ABSTRACT
In the past decades, emphasis has shifted from testing thyroid function in individuals who are likely to have clinically overt thyroid disorders to a broader population, an approach that includes also identification of so-called subclinical or mild thyroid dysfunctions. The key measurement methods used to detect thyroid dysfunction are still serum thyroid stimulating hormone (TSH) and the main circulating thyroid hormones thyroxine (T4) and triiodothyronine (T3), either as total or estimated free concentrations, and it is indeed the improved assay sensitivities and specificities that have made it possible to diagnose these milder forms. For these key variables, it is preferable for results to be interpretable in relation to population-based reference intervals to use methods that are independent of the particular laboratory assay used. This requirement is satisfied for well standardized assays for serum TSH, total T4 and total T3. However, when free T4 is estimated, assay results often need to be evaluated in relation to method-specific reference intervals or “normal ranges”. Free T3 estimates are even more subject to spurious results and high inter-method variability. This limitation of both assessments is particularly cogent during pregnancy and in the face of critical illness. The widespread search for even minor thyroid dysfunction is influenced by two key questions. How damaging are the effects of subclinical dysfunction? Does treatment confer benefit? The answers are not uniform across populations. The diagnostic imperative is now radically different for the population at large and for women who are pregnant or about to become pregnant.
INTRODUCTION
In the past decades, emphasis has shifted from testing of thyroid function in individuals who are likely to have clinically overt thyroid disorders to a broader population, an approach that identifies so-called subclinical thyroid dysfunction in up to 10% of women over fifty. The key assays that are used to detect thyroid dysfunction are serum thyroid stimulating hormone (TSH) and the main circulating thyroid hormones thyroxine (T4) and triiodothyronine (T3), either as total or estimated free concentrations. For these key variables, it is preferable for results to be interpretable in relation to population-based reference intervals that use methods that are independent of the particular assay used. This requirement is satisfied for well standardized assays for serum TSH, total T4 and total T3. However, when free T4 is estimated, assay results often need to be evaluated in relation to method-specific reference intervals or “normal ranges”. Free T3 estimates are even more subject to spurious results and high inter-method variability. This limitation of both assessments is particularly cogent during pregnancy and in the face of critical illness (see below).
The widespread search for even minor thyroid dysfunction is influenced by two key questions. How damaging are the effects of subclinical dysfunction? Does treatment confer benefit? The answers are not uniform across populations. The diagnostic imperative is now radically different for the population at large and for women who are pregnant or about to become pregnant. Some key practice points that relate to the testing of thyroid function are summarized in Table 1.
|
Table 1. Key Practice Points Related to the Testing of Thyroid Function
|
|
Many disorders are associated with increased prevalence of thyroid dysfunction; optimal testing strategy requires information on all co-existing conditions and medications.
|
|
The more widely thyroid function is tested, the greater the proportion of abnormal results that show borderline or “subclinical dysfunction”.
|
|
Testing of thyroid function is now widely advocated before and early in pregnancy, especially where fertility is impaired, assisted reproduction is used, or where pregnancy complications have occurred.
Except for “subclinical” hypothyroidism in early or impending pregnancy, intervention for subclinical thyroid dysfunction should only be considered after a sustained abnormality has been demonstrated over a minimum of three months.
|
|
There is increasing documentation of adverse effects from sustained or progressive subclinical hypothyroidism, although evidence of benefit from treatment is less clear.
|
|
The combination of raised serum TSH and positive peroxidase antibody is predictive of likely long-term progression towards overt hypothyroidism.
|
|
The relationship between serum TSH and circulating thyroid hormones gives a better index of thyroid status than any single variable. Six key assumptions underpin the diagnostic value of this relationship.
“TSH alone” first line approach to thyroid function testing has important drawbacks and limitations.
Serum TSH is a cornerstone of thyroid diagnosis, but it is not possible to define its “normal” range or reference interval for all clinical circumstances. Age, pregnancy and fertility issues, diurnal variation and pulse secretion, and associated antibody status militate against fixed cut-off points.
|
|
Both TSH and T4 show spontaneous biological fluctuations that are much greater than analytical imprecision. A serial change can be inferred with confidence with about 50% alteration in serum TSH and 25% change in the free T4 estimate.
|
|
During thyroid hormone therapy, either replacement or suppressive, the optimal target TSH range may differ from the reference interval that is used to establish a new diagnosis.
Interpretation of anomalous test results should take into account the effects of all associated medications, as well as nutraceuticals, e.g. biotin.
|
|
No estimate of circulating free thyroxine is impeccable. Especially in situations where assessment is difficult, e.g. late pregnancy and severe associated illness, there are strong arguments for re-establishing total T4 measurement as the preferred “gold standard”.
|
|
Identification of marked iodine excess, detected by urinary estimation, may identify reversible thyroid abnormalities, e.g. iodine-induced exacerbation of primary hypothyroidism, atypical thyrotoxicosis with blocked isotope uptake, or resistance to standard doses of antithyroid drugs. Some food sources (e.g. soy, sea weed) and alternative health care products can be heavily iodine-contaminated.
|
WHO SHOULD BE TESTED FOR THYROID DYSFUNCTION?
It has long been recognized that the clinical manifestations of hyperthyroidism (thyrotoxicosis), or hypothyroidism are so diverse that diagnosis based on clinical features lacks sensitivity and specificity. Hence, reliance is placed on measurements of circulating thyroid hormones and thyroid stimulating hormone (TSH) to confirm or rule out thyroid dysfunction.
After the publication of guidelines from the American College of Physicians in 1998 (1,2), testing for detection of thyroid dysfunction became widely applied, especially in women over 50, the group most likely to have either overt or subclinical thyroid dysfunction. Testing of this group is generally advocated at the time of presentation for medical care, i.e. a case finding strategy, rather than screening of a whole population group.
A normal serum TSH value in ambulatory patients without associated disease or pituitary dysfunction has a high negative predictive value in ruling out both primary hypothyroidism and hyperthyroidism (1,2), which has led to a short-cut approach in which free T4 may only be estimated if TSH is above 10 mU/l. However, this ”TSH first” strategy of thyroid function testing has important limitations (see below). If there is no suspicion of pituitary or thyroid disease, a normal TSH concentration does not need to be re-tested for about 5 years (3). To this broad indication has recently been added the still controversial recommendation that universal testing of thyroid function has a place before or as early as possible in pregnancy, although this is very much debated (4-7), and the recent official guidelines from the scientific community are still not in agreement (8,9).
In some groups (Tables 2 and 3), known to be at increased risk of thyroid dysfunction, there is a case for routine testing even in the absence of any suggestive clinical features.
Almost all developed countries now have routine neonatal screening programs for congenital hypothyroidism using heel prick filter paper blood spots (see below). The value of such programs has long been clear (10), but neonatal screening is not yet routine in numerous developing countries where nevertheless the prevalence of neonatal hypothyroidism may be high, and often associated with iodine deficiency (11) (see below). In terms of benefit from allocation of health care resources in developing countries, the establishment of neonatal screening (12) probably takes precedence over routine testing of adults, even in today’s India (13).
|
Table 2. Groups with an Increased Likelihood of Thyroid Dysfunction
|
|
Pregnancy and postpartum (14)
|
|
Previous thyroid disease or surgery
|
|
Atrial fibrillation (15)
|
|
Goiter
|
|
Associated autoimmune disease(s) (16-18)
|
|
Chromosome 18q deletions (19)
|
|
Chronic renal failure (20)
|
|
Williams syndrome (21)
|
|
Fabry disease (22)
|
|
Irradiation of head and neck (23-25)
|
|
Radical laryngeal/pharyngeal surgery
|
|
Recovery from Cushing’s syndrome (26,27)
|
|
Gout (28)
|
|
Environmental irradiation (29,30)
|
|
Thalassemia major (31)
|
|
Primary pulmonary hypertension (32)
|
|
Polycystic ovarian syndrome (33)
|
|
Morbid obesity (34)
|
|
Breast cancer (35,36)
|
|
Hepatitis C (pre-treatment) (37)
|
|
Down’s syndrome (38)
|
|
Turner’s syndrome (39)
|
|
Pituitary or cerebral irradiation (25)
|
|
Head trauma (40-42)
|
|
Very low birth weight premature infants (43,44)
|
|
Table 3. Drugs with an Increased Likelihood of Inducing Thyroid Dysfunction (45-47)
|
|
Inhibit thyroid hormone production
|
|
Antithyroid drugs, Amiodarone, Lithium, Iodide (large doses), Iodine-containing contrast media
|
|
Alter extra-thyroidal metabolism of thyroid hormone
|
|
Propylthiouracil, Glucocorticoids, Propranolol, Amiodarone, Iodine-Containing Contrast Media, Carbamazepine, Barbiturates, Rifampicin, Phenytoin, Sertraline
|
|
Alter T4/T3 binding to plasma proteins
|
|
Estrogen, Heroin, Methadone, Clofibrate, 5-Fluorouracil, Perphenazine, Glucocorticoids, Androgens, L-Asparaginase, Nicotinic Acid, Furosemide, Salicylates, Phenytoin, Fenclofenac Heparin
|
|
Induction of thyroiditis
|
|
Amiodarone, Interleukin-2, Interferon-α, Interferon-β, γ-Interferon, Sunitinib, Monoclonal antibody therapy check point inhibitors (Nivulomab, Pembrolizumab, pilimimab)
|
|
Effect on TSH secretion
|
|
Lithium, Dopamine Receptor Blockers, Dopa Inhibitors, Cimetidine, Clomiphene, Thyroid Hormone, Dopamine, L-Dopa, Glucocorticoids, Growth Hormone, Somatostatin, Octreotide
|
|
Impaired absorption of oral T4
|
|
Aluminum hydroxide, Ferrous Sulfate, Cholestyramine, Calcium Carbonate, Calcium Citrate, Calcium Acetate, Iron Sulfate, Colestipol, Sucralfate, Soya preparations, Kayexalate, Ciprofloxacin, Sevelamer, Proton pump inhibitors
|
|
Other
|
|
Thalidomide, Lenalidomide, Chemotherapy for sarcoma
|
THE BASIS FOR A CASE-FINDING STRATEGY
Routine laboratory testing of particular population groups becomes well founded if a testing strategy satisfies the following criteria:
- An abnormality cannot be identified in a reliable and timely way by standard clinical assessment.
- Dysfunction is sufficiently common to justify routine testing, either by case finding or by population screening.
- There are adverse consequences of failure to identify an abnormality, including the possibility of progression towards more severe disease.
- The laboratory test method is cost-effective and sufficiently sensitive and specific to identify those at risk of adverse consequences.
- There are no major adverse consequences of testing
- Treatment is safe and effective and prevents some or all of the adverse consequences.
- Abnormal findings can be adequately followed-up to ensure an appropriate clinical response. (An early detection program may have little value if this last requirement cannot be met.)
While the first five of the above criteria are reasonably established for thyroid dysfunction, the latter two are less secure.
Sensitivity and Accuracy of Clinical Assessment
Studies of unselected patients evaluated by primary care physicians show that clinical acumen alone lacks both sensitivity and specificity in detecting previously undiagnosed thyroid dysfunction. In up to one-third of patients evaluated for suspected thyroid dysfunction by specialists, laboratory results led to revision of the clinical assessment (48). Systematic comparison of the standard clinical features of hypothyroidism with laboratory tests (49) showed that clinical assessment identified only about 40% with overt hypothyroidism and classical signs were present only in the most severely affected individuals. Both overt hyper- and hypothyroidism can have important consequences before the usual clinical features are obvious, and clinicians may fail to recognize diagnostic features even when they are present.
Boelaert et al. (50) have recently confirmed that the typical multiple classical symptoms of hyperthyroidism become less prevalent with advancing age, with greater importance of weight loss, atrial fibrillation, and shortness of breath as presenting features (51). They proposed a low threshold for assessment of thyroid function in patients older than 60 years who have any of these features.
Clinical evaluation remains of central importance to assess severity of thyroid dysfunction, evaluate discordant results, establish the specific cause of thyroid dysfunction and monitor the response to treatment. There is little doubt that repeated laboratory confirmation of normal thyroid function can be wasteful; strategies have been suggested to improve cost-effectiveness (51-53).
However, functional disorders of the thyroid (hypothyroidism and hyperthyroidism) are common and, in many cases, managed by primary care providers. In addition to diagnosed cases, there are many patients who present to their provider seeking evaluation of their thyroid status as a possible cause of a variety of complaints including obesity, mood changes, hair loss, and fatigue. There is an ever-growing body of literature in the public domain, whether in print or internet-based, suggesting that thyroid conditions are under-diagnosed by physicians and that standard thyroid function tests are unreliable. Primary care providers are often the first to evaluate these patients and order biochemical testing. This has become a more complex process, with many patients requesting and even demanding certain biochemical tests that may not be indicated (54).
Prevalence
In considering the prevalence of thyroid dysfunction, a distinction needs to be made between so-called subclinical and overt abnormalities; paradoxically, this distinction is based on laboratory rather than clinical criteria. There is a trend to replace the term ‘subclinical hypothyroidism’ with the designation ‘mild thyroid failure’ (55,56).
In the progressive development of thyroid dysfunction, abnormal values for serum TSH generally occur before there is a diagnostic abnormality of serum T4, because of the markedly amplified relationship between serum T4 and release of TSH from the anterior pituitary (see below). For a two-fold change in serum T4, up or down from the set-point for that individual, the serum TSH will normally change up to 100-fold in the reverse direction (57,58). Thus, TSH becomes recognizably abnormal long before the serum concentrations of T4 or T3 fall outside the population-based reference interval. This was made possible by introduction of immunochemiluminometric methods for measurement of serum TSH with an increased functional sensitivity (59).
The more widespread the testing of thyroid function in the absence of suggestive clinical features, the greater the proportion of abnormal results in which only TSH is abnormal. In evaluating serum TSH, typically defined with a normal reference interval of about 0.4-4.0 mU/l, it is important to note that normal values approximate to a logarithmic distribution, with mean and median values at 1.0-1.5 mU/l (57,60-62). While values of 2-4 mU/l lie within the reference range, the likelihood of eventual hypothyroidism increases progressively for values above 2 mU/l, especially if thyroid peroxidase antibodies are present (63).
A population study in Colorado (64), of over 25,000 individuals of mean age 56 years, 56% of whom were female, showed TSH excess in 9.5 %, with a 2.2 % prevalence of suppressed TSH; over half the group with suppressed TSH were taking thyroid medication. In women, the prevalence of TSH excess increased progressively from 4% at age 18-24 to 20% over age 74 (64).
The National Health and Nutrition Examination Survey (NHANES III) (63), found hypothyroidism in 4.6% of the US population (0.3% overt and 4.3% subclinical) and hyperthyroidism in 1.3% (0.5% overt), with increasing prevalence with age in both females and males (figure 1). Abnormalities were more common in females than males. The prevalence of positive thyroid peroxidase antibodies was clearly associated with both hyper- and hypothyroidism, with important ethnic differences in antibody prevalence.

Figure 1. Percentage of the US population (NHANESIII) with abnormal serum TSH concentrations as a function of age. The disease-free population excludes those who reported thyroid disease, goiter or thyroid-related medications; the reference population excluded, in addition, those who had positive thyroid autoantibodies, or were taking medications that can influence thyroid function. Note the much higher prevalence of TSH abnormalities in the total population, than in the reference population (from reference (63)).
Prevalence data from one region do not necessarily apply in other populations, because of differences such as ethnic predisposition or variations in iodine intake. For example, in Hong Kong, where iodine intake is marginally deficient, only 1.2% of Chinese women aged over 60 years had serum TSH values > 5 mU/l, with a comparable prevalence of suppressed values indicating possible hyperthyroidism (65). Several European studies (66,67) have compared the effect of various levels of iodine intake on the prevalence of thyroid over- and underfunction.
Hypothyroidism is generally more common with abundant iodine intake, while goiter and subclinical hyperthyroidism are more common with low iodine intake (66,67). This was illustrated in a random selection of 4649 participants from the Civil Registration System in Denmark in age groups between 18 and 65 years. Thyroid dysfunction was evaluated from blood samples and questionnaires and compared with results from ultrasonography. Median iodine excretion was 53 µg/l in Aalborg and 68 µg/l in Copenhagen. Previously diagnosed thyroid dysfunction was found with the same prevalence in these regions. Serum TSH was lower in Aalborg than in Copenhagen (P=0.003) and declined with age in Aalborg, but not in Copenhagen. No previously diagnosed hyperthyroidism was found with the same overall prevalence in the two regions, but in subjects >40 years hyperthyroidism was more prevalent in Aalborg (1.3 vs 0.5%, P=0.017). No previously diagnosed hypothyroidism was found more frequently in Aalborg (0.6 vs 0.2%, P=0.03). Hyperthyroidism was more often associated with macronodular thyroid structure at ultrasound in Aalborg and hypothyroidism was more often associated with a patchy thyroid structure in Copenhagen. Thus, significant differences in thyroid dysfunction were found between the regions with a minor difference in iodine excretion. The findings are in agreement with a higher prevalence of thyroid autonomy among the elderly in the most iodine-deficient region (68).
Thus, thyroid abnormalities in populations with low iodine intake and those with high iodine intake develop in opposite directions: goiter and thyroid hyperfunction when iodine intake is relatively low, and impaired thyroid function when iodine intake is relatively high. Probably, mild iodine deficiency partly protects against autoimmune thyroid disease. Thyroid autoantibodies may be markers of an autoimmune process in the thyroid or secondary to the development of goiter (69).
These regional differences may and should influence the choice of diagnostic test and target population. For example, in an iodine replete environment, emphasis could be placed on testing younger or pregnant women for subclinical hypothyroidism by measurement of TSH and thyroid peroxidase antibodies, whereas in an iodine-deficient region there might be additional emphasis on early detection of thyroid autonomy and hyperthyroidism in older people, using a highly sensitive 3rd generation TSH assay.
SUBCLINICAL THYROID DYSFUNCTION
The proven or presumed importance of subclinical thyroid dysfunction will have a major effect on the extent to which thyroid function testing is applied in any population. The term ‘subclinical’ is used when the serum concentration of TSH is persistently abnormal (however defined), while the concentrations of T4 and T3 remain within their reference intervals. Because results can fluctuate spontaneously, a new diagnosis of subclinical thyroid dysfunction is not warranted on basis of a single laboratory sample. The following five criteria define endogenous subclinical thyroid dysfunction:
- TSH increased above or decreased below designated limits (see below)
- Normal free T4 concentration (and free T3 for hyperthyroidism)
- Abnormality is not due to medication (see below)
- There is no concurrent critical illness or pituitary dysfunction.
- A sustained abnormality is demonstrated over 3-6 months.
Apart from the situation of impending or early pregnancy, where there is clear consensus that subclinical hypothyroidism should be promptly and fully treated, the approach to subclinical thyroid dysfunction remains uncertain (70). Various authorities have expressed divergent views on the importance of detecting the mild TSH abnormalities that reflect subclinical thyroid dysfunction. Extremes of opinion can be summarized as follows. On the one hand, some take the position that subclinical thyroid dysfunction, both hypothyroidism and hyperthyroidism, are disorders that need to be treated in order to avert potential harm (71). To achieve optimal sensitivity, particularly for the diagnosis of hypothyroidism, some have advocated that the upper limit of the TSH reference interval should be lowered (72)because values in the range 2-4 mU/l, usually regarded as normal, are associated with an increased prevalence of future hypothyroidism (62). Active search for subclinical thyroid dysfunction is based on the view that treatment is usually justified, because of potential adverse outcomes, even if proof of benefit is still lacking. At this end of the opinion spectrum, there is support for general community screening for thyroid dysfunction (71), in contrast to a case-finding strategy for women over 50 when they present for medical care (1).
Others have taken the view that while there is circumstantial evidence that subclinical thyroid dysfunction can have adverse long-term effects, there is still a lack of strong evidence that treatment of thyroid dysfunction in general confers benefit (73), although more indicative evidence has emerged with time (see later). Thus, treatment of subclinical hyperthyroidism seems to improve bone health (74) and prevent atrial fibrillation (15), while evidence for treatment benefits of subclinical hypothyroidism is much weaker, at least in patients above 65 years of age (see later).
A definitive position on this dilemma should ideally emerge from long-term studies focused on outcomes, but if differences are small, studies may be under-powered and the results may still be indeterminate. Other factors to take into account in establishing an approach to widespread thyroid testing include ethnic or environmental predisposition to thyroid dysfunction in various communities, balance with other healthcare priorities that may be more compelling, cost of laboratory testing and the extent to which competent clinical assessment and therapeutic response may be overwhelmed by reliance on laboratory measurements. Yet, even with the aspect of not advocating for universal screening, some guidelines inadvertently include almost all women for testing of thyroid function, even if not stating it clearly (75). Thus, according to the American Thyroid Association (ATA) and American Association of Clinical Endocrinologists (AACE) guidelines, levothyroxine therapy would be considered for 92% of women with subclinical hypothyroidism and TSH ≤10 mU/L (75).
Adverse Consequences of Subclinical Thyroid Dysfunction
The issues that identify the clinical importance of subclinical thyroid dysfunction are summarized in Table 4; many of these adverse effects relate to the cardiovascular system (73,76). There is still conflicting evidence on whether mild thyroid abnormalities influence cardiovascular mortality and, as yet, no convincing support for the proposition that treatment of subclinical thyroid dysfunction improves survival. In a 12-year follow-up study of women over 65, neither TSH > 5 mU/l, nor <0.5 mU/l were associated with any increase in all-cause or cardiovascular mortality, although a previous history of hyperthyroidism had a minor adverse effect (77). In contrast, another survey of the relationship between serum TSH and all-cause and cardiovascular mortality over a 10-year period in individuals over 60, showed that the group with serum TSH below 0.5 mU/l had a significantly increased mortality, apparently due to cardiovascular disease (78), although an increased serum TSH was not associated with excess mortality (78). More recent studies provide strengthening evidence for adverse effects from minor degrees of thyroid dysfunction. A Scottish study (79) of over 17,000 people followed for an average of 4.5 years, correlated fatal or non-fatal first episodes of cardiovascular disease, arrhythmias, and osteoporotic fractures with ambulatory serum TSH values. With elevated TSH >4 mU/l there was an increased incidence of dysrhythmia, cardiovascular ischemic episodes, and fracture (hazard ratios 1.8-1.95) (77). With a suppressed serum TSH <0.03 mU/l, all three endpoints were also increased in frequency (hazard ratio 1.4-2.0). It is important to note that these authors distinguished between clearly suppressed TSH (<0.03 mU/l) and subnormal-detectable TSH 0.04-0.4 mU/l, the latter of which was not associated with significant adverse effects. These findings appeared to be at odds with the initial evaluation of the Wickham data that showed no adverse cardiovascular effects of subclinical hypothyroidism. However, reanalysis of the Whickham findings (80) did show an adverse effect if the beneficial effect of thyroxine treatment was excluded.
|
Table 4. Reported Effects of Subclinical Thyroid Dysfunction
|
|
Subclinical hyperthyroidism (suppressed TSH, normal free T4, and free T3 estimates)
· Exposure to iodine may precipitate severe thyrotoxicosis (81)
· Threefold increased risk of atrial fibrillation after 10 years (82)
· Abnormalities of cardiac function (15,82,83)
· Osteoporosis risk increased (84,85)
· Progression to overt hyperthyroidism (86,87)
|
|
Subclinical hypothyroidism or mild thyroid failure (increased TSH, normal free T4 estimate)
· Non-specific symptoms may improve with treatment (88)
· Progression to overt hypothyroidism (89)
· Independent risk factor for atherosclerosis (90)
· Increased risk of coronary artery disease (91-93)
· Increased frequency of congestive heart failure (91,93,94)
· Adverse effects on vascular compliance (95-97)
· Abnormal cardiac function may improve with treatment (98)
· Beneficial effect of treatment on lipids (99,100)
· Increased prevalence of depressive illness (101)
· Impaired fibrinolysis (102)
|
Subclinical Hyperthyroidism
NATURAL HISTORY OF SUBCLINICAL HYPERTHYROIDISM
Follow up studies suggest that spontaneous progression to overt hyperthyroidism is uncommon and that subnormal-detectable levels of TSH in the range 0.05-0.4 mU/l frequently return to normal within one year (87). Meyerovitch et al. (103) reported the results of sequential tests of thyroid function over a 5-year period in a large community-based cohort. During the follow-up period, test results returned to normal in 27%, 62% and 51% respectively of untreated patients whose initial serum TSH values were >10 mU/l, 5.5-10 mU/l and < 0.35 mU/l (103). While some subjects did show progression, the high chance of resolution towards normal suggests that retesting after follow-up, possibly after at least 6 months, is advisable before considering any intervention. However, quantitative conclusions from that retrospective study may be insecure, as many subjects with abnormal TSH were excluded from follow-up because they were treated after their initial result (103). If those treated were the more severely affected, whether based on symptoms, presence of goiter, or degree of TSH abnormality, the analysis may over-estimate the likelihood of resolution during follow-up. Spontaneous remission of subclinical hyperthyroidism may occur more frequently in Graves’ disease than in nodular thyroid disorders (104). However, in a recent study from UK (105), a third each of 84 patients with subclinical hyperthyroidism due to Graves’ disease progressed, normalized, or remained in the subclinically hyperthyroid state. Older people and those with positive anti-thyroperoxidase antibodies had a higher risk of progression of the disease. These data need to be verified and confirmed in larger cohorts and over longer periods of follow-up. The chance of spontaneous progression to overt hyperthyroidism appears to be no greater than 10% per year (87,104), or even lower, and also seems dependent on the cause of subclinical hyperthyroidism (Graves’ or multinodular) (106). However, it should be noted that the transition from autoimmune subclinical to overt hyperthyroidism can occur more rapidly than is generally the case in immune hypothyroidism (107).
