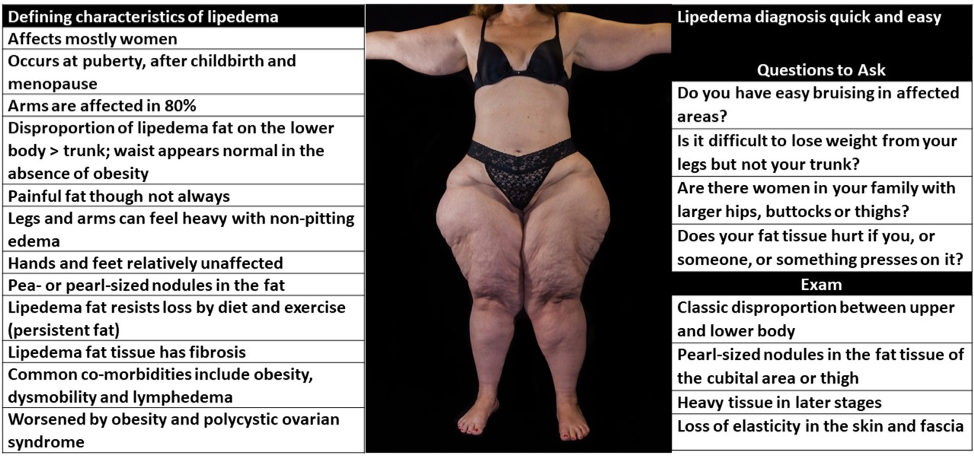
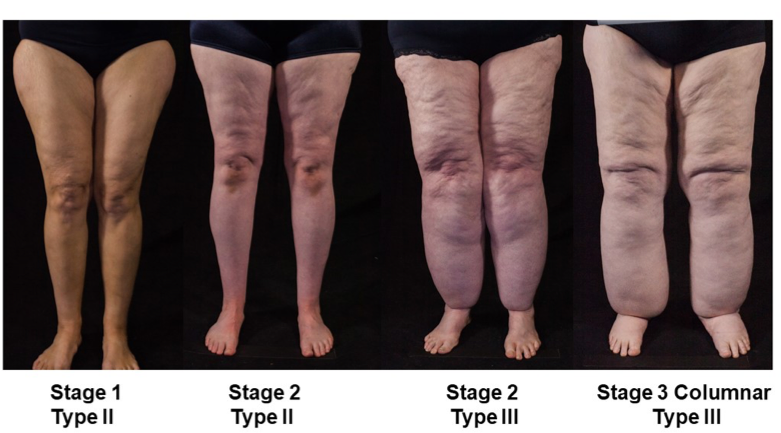
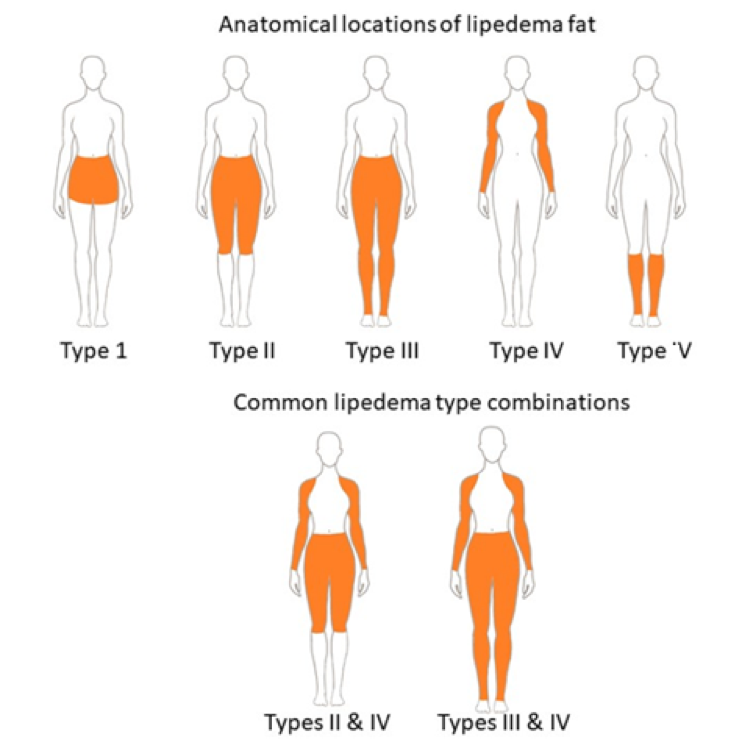
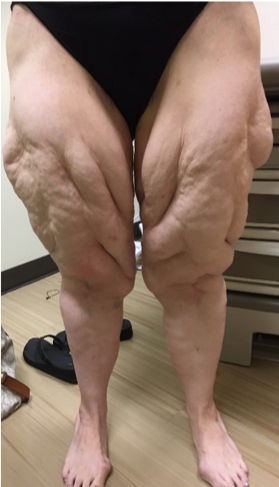
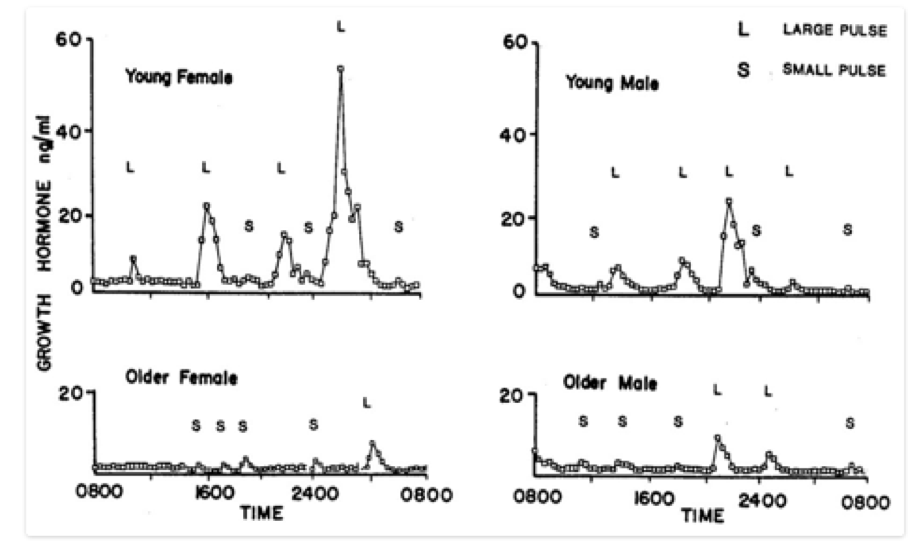
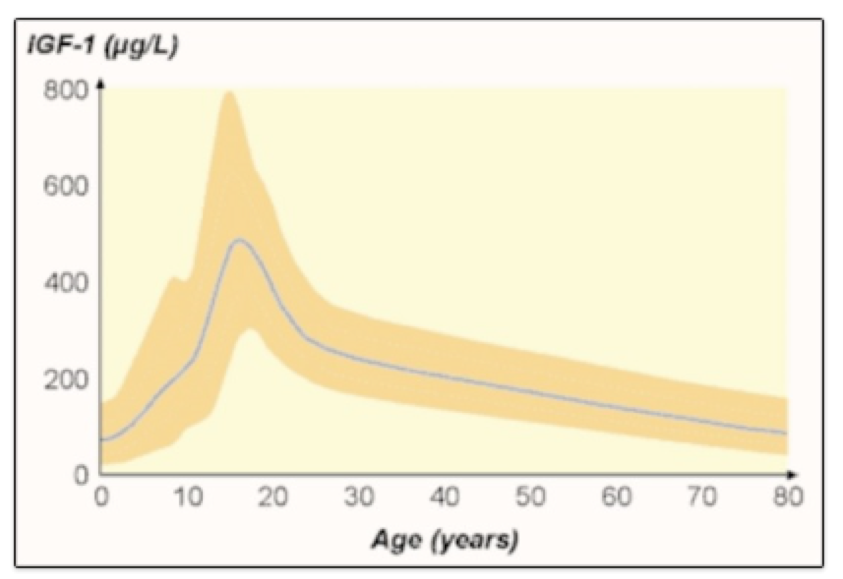
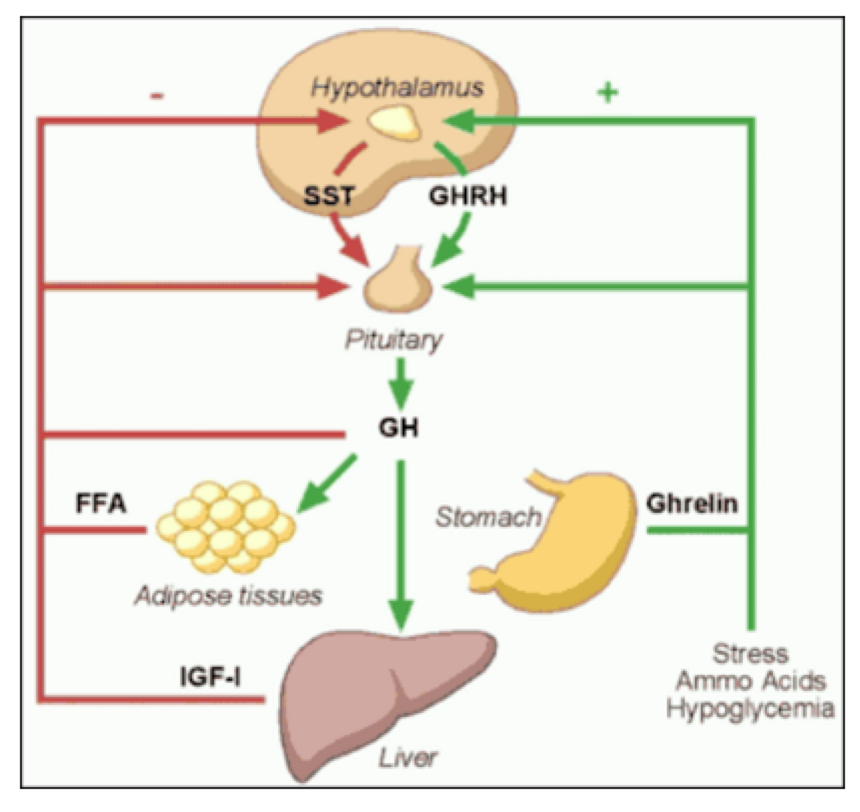
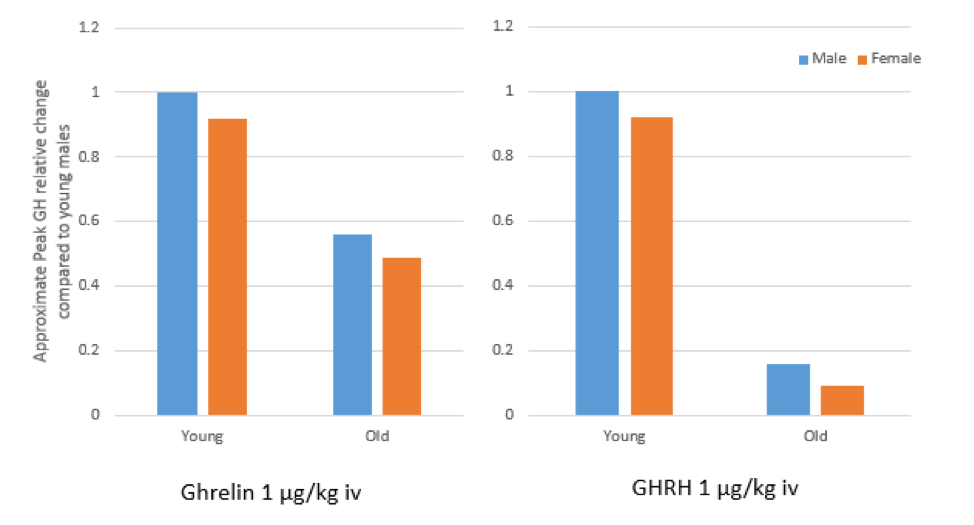
ABSTRACT
Obesity is pandemic and a multidisciplinary approach is critical for its management. Anti-obesity treatment includes lifestyle modifications combined with anti-obesity medications. Anti-obesity drugs target either central nervous system pathways, which regulate sensations of satiety and fullness, or peripheral modulators of digestion, metabolism and lipogenesis. Combined anti-obesity agents is a novel, promising field, especially the co-administration of gut hormone analogues with centrally acting molecules. Consequently, it is hoped that in the near future, individualized pharmacological management of obesity could be meaningfully achieved by targeting different pathways governing energy homeostasis and weight regulation. This chapter reviews potential molecular targets of the energy homeostasis system along with new anti-obesity drugs currently under investigation.
INTRODUCTION
The pathophysiology that leads to obesity is considered a novel field for research. Understanding human metabolism and the homeostatic mechanisms of weight regulation includes comprehension of the interaction between central nervous system and peripheral modulators of weight maintenance. Current anti-obesity molecular pharmacotherapy is based on single molecule anti-obesity drugs that act either via enhancement of satiety feeling, inhibition of hunger, or triggering of catabolism. However, on average, the weight-lowering effects of these medications are modest at best and side effects are common.
According to current clinical practice guidelines for pharmacological management of obesity published in 2015 by The Endocrine Society, if a patient’s weight is not responsive to lifestyle intervention, weight loss pharmacotherapy can be offered for a BMI ≥27kg/m2 when an obesity-related comorbidity is present, or when the BMI is ≥30kg/m2 (1). In fact, pharmacologic weight management should be considered in patients who meet these weight criteria and have any of a number of chronic conditions in which obesity is considered to play a major role, including type 2 diabetes mellitus (T2DM), cardiovascular disease, hypertension, dyslipidemia, obstructive sleep apnea, nonalcoholic fatty liver disease, certain cases of malignancies (i.e. endometrial, breast, colon) (2), osteoarthritis, depression (3), and infertility (4).
Currently, there are six anti-obesity medications that have received US Food and Drug Administration (FDA) approval: orlistat, phentermine, phentermine/topiramate extended release (ER), lorcaserin, naltrexone sustained release (SR)/bupropion SR, and liraglutide (the only injectable formulation). At the same time, the European Medicines Agency (EMA) has approved only three of these: orlistat, bupropion/naltrexone and liraglutide.
Considering the extent to which obesity impairs health alone or through expression of one or more of these comorbidities, the need for new molecular pharmaceutic agents is crucial. As detailed below, future weight-loss medications will be based on our knowledge of key regulatory sites of weight regulation and energy homeostasis so as to achieve greater efficacy while minimizing off-target side effects, characteristics that are necessary for approval by both American and European drug regulatory agencies.
TARGETS OF PHARMACOTHERAPY IN THE MANAGEMENT OF OBESITY
Novel insights provided by pathophysiology indicate the presence of a complex homeostatic system in which information about the energy reserve status and the meal quality and content is relayed from the periphery (gastrointestinal tract, pancreas, and adipose tissue) via specific orexigenic and anorexigenic peptides and hormones to the central nervous system (CNS). Peripheral peptide hormones are released postprandially and travel in the circulation to bind to their receptors in the homeostatic regulatory centers in the CNS, notably the arcuate nucleus (ARC) of the hypothalamus and the dorsal vagal complex (DVC) in the brainstem medulla. The ARC contains neurons expressing key orexigenic neurotransmitters, agouti-related peptide (AgRP) and neuropeptide Y (NPY), as well as anorexigenic neurotransmitters, proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART). Food intake is thus modulated by complementary mechanisms so as to maintain energy and weight homeostasis. New drug therapies have begun to focus on combination therapy using medications that target more than one of these central pathways, thereby achieving more favorable weight loss outcomes. In addition, combining treatments may provide a better safety profile given that lower doses of each drug when used together may achieve better weight loss than higher doses of a single agent (see Figure 1 below).
Factors That Influence Appetite
The regulation of satiety and appetite depends on the interaction of three major factors: biological systems, modern macro-environmental exposures, and micro-environmental influences. Biological systems are shaped by genetic and epigenetic influences from early-life events that govern development of orexigenic and anorexigenic neuro-hormonal pathways involved in the pathophysiology of obesity. Modern macroenvironment (food production, consumption, availability, social structure, weather influencing physical activity, television and technology, cultural norms, endocrine disruptors) and microenvironment (nutrition, exercise, sleep, stressful lifestyle, circadian rhythm) play an important role in the conformational development of cognitive and emotional brain regions, thus predisposing to the obese phenotype.
Genetic Factors of Physical Activity
Specific genes predict to what extent adults remain active. This is evidenced in a study examining identical twins in which environmental factors shared by children at age 13 accounted for 78% to 84% of sport participation, whereas genetic differences provided no contribution at all. At the age of 17 to 18 the genetic influences represented 36% of the variance in the level of participation in sports, and by age 18 to 20, genetic factors were responsible for almost all (85%) of the differences in participation in sports.
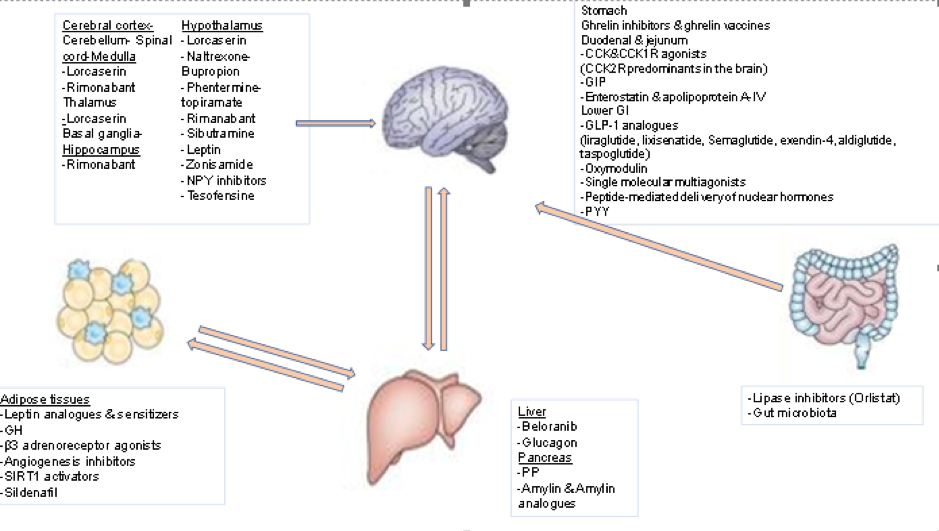
Figure 1. Sites of Action of the Most Important Anti-Obesity Drugs
CENTRALLY-ACTING ANTI-OBESITY DRUGS
Monoamine Neurotransmitter Modulators
With the exception of the glucagon-like peptide 1 (GLP-1) receptor agonist liraglutide, currently available weight loss medications act on the central nervous system to enhance dopamine, norepinephrine, and serotonin action to enhance satiety, diminish hunger, and consequently affect weight loss. Drug combinations have opened new horizons as they use multiple neural pathways, leading to better results with less adverse events. Recently, a review of fifty reports involving 43,443 subjects compared the efficacy of the central acting anti-obesity drugs lorcaserin (5HT2c receptor agonist), naltrexone-bupropion (opioid receptor antagonist combined with a norepinephrine releasing agent that stimulates POMC neuronal firing), phentermine-topiramate (a norepinephrine and dopamine modulator plus a carbonate anhydrase inhibitor), and liraglutide. It was found that the maximal mean weight loss relative to placebo was -3.06, -6.15, -7.45, and -5.5kg after 1 year with mean weight regain +0.48kg, +0.91kg, +1.27kg, +0.43kg the following year, respectively. In these studies, the one-year drop-out rate was 40.9%, 49.1%, 34.9%, 24.3%, respectively (5).
Leptin, Leptin Analogues and Leptin Sensitizers
Leptin is a protein secreted primarily by white adipose tissue (WAT). It directly stimulates anorexigenic POMC neurons and inhibits adjacent orexigenic NPY neurons in the ARC of the hypothalamus, thus promoting satiety, increasing energy expenditure, and resulting in weight loss (6). Circulating levels of leptin increase with adiposity and decline following body weight reduction; the latter might be implicated in the total and resting energy expenditure reduction seen after weight loss. The discovery of leptin in 1994 was a seminal event in obesity research. It helped to establish that body weight should be viewed as a disorder with a strong biological basis rather than simply the result of poor lifestyle choices. Studies with congenitally leptin-deficient, severely obese subjects revealed that administration of physiological doses of leptin decreased food intake and body weight (7). Obese individuals, however, are leptin-resistant and have increased circulating leptin levels. Whether administration of leptin could overcome leptin resistance and exert an anti-obesity effect was tested in a placebo-controlled study with 47 obese men and women given varying doses of recombinant human leptin (0.03 mg/kg and 0.30 mg/kg, respectively) for 24 weeks and advised to eat 500 kcal less than body requirements each day. A dose-dependent decrease in body weight was shown, ranging from -1.3 kg in the placebo group to -1.4 kg in the 0.03 mg/kg leptin-treated group, and to -7.1 kg in the 0.30 mg/kg leptin-treated group (8). These results suggested that leptin resistance can be overcome with high doses of leptin but resulting in only modest weight loss similar to currently approved medications. In addition, whether these effects can be sustained long-term is not known. Reports were similar from animal studies testing the effect of leptin sensitizers targeting the protein tyrosine phosphatase-1B (PTP1B)(9)(10) or the chemical chaperones that repair ER stress, including 4-phenyl butyric acid (PBA) and tauroursodeoxycholic acid (TUDCA) (11), each of which demonstrated reduced food intake and body weight. Like leptin treatment, sustainability of these anti-obesity effects is still not clear.
Weight loss is associated with reduction in energy expenditure, which makes long term weight loss maintenance difficult (12). Furthermore, 6 days of high fat diet in mice suffice to dramatically decrease the levels of phosphorylated signal transducer and activator of transcription 3 (p-STAT3) in the arcuate nucleus (13) while short term overfeeding of normal weight mice can lead to an increase of leptin resistance (14). Besides the inefficiency of leptin analogues as monotherapy, combinations of leptin with amylin (15), fibroblast growth factor 21(FGF21), exendin4, (16), or a GLP-1/glucagon co-agonist (17) were proposed. Only the combination with the GLP-1/glucagon co-agonist has shown improvement of leptin sensitivity (18). Apart from diet, stress of endoplasmic reticulum contributes to leptin resistance (19). Several plant-derived substances, such as celastrol (20) and withaferin (21) have been tested in diet-induced obese rodents for improvement of this pathway that leads to leptin resistance.
