime (0.25 to 0.5 mg) is usually adequate for androgen suppression in non-classical adult patients. Adequate biochemical control is assessed by measuring serum levels 17-OHP and androstenedione; serum testosterone can be used in females and prepubertal males (but not newborn males). We recommend that hormone levels are measured at a consistent time in relation to medication dosing, usually 1-2 hours after the morning corticosteroid. Titration of the dose should be aimed at maintaining 17-OHP concentrations below 1000 ng/dL and androstenedione concentrations below 200 ng/dl. Over-treatment should be avoided because it can lead to Cushing syndrome. Patients with salt wasting CAH have elevated plasma renin in response to the sodium-deficient state, and they require treatment with the salt-retaining 9α-fludrocortisone acetate. The average dose is 0.1 mg daily (0.05-0.2 mg daily). Infants should also be started on salt supplementation, as sodium chloride, at 1-2 g daily, divided into several feedings. Measurements of plasma renin and aldosterone are used to monitor the efficacy of mineralocorticoid therapy. Advancement of bone age is monitored by bone age x-rays. Growth hormone therapy, in conjunction with a GnRH analogue, has been shown to be effective in improving final adult height. Patients may also experience peripheral precocious puberty, which requires treatment with gonadotropin-releasing hormone analogues. Aromatase inhibitors and growth hormone therapy should only be used in patients with a very short predicted final stature or in clinical trials. Use of aromatase inhibitors in CAH has been shown decrease bone maturation rates and some increase in adult height but the differences were not statistically significant.
Treatment During Illness and Emergency
Adrenal crisis can present as hypotension or shock and serum electrolyte abnormalities (hypoglycemia, hyponatremia, hyperkalemia, acidosis). During adrenal crisis, an immediate bolus of hydrocortisone 50-100 mg can be given intravenously or intramuscularly followed by hydrocortisone 100 mg/m2/day given as either continuous infusion or divided at least every 6 hours. Rehydration can be started with 20ml/kg isotonic saline with D5 as rapid bolus followed by repeat boluses or continuous infusion guided by level of dehydration. Hypoglycemia may require dextrose bolus and an initial bolus of 0.5-1 gram/kg of dextrose can be given intravenously at 2-3 ml per minute. If hyperkalemia is present, cardiac monitoring should be done to monitor for EKG changes. If changes are present, hyperkalemia should be treated using insulin with glucose infusion with or without other measures.
In non-life-threatening periods of illness or physiologic stress, the corticosteroid dose should be increased to 2 or 3 times the maintenance dose for the duration of that period, divided into 3 daily doses. Each family should be given injection kits of hydrocortisone, i.e. Solu-Cortef, for emergency use, and all family members should be trained in its intramuscular administration. The injectable dose of hydrocortisone in an emergency is 25 mg for infants, 50 mg for children under 40 kg, and 100 mg for children over 40 kg and for adults. In the event of a surgical procedure, 5-10 times the daily maintenance dose of hydrocortisone is needed, with 25-100 mg hydrocortisone IM/IV administered before and during a surgical procedure (as per infant, child, adult recommendations above), followed by high doses of hydrocortisone during the first 24-48 post-operative hours; the dose can then be tapered over the following days to the normal preoperative schedule. Stress doses of dexamethasone should not be given because of the delayed onset of action. It is not necessary for increased mineralocorticoid doses during these periods of stress. It is imperative for all patients who are receiving corticosteroid replacement therapy, such as patients with CAH, to wear a Medical Alert bracelet or medallion that will enable correct and appropriate therapy in case of emergencies. It is also crucial to re-educate parents at regular intervals on the life-threatening nature of this emergency.
GUIDELINES
Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline.
J Clin Endocrinol Metab. 2018 Nov 1;103(11):4043-4088
Rodriguez A, Ezquieta B, Labarta JI, Clemente M, Espino R, Rodriguez A, et al. Recommendations for the diagnosis and treatment of classic forms of 21-hydroxylase-deficient congenital adrenal hyperplasia. An Pediatr (Barc). 2017;87(2):116 e1- e10.
REFERENCES
New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, Yuen T, Khattab A. Congenital Adrenal Hyperplasia. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2017 Mar 15
El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 2017 Nov 11;390(10108):2194-2210
Fluck CE, Miller WL. P450 oxidoreductase deficiency: a new form of congenital adrenal hyperplasia. Curr Opin Pediatr. 2006;18(4):435-41.