CARDIOVASCULAR EFFECTS
From the Framingham study it was found that undetected subclinical hyperthyroidism, defined only by suppression of TSH, carried a three-fold increased risk of atrial fibrillation within 10 years (78). As yet, there is no study that shows that treatment given on the basis of low TSH alone, modifies this risk (108), although it is clear that survival is adversely affected by atrial fibrillation (109). In a large cohort study, endogenous subclinical hyperthyroidism is associated with increased risks of total and coronary heart disease mortality, and incident atrial fibrillation, with highest risks of coronary heart disease mortality and atrial fibrillation when the TSH concentration was lower than 0.10 mIU/L (110). A recent meta-analysis of prospective cohorts found an increased risk of coronary heart disease (relative risk (RR) 1.20; 95% confidence interval (CI), 1.02-1.42), total mortality (RR = 1.27; 95% CI, 1.07-1.51), and coronary heart disease mortality (RR = 1.45; 95% CI, 1.12-1.86) from subclinical hyperthyroidism, while this was not the case for subclinical hypothyroidism (111). Another group also analyzed prospective cohorts in a systematic review and meta-analysis (112) and found that higher free T4 levels at baseline in euthyroid individuals were associated with an increased risk of atrial fibrillation in age- and sex-adjusted analyses (hazard ratio, 1.45; 95% confidence interval, 1.26-1.66, for the highest quartile versus the lowest quartile of free T4; P for trend ≤0.001 across quartiles). Estimates did not substantially differ after further adjustment for preexisting cardiovascular disease. Thus, in euthyroid individuals, higher circulating free T4 levels, but not TSH levels, were associated with increased risk of incident atrial fibrillation (112). These results, however, need verification in prospective studies. Finally, impaired left ventricular ejection fraction and reduced exercise capacity have been documented in subclinical hyperthyroidism due to high dosage thyroxine and may be alleviated by beta blockade (113).
IODINE-INDUCED THYROTOXICOSIS
Undiagnosed subclinical hyperthyroidism due to autonomous nodular thyroid disease, a condition especially prevalent in iodine deficient regions, carries the risk of progression to severe overt thyrotoxicosis after iodine exposure (114). An Australian study from a region that is not known to be iodine deficient, was suggestive of recent iodine exposure, most often from radiologic contrast agents, in up to 25% of elderly thyrotoxic patients (81). Prior knowledge of subnormal serum TSH may identify a high-risk group with thyroid autonomy in whom iodine exposure carries the risk of iatrogenic hyperthyroidism (115,116). Prophylactic drugs could be considered in high-risk populations, such as administration of perchlorate and/or a thionamide class drug to elderly patients with suppressed TSH and/or palpable goiter (115).
OSTEOPOROSIS
Notably, in a controlled trial of suppressive T4 treatment for multinodular goiter, TSH suppression without clear excess of serum T4 or T3 resulted in a mean 3.6% decrease in lumbar spine density within 2 years (74). Normalization of serum TSH resulted in normalization of the bone mineral density in postmenopausal women.
Subclinical Hypothyroidism
NATURAL HISTORY OF SUBCLINICAL HYPOTHYROIDISM
The benchmark study of thyroid epidemiology from Wickham, UK (119), showed that the likelihood of overt hypothyroidism after 20 years was directly related to the initial serum TSH concentration, even when the concentration was in the range between 2 to 4 mU/l, within the upper reference interval. The study by Meyerovitch et al. (103)showed that with the initial serum TSH in the range 5.5-10 mU/l, the chance of normalization after prolonged follow-up was greater than the chance of progression to TSH levels >10 mU/l (see above). The antibody status was unfortunately not assessed in that cohort. Huber et al. (87) followed 82 Swiss women with subclinical hypothyroidism, with normal free T4 and serum TSH >4 mU/l, for a mean of 9.2 years (Figure 2). About half of their cohort had had previous ablative treatment for Graves’ disease. The cumulative incidence of overt hypothyroidism, defined here as low free T4 with TSH >20 mU/l, was directly related to the initial serum TSH, with 55% of women with initial serum TSH >6 mU/l progressing to overt hypothyroidism. Progression was not uniform, and over half of the cohort showed no deterioration of thyroid function, but positive microsomal antibodies (corresponding to the more specific thyroperoxidase antibodies) increased the likelihood of progression. It is now clear that the opposite sequence may also occur, with spontaneous normalization of elevated TSH values (103,120). In a cardiovascular health study (121), subclinical hypothyroidism persisted for 4 years in just over half of older individuals, with high rates of reversion to euthyroidism in individuals with lower TSH concentrations and thyroperoxidase antibody negativity. It was advised that future studies should examine the impact of transitions in thyroid status on clinical outcomes (121). Rosario et al. found that most of 241 women with mild TSH elevations ranging from 4.5 to 10 mIU/l did not progress to overt hypothyroidism and even normalized their serum TSH. However, initial TSH seemed to be a more important predictor of progression than the presence of antibodies or ultrasonographic appearance (122). In a prospective study from China, patients >40 years of age, i.e. a younger mean age than most studies, with higher baseline total cholesterol or positive thyroid peroxidase antibodies, had higher risks of progression to overt hypothyroidism, while those with higher baseline creatinine, higher baseline TSH (≥7 mIU/L, p <0.001), or older age (>60 years vs. ≤50 years, p =0.012), had lower odds of reverting to euthyroidism. They concluded that thyroperoxidase antibodies and total cholesterol seemed to be more important predictors of progression to overt hypothyroidism than the initial TSH concentration, whereas high baseline TSH or creatinine were negatively correlated with reversion to euthyroidism. The prognostic value of total cholesterol and creatinine should therefore be considered in mild subclinical hypothyroidism (123).
Notably, a prospective study showed that the progression of autoimmune subclinical hypothyroidism tends to be slower than for subclinical hyperthyroidism (107). Hence, there is a need for prolonged follow-up and patient education, if the decision to treat is deferred.
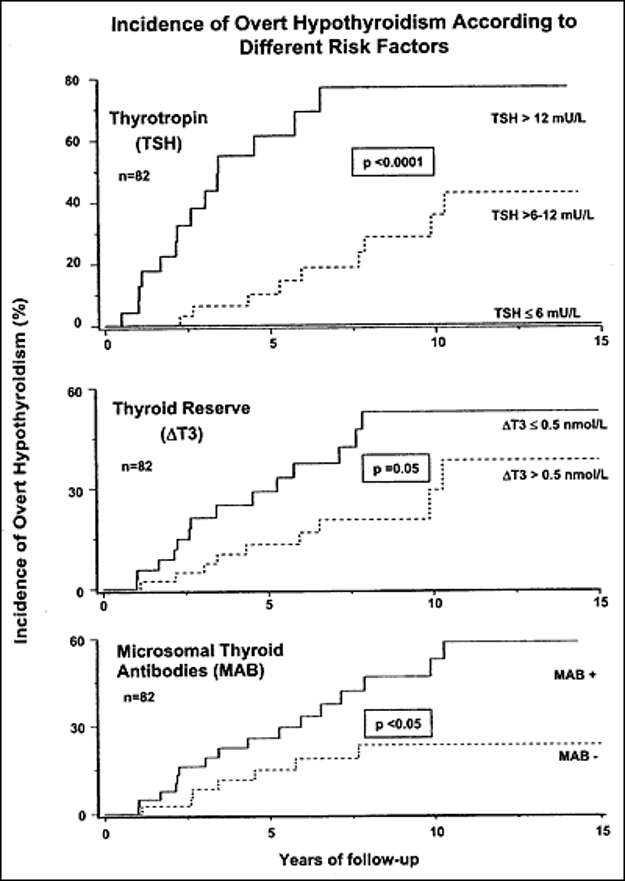
Figure 2. Kaplan-Meier estimates of the cumulative incidence of overt hypothyroidism in women with subclinical hypothyroidism (initial serum TSH >4 mU/l) as a function of initial serum TSH, thyroid secretory reserve in response to oral thyrotropin releasing hormone (TRH) and detectable microsomal antibodies. Serum TSH appears to be the strongest of these predictors (from reference (89)).
ATHEROSCLEROSIS AND VASCULAR COMPLIANCE
As mentioned earlier, subclinical hypothyroidism, or mild thyroid failure, was shown to be an independent risk factor for both myocardial infarction and radiologically-visible aortic atherosclerosis in a study of Dutch women over 55 years of age (90). This effect was independent of body mass index, total and HDL cholesterol, blood pressure, and smoking status. The attributable risk for subclinical hypothyroidism was comparable to that for the other major risk factors, hypercholesterolemia, hypertension, smoking, and diabetes mellitus. The association was slightly stronger when subclinical hypothyroidism was associated with positive peroxidase antibodies, but thyroid autoimmunity itself was not an independent risk factor (124). A systematic review and meta-analysis of 27 studies demonstrated a significant association of subclinical hypothyroidism and cardiovascular risk with arterial wall thickening and stiffening as well as endothelial dysfunction. However, sustained subclinical thyroid dysfunction did not affect the baseline or development of carotid plaques in healthy individuals (125).
In cross-sectional studies, carotid artery-intima media thickness was significantly higher in participants with subclinical hypothyroidism compared to euthyroid controls (126). Small interventional studies suggested that restoring euthyroidism in patients with subclinical hypothyroidism is associated with regression of carotid atherosclerosis (126,127). However, these trials had major limitations, with uncontrolled study designs and/or small sample sizes (the largest included only 45 participants with subclinical hypothyroidism (127).
Although well known in overt hypothyroidism (128), the finding of impaired flow-mediated, endothelium-dependent vasodilatation in subjects with borderline hypothyroidism or high-normal serum TSH values (95) was at first unexpected. Baseline artery diameter and forearm flow were comparable, but flow mediated vasodilatation during the period of reactive hyperemia was significantly impaired even in the group with serum TSH of 2-4 mU/l, compared with the group with serum TSH 0.4-2 mU/l (95). The difference could not be attributed to a difference in maximal nitrate-induced vasodilatation, age, sex, hypertension, diabetes, smoking, serum cholesterol, or levels of total T3 and T4 (91). This finding suggested that even a minor deviation from an individual’s pituitary-thyroid set point may be associated with alteration in vasodilatory response. There is no known direct action of TSH that would account for this effect. A Japanese placebo-controlled study of women aged 60-70 with subclinical hypothyroidism with mean pre-treatment serum TSH of 7.3 mU/l showed improvement in pulse wave velocity, an index of vascular stiffness, in response to TSH normalization for 2 months by progressive low dose T4 replacement only up to 37.5 ug/day (96). Thyroxine replacement for 18 months has been reported to improve blood pressure, lipids, and carotid intimal thickness in women with subclinical hypothyroidism (129). In a larger properly powered randomized placebo-controlled trial normalization of TSH with levothyroxine was associated with no difference in carotid intima-media thickness and carotid atherosclerosis in older persons with subclinical hypothyroidism (130); a meta-analysis of 12 studies showed (131) that carotid intima-media thickness was significantly higher among subjects with subclinical hypothyroidism (n=280) as compared to euthyroid controls (n=263) at baseline. This meta-analysis showed that thyroxine therapy in subjects with subclinical hypothyroidism significantly decreased carotid intimal thickness and improves lipid profiles, modifiable cardiovascular risk factors. One of the big differences between these publications was the age difference; the subjects were younger in the meta-analysis (131), while the patients in the other publications were all older and not included in the meta-analysis, which was published earlier. The same was the case in another more recent meta-analysis, including only 3 randomized clinical trials in younger patients with subclinical hypothyroidism (132). They also found a decreasing carotid intimal thickness with levothyroxine therapy. Thyroid hormone replacement in younger subjects with subclinical hypothyroidism may thus play a role in slowing down or preventing the progression of atherosclerosis (131,132), but there is still no such evidence in older individuals.
LIPIDS
Overt hypothyroidism is associated with an increase in the serum cholesterol concentration (133) and correction of overt hypothyroidism resulted in a decrease in total and low density lipoprotein (LDL) cholesterol, apolipoprotein A1, apo B and apo E, and serum triglyceride concentrations may also decrease (134,135). A defect in receptor-mediated LDL catabolism, similar to that seen in familial hypercholesterolemia, has been described in severe overt hypothyroidism (136), but there is no evidence to support such an abnormality in mild thyroid failure. At the other extreme, several large population studies reported a positive correlation between serum lipids and serum TSH across its normal range (137), a correlation also associated with increasing blood pressure (138). The clinical impact of these associations remains unknown.
The Colorado study including over 25,000 subjects showed a continuous graded increase in serum cholesterol over a range of serum TSH values from <0.3 to >60 mU/l (64). However, there is still no consensus that mild thyroid failure has an adverse effect on plasma lipids, or that T4 treatment sufficient to normalize isolated TSH elevations has a beneficial effect. A meta-analysis suggests that T4 treatment of subjects with mild thyroid failure does lower the mean total and LDL cholesterol, and is without effect on high density lipoprotein (HDL) cholesterol or triglyceride levels (99). In a prospective double-blind, placebo-controlled trial of thyroxine in subclinical hypothyroidism in which the response was carefully monitored with TSH, Meier et al. (100) reported that the decrease in LDL cholesterol was more pronounced with higher initial TSH levels >12 mU/l or with elevated baseline LDL concentrations, but also here the clinical impact remains unknown.
It remains uncertain whether the serum concentration of the highly atherogenic Lp(a) particle is increased in overt hypothyroidism and whether T4 treatment sufficient to normalize TSH has a favorable influence. Serum concentrations of Lp(a) have been found to be increased in overt hypothyroidism with normalization after treatment in some studies (139,140) while others fail to confirm this finding (141,142). Finally, in 100 women with subclinical hypothyroidism selected among 87 obese women aged between 50 and 70 years, total cholesterol, LDL-cholesterol and triglycerides concentrations as well as LDL-C/HDL-C ratio and Castelli index were higher in subclinical hypothyroidism than in controls and decreased after levothyroxine substitution. All the calculated atherosclerosisindexes showed significant positive correlations with TSH concentrations in the subclinical hypothyroidism group. Also, in this group the systolic and diastolic blood pressure decreased significantly after treatment. Thus, dyslipidemia in obese subclinical hypothyroidism women is not severe, but if untreated for many years, it is assumed to lead to atherosclerosis. Substitution therapy improved the lipid profile, changing the relations between protective and proatherogenic fractions of serum lipids, and it optimizes blood pressure (143), which by itself does not prove a positive clinical outcome.
CARDIAC FUNCTION
From echocardiographic studies, there is evidence that mild thyroid failure can significantly increase systemic vascular resistance and impair cardiac systolic and diastolic function (144), as demonstrated by decreased flow velocity across the aortic and mitral valves (98). These changes, which were associated with reduced cardiorespiratory work capacity during maximal exercise, were reversed by T4 treatment sufficient to normalize serum TSH (98). Impairment of both diastolic and systolic function was demonstrable by echocardiography in a subclinically hypothyroid group of patients with TSH in the range 4-12 mU/l (98). Thyroxine treatment sufficient to normalize TSH to a mean of 1.3 mU/l for 6 months was associated with improvement in myocardial contractility (98). Calculation of a global myocardial performance index from the echocardiographic findings also confirmed significantly higher scores in hypothyroid patients in comparison to the control group, showing that regression in global left ventricular functions is an important echocardiographic finding (145).
A very recent small study of 20 young patients with autoimmune subclinical hypothyroidism used cardiac magnetic resonance for a myocardial longitudinal relaxation time (T1) mapping technique and demonstrated significant diffuse myocardial injury (146), which may explain results of the cardiac function studies and also provide a novel method for early detection of cardiac dysfunction in subclinical hypothyroidism.
Future studies are required to determine the effects of the above finding on long-term cardiovascular outcomes and how these reversible abnormalities relate to cardiovascular prognosis.
Several studies suggested that there may be a link between insulin sensitivity, serum lipids, and thyroid function, whether assessed by serum TSH or circulating thyroid hormone levels. Bakker et al. (147) noted that while serum TSH showed no overall correlation with insulin sensitivity or serum lipids, there was a complex interaction between these variables such that the association between TSH and LDL-C was much stronger in insulin- resistant than in insulin- sensitive subjects. The same group showed that low free T4 levels within the reference range are dually associated with LDL-C and insulin resistance (148). Further results will show whether these findings account for the purported link between subclinical hypothyroidism and the metabolic syndrome (149) and whether this link contributes to increased cardiovascular risk in a subgroup of patients with subclinical hypothyroidism. Notably, there is a positive correlation between serum TSH and BMI in euthyroid obese women (9,150). The results of a population study suggest that thyroid function (also within the reference range) could be one of several factors acting in concert to determine body weight in a population. Even slightly elevated serum TSH levels were associated with an increase in the occurrence of obesity (151). Furthermore, a recent cross-sectional study in a sample of 753 subjects (46% males) aged 35-70 years who had no history of diabetes, renal, hepatic, thyroid, or coronary heart disease, and were participants of the Genetics of Atherosclerotic Disease study indicated that subclinical hypothyroidism was associated with fatty liver together with increased odds of metabolic syndrome, insulin resistance, and coronary artery calcification, independent of potential confounders (152).
Finally, subclinical hypothyroidism has been suspected to be related to polycystic ovary syndrome, since subclinical hypothyroidism is present in 10-25% of women with polycystic ovary syndrome. However, a recent meta-analysis of 12 studies found that subclinical hypothyroidism did not influence the hormonal profile of women with polycystic ovary syndrome. On the other hand, it resulted in mild metabolic abnormalities, which are, however, not clinically important in a short-term setting (153).
The value of routine thyroid testing in the above groups remains uncertain (154). The metabolic syndrome and subclinical hypothyroidism are both highly prevalent in the general population. Cross-sectional epidemiological data suggest that a mutual association exists between the two, although the cause–effect relationship remains poorly elucidated. As subclinical hypothyroidism raises cholesterol, blood pressure, and visceral fat, it is easy to understand why it associates with metabolic syndrome (155). Rather, the reasons whereby patients with metabolic syndrome are at higher risk for subclinical hypothyroidism are less apparent. Some studies have reported that subclinical hypothyroidism is itself characterized by high cardiovascular risk. Therefore, the coexistence of subclinical hypothyroidism and metabolic syndrome may identify subjects at a particularly high risk for future cardiovascular events. Recent data indicated that carotid intima-media thickness, a marker of initial atherosclerosis and a possible predictor of future events, was higher in patients with both subclinical hypothyroidism and metabolic syndrome than in the presence of each condition alone.
To date, it remains unclear whether any biological relationship between subclinical hypothyroidism and the metabolic syndrome truly exists and what the underlying mechanisms might be. Nonetheless, given the high prevalence of both conditions, and the observed associations, it is of interest to investigate whether their mutual presence confers a higher cardiovascular disease risk. If confirmed in larger studies, these results may be clinically relevant, suggesting that subclinical hypothyroidism should be investigated in patients with metabolic syndrome to better individualize therapy and counter cardiovascular risk (156).
OSTEOPOROSIS
From a previously mentioned study (118), subclinical hypothyroidism was associated with RRs of 1.34 (95% CI 1.14-1.58; I 2 =32%) for hip fracture, 1.27 (95% CI 1.02-1.58; I 2 =51.9%) for any location of fracture, and 1.25 (95% CI 1.04-1.50) for forearm fracture. The authors failed to find any associations between the change in bone mineral density and subclinical hypothyroidism but subclinical hypothyroidism was associated with an increased risk of fractures. Although subclinical hyperthyroidism was related to reduced bone mineral density, there is no evidence of a definite association between subclinical hypothyroidism and the risk of low bone mineral density.
NEUROBEHAVIORAL EFFECTS AND QUALITY OF LIFE
It is well known that overt thyroid dysfunction can include psychological or psychiatric symptomatology. A small retrospective study has shown a 2-3 fold increased frequency of previous depression in subjects with mild thyroid failure (101); T4 treatment has been reported to improve neuropsychological responses in this group (157). However, contrary to these findings, more recent controlled studies of unselected patients suggest that subclinical hypothyroidism is not associated with any consistent deficit in quality of life indices or improvement with treatment (158,159). This was very recently confirmed in the TRUST trial using the thyroid patient reported outcome (ThyPRO) questionnaire in older adults with subclinical hypothyroidism; in that study, therapy with levothyroxine did not improve symptoms or tiredness compared with placebo (160).
STUDIES IN CHILDREN
A study by Cerbone et al. (161) has shown that long-term idiopathic subclinical hypothyroidism did not appear to have an adverse effect on linear growth or intellectual development in children aged 4-18 years. More recently, a meta-analysis including nine studies demonstrated that subclinical hypothyroidism in children is a remitting process with a low risk of evolution toward overt hypothyroidism. Most of the subjects reverted to euthyroidism or remained subclinically hypothyroid, with a rate of evolution toward overt hypothyroidism ranging between 0 and 28.8%, with a rate of 50% in only one study. The initial presence of goiter and elevated thyroglobulin antibodies, the presence of celiac disease, and a progressive increase in thyroperoxidase antibodies and TSH value predict progression toward overt hypothyroidism. Replacement therapy is not indicated in children with subclinical hypothyroidism with TSH 5-10 mU/l, absence of goiter, and negative antithyroid antibodies. An increased growth velocity was observed in children treated with levothyroxine in two studies. Levothyroxine reduced thyroid volume in 25-100% of children with subclinical hypothyroidism and autoimmune thyroiditis in two studies. No effects were seen on neuropsychological functions in one study, and posttreatment evolution of subclinical hypothyroidism was reported in one study (162). Hence, it may be justifiable to follow this group without early recourse to lifelong replacement.
On the other hand, the association with Hashimoto’s thyroiditis exerted a negative influence on the evolution over time of mild subclinical hypothyroidism, irrespective of other concomitant risk factors. In children – unlike in adults - with mild and subclinical hypothyroidism related to Hashimoto’s thyroiditis, the risk of a deterioration in thyroid status over time is high (53.1%), while the probability of spontaneous TSH normalization is relatively low (21.9%). In contrast, children with mild and idiopathic subclinical hypothyroidism, had a very low risk of a deterioration in thyroid status over time (11.1%), whereas the probability of spontaneous TSH normalization was high (41.1%) (163).