METRELEPTIN
Metreleptin (MYALEPT) is an injectable human recombinant leptin analogue approved in Japan for metabolic disorders including lipodystrophy and in USA as first-line treatment for non-HIV related forms of generalized lipodystrophy (leptin deficiency, congenital/acquired lipodystrophy) (22). A previous indication for hypothalamic amenorrhea has been withdrawn (23). (see Table 1)
|
Table 1. Metreleptin (MYALEPT) |
|
|
FDA approved/Phase |
Approved in Japan for lipodystrophy disorders and in USA for non-HIV lipodystrophy |
|
Mechanism of action |
Human recombinant leptin injectable analogue |
|
Clinical Benefits |
↓blood glucose, triglycerides, hepatic fatty steatosis |
|
Adverse events |
Headache, hypoglycemia, decreased weight, abdominal pain -previous indication for hypothalamic amenorrhea discontinued |
PRAMLINTIDE/METRELEPTIN
The combination of amylin-leptin (pramlintide-metreleptin) has been shown to be effective in the treatment of obesity. The anti-obesity properties of the combined treatment with pramlintide and metreleptin (pramlintide/metreleptin) were tested and showed a significant weight reduction of 12.7 ± 0.9% (11.5 ± 0.9 kg) without plateauing in obese patients during a 20-week trial period (24). The sponsors subsequently announced positive results from a 28-week proof-of-concept study with pramlintide and metreleptin combination treatment in overweight or obese subjects. The combination treatment reduced body weight on average by 12.7%, significantly more than treatment with pramlintide alone (8.4%), which is interpreted as 10 pounds more weight loss with the combined treatment. Remarkably, subjects receiving pramlintide/metreleptin continued to lose weight until the end of the study, compared to those treated with pramlintide alone, whose weight loss had stabilized towards the end of the study. The magnitude of weight loss was found to be dose-dependent and baseline BMI-dependent. Patients with a starting BMI less than 35 kg/m2 experienced the best weight loss efficacy with the combined treatment. A year later, the results of the 52-week blinded, placebo-controlled Phase II extension study of pramlintide/metreleptin were announced. The results indicated sustained and robust weight loss through the combined treatment; again, the most robust efficacy was seen in patients with a BMI less than 35 kg/m2 (25). Although the pramlintide/metreleptin combination seemed to be the next promising anti-obesity drug to be marketed, the sponsors discontinued its development in 2011, following commercial reassessment of the program (26).
Melanocortin-4 Receptor Agonists
The melanocortin system has a highly significant role in the hypothalamic regulation of body weight and energy expenditure. Leptin inhibits the release of the orexigenic neuropeptides orexin and melanocortin-concentrating hormone (MCH) in the lateral hypothalamic area (LHA) through the release of CART and melanocyte-stimulating hormone (α-MSH). The latter derives from the cleavage of POMC by prohormone convertase-1 and acts via melanocortin-3 and -4 receptors (MC3R, MC4R) activation. α-MSH emerged as a promising novel anti-obesity drug, and intranasal administration of the melanocortin sequence MSH/ACTH4-10 to normal-weight subjects was shown to acutely increase subcutaneous WAT lipolysis (27) and decrease body fat by 1.7 kg, when administered for six weeks (28). It eventually proved not to induce any significant reduction in body weight or body fat when compared with placebo in a 12-week study of 23 overweight men.
In preclinical studies, obese primates treated for eight weeks with the MC4R agonist RM-493 (Setmelanotide) lost an average of 13.5% of their body weight, with significant improvements in both insulin sensitivity and cardiovascular function. In June 2014, the results from the first human Phase II trial were released, testing the hypothesis that an MC4R agonist increases resting energy expenditure in obese subjects. A total of 12 obese but otherwise healthy individuals were randomized and completed both RM-493 and placebo periods in this double-blind, placebo-controlled, two-period crossover study. Analysis of the data indicates that short-term treatment with RM-493 increased resting energy expenditure significantly (by 6.4% vs placebo), thus suggesting RM-493 may be clinically effective for treating obesity. In 2015, administration of Setmelanotide to obese individuals for a limited time increased resting energy expenditure (REE) by 6.4% and shifted substrate oxidation to fat (29). Currently, Setmelanotide is being tested as a therapeutic option for rare genetic disorders of obesity such as POMC deficiency, heterozygous deficiency obesity, and POMC epigenetic disorders (30-32). (see Table 2)
|
Table 2. Setmelanotide (RM-493) |
|
|
FDA approved/Phase |
Phase II |
|
Mechanism of action |
MC4R-agonist |
|
Weight loss vs placebo |
13.5% |
|
Clinical Benefits |
↑insulin sensitivity, cardiovascular function, energy expenditure, ↓ body weight -tested for POMC deficiency, heterogenous deficiency obesity, POMC epigenetic disorders |
|
Adverse events |
Headache, arthralgia, nausea, spontaneous penile erection, female genital sensitivity |
Melanin-Concentrating Hormone (MCH) Antagonists
The melanocortin-concentrating hormone (MCH) is an important orexigenic neuropeptide in the LHA. Its release is stimulated by NPY and inhibited by leptin, exerting its orexigenic effects through the MCH1 receptor (MCHR1) (33). Like NPY, MCH exerts pleiotropic effects on locomotor activity, sensory processing, anxiety, aggression, and learning. Thus, despite the role of MCH in hunger stimulation, MCHR1 blockade as an anti-obesity target is questionable because such inhibition could elicit undesirable side effects. In animal models, MCH antagonists have consistently demonstrated efficacy in reducing food intake acutely and in inhibiting body weight gain when given chronically (34). Five compounds have reached testing in human subjects. Although they were reported as well-tolerated, none has proceeded to Phase II studies. A major issue with many lead compounds is increased cardiovascular risk due to drug-induced QTc prolongation (35). Among others, the MCHR1 antagonist AMG 076 entered Phase I safety and tolerability testing in 2004, but there have been no subsequent reports of its status since 2005. The MCHR1 antagonist GW-856464 also entered Phase I studies in 2004; however, in 2010 it was reported that low bioavailability precluded further development. The MCHR1 antagonist NGD-4715 was safe and well-tolerated in a Phase I clinical trial, but its development ceased in 2013. Similarly, despite the reported tolerability and indication of efficacy of the MCHR1 antagonist ALB-127158, its development was terminated before the initiation of Phase II studies. Finally, the longest (28-day) Phase I study with BMS-830216, a pharmacological antagonist of MCH signaling (36) produced no indications of weight loss or reduced food intake and the compound did not proceed to Phase II studies.
Subtype-Selective Serotonin-Receptor Agonists
Central serotonin participates in feeding behavior and energy balance modulation, reducing food intake in animals and human beings. This finding was supported by reports of two selective serotonin reuptake inhibitors (SSRIs) developed to treat depression, fluoxetine and sertraline, being associated with non-sustained weight loss in obese subjects. Thus, agonists to appropriate serotonin receptors are potentially valuable drugs. The serotonin (5-HT) system directly modulates the hypothalamic POMC (anorexigenic) and NPY (orexigenic) networks, enhancing satiety and causing hypophagia. These effects are mediated by 5-HT2C and 5-HT1B receptors, located on hypothalamic POMC and NPY neurons, respectively. Through the 5-HT1B receptors, serotonin inhibits the NPY/Agrp neurons, thereby decreasing the GABAergic inhibitory input to POMC cells; while through the 5-HT2C receptors it directly activates the anorexigenic POMC neurons. Via these actions, serotonin increases α-MSH and decreases AgRP release into the hypothalamic melanocortin system, promoting satiety. Between 1973 and 2000 there was an explosion in the pharmaceutic industry regarding central acting anti-obesity drugs. Three non-selective serotonin-receptor agonists were approved by FDA: fenfluramine (1973-1997), the combination phentermine-fenfluramine (1992-1997), and dexfenfluramine (1996-1997). These were all 5-HT1b agonists characterized for their ability to inhibit food consumption, but also had effects on other serotonin receptors that lead to unacceptable side effects (cardiac valvular thickening) and were voluntarily withdrawn from the market.
In 1997, when fenfluramine and dexfenfluramine were discontinued by the manufacturer, sibutramine, a serotonin and norepinephrine reuptake inhibitor emerged. Sibutramine has only little clinical relevance as an antidepressant but enhances weight loss due to an increase in energy expenditure and inhibition of food intake (37). In addition to weight loss, sibutramine was found to improve fasting levels of insulin, triglycerides, and high-density lipoprotein cholesterol. Sibutramine was also associated with increase of blood pressure, cardiovascular events, and cardiac arrhythmias (38). For these reasons, FDA withdrew it in 2010.
LORCASERIN
As activation of the 5-HT1B receptor has been implicated in both primary pulmonary hypertension (39) and valvopathy (40), the 5-HT2C receptor subtype has been proposed as a target for therapeutic intervention to allow weight loss. Several potent and selective 5-HT2Creceptor agonists proved to be effective in suppressing food intake and inducing weight loss in rodents, including WAY-163909 (41), CP-809101 (42), and vabicaserin (43). However, only lorcaserin (APD356) moved into clinical testing. Lorcaserin (Belviq) is a selective 5-HT2c receptor agonist, which belongs in the third generation of 5-HT-based anti-obesity drugs (44). It activates hypothalamic POMC neurons to induce satiety and decrease food intake but does not affect energy expenditure. Through actions on midbrain dopaminergic tone, it has been shown to suppress binge- food behaviors. Its action in addictive disorders is currently under investigation (45). Based on the outcome of the BLOOM (46) and BLOSSOM trials (47), in 2012 the FDA approved lorcaserin as an addition to a reduced-calorie diet and exercise for eligible patients (48). The efficacy of lorcaserin appears similar to that of orlistat (mean difference in weight loss between active and placebo treated groups approximately 3 to 4 kg) and perhaps slightly less than that of phentermine-topiramate. The impact of lorcaserin in patients with T2DM and BMI: 27-45kg/m2 was examined in the BLOOM-DM trial which showed a reduction of body weight by approximately 5kg versus 1.6kg in the placebo group, as well as significant decreases in heart rate, HDL levels, and waist circumference. Valvopathy was shown not to occur in excess with treatment and lorcaserin was generally well tolerated, with a low incidence of side effects such as headache, dizziness, fatigue, nausea. After the results of BLOOM-DM trial, a potential combination of GLP-1RA and 5-HT2A/C is now under investigation (49).
In a multicenter, randomized, double-blind, placebo-controlled, parallel-group study involving12,000 overweight and obese patients with cardiovascular disease or multiple cardiovascular risk factors (CAMELLIA-TIMI 61), the effect of long-term treatment with lorcaserin on major cardiovascular events and conversion to T2DM over a 5-year period were examined. After one year of treatment, 5% weight loss was observed in 38.7% and 17.4% in the lorcaserin and the placebo groups, respectively. Regarding cardiac risk, the lorcaserin group was non-inferior to the placebo group with slightly better values in cardiac risk factors (blood pressure, heart rate, glycemic control, lipid profile). Adverse events were rare in both groups, apart from the incidence of serious hypoglycemia in the lorcaserin group in those with diabetes managed using insulin or sulfonylureas (50, 51). In addition, lorcaserin administration decreased the incidence of T2DM by 19% in patients with prediabetes and by 23% in patients without diabetes. In patients with T2DM, lorcaserin resulted in a reduction of 0.33% in HbA1c compared with placebo at 1 year from a mean baseline of 7.0%. (see Table 3, 4)
|
Table 3. Lorcaserin (Belviq) |
|
|
FDA approved/Phase |
2012 |
|
Mechanism of action |
Selective Serotonin 2C agonist |
|
Weight loss vs placebo |
3-4kg |
|
Clinical Benefits |
↓food intake, heart rate, HDL levels, waist circumference, HbA1c |
|
Adverse events |
Headache, dizziness, fatigue, nausea, dry mouth, constipation, heart valvopathy -In diabetics: hypoglycemia, headache, back pain, cough, fatigue, risk of serotonin syndrome/neuroleptic malignant syndrome, valvular heart disease |
|
Table 4. Clinical Trials of Lorcaserin |
|||||
|
Clinical trial |
Patients |
Dose |
Treatment, placebo from baseline |
% of patients losing ≥5% of baseline weight |
Comment
|
|
BLOSSOM 1-year randomized, double-blind, placebo-controlled trial (2011) |
4008 patients (18-65 y.o., BMI- 30-45kg/m2 or 27-29.9kg/m2 with comorbidity) randomized in a 2:1:2 ratio |
i.10mg x2 po
ii.10mg x1 po
iii.placebo |
i.-5.8kg
ii.-4.7kg
iii.-2.9kg |
i.47.2%
ii.40.2%
iii.25% |
Exclusion criteria: recent cardiovascular events, diabetes mellitus, BP >150/95mmHg |
|
BLOOM 2-year randomized, double-blind, placebo-controlled trial (2010)
|
3182 adults (mean BMI-36.2kg/m2) randomized to lorcaserin twice daily or placebo group. After 52 weeks, the placebo group continued placebo and lorcaserin group selected placebo or lorcaserin for 52 weeks |
i.10mg x2 po
ii. placebo |
i.-5.8kg
ii.-2.2kg |
i.47.5%
ii.20.3% |
Weight loss was greater in the group which continued lorcaserin for the second year |
|
BLOOM-DM 1-year randomized, double-blind, placebo-controlled trial (2012) |
604 patients (HbA1c: 7-10%, BMI-27-45kg/m2, treatment with metformin, sulfonylurea or both) |
i.10mg x2 po
ii.10mg x1 po
iii.placebo |
i.-4.7kg
ii.-5.0kg
iii.-1.6kg |
i.37.5%
ii.44.7%
iii.16.1% |
↓heart rate, HDL levels, waist circumference in lorcaserin treated groups NO valvopathy was statistically significant |
|
CAMELLIA-TIMI 61 3.3-year randomized, placebo-controlled trial (2018)
|
12,000 patients overweight/obese-three subgroups A. diabetes B. prediabetes C. normoglycemic |
i.10mg x2/day
ii. placebo |
At 1 year the mean treatment difference: A: -2.6kg B: -2.8kg C: -3.3kg
|
At 1 year compared with placebo: A: 37.4% B: 39.7% C: 42.3% |
↓ BMI, waist circumference, waist-to-hip ratio, HbA1c, reduced microvascular complications |
Bupropion
Bupropion is a dopamine and norepinephrine-reuptake inhibitor that has been marketed as an anti-depressant and for smoking cessation. Previous animal studies have clearly shown a dose-dependent satiety effect of bupropion following intraperitoneal injection (52). The acute effects of dopamine and noradrenaline reuptake inhibition on energy homeostasis demonstrated their additive effects on short-term food intake (53). Bupropion increases dopamine activity and POMC neuronal activation, thereby reducing appetite and increasing energy expenditure (54). Whether the acute meal terminating effects of bupropion documented in animal studies could be translated into long-term weight loss efficacy in humans was addressed by three clinical trials with overweight and obese adults (55, 56, 57) using different treatment doses (100 to 400 mg/d) and duration (up to 24 weeks). They have all shown bupropion to have dose-dependent modest weight reducing efficacy, plus a safe profile. One study that assessed the anti-obesity efficacy of bupropion over two years reported maintenance of weight loss during the continuation phase, while another demonstrated its efficacy even in depressed patients. Although the weight loss effect of bupropion was superior in non-depressed patients compared to those suffering from depression, the fact that bupropion was well-tolerated and effective in this group of patients provides a potential valuable adjunctive therapy to elevate mood in depressed subjects in whom weight gain secondary to antidepressant therapy is an issue. Cardiovascular effects, such as a rise in blood pressure and tachycardia, were usually mild, while the risk of seizure, which was high with the original bupropion formulation, has been significantly reduced with the advent of bupropion-SR and bupropion-ER.
An interesting finding of the previous studies was that the rather modest weight loss effect of bupropion reached a plateau by 24 weeks of treatment. This could be explained by the molecular pathophysiology of the weight reducing effects of bupropion, which directly stimulates the hypothalamic POMC neurons that in turn release α-MSH and β-endorphin. α-MSH mediates the anorectic effect of POMC activation, whereas β-endorphin exerts negative feedback on POMC neurons via opioid receptors (58). The latter possibly points to one of the compensatory mechanisms that limits long-term efficacy of bupropion and other weight loss modalities.
Naltrexone
Naltrexone is an opioid receptor antagonist. By blocking opioid receptors on the POMC neurons, feedback inhibition is prevented further increasing POMC activity. Monotherapy with opioid antagonists to decrease short-term food intake has been tested (59). Naltrexone failed to produce consistent or clinically meaningful weight loss, even at large doses (300 mg/d) (60), implying that a single opioid mechanism is unlikely to explain all aspects of ingestive behavior.
Bupropion/Naltrexone Sustained Release (SR)
The combined bupropion/naltrexone (NB) therapy induced significantly greater weight loss on a diet and exercise program over 56 weeks compared to monotherapy and placebo (61). In 2014, the FDA approved this combination (Contrave, Mysimba) for body weight management in adults who are overweight and obese. This combined therapy of opioid antagonist and aminoketone antidepressant is titrated over four weeks to the maximum dose. NB has shown remarkable benefit in patients with binge-eating disorder (BED) and concomitant alcohol abuse, but this result needs further evaluation (62). Four major 56-week phase III randomized, double-blind, placebo-controlled trials have shown the therapeutic effect of ΝΒ SR (COR-I, COR-II, COR-BMOD, COR-DIABETES) in different dosage combinations (see Table 6). In COR-I, the weight loss ratio on NB 16/360mg, NB 32/360mg or placebo was -5.0%, -6.1%, -1.3% (P<0.00) respectively. In COR-II, the weight loss ratio on NB 32/360mg or placebo was -6.4%, -1.2% (P<0.001) (63). In COR-BMOD, NB SR 32/360mg plus intensive behavioral modification was compared with the behavioral modification alone as a therapeutic option. The weight loss ratio was -11.5% versus -7.3% (P<0.001), respectively (64). Recently, COR-Diabetes has included patients with T2DM with or without antidiabetic treatment. The NB SR 32/360mg treatment resulted in -5.1% weight loss versus -1.8% in the placebo group (P<0.001). NB treatment resulted in a HbA1c reduction, cardiovascular benefit, and lipid profile improvement (65). Due to FDA request for further investigation of the effect of NB on major cardiovascular events, the LIGHT trial was created. Unfortunately, this trial terminated early following recommendation by the academic leadership of the study because confidential interim data were publicly released by the sponsor (66). (See Table 5, 6)
|
Table 5. Bupropion/Naltrexone Sustained Release (Contrave, Mysimba+ |
|
|
FDA approved/Phase |
2014 |
|
Mechanism of action |
Aminoketone antidepressant/Opioid antagonist |
|
Weight loss vs placebo |
4.8kg |
|
Clinical Benefits |
↓ appetite |
|
Adverse events |
Nausea, constipation, headache, vomiting, dizziness, insomnia, dry mouth, suicidal ideation, increase blood pressure/heart rate, hepatotoxicity, angle-closure glaucoma Uncontrolled hypertension, seizures, anorexia nervosa/bulimia, chronic opioid use, coadministration with MAO inhibitors |
|
Table 6: Clinical trials of Naltrexone/Bupropion SR |
|||||
|
Clinical trial |
Patients |
Dose |
Treatment, placebo from baseline |
% of patients losing ≥5% of baseline weight |
Comment
|
|
COR I 1-year randomized, double-blind, placebo-controlled trial (2010) |
1742 patients randomly categorized in a 1:1:1 ratio |
i.16/360mg po
ii.32/360mg po
iii. placebo |
i.-5.0%
ii.-6.1%
iii.-1.3%
|
i.39%
ii.48%
iii.16% |
|
|
COR II 1-year randomized, double-blind, placebo-controlled trial (2013) |
1496 patients randomly categorized in a 2:1 ratio to NB 32/360mg or placebo; patients on NB with <5% weight loss in 28-44 week were reassigned to continue 32/360mg or increase daily dose to NB 48/360mg |
i.32/360mg (or increased daily dose 48/360mg)
ii.placebo |
i.-6.4%
ii.-1.2%
|
i.50.5%
ii.17.1% |
Random reassignment to higher dose did not change weight loss results |
|
COR-BMOD 1-year randomized, double-blind, placebo-controlled trial (2011) |
793 patients with obesity randomly categorized in a 1:3 ratio |
i. BMOD+ NB (32/350mg)
ii. BMOD+ placebo |
i.-11.5%
ii.-7.3% |
i.66.4%
ii.42.5% |
The efficacy of NB is obvious, and a lifestyle change can increase weight loss |
|
COR-DIABETES 1-year randomized, double-blind, placebo-controlled trial (2013) |
505 patients overweight/obese and T2DM with/without oral anti-hypoglycemic agents randomly categorized in a 2:1 ratio |
i.32/360mg
ii. placebo |
i.-5.0%
ii.-1.8% |
i.44.5% ii.18.9% |
↓HbA1c, certain improvements in CVD risk factors. ↑ nausea, constipation, vomiting |
Zonisamide
Given the pathophysiology behind the anti-obesity efficacy of the selective serotonin-receptor agonists and the dopamine-reuptake inhibitors, an ideal drug would combine serotonergic and dopaminergic activity. This is exactly the case of Zonisamide, a marketed antiepileptic drug that exerts dose-dependent biphasic dopaminergic (67) and serotonergic (68) activity. Its weight loss efficacy was investigated by a double-blind, placebo-controlled trial which reported a 32-week mean weight loss of 9.2 kg (1.7 kg) (9.4% loss) for the Zonisamide group (dose administered up to 600 mg/d) compared with 1.5 kg (0.7 kg) (1.8% loss) for the placebo group (P<0.001); Zonisamide was generally well-tolerated with only a few adverse effects (69). The findings were similar when the long-term effectiveness and tolerability of Zonisamide for weight control was examined in psychiatric outpatients using various psychotropic medications; the mean BMI reduction achieved was 0.8±1.7 kg/m2 and ranged from -2.9 kg/m2 to 4.7 kg/m2 (p<0.001), while the drug was generally safe and well-tolerated (70). Zonisamide was also assessed in the treatment of binge-eating (BE) disorder where it proved to be effective in reducing binge-eating frequency, severity of illness, and weight; however, the reports regarding its tolerability were conflicting (71). (see Table 7).