Yilmaz R, Sahin D, Aghayev A, Erol OB, Poyrazoglu S, Saka N, et al. Sonography and Magnetic Resonance Imaging Characteristics of Testicular Adrenal Rest Tumors. Pol J Radiol. 2017;82:583-8.
New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2013;110(7):2611-6.
Balsamo A, Baldazzi L, Menabo S, Cicognani A. Impact of molecular genetics on congenital adrenal hyperplasia management. Sex Dev. 2010;4(4-5):233-48.
New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99(6):E1022-30.
Lin-Su K, Harbison MD, Lekarev O, Vogiatzi MG, New MI. Final adult height in children with congenital adrenal hyperplasia treated with growth hormone. J Clin Endocrinol Metab. 2011;96(6):1710-7.
Disorders of Sexual Differentiation in Newborns
INTRODUCTION
In 2006, new nomenclature for conditions previously referred to as intersex was proposed in a consensus statement from the Lawson Wilkins Pediatric Endocrine Society and European Society of Pediatric Endocrinology in response to advanced identification of molecular genetic causes of sex. Disorders of sexual differentiation (DSD) are congenital conditions within which the development of chromosomal, gonadal and phenotypic sex is atypical. These disorders have a broad differential including variations in sex chromosomes, variations in genes involved in gonadal and genital development, disorders in steroidogenesis within the gonads and adrenals, maternal factors, and endocrine disruptors. Classification of these disorders is based on sex chromosomes as such 46XX DSD, 46XY DSD, Ovotesticular DSD, and 46XX testicular DSD.
CLINICAL RECOGNITION
Given the potential association with glucocorticoid and mineralocorticoid deficiencies in CAH, the birth of a child with atypical genitalia constitutes a medical emergency requiring immediate evaluation. Further, the parents’ reaction to the birth of a child with atypical genitalia is one of shock and concern about which gender to assign, whether or not to decide for early genital surgery, and what to expect regarding the long-term outcome in terms of gender, sexual function, fertility, and general quality of life. In order to provide appropriate counseling to the family, there is an urgency to determine the etiology.
PATHOPHYSIOLOGY
The phenotypic sex of a newborn is the result of external genital development that is under the influence of sex-determining genes as well as both endogenous and exogenous hormone exposures.
The commitment of the bipotential primordial gonads to become testes or ovaries begins at 6 weeks and is fully achieved at 13-14 weeks. Gonadal differentiation is controlled by a number of time and dosage-sensitive genes including the SRY gene on the Y chromosome, SOX9, and WNT4 genes. The expression SRY and SOX9 and suppression of WNT4 expression is crucial to testicular differentiation. The expression of WNT4 in the absence of SRY and SOX9 expression allows for ovarian differentiation. Leydig cells produce insulin like factor 3 (INSL3) which is responsible for transabdominal phase of testicular descent.
Fetal productions of androgens from the Leydig cells within the testes and from the adrenal glands begins at approximately 8-9 weeks. External genitalia develop concurrently around the 9th week of gestation under the influence of androgens, mainly dihydrotestosterone (DHT). Testosterone is the principal hormone produced by the testes and is required for the onset of virilization and promotion of Wolffian ducts. Testosterone is converted to DHT by 5-alpha reductase. DHT leads to the development of the prostate, scrotum and phallus.
Anti-Mullerian Hormone (AMH) produced from Sertoli cells in the testes is required to support the development of Wolffian ducts including vas deferens, epididymis and seminal tubules in males. In females, Mullerian ductal structures including the uterus, fallopian tubes and cervix develop in the absence of AMH.
Disorders of Sexual Differentiation
46XX DSD
Patients with 46XX DSD are genotypic females with virilized characteristics. In 46XX DSD, the degree of genital virilization can be classified into five Prader stages. Stage 1, with the mildest degree of virilization, is characterized by clitoromegaly without labial fusion. Stage 5, with the highest degree of virilization, is characterized by clitoromegaly with the urethral meatus at the tip, labial fusion, and scrotal-like appearance of the labia.
46XX DSD can result from exogenous androgen exposure, endogenous adrenal androgen production or placental aromatase deficiency. Congenital adrenal hyperplasia (CAH) is the most common cause of 46XX DSD. The most common enzyme defects leading to CAH are 21-hydroxylase deficiency and 11-hydroxylase deficiency. In a very rare form of CAH owing to p450 oxidoreductase deficiency, there is a mutation in the P450 oxidoreductase (POR) enzyme which causes partial deficiency of 21-hydroxylase and 17a-hydroxylase/17,20 lyase activities. Affected females can present with virilization of the external genitalia, glucocorticoid deficiency, and skeletal malformations such as craniosynostosis.