Safety and Effectiveness of Treatment of Subclinical Thyroid Dysfunction
The benefits of early diagnosis and treatment are self-evident from the obvious decline in hospitalization and mortality rates for severe thyroid dysfunction over the past decades. Before reliable tests of thyroid function became widely used, severe hyperthyroidism approaching thyroid storm and hypothyroidism with impending myxedema coma occurred quite regularly, but these presentations are now very uncommon (164,165). While the arguments for seeking and treating mild thyroid dysfunction are less compelling, there may be potential benefits for large numbers of people.
The points in favor of treating mild thyroid dysfunction relate directly to the adverse consequences listed in Table 4, but for many of these adverse outcomes there is still a lack of long-term studies that show benefit as illustrated in a very recent randomized, double-blind placebo-controlled trial nested within the TRUST trial, which found that normalization of TSH with levothyroxine in people >65 years was associated with no difference in carotid-intima media thickness and carotid atherosclerosis by ultrasound in older persons with subclinical hypothyroidism (130). On the basis of potential benefit from simple straightforward treatment and absence of adverse effects, the argument for active treatment is generally stronger for mild thyroid failure than for subclinical hyperthyroidism. Conservative T4 therapy aimed at normalizing TSH is simple, inexpensive and generally safe (166), although replacement may not be warranted in the older adults with very advanced age (167). Where cardiovascular disease precludes full thyroid hormone replacement, detailed evaluation of the cardiac abnormality is appropriate (168,169). In contrast, treatment of subclinical hyperthyroidism needs to be evaluated in relation to adverse drug effects and the potential for hypothyroidism. It is, however, prudent to take into consideration an effect and consequence of suppressed serum TSH on atrial fibrillation, bone loss, depression, quality of life and mortality when counseling individual patients.
In younger patients there may be a benefit of early treatment of subclinical hypothyroidism (170) although the studies supporting this approach were not placebo-controlled and only used surrogate endpoints in small cohorts.
USE OF LABORATORY ASSAYS FOR CASE FINDING AND SCREENING
If the identification of abnormal thyroid function is to be based on laboratory testing, it is desirable that population reference intervals should not vary between methods. As recently emphasized, serum T3, T4 and serum TSH concentrations are among the many hormone variables where between-assay standardization is crucial to ensure optimal assay specificity (171). At present, that aim is not satisfactory for free T4 estimates, a problem that is particularly troublesome during pregnancy (see below). Analytically, serum TSH, total T4 and total T3 are well standardized, so that considerations of so-called normal ranges relate to the clinically relevant issues. By contrast, the diverse, ingenious manoeuvres involved in the estimation of free T4 and T3 lead to poor standardization between methods, so that method-specific reference intervals need to be used, both for individual clinical diagnosis and population studies. Between-method free T4 variations are especially troublesome in pregnancy and critical illness (see below).
TSH Reference Interval or “Normal Range”
For serum TSH, arguably the most important variable for the diagnosis of primary thyroid dysfunction, no firm consensus range has been agreed, despite reliable standardization between methods. Firstly, a reference interval for the diagnosis of hypothyroidism in adults may be far too broad and lenient to identify women whose fertility or pregnancy outcome might be improved by thyroid supplementation. Second, it has been shown that the reference TSH set-point for each individual can be defined with a narrow band of the broad population range (172,173) (see below). Third, criteria for the new diagnosis of thyroid dysfunction may not be the same as those required for optimal adjustment of therapy. Fourth, for population studies, whether screening or case-finding, a reference interval with higher sensitivity will have lower specificity.
Thus, the controversies as to whether the standard TSH reference interval of about 0.4–4.0 mU/l should be narrowed, with lowering of the upper limit (70), or retained (174-176) do not address the needs of individuals. Population studies to define the upper limit of the “normal range” for serum TSH will be influenced by whether those with positive thyroperoxidase antibodies are excluded (see below) (63,177,178). However, even after exclusion of individuals with clinical, antibodies, or sonographic evidence of any thyroid disorder, Hamilton et al. supported an upper reference limit at about 4 mU/l (176), although quite different criteria may apply around pregnancy (see below). Ethnic differences (179), as well as differences and time of sampling in relation to diurnal variation are also important.
Terminology for abnormal TSH values has also become inconsistent, as for example in the use of the term suppressed to describe lower-than-normal TSH values. In some studies (180,181) any subnormal value is classified as suppressed, while others reserve this term for lower levels (63) that allow a distinction between undetectable (e.g. <0.03 mU/l) and a subnormal-detectable range below about 0.4 mU/l (76). These two categories may have different diagnostic and prognostic significance. Based on follow-up studies of the probability of progression to overt thyroid dysfunction, there is a strong case for regarding TSH values in the subnormal-detectable range, arbitrarily 0.05-0.4 mU/l, as distinct from the even lower levels that are typical of hyperthyroidism. It is a personal view that the term “suppressed” should be avoided in describing subnormal-detectable values, a semantic point that may affect up to 1% of the population. Since the gradation from normality to severe thyroid dysfunction is a continuum, studies of adverse outcomes or benefits from intervention will be critically dependent on uniform terminology.
Choice of Initial Test
The definitive diagnosis of thyroid dysfunction should always be made using the typical relationships between trophic hormone and target gland secretion that define endocrine dysfunction. In contrast, case-finding studies of untreated subjects may begin with measurement of TSH alone (182), with T4 and T3 assays added only if TSH is abnormal, or if an abnormality of TSH secretion is suspected. It is self-evident that serum TSH loses its diagnostic value when pituitary function is abnormal (183-186).
In the absence of associated disease, a normal serum TSH concentration by a so-called third generation assay (a functional lower limit of sensitivity of about 0.03 mU/l) (57,58), has a high negative predictive value in ruling out primary hypothyroidism and hyperthyroidism. Such immunometric assays, which use two antibodies against different epitopes of the TSH molecule, give a wide separation between the lower limit of the normal reference range at about 0.4 mU/l and the typical suppressed TSH values found in hyperthyroidism. While some subjects with subnormal-detectable TSH values do progress to overt hyperthyroidism, values in this range may also revert to normal (see above).
There are some clinical situations in which assessment of thyroid function will give a high prevalence of abnormalities that cannot be interpreted with certainty. Notably, glucocorticoids and dopaminergic agents have a potent effect to suppress TSH secretion (45,187), while TSH is also frequently subnormal in starvation or caloric deprivation (188). Transient increases to above normal can occur in euthyroid subjects during recovery from critical illness (178,189-191). The finding that 33% of serum TSH values fell more than 2 standard deviations (SD) from the geometric mean in acutely hospitalized patients, with 17% of values more than 3 SD from the mean value, indicates that TSH concentrations lack diagnostic specificity in this setting (192). A serum free T4 estimate will generally follow from an abnormal TSH concentration, but during critical illness, free T4 estimates often show non-specific abnormalities (see below) (193). Lack of specificity was the basis for a recommendation against routine assessment of serum TSH and free T4 during acute critical illness in the absence of risk factors, or clinical features suggestive of a thyroid disorder (194).
Testing of thyroid function is appropriate in a wide range of psychiatric disorders, but diagnostic specificity is limited by a high prevalence of transient non-specific abnormalities at the time of acute psychiatric admission (195). Thus, laboratory evaluation should be delayed for 2-3 weeks after acute presentation, unless there are specific risk factors for thyroid dysfunction (196).
Potential Adverse Effects of Testing
In terms of potential for adverse effects, there may be important differences between screening of unselected populations and the case-finding strategy that is now recommended for thyroid dysfunction. There are no reports of a “labelling effect” (i.e. perception of chronic illness in previously asymptomatic subjects), described in hypertension screening programs (197), when testing for thyroid dysfunction is done at the time of presentation for medical care. Nevertheless, further attention needs to be given to the potential for unwarranted treatment based on false positive results, as well as the cost of follow-up investigations for perceived abnormalities that may not warrant treatment at any stage. In particular, there is a need for clinical consensus as to how marginally abnormal results should be classified. Widespread laboratory testing will lead to an increase in the number of false positive results; the potential for a diagnostic method to give misleading variations from normal may not become known for some years until the full diversity of the non-diseased population is documented (198).
Follow-up of Abnormal Results
If serum TSH is used as a single initial test for case-finding, a value outside the reference interval should lead to estimation of serum free T4 on the same sample, if possible, without recall of the patient. This requires an algorithm-based testing protocol, which should also include measurement of serum free T3 if TSH is suppressed, to identify T3 toxicosis. It may also be relevant to measure thyroid peroxidase antibodies if TSH is increased, so as to define an autoimmune mechanism of hypothyroidism, particularly as a raised level of these antibodies is associated with an increased likelihood of progression to overt hypothyroidism (61,178).
For screening or case-finding to be effective, patients with unsuspected overt thyroid dysfunction should be actively traced because they will benefit most from treatment and have the most to lose if the abnormal finding is ignored. For mild thyroid dysfunction, a practitioner who has continuing contact with the patient should evaluate the assay result in clinical context and initiate any necessary follow-up. However, there is currently no consensus as to how an appropriate clinical response to abnormal laboratory findings can be assured.
A transition towards identification of thyroid dysfunction by laboratory measurement, rather than on clinical criteria, modifies, but does not diminish the role of the clinician. The severity of thyroid dysfunction cannot be judged from the extent of the laboratory abnormality (198,199), which indicates neither the duration of exposure, nor individual susceptibility. Whether a decision is made to treat or to observe, patient education is crucial in establishing effective compliance and rational cost-effective long-term follow-up. Computer based programs can identify affected individuals, but do not replace direct involvement of both patient and clinician. There may be potential medicolegal consequences of failure to respond to abnormal results, if widespread laboratory testing is initiated without an established follow-up plan.
INTERPRETATION OF TSH AND T4 ASSAYS
The TSH-T4 Relationship
Definitive assessment of thyroid status requires both a sensitive serum TSH assay and a valid T4 estimate, with interpretation based on the relationship between these two values. It is generally assumed that inverse TSH responses to changes in free T4 are approximately logarithmic (59) (Figure 3). While this concept is useful, recent data suggests that the relationship varies with thyroid status, age, sex, smoking and thyroid autoantibody status (200-203) with progressively greater amplification of the TSH response as thyroid function declines. Diurnal variation in serum TSH, with amplitude about 50% with higher levels between 2100 and 0600, is superimposed on rapid pulse secretion with amplitude about 10%. Basal secretion and pulsatility are both increased in primary hypothyroidism, with retention of diurnal variation (204).
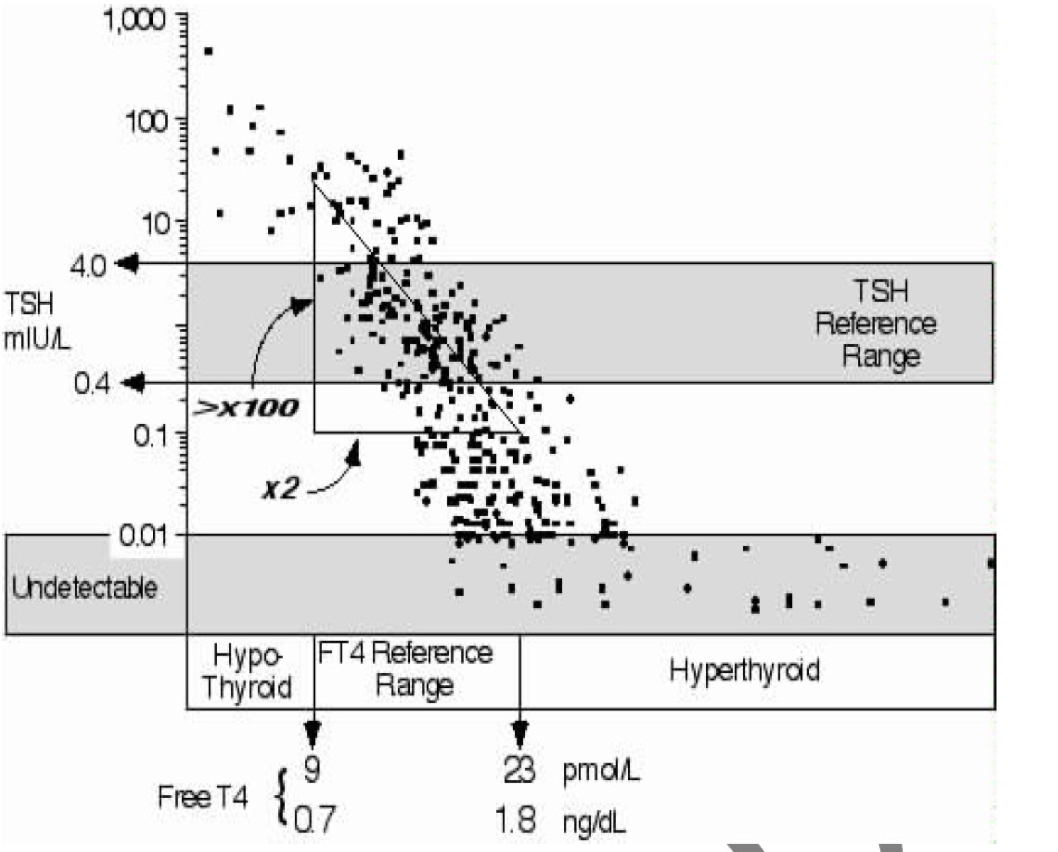
Figure 3. The relationship between serum TSH and free T4 estimate in ambulatory individuals with stable thyroid function and normal hypothalamic-pituitary-thyroid function (adapted from reference (178) with permission).
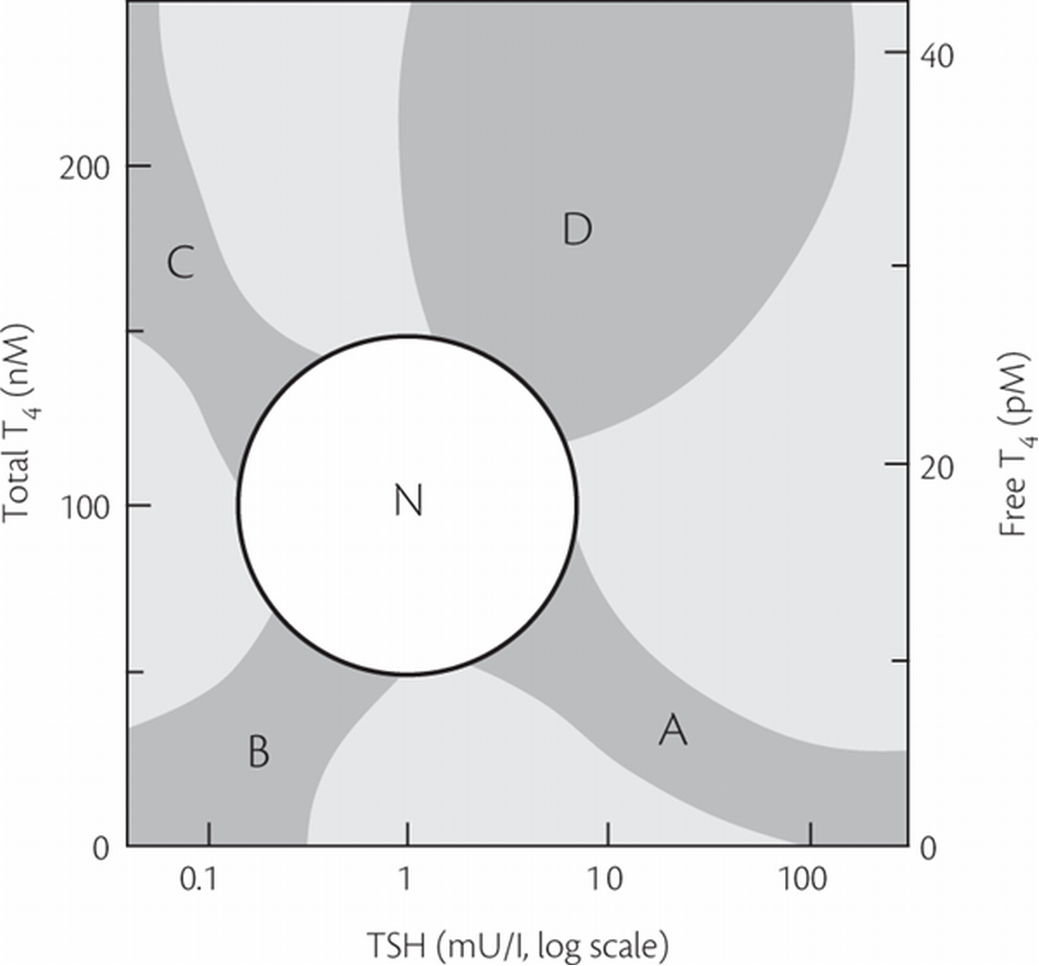
Figure 4. The relationship between serum TSH and free T4 concentration is shown for normal subjects (N) and in the typical abnormalities of thyroid function: A, primary hypothyroidism; B, central or pituitary-dependent hypothyroidism; C, hyperthyroidism due to autonomy or abnormal stimulation of the gland; D, TSH-dependent hyperthyroidism or thyroid hormone resistance. Note that linear changes in the concentration of T4 correspond approximately to logarithmic changes in serum TSH (178).
The T4-TSH Setpoint
Small changes in serum T4 and T3 concentrations, within the normal range, alter the serum TSH concentration, indicating that the inverse feedback relationship between serum free T4 and TSH applies across their normal ranges, as well as in disease states (205,206). Studies of normal subjects demonstrate significant individual variation, independent of sex and age, in the setpoint of the pituitary-thyroid axis (172,173,207), which suggests that the TSH set-point for a particular serum free T4 or free T3 concentration is an individual characteristic. It follows that the concept of “normality” for an individual may be narrower than for a population at-large. Studies of monozygotic and dizygotic twin pairs also suggest that genetic factors influence the serum concentrations of total and free T4 within the normal range (208), as well as the relationship between TSH and free T3 and T4 (209,210). The recent demonstration (211) that multiple genetic factors can influence inter-individual differences in the TSH response to circulating thyroid hormones may ultimately increase the precision and the complexity of interpreting thyroid function are also reviewed recently (212,213).
Andersen et al. (173), in a study of normal subjects whose samples were taken monthly between 09:00 and 12:00 for a year, showed that individual references ranges for T4 and T3 were only about half the width of the population reference ranges, indicating that a test result within the population range is not necessarily normal for that individual. Serum TSH showed greater between-sample variation for each individual than serum T4 or T3. Based on the degree of individual variation, it was estimated that a normal serum TSH concentration needed to change by 0.2-1.6 mU/l to be confident of a serial change in thyroid status. Based on this analysis, it was estimated that a single morning sample defined serum T4 and T3 to within 25%, and serum TSH only to within 50%. Since TSH shows diurnal variation and pulsatile secretion in both normal and hypothyroid subjects (204,214), random samples are likely to show even greater variation. This concept can also be described as individuality index (166).
It has been suggested that some healthy older adults have normal serum TSH concentrations despite having low serum free T4 values, attributed to resetting of the threshold for TSH inhibition (215). In a large cohort of Danish patients with newly diagnosed hypothyroidism, the increase in serum TSH for a given degree of lowering of serum free T4 was less in the elderly, suggesting that equivalent TSH increases in the elderly may be accompanied by more severe thyroid hormone deficiency (216). It is notable that a higher level of serum free T4 was necessary to normalize serum TSH in children with congenital hypothyroidism than in adults with acquired autoimmune hypothyroidism (217). It is uncertain whether this is a further reflection of age-related difference, or whether this difference reflects a perinatal shift in the central setpoint for regulation of TSH secretion.
The TSH-T4 Relationship: Diagnostic Assumptions
The precise diagnosis of thyroid dysfunction can generally be established from a single serum sample from the relationship shown in Figure 4, subject to six key assumptions (Table 5). It should be noted that only the last three of these assumptions can be validated in the laboratory; the first three are best verified clinically.
|
Table 5. Assumptions Inherent to Diagnostic Use of the T4 -TSH Relationship
(Conditions that may breach these assumptions are shown in italics)
|
|
1. Steady-state conditions (note differences in the half-lives of TSH and T4)
· Early treatment with antithyroid drugs (218)
· Early response to T4 therapy
· Evolution of transient thyroid dysfunction (178)
· Recovery from severe illness (190,191)
|
|
2. Normal trophic-target hormone relationship
· Alternative thyroid stimulators
· Immunoglobulins (219)
· Chorionic gonadotrophin (220)
· Medications that influence TSH secretion (45)
· T3, triiodothyroacetic acid (221)
· Other thyroid hormone analogues (222)
· Glucocorticoids (223)
· Dopamine (224)
· Amiodarone (45)
· Recent hyperthyroidism (218)
· Recent longstanding hypothyroidism
· Treated congenital hypothyroidism (225)
· TSH receptor mutations (226)
· Variable individual setpoint (173,207,208,213,215,216)
|
|
3. Tissue responses proportional to hormone concentration
· Hormone resistance syndromes (227)(see below)
· Slow onset/offset of thyroid hormone action
· Drug effects (45)
· Amiodarone (45)
· Phenytoin (228)
|
|
4. The assay measurement represents the active hormone
· Unmeasured agonist in excess (e.g. T3, triiodothyroacetic acid, human choriogonadotropin hormone (178)
· TSH of altered biologic activity (229,230)
· Spurious immunoassay results (178)
· TSH (178)
· Heterophilic antibodies (231,232)
· Free T4
· Abnormal serum binding proteins (233)
· Autoantibodies (234)
· Medications that inhibit protein binding (233,235)(227, 229)
· Heparin artefact (236,237)
|
|
5. The assay can reliably distinguish low from normal values
· Lack of precision at the limit of detection (178,238)
|
|
6. Reference ranges are appropriate
· Influence of age (239,240)(see also above)
· Associated illness (178)( see below)
|
STEADY-STATE CONDITIONS
This first assumption should be questioned whenever anomalous results occur during associated illness, or with medications that perturb the pituitary-thyroid axis. The half-lives of plasma TSH (approximately 1 hour) and plasma T4 (approximately 1 week) differ so widely that acute perturbation of the pituitary-thyroid axis will often result in transient nonsteady state conditions (Figure 5). Due to its much shorter half-life, serum TSH deviates more rapidly from steady state. Other common deviations from steady state relate to short-term pulsatile or diurnal fluctuations in hormone secretion, responses to treatment and spontaneous evolution of disease, as can occur in subacute thyroiditis or postpartum thyroid dysfunction.
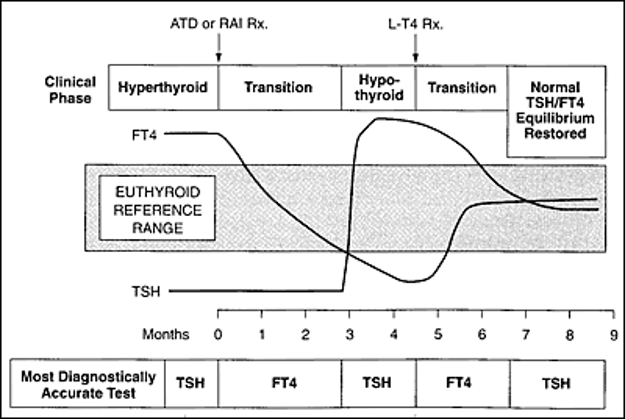
Figure 5. Measurement of serum T4, rather than serum TSH, is the more reliable single test of thyroid function when steady state conditions do not apply, as in the early phase of treatment for hyperthyroidism or hypothyroidism. (adapted from reference (178)
NORMAL TROPIC-TARGET HORMONE RELATIONSHIP
During treatment of prolonged hyperthyroidism, TSH secretion may remain low for several months after serum free T4 becomes normal (218). Conversely, after severe prolonged hypothyroidism, or in some children treated for congenital hypothyroidism (217), TSH hypersecretion may persist despite normalization of serum T4. Serum TSH will then give an inaccurate indication of thyroid status, with the potential for over-treatment if this variable alone is used to assess therapy.
TISSUE RESPONSES PROPORTIONAL TO THE HORMONE CONCNETRATION
The active or free concentrations of T3 and T4 generally correlate well with clinical features. However, in generalized thyroid hormone resistance due to mutations in the thyroid hormone receptor, serum free T3 and T4 concentrations are elevated and the TSH is inappropriately normal or elevated due to the impaired feedback at the level of the pituitary thyrotrophs.