|
Table 7. Zonisamide |
|
|
Mechanism of action |
Selective serotonin-receptor agonist and dopamine-reuptake inhibitor |
|
Weight loss vs placebo |
7.8kg |
|
Clinical Benefits |
Assess in the treatment of binge-eating disorder |
|
Adverse events |
Nausea, headache, insomnia |
Zonisamide/Bupropion SR
Whether the anti-obesity efficacy of Zonisamide is increased when combined with bupropion (dopamine and norepinephrine -reuptake inhibitor) has been evaluated in a few Phase II clinical trials with different combined doses; the bupropion SR/Zonisamide SR combination is marketed under the trade name Empatic. In its 24-week, double-blind, placebo-controlled Phase IIb trial (72), patients completing 24 weeks of bupropion SR 360 mg/Zonisamide SR 360 mg therapy lost 9.9% of their baseline body weight, or 22 pounds, compared to 1.7% for placebo patients (p<0.001). Of patients who completed 24 weeks of therapy, 82.6% lost at least 5% of their baseline body weight and 47.7% lost at least 10% of their baseline body weight compared to 18.9% and 5.7% of placebo patients, respectively (p<0.001 for both). Patients experienced significant weight loss as early as by their first post-baseline visit at week four. Importantly, patients continued to lose weight until the end of the trial period with no evidence of a weight loss plateau. Early results showed that patients lost an average of 14% over 48 weeks. Improvements were observed in key markers of cardiometabolic risk such as waist circumference, triglycerides, fasting insulin, and blood pressure. The most commonly reported adverse events for all patients were headache, insomnia, and nausea. The most common adverse events leading to discontinuation were insomnia, headache, and urticaria (hives). There were no serious adverse events attributed by investigators to the study drug. There were no statistically or clinically meaningful differences between the drug and placebo on measures of cognitive function, depression, suicidality or anxiety. These reports revealed a significant weight-reduction effect for the combination Bupropion/Zonisamide. However, the safety concerns (73) will need to be addressed in the upcoming Phase III studies before firm conclusions about its safety profile can be drawn. (see Table 8)
|
Table 8. Zonisamide/Bupropion (Empatic) |
|
|
FDA approved/Phase |
Phase II completed |
|
Mechanism of action |
Selective serotonin-receptor agonist and dopamine-reuptake inhibitor/dopamine and norepinephrine reuptake inhibitor |
|
Weight loss vs placebo |
9.9% of their baseline weight |
|
Clinical Benefits |
↓cardiometabolic risk |
|
Adverse events |
Headache, insomnia, nausea, urticaria |
Topiramate
Topiramate is another anticonvulsant agent associated with weight loss. It is a sulphamate-substituted fructose that is approved as an antiepileptic/antimigraine agent and has multiple effects on the CNS, including action on the orexigenic GABA systems causing appetite suppression (74). A 6-month dose-ranging study in obese human subjects addressing its anti-obesity efficacy at doses of 64, 96, 192, and 384 mg/day (in divided twice-daily dosing) concluded that all doses produced significantly greater weight loss compared to placebo, and that weight loss in the 192 mg/day group was similar to the 384 mg/day group (75). This is important as topiramate has been associated with several neuropsychiatric effects, especially when administered at high doses (of 192 mg/d or more). Another study investigating the weight loss efficacy and safety of topiramate doses of 96, 192, and 256 mg/day over a 1-year period in obese subjects using the immediate release form tablets (before the development of the controlled-release formulation). Clinically significant weight loss (7.0, 9.1, and 9.7% of their baseline body weight for the doses of 96, 192, and 256 mg/day, respectively), was reported compared to 1.7% body weight loss in the placebo group (P<0.001) plus improvements in blood pressure and glucose tolerance (76). Finally, several other studies investigated the therapeutic effect of topiramate in patients with BED and bulimia (77) that are both associated with obesity; the results were very promising regarding control of symptoms in both disorders. (see Table 9)
|
Table 9. Topiramate |
|
|
FDA approved/Phase |
Phase II completed |
|
Mechanism of action |
Sulphamate-substituted fructose acts on GABA system |
|
Weight loss vs placebo |
7.0%(96mg),9.1%(192mg), 9.7% (256mg/day) |
|
Clinical Benefits |
Assess in the treatment of binge-eating, bulimia |
|
Adverse events |
Headache, insomnia, nausea, urticaria |
Phentermine
Phentermine is a sympathomimetic amine, which has anorexigenic action, that also releases insignificant quantities of dopamine. Thus, it is characterized by lower abuse potential (78). Its main mechanism of action involves catecholamine release in the hypothalamus resulting in enhanced satiety feeling and reduction of food intake (79). The most common side effects of phentermine as a sympathomimetic drug is heart rate increase, hypertension, dizziness, dry mouth, insomnia, irritability, and gastrointestinal disorders (80). Phentermine was the first FDA approved anti-obesity drug in 1959 for those aged >16 years old, but for only short-term use (maximum 3 months). The reason for the time limit is because the pharmaceutic industry had not updated labeling since 1959. In 1968, in a double-blind, placebo-controlled trial, 108 overweight or obese women were categorized into three groups that received continuously or intermittently (every 4 weeks) dosed phentermine or placebo, respectively. The weight loss was -12.2kg, -13.0kg or -4.8kg, respectively (81).
Currently, the off-label long-term use of phentermine is indicated only if there is clinical benefit, stable blood pressure and pulse rate in the absence of cardiovascular history or substance abuse disorders. In a recently published retrospective cohort study, 13,972 patients were observed for 6, 12 and 24 months after phentermine initiation. They were categorized in five groups based on the time of phentermine administration: short-term use, short-term intermittent use, medium-term continuous use, medium-term intermittent use, long-term continuous use. Weight-loss, changes in blood pressure, heart rate, and incidence of cardiovascular events (myocardial infarction, stroke, angina, coronary artery bypass grafting, carotid artery intervention, death) were examined. Weight loss was greater among off-label groups than referent group of short-term use with results depending on the duration of phentermine initiation. Specifically, at six months, short-term intermittent patients lost 1.8% further body weight relative to short-term single patients and medium-term intermittent patients lost 5.6% further body weight relative to short-term single patients. At twelve months, the medium-term intermittent use group lost further 5.6% body weight relatively to the short-term use group. At twenty-four months, long-term the continuous use group lost 7.4% additional body weight in comparison with the short-term use group. Concerning safety of phentermine, changes in heart rate and diastolic blood pressure were insignificant at six, twelve, and twenty-four months. Interestingly, although the referent group showed a slight increase in systolic blood pressure (+0.5-3.2 mmHg) at twenty-four months, all groups had slightly lower systolic blood pressure than the referent group at twelve- and twenty-four-months follow-up period. Lastly, the incidence of major cardiovascular events was low. So, it was shown that the off-label over three months therapy with phentermine was superior to short–term administration with greater weight-loss effect and cardiovascular safety. More studies with fewer limitations should follow in order to support these findings (82). In 2013, a clinical trial comparing phentermine as monotherapy or as part of a combined therapy, took place and resulted in a weight loss of 5.1% at 28 weeks follow-up period in favor of the combined phentermine/topiramate group.(see Table 10)
|
Table 10. Phentermine |
|
|
FDA approved/Phase |
1959 |
|
Mechanism of action |
Norepinephrine release and minor dopamine release |
|
Weight loss vs placebo |
0.23kg/week |
|
Clinical Benefits |
Lower abuse potential |
|
Adverse events |
Stimulation, insomnia, dry mouth, constipation, primary pulmonary hypertension Contraindicated in cardiovascular disease, coadministration with MAO inhibitors, hyperthyroidism, glaucoma, drug abuse |
Phentermine/Topiramate ER
Because of dose-related side effects seen with topiramate treatment including suicidality, metabolic acidosis, acute myopia, and secondary angle closure glaucoma, a lower dose of topiramate was used (in a special controlled release formulation) in a novel anti-obesity drug called Qsymia. The main mechanism of action of Phentermine/Topiramate extended release(ER) is possibly the alteration of various neurotransmitters, including inhibition of voltage-dependent sodium channels, glutamate receptors, and carbonic anhydrase as well as potentiation of γ-aminobutyrate activity (83).Two large randomized, double-blind, placebo-controlled trials took place (EQUIP and CONQUER) followed by a 2-year extension trial (SEQUEL). In the EQUIP trial 1,267 patients with BMI>35kg/m2were allocated in two groups and received phentermine/topiramate ER 3.75/23mg and 15/92mg, respectively, once daily. With 59.9% of the patients discontinuing, this trial found no statistically significant difference between the two groups regarding weight reduction (84). In the CONQUER trial 2,487 patients were allocated in three groups and received phentermine/topiramate ER 7.5/46mg, phentermine/topiramate ER 15/92mg, and placebo, respectively. The results were in favor of the combined therapy while the greater dosage resulted in greater weight loss with mean weight loss -7.8kg, -9.8kg, and -1.2kg in the three respective groups (85). Patients who completed the CONQUER trial entered the SEQUEL trial for 52 weeks. The weight loss as percentage of the initial weight was -9.3%, -10.5% and -1.8% in the three respective groups. A statistically significant improvement of lipid profile, glycemic control, and waist circumference in the phentermine/topiramate ER groups was reported (86). Based on the positive results from three Phase III studies, in 2012 FDA approved topiramate/phentermine extended-release as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in eligible adults. Meanwhile however, approval was denied by European regulatory authorities, who cited potential risk to the heart and blood vessels, psychiatric side effects, and cognitive side effects in explaining their decision (see Table 11, 12).
|
Table 11. Topiramate/Phentermine Extended Release (ER) (Qsymia) |
|
|
FDA approved/Phase |
2012 |
|
Mechanism of action |
Norepinephrine release, GABA modulation, voltage-gated ion channel modulation, stop of AMPA/kainite excitatory glutamate receptors and carbonic anhydrase |
|
Weight loss vs placebo |
6,6kg |
|
Clinical Benefits |
↓ lipid profile, HbA1c, waist circumference |
|
Adverse events |
Paresthesia, dizziness, dysgeusia, insomnia, constipation, dry mouth, fetal toxicity, metabolic acidosis, cognitive impairment Contraindicated in: Glaucoma, hyperthyroidism, coadministration with MAO inhibitors |
|
Table 12. Clinical Trials of Phentermine/Topiramate ER |
|||||
|
Clinical trial |
Administration |
N |
Treatment, placebo from baseline |
% of patients losing ≥5% of baseline weight |
Comment
|
|
CONQUER Double-blind, placebo-controlled trial over 1 year (2011) |
4-week titration+ 52 weeks of treatment: 15/92mg po or 7.5/46mg po |
2487 patients (BMI:27-45kg/m2 with 2+ risk factors i.15/92mg
ii.7.5/46mg
iii. placebo |
i.-9.8kg
ii.-7.8kg
iii.-1.2kg |
i.70%
ii.62%
iii.21% |
↑improvement in blood pressure, waist circumference, lipid levels, fasting glucose and insulin |
|
SEQUEL 2-year study overall;1-year extension of CONQUER (2012) |
227 patients completed the original blinded treatment
|
i.15/92mg
ii.7.5/46mg
iii. placebo |
i.-10.9kg
ii.-9.6kg
iii.-2.1kg |
i.79.3%
ii.75.2%
iii.30% |
|
Neuropeptide Y (NPY) Inhibitors
The ARC NPY neurons inhibit the anorexigenic POMC neurons (via NPY Y1 and Y5 receptors) and promote the release of the orexigenic neuropeptides orexin and MCH in the LHA, thus promoting food intake. Therefore, NPY blockade could be a promising target for body weight management. Animal experiments (in mice) have shown that pharmacologic blockade or genetic deletion of the Y1- and Y5-receptors reduces food intake and weight, with Y1-receptor signaling appearing to be the major mediator of the orexigenic effects of NPY. However, NPY is the most abundant central neuropeptide and regulates many functions beyond feeding; thus, targeting NPY neurons/Y receptors specifically for obesity is not easy and could result in unacceptable side effects. In addition, experimental medical blockade of NPY signaling with the Y5-receptor antagonist MK-0577 failed to cause any significant weight loss in a 1-year clinical trial (87). On the other hand, the oral, once-daily, centrally acting selective Y5-receptor antagonist velneperit, previously known as S-2367, induced a mean placebo-adjusted weight loss of 5.0% from initial weight (p <0.0001) over 54 weeks of therapy and was accompanied by improvement of lipid profile and waist circumference reduction (88).Nevertheless, velneperit did not proceed in markets due to disappointing results in phase IIb trials. However, the combined Y1/Y5-receptor antagonism may prove more effective, though we are not aware of any Y1/Y5-receptor antagonist in development to date. In contrast to Y1 and Y5, the Y2- and Y4-receptors are the targets of the satiety hormones PYY and pancreatic polypeptide (PP), respectively, and, as mentioned below, two drugs, a Y2/Y4-receptor agonist (obinepitide and a selective Y4-receptor agonist (TM30339)) are in Phase I/II clinical trials and are yielding results that appear quite promising as regards weight loss. A combined anti-obesity medication of velneperit/orlistat is under way (phase II clinical trial), also with promising results (89).
Dopamine antagonists
The mesolimbic dopamine system was proven to play a critical role in compulsive overeating or binge eating, which is one of the main reasons why people become overweight or obese. There is some evidence that blocking the action of dopamine in animals can reduce food intake, particularly of foods that are high in fat and sugar. GSK 598809 is a D3 antagonist that blocks dopamine. Preliminary data from human studies failed to show any significant effect on body weight control (90).
Tesofensine
Tesofensine (TE) is a presynaptic inhibitor of norepinephrine, dopamine, and serotonin. Like sibutramine, it suppresses appetite and may result in significant weight loss, as this was shown when given for the treatment of Parkinson’s disease, but also in a multi-dose, dose-ranging trial where 203 obese patients were randomly assigned to Tesofensine (0.25, 0.5, and 1.0 mg) or placebo once daily. Phase II testing of the drug has been completed. After 24 weeks, mean weight reduction was greater in the Tesofensine groups (-6.7, -11.3, -12.8 kg, for the three doses, respectively) compared with placebo (-2.2 kg). Additionally, an improvement in lipid profile and glycemic control was observed. A dose-dependent increase in blood pressure was observed along with a 7.4bpm increase in pulse rate in the 0.5mg/day group. Adverse events such as headache and mood alterations were also present in all groups especially in the 1mg/day group (91). In another trial, 32 males were allocated in two groups and received 2mg/day Tesofensine and placebo, respectively. The interesting point in this trial was that the patients were free to consume their usual amounts levels of food and exercise as usual. However, in the Tesofensine group they lost 1.8kg over 2 weeks because Tesofensine increased visual analog scale ratings of satiety and 24h fat oxidation in comparison with placebo. Even if an increase in total energy expenditure was not observed, an increase in sleeping energy expenditure was found. Altogether, Tesofensine induces weight loss by promoting the satiety feeling and slightly increasing metabolic rate (92). The effect of Tesofensine in appetite sensations was evaluated in another phase II trial, in which patients were allocated in 4 groups and received 0.25mg, 0.5mg, 1mg and placebo, respectively, for 24 weeks. For the first 12 weeks, a dose-dependent increase in the satiety feeling was noticed even though this feeling faded away as the trial was in progress (93). In 2010, a study on the abuse effect of Tesofensine, bupropion, atomoxetine, and placebo in comparison to d-amphetamine took place and concluded that the studied substances had no abusive action (94). Tesofensine has been shown to increase both blood pressure and pulse rate. In 2018, a phase III clinical trial was powered by the pharmaceutic industry producing Tesofensine. In this study 372 patients were allocated in three groups and received Tesofensine 0.25mg, 0.5mg and placebo. Furthermore, a combination of Tesofensine/metoprolol is recently being examined against hypothalamic injury-induced obesity and Prader-Willi syndrome (95). (see Table 13)
|
Table 13. Tesofensine |
|
|
FDA approved/Phase |
Phase III |
|
Mechanism of action |
Triple monoamine reuptake inhibitor of dopamine, norepinephrine, serotonin |
|
Weight loss vs placebo |
4.5-10.6% |
|
Clinical Benefits |
Pharmacological similar to sibutramine ↓ appetite, body weight, lipid profile, blood glucose |
|
Adverse events |
Headache, mood alterations, potentially increase heart rate, blood pressure, psychiatric disorders |
.
Lisdexamfetamine dimesylate
Another sympathomimetic, Lisdexamfetamine dimesylate, at certain doses appears effective in decreasing binge-eating days in patients with BED compared with placebo, according to a study published online by JAMA Psychiatry (96). The study included 259 and 255 adults with BED in safety and intention-to-treat analyses, respectively. Patients received lisdexamfetamine 30, 50 or 70 mg/day or placebo. BE days per week decreased in the 50 mg and 70 mg groups but not in the 30 mg group compared with placebo. Confirmation of these findings in ongoing clinical trials may result in improved pharmacologic treatment for moderate to severe BED.
Cannabinoid-1 Receptor (CB1) Antagonists
Among other neurotransmitter systems, the cannabinoid system modulates the hypothalamic melanocortin and NPY feeding networks. It has been shown that administration of cannabinoid-1 receptor (CB1) agonists and antagonists induces hyperphagia and hypophagia, respectively. These observations led to development of rimonabant, a cannabinoid-1 receptor antagonist, for the treatment of obesity, which was shown quite effective in promoting weight loss; however, it increased the incidence of mood-related disorders (97). As a result, in 2009, rimonabant was withdrawn from the market and the development of other cannabinoid-1 receptor antagonists for the treatment of obesity has also been discontinued. Before withdrawal, rimonabant was shown to have advantages in glycemic control and cardiovascular events (98). In 2010, another CB1 antagonist (AM6545) was found to have less psychological side effects and to induce satiety feeling and weight loss in animal studies (99). (see Table 14)
|
Table 14. Cannabinoid Type-1 Receptor Antagonists (SR141716, AM251, AM6545) |
|
|
Mechanism of action |
Antagonism of cannabinoid type-1 receptors stimulates anorexigenic signaling |
|
Clinical Benefits |
↓ body weight, blood glucose, cardiovascular events -AM6545: has limited CNS penetration |
|
Adverse events |
Mood alterations |
Human Chorionic Gonadotropin (hCG)
Human chorionic gonadotropin (hCG) in the form of subcutaneous injection and oral or sublingual diet drops has been advertised as aiding in weight loss of one to two pounds daily, absence of hunger, and maintenance of muscle tone. Clinical trials, however, failed to support this claim (100). In fact, FDA recommended avoiding buying over-the-counter weight loss products which contain hCG. One might ask why the hCG diet has so many enthusiastically supporting it. The reason may be that this diet needs to be accompanied by severe calorie restriction, to only 500-800 calories per day. Anyone following such recommendations is bound to lose weight, if only short-term. Most crucially, though, since hCG has been reported to induce serious side effects, this drug should not be used for the treatment of obesity. In addition, very low-calorie diets have not been shown to be superior to conventional diets for long-term weight loss, plus they have risks, such as gallstone formation, irregular heartbeat, and an imbalance of electrolytes. Therefore, if weight loss is the goal, there are safer ways to make it happen.
Nesfatin-1
Nesfatin-1 is a satiety molecule, which was first described in rats and is derived from its precursor molecule nucleobindin2 (NUCB2) (101). It is expressed both centrally in hypothalamic food intake-regulatory nuclei, the nucleus paraventricular and the arcuate nucleus, and peripherally, in the stomach, pancreas, adipose tissue, and testis. In the gastric oxyntic mucosa, nesfatin-1 is co-expressed with the orexigenic peptide ghrelin in X/A-like cell in rats and humans. The anorexigenic action of nesfatin-1 is based on its ability to cross the blood-brain barrier. It is notable that NUCB2/nesfatin-1 not only decreases food intake, gastric emptying, and small intestine motility, but also reduces glucose and increases insulin levels (102). Intracerebroventricular (icv) injection of full length nesfatin-1 caused a significant reduction of food intake in rats and mice (103). These findings suggest that downstream signaling might be altered, a hypothesis to be further investigated. The fact that nesfatin-1 acts in a leptin-independent way, indicates that it might be a new molecular target in the pharmacotherapy of obesity. The identification of the yet unknown nesfatin-1 receptor will allow the development of nesfatin-1 agonists and antagonists. Whether peripheral nesfatin-1 is primarily involved in the regulation of food intake is questionable and should be further investigated.
GASTROINTESTINAL AND PANCREATIC PEPTIDES THAT REGULATE FOOD INTAKE
The gut-brain axis plays an important role in food consumption regulation. During food intake, information regarding meal quality and content and short-term alterations in nutrient status is relayed from the gastrointestinal (GI) tract and pancreas to the brain which in turn determines meal size. Apart from feeding, a few satiation signals optimize these processes by influencing gastrointestinal motility and secretion. Several peptides have been identified that mediate this GI system-brain communication including satiety signals such as gastrin releasing peptide (GRP), cholecystokinin (CCK), peptide YY (PYY), glucagon-like peptide-1 (GLP-1), pancreatic polypeptide, glucagon, and amylin, as well as the orexigenic peptide ghrelin. While the anorexigenic peptides are secreted during feeding, ghrelin is secreted before meals and acts to increase hunger and meal initiation. Some of the GI and pancreatic peptides implicated in the regulation of food intake act directly on regions of the brain involved in the regulation of food intake, including the ARC in the hypothalamus and the area postrema, while others act outside of the CNS. For example, modulating the activity of neurons such as the vagus nerve, which projects to the nucleus of the solitary tract in the brain stem.
CCK and CCK1R Agonists
CCK is the first described intestinal satiation peptide (104). It is produced by the mucosal I cell (105) of the duodenum and jejunum, and the enteric nervous system, in response to luminal nutrients, especially lipids and proteins. Through endocrine and/or neural mechanisms, CCK regulates numerous GI functions, including satiation, by acting on two CCK-specific receptors: the CCK receptor 1 (CCK1R), expressed mainly in the GI system, and the CCK2R that predominates in the brain. The vagus nerve plays a critical role in CCK-induced satiation as it contains CCK1R, indicating the afferent pathway through which CCK relays satiation signals from the GI to the hindbrain region. Corroborating this hypothesis is the well-documented attenuation of CCK-induced satiation following abdominal subdiaphragmatic vagotomy (106). In addition, CCK inhibits gastric emptying, thereby augmenting gastric distention and mechanoreceptor stimulation, which in turn augments the anorectic effects of CCK (107). Despite the satiety effect of CCK, its potential as an anti-obesity target is questionable. Human studies with intravenously infused CCK carboxy-terminal octapeptide (CCK-8) have shown decreases in meal size and duration in a dose-dependent manner (108). However, the CCK satiating effects were very short-lived, usually not lasting more than 30 minutes, which raises issues as to its importance in long-term body weight regulation. In an animal study, chronic CCK administration with up to 20 peripheral injections per day, although reducing meal size, was associated with increased meal frequency, leaving body weight unaffected (109). Finally, the reports from trials testing CCK1R agonists as potential anti-obesity drugs were disappointing (110). It is currently suggested that there might be a role for CCK in body weight regulation not as a monotherapy but possibly as an adjunctive/synergistic therapy to long-term adiposity signals, such as leptin (111).