Maternal hyperandrogenism during gestation can cause virilization of the external genitalia in females when the placental aromatase is overwhelmed. The hyperandrogenism can be due to luteomas, androgen producing tumors and exogenous exposure.
Maternal aromatase deficiency leads to decreased production of estrogen from androgen precursors. This leads to conversion of fetal DHEAS to androstenedione and testosterone by placental 3-beta hydroxysteroid dehydrogenase and virilization of female fetus.
The majority of 46XX testicular DSD cases are caused by translocation between the X and Y chromosome, involving the SRY gene.
46XY DSD
Patients with 46XY DSD are genotypic males with under-virilization. Micropenis is defined as a penile length less than 2.5 standard deviations below the mean penile length (<2.5 cm in a full-term newborn). The severity of hypospadias is based on the distance of the urethral opening from its normal position at the tip of the phallus. Lack of testicular palpation in the scrotum may signify cryptorchidism, vanishing testes, or gonadal dysgenesis.
46XY DSD can be caused by atypical testicular formation, low testosterone or dihydrotesterone production, or defects in the androgen receptor. In complete gonadal dysgenesis, there is no testicular development and patients present as phenotypic female with delayed puberty or amenorrhea. Up to 20% of these cases occur due to deletion or mutation of the SRY gene.
Defects in androgen biosynthesis can lead to under-virilization in a 46XY patient. These defects can occur at various points along the production pathway of testosterone from cholesterol. Adrenal dysfunction is associated with defects in steroidogenic enzymes such as steroidogenic acute regulatory protein (StAR), p450 side chain cleavage enzyme, 3 beta HSD type 2, 17 alpha hydroxylase/17,20 lyase. Other defects of testosterone production can occur in the following enzymes: 7-dehydrocholesterol reductase causing Smith Lemli Opitz syndrome and 17 beta hydroxysteroid dehydrogenase. Affected males with 5-alpha-reductase deficiency have atypical genitalia (small phallus and perineal hypospadias). With rises in testosterone at puberty, progressive virilization with phallic enlargement and testicular descent is seen.
Androgen insensitivity syndrome has been reported to be the main cause of 46XY DSD and is due to mutation in the androgen receptor. In complete androgen insensitivity, the androgenic effects of testosterone and dihydrotestosterone are abolished and patients have unambiguously female appearing external genitalia. In partial androgen insensitivity, the androgenic effects are attenuated and patients can present on a spectrum of under-virilization.
Mutations in genes responsible for sex determination such as SRY, SOX9, and SF1 lead to 46XY complete gonadal dysgenesis. Duplication of the DAX1 gene is associated with male to female sex reversal.
Endocrine disruptors with anti-androgenic effects such as diethylstilbestrol or phthalates can also lead to atypical genitalia in males.
|
Table 1. Laboratory Values to Differentiate Between Etiologies of Ambiguous Genitalia in Newborns with a 46, XY Chromosomal Complement. |
|||
|
Diagnosis |
T |
DHT |
MIS |
|
Androgen Insensitivity |
Normal/up |
Normal/up |
Normal |
|
5α-Reductase Deficiency |
Normal/up |
Low |
Normal |
|
Testosterone Biosynthetic Defect orLeydig Cell Hypoplasia |
Low |
Low |
Normal |
|
Gonadal Dysgenesis |
Low |
Low |
Low |
T=testosterone, DHT=dihydrotestosterone and MIS=müllerian inhibiting substance.
OVOTESICULAR DSD
Ovotesticular DSD, one of the rarest forms of DSD, describes patients that were previously categorized as true hermaphrodites. The gonads of patients with ovotesticular DSD contain both ovarian and testicular tissue. Thus, the presentation of genital ambiguity can be variable. In ovotesticular DSD in which the gonads contain both ovarian and testicular tissue, the majority have an XX chromosomal constitution. Complex mosaicism (XX/XY) are seen in approximately 10% of cases. Patients can present with a wide variety of genital ambiguity as well as a mixture of Wolffian and Mullerian structures.