The onset and offset of genomic thyroid hormone action are relatively slow, so that tissue responses may lag behind changes in serum concentrations of free T4 and T3. There is a notable lack of convenient, sensitive, specific, objective indices of thyroid hormone action (see later), so that assessment remains predominantly clinical. Corroborative measurements that can be useful, especially in following the response of individuals to therapy, include measurement of oxygen consumption (241), sex hormone binding globulin (242), and angiotensin converting enzyme (243), as well as several indices of cardiac contractility, although none of them is a very accurate nor specific biomarker for changes in thyroid function.
THE ASSAY MEASUREMENT REFLECTS THE ACTIVE HORMONE(S)
TSH and iodothyronine assays make comparative, rather than absolute, measurements of hormone concentrations, based on the premise that samples and assay standards differ only in their concentration of the analyte. This assumption fails if there is any other difference between a serum sample and assay standards that influence the measured variable, as, for example, dissimilar protein binding of tracer (244), the presence of binding competitors (235,245), or possible nonspecific interference with enzymatic, fluorescent, or chemiluminescent detection systems. Circulating T3 and T4 autoantibodies may invalidate immunoassays by sequestering the assay tracer (246), while heterophile mouse or sheep (231,232) antibodies and rheumatoid factor can interfere with immunoglobulin aggregation, or with cross linking of the signal and capture antibodies (247,248).
If the biologic activity of circulating immunoreactive TSH is increased or decreased, the normal relationship between measured serum TSH and free T4 may be altered. Secreted immunoreactive TSH is heterogeneous, due to differences in its three oligosaccharide side chains. In hypothalamic hypothyroidism, the secreted TSH has decreased bioactivity (229,230), whereas activity may be enhanced in thyroid hormone resistance, primary hypothyroidism, and in some TSH-producing tumors (249,250).
THE ASSAY CAN RELIABLY DISTINGUISH LOW FROM NORMAL LEVELS
Assay precision inevitably deteriorates as the limit of detection is approached; this characteristic is crucial in evaluating TSH assays that are used to distinguish hyperthyroidism from normal (57,178). The lower working limit of a TSH assay should be defined in terms of its between-assay reproducibility, defined as functional sensitivity, rather than by the analytical sensitivity of individual assay runs (57,178).
REFERENCE RANGES ARE APPROPRIATE
Since reference ranges of the thyroid related hormone measurements vary substantially according to the methods of measurement, it does not make much sense to provide typical ranges. However, reference ranges show little change with advancing age, except for a possible decline in normal serum T3 with age (215,240). Serum T3 is significantly higher in children (239) and probably also in young adults. For statistical analyses, TSH reference ranges are generally evaluated after logarithmic transformation; the geometric mean is then used to define a realistic lower normal limit. There may be an age-related change in the central setpoint for TSH secretion, with a progressive decline in the TSH setpoint or response to decreased levels in serum T4 (215,216). Associated illness, nutritional changes, and medications frequently cause assay results to fall outside the normal reference ranges as defined in healthy subjects, so that it may be relevant to use wider than normal reference ranges in the face of associated illness (178). Thus, reference ranges are only appropriate and useful in the context of employing the same biochemical laboratory method in establishing the reference range as the one used for routine measurement in patients. Since automated laboratory methods have been extensively implemented globally, where most clinicians rely entirely on the manufacturers’ information on the method quality (sensitivity, accuracy and imprecision), as well as the reference interval, above requirement for one’s own local laboratory to perform these assessments in order to improve local laboratory quality of results has currently left much to desire.
APPLICATION AND INTERPRETATION OF INDIVIDUAL SERUM ASSAYS
Serum TSH as the Initial Test of Thyroid Function
Either serum TSH or free T4 can be used as the initial test of thyroid function, with TSH previously assessed as giving better first-line discrimination at slightly higher cost (182). A normal serum TSH concentration has high negative predictive value in ruling out primary thyroid disease, and this assay has become increasingly used as the single initial test of thyroid function, with no further assays done routinely if serum TSH is normal. If serum TSH is increased, free T4 is measured on the same sample to distinguish between overt and subclinical hypothyroidism (Figure 6). If serum TSH is suppressed, i.e.<0.05 mU/l, both free T4 and free T3 should be assessed to distinguish between overt hyperthyroidism, T3-thyrotoxicosis and subclinical hyperthyroidism. The interpretation of subnormal TSH values is influenced by the functional sensitivity of the particular assay (see below).
The demonstrated slightly higher costs are probably eliminated by the much better interlaboratory robustness of the TSH assays compared to free T4 estimates, although this has never been properly verified.
Figure 6. An algorithm for the initial assessment of thyroid function, based on initial assay of serum TSH. The limitations of this strategy are summarized in table 5. TPOAb=thyroperoxidase antibodies; TRAb=Thyrotropin receptor antibodies; FT4 and 3=Free thyroxine and -triiodothyronine.
LIMITATIONS OF THE “TSH FIRST” TESTING STRATEGY
The rationale for using TSH alone as a first-line test of thyroid function rests on the assumptions that thyroprivic, or primary hypothyroidism is far more common than central or secondary hypothyroidism, and on the fact that serum TSH deviates outside the reference range early in the natural history of thyroid over-function or progressive thyroid failure. The major weaknesses of this approach are the likelihood of missing secondary hypothyroidism (in which the concentration of immunoreactive serum TSH is frequently normal rather than low), and the frequency of abnormal TSH concentrations in the absence of thyroid dysfunction, especially in patients with an associated illness or interference in the TSH assays (Table 6).
Beckett and Toft (184) have pointed out the adverse consequences of missing secondary hypothyroidism due to pituitary failure by relying on normal serum TSH to rule out thyroid dysfunction. The diagnosis of this disorder is often difficult, with diverse presentations to a wide range of practitioners who may not be attuned to key clinical features. They estimated that as many as 1500 cases per year of central hypothyroidism could be missed in the UK if the “TSH first” policy were followed inflexibly. This potentially serious diagnostic problem was shown by Wardle et al. (183) who reported an analysis of 56,000 requests for thyroid function testing over 12 months from a population of 471,000. Serum TSH was normal in association with subnormal total and free T4 in 15 patients who, on further investigation, had probable hypopituitarism that would have been missed by assessment of serum TSH alone. Cost benefit analyses would therefore need to balance the savings from not measuring serum T4 routinely, against the cost of further investigation and medical care for the missed diagnoses, in addition to a component for burden of suffering and potential litigation. If advances in technology can reduce the unit cost of measuring T4, the “TSH first” approach may be abandoned.
|
Table 6. Situations in Which Serum TSH Alone can Give a False or Uncertain Indication of Thyroid Status.
|
|
Condition
|
TSH
|
fT4
|
fT3
|
|
Primary abnormality of TSH secretion
|
|
Pituitary-hypothalamic abnormality
|
L- N
|
L
|
|
|
Extremely premature infants
|
L- N
|
L
|
L
|
|
Central TSH excess
|
N -H
|
H
|
H
|
|
Hyperthyroidism
|
|
T3 toxicosis
|
U
|
N
|
H
|
|
Subclinical
|
U
|
N
|
N
|
|
Early Treatment
|
U
|
H-N-L
|
H-N-L
|
|
TSH assay artefact
|
L- N -H
|
H
|
H
|
|
Hypothyroidism
|
|
Subclinical
|
H
|
N
|
|
|
Early Treatment
|
H
|
L-N
|
|
|
Thyroid hormone resistance
|
N -H
|
H
|
H
|
|
Medications
|
|
Dopamine
|
L
|
N
|
N
|
|
Glucocorticoids
|
L
|
N
|
L-N
|
|
Amiodarone (acute)
|
H
|
N-H
|
L
|
N: normal; L: low; H: high; U: undetectable.
Serum Free and Total T4
Estimation of free T4, or total T4 linked to a measurement of the thyroid hormone binding ratio, in association with serum TSH, has now become the standard method of assessing thyroid function, except in settings where the TSH-first strategy persists. Assays for total T4 and T3 in unextracted serum include a reagent such as 8-anilinonaphthalene sulfonic acid that blocks T4 and T3 binding to serum proteins, so that total hormone is available for competition with the assay antibody. Assays for free T4 or T3 omit this blocking reagent and use a wide variety of manoeuvres to isolate a moiety that reflects the free hormone concentration. The theoretical basis, practical utility, and validity of the many different approaches to the estimation of serum free T4 and T3, have been considered in detail (251) (see below).
Two key assumptions in any method of free T4 estimation are (i) that the dissociation of bound hormone with sample dilution is similar in samples and standards, and (ii) that samples and standards show identical protein binding of the assay tracer. If either of these conditions is breached, the assay is likely to give inaccurate results. Serum free T4 and free T3 can be estimated either by two-step methods that separate a fraction of the free hormone pool from the binding proteins before the assay incubation, or by one-step methods in which the free hormone concentration is measured in the presence of binding proteins (251). Many of the one-step methods become invalid when the sample and standard differ in their binding of assay tracer, but the two-step methods are less prone to non-specific artefacts. Almost all techniques of estimating free T4 give a useful correction for moderate variations in serum thyroxine binding globulin concentration, but no method can yet accommodate the effect of circulating competitors for T4 binding (193,235,252).
Indications for Measurement of Serum T3
Measurement of serum T3 is indicated, in addition to serum T4, as follows:
- In suspected hyperthyroidism with suppressed TSH and normal serum T4, to identify T3-thyrotoxicosis and distinguish this entity from subclinical thyrotoxicosis.
- During antithyroid drug therapy to identify persistent T3 excess, despite normal or low serum T4 values (253).
- For diagnosis of amiodarone-induced hyperthyroidism, which should not be based on T4 excess alone because of the frequency of euthyroid hyperthyroxinemia during amiodarone treatment (254,255).
- To assess the extent of T3 excess in individuals treated with thyroid extracts of animal origin, to assess a potentially damaging hormone excess that is not reflected by the level of T4.
- To identify T3-predominant thyrotoxicosis, an entity that is less likely to achieve remission (see below). In some studies this entity is been reported to respond poorly to radioiodine (256), a phenomenon that may relate to rapid iodine turnover within the gland (257).
Serum T3 measurements may also be useful:
- For estimation of the serum T3-T4 ratio. A high ratio (>0.024 on a molar basis or >20 calculated as ng/µg) that persists during antithyroid drug treatment suggests that patients with hyperthyroid Graves’ disease are unlikely to achieve remission (258). This ratio usually is lower in iodide-induced hyperthyroidism (259) or hyperthyroidism caused by thyroiditis (260) than in Graves’ disease.
- To detect early recurrence of hyperthyroidism after cessation of antithyroid drug therapy.
- To establish the extent of T3 excess during high-dose replacement or suppressive therapy with T4, or after an accidental or intentional T4 overdose.
Low serum T3 concentrations have little specificity or sensitivity for the diagnosis of hypothyroidism. Many patients with nonthyroidal illness have low values, and the serum T3 concentration can remain in the reference range until hypothyroidism is severe. Serum T3 concentrations are usually interpreted together with the concentrations of T4 (Table 7).
|
Table 7. Relationship Between Serum T4 and T3 in Various Disorders*
|
|
|
Serum T4
|
|
Serum T3
|
Low
|
Normal
|
High
|
|
Low
|
Severe hypothyroidism
TBG deficiency #
Severe nonthyroidal illness
Euthyroid hypo-thyroxinemia
|
Nonthyroidal illness
Medications
Fetus
Restricted nutrition
|
Hyperthyroidism with
severe nonthyroidal illness
Amiodarone
|
|
Normal
|
Iodine deficiency
T3 treatment
Hypothyroidism
|
|
T4 treatment
Euthyroid hyper- thyroxinemia
Hyperthyroidism with
nonthyroidal illness
T4 binding autoantibodies
|
|
High
|
Iodine deficiency
T3 treatment
Antithyroid drugs
|
T3 toxicosis
T3 binding autoantibodies
|
Hyperthyroidism
Excess T4 ingestion
Hormone resistance
TBG excess #
|
* Excludes short term changes related to initiation or cessation of therapy
# Effect on total hormone concentration; free hormone concentration remains normal
TBG, thyroxine binding globulin
Indications for TRH Testing
The development of high-sensitivity TSH assays has almost eliminated the need for TRH testing in clinical practice. With intact hypothalamic-pituitary function, there is a close correlation between the TSH response to TRH and the basal level of serum TSH, when measured by a highly sensitive assay (261,262). Hence, TRH testing now offers little diagnostic advantage over accurate measurement of the basal serum TSH concentration in the detection of hyperthyroidism (263). However, measurement of serum TSH 20 to 30 minutes after intravenous injection of 200 to 500 µg TRH remains useful for several purposes:
- To assess patients whose basal serum TSH values are out of context, in order to identify assay artefacts. For example, a detectable serum TSH concentration that is unresponsive to TRH in a thyrotoxic patient suggests either non-specific assay interference (263), or the presence of a TSH-secreting pituitary tumor where serum TSH is often unresponsive to TRH (250).
- To distinguish between cases of thyroid hormone resistance and pituitary-dependent hyperthyroidism. In most instances of hyperthyroidism caused by TSH-secreting pituitary tumors there is no increase in serum TSH in response to TRH (250), while an increased serum TSH response to TRH is seen in the thyroid hormone resistance (227).
Serum Thyroglobulin
This iodinated 660 kDa dimeric glycoprotein, which is the scaffold for thyroid hormone synthesis, is normally detectable in serum and is released in excess in a wide variety of situations where thyroid tissue is overactive, inflamed, or proliferating (264). Undetectable serum levels suggest absence or suppression of thyroid tissue. In the past decade functional assay sensitivity has improved almost ten-fold from about 1 µg/L to around 0.1 µg/L. As a consequence, thyroglobulin (Tg) is now detectable in serum without TSH stimulation in a larger proportion of patients and has greater diagnostic sensitivity during continuing T4 therapy (265). Nevertheless, undetectable serum Tg in the presence of high TSH remains the benchmark for proof of successful ablation after treatment of differentiated thyroid cancer.
Standard indications for measurement of serum Tg are as follows:
- Follow-up of treatment for differentiated thyroid cancer to identify or rule out the presence of residual thyroid tissue, whether normal or metastatic. An undetectable serum thyroglobulin concentration in the presence of high TSH, whether achieved by thyroxine withdrawal, or injection of recombinant TSH, is presumptive evidence that differentiated thyroid tissue has been ablated (266). In contrast, the level of serum Tg that persists when serum TSH is suppressed, can give a useful index of tumor burden (201). In a 16-year follow-up study in differentiated carcinoma, post-ablative Tg concentration was a strong predictor of disease recurrence (267). However, serum Tg measurement is a technically challenging assay and criteria to define a 'highly sensitive' assay may be different, a good knowledge of the technical difficulties and interpretation criteria is of paramount importance for both clinical thyroidologists, laboratory physicians, and scientists involved in the care of differentiated thyroid cancer patients (268). Concurrent measurement of serum TSH and Tg antibody is always required for interpretation of serum Tg values.
- Investigation of atypical hyperthyroidism, where there is a suspicion of thyrotoxicosis factitia in which serum Tg is generally very low or undetectable (269).
- Assay of Tg in the needle washes after biopsy of extrathyroidal neck masses is useful in identifying metastatic thyroid tissue (270). This technique has higher specificity and sensitivity than cytology from suspect lymph nodes (271).
- Serum Tg may serve as a sensitive if non-specific marker of iodine deficiency or ineffective synthesis of thyroid hormone (272).
Because elevation of serum Tg concentration occurs in a wide range of thyroid disorders, interpretation of results always requires a knowledge of (i) the clinical context, (ii) the serum TSH concentration and (iii) whether Tg antibodies are present.
Interactions between serum Tg and circulating antibodies have the potential to cause false-negative assay results, as discussed below. However, measurement of Tg antibodies in the follow up protocol of treated differentiated thyroid carcinoma can also function as surrogate biomarkers for relapse of the thyroid carcinoma (273-275).
Thyroglobulin Antibodies (TgAb)
Assessment of thyroglobulin antibodies (TgAb) in serum is indicated:
- As an essential component of the interpretation of assays for serum Tg, to establish whether endogenous antibodies could be responsible for spuriously low Tg values by interfering with assay separation, particularly in immunometric assays. It is cause for concern that current assays for TgAb may not give congruent results when cross-matched in the assessment of Tg in antibody-positive samples (178,276,277) (See below).
- As secondary follow-up criterion in differentiated thyroid cancer, where a progressive decline in antibody concentration may follow successful ablation (273,274).
- In pregnant populations where positive anti-thyroperoxidase antibodies may not reflect the woman’s autoimmune status in all situations (278), perhaps in particular in iodine fortification programs. It may, however, turn out to be more important in future practice than previously thought (278), when previous advice of routine measurements of both thyroperoxidase/microsomal antibodies and TgAb were abandoned (279).
Thyroid Peroxidase Antibodies
This entity, previously termed antimicrosomal antibody, is closely linked with autoimmune thyroid disease, in particular Hashimoto’s thyroiditis. Measurement of thyroid peroxidase antibody is indicated as follows (278):
- To identify an autoimmune cause for primary hypothyroidism.
- In individuals with the TSH excess of mild thyroid failure, in whom a positive thyroperoxidase antibody indicates an approximately two-fold increase in risk of progression to overt hypothyroidism (280). A strongly positive result in the presence of mild thyroid failure may influence the decision to commence replacement.
- In screening for immune thyroid dysfunction or potential hypothyroidism before or early in pregnancy, especially in high-risk groups and as a predictor of the potential postpartum thyroid dysfunction (see below).
- In order to establish whether there is an immune basis for euthyroid goiter, and to evaluate the risk of future hypothyroidism at a stage before there is any increase in serum TSH
- In cases suspected of polyautoimmunity, where thyroid autoimmunity is by far the most prevalent autoimmune manifestation (16)
TSH Receptor Antibodies (TRAb)
Antibodies that interact with the TSH receptor can be measured either by bioassay, or competitive binding techniques. Bioassays that use thyroid cells of human or animal origin, or cells with transfected TSH receptor, generally depend on tissue production of adenylate cyclase for quantitation. These assays allow a distinction to be made between antibodies with thyroid stimulating (TSAb) and blocking (TBAb) activity. Radioreceptor (thyroid binding inhibitor immunoglobulin, TBII) assays that measure competition by circulating immunoglobulin for specific binding of labelled TSH, are widely available, but do not distinguish between blocking and stimulating activity.
A competitive binding assay using a recombinant human TSH receptor, showed 98-99% positivity in active Graves’ disease, with positive results in <1% of subjects with nonautoimmune thyroid diseases (281). Impressive sensitivity and specificity have been reported with an assay that uses solubilized native porcine TSH receptor with a biotinylated anti-porcine TSH receptor monoclonal capture antibody (282).
Indications for the measurement of TRAb vary in different practice environments, but are clearly applicable to the following situations:
- During pregnancy in women with active or previous autoimmune thyroid disease, to assess the risk of neonatal thyroid dysfunction due to transplacental passage of TRAb. Guidelines from the European Thyroid Association (ETA) (283) suggest that TRAb measurements should be made early in pregnancy in women who have received previous ablative treatment, and in the last trimester in women receiving drug treatment for active Graves’ disease. Antibody measurement during pregnancy was not considered necessary for Graves’ disease in remission. Recent guidelines from ETA, however, state that all patients with a history of autoimmune thyroid disease should have their TRAb serum concentrations measured at the first presentation of pregnancy using either a sensitive binding or a functional cell-based bio-assay and if they are elevated, again at 18-22 weeks of gestation (284).
- In the differential diagnosis of atypical hyperthyroidism that may be due to Graves’ disease.
- In atypical eye disease that may be due to Graves’ orbitopathy.
- In Graves’ orbitopathy for risk assessment of deterioration after radioiodine therapy for the hyperthyroidism.
- To assess the chance of achieving a drug-induced remission of Graves’ disease and to predict whether an apparent remission of Graves’ disease is likely to be sustained, or whether relapse should be anticipated, although this is still controversial (see below)(285-287).
TSH Alpha Subunit
Assay of the TSH alpha subunit is indicated where hyperthyroidism appears to be the result of central autonomous TSH excess to distinguish this entity from thyroid hormone resistance (250). Most TSH producing pituitary adenomas show an increase in alpha subunit (250), and this assay may serve as a useful tumor marker after treatment. By contrast, levels generally remain normal in thyroid hormone resistance (227). Values are also high in postmenopausal women, in men with hypogonadism, and in gonadotrophin-producing pituitary tumors; both thyrotrophs and gonadotrophs secrete this subunit.
Serum Reverse T3
rT3 levels are altered in some rare forms of impaired sensitivity to thyroid hormone: it is significantly reduced in patients with SBP2 mutations, and substantially increased in patients with MCT8 mutations. However, these syndromes are rare (288). Previous suggestions that it might be useful in distinguishing true hypothyroidism from the hypothyroxinemia of severe illness have not been confirmed (289).
Urinary Iodine Estimation
Both iodine deficiency and excess can lead to important abnormalities of thyroid function. In contrast to the wide-reaching effects of iodine deficiency, especially in relation to pregnancy and the neonate (see below), possible iodine excess should be considered as a precipitating cause of hyperthyroidism in the following situations (290):
- Atypical hyperthyroidism with blocked isotope thyroid uptake, with features similar to those of excess thyroid hormone ingestion, or subacute thyroiditis without the expected systemic features.
- High dose requirement for antithyroid drug, or failure to respond to these medications. (In some instances, iodine-induced thyrotoxicosis may be extremely resistant to standard therapy, sufficient to require emergency thyroidectomy (291)).
- Rapid progression from subclinical to overt hyperthyroidism, especially in patients with autonomous multinodular goiter.
Thyrotoxicosis due to iodine excess, which may ultimately be self-limiting, may result from iodine-containing radiographic contrast media or medications, alternative or “natural” health care products and contaminated food products, as observed with soy milk preparations (292). In contrast to subtleties related to urine volume and concentration that are important in assessing urinary iodine excretion in population studies of iodine deficiency, the possibility of marked iodine excess can be assessed from a single urine sample (293). The urinary iodine concentration that can be regarded as excessive is ill-defined and may vary in different populations. Urinary iodine concentrations >1000 ug/l are certainly abnormal, but iodine-induced thyrotoxicosis may also occur at lower levels.
THYROID TESTING STRATEGIES IN SPECIAL SITUATIONS
Subclinical Dysfunction
Follow-up studies suggest that it is inappropriate to commence treatment for subclinical thyroid dysfunction on the basis of a single laboratory result, or even a confirmed assay result within a short period of time (87,103,168,294) (see above). Because both free T4 and TSH show spontaneous fluctuation, it has been suggested that frequent testing may increase the likelihood of an apparent change (295).
Hyperthyroidism
INITIAL DIAGNOSIS
The initial diagnosis of hyperthyroidism is securely established when excess of free T4 and free T3 is associated with an undetectable serum TSH concentration in an assay of appropriate sensitivity. However, the condition can be present without any one of these three criteria. T3-thyrotoxicosis, in which serum T4 remains normal, is more prevalent in iodine deficient regions (296) and can be a premonitory stage of typical hyperthyroidism (297). When hyperthyroidism coexists with another severe illness, serum T3 may be transiently normal or even subnormal (298). Detectable or increased serum TSH concentrations in the face of hyperthyroidism can be due to laboratory artefacts (263), or to autonomous over-secretion of TSH (250). The increase in free T4 and free T3 estimates is usually more marked than the increase in total hormone concentration. Progressive increases in serum total T4, will approach and eventually exceed the limited T4 binding capacity of thyroxine binding globulin (about 300 nmol/l or 24 ug/dl), leading to disproportionate increases in the free serum concentrations of T4 (299) and T3 (300), relative to their total concentrations.