Glucagon-Like Peptide-1 Analogues
The dominant role of GI in satiation (112) is mediated not only by the gastric mechanoreceptors and upper intestinal neuropeptides such as CCK, but also by gut satiation peptides that are secreted from lower-intestine enteroendocrine cells in response to ingested food. They in turn diffuse through interstitial fluids to activate nearby nerve fibers and/or enter the bloodstream to function as hormones and augment the perception of GI fullness by acting in specific parts of the CNS. There is a well-defined duodenal-ileal communication (the ileal brake) via which the proximal intestine informs the distal intestine as to meal quality and content so that the latter modulates/restricts feeding duration, proximal GI motility, and gastric emptying, while it also regulates metabolic responses to nutrient intake. GLP-1 appears to engage such a mechanical and behavioral brake effect on eating and is produced primarily by L cells in the distal small intestine and colon. Along with glucagon and oxyntomodulin, GLP-1 is cleaved from proglucagon, which is expressed in the gut, pancreas, and brain. The GLP-1 equipotent bioactive forms GLP17–36 and GLP17-37 are rapidly inactivated in the circulation by dipeptidyl peptidase-4 (DPP4). Therefore, GLP-1 analogues that have a slightly different molecular structure, but a significantly longer duration of action compared to wild GLP-1 have been used for therapeutic interventions in patients with diabetes, in whom they significantly improved glycemic control, fasting plasma glucose, β-cell function, and probably β-cell regeneration. Currently, the GLP-1 analogues used in clinical practice for diabetes control are exenatide, lixisenatide, dulaglutide, liraglutide, and semaglutide. Beyond the improved glycemic control achieved, clinical studies have also demonstrated anorectic effects and significant weight loss via these agents (113, 114). Although the exact mechanisms by which GLP1 induces anorexia are not yet fully known, it is suggested that vagal and possibly direct central pathways are involved (115). The GLP-1 receptor R (GLP1R) is the principle mediator of the anorectic effects of GLP-1 (116) and is expressed by the gut, pancreas, brainstem, hypothalamus, and vagal-afferent nerves (117).
LIRAGLUTIDE
Its mechanism of action is both central and peripheral targeting satiety centers of the brain and regulating glucose metabolism. It is the only injectable medication for obesity and is titrated from 0.6mg to 3.0mg over 4 weeks. The most common side effects of liraglutide and generally of GLP1 analogues are gastrointestinal (nausea, diarrhea, constipation, vomiting, dyspepsia, abdominal pain) and rarely pancreatitis. The product has a boxed warning stating that thyroid C-cell tumors have been seen in rodents but the relevance of this in humans is uncertain. The drug should not be used in patients with a personal or family history of medullary thyroid carcinoma (MTC) or in patients with multiple endocrine neoplasia syndrome type 2. Three major trials, SCALE-Obesity, Prediabetes, SCALE-Diabetes, SCALE-Maintenance, have established the therapeutic benefit of liraglutide for weight loss. The SCALE-Obesity, Prediabetes evaluated liraglutide in patients who were overweight and obese but did not have diabetes. The study included 3,731 individuals who were assigned to treatment with liraglutide 3 mg or a placebo. Patients were also counseled on diet and exercise. At the end of the 56-week trial, the liraglutide group lost an average 8% (7.2kg) of their body weight compared to 2.6% (2.8kg) in the placebo group (118); net weight loss was 4.4kg. In the SCALE-Diabetes trial, 846 adults who were overweight or obese and had T2DM were allocated to receive either daily 3.0mg liraglutide or placebo, with mean weight loss -6.0% and -2.0%, respectively (119). In the SCALE-Maintenance trial, 422 adults who were overweight or obese and had lost >5% of initial body weight with a calorie restriction diet were allocated to receive either liraglutide or placebo, respectively, with mean weight loss -6.2% and -0.2%, respectively (120).Recently, Saxenda (liraglutide 3.0mg) has asked for a label update based on the results of the LEADER trial, which studied the effects of the lower dose version of liraglutide (1.8 mg) used to treat diabetes. According to this trial, which examined a population with T2DM and established cardiovascular disease,1.8mg liraglutide daily showed statistically significant reduction of cardiovascular death, of non-fatal myocardial infarction (heart attack), and of non-fatal stroke by 13% versus placebo, when added to standard care. (121) (see Table 15, 16).
|
Table 15. Liraglutide (Saxenda) |
|
|
FDA approved/Phase |
2014 |
|
Mechanism of action |
Glucagon-like peptide-1 agonist |
|
Weight loss vs placebo |
4.4kg |
|
Clinical Benefits |
↓cardiovascular death, non-fatal myocardial infarction, non-fatal stroke |
|
Adverse events |
Nausea, hypoglycemia (serious if co-administrated with insulin), gastrointestinal disorders, fatigue, dizziness, abdominal pain, increased lipase, acute pancreatitis, acute gallbladder disease, increase heart rate, suicidal ideation, thyroid c-cell tumors seen in mice Contraindicated in: History of medullary thyroid carcinoma or multiple endocrine neoplasia 2 |
|
Table 16. Clinical Trials of Liraglutide |
|||||
|
Clinical trial |
Patients |
Dose |
Treatment, placebo from baseline |
% of patients losing ≥5% of baseline weight |
Comment
|
|
SCALE-Obesity+Prediabetes 1-year randomized, double-blind, placebo-controlled trial (2015) |
3731 patients overweight/obese without DM (61.2% had prediabetes) randomly divided into two groups |
i.3.0mg sc once daily
ii. placebo |
i.-8.4kg
ii.-2.8kg |
i.63.2%
ii.27.1% |
Improvement of body weight, glycemic index, blood pressure, waist circumference |
|
SCALE-Diabetes 1-year randomized, double-blind, placebo-controlled trial (2015) |
846 adults with T2DM overweight/obese |
i.3.0mg sc once daily
ii. placebo |
i.-6.0%
ii.-2.0% |
i.54.2%
ii.21.4% |
More GI disorders in the liraglutide group. No pancreatitis was reported |
|
SCALE-Maintenance 1-year randomized, double-blind, placebo-controlled trial (2013) |
422 adults overweight/obese who had lost ≥5% of initial body weight during a calorie-restriction period were randomized |
i.3.0mg sc once daily
ii. placebo |
i.-6.2%
ii.-0.2% |
i.81.4%
ii.48.9% |
A combination of liraglutide, diet, exercise induced further weight loss and improvement in certain cardiovascular risk factors |
SEMAGLUTIDE
Semaglutide is a novel long-acting GLP1 analogue indicated for T2DM and awaiting approval for obesity at higher doses. The efficacy of this anti-obesity drug has been proven by the SUSTAIN 1-6 trials. In these trials, patients who were overweight or obese, with and without T2DM, with or without antidiabetic medications, were allocated in groups which received semaglutide in two different dosages (0.5mg or 1.0mg) or placebo or another anti-diabetic therapy. The superiority of semaglutide 1.0mg against semaglutide 0.5mg or placebo or another anti-diabetic agent was obvious (122). In SUSTAIN 7, Semaglutide administered in subcutaneous injections once weekly was compared with Dulaglutide. Mean weight loss was greater in the group which received 1.0mg semaglutide (-4.9kg) vs the groups that received 0.5mg semaglutide (-3.6kg), 1.5mg Dulaglutide (-3kg), and0.75mg Dulaglutide (-2.3kg). Additionally, oral semaglutide is currently approved for the treatment of T2DM. In order to avoid malabsorption, semaglutide is administrated 30 minutes before breakfast. Apart from semaglutide, other oral GLP-1 agonists, such as TTP054/TTP-054 and ZYOG1, are under investigation (122). Two other trials, STEP, which studies the effects of semaglutide in patients with obesity, and SELECT, which investigates the cardiovascular effects of semaglutide in patients with obesity are currently underway (123). PIONEER, which examines the cardiovascular safety of oral administration of semaglutide in patients with T2DM, recently showed the non-inferiority of this medication to placebo (124). (see Table 17).
|
Table 17. Clinical Trials of Semaglutide |
||||
|
Clinical trial |
Study Design |
Dose |
Treatment, placebo from baseline |
% of patients losing ≥5% of baseline weight |
|
SUSTAIN 1 |
Double-blinded For 30 weeks |
i.0.5mg sc once weekly ii.1.0mg sc once weekly iii. placebo |
i. -3.7kg ii. -4.5kg iii. -1.0kg |
i.37% ii.45% iii.7% |
|
SUSTAIN 2 |
Double-blinded Duration: 56 weeks |
i.0.5mg sc once weekly ii.1.0mg sc once weekly iii. sitagliptin 100mg per po once daily |
i.-4.3kg ii.-6.1kg iii.-1.9kg |
i.46% ii.62% iii.18% |
|
SUSTAIN 3 |
Open-label Duration:56 weeks |
i.1.0mg sc once weekly ii. exenatide extended release 2.0mg |
i.-5.6kg ii.-1.9kg |
i.52% ii.17% |
|
SUSTAIN 4 |
Open-label Duration: 30 weeks |
i.0.5mg sc once weekly ii.1.0mg sc once weekly iii. insulin glargine |
i.-3.5kg ii.-5.2kg iii.+1.2kg |
i.37% ii.51% iii.5% |
|
SUSTAIN 5 |
Double-blinded Duration:30 weeks |
ii.0.5mg sc once weekly ii.1.0mg sc once weekly iii. placebo |
i.-3.7kg ii.-6.4kg iii.-1.4kg |
i.42% ii.66% iii.11% |
|
SUSTAIN 7 |
Open-label Duration: 40 weeks |
i.0.5mg sc once weekly ii.0.75mg dulaglutide sc once weekly iii.1.0mg sc once weekly iv.1.5mg dulaglutide sc once weekly |
i.-4.6kg ii.-2.3kg
iii.-6.5kg iv.-3.0kg |
i.44% ii.23%
iii.63% iv.30% |
|
SUSTAIN 6 (CVD outcomes) |
Double-blinded Duration:104 weeks |
i. 0.5mg sc once weekly ii.1.0mg sc once weekly iii. placebo 0.5mg iv. placebo 1.0mg |
i.-3.6kg ii.-4.9kg iii.-0.7kg iv.-0.5kg |
Non inferior |
|
SUSTAIN 8 |
Phase 3b Semaglutide vs canagliflozin |
|
|
|
|
SUSTAIN 9 |
Semaglutide as an add-on to SGLT2 monotherapy or in combination with either metformin or sulfonylurea |
|
|
|
OTHER LONG-ACTING GLP-1 ANALOGUES
Other long-acting GLP-1 analogues are currently being investigated for weight loss in addition to diabetes treatment. Once-daily 13-week treatment with 20 μg or 30 μg of lixisenatide reduced body weight significantly more compared to placebo (-3 kg for lixisenatide 20 μg; p<0.01, -3.47 kg for lixisenatide 30 μg; p<.01, -1.94 kg for placebo) (125). Current findings regarding CJC-1134-PC, which is a conjugate of exendin-4 and recombinant human albumin and represents a once-weekly glucagon-like peptide-1 receptor agonist, suggest that it provides similar reduction in body weight compared with exenatide twice-daily. It may have a more favorable adverse event profile which might improve patient compliance and probably total weight loss in the long-term (126). Finally, albiglutide and taspoglutide are two novel GLP-1 analogues currently being investigated. A recent review that examined the efficacy, safety, and perspective for the future of the once-weekly GLP-1 receptor agonists exenatide, taspoglutide, albiglutide, LY2189265 and CJC-1134-PC, and compared them to the currently available agonists, exenatide BID and liraglutide QD, concluded that the long-acting agonists are not superior compared to the currently used exenatide BID and liraglutide QD regarding weight loss (127).In a separate development, an orally administered PYY3-36 and GLP-1 combination has been formulated using a sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC) carrier (127). Early studies revealed that the neuropeptides delivered orally in this way had a pharmacodynamic profile consistent with the reported pharmacology, were rapidly absorbed by the gastrointestinal tract, and reached concentrations several-fold higher than those seen naturally postprandially (128). Oral GLP-1 (2-mg tablet) alone and in combination with PYY3-36 (1-mg tablet) showed enhanced fullness at meal onset and induced a significant reduction in energy intake. Exenatide-CCK (129) and Liraglutide-Setmelanotide (130) have been also introduced as different combined anti-obesity therapies which act synergistically on POMC-deficient patients.
Single Molecule Multi-Agonists
The main therapeutic idea of this category is based on the concept that a single molecule could target multiple receptors (at least two; multi-agonist), thus allowing synergistic action of both pharmaceutical agents.
GLUCAGON-LIKE PEPTIDE 1/GLUCAGON
As mentioned before, GLP-1 analogues are effective anti-obesity medications and improve glucose intolerance. Glucagon has direct action on the liver by stimulating gluconeogenesis and glycogenolysis (131). It can even result in hyperglycemia and T2DM. Of note, patients with T2DM are characterized by impaired glucagon secretion. However, glucagon in CNS decreases food intake, increases energy expenditure via brown fat thermogenesis, decreases fat accumulation via lipolysis and lipid synthesis inhibition, improves cardiac performance, inhibits gastric motility, and stimulates autophagy. In 2009, the first human study announced that low-dose co-infusion of GLP-1 and glucagon could decrease food intake and increase energy expenditure (132). Therapy with a GLP-1/glucagon multi-agonist was created when amino acids 17, 18, 20, 21, 23 of glucagon were substituted in the glucagon molecule by the respective GLP-1 residues (133). The alanine at position 2 of the peptide was substituted with Aminoisobuturic acid (Aib) to protect the molecule from DDP-IV inactivation, and a lactam bridge was introduced between amino acids 16 and 20 to stabilize the secondary structure to ensure glucagon receptor potency. Once weekly administration of this pharmaceutical agent, for 4 weeks, in diet-induced obesity in mice, resulted in improvement of obesity, hepatic steatosis, glucose control, and lipid profile. Increase in energy expenditure was observed only with the multi-agonist therapy, but not with the glucagon monotherapy. Moreover, it was found that therapy with the multi-agonist improved leptin sensitivity in DIO mice (134). Different GLP-1/glucagon multi-agonists are currently under investigation (135). Interestingly, an oxyntomodulin multi-agonist was under investigation concurrently with the GLP1/glucagon multi-agonist.
OXYNTOMODULIN
Oxyntomodulin (OXM) is a 37-amino acid anorexigenic peptide hormone produced in the L-cells of the distal small intestine and colon, where it co-localizes with GLP-1 and PYY. Animal studies have shown weight reduction and improved glucose metabolism following chronic OXM injections beyond that explained by food intake restriction, suggesting an additional effect of OXM on energy expenditure. Just like GLP1, OXM is a product of proglucagon gene believed to modulate energy homeostasis at least in part via GLP1R, although its GLP1R binding affinity is about 100 times lower than that of GLP1 (136). Centrally however, GLP1 and OXM have different targets, as OXM activates neurons in the hypothalamus (137), whereas GLP1 acts in the hindbrain and other autonomic control areas (138). In human studies, acute anorectic effect of OXM was demonstrated by intravenously infused OXM (139). A reduction in food intake was also seen and retained during chronic administration in a 4-week trial with OXM injections three times a day 30 minutes before meals in a group of overweight and obese volunteers (n = 14). OXM reduced nutrient intake (35% ± 9%) resulting in significant weight loss compared to placebo (2.3 ± 0.4 kg vs 0.5 ± 0.5 kg, respectively). The findings of another study with twelve overweight or obese human volunteers who underwent a randomized, double-blinded, placebo-controlled study were similar; an ad libitum test meal was used to measure energy intake during intravenous infusions of either PYY3-36 or OXM or combined PYY3-36/OXM. Again, OXM significantly reduced energy intake compared to placebo, although the combined treatment had superior effects compared to PYY3-36 or OXM monotherapy. Human studies have also clearly demonstrated the direct effect of OXM on energy expenditure (140); this effect was later confirmed by indirect calorimetry (141). These modest but favorable results suggest significant promise for OXM-based therapies for obesity. In addition to the established action of OXM on appetite, another mechanism that potentially plays a role in energy intake and glucose metabolism is gastric emptying. Intravenous infusion of OXM reduced gastric emptying in humans (142). Whether reduction in gastric emptying is involved in the acute and long-term metabolic effects of OXM is not yet clear. Nevertheless, the immediate future will reveal OXM’s role in obesity management. However, as for other peptide hormones, their clinical application is limited by their short circulatory half-life, a major component of which is cleavage by DPP-IV. Therefore, structurally modified analogues with an altered OXM pharmacological profile have been produced with longer duration of action, good safety profile, and positive effects on body weight (and glucose metabolism) management in animal studies (143). These findings bring closer their usage in human clinical trials. Furthermore, the crystal structure of OXM has been determined, and this advance should facilitate the rational design of oxyntomodulin peptidomimetics to be tested as oral anti-obesity pharmaceuticals. Even so, despite the promising weight reduction efficacy of OXM, only a small number of development projects appears to be at an advanced stage. TKS1225 is an OXM analogue. The present status of this molecule is unknown. OXY-RPEG has been engineered via its proprietary reversible pegylation technology to increase its half-life and increased potency. In preclinical testing, OXY-RPEG was significantly superior to twice daily injections of OXM in the reduction of food intake and the degree and durability of weight loss. In 2009, an oxyntomodulin-based multi-agonist peptide with glucagon and GLP-1 agonistic actions were created. This multi-agonist had advanced action comparing to the one that Day et al had introduced at the same year (144). A 2-weeks trial in DIO mice showed weight loss and glucose control improvement. This beneficial action was obvious even in mice without GLP1 or glucagon receptor confirming the superiority of this analogue. Oxyntomodulin functions endogenously as a physiologic co-agonist, but regarding its small bioactivity, it is mainly characterized by its function as biosynthetic precursor to glucagon.
GLUCAGON-LIKE PEPTIDE 1/AMYLIN
In 2010, salmon calcitonin-exendin-4 combined therapy achieved reduction of food intake and weight in non-human primates (145). Of note, the human amylin receptor subtypes consist of calcitonin receptor and receptor activity-modifying proteins. This observation was the first step in the development of multi-agonist molecules targeting GLP-1 and Amylin (146). Two of these peptide hybrids (phybrids) had a C-terminally truncated Exenatide, which was covalently linked to the N-terminus of an amylin analogue (Davalintide) through either a repeat β-ala-β-ala dipeptide, or through triple-glycine linear repeat. Administration of phybrids resulted in greater weight loss in non-human primates than monotherapy, although similar to that achieved by a physical commixture of the single hormones. Another GLP1/Amylin phybrid was introduced, which used a full-length Exenatide sequence linked to Davalintide viaan intervening 40-kDa PEG. This phybrid reduced both blood glucose and body weight in a dose-dependent fashion.
GLUCAGON-LIKE PEPTIDE 1/GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE
This single-molecule multi-agonist was quite controversial. Glucose-Dependent Insulinotropic Polypeptide (GIP), is a 42-amino acid peptide, produced by K-cells in the duodenum and jejunum and released into the general circulation upon stimulation by dietary lipids (147). The investigation following the discovery of this new peptide, showed that GIP is the first incretin hormone. It acts directly on the pancreas augmenting glucose-stimulated insulin secretion (148). It is worth mentioning that GIP has the ability to enhance both insulin secretion in hyperglycemia and glucagon release in hypoglycemia (149). A few years later, the role of GIP in obesity development became apparent. GIP acts on adipocytes enhancing adipogenesis, inhibition of lipolysis, stimulation of de novo lipogenesis (150) and on chylomicrons stimulating triglyceride release. It also affects adipocyte glucose and fatty acid uptake and adipocyte lipoprotein lipase enzyme activity (151). It is remarkable that although GIP was regarded as an obesogenic hormone, mice overexpressing GIP showed improved β-cell function and improved glycemic control and were resistant to DIO (152). Additionally, in studies with mice, it was shown that the chronic GIP agonist administration improves glucose metabolism without body weight changes (153). In 2013, two single-molecule multi-agonists GLP1/GIP were introduced, whose action was based on the insulinotropic action of both components (153). GIP agonist enhanced GLP1 action upon glucose metabolism and GLP1 could mitigate obesogenic effect of GIP via its anorectic effect. The biochemical structure of multi-peptide was similar to GLP1/glucagon multi-agonist i.e. a single peptide with potency at both receptors (GIP residues were introduced in the median and the C-terminal part of peptide; certain modifications that increased activity on the glucagon receptor were removed; the C-terminus of the peptide ended with the nine amino acid extension found in exendin-4 and an Aib was added at position 2 to protect against DPP-IV inactivation) (154). Several clinical trials in mice, rodent models, non-human primates and humans were performed, concluding that the GLP1/GIP multi-agonist therapy reduced food intake, and consequently body weight, improved glycemic control, lipid profile and lipolysis but without any improvement in energy expenditure.
GLUCAGON LIKE PEPTIDE 1/GLUCAGON/GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE
The creation of this single-molecule multi-agonist was based on the biochemical structure of GLP1/GIP and GLP1/glucagon multi-peptides. An Aib at position 2 both protected the molecule from DPP-IV inactivation and decreased its potency at the glucagon receptor; an amino acid lysine at position 10 was fatty-acylated via a γ-glutamic acid linker to palmitic acid; amino acids at positions 16,17, 20, 27, 28 replaced balanced glucagon bioactivity; a C-terminal exendin-4 extension sequence (CEX) succeeds agonism at all three receptors 10-fold greater than native hormones (155). The main mechanism of action is based on the combination of the anorectic effect of GLP-1, the lipolytic and thermogenic characteristics of glucagon, and the action of GIP on β-cell function and glycemic control. Contrary to GLP-1/GIP multi-agonist, which doesn’t affect energy expenditure, this triple agonist increases energy expenditure. (see Table 18)
|
Table 18. Single Molecule Multi-Agonists |
||
|
Drug name |
Clinical benefits |
Adverse events |
|
Glucagon-like peptide 1/glucagon oxyntomodulin, MED10382, G530S (Glucagon analogue/Semaglutide), GC-co-agonist 1177 |
↓ food intake, obesity, hepatic steatosis, HbA1c, lipid profile ↑energy expenditure |
|
|
Glucagon-like peptide1/amylin co-agonism |
↓ blood glucose and body weight dose-dependently |
|
|
Glucagon-like peptide 1/glucose-dependent insulinotropic polypeptide |
↓ blood glucose, lipid profile, food intake, body weight, ↑ lipolysis |
No improvement in energy expenditure |
|
Glucagon-like peptide 1/glucagon/glucose-dependent insulinotropic polypeptide |
↓body weight, HbA1c, hepatosteatosis, cholesterol, ↑energy expenditure, lipolysis |
|
Peptide-Mediated Delivery of Nuclear Hormones
The use of nuclear hormones as an agent of GLP-1 and Glucagon is a novel promising therapy in the treatment of obesity. Nuclear hormones are characterized by high potency and pleiotropic action as well as unwanted adverse effects. The basic idea involves a linkage of a nuclear hormone to a peptide, usually through a linker that would allow metabolism of the nuclear hormone only within the targeted cell reducing the undesirable effects in other tissues. However, in the cell types that possess the specific peptide receptor, its activation should lead to internalization of the ligand-nuclear hormone receptor complex. In this case, the peptide receptor plays the role of a gateway into the cell. Upon internalization, biological processing of a suitably designed linker would release the nuclear hormone and allow activation of its intracellular receptor. Although a promising option, not all nuclear hormones can be used as peptide-mediated agents. They should have high tissue selectivity, ability to be internalized and compatibility to peptide wanted. Estrogens, tri-iodothyronine, and dexamethasone are the nuclear hormones that have been tested.