DIAGNOSIS
Determination of chromosomal sex by karyotype with FISH analysis for SRY and pelvic ultrasound to evaluate for the presence of a uterus should be performed immediately. Currently, the only newborn screening test for steroid disorders is the measurement of 17-hydroxyprogesterone for 21-hydroxylase deficiency. Further laboratory evaluation to accurately diagnose the specific underlying defect should be directed by a pediatric endocrinologist. If CAH is suspected, measurement of adrenal hormones, ACTH stimulation testing, and molecular genetic testing can elucidate the form of CAH. Each form of CAH has its own unique hormonal profile, consisting of elevated levels of precursors and elevated or diminished levels of adrenal steroid products. HCG stimulation testing to assess testosterone and dihydrotestosterone response may be particularly helpful in 46XY DSD to assess testicular androgen production. Molecular genetic evaluations should be guided by chromosomal and hormonal evaluations.
Chromosomal sex can be determined prenatally invasively by chorionic villus sampling and amniocentesis and noninvasively via free fetal DNA in the maternal blood. Thus, DSD may be suspected in utero if the phenotype on prenatal ultrasonogram is discordant with the chromosomal sex.
THERAPY
When considering the gender of rearing, the prognosis for masculinization of brain and behavior, the anatomic and physiologic character of the reproductive tract with its potential for development and function in regard to both sexuality and fertility, and the social environment of the infant should be taken into account along with the genetic sex. Both male and female gender assignment should be thoroughly considered.
Sex hormone replacement is needed to induce pubertal development. Testosterone is used in the treatment of patients with testosterone deficiency (46XY DSD). Different forms of testosterone (topical and intramuscular) are available and treatment will vary depending on what is best for the patient. A short course of testosterone can be given during infancy to induce penile growth prior to surgical correction. For 46XY DSD patients with functioning Sertoli cells, HCG can be used to stimulate testicular production. Estrogen is used in the treatment of those reared female. Estrogen is available as an oral tablet or transdermal patch. Estrogen doses should be initiated at the lowest dose possible and slowly increased to a maximum of 0.625 mg/day of conjugated estrogen to allow for gradual breast development. Progesterone supplementation with estrogen is recommended in patients with a uterus.
Glucocorticoids are needed to treat congenital adrenal hyperplasia. They suppress the pituitary glands oversecretion of adrenocorticotropic hormone and thus decrease the production of precursor hormones. This also leads to a decrease in adrenal androgen production in forms of CAH associated with 46XX DSD.
The aim of surgical repair in patients with atypical genitalia reared in the female gender is generally to remove the redundant erectile tissue, preserve the sexually sensitive glans clitoris, and provide a normal vaginal orifice. A medical indication for early surgery other than for sex assignment is recurrent urinary tract infections as a result of pooling of urine in the vagina or urogenital sinus. In the past, it was routine to recommend early corrective surgery for neonates born with ambiguous genitalia. However, in recent years, the implementation of early corrective surgery has become increasingly controversial due to lack of data on long-term functional outcome. It is advised that all surgical decisions remain the prerogative of families in conjunction with experienced surgical consultants.
The process of assigning and accepting a gender of rearing for a child with ambiguous genitalia and of deciding the necessity of genital surgery is challenging. A team approach that combines the insights of the DSD-experienced pediatrician, endocrinologist, psychologist/psychiatrist, surgeon, and the child’s parents or guardian is essential. Although there is no consensus as to the appropriate age to disclose a condition, it is recommended to proceed gradually in line with the child’s cognitive and psychological development.
GUIDELINES
Audi L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus PM, Greenfield A, Bashamboo A, Hiort O, Wudy SA, McGowan R, The EUCA: GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 'DSDnet'. Eur J Endocrinol 2018;179:R197-R206.
Lee PA, Houk CP, Ahmed SF, Hughes IA, International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine S, the European Society for Paediatric E: Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 2006;118:e488-500.
Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC: Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2018;103:4043-4088.
REFERENCES
Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH, 3rd, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S, Global DSDUC: Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm Res Paediatr 2016; 85:158-180.
Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, Lauzanne K, Lee E: How sexually dimorphic are we? Review and synthesis. Am J Hum Biol 2000; 12:151-166.
Krishnan S W, AB: Ambiguous Genitalia in Newborns; in New MI LO, Parsa A, Yuen T, O'Malley B, Hammer G (ed) Genetic Steroid Disorders. San Diego, CA, Elsevier, 2014, pp 87-97.
New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, Yuen T, Khattab A. Congenital Adrenal Hyperplasia. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 Mar 15.
Domenice S, Arnhold IJP, Costa EMF, Mendonca BB. 46,XY Disorders of Sexual Development. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 May 3.