TREATED HYPERTHYROIDISM
In the early drug treatment of hyperthyroidism, measurements of serum T4 and T3 are required for dose adjustment because suppression of TSH may persist for months after correction of longstanding hyperthyroidism (298). Hence, failure to decrease thionamide dosage while TSH suppression persists, can result in serious over-treatment during the early phase of therapy. During thionamide therapy, hyperthyroidism may persist due solely to T3 excess (253); assessment of therapy based on serum T4 alone can then result in under-treatment. A daily dose of 15 mg methimazole can result in hypothyroxinemia in about 10 % of previously thyrotoxic subjects within 4 weeks (301); hence a reassessment of both serum free T3 and free T4 is timely after about 3 weeks to allow appropriate dose-adjustment. In contrast to these discrepancies during early treatment, serum TSH generally gives a reliable index of therapy during the long-term drug treatment of hyperthyroidism. The rate of change in serum free T4 and free T3 during treatment of hyperthyroidism with thionamide gives a valuable guide to dose adjustment, even while levels remain increased and serum TSH is still undetectable. For example, if serum T4 decreases by half in the first four weeks of treatment, it is appropriate to decrease the initial thionamide dosage by about half to minimize the possibility of over-treatment.
Davies et al. (302) assessed the prognostic significance of various serum TSH levels in a large cohort of patients treated with radioiodine 2-35 years previously, who were receiving no other treatment. After a further two years of follow-up, 83% of those with normal TSH had not changed their diagnostic category, although there was a trend for TSH to increase. An increased TSH was associated with a 14.5 % incidence of hypothyroidism after one year. Notably, spontaneous normalization of subnormal or undetectable TSH values during the follow-up period was more common than recurrence of overt hyperthyroidism (302). Hence, while an increased TSH value might be a pointer towards T4 treatment, there appears to be no basis for further radioiodine therapy solely because TSH remains suppressed. These treatment issues have also been described in guidelines from the ATA (303) and ETA (304).
Hypothyroidism
INITIAL DIAGNOSIS
The initial diagnosis of overt primary hypothyroidism is established by an increase in serum TSH with subnormal free T4; serum free T3 may remain normal except in severe cases. Subclinical hypothyroidism or mild thyroid failure is defined by persistent elevation of serum TSH, while free T4 remains normal (see above). The precise upper limit of the reference range for TSH is difficult to define, because of the approximately logarithmic distribution of this variable. The upper “tail” of normal TSH in the range 2-4 mU/l is thin and subjects with TSH concentrations in this range have an increased likelihood of future hypothyroidism, especially if thyroperoxidase antibodies are positive (280).
Serum TSH is the key to the distinction between primary and secondary hypothyroidism. A low serum free T4 in the absence of TSH elevation should always raise the possibility of a pituitary or hypothalamic abnormality, although this combination of findings is also frequent in a number of other situations (Table 8), especially during critical illness (190). It should be noted also that immunoreactive serum TSH is often detectable in secondary or central thyroid deficiency, a phenomenon that appears to result from dissociation between biological and immunological TSH activity (249). Hence, normal or even elevated serum TSH should not be interpreted as conclusive evidence againsthypopituitarism (305).
|
Table 8. Causes of Subnormal Free T4 without TSH Excess
|
|
Secondary or central hypothyroidism
Impaired biological activity of TSH
|
|
Critical illness
|
|
Falsely low free T4 estimate (method-dependent)
Dilution-dependent artefact (see below)
Effect of medications that compete for T4 binding
|
|
Impaired TSH response to hypothyroxinemia
Effect of severe illness
|
|
Medications e.g. dopamine, glucocorticoids
|
RESPONSE TO TREATMENT
A serum TSH value in the low-normal range between 0.5 and 1.5 mU/l, close to the geometric mean, is probably the best single indicator of appropriate thyroxine dosage during standard replacement therapy. In a study of ambulatory patients attending a thyroid clinic, hypothyroid patients taking T4 replacement seldom needed a serum free T4 measurement if the serum TSH was greater than 0.05 mU/L, although at lower TSH values, the magnitude of T4 excess did influence management (306). Numerous studies show that patients taking exogenous thyroxine show higher levels of serum total and free T4 for equivalent levels of serum TSH and T3 when compared with untreated euthyroid control subjects (307,308).
It should be noted that in some situations, for example patients with both ischemic heart disease and hypothyroidism, or in very old subjects, the appropriate dose of T4 should be influenced by clinical judgment as well as laboratory findings. The assessment of optimal levothyroxine dosage in patients with secondary or central hypothyroidism remains a challenge because serum TSH does not serve as a reliable marker of under-replacement. Serum TSH may in this context diminish during thyroxine replacement in hypopituitarism (309); while not of primary diagnostic value, the concentration may serve as an index of individual progressive response (186,310)). Since there is no readily available alternative variable of thyroid hormone action, it is generally appropriate to assess replacement clinically and on the basis of both serum T4 and T3. Measured levels of free and total T4 are influenced by the interval between tablet ingestion and blood sampling. In athyreotic subjects who took 0.15-0.2 mg T4 orally, the serum free and total T4 concentrations were increased by about 20% one to four hours later, with return to baseline about nine hours after T4 ingestion; serum TSH and T3 levels showed no time-dependent variation in relation to timing of thyroxine dosage (310).
In some situations, the regulation of T4 replacement is both unreliable, unpredictable and very variable. Excluding low patient compliance, this is often due to variable absorption of T4 from the gastrointestinal tract due to e.g. gastric or bowel disease (311), pernicious anemia with achlorhydria (312), a variety of over the counter medications such as antacids (313), laxatives (313), calcium (314), coffee (315), and many other substances. This untoward situation for the patient who is most often very negatively affected by the variable and irregular thyroid function can sometimes be alleviated by changing treatment formulation to a soluble form or soft gel capsules rather than tablets (312).
SUPPRESSIVE TREATMENT WITH T4
In line with recent guidelines for the stratification of risk in patients with differentiated thyroid cancer (316), there is no single TSH target during long-term postoperative thyroxine therapy (317,318). In high risk patients there may be benefit from adjusting T4 dosage to suppress TSH to undetectable levels (e.g. <0.03 mU/l). However, in many situations it may be appropriate to aim for a TSH target in the lower normal range in order to minimize adverse effects on bone and cardiovascular system. It is also worth noting that the initial ablative treatment of differentiated thyroid carcinoma tends to be less radical with classification into low and high-risk groups according to the guidelines. This may influence the long-term outcome which however remains to be seen in future long-term follow-up studies. When the aim of T4 suppressive therapy is regression of benign thyroid tissue, it may be adequate to give sufficient T4 to reduce serum TSH to 0.1 to 0.3 mU/L (319).
TREATMENT WITH T3
If currently available T3 formulations are used for replacement, it is difficult to monitor the effectiveness of treatment. Owing to its short plasma half-life, the serum concentration is highly dependent on the interval between dosage and sampling (320). There is also doubt as to whether TSH serves as an accurate index of thyroid hormone action during continuing T3 therapy, with the suggestion that doses of T3 required to normalize TSH could produce tissue hyperthyroidism (321). The difficulty of monitoring T3 replacement by currently available techniques is one of the arguments against the routine use of T3 or thyroid extract (321-323).
If the suggestion is confirmed that some genetically-identifiable individuals within the hypothyroid population are better served by combined T4-T3 therapy on the basis of polymorphism of type 2 deiodinase (324,325), or other variations in T4 transport and metabolism, the use of T4-T3 combination therapy will increase. That will require re-evaluation and refinement of monitoring strategies for combined replacement. The development of sustained-release T3 formulations will be advantageous, so that the interval between dosage and sampling can be minimized as a variable (326,327), and the treatment can thereby become more stable.
REPLACEMENT WITH UNCERTAIN INDICATION
The recent demonstration that fertility and pregnancy outcome may be adversely affected by minor degrees of thyroid dysfunction (see below) has led to the initiation of thyroxine treatment in women of reproductive age, who may lack conclusive criteria for lifelong therapy. It may later be questioned whether continuing replacement is required. Positive thyroperoxidase antibodies may be a basis for continuing therapy, but in the absence of this marker, measurement of serum TSH after 3-4 weeks withdrawal of treatment is likely to be definitive. The alternative of following the TSH response during partial replacement with T4, 50 ug daily, may be preferred. Similar approaches apply where no documented indication exists. Because of the difficulty in establishing the need for treatment during optimal replacement, it is an advantage for hypothyroid patients to retain a permanent record of pre-treatment documentation.
Assessment of Thyroid Function During Non-Thyroidal Illness (NTI)
Several distinct issues need to be considered when assessing thyroid function during critical illness. First, there is the possibility that an underlying thyroid abnormality might be missed, and second, prolonged severe illness per se may be associated with an abnormality of thyroid hormone secretion or action that may benefit from treatment (328,329). Third, some of the observed abnormalities will reflect the effect of medications or may be methodological artefacts.
It is difficult to rule out previously unrecognized or recent thyroid dysfunction by clinical assessment in critically ill patients; current laboratory tests often do little to resolve the problem. During severe illness, one or more of the assumptions that underpin the diagnostic use of the TSH-T4 relationship (see above) may not be valid. For example, acute fluctuations from the steady-state can lead to an anomalous T4-TSH relationship simply because of the marked difference in the plasma half-lives of T4 and TSH.
In general, the same sample assayed for TSH by various methods will give similar results during critical illness, while various estimates of free T4 may give widely divergent results (see below). When TSH and T4 changes are considered together, the abnormal results rarely correspond to standard criteria for diagnosis of primary hypothyroidism or hyperthyroidism. In contrast, persistent hypothyroxinemia without the corresponding anticipated rise in serum TSH is a common finding that suggests secondary or central hypothyroidism. These changes appear to be part of a broad neuroendocrine response that also involves the pituitary-adrenal axis, the pituitary-gonadal axis and the IGF binding proteins (329,330). Further study of these responses may eventually lead to therapy that could extend beyond thyroid hormone replacement, for example to substitution of hypothalamic releasing hormones (330).
The only reasonably robust method for distinguishing central hypothyroidism from thyroid function variables during critical illness is measurement of the T3 uptake test, which will be high (as opposite to low in central hypothyroidism) (331) and independent of the free T4 estimate which can be affected by a number of drugs used in intensive care patients (see below), and are anyway most often ‘false’ in the extreme situations (here low) where the automated methods for free thyroid hormones are unable to correct properly for binding protein abnormalities, or for abnormal binding to the binding proteins (331) (Figure 7).
EFFECTS OF MEDICATION
Interpretation of thyroid function tests during critical illness can be influenced by multiple medications (Table 3) (45), in particular dopaminergics and glucocorticoids, which inhibit TSH secretion, and a wide range of inhibitors of T4 and T3 binding to circulating thyroxine binding globulin (332). Thus, dopamine, dobutamine, glucocorticoids, octreotide, bexarotene, and metformin all inhibit pituitary TSH secretion, while an iodine load such as by using contrast agents, amiodarone or topical iodine preparations modifies hormone synthesis and release in a dual fashion. Lithium, glucocorticoids, and aminoglutethimide are inhibitors of both synthesis and release. Both amiodarone, glucocorticoids and beta-blockers, as well as hepatic contrast agents, inhibit T4-T3 5′ deiodination, and immune function modifications are seen by use of interleukin 1, interferon alpha, interferon beta and monoclonal antibody therapies.
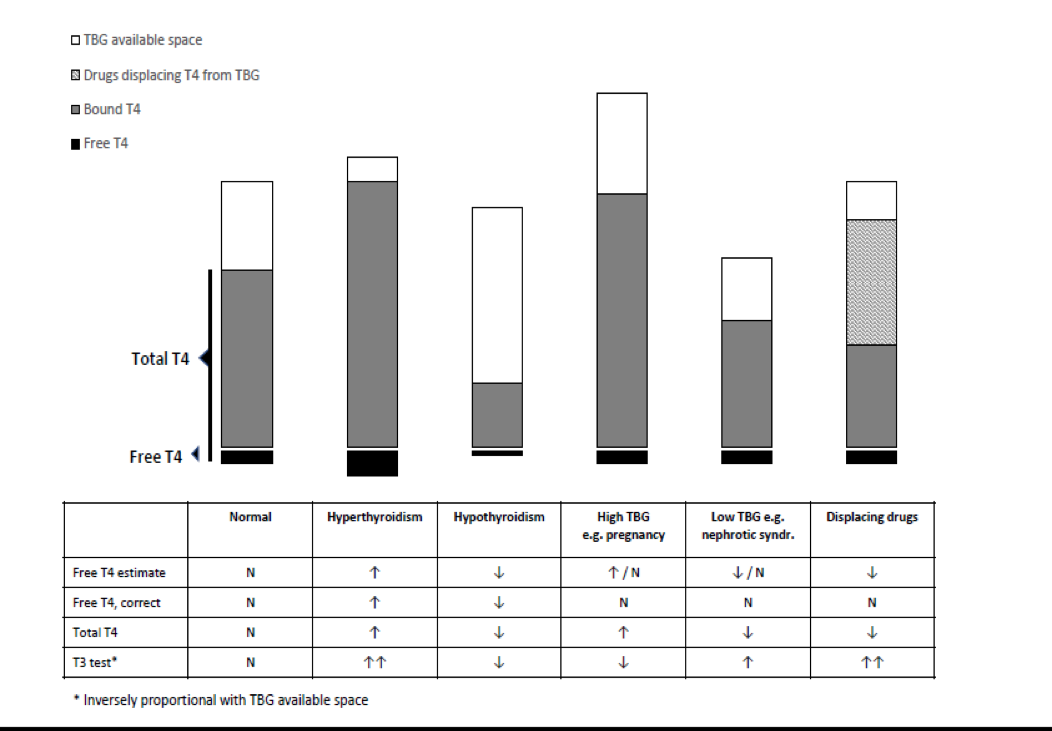
Figure 7. Plots of peripheral thyroid hormone measurements (here T4) in various situations of thyroid dysfunction, protein binding abnormalities and drugs displacing the thyroid hormone from the binding proteins. The degree of distortion of the free T4 estimate depends on the assay methods (measurement platforms) as well as the degree of binding protein abnormalities. T3-test is able to distinguish central hypothyroidism from the effects of critical illness and displacement of the hormone by medicaments.
A number of drugs modify the binding of T4 and T3 to plasma proteins. e.g. estrogen, heroin, methadone, clofibrate, 5-fluouracil, perphenazine, tamoxifen, raloxifene, 5-fluouracil, and perphenazine, increase the concentration of thyroid hormone binding globulin, while glucocorticoids, androgens and l-asparaginase decrease the concentration. A number of drugs may displace T4 and T3 from binding proteins or displace T4 from the tissue pool and others increase the clearance of T4 and T3. A major issue in the serum free T4 measurement is drug and other interference in T4 replacement when absorption of T4 is impaired (333,334). This can happen in cases with impaired gastric activity (311), and coffee intake (315), but also any pharmaceutical drugs (335), many of them sold over the counter (313).
METHODOLOGICAL DISCREPANCIES IN NON-THYROIDAL ILLNESS
The Heparin Artifact and Free T4
The effect of heparin to increase serum free T4 is an important in vitro phenomenon that can lead to spuriously high estimates of circulating free T4 (236). In the presence of a normal serum albumin concentration, non-esterified fatty acid concentrations >3 mmol/l are required to increase free T4 by displacement from thyroxine binding globulin, but these concentrations are uncommon in vivo. However, in samples from heparin-treated patients, serum non-esterified fatty acids may increase to these levels during in vitro sample storage or incubation as a result of heparin-induced lipase activity (236) (Figure 8). This effect is accentuated by incubation of serum at 37ºC and by increased serum triglyceride or low serum albumin concentrations, particularly if the sample is pre-diluted. If heparin is given in vivo and the sample is then incubated at 37ºC, doses of heparin as low as 10 units may result in non-esterified fatty acid-induced increases in the apparent concentration of serum free T4 (235,237,336). The assay result is analytically correct but does not reflect the in vivo concentration of free T4. Low molecular weight heparin preparations have a similar effect (336).
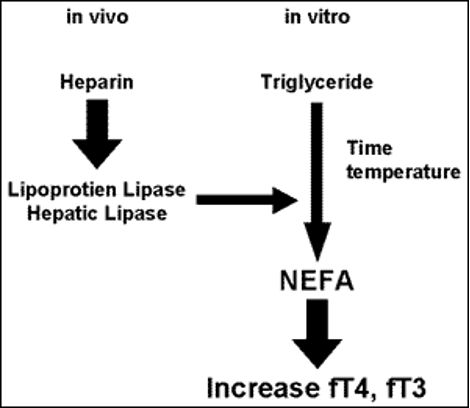
Figure 8. Heparin-induced release of lipase in vivo can lead to in vitro generation of non-esterified fatty acids during sample incubation or storage. An increase in serum non-esterified fatty acid (NEFA) concentrations to >3 mmol/l is sufficient to displace T4 from thyroxine binding globulin, but such values are uncommon in vivo. This artefact is accentuated by high triglyceride or by low albumin concentrations.
Competitors for Plasma Protein Binding
The accuracy of virtually all methods of free T4 estimation is compromised by medications that displace T4 and T3 from thyroxine binding globulin. Current methods tend to underestimate the concentration of free T4 in the presence of binding competitors because of dilution-related artefacts. Binding competitors are usually less protein-bound than T4 itself so that progressive sample dilution leads to a fall in the free concentration of competitor before the free T4 concentration alters (235). (For a hormone such as T4, with a free fraction in serum of about 1:4000, progressive dissociation will sustain the free T4 concentration up to at least 1:100 dilution. In contrast, 1:10 dilution of serum will result in a marked decrease in the free concentration of a drug that is 98% bound, i.e. has a free fraction in serum of 1:50). Because displacement depends on the relative free concentrations of primary ligand and competitor, the underestimate of free T4 will be greatest in assays with the highest sample dilution. This important dilution-dependent difference between various free T4 methods was shown by the relative ability of three commercial free T4 assays to detect the T4-displacing effect of therapeutic concentrations of furosemide (Figure 9) (337).
Similarly, therapeutic concentrations of phenytoin and carbamazepine increase the free concentration of T4 by 40-50% using ultrafiltration of serum that had not been diluted, while the free hormone estimate was spuriously low using a commercial single-step free T4 assay after 1:5 serum dilution (245).
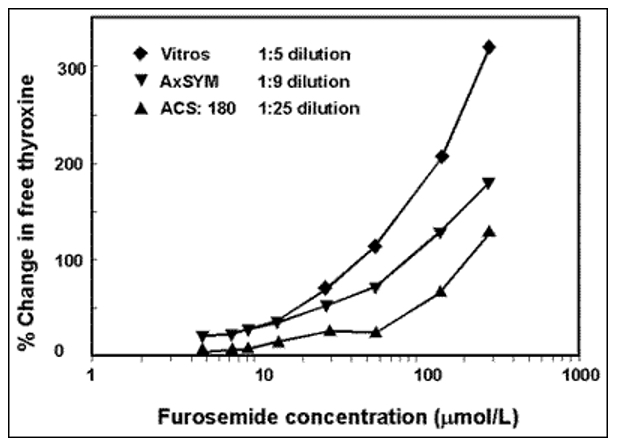
Figure 9. Influence of increasing serum concentrations of added furosemide on estimates of serum free T4 using three commercial free T4 methods that involve varying degrees of sample dilution. The effect of the competitor is progressively obscured with increasing sample dilution (redrawn from (337)).
It is possible that methodologic artefacts have influenced previous descriptions of free T4 changes during critical illness. On the one hand, an apparent increase in free T4 may arise from heparin-induced in vitro generation of free fatty acids during sample incubation (236). On the other hand, estimates of free T4 may be spuriously low in assays that use diluted serum (235,245).
Divergent Estimates of Free T4
That estimates of free T4 may show opposite discrepancies by different methods was shown by Sapin et al. (338) in a prospective study of bone marrow transplant recipients. Twenty previously euthyroid subjects were studied on the seventh day after bone marrow transplantation using six commercial free T4 kits, during multiple drug therapy, including heparin and glucocorticoids (Figure 10). Free T4 methods that involved sample incubation at 37ºC showed supranormal free T4 values in 20-40% of these subjects (see heparin effect above), while analog tracer methods that are influenced by tracer binding to albumin gave subnormal estimates of free T4 in 20-30%. By contrast, total T4 was normal in 19 of these 20 subjects. Serum TSH was <0.1 mU/l in half the subjects, independent of the method that was used. Thus, there was the possibility that an erroneous diagnosis of either hyperthyroidism or secondary or central hypothyroidism could be considered, solely as a result of variations in free T4 methodology.
Figure 10. Free T4 estimated by six different kit methods in 20 previously euthyroid patients on the seventh day after bone marrow transplantation. There was a high proportion of abnormal values, either increased or decreased, depending on the type of free T4 method used (see text). Therapy included heparin and glucocorticoids at the time of sampling. The mean for each method has been normalized to 100%, with the limits of the range shown by the box. Serum total T4 remained normal in 19 of the 20 study subjects, while serum TSH was subnormal in 11, independent of assay method (redrawn from (338))
SERUM TSH IN CRITICAL ILLNESS
Serum TSH assessment in severe non-thyroidal illness depends on the sensitivity of the particular method. The “third generation” assays with a functional sensitivity below 0.01 mU/L are generally sufficiently sensitive to distinguish the very low values in most thyrotoxic patients from the subnormal but somewhat higher TSH levels of non-thyroidal illness (59,339). Among a group of patients with low serum TSH values (<0.1 mU/L), almost all thyrotoxic patients had values less than 0.01 mU/L, when assessed with a highly sensitive assay, whereas most critically ill euthyroid patients had values between 0.01 and 0.1 mU/L (59). However, in another study, about 4% of patients with non-thyroidal illness had values below the functional sensitivity of a “third generation” assay, indicating that an absolute distinction cannot be made on the basis of TSH alone (339).
Differentiated Thyroid Cancer
There is now consensus that not all patients with differentiated thyroid cancer require T4 treatment in doses that achieve complete TSH suppression. Thus, based on a general assessment of risk (316,317,340), a TSH target should be determined as a guide to T4 dosage. In the follow-up of differentiated thyroid cancer the interpretation of TSH and serum Tg is inter-dependent. For studies done after initial near-total thyroidectomy, following withdrawal or temporary reduction of suppressive therapy (341) (or with the use of recombinant human TSH (342), serum TSH levels in excess of 30 mU/l appear to achieve adequate stimulation of potential Tg production (343). The failure of endogenous TSH to increase into this range suggests too short a period of T4 withdrawal, a compliance problem, or the presence of a substantial amount of active thyroid tissue.
Serum Tg measurement and whole body radioiodine scanning have generally been used in a complementary fashion, but there is now good evidence that current assays for Tg have greater sensitivity than follow-up whole body scanning with 2mCi 131I (316). There has been a clear trend to place greater emphasis on measurements of Tg and to move away from repeated low dose diagnostic whole-body radioiodine scanning, a procedure that has limited sensitivity (344,345). Management of low risk patients now tends to be based on assessment of serum Tg after recombinant TSH stimulation, without the need for diagnostic scanning (345). However, based on reported data the current fashion of obviating RAI based on a negative post-operative US and low serum Tg value is not yet supported by the literature and should be investigated using well designed prospective studies with clear-cut endpoints (346). In the opinion of these authors, the evolution of thyroid cancer management cannot pass through a substitution of RAI by serum Tg measurement and neck US but by an appropriate use of radioiodine theranostics. This will also need to develop basic and clinical research programs that bring together physicians from various specialties.
Undetectable serum Tg in the years after ablative treatment has been shown to be a reliable index in ruling out persistent or recurrent disease that requires further evaluation and treatment. Detectable serum Tg, together with detailed ultrasonography for localization has become the mainstay of further investigation (347). There has also been a tendency to deescalate serum Tg measurements by using only unstimulated sensitive serum Tg measurement and not rTSH stimulated assessments (348). Based on ATA’s most recent 2015 guidelines (316), Sunny et al. (349)performed a systematic study of 650 patients followed for papillary thyroid carcinoma who had total thyroidectomy performed and compared the evolution of stimulated and unstimulated serum Tg concentrations. The concentrations were corroborated with tumor burden as determined by additional clinical, ultrasonography neck, and whole-body scintigraphy. The study highlighted the superiority of sSTg over uSTg in the follow-up of papillary thyroid cancerpatients. Follow-up with uSTg alone may result in underestimating the tumor burden.