GLUCAGON-LIKE PEPTIDE 1-MEDIATED DELIVERY OF ESTROGEN
Glucagon-Like Peptide 1-mediated delivery of estrogen was first introduced in 2012. The use of estrogens was indicated by the fact that estrogen replacement therapy in postmenopausal women improved multiple cardio metabolic parameters (156). Furthermore, estrogens have anabolic, insulinotropic, and anorectic effects (157). The combination of estrogen and GLP-1 was found to improve body weight and glycemic control in rodent models with the metabolic syndrome (158). The weight-lowering effect was due to appetite suppression, while the GLP-1/E2 combination showed greater potency comparing to GLP-1 analog or E2 alone. Further clinical trials enhanced this finding showing an influence on feeding behavior. Additive contribution of GLP-1/E2 on pancreatic islet function, cytoarchitecture and protection from deleterious insults such as lipotoxicity was found in 2015 (159). Despite the powerful metabolic benefits associated with estrogen action, effects on the reproductive endocrine system and oncogenic potential have restricted their clinical use in postmenopausal women. Furthermore, many aspects of molecular pharmacology and mechanism of action remain unresolved. Specifically, neither the precise intracellular processing of the GLP-1/E2 conjugate, which results in active estrogen cargo release, nor the molecular identity that delivers estrogen activity, have been determined. It is possible that estrogens enhance brain penetration and alter the bio-distribution of the conjugate to more privileged sites for central nervous action.
GLUCAGON-MEDIATED DELIVERY OF TRI-IODOTHYRONINE
Glucagon and thyroid hormone can separately lower body weight and LDL cholesterol in humans. Thyroid hormones act both on liver, regulating hepatic lipid metabolism and hepatosteatosis, and in adipose cells, increasing energy expenditure and enhancing lipolysis (160, 161). On the other hand, they can cause cardiac hypertrophy, tachycardia, muscle catabolism, and bone deterioration. Glucagon receptors are highly concentrated not only in the liver, which is the preferred site for T3 action, but also in adipose tissues, kidney, and the cardiovascular system resulting in metabolic enhancement along with toxicity risk. Considering all of the above, a glucagon/T3 conjugate was created. A native T3 combined with a DPP4-protected C-terminally extended glucagon analog via a peptide spacer (162). Several control compounds were also generated to permit appropriate pharmacological comparisons. These additional peptides included a conjugate with selective chemical substitution in the peptide to suppress glucagon activity, a compound with a linker that proved metabolically stable and was incapable of intracellular T3 release, and a third control conjugate that carried a metabolically-inert thyroid hormone. Finan found that the conjugate glucagon/T3 corrected lipid metabolism in rodent models with dietary-induced metabolic syndrome. The above findings showed that the body-weight effect of the conjugate can partially be governed by actions in adipose depots because glucagon receptors exist in rodent adipocytes, less than in liver. Moreover, the glucagon/T3 conjugate effect is supported by the uncoupling protein 1-mediated thermogenesis, enhanced FGF21 secretion and biased by PGC-1 cofactor signaling. Interestingly, the combination of glucagon/T3 seems to decrease arterial plaque area in LDL receptor -/- mice and fibrosis in mice with advanced fatty liver disease. Although the above data demonstrate the cardiovascular benefit of this conjugate, further chemical improvements should be made in order to be safe for chronic use in higher mammals and especially humans.
GLUCAGON-LIKE PEPTIDE 1-MEDIATED DELIVERY OF DEXAMETHASONE
It is widely known that dietary-induced obesity causes chronic peripheral and central inflammation (163). Glucocorticoids are widely known for their anti-inflammatory characteristics, but due to their ubiquitous action profile, their therapeutic use can lead to off-target effects. In 2017, DiMarchi and Tschop created a GLP1/dexamethasone conjugate which managed to improve body weight in DIO mice, in a superior way to GLP1 or dexamethasone alone. This combination improved hypothalamic inflammation, astrocytosis, microgliosis, and insulin sensitivity. The targeted delivery of dexamethasone to GLP1R-positive cells prevented typical dexamethasone off–target effects on glucose metabolism, bone density, and the hypothalamus-pituitary-adrenal axis activity (164). (see Table 19)
|
Table 19. Peptide-Mediated Delivery of Nuclear Hormones |
|
|
Drug name |
Clinical benefits |
|
Glucagon-like peptide 1/estrogen |
↓ food intake, body weight, HbA1c |
|
Glucagon/tri-iodothyronine |
↓ lipid profile, arterial plaque and fibrosis in advanced fatty liver disease |
|
Glucagon-like peptide 1/dexamethasone |
↓ hypothalamic inflammation, astrocytosis, microgliosis, ↑insulin sensitivity |
Peptide Y (PYY)
PYY is a 36-amino acid anorexigenic peptide with a hairpin-like U-shaped fold secreted from the entero-endocrine L-cells of the ileum and colon in response to feeding. PYY presents in two major forms, PYY1-36 and PYY3-36. More specifically, PYY is a member of the pancreatic polypeptide-fold (PP-fold) family which also includes NPY and PP and interacts with a family of receptors (mainly Y2R). It is produced postprandially, in response and proportionally to caloric load, by the distal-intestinal L cells along with oxyntomodulin (OXM) and GLP-1. Just like GLP-1 and OXM, PYY1-36 is rapidly proteolyzed by DPP4. However, unlike the other two neuropeptides, the cleaved product PYY3-36, is bioactive. Human studies have shown that PYY delays gastric emptying and promotes satiety (165), while short-term intravenous administration of PYY3-36 , at doses generating physiologic postprandial blood excursions, was shown to decrease calorie intake by approximately 30% in lean and obese subjects, without causing nausea, affecting food palatability, or altering fluid intake, nor was it followed by compensatory hyperphagia (166). Another study confirmed the above findings, reporting dose-dependent reductions of food intake (maximal inhibition, 35%; P<0.001 vs control) and calorie intake (32%; P<0.001) after intravenous infusions of several different concentrations of PYY3-36 (167). Sloth et al. first showed the significantly higher energy expenditure following PYY3-36 intravenous infusion compared with PYY1-36 or control. In a recent study, the effect of infused PYY3-36 on energy intake was compared to that of OXM or the combined PYY3-36/OXM treatment; the results demonstrated that energy intake was significantly less with the combined treatment compared to PYY3-36 or OXM monotherapy (168). Whether these findings pointed to a weight loss efficacy of PYY was evaluated in a 12-week trial of 133 obese patients who were randomly assigned to intranasal PYY3-36 (200 or 600 mcg three times a day before meals) or placebo, in conjunction with diet and exercise. At the 200 mcg dose, PYY3-36 failed to reduce body weight, while 60% of patients treated with the high PYY3-36 dose (600 mcg three times a day) dropped out due to nausea and vomiting, so that no meaningful inference could be drawn from the few patients who completed the study on 600 mcg. These findings contrast with those in rodents (169, 170) and nonhuman primates (171) where PYY3-36 preparations reduce body weight. One suggested explanation is that the PYY3-36 effect is critically modulated by the time of injection. As the main anorexigenic effect of PYY is by Y2R-mediated NPY inhibition, PYY is obviously more effective at times that the orexigenic NPY is increased. In accordance with this theory is the reported weight loss effect of PYY3-36 when injected in rodents in the fasting state or in the early dark cycle — times when NPY is naturally induced (172).
PYY3-36 is structurally similar to pancreatic polypeptide (PP); PYY3-36 acts mainly through Y2R, while PP acts through Y4R. Obinepitide (TM30338), a synthetic dual-analogue of PYY3-36 and PP that stimulates both Y2/Y4-receptors, has been developed. Pre-clinical studies have shown that obinepitide efficiently reduces weight in obese mice. Furthermore, initial studies in humans have shown that once-a-day subcutaneous administration of obinepitide in obese human subjects inhibited food intake, at a statistically significant level, up to at least nine hours after dosing (173). Various PYY analogues have been created including intravenous, oral or nasal formulations. Interestingly, the combined therapy of PYY3-36 and GLP-1 receptor agonist (exendin-4) was found to decrease food intake and body weight in an additive manner in animal models and humans. Specifically, this synergistic result was attributed to the enhancement of c-fos reactivity in special cerebral nuclei (174). (see Table 20)
|
Table 20. PYY |
|
|
Mechanism of action |
Anorexigenic peptide which decreases gastric motility, increases satiety, inhibits NPY receptors |
|
Clinical Benefits |
↓ appetite, decreases food intake, ghrelin levels |
|
Adverse events |
Short-time action |
Ghrelin Vaccines and Ghrelin Inhibitors
Ghrelin is a 28-amino acid peptide produced primarily by the stomach and proximal small intestine (175). It is the only known circulating orexigenic hormone and signals both on vagal afferents and in the arcuate nucleus where it powerfully enhances NPY orexigenic signaling (176, 177). Its levels increase before meals and are suppressed by ingested nutrients, with carbohydrates being the most effective ones (compared to proteins and lipids). Ghrelin’s suppression results from neutrally transmitted (non-vagal) intestinal signals, augmented by insulin. An experimental ghrelin vaccine, CYT009-GhrQb, was discontinued in 2006 as it did not have the expected effects on weight loss. A novel one conjugated to the hapten, keyhole limpet hemocyanin (KLH), tested in rodent models, was shown to decrease feeding and induce weight loss (178). NOX-B11 is a ghrelin-neutralizing RNA spiegelmer that attaches to the active form of ghrelin and blocks its ability to bind to its receptor thus blocking the orexigenic activity of exogenously administrated ghrelin in rats (179). However, NOX-B11 did not affect basal food intake in nonfood-deprived rats, thus this treatment may only be efficacious when plasma ghrelin levels are high, such as before a meal or during times of food restriction (dieting).Since the discovery that the effects of ghrelin are primarily mediated by the GH secretagogue receptor (GHSR) 1a, there have been multiple potent, selective, and orally bioavailable ghrelin antagonists produced with good pharmacokinetic (PK) profiles that are currently in preclinical testing. An amide derivative 13d (Ca2+ flux IC50 = 188 nM, [brain]/[plasma] = 0.97 @ 8 h in rat), for example, showed a 10% decrease in 24-hour food intake in rats, and over 5% body weight reduction after 14-day oral treatment in diet-induced obese (DIO) mice (180).
Moreover, the discovery of ghrelin O-acyltransferase (GOAT) as the enzyme that catalyzes ghrelin octanoylation, revealed several therapeutic possibilities including the design of drugs that inhibit GOAT and block the attachment of the octanoyl group to the ghrelin third serine residue; such GOAT inhibitors could potentially prevent or treat obesity (181). Octanolyation of ghrelin by GOAT on its third amino acid (serine-3) is necessary for the hormone’s biological functions. Octanoylated ghrelin enhances hyperphagia and increases gastrointestinal motility. Furthermore, it reduces insulin secretion causing glucose dysfunction, enhances thermogenesis, adipogenesis and liver lipogenesis, limiting lipolysis at the same time (182). So, inhibiting GOAT could impede the production of acyl-ghrelin and increase desacyl-ghrelin, thus improving glucose homeostasis. In 2010, GO-CoA-Tat was created. A peptide-based bi-substrate analog which inhibited GOAT activity. The chronic treatment with GO-CoA-Tat, resulted in body weight stabilization in vehicle-treated mice fed MCT-rich HFD. Additionally, a decrease of fat mass was shown, but not of lean mass (183). Another study on Siberian hamsters also resulted in improvement in ingestive behavior. Remarkably, after 48h food deprivation, GO-CoA-Tat attenuated food foraging, food intake, and food hoarding post-refeeding relative to animals treated with saline. GO-CoA-Tat treated mice improved their blood glucose (184).
Another promising anti-obesity agent against ghrelin is a brain penetrant CAMKK2 inhibitor. Generally, CAMKK2 has been identified as the hypothalamic AMPK kinase that transduces Ca2+-mediated ghrelin signaling, inhibiting selectively hypothalamic AMPK and NPY’s downstream orexigenic effect. 4t, a 2,4-diaryl 7-azaindole, was created in order to inhibit AMPK phosphorylation in a hypothalamus-derived cell line. When this agent was tested in rodents, it managed to reduce ghrelin-induced food intake (185) (see Table 21).
|
Table 21. Ghrelin Vaccine (NOX-B11) |
|
|
Mechanism of action |
Ghrelin vaccine |
|
Clinical Benefits |
↓ food intake, hypothalamic orexigenic signals, ↑energy expenditure |
|
Adverse events |
No weight loss seen in human trials |
Fat-Specific Satiation Peptides
ENTEROSTATIN AND APOLIPOPROTEIN A-IV
Enterostatin and apolipoprotein A-IV appear to be GI peptides that are specifically stimulated by fat ingestion and subsequently regulate intake and/or metabolism of lipids. Although peripheral and central enterostatin administration decreases dietary fat intake in animals (while enterostatin-receptor antagonists did the opposite) (186), its administration to humans has shown no effects on food intake, appetite, energy expenditure, or body weight (187). Similarly, apolipoprotein A-IV, which is synthesized and secreted exclusively by the small intestine (primarily by the jejunum, but also by the duodenum and ileum), acts as a satiety factor that is downregulated by leptin (188) and upregulated by insulin and PYY in both rodents and humans (189). Although exogenous administration of apolipoprotein A-IV was quite effective concerning meal size, food intake, and weight gain reduction in rats (190), data is lacking regarding apo A-IV therapeutic administration in humans and its effects on body weight.
Pancreatic Satiation Peptides
PANCREATIC POLYPEPTIDE (PP)
Pancreatic polypeptide (PP) is a 36-amino acid peptide that is structurally similar to PYY. It is primarily produced in the pancreas in response to ingestion of food and in proportion to caloric load (191). Animal studies have shown that peripheral administration of PP decreases feeding (through Y4R in the area postrema), whereas centrally administrated PP increases it (through Y5R deeper in the brain) (192). In humans, intravenous infusion of PP (10 pmol/kg/min) (supra-physiological levels of PP) in ten healthy volunteers (men and women of normal body weight) caused a sustained decrease in both appetite and cumulative 24-hour energy intake by 25.3 +/- 5.8% (193). The findings of another study studying the anorexigenic effect of a lower infusion rate of PP (5 pmol/kg/min) in lean fasted volunteers were similar, holding promise for potential use as an anti-obesity agent (194). Another trial studying whether combined treatment with PP/PYY3-36 is superior regarding weight loss compared to either agent alone concluded that PP and PYY3-36 do not inhibit feeding additively in humans (195). Again, this study was conducted on lean subjects. Conversely, as previously mentioned, a synthetic analogue (TM30338) of both PYY3-36 and PP, which acts as an agonist of both the Y2 and Y4 receptors, yielded very promising results as concerns early meal termination when administered once-a-day subcutaneously in obese human subjects. Similarly, initial reports of a selective Y4-receptor agonist (TM30339) currently under development were also quite promising inducing reduction of food intake and promoting weight loss.
AMYLIN AND AMYLIN ANALOGUES
Amylin is a 37-amino acid neuroendocrine peptide hormone co-secreted postprandially with insulin by pancreatic β-cells. Among other properties, amylin is characterized by centrally mediated glucoregulatory and anorexigenic actions (196). It inhibits gastric emptying and glucagon secretion as well as decreases meal size and calorie intake (fat specific) (197) in a dose-dependent manner. These are vagus-independent actions and are exerted via binding to specific amylin receptors in the hindbrain area postrema (198), which is in contrast with the peripheral neural mechanisms engaged by most other gut peptides involved in energy homeostasis system regulation. The anorectic efficacy of amylin along with its glucoregulatory actions were investigated in human studies with the usage of pramlintide, a subcutaneous injectable amylin analogue which differs from amylin by only three amino acids. Studies in patients with type 1 and type 2 diabetes have shown great improvement in glycemic control plus sustained reductions in food intake and meal size, as well as mild progressive weight loss, following acute and long-term adjunctive pramlintide treatment (120 μg) (199). The most common adverse event associated with pramlintide usage was transient, mild-to-moderate nausea. This weight loss is noteworthy because it occurred in subjects with type 2 diabetes, on concomitant insulin therapy, and in the face of a significant A1C reduction, factors that all favor weight gain. Similar to the GLP-1 analogues discussed previously, pramlintide is currently approved for the treatment of type 1 and type 2 diabetes.
Whether pramlintide could constitute a potent anti-obesity agent was investigated in well-designed trials addressing this issue. In such a study (16-week randomized, double-blind, placebo-controlled), 204 individuals with obesity but not diabetes were treated with self-administered subcutaneous injections of pramlintide (nonforced dose escalation ≤ 240 μg) or placebo three times a day, 15 minutes before meals without concomitant lifestyle intervention (200). Pramlintide was generally well-tolerated and approximately 90% of the pramlintide-treated subjects were able to escalate to the highest dose of 240 μg three times a day. In contrast to the placebo-treated subjects who experienced minimal changes in body weight over the 16-week treatment period, the pramlintide-treated subjects attained significant weight loss from baseline as early as week 2, which was progressive up to week 16, with no evidence of a plateau. At week 16, the placebo-corrected reduction in body weight after pramlintide treatment was statistically significant compared with placebo (3.7 ± 0.5%, P < 0.001; 3.6 ± 0.6 kg, P < 0.001). Furthermore, the reduction in weight in pramlintide-treated subjects was accompanied by a significant reduction in waist circumference compared with placebo-treated subjects after 16 weeks of treatment (evaluable 4.3 ± 0.6 vs. 0.7 ± 0.9 cm, P < 0.01). At the end of the 16-week trial, 31% of the subjects treated with pramlintide achieved ≥ 5% weight loss compared to just 2% of the placebo group (P < 0.001). Interestingly, 8 weeks after treatment cessation, the pramlintide-treated subjects had on average regained one third of the overall weight loss observed by week 16. These findings constitute a proof of concept that pramlintide may have therapeutic use as an anti-obesity agent. Remarkably, at this higher dose (240 μg three times a day), the mean reduction in body weight with pramlintide treatment over 16 weeks was approximately twice that previously observed over a similar time-frame in insulin-treated subjects with type 2 diabetes who were treated with lower pramlintide doses (120 μg). This could suggest that higher doses of pramlintide might be necessary to achieve significant weight loss, although it is not yet clear whether concurrent insulin treatment was the main cause of that difference.
AMYLIN/PRAMLINTIDE COMBINATIONS
Previous animal studies have shown that amylin treatment significantly enhanced hypothalamic anorexigenic leptin signaling, while the combination treatment with amylin and leptin led to marked, synergistic reductions in food intake (up to 45%) and fat-specific weight loss (up to 15%). Recently, the weight-lowering effect of combined amylin/leptin agonism in human obesity was evaluated using the analogues pramlintide/metreleptin, respectively. As previously discussed, (see leptin), three trials addressing the weight loss efficacy of the combined treatment over 20, 28, and 52 weeks, respectively) reported sustained and robust weight loss by the combined treatment. Development was discontinued following commercial reassessment of the program. A Phase II study of davalintide, a second-generation analogue of amylin, for the treatment of obesity has also completed. In this study however, the weight loss efficacy and tolerability profile of davalintide was not superior to pramlintide, and was inferior to the pramlintide/metreleptin combination, thus resulting in deciding to halt further development of davalintide.
The anti-obesity effect of the combined treatment amylin/PYY3-36 was evaluated in an animal study, given that they both may have the potential for short-term signals of meal termination with anorexigenic and weight-reducing effects (201, 202). Statistical analyses revealed that food intake suppression with the combined treatment was synergistic, whereas body weight reduction was additive; this combination has not yet been studied in humans. Additional preclinical studies looking at the safety and efficacy of the combined treatment with pramlintide/phentermine and pramlintide/sibutramine was evaluated in a randomized placebo-controlled study with 244 obese or overweight nondiabetic subjects (203). The results suggested that the weight loss achieved at week 24 with either combination treatment was greater than with pramlintide alone or placebo (P < 0.001; 11.1 +/- 1.1% with pramlintide + sibutramine, 11.3 +/- 0.9% with pramlintide + phentermine, -3.7 +/- 0.7% with pramlintide; -2.2 +/- 0.7% with placebo; mean +/- s.e.), without any major adverse events.
As mentioned above, the human amylin receptor subtypes consist of calcitonin receptor and receptor activity-modifying proteins. Because of their mechanism of action, amylin mimetics coupled with calcitonin receptor agonists, are known as dual action amylin and calcitonin receptor agonists (DACRA). DACRA KBP-088 showed greater efficacy relative to davalintide regarding in vitro receptor pharmacology and in vivo efficacy of food intake and body weight (204). DACRA KBP-088 and KBP-042 improved body weight, glycemic control and adipose hypertrophy in high-fat diet-fed rats (205). A long acting amylin analogue is also in phase I clinical trial as a once daily anti-obesity treatment (206). (Table 22)
|
Table 22. Amylin/Pramlintide Combinations |
||||
|
Drug name |
FDA approved/Phase |
Mechanism of action |
Clinical Benefits |
Adverse events |
|
pramlintide |
Approved for DM1, DM2 |
Amylin analogue |
-in DM1, DM2: ↓ blood glucose, food intake, body weight, waist circumference |
Nausea |
|
Davalintide (AC2307) |
Phase II |
Amylin analogue |
↓food intake, body weight, HbA1c |
hypoglycemia |
|
DACRA KBP-088, KBP-042 |
|
Dual amylin and calcitonin receptor agonist |
↓body weight, glycemic control, adipose hypertrophy |
|
PERIPHERAL MODULATORS OF THE EFFICIENCY OF DIGESTION, METABOLISM, AND LIPOGENESIS
Lipase Inhibitors
Apart from early termination of food intake augmented by the centrally acting appetite suppressants, another potential therapeutic anti-obesity approach is the induction of a negative energy balance through the inhibition of nutrient, particularly fat, absorption. Lipase inhibitors inhibit gastric and pancreatic lipases in the lumen of the gastrointestinal tract that decrease systemic absorption of dietary fat. Orlistat is currently the only marketed anti-obesity drug of this category licensed for the treatment of obesity (including weight loss and weight maintenance). Additionally, it has been proven to improve glucose metabolism and nonalcoholic fatty liver disease. The most common adverse events are gastrointestinal system and include oily spotting, flatus with discharge, diarrhea, fecal urgency, and vitamin malabsorption (207).