It should not be forgotten, that serum Tg can only be reliably measured in TgAb negative samples, even if measured by high sensitive Tg assays (268,269) so they need to be assessed each time a serum Tg is measured, even if the patient has previously been negative for the antibodies (268,269,350). It also should not be forgotten that measurement of serum TgAb can themselves be used as surrogate markers of relapse/metastases from differentiated thyroid carcinoma (274-276,351).
Assay of thyroglobulin on needle washes of suspect lymph nodes has a high degree of sensitivity and specificity, apparently superior to cytological examination (271,272). During long-term follow-up, it is crucial that clinicians are kept informed of changes in serum thyroglobulin methodology that may give a false impression of remission or recurrence (see above).
Psychiatric Illness
An unusual variety of euthyroid hyperthyroxinemia occurs in some patients hospitalized with acute psychiatric illness (195). Serum T4 is increased, but serum T3 is less frequently elevated; serum TSH is generally normal or slightly high (352). These abnormalities, presumed to be due to central activation of the hypothalamic-pituitary-thyroid axis, often resolve in several weeks (195).
Assessment of Thyroid Function Before, During and After Pregnancy
Recent advances in the understanding of the importance of optimal maternal thyroid function for fetal brain development in early pregnancy, as well as the influence of thyroid immunity, or thyroid status, on numerous pregnancy outcomes, has greatly increased the frequency of thyroid testing before, during, and after pregnancy. Several guidelines have been published from The Endocrine Society (353), American Thyroid Association (8) – and for subclinical hypothyroidism from European Thyroid Association (9). Projecting this to fertility issues even complicate matters further, and no consensus can currently be obtained (354). There is thus also still controversy regarding whether to treat subtle abnormalities of thyroid dysfunction in the infertile female patient. This guideline document reviews the risks and benefits of treating subclinical hypothyroidism in female patients with a history of infertility andmiscarriage, as well as obstetrical and neonatal outcomes in this population.
Clinical guidelines from The Endocrine Society (353) suggest consensus, but significant controversies remain (355). Evidence exists that obstetricians struggle with the diagnosis and treatment of hypothyroidism. According to recent surveys, the management of hypothyroidism during pregnancy is the number 1 endocrine topic of interest for obstetricians. A synopsis of recently published subspecialty guidelines is timely but has not really been done yet. The reproductive effects of abnormal thyroid function and its immune associations are considered in other Endotext chapters. This section will consider the issues from the point of view of testing strategy and assay methodology. From studies of pregnancy outcome, some evidence has become persuasive that miscarriage rate and prematurity (6,356-358) are favorably influenced by maternal thyroid hormone replacement, even when hypothyroidism would be regarded as “subclinical” by standard criteria. Some clinicians advocate early and liberal screening and treatment (356,358), but a recent Cochrane review, however, did not find any clear benefit for universal screening rather than case finding (6).They found based on the existing evidence, that though universal screening for thyroid dysfunction in pregnancy increases the number of women diagnosed with hypothyroidism who can be subsequently treated, it does not clearly impact (benefit or harm) maternal and infant outcomes. While universal screening versus case finding for thyroid dysfunction increased diagnosis and subsequent treatment, they found no clear differences for the primary outcomes: pre-eclampsia or preterm birth. No clear differences were seen for secondary outcomes, including miscarriage and fetal or neonatal death; data were lacking for the primary outcome: neurosensory disability for the infant as a child, and for many secondary outcomes. Though universal screening versus no screening for hypothyroidism similarly increased diagnosis and subsequent treatment, no clear difference was seen for the primary outcome: neurosensory disability for the infant as a child (IQ <85 at three years); data were lacking for the other primary outcomes: pre-eclampsia and preterm birth, and for the majority of secondary outcomes. For outcomes assessed using the GRADE approach the evidence was considered to be moderate or high quality, with any downgrading of the evidence based on the presence of wide confidence intervals crossing the line of no effect. More evidence is needed to assess the benefits or harms of different screening methods for thyroid dysfunction in pregnancy, on maternal, infant and child health outcomes. Future trials should assess impacts on use of health services and costs and be adequately powered to evaluate the effects on short- and long-term outcomes (6).
A key consideration is therefore still whether thyroid function should be universally tested (357) and if so whether it should be done before or early in pregnancy, in view of the fact that women at risk of adverse outcome cannot be identified reliably from their clinical history, even if this is assessed in full detail (359). A recent review (358) has concluded that current guidelines agree that overt hyperthyroidism and hypothyroidism need to be promptly treated and that as potential benefits outweigh potential harm, subclinical hypothyroidism also requires substitutive treatment. The chance that women with thyroid autoimmunity may benefit from levothyroxine treatment to improve obstetric outcome is intriguing, but adequately powered randomized controlled trials are needed. The issue of universal thyroid screening at the beginning of pregnancy is still a matter of debate, and aggressive case-finding is supported. Careful discussion between the physician and patient concerning benefits on the one hand and unnecessary disease burden should be carried out (359).
METHODOLOGICAL ISSUES
Some methodological issues are relevant before the application of tests is considered (360). There has been almost universal acceptance of free T4 estimates in preference to total T4 measurement in pregnancy because of the well-known estrogen effect to increase thyroxine binding globulin and hence total T4. It is clear that normal pregnancy is associated with a marked increase of about 40% in total T4 concentration, but free T4 is maintained close to normal non-pregnant levels (360). Previous suggestions that free T4 actually declines significantly in late pregnancy (361) are uncertain because of wide method-dependent variations, with strong negative bias in some methods (362-364). In a comparative study of free T4 by seven commercial methods in 23 euthyroid women at term, Roti et al. found that albumin-dependent methods show marked negative bias, with up to 50% subnormal values, while other methods gave values above their non-pregnant reference interval (362). Notably, particularly during pregnancy, methods of free T4 estimation fall far short of desirable assay standardization criteria that aim to minimize between-assay variation (171).
A study by Lee et al. confirmed marked negative bias in free T4 estimates during pregnancy and questioned the basis for continuing to rely on free thyroxine estimates during pregnancy (365). In contrast to two kit free T4 methods, totalserum T4 and its derivative, the free T4 index, showed the anticipated inverse relationship with serum TSH, with historically consistent results in numerous reports (365). Thus, because of consistency between methods, total T4 measurement may be superior to free T4 estimation as a guide to therapy during pregnancy, provided that reference values take account of the normal estrogen-induced increase in thyroxine binding globulin during pregnancy (360,366).
Serum TSH is generally analytically reliable during pregnancy, but the reference interval is lower at the end of the first trimester, associated with the effect of human choriogonadotropin as a surrogate thyroid stimulator (360). Hence, the general advice that tests of thyroid function during pregnancy should relate to trimester-specific reference intervals (360,367). In the case of free T4, ranges also need to be method-specific (362-365,367). The magnitude of method-dependent variations between non-pregnant and third trimester free T4 estimates is shown in Figure 11.
Figure 11. Free T4 estimates in the third trimester of pregnancy show large method-dependent differences from the reference intervals for non-pregnant subjects. Redrawn from ref. (363) and (364) which identify each method studied. The left panel shows values measured by equilibrium dialysis (ED). The solid bars indicate third trimester free T4 estimates, the grey bars, non-pregnant results.
It has recently been demonstrated that mass spectrometry measurement of free thyroid hormones may alleviate the problems of flaws in the usual free thyroid hormone measurements in pregnancy (368), although these results need to be confirmed on the one hand, and a solution to the must higher costs of this type of methodology has to be solved on the other hand. Until then a pragmatic approach is to use total thyroid hormone measurements multiplied by 1.5 as useful reference interval during pregnancy.
There is also during pregnancy, as in the non-pregnant state, a very high individuality index i.e. the intraindividual variability of pregnant women is much higher than the interindividual population- and even trimester-based reference range (369), which further complicates thyroid function assessment in cross sectional measurements. The function variables become much more clinically significant when measurements are used in longitudinal assessments e.g. during pregnancy.
BEFORE PREGNANCY OR WHEN PREGNANCY IS CONFIRMED
Because of the adverse effects of maternal thyroid deficiency, there is agreement that pre-pregnancy testing is now mandatory to optimize thyroxine replacement and to assess thyroid status in women who have an increased risk of thyroid dysfunction (8,9,353,370-372), for example a history of recurrent miscarriage, impaired fertility requiring assisted reproductive technology, type 1 diabetes, or any other factors known to be associated with increased risk of thyroid dysfunction (Table 2). Notably, in one key study, about a third of women with subclinical or overt hypothyroidism in the first trimester would have been missed if testing had been confined to those assessed as being at higher risk, based on detailed questioning about potential risk factors, in particular a personal or family history of thyroid or other autoimmune disorder, or previous thyroid-related treatment (373). A further study confirms that about half of the women at risk for pregnancy-related thyroid dysfunction would be missed by testing confined to the group assessed to be at high risk (374). Hence, the view that routine testing may now be warranted (14). Because the major influence of thyroid hormone on fetal brain development is early in pregnancy and the onset of thyroid hormone action is slow, effective testing needs to be done earlier than would follow the first routine obstetric visit at 10-12 weeks.
While there is now good evidence that any level of TSH elevation that suggests maternal hypothyroidism should be treated, there is currently no consensus about responses to other test abnormalities. An important finding is that thyroxine treatment of peroxidase antibody-positive euthyroid women from about the end of the first trimester may improve obstetrical outcome, as judged by rates of miscarriage and premature delivery (375). A recent meta-analysis confirms that thyroid autoantibodies are associated with miscarriage and preterm delivery and suggests that thyroxine treatment diminishes these risks (376). Pre-partum assessment of antibody status may also provide information that improves prediction of post-partum thyroid dysfunction (377).
A different aspect is related to women already on thyroxine replacement therapy before pregnancy. It has been shown that almost 50% of thyroxine treated women were not optimally replaced in early pregnancy (TSH <0.4 or >4.0 mU/l) with the potential for significant adverse effect on pregnancy outcome (370). In thyroxine-treated women it should now be standard practice to assess optimal replacement prior to pregnancy and to re-evaluate this when pregnancy is confirmed, with a prompt 25-30% increase in replacement dosage (371), with two extra daily doses per week as an approximation. The required increase is larger in women who have had thyroid ablation than in those with spontaneous hypothyroidism (371).
A special situation arises in thyroxine-treated women who have in vitro fertilization. Ovulation-induction cycles, even in the absence of ongoing pregnancy, can be associated with 2-10 fold increases in serum TSH (378), indicating that previously adequate replacement may not accommodate estrogen-induced thyroxine requirements induced by a very rapid increase of thyroid hormone binding proteins. This suggests that women who lack endogenous thyroxine should receive increased dosage from the time of ovulation induction, to accommodate increased thyroxine requirement, whether associated with a subsequent pregnancy, or not. The need to routinely test thyroid function before in vitro fertilization and institute or increase thyroxine treatment seems self-evident.
ASSESSMENT DURING PREGNANCY
In women treated for hypothyroidism, some guidelines still recommend that replacement thyroxine dosage should be progressively adjusted in pregnancy in response to increasing serum TSH, a response that is likely to be too late to assure optimal first trimester maternal T4 levels for optimal fetal brain development. Hence, the advice to increase dosage by two daily doses per week as soon as pregnancy is confirmed, followed by a check of serum TSH about 4 weeks later (353,371).
In women treated for Graves’ disease with antithyroid drug, dose reduction should be the aim, keeping TSH in the lower normal range. In contrast to the adverse effects of even the mildest grades of hypothyroidism, an extensive study has shown no adverse effect of mild overactivity during pregnancy (379). It is thus often possible to cease antithyroid drug late in pregnancy, with prospective follow-up to detect post-partum recurrence. In contrast to immune thyroid disorders, nodular thyroid disorders do not appear to show marked fluctuation during and after pregnancy. Recent guidelines recommend the use of propyluracil in the first trimester (380), and thiamazole (methimazole) thereafter due to the risk of birth defects by thiamazole (380,381), and general side effects to propylthiouracil (382,383).
In the event of uncontrolled or severe maternal hyperthyroidism, careful assessment should be made of fetal growth and thyroid size followed by fetal blood sampling in some cases as a guide to optimal therapy (384,385). Guidelines for the assessment of TRAb to gauge the likelihood of neonatal thyrotoxicosis have been discussed above (283).
POSTPARTUM ASSESSMENT
Among women who were euthyroid prior to pregnancy, autoimmune thyroid dysfunction not due to Graves' disease occurs in approximately 5–8% in the first year post-partum (386). Postpartum thyroiditis is an autoimmune disorder that causes lymphocytic inflammation of the thyroid. It is more frequent in women with elevated first-trimester serum thyroperoxidase antibody concentrations (387) and in women with other autoimmune disorders, such as type 1 diabetes mellitus (388,389).
The thyroid inflammation in postpartum thyroiditis initially leads to transient thyrotoxicosis as preformed thyroid hormones are released from the damaged gland. This phase usually occurs 1–3 months post-partum and lasts for 6–9 weeks. Symptoms are typically mild, but rarely florid, transient thyrotoxicosis can be seen. Serum T4 levels are proportionally higher than T3 levels, reflecting the ratio of stored hormone in the thyroid gland (in contrast to Graves' disease where T3 is often preferentially elevated) (390).
In prospective studies there is a high prevalence of thyroid dysfunction during the post-partum period (8,353), and a high percentage of those women with subclinical postpartum thyroiditis proceeded to permanent thyroid failure (391,392). In an Australian study, this effect was most pronounced in women with low iodine intake (393). Hence, women with postpartum hypothyroidism should be treated on the basis of confirmed TSH elevation even if the indication for lifelong replacement has not been firmly established. This strategy is especially important because untreated maternal hypothyroidism has critical implications for fetal development in subsequent pregnancies. Also, recent findings of psychiatric disorders coinciding with autoimmune thyroid disorders in the postpartum period (394)indicates that these women should be screened for thyroid dysfunction, and very likely both conditions should be followed on a longer term basis if significant in the postpartum period. By contrast, almost all women with suppressed or subnormal TSH values 6 months postpartum, showed normal TSH 12 and 18 months later, indicating that postpartum hyperthyroidism is likely to be a temporary phenomenon (8,353).
In women with established hypothyroidism, replacement dosage of thyroxine can generally be decreased to the pre-pregnancy dose with testing deferred for several months to review dosage. In Graves’ hyperthyroidism, where antithyroid drug has been ceased during pregnancy, resumption of treatment is generally deferred, based on symptoms and the results of testing at two-three monthly intervals. Where thyroperoxidase antibody testing early in pregnancy has been positive in the absence of frank thyroid dysfunction, prospective testing may be warranted in the 12 months post-partum.
Knowledge of maternal thyroid status should be taken into account in assessing the significance of neonatal screening for congenital hypothyroidism, because of reports of temporary effects of maternal blocking antibody in the infant.
IODINE STATUS
The critical importance of adequate iodine nutrition during pregnancy is widely acknowledged. The increased pool of thyroxine that follows from the increase in serum thyroxine binding globulin, as well as increase in urinary iodine excretion that will aggravate any degree of iodine deficiency, with potential for adverse effects on fetal development. While assessment of iodine status by measurement of urinary iodine excretion in individual women does not have wide support, data on iodine nutrition within each geographical area should be an important public health issue, with appropriate supplementation to achieve estimated urinary iodine excretion in the range 150-300 microgram daily in pregnant women.
THE PHYSICIAN-LABORATORY INTERFACE
Because of the diverse clinical presentations of thyroid dysfunction, initial requests for assessment of thyroid function are often made by clinicians who, while alert to the possibility of thyroid dysfunction, may not be familiar with the limitations of current assays, or with medication effects. Clinical decisions can be assisted by comments from the laboratory, based on detailed knowledge of current immunoassay limitations (263,395-398). The quality of this assistance will depend on two key components: the training and experience of the reporter and the available clinical information. It is therefore paramount that clinicians and clinical biochemistry specialist communicate, since a practical and useful approach to optimal patient management can only be achieved by collaboration (178).
Historical
The competitive binding assays that are used for thyroid diagnosis were initially developed 30-35 years ago by clinical investigators who used ‘in house’ assay reagents and were often closely involved in patient care. This nexus between laboratory and clinical investigation allowed deficiencies in early assays to be readily appreciated, but advanced diagnostic technology was available to only a few practitioners, with results sometimes available only after long delay. In recent decades there has been a strong trend away from ‘in house’ reagents which have been replaced by kits that incorporate highly sophisticated standardized reagents and automated instrumentation (e.g. solid phase antibodies, magnetic separation systems, chemiluminescent detection systems). Assay turnaround is much faster and up-to-date techniques are widely available to almost all practitioners, most of whom are inexperienced in endocrinology or laboratory methodology. Non-specialist users of endocrine assays are most likely to benefit from laboratory assistance in the interpretation of results, but as assay automation has increased, laboratory professionals have become more distant from the bedside. As clinicians receive less assistance, they tend to provide less relevant information and vice versa. Laboratory personnel in turn, see results that are uninterpretable or ambiguous without the relevant clinical background. Potential assay imperfections may be ignored simply because clinical correlation is not possible (see below).
Diagnostic Approach to Discordant or Apparently Anomalous Results
The relationship between laboratory results and clinical findings may be either concordant or discordant. With discordant results, a distinction needs to be made between a previously unsuspected diagnosis, subclinical disease, and anomalous assay results. If the assay result is confirmed, the fundamental assumptions that underpin the diagnostic use of tropic-target hormone relationships should be considered (see above).
The following steps may be helpful in evaluating anomalous assay results:
- Re-evaluation of the clinical context, with particular attention to pre-diagnostic probability, long-term features suggestive of thyroid disease and medication history.
- Assessment of whether serum TSH is markedly suppressed (<0.05 mU/l) or simply in the subnormal-detectable range.
- Further sampling to establish whether the anomalous result is persistent or transient.
- Estimation of serum free T4 by an alternative method, preferably a two-step technique that removes binding proteins from the assay system before quantitation.
- If hyperthyroidism is suspected, measurement of the serum free T3, or total T3 with appropriate binding correction.
- Measurement of serum total T4 to establish whether the serum free T4 is disproportionately high or low, possibly due to a preanalytical or method-dependent artefact.
- Possible evaluation of propositus and family members for evidence of unusual binding abnormalities or hormone resistance.
In some circumstances, the cause of an apparently anomalous or unusual laboratory result may become obvious from the clinical context (table 10). By taking account of drug therapy, or acute perturbation of the pituitary-thyroid axis, assay validity can be affirmed and unnecessary further investigation may be avoided.
|
Table 10. Situations in Which Interpretation of Unusual Laboratory Results Depends on Relevant Clinical Information
|
|
Clinical background
|
Assay Results
|
|
fT4
|
fT3
|
TSH
|
|
Pregnancy
|
L*
|
L,N
|
N
|
|
Antithyroid drug treatment, initial months
|
H,N,L
|
H,N,L
|
U
|
|
Recent T4 commencement for hypothyroidism
|
N
|
N
|
H
|
|
Hypothyroidism, appropriate T4 dose
|
H
|
N
|
N
|
|
Hypothyroidism, intermittent compliance
|
H,N
|
|
H
|
|
Appropriate T4 suppressive therapy
|
H
|
N
|
U,L
|
|
Recombinant TSH, suppressive T4
|
H
|
N
|
H
|
|
Hypopituitarism
|
L
|
|
L,N
|
|
Phenytoin
|
L*
|
|
L,N
|
|
Critical illness
|
L*
|
L
|
L
|
|
Heparin effect in critical illness
|
L,N,H*
|
L
|
L
|
|
Recovery phase of critical illness
|
L,N
|
|
H
|
|
Drugs that inhibit T4/T3 binding to TBG
|
L*
|
L*
|
L,N
|
|
Amiodarone effect in euthyroid subject
|
H
|
L
|
N
|
|
Acute T4 overdose
|
HH
|
H,N
|
N
|
|
Rheumatoid arthritis
|
N
|
N
|
H
|
U undetectable; L low; N normal; H high; * effect dependent on assay method
Clinical Feedback, Quality Assurance, and Cost Effectiveness
Clinical feedback will remain a key aspect of quality assurance in laboratory testing. While assay precision or reproducibility can be evaluated solely in the laboratory, diagnostic accuracy requires clinical correlation. As with any diagnostic test, the non-specificity of a procedure may not become apparent until the full diversity of the non-diseased population is appreciated. Premarketing evaluation of assays may fail to include the critical samples that probe the diagnostic accuracy of an assay, so that non-specific interference is not appreciated until procedures have been in use for some time.
In studies of “cost effectiveness” it is hard to evaluate the human and financial costs that result from needless duplication, unnecessary testing and inappropriate management. Diagnostic inaccuracy of immunoassays may account for substantial unnecessary expenditure on laboratory resources (263,397,398). Dilution effects and binding protein abnormalities that affect free T4 estimation, the multiple effects of medications in non-thyroidal illness and the problems associated with thyroglobulin autoantibodies will continue to challenge both clinical chemists and clinicians; further technological development will not substitute for collaboration between these two groups.
Clinical issues may influence the selection of thyroid tests that will best serve a particular population. For example, the effects of severe illness on free T4 estimates may be of minor importance for a laboratory that serves predominantly ambulatory patients. A different assay profile, with emphasis on measurement of total hormone as the “gold standard” for T4 assessment may be required in a laboratory that evaluates thyroid function during critical illness (328-330) or in obstetric practice (8,353). In the future, hopefully a more sophisticated methodology such as tandem mass spectrometry may be able to eliminate the many confounders in thyroid hormone measurements.
ACKNOWLEDGEMENTS
The chapter was updated from a previous comprehensive version by the late Jim Stockigt M.D., FRACP, FRCPA, Monash University and Alfred and Epworth Hospitals, Melbourne, Australia.