The only other pancreatic and gastrointestinal lipase inhibitor currently in clinical development is Cetilistat (ATL-962). A short-term (12-week) randomized, placebo-controlled study of weight reduction addressing the efficacy, safety, and tolerability of Cetilistat in obese patients reported that Cetilistat produced a clinically and statistically significant weight loss in obese patients to similar extents at all doses examined compared to placebo (60 mg t.i.d. 3.3 kg, P<0.03; 120 mg t.i.d. 3.5 kg, P=0.02; 240 mg t.i.d. 4.1 kg, P<0.001), plus it significantly improved other obesity-related parameters including waist circumference, serum cholesterol and low-density lipoprotein cholesterol levels. Cetilistat treatment was also well-tolerated and the common orlistat-induced GI adverse events, such as flatus with discharge and oily spotting, occurred in only 1.8-2.8% of subjects in the Cetilistat-treated group (208). The combined results from three Phase I clinical studies designed to investigate the efficacy, pharmacodynamics, and tolerability of a range of Cetilistat doses [50 mg t.i.d. (n = 7), 60 mg t.i.d. (n = 9), 100 mg t.i.d. (n = 7), 120 mg t.i.d. (n = 9), 150 mg t.i.d. (n = 16), 240 mg t.i.d. (n = 9) and 300 mg t.i.d. (n = 9)] compared with placebo or orlistat [120 mg t.i.d. (n = 9)] in healthy volunteers were published (209). They reported that Cetilistat is equipotent with orlistat regarding fecal fat excretion; it however achieves a much better tolerance profile, as the number of episodes of steatorrhea per subject in the orlistat group (4.11) was 2.5-fold greater than that in the Cetilistat-treated group. The different tolerance profile between the two lipase inhibitors, seems to be related to the physical form of the fat in the intestine (rather than the amount of fat) resulting from each medication. Thus, Cetilistat acts more like a detergent, whereas orlistat may promote the coalescence of micelles, leading to oil-drops and increased gastrointestinal adverse events. Finally, a 12-week trial compared the efficacy and safety of Cetilistat (40, 80 or 120 mg three times daily) and orlistat (120 mg t.i.d.) relative to placebo in obese patients with type 2 diabetes on metformin (210). In this study similar reductions in body weight were observed in patients receiving Cetilistat (80 or 120 mg t.i.d.) or orlistat; these reductions were significant compared to placebo (3.85 kg, P = 0.01; 4.32 kg, P = 0.0002; 3.78 kg, P = 0.008). Furthermore, treatment with Cetilistat (80 or 120 mg t.i.d.) or with orlistat significantly improved glycemic control relative to placebo; again, Cetilistat was well-tolerated and showed fewer discontinuations due to adverse events than in the placebo and orlistat groups. Based on the above findings, this novel lipase inhibitor is currently at the furthest stage in the clinical development of new drugs of this class (see Table 23).
|
Table 23. Lipase Inhibitors |
|||||
|
Drug name |
FDA approved |
Mechanism of action |
Weight loss vs placebo |
Clinical Benefits |
Adverse events |
|
Orlistat (Xenical) |
1999 |
Lipase inhibitor |
2.6% |
↓ HbA1c, nonalcoholic fatty liver disease |
Gastrointestinal side effects, vitamin malabsorption Contraindicated in:Chronic malabsorption syndrome, cholestasis |
|
Cetilistat (ATL-962) |
|
Pancreatic and gastric lipase inhibitor |
|
↓body weight, lipid profile, waist circumference |
Gastrointestinal (less than orlistat) |
Growth Hormone (GH) and GH Lipolytic Domain Synthetic Analogues
Besides its growth effects, GH also possesses significant metabolic properties, including lipolysis induction. On the other hand, GH dynamics change with increasing adiposity and GH circulating levels and response to stimuli are repressed in obesity (211, 212). Taken together, it could be hypothesized that GH administration is an effective therapeutic option for weight loss and fat mass reduction in obese individuals. However, the majority of the 16 clinical trials of GH administration in obesity indicated little or no beneficial effects of GH treatment on body weight (213). There is a report from an Australia-based biotechnology company of the development of a modified fragment of amino acids 177-191 of GH (hGH177-191) (AOD-9604) that mimics the lipolytic effects of GH without producing growth effects. AOD-9604 however failed to induce significant weight loss in a 24-week trial of 536 subjects and its development as an anti-obesity agent was terminated (214). In 2018, it was announced that GH not only promotes lipolysis, but also enhances the creation of beige adipose tissue through activation of STAT5 and induction of ADRB3. Consequently, it promotes the adrenergic action of WAT.
β3-Adrenoreceptor Agonists
The β3-adrenergic receptor is expressed in adipocytes; its activation by cognate β-agonists cause lipolysis and increase thermogenesis. Thyroid hormones increase thermogenesis via the thyroid hormone receptor β subtype; however, to date, every attempt to develop selective thyroid hormone receptor agonists which are effective in adipose tissue without systemic side-effects has failed. In 2000, a selective human β3-agonist, L-796568, was developed (215). Although its acute (4-hour period) administration in overweight human subjects was associated with significant increase in energy expenditure (by ~8%) (216), a 28-day clinical trial investigating the efficacy of chronic use of L-796568 in overweight and obese non-diabetic men receiving the drug (350 mg/d) failed to display any significant changes in body composition or 24-hour energy expenditure (217). The ineffectiveness of β3-adrenreceptor activation to induce significant and sustained lipolysis in humans may be explained by the fact that human WAT expresses minimal levels of β3-adrenoreceptors; similarly, their expression is also low within human brown adipose tissue.
11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitors
Previous studies have shown enhanced conversion of inactive cortisone to active cortisol through the expression of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) in cultured omental adipose stromal cells (218); the autocrine action of cortisol may be crucial in the pathogenesis of central obesity and features of the metabolic syndrome, such as insulin resistance. The reports relating to effectiveness of carbenoxolone (nonselective 11β-HSD inhibitor) in reducing central obesity are conflicting (219). Currently, several pharmaceutical companies are developing selective 11β-HSD1 inhibitors that are effective in adipose tissue and may be more effective in improving insulin sensitivity and reducing body weight. Preliminary data from animal studies evaluating the weight-loss benefit of T-BVT, a new 11β-HSD1 pharmacological inhibitor with specificity for WAT, are very promising regarding its anti-obesity effectiveness and amelioration of multiple metabolic syndrome parameters (220). CNX-010-49, is another selective tissue-acting 11β-HSD1 inhibitor under investigation. Animal studies showed that this inhibitor acts on glucocorticoids and isoproterenol resulting in lipolysis in mature 3T3-L1 adipocytes. It not only enhances muscle glucose oxidation and mitochondrial biogenesis, but also reduces proteolysis and gluconeogenesis in primary mouse hepatocytes. As a result, it improves glucose control, lipid metabolism, and inhibits body weight gain without affecting feed consumption. A potential cardiovascular benefit was found because of the action of CNX-010-49 on plasminogen activator inhibitor-1 (PAI-1), interleukin-6 (IL-6), and fetuin-A (221). (see Table 24)
|
Table 24. 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitors |
||
|
Drug name |
Mechanism of action |
Clinical Benefits |
|
T-BVT |
Selective to white adipose tissue 11β-HSD1 |
|
|
CNX-010-49 |
Selective to white adipose tissue 11β-HSD1 |
↑lipolysis, ↓ HbA1c, lipid metabolism, inhibits body weight gain without affecting feed consumption |
Angiogenesis Inhibitors
Increasing adiposity is associated with expansion of the adipose capillary bed. Several vascular growth factors are produced by enlarged adipocytes, for example, vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and angiogenin, which may in turn facilitate the expansion of adipose tissue. Thus, anti-angiogenesis may eventually participate in the treatment of obesity. This hypothesis is strengthened by studies where the experimental administration of anti-angiogenic agents in mice from different obesity models resulted in significant weight reduction and adipose tissue loss (222). Remarkably, there were benefits on food intake, metabolic rate, and preferred energy substrate. These findings appeared to modulate fat tissue by altering vasculature. Although there are many foods and beverages containing naturally occurring inhibitors of angiogenesis (e.g. green tea, oranges, strawberries, lemons, red wine, ginseng, garlic, tomato, olive oil, etc.), no convincing clinical trials have been conducted investigating their anti-obesity effect so far. Currently, a Phase II trial using the anti-angiogenic/anti-MMP drug ALS-L1023 for the treatment of obesity is underway (223). Similarly, endostatin was found to have both anti-adipogenic and anti-angiogenic action protecting mice against dietary-induced obesity (224).
Sirtuin 1 (SIRT1) Activators
Sirtuin 1 (SIRT1) is a member of the Sirtuin family of proteins that comprises seven members in mammals (SirT1-T7). Sirtuin proteins have gained considerable attention due to their importance as physiological targets for treating diseases associated with aging. They contribute to cellular regulation interacting with metabolic pathways and may serve as entry points for drugs. SIRT1 has gained popularity as it has been linked with the French Paradox and the calorie restriction-mediated longevity and delayed incidence of several diseases associated with aging, such as cancer, atherosclerosis, and diabetes. The calorie restriction-induced modulations have been demonstrated in organisms ranging from yeast to mammals. White adipose tissue seems to be a primary factor in the longevity brought about through calorie restriction, as mice engineered to have reduced levels of WAT live longer (224). Corroborating this, it was found that food withdrawal is followed by SIRT1 binding and repression of genes controlled by the fat regulator PPAR-γ (peroxisome proliferator-activated receptor-γ), including genes mediating fat storage. This, in turn, activates fat mobilization and lipolysis and reduces WAT mass (225). In addition to PPAR-γ, SIRT1 also interacts with PGC-1α, inducing the expression of mitochondrial genes involved in oxidative metabolism and fatty acid oxidation, while it also enhances leptin sensitivity by repressing PTP1B. The weight restricting effects of SIRT1 were further supported by experiments with resveratrol (RSV), a potent allosteric SIRT1 activator, which was shown to protect mice from diet-induced obesity (226). Furthermore, mice treated with SRT1720, a potent, selective synthetic activator of SIRT1, were resistant to diet-induced obesity due to enhanced oxidative metabolism in skeletal muscle, liver, and brown adipose tissue, indicating the positive metabolic consequences of specific SIRT1 activation (227). Currently, several pharmaceutical companies are investigating specific SIRT1 activators in Phase I and Phase II trials for the treatment of type II diabetes and obesity (228) to define their utility in the treatment of obesity and metabolic diseases.
Cyclic-GMP Signaling in Anti-Obesity Pharmacotherapy
Cyclic nucleotides, including 3-5-cyclic guanosine monophosphate (cGMP) and 3-5-cyclic adenosine monophosphate (cAMP), are second messengers important in many biological processes. Knowledge of the role of cAMP in the regulation of energy homeostasis has been extended, thanks to its intimate relationship with AMPK (AMP-activated protein kinase) signaling; intracellular cAMP activates the AMPK signaling pathway. AMPK regulates energy balance at both cellular and whole-body levels (229). Activation of AMPK facilitates fatty acid oxidation and mitochondria biogenesis, which promotes energy expenditure (230). Interestingly, activation of AMPK in the hypothalamus promotes food intake behavior (231). e.g. physiologic processes in the same direction and induces weight loss by mutual reinforcement. Moreover, off-the-shelf approaches might be possible, given the existence of an established market for medications targeting cGMP pathways, with FDA- and EMA-approved drugs such as sildenafil and linaclotide. Sildenafil acts on adipocytes, possibly through cGMP-dependent protein kinase I and mechanistic/mammalian target of rapamycin (mTOR) signaling pathways, browning subcutaneous white fat, thus increasing energy expenditure (232).
Beloranib
Beloranib is an analogue of the natural chemical compound fumagillin and is a methionine aminopeptidase 2 (MetAP2) inhibitor acting to reduce production of new fatty acid molecules by the liver and converting stored fats into useful energy (233). It was first tested in 31 obese women, who were divided into four groups (0.1mg, 0.3mg, 0.9mg, or placebo twice weekly). A dose-dependent weight loss was shown after four weeks of 0.9mg Beloranib administration with mean 3.8kg loss vs 0.6kg in the placebo group. It also improved lipid metabolism and lowered C-reactive protein and adiponectin. A phase II double-blinded, randomized clinical trial examined the efficacy and safety of Beloranib administration (234).147 obese patients were divided into four groups: 0.6, 1.2, 2.4 mg subcutaneous injection or placebo. After twelve weeks of administration, a dose-dependent weight loss of -5.5, -6.9, -10kg, respectively, was reported, vs -0.4kg in the placebo group. The main adverse events were sleep disturbance and gastrointestinal abnormalities. Beloranib may also cause robust weight loss and hypophagia in rats with hypothalamic and genetic obesity (235). In 2015, however, a phase III clinical trial for Prader-Willi was stopped after a second patient death (236). (see Table 25)
|
Table 25. Beloranib |
|
|
FDA approved/Phase |
Phase III aborted in 2015 after second patient death in Prader-Willi trial |
|
Mechanism of action |
Fumagillin analogue with methionine aminopeptidase 2 inhibition that reduced fatty acid synthesis in the liver and converted stored fat into useful energy; originally designed as an angiogenesis inhibitor |
|
Clinical Benefits |
↑ weight loss, hypophagia, ↓ lipid metabolism, CRP, adiponectin, cardiovascular factors |
|
Adverse events |
Sleep disturbance, gastrointestinal abnormalities |
Fibroblast Growth Factor (FGF21)
Fibroblast growth factor (FGF) 21, expressed primarily in the liver, but also found in adipose tissue, skeletal muscle, and pancreas, is a member of the FGF family and acts as a metabolic regulator of body weight, glucose metabolism, and lipid metabolism (237). In WAT, FGF21 induces glucose uptake and adiponectin secretion with browning of white adipose tissue. In brown adipose tissue, it stimulates glucose uptake and thermogenesis, thus increasing energy expenditure. In the liver, it blocks GH signaling, regulates fatty acid oxidation both in the fasted state and in mice consuming high-fat, low-carbohydrate ketogenic diet and it maintains lipid homeostasis (238). FGF21 is characterized by anti-inflammatory, anti-oxidative stress properties with its circulating concentration increasing during periods of muscle activity or critical stress (239). Although, it is an attractive anti-obesity and anti-diabetes target, FGF21 levels are increased in obese ob/ob and db/db mice and correlate positively with BMI in humans. Exogenous administration of FGF21 in DIO in mice show virtually no beneficial effects on glucose tolerance and lipid metabolism, suggesting that the obesity state is FGF21-resistant (240).
ALTERNATIVE AND COMPLEMENTARY TYPES OF TREATMENT OF OBESITY
Gut Microbiota
Recently, a major shift in research has occurred towards the investigation of gut microbiota effects on energy expenditure and metabolism. Gut microbiota are responsible for a significant amount of the interaction between the host and the nutritional environment. Soluble fiber such as galacto-oligosaccharides and fructo-oligosaccharides (FOS), are fermented by the gut microbiota into short-chain fatty acids (SCFAs) acetate, propionate and butyrate (241). This mechanism provides to host 10% of its daily energy requirement (242). These SCFAs are an energy source for colonic epithelium, liver, and peripheral tissues (243). By fermenting nondigestible dietary fibers, host metabolism is enhanced. In mice with DIO, SCFAs improved glucose metabolism, insulin resistance, and obesity. In other animal studies, butyrate-producing bacteria (F. prausnitzii) induced secretion of glucagon-like peptide 1 (GLP1) from colonic L cells through the fatty acid receptor FFAR2(244). Furthermore, butyrate and propionate activate intestinal gluconeogenesis. Butyrate, through a cAMP-dependent mechanism, promotes the gene expression involved in intestinal gluconeogenesis. Propionate, itself a substrate for intestinal gluconeogenesis, activates its expression viaa gut-brain neural circuit involving the fatty acid receptor FFAR3 (245).
Given the key role played by microbiota in host nutrient processing and metabolism, it is not surprising that data points to a strong relation between gut microbiota and obesity and diabetes in humans. A reduced gut microbial diversity and altered microbiota composition is observed in obese individuals. There is also a low rate of gut microbial richness and specific bacterial groups are enriched or decreased in obese patients in comparison with lean people (246). Moreover, chronic diseases, such as obesity, diabetes, and HIV are associated with chronic low-grade inflammation. Gut microbiota regulates this inflammation through several mechanisms. Lipopolysaccharides (LPSs) from the outer membrane of Gram-negative bacteria may translocate through the intestinal border and cause subsequent systemic inflammation (247). Indeed, the intestinal barrier of obese patients is more permeable compared with that of lean individuals. Bile acids are characterized by a strong relation with gut microbiota affecting host’s body-weight homeostasis. Bile acids are microbially altered metabolites that are first endogenously produced by the liver and further metabolized by the gut microbiota (248). FXR signaling is an important pathway connecting gut microbiota and bile acids.
Based on the above knowledge, several interventions involving manipulation of the microbiome have been proposed as anti-obesity treatment. A diet which contains soluble fiber, prebiotics and/or probiotics could enhance the growth of beneficial gut microbiota and boost host metabolism. Lately, there has been interest in berberine administration in T1D, T2D, gestational diabetes, and prediabetes. The early reports of interventions using probiotics appear successful (249). Fecal microbiota transplant (FMT), the transfer of fecal suspension from a healthy (lean) donor into the gastrointestinal tract of an individual with disease (obesity) in order to restore a healthy gut is a potentially novel option to treat obesity. However, there is not enough data about the safety of this method, that is why it is only FDA approved for recurrent Clostridium difficile infection.
Anti-Obesity Vaccines (Ghrelin, Somatostatin, Ad36)
The idea of a vaccination against obesity is also intriguing. The main action of these vaccines would be based on suppressing appetite-stimulating hormones or blocking food absorption. Three vaccines have been tested so far:
- An anti-ghrelin vaccine was found not only to reduce appetite by decreasing hypothalamic orexigenic signals but also to increase energy expenditure in rodent and pigs (250). Despite the promising results in rodents, clinical trials in humans showed no weight loss despite the development of ghrelin autoantibodies after four injections of anti-ghrelin vaccine (251). Another study, however, showed that IgG anti-ghrelin autoantibodies could protect ghrelin from degradation, suggesting that an autoimmune response may be involved in the orexigenic effects of ghrelin (252).
- An anti-somatostatin vaccine. Somatostatin is a peptide hormone which is produced, mainly, in the hypothalamus as well as other tissues, such as the gastrointestinal system. Somatostatin has the ability to suppress GH and insulin-like growth factor 1 (IGF-1) secretion. Reduced GH is associated with obesity and increased adiposity. So, the somatostatin vaccine could increase the secretion of GH and IGF-1(253). However, clinical trials in mice failed to reduce food intake, though a 10% improvement of body weight was observed (254).
- A live adenovirus 36 (Ad36) vaccine. Adenovirus 36 increases the risk of obesity in humans, characterized by increased inflammation and adiposity (255). Mice were injected with live Ad36 vaccine and compared to the control group (unvaccinated) after 14 weeks. The control group had 17% greater body weight and 20% more epididymal fats versus the vaccinated group, which also had decreased inflammatory cytokines and macrophages in fat tissue (256). (see Table 26)
|
Table 26. Anti-Obesity Vaccines |
|||
|
Drug name |
Mechanism of action |
Weight loss vs placebo |
Clinical Benefits |
|
Anti-obesity vaccine: somatostatin vaccine |
Increases the secretion of GH, IGF-1 |
10% |
|
|
Adenovirus 36 |
Live adenovirus36 |
|
Decreases body weight, epididymal fat in mice, inflammatory cytokines and macrophages |
Nanomedicine
The introduction of nanomedicine in the field of obesity treatment is highly novel (257). Nanoparticles can achieve targeted drug delivery along with minimized side effects. The poor water-solubility of anti-obesity drugs can be overcome via nano-encapsulation. More specifically, nanoemulsion of orlistat has been tried in order to overcome its high lipophilicity, to improve its dissolution and to avoid the pancreatic lipase inhibition caused by this pharmaceutical agent in vivo (258). Additionally, a conjugated polymer-nanocarrier was created in order to reduce the side effects of orlistat (259). In 2014, the ability of mesoporous silica particles to reduce body weight was investigated (260). They found that the silica particles embedded in food could sequestrate lipase in their small pores through a lipase-specific interaction, leading to decreased absorption of fat.
Appetite suppression is an alternative method to decrease food intake and impact energy homeostasis (261). As mentioned above, however, anti-ghrelin vaccine was formed using virus-like particles for obesity treatment. The passive delivery of anti-ghrelin antibodies did not lead to long-term inhibition of food intake. So, to solve this problem, investigators immunoconjugated ghrelin with virus proteins to create a vaccine that was able to trigger an immune response leading to generation of specific anti-ghrelin antibodies. This anti-ghrelin vaccine played an important role in maintaining energy homeostasis in a DIO murine model.
In other examples, nanomedicine has enhanced the action of antiangiogenic agents in the treatment of obesity. Detailed above, antiangiogenic therapy inhibits the progression of adipocyte hyperplasia and reduces weight gain. A targeting nanoparticle was created in order to enhance the accumulation of the antiangiogenic drug in WATs by delivering it to vascular endothelial cells. Unlike WAT, brown adipose tissue (BAT) is full of mitochondria and a robust vascular structure helps to induce thermogenesis, increasing energy expenditure, and decreasing body weight. Thus, two nanoparticle platforms delivering browning agents to adipose tissue vasculature were formed (262). PPARγ nuclear receptor agonists (including rosiglitazone) have been shown to be characterized by anti-inflammatory properties against obesity and atherosclerosis. However, they are associated with severe side effects that limit their therapeutic use (263). In another, a mitochondria-targeted nanoparticle delivers the proposed anti-obesity compound PLGA-bPEG-triphenylphosphonium (TPP) polymer (264). The PEG shell extends the circulation time of nanoparticles, and TPP could facilitate the internalization into the matrix space of mitochondria to achieve targeted drug delivery.
Instead of targeted delivery, a localized and sustained release of a browning agent is a promising alternative for facilitation of WAT browning. Two nanoparticles, one injectable (265) and one in a painless microneedle array patch (266) were introduced. In vivo studies revealed successful delivery of the model drug into the human adipose tissue followed by ~15% decrease of weight gain after a four-week treatment.
CONCLUSION
The field of anti-obesity molecular pharmacotherapy is expanding. The homeostasis of body weight and metabolism are tightly linked to the central nervous system. The latter is characterized by centers that send orexigenic and anorexigenic signals regulating starvation and satiety, reducing and increasing energy expenditure, respectively. Pharmaceutical multi-agents in single compounds containing active portions of two or more drugs may allow for simultaneous effects on several synergistic pathways affecting appetite control and energy expenditure. Such medications could achieve increased weight loss with fewer side effects. Furthermore, the possibility of improved formulations (e.g., injectable forms of anti-obesity drugs and or once weekly verses daily administration) serve to enhance compliance. Considering that obesity is a multifactorial disease, it needs multimodal treatment. In an era where a variety of different therapeutic options is the norm for the management of chronic diseases such as type 2 diabetes and hypertension, the hope is that this process will led to better personalized anti-obesity treatments, focusing on the special characteristics, needs, and comorbidities of each patient and the effectiveness and safety of the recommended therapy. Thus, before starting any therapy, it will be important to record the detailed medical profile of the patient. Hereditary or acquired diseases, lifestyle parameters, and psychiatric history have to be taken into account when anti-obesity treatment is tailored for each patient. Further on the therapeutic horizon and still in much need of research are the place for altering gut microbiota balance and development of anti-obesity vaccines, novel peptide-mediated delivery of nuclear hormones, single molecular multi-agonists, and nanotechnologies that improve drug delivery and hold promise in the future of molecular pharmacotherapy of obesity.