REFERENCES
- Clinical guideline, part 1. Screening for thyroid disease. American College of Physicians. Annals of internal medicine 1998; 129:141-143
- Helfand M, Redfern CC. Clinical guideline, part 2. Screening for thyroid disease: an update. American College of Physicians. Annals of internal medicine 1998; 129:144-158
- Danese MD, Powe NR, Sawin CT, Ladenson PW. Screening for mild thyroid failure at the periodic health examination: a decision and cost-effectiveness analysis. Jama 1996; 276:285-292
- Casey B, de Veciana M. Thyroid screening in pregnancy. American journal of obstetrics and gynecology 2014; 211:351-353.e351
- Velasco I, Taylor P. Identifying and treating subclinical thyroid dysfunction in pregnancy: emerging controversies. European journal of endocrinology 2018; 178:D1-d12
- Spencer L, Bubner T, Bain E, Middleton P. Screening and subsequent management for thyroid dysfunction pre-pregnancy and during pregnancy for improving maternal and infant health. The Cochrane database of systematic reviews 2015:Cd011263
- Stagnaro-Green A. Clinical guidelines: Thyroid and pregnancy - time for universal screening? Nature reviews Endocrinology 2017; 13:192-194
- Alexander EK, Pearce EN, Brent GA, Brown RS, Chen H, Dosiou C, Grobman WA, Laurberg P, Lazarus JH, Mandel SJ, Peeters RP, Sullivan S. 2017 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease During Pregnancy and the Postpartum. Thyroid 2017; 27:315-389
- Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaidya B. 2014 European thyroid association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. European thyroid journal 2014; 3:76-94
- LaFranchi S. Congenital hypothyroidism: etiologies, diagnosis, and management. Thyroid 1999; 9:735-740
- Delange F. Neonatal screening for congenital hypothyroidism: results and perspectives. Hormone research 1997; 48:51-61
- Bhatara V, Sankar R, Unutzer J, Peabody J. A review of the case for neonatal thyrotropin screening in developing countries: the example of India. Thyroid 2002; 12:591-598
- Gupta R. Nobody's children. The lancet Diabetes & endocrinology 2015; 3:486
- Alexander EK. Here's to you, baby! A step forward in support of universal screening of thyroid function during pregnancy. The Journal of clinical endocrinology and metabolism 2010; 95:1575-1577
- Selmer C, Faber J. Mild Thyroid Dysfunction: A Potential Target in Prevention of Atrial Fibrillation? Circulation 2017; 136:2117-2118
- Bliddal S, Nielsen CH, Feldt-Rasmussen U. Recent advances in understanding autoimmune thyroid disease: the tallest tree in the forest of polyautoimmunity. F1000Research 2017; 6:1776
- Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid 2002; 12:977-988
- Wiebolt J, Achterbergh R, den Boer A, van der Leij S, Marsch E, Suelmann B, de Vries R, van Haeften TW. Clustering of additional autoimmunity behaves differently in Hashimoto's patients compared with Graves' patients. European journal of endocrinology 2011; 164:789-794
- Schaub RL, Hale DE, Rose SR, Leach RJ, Cody JD. The spectrum of thyroid abnormalities in individuals with 18q deletions. The Journal of clinical endocrinology and metabolism 2005; 90:2259-2263
- Kaptein EM. Thyroid hormone metabolism and thyroid diseases in chronic renal failure. Endocrine reviews 1996; 17:45-63
- Stagi S, Bindi G, Neri AS, Lapi E, Losi S, Jenuso R, Salti R, Chiarelli F. Thyroid function and morphology in patients affected by Williams syndrome. Clinical endocrinology 2005; 63:456-460
- Faggiano A, Pisani A, Milone F, Gaccione M, Filippella M, Santoro A, Vallone G, Tortora F, Sabbatini M, Spinelli L, Lombardi G, Cianciaruso B, Colao A. Endocrine dysfunction in patients with Fabry disease. The Journal of clinical endocrinology and metabolism 2006; 91:4319-4325
- Sinard RJ, Tobin EJ, Mazzaferri EL, Hodgson SE, Young DC, Kunz AL, Malhotra PS, Fritz MA, Schuller DE. Hypothyroidism after treatment for nonthyroid head and neck cancer. Archives of otolaryngology--head & neck surgery 2000; 126:652-657
- Sklar C, Whitton J, Mertens A, Stovall M, Green D, Marina N, Greffe B, Wolden S, Robison L. Abnormalities of the thyroid in survivors of Hodgkin's disease: data from the Childhood Cancer Survivor Study. The Journal of clinical endocrinology and metabolism 2000; 85:3227-3232
- Schmiegelow M, Feldt-Rasmussen U, Rasmussen AK, Poulsen HS, Muller J. A population-based study of thyroid function after radiotherapy and chemotherapy for a childhood brain tumor. JClinEndocrinolMetab 2003; 88:136-140
- Colao A, Pivonello R, Faggiano A, Filippella M, Ferone D, Di Somma C, Cerbone G, Marzullo P, Fenzi G, Lombardi G. Increased prevalence of thyroid autoimmunity in patients successfully treated for Cushing's disease. Clinical endocrinology 2000; 53:13-19
- Niepomniszcze H, Pitoia F, Katz SB, Chervin R, Bruno OD. Primary thyroid disorders in endogenous Cushing's syndrome. European journal of endocrinology 2002; 147:305-311
- Erickson AR, Enzenauer RJ, Nordstrom DM, Merenich JA. The prevalence of hypothyroidism in gout. The American journal of medicine 1994; 97:231-234
- Eheman CR, Garbe P, Tuttle RM. Autoimmune thyroid disease associated with environmental thyroidal irradiation. Thyroid 2003; 13:453-464
- Agate L, Mariotti S, Elisei R, Mossa P, Pacini F, Molinaro E, Grasso L, Masserini L, Mokhort T, Vorontsova T, Arynchyn A, Tronko MD, Tsyb A, Feldt-Rasmussen U, Juul A, Pinchera A. Thyroid autoantibodies and thyroid function in subjects exposed to Chernobyl fallout during childhood: evidence for a transient radiation-induced elevation of serum thyroid antibodies without an increase in thyroid autoimmune disease. The Journal of clinical endocrinology and metabolism 2008; 93:2729-2736
- Zervas A, Katopodi A, Protonotariou A, Livadas S, Karagiorga M, Politis C, Tolis G. Assessment of thyroid function in two hundred patients with beta-thalassemia major. Thyroid 2002; 12:151-154
- Chu JW, Kao PN, Faul JL, Doyle RL. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest 2002; 122:1668-1673
- Janssen OE, Mehlmauer N, Hahn S, Offner AH, Gartner R. High prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. European journal of endocrinology 2004; 150:363-369
- Rotondi M, Leporati P, La Manna A, Pirali B, Mondello T, Fonte R, Magri F, Chiovato L. Raised serum TSH levels in patients with morbid obesity: is it enough to diagnose subclinical hypothyroidism? European journal of endocrinology 2009; 160:403-408
- Rasmusson B, Feldt-Rasmussen U, Hegedus L, Perrild H, Bech K, Hoier-Madsen M. Thyroid function in patients with breast cancer. European journal of cancer & clinical oncology 1987; 23:553-556
- Giustarini E, Pinchera A, Fierabracci P, Roncella M, Fustaino L, Mammoli C, Giani C. Thyroid autoimmunity in patients with malignant and benign breast diseases before surgery. European journal of endocrinology 2006; 154:645-649
- Antonelli A, Ferri C, Fallahi P, Ferrari SM, Ghinoi A, Rotondi M, Ferrannini E. Thyroid disorders in chronic hepatitis C virus infection. Thyroid 2006; 16:563-572
- Goday-Arno A, Cerda-Esteva M, Flores-Le-Roux JA, Chillaron-Jordan JJ, Corretger JM, Cano-Perez JF. Hyperthyroidism in a population with Down syndrome (DS). Clinical endocrinology 2009; 71:110-114
- Elsheikh M, Wass JA, Conway GS. Autoimmune thyroid syndrome in women with Turner's syndrome--the association with karyotype. Clinical endocrinology 2001; 55:223-226
- Garrahy A, Sherlock M, Thompson CJ. MANAGEMENT OF ENDOCRINE DISEASE: Neuroendocrine surveillance and management of neurosurgical patients. EurJEndocrinol 2017; 176:R217-R233
- Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: a systematic review. JAMA 2007; 298:1429-1438
- Klose M, Feldt-Rasmussen U. Chronic endocrine consequences of traumatic brain injury - what is the evidence? NatRevEndocrinol 2018; 14:57-62
- Rooman RP, Du Caju MV, De Beeck LO, Docx M, Van Reempts P, Van Acker KJ. Low thyroxinaemia occurs in the majority of very preterm newborns. European journal of pediatrics 1996; 155:211-215
- Fisher DA. The hypothyroxinemia [corrected] of prematurity. The Journal of clinical endocrinology and metabolism 1997; 82:1701-1703
- Feldt-Rasmussen U, Rasmussen AK. Drug effects and thyroid function. In: Huhtaniemi I, ed. Encyclopedia of Endocrine Diseases. 2nd ed: Academic Press; 2018.
- Figaro MK, Clayton W, Jr., Usoh C, Brown K, Kassim A, Lakhani VT, Jagasia S. Thyroid abnormalities in patients treated with lenalidomide for hematological malignancies: results of a retrospective case review. American journal of hematology 2011; 86:467-470
- Clement SC, Schoot RA, Slater O, Chisholm JC, Abela C, Balm AJM, van den Brekel MW, Breunis WB, Chang YC, Davila Fajardo R, Dunaway D, Gajdosova E, Gaze MN, Gupta S, Hartley B, Kremer LCM, van Lennep M, Levitt GA, Mandeville HC, Pieters BR, Saeed P, Smeele LE, Strackee SD, Ronckers CM, Caron HN, van Santen HM, Merks JHM. Endocrine disorders among long-term survivors of childhood head and neck rhabdomyosarcoma. European journal of cancer (Oxford, England : 1990) 2016; 54:1-10
- Jarlov AE, Nygaard B, Hegedus L, Hartling SG, Hansen JM. Observer variation in the clinical and laboratory evaluation of patients with thyroid dysfunction and goiter. Thyroid 1998; 8:393-398
- Zulewski H, Muller B, Exer P, Miserez AR, Staub JJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. JClinEndocrinolMetab 1997; 82:771-776
- Boelaert K, Torlinska B, Holder RL, Franklyn JA. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: a large cross-sectional study. The Journal of clinical endocrinology and metabolism 2010; 95:2715-2726
- Toubert ME, Chevret S, Cassinat B, Schlageter MH, Beressi JP, Rain JD. From guidelines to hospital practice: reducing inappropriate ordering of thyroid hormone and antibody tests. European journal of endocrinology 2000; 142:605-610
- Schectman JM, Elinsky EG, Pawlson LG. Effect of education and feedback on thyroid function testing strategies of primary care clinicians. Archives of internal medicine 1991; 151:2163-2166
- Lopez-Achigar E, Collazo C, Blanco R, Raymondo S. Suporting the cost-effectiveness of a thyroid testing algorithm. Clinica chimica acta; international journal of clinical chemistry 2000; 296:213-215
- Sheehan MT. Biochemical Testing of the Thyroid: TSH is the Best and, Oftentimes, Only Test Needed - A Review for Primary Care. Clinical medicine & research 2016; 14:83-92
- McDermott MT, Ridgway EC. Subclinical hypothyroidism is mild thyroid failure and should be treated. The Journal of clinical endocrinology and metabolism 2001; 86:4585-4590
- Biondi B. Natural history, diagnosis and management of subclinical thyroid dysfunction. Best practice & research Clinical endocrinology & metabolism 2012; 26:431-446
- Nicoloff JT, Spencer CA. Clinical review 12: The use and misuse of the sensitive thyrotropin assays. The Journal of clinical endocrinology and metabolism 1990; 71:553-558
- Ercan-Fang S, Schwartz HL, Mariash CN, Oppenheimer JH. Quantitative assessment of pituitary resistance to thyroid hormone from plots of the logarithm of thyrotropin versus serum free thyroxine index. The Journal of clinical endocrinology and metabolism 2000; 85:2299-2303
- Spencer CA, LoPresti JS, Patel A, Guttler RB, Eigen A, Shen D, Gray D, Nicoloff JT. Applications of a new chemiluminometric thyrotropin assay to subnormal measurement. The Journal of clinical endocrinology and metabolism 1990; 70:453-460
- Hershman JM, Pekary AE, Berg L, Solomon DH, Sawin CT. Serum thyrotropin and thyroid hormone levels in elderly and middle-aged euthyroid persons. Journal of the American Geriatrics Society 1993; 41:823-828
- Morley JE. Neuroendocrine control of thyrotropin secretion. Endocrine reviews 1981; 2:396-436
- Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, Grimley EJ, Hasan DM, Rodgers H, Tunbridge F. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. ClinEndocrinol(Oxf) 1995; 43:55-68
- Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, Braverman LE. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). The Journal of clinical endocrinology and metabolism 2002; 87:489-499
- Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Archives of internal medicine 2000; 160:526-534
- Kung AW, Janus ED. Thyroid dysfunction in ambulatory elderly Chinese subjects in an area of borderline iodine intake. Thyroid 1996; 6:111-114
- Szabolcs I, Podoba J, Feldkamp J, Dohan O, Farkas I, Sajgo M, Takats KI, Goth M, Kovacs L, Kressinszky K, Hnilica P, Szilagyi G. Comparative screening for thyroid disorders in old age in areas of iodine deficiency, long-term iodine prophylaxis and abundant iodine intake. Clinical endocrinology 1997; 47:87-92
- Aghini-Lombardi F, Antonangeli L, Martino E, Vitti P, Maccherini D, Leoli F, Rago T, Grasso L, Valeriano R, Balestrieri A, Pinchera A. The spectrum of thyroid disorders in an iodine-deficient community: the Pescopagano survey. The Journal of clinical endocrinology and metabolism 1999; 84:561-566
- Knudsen N, Bulow I, Jorgensen T, Laurberg P, Ovesen L, Perrild H. Comparative study of thyroid function and types of thyroid dysfunction in two areas in Denmark with slightly different iodine status. European journal of endocrinology 2000; 143:485-491
- Laurberg P, Pedersen KM, Hreidarsson A, Sigfusson N, Iversen E, Knudsen PR. Iodine intake and the pattern of thyroid disorders: a comparative epidemiological study of thyroid abnormalities in the elderly in Iceland and in Jutland, Denmark. The Journal of clinical endocrinology and metabolism 1998; 83:765-769
- Gharib H, Tuttle RM, Baskin HJ, Fish LH, Singer PA, McDermott MT. Subclinical thyroid dysfunction: a joint statement on management from the American Association of Clinical Endocrinologists, the American Thyroid Association, and the Endocrine Society. The Journal of clinical endocrinology and metabolism 2005; 90:581-585; discussion 586-587
- Surks MI, Ortiz E, Daniels GH, Sawin CT, Col NF, Cobin RH, Franklyn JA, Hershman JM, Burman KD, Denke MA, Gorman C, Cooper RS, Weissman NJ. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. Jama 2004; 291:228-238
- Wartofsky L, Dickey RA. The evidence for a narrower thyrotropin reference range is compelling. The Journal of clinical endocrinology and metabolism 2005; 90:5483-5488
- Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. NEnglJMed 2001; 344:501-509
- Faber J, Jensen IW, Petersen L, Nygaard B, Hegedus L, Siersbaek-Nielsen K. Normalization of serum thyrotrophin by means of radioiodine treatment in subclinical hyperthyroidism: effect on bone loss in postmenopausal women. Clinical endocrinology 1998; 48:285-290
- Rosario PW, Calsolari MR. How selective are the new guidelines for treatment of subclinical hypothyroidism for patients with thyrotropin levels at or below 10 mIU/L? Thyroid 2013; 23:562-565
- Kahaly GJ. Cardiovascular and atherogenic aspects of subclinical hypothyroidism. Thyroid 2000; 10:665-679
- Bauer DC, Rodondi N, Stone KL, Hillier TA. Thyroid hormone use, hyperthyroidism and mortality in older women. The American journal of medicine 2007; 120:343-349
- Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 2001; 358:861-865
- Flynn RW, Bonellie SR, Jung RT, MacDonald TM, Morris AD, Leese GP. Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. The Journal of clinical endocrinology and metabolism 2010; 95:186-193
- Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham Survey cohort. The Journal of clinical endocrinology and metabolism 2010; 95:1734-1740
- Martin FI, Tress BW, Colman PG, Deam DR. Iodine-induced hyperthyroidism due to nonionic contrast radiography in the elderly. The American journal of medicine 1993; 95:78-82
- Sawin CT, Geller A, Wolf PA, Belanger AJ, Baker E, Bacharach P, Wilson PW, Benjamin EJ, D'Agostino RB. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. The New England journal of medicine 1994; 331:1249-1252
- Biondi B, Fazio S, Carella C, Amato G, Cittadini A, Lupoli G, Sacca L, Bellastella A, Lombardi G. Cardiac effects of long term thyrotropin-suppressive therapy with levothyroxine. The Journal of clinical endocrinology and metabolism 1993; 77:334-338
- Wesche MF, Tiel VBMM, Lips P, Smits NJ, Wiersinga WM. A randomized trial comparing levothyroxine with radioactive iodine in the treatment of sporadic nontoxic goiter. The Journal of clinical endocrinology and metabolism 2001; 86:998-1005
- Bauer DC, Ettinger B, Nevitt MC, Stone KL. Risk for fracture in women with low serum levels of thyroid-stimulating hormone. Annals of internal medicine 2001; 134:561-568
- Stott DJ, McLellan AR, Finlayson J, Chu P, Alexander WD. Elderly patients with suppressed serum TSH but normal free thyroid hormone levels usually have mild thyroid overactivity and are at increased risk of developing overt hyperthyroidism. The Quarterly journal of medicine 1991; 78:77-84
- Parle JV, Franklyn JA, Cross KW, Jones SC, Sheppard MC. Prevalence and follow-up of abnormal thyrotrophin (TSH) concentrations in the elderly in the United Kingdom. Clinical endocrinology 1991; 34:77-83
- Nystrom E, Caidahl K, Fager G, Wikkelso C, Lundberg PA, Lindstedt G. A double-blind cross-over 12-month study of L-thyroxine treatment of women with 'subclinical' hypothyroidism. Clinical endocrinology 1988; 29:63-75
- Huber G, Staub JJ, Meier C, Mitrache C, Guglielmetti M, Huber P, Braverman LE. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. The Journal of clinical endocrinology and metabolism 2002; 87:3221-3226
- Hak AE, Pols HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam Study. Annals of internal medicine 2000; 132:270-278
- Rodondi N, Newman AB, Vittinghoff E, de Rekeneire N, Satterfield S, Harris TB, Bauer DC. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Archives of internal medicine 2005; 165:2460-2466
- Volzke H, Schwahn C, Wallaschofski H, Dorr M. Review: The association of thyroid dysfunction with all-cause and circulatory mortality: is there a causal relationship? The Journal of clinical endocrinology and metabolism 2007; 92:2421-2429
- Rodondi N, Aujesky D, Vittinghoff E, Cornuz J, Bauer DC. Subclinical hypothyroidism and the risk of coronary heart disease: a meta-analysis. The American journal of medicine 2006; 119:541-551
- Cappola AR, Fried LP, Arnold AM, Danese MD, Kuller LH, Burke GL, Tracy RP, Ladenson PW. Thyroid status, cardiovascular risk, and mortality in older adults. Jama 2006; 295:1033-1041
- Lekakis J, Papamichael C, Alevizaki M, Piperingos G, Marafelia P, Mantzos J, Stamatelopoulos S, Koutras DA. Flow-mediated, endothelium-dependent vasodilation is impaired in subjects with hypothyroidism, borderline hypothyroidism, and high-normal serum thyrotropin (TSH) values. Thyroid 1997; 7:411-414
- Nagasaki T, Inaba M, Yamada S, Shirakawa K, Nagata Y, Kumeda Y, Hiura Y, Tahara H, Ishimura E, Nishizawa Y. Decrease of brachial-ankle pulse wave velocity in female subclinical hypothyroid patients during normalization of thyroid function: a double-blind, placebo-controlled study. European journal of endocrinology 2009; 160:409-415
- Owen PJ, Rajiv C, Vinereanu D, Mathew T, Fraser AG, Lazarus JH. Subclinical hypothyroidism, arterial stiffness, and myocardial reserve. The Journal of clinical endocrinology and metabolism 2006; 91:2126-2132
- Monzani F, Di Bello V, Caraccio N, Bertini A, Giorgi D, Giusti C, Ferrannini E. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. The Journal of clinical endocrinology and metabolism 2001; 86:1110-1115
- Danese MD, Ladenson PW, Meinert CL, Powe NR. Clinical review 115: effect of thyroxine therapy on serum lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. The Journal of clinical endocrinology and metabolism 2000; 85:2993-3001
- Meier C, Staub JJ, Roth CB, Guglielmetti M, Kunz M, Miserez AR, Drewe J, Huber P, Herzog R, Muller B. TSH-controlled L-thyroxine therapy reduces cholesterol levels and clinical symptoms in subclinical hypothyroidism: a double blind, placebo-controlled trial (Basel Thyroid Study). The Journal of clinical endocrinology and metabolism 2001; 86:4860-4866
- Haggerty JJ, Jr., Stern RA, Mason GA, Beckwith J, Morey CE, Prange AJ, Jr. Subclinical hypothyroidism: a modifiable risk factor for depression? The American journal of psychiatry 1993; 150:508-510
- Chadarevian R, Bruckert E, Leenhardt L, Giral P, Ankri A, Turpin G. Components of the fibrinolytic system are differently altered in moderate and severe hypothyroidism. The Journal of clinical endocrinology and metabolism 2001; 86:732-737
- Meyerovitch J, Rotman-Pikielny P, Sherf M, Battat E, Levy Y, Surks MI. Serum thyrotropin measurements in the community: five-year follow-up in a large network of primary care physicians. Archives of internal medicine 2007; 167:1533-1538
- Woeber KA. Observations concerning the natural history of subclinical hyperthyroidism. Thyroid 2005; 15:687-691
- Zhyzhneuskaya S, Addison C, Tsatlidis V, Weaver JU, Razvi S. The Natural History of Subclinical Hyperthyroidism in Graves' Disease: The Rule of Thirds. Thyroid 2016; 26:765-769
- Abdul Shakoor SA, Hawkins R, Kua SY, Ching ME, Dalan R. Natural history and comorbidities of subjects with subclinical hyperthyroidism: analysis at a tertiary hospital setting. Annals of the Academy of Medicine, Singapore 2014; 43:506-510
- Effraimidis G, Strieder TG, Tijssen JG, Wiersinga WM. Natural history of the transition from euthyroidism to overt autoimmune hypo- or hyperthyroidism: a prospective study. European journal of endocrinology 2011; 164:107-113
- Faber J, Selmer C. Cardiovascular disease and thyroid function. Frontiers of hormone research 2014; 43:45-56
- Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 1998; 98:946-952
- Collet TH, Gussekloo J, Bauer DC, den Elzen WP, Cappola AR, Balmer P, Iervasi G, Asvold BO, Sgarbi JA, Volzke H, Gencer B, Maciel RM, Molinaro S, Bremner A, Luben RN, Maisonneuve P, Cornuz J, Newman AB, Khaw KT, Westendorp RG, Franklyn JA, Vittinghoff E, Walsh JP, Rodondi N. Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. ArchInternMed 2012; 172:799-809
- Sun J, Yao L, Fang Y, Yang R, Chen Y, Yang K, Tian L. Relationship between Subclinical Thyroid Dysfunction and the Risk of Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. International journal of endocrinology 2017; 2017:8130796
- Baumgartner C, da Costa BR, Collet TH, Feller M, Floriani C, Bauer DC, Cappola AR, Heckbert SR, Ceresini G, Gussekloo J, den Elzen WPJ, Peeters RP, Luben R, Volzke H, Dorr M, Walsh JP, Bremner A, Iacoviello M, Macfarlane P, Heeringa J, Stott DJ, Westendorp RGJ, Khaw KT, Magnani JW, Aujesky D, Rodondi N. Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 2017; 136:2100-2116
- Biondi B, Fazio S, Cuocolo A, Sabatini D, Nicolai E, Lombardi G, Salvatore M, Sacca L. Impaired cardiac reserve and exercise capacity in patients receiving long-term thyrotropin suppressive therapy with levothyroxine. The Journal of clinical endocrinology and metabolism 1996; 81:4224-4228
- Dunn JT, Semigran MJ, Delange F. The prevention and management of iodine-induced hyperthyroidism and its cardiac features. Thyroid 1998; 8:101-106
- Iakovou I, Zapandiotis A, Mpalaris V, Goulis DG. Radio-contrast agent-induced hyperthyroidism: case report and review of the literature. Archives of endocrinology and metabolism 2016; 60:287-289
- Basaria S, Cooper DS. Amiodarone and the thyroid. The American journal of medicine 2005; 118:706-714
- Ross DS. Hyperthyroidism, thyroid hormone therapy, and bone. Thyroid 1994; 4:319-326
- Ding B, Zhang Y, Li Q, Hu Y, Tao XJ, Liu BL, Ma JH, Li DM. Low Thyroid Stimulating Hormone Levels Are Associated with Low Bone Mineral Density in Femoral Neck in Elderly Women. Archives of medical research 2016; 47:310-314
- Tunbridge WM, Evered DC, Hall R, Appleton D, Brewis M, Clark F, Evans JG, Young E, Bird T, Smith PA. The spectrum of thyroid disease in a community: the Whickham survey. Clinical endocrinology 1977; 7:481-493
- Diez JJ, Iglesias P, Burman KD. Spontaneous normalization of thyrotropin concentrations in patients with subclinical hypothyroidism. The Journal of clinical endocrinology and metabolism 2005; 90:4124-4127
- Somwaru LL, Rariy CM, Arnold AM, Cappola AR. The natural history of subclinical hypothyroidism in the elderly: the cardiovascular health study. The Journal of clinical endocrinology and metabolism 2012; 97:1962-1969
- Rosario PW, Carvalho M, Calsolari MR. Natural history of subclinical hypothyroidism with TSH </=10 mIU/l: a prospective study. Clinical endocrinology 2016; 84:878-881
- Li X, Zhen D, Zhao M, Liu L, Guan Q, Zhang H, Ge S, Tang X, Gao L. Natural history of mild subclinical hypothyroidism in a middle-aged and elderly Chinese population: a prospective study. Endocrine journal 2017; 64:437-447
- Kim H, Kim TH, Kim HI, Park SY, Kim YN, Kim S, Kim MJ, Jin SM, Hur KY, Kim JH, Lee MK, Min YK, Chung JH, Kang M, Kim SW. Subclinical thyroid dysfunction and risk of carotid atherosclerosis. PloS one 2017; 12:e0182090
- Yao K, Zhao T, Zeng L, Yang J, Liu Y, He Q, Zou X. Non-invasive markers of cardiovascular risk in patients with subclinical hypothyroidism: A systematic review and meta-analysis of 27 case control studies. Scientific reports 2018; 8:4579
- Kim SK, Kim SH, Park KS, Park SW, Cho YW. Regression of the increased common carotid artery-intima media thickness in subclinical hypothyroidism after thyroid hormone replacement. Endocrine journal 2009; 56:753-758
- Monzani F, Caraccio N, Kozakowa M, Dardano A, Vittone F, Virdis A, Taddei S, Palombo C, Ferrannini E. Effect of levothyroxine replacement on lipid profile and intima-media thickness in subclinical hypothyroidism: a double-blind, placebo- controlled study. The Journal of clinical endocrinology and metabolism 2004; 89:2099-2106
- Clausen P, Mersebach H, Nielsen B, Feldt-Rasmussen B, Feldt-Rasmussen U. Hypothyroidism is associated with signs of endothelial dysfunction despite 1-year replacement therapy with levothyroxine. ClinEndocrinol(Oxf) 2009; 70:932-937
- Adrees M, Gibney J, El-Saeity N, Boran G. Effects of 18 months of L-T4 replacement in women with subclinical hypothyroidism. Clinical endocrinology 2009; 71:298-303
- Blum MR, Gencer B, Adam L, Feller M, Collet TH, da Costa BR, Moutzouri E, Dopheide J, Depairon M, Sykiotis GP, Kearney P, Gussekloo J, Westendorp R, Stott DJ, Bauer DC, Rodondi N. Impact of Thyroid Hormone Therapy on Atherosclerosis in the Elderly With Subclinical Hypothyroidism: A Randomized Trial. The Journal of clinical endocrinology and metabolism 2018; 103:2988-2997
- Aziz M, Kandimalla Y, Machavarapu A, Saxena A, Das S, Younus A, Nguyen M, Malik R, Anugula D, Latif MA, Humayun C, Khan IM, Adus A, Rasool A, Veledar E, Nasir K. Effect of Thyroxin Treatment on Carotid Intima-Media Thickness (CIMT) Reduction in Patients with Subclinical Hypothyroidism (SCH): a Meta-Analysis of Clinical Trials. Journal of atherosclerosis and thrombosis 2017; 24:643-659
- Zhao T, Chen B, Zhou Y, Wang X, Zhang Y, Wang H, Shan Z. Effect of levothyroxine on the progression of carotid intima-media thickness in subclinical hypothyroidism patients: a meta-analysis. BMJ open 2017; 7:e016053
- Friis T, Pedersen LR. Serum lipids in hyper- and hypothyroidism before and after treatment. ClinChimActa 1987; 162:155-163
- Delitala AP, Fanciulli G, Maioli M, Delitala G. Subclinical hypothyroidism, lipid metabolism and cardiovascular disease. European journal of internal medicine 2017; 38:17-24
- Franklyn JA. Metabolic changes in hypothyroidism. In: Braverman LE, Utiger RG, eds. The Thyroid. Philadelphia: Lippincott, Williams & Wilkens; 2000:833-836.