REFERENCES
- Apovian CM, Aronne LJ, Bessesen DH, McDonnell ME, Murad MH, Pagotto U, et al. Pharmacological Management of Obesity: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2015;100(2):342-62.
- Kopelman P. Health risks associated with overweight and obesity. Obesity Reviews. 2007;8(s1):13-7.
- de Wit L, Luppino F, van Straten A, Penninx B, Zitman F, and Cuijpers P. Depression and obesity: A meta-analysis of community-based studies. Psychiatry Research. 2010;178(2):230-5.
- Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg. 1995;222(3):339-52.
- Dong Z, Xu L, Liu H, Lv Y, Zheng Q, and Li L. Comparative efficacy of five long-term weight loss drugs: quantitative information for medication guidelines. Obes Rev. 2017;18(12):1377-85.
- Ahima RS, and Flier JS. Leptin. Annu Rev Physiol. 2000;62:413-37.
- Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903-8.
- Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Jama. 1999;282(16):1568-75
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544-8
- Picardi PK, Calegari VC, Prada PO, Moraes JC, Araujo E, Marcondes MC, et al. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology. 2008;149(8):3870-80.
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35-51.
- Vasselli JR. The Role of Dietary Components in Leptin Resistance. Advances in Nutrition. 2012;3(5):736-8.
- Munzberg H, Flier JS, and Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145(11):4880-9.
- Wang J, Obici S, Morgan K, Barzilai N, Feng Z, and Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50(12):2786-91.
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105(20):7257-62.
- Müller TD, Sullivan LM, Habegger K, Yi C-X, Kabra D, Grant E, et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. Journal of Peptide Science. 2012;18(6):383-93.
- Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Kuchler D, et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2014;63(4):1422-7.
- Trevaskis JL, Wittmer C, Athanacio J, Griffin PS, Parkes DG, and Roth JD. Amylin/leptin synergy is absent in extreme obesity and not restored by calorie restriction-induced weight loss in rats. Obes Sci Pract. 2016;2(4):385-91.
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35-51.
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, and Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161(5):999-1011.
- Lee J, Liu J, Feng X, Salazar Hernandez MA, Mucka P, Ibi D, et al. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat Med. 2016;22(9):1023-32.
- Meehan CA, Cochran E, Kassai A, Brown RJ, and Gorden P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev Clin Pharmacol. 2016;9(1):59-68.
- Gordon CM, Ackerman KE, Berga SL, Kaplan JR, Mastorakos G, Misra M, et al. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2017;102(5):1413-39.
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin r responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105(20):7257-62.
- Aronne LJ, Halseth AE, Burns CM, Miller S, and Shen LZ. Enhanced weight loss following coadministration of pramlintide with sibutramine or phentermine in a multicenter trial. Obesity (Silver Spring). 2010;18(9):1739-46.
- Amylin and Takeda Discontinue Development of Pramlintide/Metreleptin Combination Treatment for Obesity Following Commercial Reassessment of the Program. 2011.
- Wellhoner P, Horster R, Jacobs F, Sayk F, Lehnert H, and Dodt C. Intranasal application of the melanocortin 4 receptor agonist MSH/ACTH(4-10) in humans causes lipolysis in white adipose tissue. Int J Obes (Lond). 2012;36(5):703-8.
- Fehm HL, Smolnik R, Kern W, McGregor GP, Bickel U, and Born J. The melanocortin melanocyte-stimulating hormone/adrenocorticotropin(4-10) decreases body fat in humans. J Clin Endocrinol Metab. 2001;86(3):1144-8.
- Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015;100(4):1639-45.
- Low MJ. Neuroendocrinology: New hormone treatment for obesity caused by POMC-deficiency. Nat Rev Endocrinol. 2016;12(11):627-8.
- Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. New England Journal of Medicine. 2016;375(3):240-6.
- An investigational, melanocortin-4 receptor (MC4R) agonist in clinical development for the treatment of rare genetic disorders of obesity. https://www.rhythmtx.com/our-pipeline/.
- Pissios P, Bradley RL, and Maratos-Flier E. Expanding the scales: The multiple roles of MCH in regulating energy balance and other biological functions. Endocr Rev. 2006;27(6):606-20.
- Ito M, Ishihara A, Gomori A, Egashira S, Matsushita H, Mashiko S, et al. Melanin-concentrating hormone 1-receptor antagonist suppresses body weight gain correlated with high receptor occupancy levels in diet-induced obesity mice. Eur J Pharmacol. 2009;624(1-3):77-83.
- Mendez-Andino JL, and Wos JA. MCH-R1 antagonists: what is keeping most research programs away from the clinic? Drug Discov Today. 2007;12(21-22):972-9.
- trials.gov C. Safety, Pharmacokinetics and Pharmacodynamics Study to Evaluate BMS-830216 in Obese Subjects. https://clinicaltrials.gov/ct2/show/NCT00909766?term=BMS-830216&draw=2&rank=1.
- Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, and Cheetham SC. Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine. Int J Obes Relat Metab Disord. 1998;22 Suppl 1:S18-28; discussion S9.
- Wooltorton E. Obesity drug sibutramine (Meridia): hypertension and cardiac arrhythmias. CMAJ. 2002;166(10):1307-8.
- Launay JM, Herve P, Peoc'h K, Tournois C, Callebert J, Nebigil CG, et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med. 2002;8(10):1129-35.
- Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol. 2000;57(1):75-81.
- Dunlop J, Sabb AL, Mazandarani H, Zhang J, Kalgaonker S, Shukhina E, et al. WAY-163909 [(7bR, 10aR)-1,2,3,4,8,9,10,10a-octahydro-7bH-cyclopenta-[b][1,4]diazepino[6,7,1hi]indol e], a novel 5-hydroxytryptamine 2C receptor-selective agonist with anorectic activity. J Pharmacol Exp Ther. 2005;313(2):862-9.
- Siuciak JA, Chapin DS, McCarthy SA, Guanowsky V, Brown J, Chiang P, et al. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology. 2007;52(2):279-90.
- John Dunlop SW, James E. Barrett, Joseph Coupet, Boyd Harrison, Hossein, Mazandarani SN, Menelas N. Pangalos, Siva Ramamoorthy, Lee, Schechter DS, Gary Stack, Jean Zhang, Guoming Zhang and Sharon, and Rosenzweig-Lipson. Characterization of Vabicaserin (SCA-136), a Selective 5-HT2C Receptor Agonist ASPET Journals. 2011,March 14.
- Burke LK, and Heisler LK. 5-Hydroxytryptamine Medications for the Treatment of Obesity. Journal of Neuroendocrinology. 2015;27(6):389-98.
- Higgins GA, and Fletcher PJ. Therapeutic Potential of 5-HT2C Receptor Agonists for Addictive Disorders. ACS Chem Neurosci. 2015;6(7):1071-88.
- Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-56.
- Fidler MC, Sanchez M, Raether B, Weissman NJ, Smith SR, Shanahan WR, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-77.
- FDA approves Belviq to treat some overweight or obese adults. Home Healthc Nurse. 2012;30(8):443-4.
- Anderberg RH, Richard JE, Eerola K, Lopez-Ferreras L, Banke E, Hansson C, et al. Glucagon-Like Peptide 1 and Its Analogs Act in the Dorsal Raphe and Modulate Central Serotonin to Reduce Appetite and Body Weight. Diabetes. 2017;66(4):1062-73.
- Bohula EA, Wiviott SD, McGuire DK, Inzucchi SE, Kuder J, Im K, et al. Cardiovascular Safety of Lorcaserin in Overweight or Obese Patients. New England Journal of Medicine. 2018;379(12):1107-17.
- Bohula EA, Scirica BM, Inzucchi SE, McGuire DK, Keech AC, Smith SR, et al. Effect of lorcaserin on prevention and remission of type 2 diabetes in overweight and obese patients (CAMELLIA-TIMI 61): a randomised, placebo-controlled trial. Lancet. 2018;392(10161):2269-79.
- Zarrindast MR, and Hosseini-Nia T. Anorectic and behavioural effects of bupropion. Gen Pharmacol. 1988;19(2):201-4.
- Billes SK, and Cowley MA. Inhibition of dopamine and norepinephrine reuptake produces additive effects on energy balance in lean and obese mice. Neuropsychopharmacology. 2007;32(4):822-34.
- Greenway FL, Whitehouse MJ, Guttadauria M, Anderson JW, Atkinson RL, Fujioka K, et al. Rational design of a combination medication for the treatment of obesity. Obesity (Silver Spring). 2009;17(1):30-9.
- Anderson JW, Greenway FL, Fujioka K, Gadde KM, McKenney J, and O'Neil PM. Bupropion SR enhances weight loss: a 48-week double-blind, placebo- controlled trial. Obes Res. 2002;10(7):633-41.
- Jain AK, Kaplan RA, Gadde KM, Wadden TA, Allison DB, Brewer ER, et al. Bupropion SR vs. placebo for weight loss in obese patients with depressive symptoms. Obes Res. 2002;10(10):1049-56.
- Gadde KM, Parker CB, Maner LG, Wagner HR, 2nd, Logue EJ, Drezner MK, et al. Bupropion for weight loss: aninvestigation of efficacy and tolerability in overweight and obese women. Obes Res. 2001;9(9):544-51.
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480-4.
- Yeomans MR, and Gray RW. Opioid peptides and the control of human ingestive behaviour. Neurosci Biobehav Rev. 2002;26(6):713-28.
- Mitchell JE, Morley JE, Levine AS, Hatsukami D, Gannon M, and Pfohl D. High-dose naltrexone therapy and dietary counseling for obesity. Biol Psychiatry. 1987;22(1):35-42.
- Greenway FL, Fujioka K, Plodkowski RA, Mudaliar S, Guttadauria M, Erickson J, et al. Effect of naltrexone plusbupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
- Calderone A, Calabro PF, Lippi C, Jaccheri R, Vitti J, and Santini F. Psychopathological Behaviour and Cognition in Morbid Obesity. Recent Pat Endocr Metab Immune Drug Discov. 2017;10(2):112-8.
- Apovian CM, Aronne L, Rubino D, Still C, Wyatt H, Burns C, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013;21(5):935-43.
- Wadden TA, Foreyt JP, Foster GD, Hill JO, Klein S, O'Neil PM, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring). 2011;19(1):110-20.
- Hollander P, Gupta AK, Plodkowski R, Greenway F, Bays H, Burns C, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-9.
- Nissen SE, Wolski KE, Prcela L, Wadden T, Buse JB, Bakris G, et al. Effect of Naltrexone-Bupropion on Major Adverse Cardiovascular Events in Overweight and Obese Patients with Cardiovascular Risk Factors: A Randomized Clinical Trial. Jama. 2016;315(10):990-1004.
- Okada M, Kaneko S, Hirano T, Mizuno K, Kondo T, Otani K, et al. Effects of zonisamide on dopaminergic system. Epilepsy Res. 1995;22(3):193-205.
- Okada M, Hirano T, Kawata Y, Murakami T, Wada K, Mizuno K, et al. Biphasic effects of zonisamide on serotonergic system in rat hippocampus. Epilepsy Res. 1999;34(2-3):187-97.
- Gadde KM, Franciscy DM, Wagner HR, 2nd, and Krishnan KR. Zonisamide for weight loss in obese adults: a randomized controlled trial. Jama. 2003;289(14):1820-5.
- Lim J, Ko YH, Joe SH, Han C, Lee MS, and Yang J. Zonisamide produces weight loss in psychotropic drug-treated psychiatric outpatients. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(8):1918-21.
- McElroy SL, Kotwal R, Hudson JI, Nelson EB, and Keck PE. Zonisamide in the treatment of binge-eating disorder: an open-label, prospective trial. J Clin Psychiatry. 2004;65(1):50-6.
- Gadde KM, Yonish GM, Foust MS, and Wagner HR. Combination therapy of zonisamide and bupropion forweight reduction in obese women: a preliminary, randomized, open-label study. J Clin Psychiatry.2007;68(8):1226-9.
- Ohtahara S, and Yamatogi Y. Safety of zonisamide therapy: prospective follow-up survey. Seizure. 2004;13:S50-S5.
- Rosenfeld WE. Topiramate: a review of preclinical, pharmacokinetic, and clinical data. Clin Ther. 1997;19(6):1294-308.
- Bray GA, Hollander P, Klein S, Kushner R, Levy B, Fitchet M, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res. 2003;11(6):722-33.
- Wilding J, Van Gaal L, Rissanen A, Vercruysse F, and Fitchet M. A randomized double-blind placebo-controlledstudy of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int J Obes Relat Metab Disord. 2004;28(11):1399-410.
- McElroy SL, Arnold LM, Shapira NA, Keck PE, Jr., Rosenthal NR, Karim MR, et al. Topiramate in the treatmentof binge eating disorder associated with obesity: a randomized, placebo-controlled trial. Am J Psychiatry. 2003;160(2):255-61.
- Hendricks EJ, Srisurapanont M, Schmidt SL, Haggard M, Souter S, Mitchell CL, et al. Addiction potential of phentermine prescribed during long-term treatment of obesity. Int J Obes (Lond). 2014;38(2):292-8.
- Samanin R, and Garattini S. Neurochemical mechanism of action of anorectic drugs. Pharmacol Toxicol. 1993;73(2):63-8
- Yanovski SZ, and Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. Jama. 2014;311(1):74-86.
- Munro JF, MacCuish AC, Wilson EM, and Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J. 1968;1(5588):352-4.
- Lewis KH, Fischer H, Ard J, Barton L, Bessesen DH, Daley MF, et al. Safety and Effectiveness of Longer-Term Phentermine Use: Clinical Outcomes from an Electronic Health Record Cohort. Obesity (Silver Spring). 2019;27(4):591-602.
- Antel J, and Hebebrand J. Weight-reducing side effects of the antiepileptic agents topiramate and zonisamide. Handb Exp Pharmacol. 2012(209):433-66.
- Allison DB, Gadde KM, Garvey WT, Peterson CA, Schwiers ML, Najarian T, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-42.
- Gadde KM, Allison DB, Ryan DH, Peterson CA, Troupin B, Schwiers ML, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-52.
- Garvey WT, Ryan DH, Look M, Gadde KM, Allison DB, Peterson CA, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
- Erondu N, Gantz I, Musser B, Suryawanshi S, Mallick M, Addy C, et al. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metab. 2006;4(4):275-82.
- Sargent BJ, and Moore NA. New central targets for the treatment of obesity. Br J Clin Pharmacol. 2009;68(6):852-60.
- Double-Blind, Multi-Center, Randomized Study to Assess the Efficacy and Safety of Velneperit (S-2367) and Orlistat Administered Individually or Combined with a Reduced Calorie Diet (RCD) in Obese Subjects. https://clinicaltrials.gov/ct2/show/NCT01126970.
- Nathan PJ, O'Neill BV, Mogg K, Bradley BP, Beaver J, Bani M, et al. The effects of the dopamine D(3) receptor antagonist GSK598809 on attentional bias to palatable food cues in overweight and obese subjects. Int J Neuropsychopharmacol. 2012;15(2):149-61.
- Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, and Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9653):1906-13.
- Sjodin A, Gasteyger C, Nielsen AL, Raben A, Mikkelsen JD, Jensen JK, et al. The effect of the triple monoamine reuptake inhibitor tesofensine on energy metabolism and appetite in overweight and moderately obese men. Int J Obes (Lond). 2010;34(11):1634-43.
- Gilbert JA, Gasteyger C, Raben A, Meier DH, Astrup A, and Sjodin A. The effect of tesofensine on appetite sensations. Obesity (Silver Spring). 2012;20(3):553-61.
- Schoedel KA, Meier D, Chakraborty B, Manniche PM, and Sellers EM. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharmacol Ther. 2010;88(1):69-78.
- Effect of Tesofensine on Weight Reduction in Patients with Obesity. . Updated April 22, 2013.
- McElroy SL, Mitchell JE, Wilfley D, Gasior M, Ferreira-Cornwell MC, McKay M, et al. LisdexamfetamineDimesylate Effects on Binge Eating Behaviour and Obsessive-Compulsive and Impulsive Features in Adults with Binge Eating Disorder. Eur Eat Disord Rev. 2016;24(3):223-31.
- Greenway FL, and Bray GA. Human chorionic gonadotropin (HCG) in the treatment of obesity: a critical assessment of the Simeons method. West J Med. 1977;127(6):461-3.
- Despres JP, Golay A, and Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353(20):2121-34.
- Randall PA, Vemuri VK, Segovia KN, Torres EF, Hosmer S, Nunes EJ, et al. The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmacol Biochem Behav. 2010;97(1):179-84.
- Bosch B, Venter I, Stewart RI, and Bertram SR. Human chorionic gonadotrophin and weight loss. A double-blind, placebo-controlled trial. S Afr Med J. 1990;77(4):185-9.
- Oh IS, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature. 2006;443(7112):709-12.
- Gonzalez R, Reingold BK, Gao X, Gaidhu MP, Tsushima RG, and Unniappan S. Nesfatin-1 exerts a direct, glucose-dependent insulinotropic action on mouse islet beta- and MIN6 cells. J Endocrinol. 2011;208(3):R9-r16.
- Prinz P, Teuffel P, Lembke V, Kobelt P, Goebel-Stengel M, Hofmann T, et al. Nesfatin-130-59 Injected Intracerebroventricularly Differentially Affects Food Intake Microstructure in Rats Under Normal Weight and Diet-Induced Obese Conditions. Front Neurosci. 2015;9:422.
- Gibbs J, Young RC, and Smith GP. Cholecystokinin elicits Satiety in Rats with Open Gastric Fistulas. Nature. 1973;245(5424):323-5.
- Moran TH, and Kinzig KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286(2):G183-8.
- Moran TH, Baldessarini AR, Salorio CF, Lowery T, and Schwartz GJ. Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am J Physiol. 1997;272(4 Pt 2):R1245-51.
- Kissileff HR, Carretta JC, Geliebter A, and Pi-Sunyer FX. Cholecystokinin and stomach distension combine to reduce food intake in humans. Am J Physiol Regul Integr Comp Physiol. 2003;285(5):R992-8.
- Muurahainen N, Kissileff HR, Derogatis AJ, and Pi-Sunyer FX. Effects of cholecystokinin-octapeptide (CCK-8) on food intake and gastric emptying in man. Physiol Behav. 1988;44(4-5):645-9.
- West DB, Fey D, and Woods SC. Cholecystokinin persistently suppresses meal size but not food intake in free-feeding rats. Am J Physiol. 1984;246(5 Pt 2):R776-87.
- Castillo EJ, Delgado-Aros S, Camilleri M, Burton D, Stephens D, O'Connor-Semmes R, et al. Effect of oral CCK-1 agonist GI181771X on fasting and postprandial gastric functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol. 2004;287(2):G363-9.
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, and Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289-95.
- Woods SC, Lutz TA, Geary N, and Langhans W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos Trans R Soc Lond B Biol Sci. 2006;361(1471):1219-35.
- Verdich C, Flint A, Gutzwiller JP, Naslund E, Beglinger C, Hellstrom PM, et al. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. 2001;86(9):4382-9.
- Buse JM, Leigh; Stonehouse, Anthony; Guan, Xuesong; Malone, James; Okerson, Ted; Maggs, David; Kim, Dennis. Exenatide Maintained Glycemic Control with Associated Weight Reduction Over Three Years in Patients with Type 2 Diabetes. Diabetes. June 2007; Vol. 56, pA73.
- Abbott CR, Monteiro M, Small CJ, Sajedi A, Smith KL, Parkinson JR, et al. The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res. 2005;1044(1):127-31.
- Baggio LL, Huang Q, Brown TJ, and Drucker DJ. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127(2):546-58.
- Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3(3):153-65.
- Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. New England Journal of Medicine. 2015;373(1):11-22.
- Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjoth TV, et al. Efficacy of Liraglutide for Weight Loss Among Patients with Type 2 Diabetes: The SCALE Diabetes Randomized Clinical Trial. Jama. 2015;314(7):687-99.
- Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-51.
- Aroda VR, Ahmann A, Cariou B, Chow F, Davies MJ, Jodar E, et al. Comparative efficacy, safety, and cardiovascular outcomes with once-weekly subcutaneous semaglutide in the treatment of type 2 diabetes: Insights from the SUSTAIN 1-7 trials. Diabetes Metab. 2019;45(5):409-18.
- Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2019;381(9):841-51.
- Fosgerau K, and Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122-8.
- Semaglutide Effects on Heart Disease and Stroke in Patients with Overweight or Obesity (SELECT). https://clinicaltrials.gov/ct2/show/NCT03574597. Updated 2019.
- Barnett AH. Lixisenatide: evidence for its potential use in the treatment of type 2 diabetes. Core Evid. 2011;6:67-79.
- Pinelli NR, and Hurren KM. Efficacy and safety of long-acting glucagon-like peptide-1 receptor agonistscompared with exenatide twice daily and sitagliptin in type 2 diabetes mellitus: a systematic review and meta-analysis. Ann Pharmacother. 2011;45(7-8):850-60.
- Madsbad S, Kielgast U, Asmar M, Deacon CF, Torekov SS, and Holst JJ. An overview of once-weekly glucagon-like peptide-1 receptor agonists-available efficacy and safety data and perspectives for the future. Diabetes Obes Metab. 2011;13(5):394-407
- Steinert RE, Poller B, Castelli MC, Drewe J, and Beglinger C. Oral administration of glucagon-like peptide 1 orpeptide YY 3-36 affects food intake in healthy male subjects. Am J Clin Nutr. 2010;92(4):810-7.
- Trevaskis JL, Sun C, Athanacio J, D'Souza L, Samant M, Tatarkiewicz K, et al. Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes Obes Metab. 2015;17(1):61-73.
- Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, et al. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015;7(3):288-98.
- Muller TD, Finan B, Clemmensen C, DiMarchi RD, and Tschop MH. The New Biology and Pharmacology of Glucagon. Physiol Rev. 2017;97(2):721-66.
- Cegla J, Troke RC, Jones B, Tharakan G, Kenkre J, McCullough KA, et al. Coinfusion of low-dose GLP-1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63(11):3711-20.
- Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A new glucagon and GLP-1 co-agonist eliminate obesity in rodents. Nat Chem Biol. 2009;5(10):749-57.
- Clemmensen C, Chabenne J, Finan B, Sullivan L, Fischer K, Kuchler D, et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2014;63(4):1422-7.
- Brandt SJ, Gotz A, Tschop MH, and Muller TD. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides. 2018;100:190-201.
- 136. Dakin CL, Gunn I, Small CJ, Edwards CM, Hay DL, Smith DM, et al. Oxyntomodulin inhibits food intake in the rat. Endocrinology. 2001;142(10):4244-50.
- Dakin CL, Small CJ, Batterham RL, Neary NM, Cohen MA, Patterson M, et al. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145(6):2687-95.
- Yamamoto H, Lee CE, Marcus JN, Williams TD, Overton JM, Lopez ME, et al. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J Clin Invest. 2002;110(1):43-52.
- Cohen MA, Ellis SM, Le Roux CW, Batterham RL, Park A, Patterson M, et al. Oxyntomodulin suppresses appetite and reduces food intake in humans. J Clin Endocrinol Metab. 2003;88(10):4696-701.
- Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond). 2006;30(12):1729-36.
- Baggio LL, Huang Q, Brown TJ, and Drucker DJ. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127(2):546-58.
- Schjoldager B, Mortensen PE, Myhre J, Christiansen J, and Holst JJ. Oxyntomodulin from distal gut. Role in regulation of gastric and pancreatic functions. Dig Dis Sci. 1989;34(9):1411-9.
- Santoprete A, Capito E, Carrington PE, Pocai A, Finotto M, Langella A, et al. DPP-IV-resistant, long-actingoxyntomodulin derivatives. J Pept Sci. 2011;17(4):270-80.
- Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58(10):2258-66.
- Bello NT, Kemm MH, Ofeldt EM, and Moran TH. Dose combinations of exendin-4 and salmon calcitonin produce additive and synergistic reductions in food intake in nonhuman primates. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R945-52.
- Sun C, Trevaskis JL, Jodka CM, Neravetla S, Griffin P, Xu K, et al. Bifunctional PEGylated exenatide-amylinomimetic hybrids to treat metabolic disorders: an example of long-acting dual hormonal therapeutics. J Med Chem. 2013;56(22):9328-41.
- Takeda J, Seino Y, Tanaka K, Fukumoto H, Kayano T, Takahashi H, et al. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc Natl Acad Sci U S A. 1987;84(20):7005-8
- Dupre J, Ross SA, Watson D, and Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37(5):826-8.
- Pederson RA, and Brown JC. Interaction of gastric inhibitory polypeptide, glucose, and arginine on insulin and glucagon secretion from the perfused rat pancreas. Endocrinology. 1978;103(2):610-5.
- Oben J, Morgan L, Fletcher J, and Marks V. Effect of the entero-pancreatic hormones, gastric inhibitory polypeptide and glucagon-like polypeptide-1(7-36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol. 1991;130(2):267-72.
- Eckel RH, Fujimoto WY, and Brunzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes. 1979;28(12):1141-2.
- Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, and McIntosh CH. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS One. 2012;7(7):e40156.
- Martin CM, Irwin N, Flatt PR, and Gault VA. A novel acylated form of (d-Ala(2))GIP with improved antidiabetic potential, lacking effect on body fat stores. Biochim Biophys Acta. 2013;1830(6):3407-13.
- Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151.
- Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27-36.
- Mauvais-Jarvis F, Manson JE, Stevenson JC, and Fonseca VA. Menopausal Hormone Therapy and Type 2 Diabetes Prevention: Evidence, Mechanisms, and Clinical Implications. Endocr Rev. 2017;38(3):173-88.
- Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic estrogen mimics leptin's effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007;13(1):89-94.
- Finan B, Yang B, Ottaway N, Stemmer K, Muller TD, Yi CX, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18(12):1847-56.
- Schwenk RW, Baumeier C, Finan B, Kluth O, Brauer C, Joost HG, et al. GLP-1-oestrogen attenuates hyperphagia and protects from beta cell failure in diabetes-prone New Zealand obese (NZO) mice. Diabetologia. 2015;58(3):604-14.
- Angelin B, and Rudling M. Lipid lowering with thyroid hormone and thyromimetics. Curr Opin Lipidol. 2010;21(6):499-506.
- Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16(9):1001-8.
- Finan B, Clemmensen C, Zhu Z, Stemmer K, Gauthier K, Muller L, et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell. 2016;167(3):843-57.e14.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860-7.
- Quarta C, Clemmensen C, Zhu Z, Yang B, Joseph SS, Lutter D, et al. Molecular Integration of Incretin and Glucocorticoid Action Reverses Immunometabolic Dysfunction and Obesity. Cell Metab. 2017;26(4):620-32.e6.
- Pironi L, Stanghellini V, Miglioli M, Corinaldesi R, De Giorgio R, Ruggeri E, et al. Fat-induced ileal brake in humans: a dose-dependent phenomenon correlated to the plasma levels of peptide YY. Gastroenterology. 1993;105(3):733-9.
- Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS, et al. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med. 2003;349(10):941-8.
- Degen L, Oesch S, Casanova M, Graf S, Ketterer S, Drewe J, et al. Effect of peptide YY3-36 on food intake in humans. Gastroenterology. 2005;129(5):1430-6.
- Field BC, Wren AM, Peters V, Baynes KC, Martin NM, Patterson M, et al. PYY3-36 and oxyntomodulin can beadditive in their effect on food intake in overweight and obese humans. Diabetes. 2010;59(7):1635-9.
- Talsania T, Anini Y, Siu S, Drucker DJ, and Brubaker PL. Peripheral exendin-4 and peptide YY(3-36) synergistically reduce food intake through different mechanisms in mice. Endocrinology. 2005;146(9):3748-56.
- Scott V, Kimura N, Stark JA, and Luckman SM. Intravenous peptide YY3-36 and Y2 receptor antagonism in the rat: effects on feeding behaviour. J Neuroendocrinol. 2005;17(7):452-7.
- Moran TH, Smedh U, Kinzig KP, Scott KA, Knipp S, and Ladenheim EE. Peptide YY(3-36) inhibits gastric emptying and produces acute reductions in food intake in rhesus monkeys. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R384-8.
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418(6898):650-4.
- 7TM Pharma Initiates Phase II Clinical Study with the Drug Candidate Obinepitide for the Treatment of Obesity. https://www.biospace.com/article/releases/7tm-pharma-initiates-phase-ii-clinical-study-with-the-drug-candidate-obinepitide-for-the-treatment-of-obesity-/.
- Kjaergaard M, Salinas CBG, Rehfeld JF, Secher A, Raun K, and Wulff BS. PYY(3-36) and exendin-4 reduce food intake and activate neuronal circuits in a synergistic manner in mice. Neuropeptides. 2019;73:89-95.
- Cummings DE, Foster-Schubert KE, and Overduin J. Ghrelin and energy balance: focus on current controversies. Curr Drug Targets. 2005;6(2):153-69.
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, et al. The role of the gastric afferent vagalnerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123(4):1120-8.
- Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86(12):5992.
- Zorrilla EP, Iwasaki S, Moss JA, Chang J, Otsuji J, Inoue K, et al. Vaccination against weight gain. Proc Natl Acad Sci U S A. 2006;103(35):13226-31.
- Moran TH, and Dailey MJ. Minireview: Gut peptides: targets for antiobesity drug development? Endocrinology. 2009;150(6):2526-30.
- Xin Z, Serby MD, Zhao H, Kosogof C, Szczepankiewicz BG, Liu M, et al. Discovery and pharmacologicalevaluation of growth hormone secretagogue receptor antagonists. J Med Chem. 2006;49(15):4459-69.
- Yang J, Zhao TJ, Goldstein JL, and Brown MS. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci U S A. 2008;105(31):10750-5.
- Li Z, Mulholland M, and Zhang W. Ghrelin O-acyltransferase (GOAT) and energy metabolism. Science China Life Sciences. 2016;59(3):281-91.
- Barnett BP, Hwang Y, Taylor MS, Kirchner H, Pfluger PT, Bernard V, et al. Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor. Science. 2010;330(6011):1689-92.
- Teubner BJ, Garretson JT, Hwang Y, Cole PA, and Bartness TJ. Inhibition of ghrelin O-acyltransferase attenuates food deprivation-induced increases in ingestive behavior. Horm Behav. 2013;63(4):667-73.
- Price DJ, Drewry DH, Schaller LT, Thompson BD, Reid PR, Maloney PR, et al. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg Med Chem Lett. 2018;28(10):1958-63.
- Okada S, York DA, Bray GA, Mei J, and Erlanson-Albertsson C. Differential inhibition of fat intake in two strains of rat by the peptide enterostatin. Am J Physiol. 1992;262(6 Pt 2):R1111-6.
- Kovacs EM, Lejeune MP, and Westerterp-Plantenga MS. The effects of enterostatin intake on food intake and energy expenditure. Br J Nutr. 2003;90(1):207-14.
- Doi T, Liu M, Seeley RJ, Woods SC, and Tso P. Effect of leptin on intestinal apolipoprotein AIV in response to lipid feeding. Am J Physiol Regul Integr Comp Physiol. 2001;281(3):R753-9.
- Attia N, Touzani A, Lahrichi M, Balafrej A, Kabbaj O, and Girard-Globa A. Response of apolipoprotein AIV and lipoproteins to glycaemic control in young people with insulin-dependent diabetes mellitus. Diabet Med. 1997;14(3):242-7.
- Fujimoto K, Machidori H, Iwakiri R, Yamamoto K, Fujisaki J, Sakata T, et al. Effect of intravenous administration of apolipoprotein A-IV on patterns of feeding, drinking and ambulatory activity of rats. Brain Res. 1993;608(2):233-7.
- Katsuura G, Asakawa A, and Inui A. Roles of pancreatic polypeptide in regulation of food intake. Peptides. 2002;23(2):323-9.
- Asakawa A, Inui A, Ueno N, Fujimiya M, Fujino MA, and Kasuga M. Mouse pancreatic polypeptide modulatesfood intake, while not influencing anxiety in mice. Peptides. 1999;20(12):1445-8.
- Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, et al. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab. 2003;88(8):3989-92.
- Jesudason DR, Monteiro MP, McGowan BM, Neary NM, Park AJ, Philippou E, et al. Low-dose pancreaticpolypeptide inhibits food intake in man. Br J Nutr. 2007;97(3):426-9.
- Neary NM, McGowan BM, Monteiro MP, Jesudason DR, Ghatei MA, and Bloom SR. No evidence of an additive inhibitory feeding effect following PP and PYY 3-36 administration. Int J Obes (Lond). 2008;32(9):1438-40.
- Lutz TA, Althaus J, Rossi R, and Scharrer E. Anorectic effect of amylin is not transmitted by capsaicin-sensitive nerve fibers. Am J Physiol. 1998;274(6):R1777-82.
- Roth JD, Hughes H, Kendall E, Baron AD, and Anderson CM. Antiobesity effects of the beta-cell hormone amylin in diet-induced obese rats: effects on food intake, body weight, composition, energy expenditure, and gene expression. Endocrinology. 2006;147(12):5855-64.
- Rushing PA, Hagan MM, Seeley RJ, Lutz TA, and Woods SC. Amylin: a novel action in the brain to reduce body weight. Endocrinology. 2000;141(2):850-3.
- Chapman I, Parker B, Doran S, Feinle-Bisset C, Wishart J, Strobel S, et al. Effect of pramlintide on satiety and food intake in obese subjects and subjects with type 2 diabetes. Diabetologia. 2005;48(5):838-48.
- Aronne L, Fujioka K, Aroda V, Chen K, Halseth A, Kesty NC, et al. Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo-controlled, dose-escalation study. J Clin Endocrinol Metab. 2007;92(8):2977-83.
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin responsiveness restored by amylinagonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105(20):7257-62.
- Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, et al. Amylin-mediated restoration of leptinresponsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology. 2008;149(11):5679-87.
- Aronne LJ, Halseth AE, Burns CM, Miller S, and Shen LZ. Enhanced weight loss following coadministration ofpramlintide with sibutramine or phentermine in a multicenter trial. Obesity (Silver Spring). 2010;18(9):1739-46.
- Gydesen S, Andreassen KV, Hjuler ST, Christensen JM, Karsdal MA, and Henriksen K. KBP-088, a novel DACRA with prolonged receptor activation, is superior to davalintide in terms of efficacy on body weight. Am J Physiol Endocrinol Metab. 2016;310(10):E821-7.
- Hjuler ST, Andreassen KV, Gydesen S, Karsdal MA, and Henriksen K. KBP-042 improves bodyweight and glucose homeostasis with indices of increased insulin sensitivity irrespective of route of administration. Eur J Pharmacol. 2015;762:229-38.
- Long-acting amylin analog (obesity / diabetes). https://www.zealandpharma.com/longacting-amylin-analog
- Drent ML, Larsson I, William-Olsson T, Quaade F, Czubayko F, von Bergmann K, et al. Orlistat (Ro 18-0647), a lipase inhibitor, in the treatment of human obesity: a multiple dose study. Int J Obes Relat Metab Disord. 1995;19(4):221-6
- Kopelman P, Bryson A, Hickling R, Rissanen A, Rossner S, Toubro S, et al. Cetilistat (ATL-962), a novel lipase inhibitor: a 12-week randomized, placebo-controlled study of weight reduction in obese patients. Int J Obes (Lond). 2007;31(3):494-9.
- Bryson A, de la Motte S, and Dunk C. Reduction of dietary fat absorption by the novel gastrointestinal lipase inhibitor cetilistat in healthy volunteers. Br J Clin Pharmacol. 2009;67(3):309-15.
- Kopelman P, Groot Gde H, Rissanen A, Rossner S, Toubro S, Palmer R, et al. Weight loss, HbA1c reduction, and tolerability of cetilistat in a randomized, placebo-controlled phase 2 trial in obese diabetics: comparison with orlistat (Xenical). Obesity (Silver Spring). 2010;18(1):108-15.
- Nelson CN, List EO, Ieremia M, Constantin L, Chhabra Y, Kopchick JJ, et al. Growth hormone activated STAT5is required for induction of beige fat in vivo. Growth Horm IGF Res. 2018;42-43:40-51.
- Scacchi M, Pincelli AI, and Cavagnini F. Growth hormone in obesity. Int J Obes Relat Metab Disord. 1999;23(3):260-71.
- Shadid S, and Jensen MD. Effects of growth hormone administration in human obesity. Obes Res. 2003;11(2):170-5.
- Valentino MA, Lin JE, and Waldman SA. Central and peripheral molecular targets for antiobesity pharmacotherapy. Clin Pharmacol Ther. 2010;87(6):652-62.
- Mathvink RJ, Tolman JS, Chitty D, Candelore MR, Cascieri MA, Colwell LF, Jr., et al. Discovery of a potent, orally bioavailable beta(3) adrenergic receptor agonist, (R)-N-[4-[2-[[2-hydroxy-2-(3-pyridinyl)ethyl]amino]ethyl]phenyl]-4-[4 -[4-(trifluoromethyl)phenyl]thiazol-2-yl]benzenesulfonamide. J Med Chem. 2000;43(21):3832-6.
- van Baak MA, Hul GB, Toubro S, Astrup A, Gottesdiener KM, DeSmet M, et al. Acute effect of L-796568, a novel beta 3-adrenergic receptor agonist, on energy expenditure in obese men. Clin Pharmacol Ther. 2002;71(4):272-9.
- Larsen TM, Toubro S, van Baak MA, Gottesdiener KM, Larson P, Saris WH, et al. Effect of a 28-d treatment with L-796568, a novel beta(3)-adrenergic receptor agonists, on energy expenditure and body composition in obese men. Am J Clin Nutr. 2002;76(4):780-8.
- Bujalska IJ, Kumar S, and Stewart PM. Does central obesity reflect "Cushing's disease of the omentum"? Lancet. 1997;349(9060):1210-3.
- Sandeep TC, Andrew R, Homer NZ, Andrews RC, Smith K, and Walker BR. Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes. 2005;54(3):872-9.
- Liu J, Wang L, Zhang A, Di W, Zhang X, Wu L, et al. Adipose tissue-targeted 11beta-hydroxysteroid dehydrogenase type 1 inhibitor protects against diet-induced obesity. Endocr J. 2011;58(3):199-209.
- Anil TM, Dandu A, Harsha K, Singh J, Shree N, Kumar VS, et al. A novel 11beta-hydroxysteroid dehydrogenase type1 inhibitor CNX-010-49 improves hyperglycemia, lipid profile and reduces body weight in diet induced obese C57B6/J mice with a potential to provide cardio protective benefits. BMC Pharmacol Toxicol. 2014;15:43.
- Kim YM, An JJ, Jin YJ, Rhee Y, Cha BS, Lee HC, et al. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007;38(4):455-65.
- 223. The Company is developing an anti-abdominal obesity botanical drug, ALS-L1023, which is in phase III clinical trial in Korea. The company has already commercialized anti-abdominal obesity dietary supplement (Ob-X). https://www.pharmalicensing.com/detail.php?uid=64551.
- Bluher M, Kahn BB, and Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299(5606):572-4.
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429(6993):771-6.
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127(6):1109-22.
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8(5):347-58.
- Huynh FK, Hershberger KA, and Hirschey MD. Targeting sirtuins for the treatment of diabetes. Diabetes Manag (Lond). 2013;3(3):245-57.
- Hardie DG, and Frenguelli BG. A neural protection racket: AMPK and the GABA(B) receptor. Neuron. 2007;53(2):159-62.
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134(3):405-15.
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569-74.
- Li S, Li Y, Xiang L, Dong J, Liu M, and Xiang G. Sildenafil induces browning of subcutaneous white adipose tissue in overweight adults. Metabolism. 2018;78:106-17.
- Chun E, Han CK, Yoon JH, Sim TB, Kim YK, and Lee KY. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005;114(1):124-30.
- Kim DD, Krishnarajah J, Lillioja S, de Looze F, Marjason J, Proietto J, et al. Efficacy and safety of beloranib for weight loss in obese adults: a randomized controlled trial. Diabetes Obes Metab. 2015;17(6):566-72.
- Elfers CT, and Roth CL. Robust Reductions of Excess Weight and Hyperphagia by Beloranib in Rat Models of Genetic and Hypothalamic Obesity. Endocrinology. 2017;158(1):41-55.
- McCandless SE, Yanovski JA, Miller J, Fu C, Bird LM, Salehi P, et al. Effects of MetAP2 inhibition on hyperphagia and body weight in Prader-Willi syndrome: A randomized, double-blind, placebo-controlled trial. Diabetes, obesity & metabolism. 2017;19(12):1751-61.
- Giralt M, Gavalda-Navarro A, and Villarroya F. Fibroblast growth factor-21, energy balance and obesity. Mol Cell Endocrinol. 2015;418 Pt 1:66-73.
- Fisher FM, and Maratos-Flier E. Understanding the Physiology of FGF21. Annu Rev Physiol. 2016;78:223-41.
- Gomez-Samano MA, Grajales-Gomez M, Zuarth-Vazquez JM, Navarro-Flores MF, Martinez-Saavedra M, Juarez-Leon OA, et al. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 2017;11:335-41.
- Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, et al. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes. 2010;59(11):2781-9.
- Flint HJ, Scott KP, Louis P, and Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577-89.
- Rosenbaum M, Knight R, and Leibel RL. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol Metab. 2015;26(9):493-501.
- Rooks MG, and Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16(6):341-52.
- Christiansen CB, Gabe MBN, Svendsen B, Dragsted LO, Rosenkilde MM, and Holst JJ. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am J Physiol Gastrointest Liver Physiol. 2018;315(1):G53-g65.
- De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014;156(1-2):84-96.
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500(7464):585-8.
- Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58(8):1091-103.
- de Aguiar Vallim TQ, Tarling EJ, and Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17(5):657-69.
- Stefanaki C, Peppa M, Mastorakos G, and Chrousos GP. Examining the gut bacteriome, virome, and mycobiome in glucose metabolism disorders: Are we on the right track? Metabolism. 2017;73:52-66.
- Altabas V, and Zjacic-Rotkvic V. Anti-ghrelin antibodies in appetite suppression: recent advances in obesity pharmacotherapy. Immunotargets Ther. 2015;4:123-30.
- Colon-Gonzalez F, Kim GW, Lin JE, Valentino MA, and Waldman SA. Obesity pharmacotherapy: what is next? Mol Aspects Med. 2013;34(1):71-83.
- Takagi K, Legrand R, Asakawa A, Amitani H, Francois M, Tennoune N, et al. Anti-ghrelin immunoglobulins modulate ghrelin stability and its orexigenic effect in obese mice and humans. Nat Commun. 2013;4:2685.
- Monteiro MP. Obesity vaccines. Hum Vaccin Immunother. 2014;10(4):887-95.
- Haffer KN. Effects of novel vaccines on weight loss in diet-induced-obese (DIO) mice.J Anim Sci Biotechnol. 2012;3(1):21.
- Yamada T, Hara K, and Kadowaki T. Association of adenovirus 36 infection with obesity and metabolic markers in humans: a meta-analysis of observational studies. PLoS One. 2012;7(7):e42031.
- Na HN, and Nam JH. Proof-of-concept for a virus-induced obesity vaccine; vaccination against the obesity agent adenovirus 36. Int J Obes (Lond). 2014;38(11):1470-4.
- Zhang Y, Yu J, Qiang L, and Gu Z. Nanomedicine for obesity treatment. Sci China Life Sci. 2018;61(4):373-9.
- Sangwai M, Sardar S, and Vavia P. Nanoemulsified orlistat-embedded multi-unit pellet system (MUPS) with improved dissolution and pancreatic lipase inhibition. Pharm Dev Technol. 2014;19(1):31-41.
- Chen YL, Zhu S, Zhang L, Feng PJ, Yao XK, Qian CG, et al. Smart conjugated polymer nanocarrier for healthy weight loss by negative feedback regulation of lipase activity. Nanoscale. 2016;8(6):3368-75.
- Kupferschmidt N, Csikasz RI, Ballell L, Bengtsson T, and Garcia-Bennett AE. Large pore mesoporous silica induced weight loss in obese mice. Nanomedicine (Lond). 2014;9(9):1353-62.
- Mun EC, Blackburn GL, and Matthews JB. Current status of medical and surgical therapy for obesity. Gastroenterology. 2001;120(3):669-81.
- Xue Y, Xu X, Zhang XQ, Farokhzad OC, and Langer R. Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc Natl Acad Sci U S A. 2016;113(20):5552-7.
- Di Mascolo D, C JL, Aryal S, Ramirez MR, Wang J, Candeloro P, et al. Rosiglitazone-loaded nanospheres for modulating macrophage-specific inflammation in obesity. J Control Release. 2013;170(3):460-8.
- Marrache S, and Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(40):16288-93.
- Jiang C, Cano-Vega MA, Yue F, Kuang L, Narayanan N, Uzunalli G, et al. Dibenzazepine-Loaded Nanoparticles Induce Local Browning of White Adipose Tissue to Counteract Obesity. Mol Ther. 2017;25(7):1718-29.
- Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D, et al. Microneedle-array patches loaded with hypoxia-sensitive vesicles provide fast glucose-responsive insulin delivery. Proc Natl Acad Sci U S A. 2015;112(27):8260-5.