- Thompson GR, Soutar AK, Spengel FA, Jadhav A, Gavigan SJ, Myant NB. Defects of receptor-mediated low density lipoprotein catabolism in homozygous familial hypercholesterolemia and hypothyroidism in vivo. Proceedings of the National Academy of Sciences of the United States of America 1981; 78:2591-2595
- Asvold BO, Vatten LJ, Nilsen TI, Bjoro T. The association between TSH within the reference range and serum lipid concentrations in a population-based study. The HUNT Study. European journal of endocrinology 2007; 156:181-186
- Waterhouse DF, McLaughlin AM, Walsh CD, Sheehan F, O'Shea D. An examination of the relationship between normal range thyrotropin and cardiovascular risk parameters: a study in healthy women. Thyroid 2007; 17:243-248
- de Bruin TW, van Barlingen H, van Linde-Sibenius Trip M, van Vuurst de Vries AR, Akveld MJ, Erkelens DW. Lipoprotein(a) and apolipoprotein B plasma concentrations in hypothyroid, euthyroid, and hyperthyroid subjects. The Journal of clinical endocrinology and metabolism 1993; 76:121-126
- Tzotzas T, Krassas GE, Konstantinidis T, Bougoulia M. Changes in lipoprotein(a) levels in overt and subclinical hypothyroidism before and during treatment. Thyroid 2000; 10:803-808
- Klausen IC, Nielsen FE, Hegedus L, Gerdes LU, Charles P, Faergeman O. Treatment of hypothyroidism reduces low-density lipoproteins but not lipoprotein(a). Metabolism: clinical and experimental 1992; 41:911-914
- Arem R, Escalante DA, Arem N, Morrisett JD, Patsch W. Effect of L-thyroxine therapy on lipoprotein fractions in overt and subclinical hypothyroidism, with special reference to lipoprotein(a). Metabolism: clinical and experimental 1995; 44:1559-1563
- Adamarczuk-Janczyszyn M, Zdrojowy-Welna A, Rogala N, Zatonska K, Bednarek-Tupikowska G. Evaluation of Selected Atherosclerosis Risk Factors in Women with Subclinical Hypothyroidism Treated with L-Thyroxine. Advances in clinical and experimental medicine : official organ Wroclaw Medical University 2016; 25:457-463
- Biondi B, Kahaly GJ. Heart in Hypothyroidism. In: Luster M, Duntas L, Wartofsky L, eds. The Thyroid and Its Diseases. A Comprehensive Guide for the Clinician. : Springer International Publishing AG, part of Springer Nature; 2019:129-160.
- Karabulut A, Dogan A, Tuzcu AK. Myocardial Performance Index for Patients with Overt and Subclinical Hypothyroidism. Medical science monitor : international medical journal of experimental and clinical research 2017; 23:2519-2526
- Yao Z, Gao X, Liu M, Chen Z, Yang N, Jia YM, Feng XM, Xu Y, Yang XC, Wang G. Diffuse Myocardial Injuries are Present in Subclinical Hypothyroidism: A Clinical Study Using Myocardial T1-mapping Quantification. Scientific reports 2018; 8:4999
- Bakker SJ, ter Maaten JC, Popp-Snijders C, Slaets JP, Heine RJ, Gans RO. The relationship between thyrotropin and low density lipoprotein cholesterol is modified by insulin sensitivity in healthy euthyroid subjects. The Journal of clinical endocrinology and metabolism 2001; 86:1206-1211
- Roos A, Bakker SJ, Links TP, Gans RO, Wolffenbuttel BH. Thyroid function is associated with components of the metabolic syndrome in euthyroid subjects. The Journal of clinical endocrinology and metabolism 2007; 92:491-496
- Uzunlulu M, Yorulmaz E, Oguz A. Prevalence of subclinical hypothyroidism in patients with metabolic syndrome. Endocrine journal 2007; 54:71-76
- Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Leonetti F. Relationship of thyroid function with body mass index, leptin, insulin sensitivity and adiponectin in euthyroid obese women. Clinical endocrinology 2005; 62:487-491
- Knudsen N, Laurberg P, Rasmussen LB, Bulow I, Perrild H, Ovesen L, Jorgensen T. Small differences in thyroid function may be important for body mass index and the occurrence of obesity in the population. The Journal of clinical endocrinology and metabolism 2005; 90:4019-4024
- Posadas-Romero C, Jorge-Galarza E, Posadas-Sanchez R, Acuna-Valerio J, Juarez-Rojas JG, Kimura-Hayama E, Medina-Urrutia A, Cardoso-Saldana GC. Fatty liver largely explains associations of subclinical hypothyroidism with insulin resistance, metabolic syndrome, and subclinical coronary atherosclerosis. European journal of endocrinology 2014; 171:319-325
- Pergialiotis V, Konstantopoulos P, Prodromidou A, Florou V, Papantoniou N, Perrea DN. MANAGEMENT OF ENDOCRINE DISEASE: The impact of subclinical hypothyroidism on anthropometric characteristics, lipid, glucose and hormonal profile of PCOS patients: a systematic review and meta-analysis. European journal of endocrinology 2017; 176:R159-r166
- Franca MM, Nogueira CR, Hueb JC, Mendes AL, Padovani CR, Mazeto GM. Higher Carotid Intima-Media Thickness in Subclinical Hypothyroidism Associated with the Metabolic Syndrome. Metabolic syndrome and related disorders 2016; 14:381-385
- Bonora BM, Fadini GP. Subclinical Hypothyroidism and Metabolic Syndrome: A Common Association by Chance or a Cardiovascular Risk Driver? Metabolic syndrome and related disorders 2016; 14:378-380
- Cheserek MJ, Wu G, Shen L, Shi Y, Le G. Evaluation of the relationship between subclinical hypothyroidism and metabolic syndrome components among workers. International journal of occupational medicine and environmental health 2014; 27:175-187
- Monzani F, Del Guerra P, Caraccio N, Pruneti CA, Pucci E, Luisi M, Baschieri L. Subclinical hypothyroidism: neurobehavioral features and beneficial effect of L-thyroxine treatment. The Clinical investigator 1993; 71:367-371
- Bell RJ, Rivera-Woll L, Davison SL, Topliss DJ, Donath S, Davis SR. Well-being, health-related quality of life and cardiovascular disease risk profile in women with subclinical thyroid disease - a community-based study. Clinical endocrinology 2007; 66:548-556
- Jorde R, Waterloo K, Storhaug H, Nyrnes A, Sundsfjord J, Jenssen TG. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. The Journal of clinical endocrinology and metabolism 2006; 91:145-153
- Stott DJ, Rodondi N, Kearney PM, Ford I, Westendorp RGJ, Mooijaart SP, Sattar N, Aubert CE, Aujesky D, Bauer DC, Baumgartner C, Blum MR, Browne JP, Byrne S, Collet TH, Dekkers OM, den Elzen WPJ, Du Puy RS, Ellis G, Feller M, Floriani C, Hendry K, Hurley C, Jukema JW, Kean S, Kelly M, Krebs D, Langhorne P, McCarthy G, McCarthy V, McConnachie A, McDade M, Messow M, O'Flynn A, O'Riordan D, Poortvliet RKE, Quinn TJ, Russell A, Sinnott C, Smit JWA, Van Dorland HA, Walsh KA, Walsh EK, Watt T, Wilson R, Gussekloo J. Thyroid Hormone Therapy for Older Adults with Subclinical Hypothyroidism. The New England journal of medicine 2017; 376:2534-2544
- Cerbone M, Bravaccio C, Capalbo D, Polizzi M, Wasniewska M, Cioffi D, Improda N, Valenzise M, Bruzzese D, De Luca F, Salerno M. Linear growth and intellectual outcome in children with long-term idiopathic subclinical hypothyroidism. European journal of endocrinology 2011; 164:591-597
- Monzani A, Prodam F, Rapa A, Moia S, Agarla V, Bellone S, Bona G. Endocrine disorders in childhood and adolescence. Natural history of subclinical hypothyroidism in children and adolescents and potential effects of replacement therapy: a review. European journal of endocrinology 2013; 168:R1-r11
- Aversa T, Valenzise M, Corrias A, Salerno M, De Luca F, Mussa A, Rezzuto M, Lombardo F, Wasniewska M. Underlying Hashimoto's thyroiditis negatively affects the evolution of subclinical hypothyroidism in children irrespective of other concomitant risk factors. Thyroid 2015; 25:183-187
- Feldt-Rasmussen U, Emerson CH. Further thoughts on the diagnosis and diagnostic criteria for thyroid storm. Thyroid 2012; 22:1094-1095
- Salomo LH, Laursen AH, Reiter N, Feldt-Rasmussen U. Myxoedema coma: an almost forgotten, yet still existing cause of multiorgan failure. BMJ CaseRep 2014; 2014
- Weetman AP. Hypothyroidism: screening and subclinical disease. BMJ 1997; 314:1175-1178
- Hennessey JV, Espaillat R. Diagnosis and Management of Subclinical Hypothyroidism in Elderly Adults: A Review of the Literature. Journal of the American Geriatrics Society 2015; 63:1663-1673
- Pearce SH, Brabant G, Duntas LH, Monzani F, Peeters RP, Razvi S, Wemeau JL. 2013 ETA Guideline: Management of Subclinical Hypothyroidism. European thyroid journal 2013; 2:215-228
- Pearce SH, Vaisman M, Wemeau JL. Management of subclinical hypothyroidism: the thyroidologists' view. European thyroid journal 2012; 1:45-50
- Pandrc MS, Ristic A, Kostovski V, Stankovic M, Antic V, Milin-Lazovic J, Ciric J. The Effect of Early Substitution of Subclinical Hypothyroidism on Biochemical Blood Parameters and the Quality of Life. Journal of medical biochemistry 2017; 36:127-136
- Wartofsky L, Handelsman DJ. Standardization of hormonal assays for the 21st century. The Journal of clinical endocrinology and metabolism 2010; 95:5141-5143
- Feldt-Rasmussen U, Hyltoft PP, Blaabjerg O, Horder M. Long-term variability in serum thyroglobulin and thyroid related hormones in healthy subjects. Acta Endocrinol(Copenh) 1980; 95:328-334
- Andersen S, Pedersen KM, Bruun NH, Laurberg P. Narrow individual variations in serum T(4) and T(3) in normal subjects: a clue to the understanding of subclinical thyroid disease. JClinEndocrinolMetab 2002; 87:1068-1072
- Brabant G, Beck-Peccoz P, Jarzab B, Laurberg P, Orgiazzi J, Szabolcs I, Weetman AP, Wiersinga WM. Is there a need to redefine the upper normal limit of TSH? European journal of endocrinology 2006; 154:633-637
- Surks MI, Goswami G, Daniels GH. The thyrotropin reference range should remain unchanged. The Journal of clinical endocrinology and metabolism 2005; 90:5489-5496
- Hamilton TE, Davis S, Onstad L, Kopecky KJ. Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. The Journal of clinical endocrinology and metabolism 2008; 93:1224-1230
- Spencer CA, Hollowell JG, Kazarosyan M, Braverman LE. National Health and Nutrition Examination Survey III thyroid-stimulating hormone (TSH)-thyroperoxidase antibody relationships demonstrate that TSH upper reference limits may be skewed by occult thyroid dysfunction. The Journal of clinical endocrinology and metabolism 2007; 92:4236-4240
- Baloch Z, Carayon P, Conte-Devolx B, Demers LM, Feldt-Rasmussen U, Henry JF, LiVosli VA, Niccoli-Sire P, John R, Ruf J, Smyth PP, Spencer CA, Stockigt JR. Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid 2003; 13:3-126
- Surks MI, Boucai L. Age- and race-based serum thyrotropin reference limits. The Journal of clinical endocrinology and metabolism 2010; 95:496-502
- Cooper DS. Subclinical thyroid disease: consensus or conundrum? Clinical endocrinology 2004; 60:410-412
- Lazarus JH. Aspects of treatment of subclinical hypothyroidism. Thyroid 2007; 17:313-316
- Nordyke RA, Reppun TS, Madanay LD, Woods JC, Goldstein AP, Miyamoto LA. Alternative sequences of thyrotropin and free thyroxine assays for routine thyroid function testing. Quality and cost. Archives of internal medicine 1998; 158:266-272
- Wardle CA, Fraser WD, Squire CR. Pitfalls in the use of thyrotropin concentration as a first-line thyroid-function test. Lancet 2001; 357:1013-1014
- Beckett GJ, Toft AD. First-line thyroid function tests -- TSH alone is not enough. ClinEndocrinol(Oxf) 2003; 58:20-21
- Feldt-Rasmussen U, Klose M. Central hypothyroidism and its role for cardiovascular risk factors in hypopituitary patients. Endocrine 2016; 54:15-23
- Persani L, Brabant G, Dattani M, Bonomi M, Feldt-Rasmussen U, Fliers E, Gruters A, Maiter D, Schoenmakers N, van Trotsenburg AS. 2018 European Thyroid Association (ETA) Guidelines on the Diagnosis and Management of Central Hypothyroidism. European thyroid journal 2018;
- Surks MI, Sievert R. Drugs and thyroid function. The New England journal of medicine 1995; 333:1688-1694
- Borst GC, Osburne RC, O'Brian JT, Georges LP, Burman KD. Fasting decreases thyrotropin responsiveness to thyrotropin-releasing hormone: a potential cause of misinterpretation of thyroid function tests in the critically ill. The Journal of clinical endocrinology and metabolism 1983; 57:380-383
- Hamblin PS, Dyer SA, Mohr VS, Le Grand BA, Lim CF, Tuxen DV, Topliss DJ, Stockigt JR. Relationship between thyrotropin and thyroxine changes during recovery from severe hypothyroxinemia of critical illness. The Journal of clinical endocrinology and metabolism 1986; 62:717-722
- Van den Berghe G. Non-thyroidal illness in the ICU: a syndrome with different faces. Thyroid 2014; 24:1456-1465
- Van den Berghe G. The 2016 ESPEN Sir David Cuthbertson lecture: Interfering with neuroendocrine and metabolic responses to critical illness: From acute to long-term consequences. Clinical nutrition (Edinburgh, Scotland) 2017; 36:348-354
- Spencer C, Eigen A, Shen D, Duda M, Qualls S, Weiss S, Nicoloff J. Specificity of sensitive assays of thyrotropin (TSH) used to screen for thyroid disease in hospitalized patients. Clinical chemistry 1987; 33:1391-1396
- Stockigt JR. Free thyroid hormone measurement. A critical appraisal. Endocrinology and metabolism clinics of North America 2001; 30:265-289
- Stockigt JR. Guidelines for diagnosis and monitoring of thyroid disease: nonthyroidal illness. Clinical chemistry 1996; 42:188-192
- Hein MD, Jackson IM. Review: thyroid function in psychiatric illness. General hospital psychiatry 1990; 12:232-244
- White AJ, Barraclough B. Thyroid disease and mental illness: a study of thyroid disease in psychiatric admissions. Journal of psychosomatic research 1988; 32:99-106
- Tikhonoff V, Hardy R, Deanfield J, Friberg P, Muniz G, Kuh D, Pariante CM, Hotopf M, Richards M. The relationship between affective symptoms and hypertension-role of the labelling effect: the 1946 British birth cohort. Open heart 2016; 3:e000341
- Hyltoft Petersen P, Klee GG. Influence of analytical bias and imprecision on the number of false positive results using Guideline-Driven Medical Decision Limits. Clinica chimica acta; international journal of clinical chemistry 2014; 430:1-8
- Meier C, Trittibach P, Guglielmetti M, Staub JJ, Muller B. Serum thyroid stimulating hormone in assessment of severity of tissue hypothyroidism in patients with overt primary thyroid failure: cross sectional survey. Bmj 2003; 326:311-312
- Hoermann R, Eckl W, Hoermann C, Larisch R. Complex relationship between free thyroxine and TSH in the regulation of thyroid function. European journal of endocrinology 2010; 162:1123-1129
- Fitzgerald SP, Bean NG. The Relationship between Population T4/TSH Set Point Data and T4/TSH Physiology. Journal of thyroid research 2016; 2016:6351473
- Brown SJ, Bremner AP, Hadlow NC, Feddema P, Leedman PJ, O'Leary PC, Walsh JP. The log TSH-free T4 relationship in a community-based cohort is nonlinear and is influenced by age, smoking and thyroid peroxidase antibody status. Clinical endocrinology 2016; 85:789-796
- Hadlow NC, Rothacker KM, Wardrop R, Brown SJ, Lim EM, Walsh JP. The relationship between TSH and free T(4) in a large population is complex and nonlinear and differs by age and sex. The Journal of clinical endocrinology and metabolism 2013; 98:2936-2943
- Roelfsema F, Pereira AM, Adriaanse R, Endert E, Fliers E, Romijn JA, Veldhuis JD. Thyrotropin secretion in mild and severe primary hypothyroidism is distinguished by amplified burst mass and Basal secretion with increased spikiness and approximate entropy. The Journal of clinical endocrinology and metabolism 2010; 95:928-934
- Snyder PJ, Utiger RD. Inhibition of thyrotropin response to thyrotropin-releasing hormone by small quantities of thyroid hormones. The Journal of clinical investigation 1972; 51:2077-2084
- Vagenakis AG, Rapoport B, Azizi F, Portnay GI, Braverman LE, Ingbar SH. Hyperresponse to thyrotropin-releasing hormone accompanying small decreases in serum thyroid hormone concentrations. The Journal of clinical investigation 1974; 54:913-918
- Meier CA, Maisey MN, Lowry A, Muller J, Smith MA. Interindividual differences in the pituitary-thyroid axis influence the interpretation of thyroid function tests. Clinical endocrinology 1993; 39:101-107
- Meikle AW, Stringham JD, Woodward MG, Nelson JC. Hereditary and environmental influences on the variation of thyroid hormones in normal male twins. The Journal of clinical endocrinology and metabolism 1988; 66:588-592
- Hansen PS, Brix TH, Sorensen TI, Kyvik KO, Hegedus L. Major genetic influence on the regulation of the pituitary-thyroid axis: a study of healthy Danish twins. The Journal of clinical endocrinology and metabolism 2004; 89:1181-1187
- Panicker V, Wilson SG, Spector TD, Brown SJ, Falchi M, Richards JB, Surdulescu GL, Lim EM, Fletcher SJ, Walsh JP. Heritability of serum TSH, free T4 and free T3 concentrations: a study of a large UK twin cohort. ClinEndocrinol(Oxf) 2008; 68:652-659
- Medici M, Visser TJ, Peeters RP. Genetics of thyroid function. Best practice & research Clinical endocrinology & metabolism 2017; 31:129-142
- Medici M, Visser WE, Visser TJ, Peeters RP. Genetic determination of the hypothalamic-pituitary-thyroid axis: where do we stand? Endocrine reviews 2015; 36:214-244
- Medici M, van der Deure WM, Verbiest M, Vermeulen SH, Hansen PS, Kiemeney LA, Hermus AR, Breteler MM, Hofman A, Hegedus L, Kyvik KO, den Heijer M, Uitterlinden AG, Visser TJ, Peeters RP. A large-scale association analysis of 68 thyroid hormone pathway genes with serum TSH and FT4 levels. European journal of endocrinology 2011; 164:781-788
- Eisenberg MC, Santini F, Marsili A, Pinchera A, DiStefano JJ, 3rd. TSH regulation dynamics in central and extreme primary hypothyroidism. Thyroid 2010; 20:1215-1228
- Lewis GF, Alessi CA, Imperial JG, Refetoff S. Low serum free thyroxine index in ambulating elderly is due to a resetting of the threshold of thyrotropin feedback suppression. The Journal of clinical endocrinology and metabolism 1991; 73:843-849
- Carle A, Laurberg P, Pedersen IB, Perrild H, Ovesen L, Rasmussen LB, Jorgensen T, Knudsen N. Age modifies the pituitary TSH response to thyroid failure. Thyroid 2007; 17:139-144
- Kempers MJ, van Trotsenburg AS, van Tijn DA, Bakker E, Wiedijk BM, Endert E, de Vijlder JJ, Vulsma T. Disturbance of the fetal thyroid hormone state has long-term consequences for treatment of thyroidal and central congenital hypothyroidism. The Journal of clinical endocrinology and metabolism 2005; 90:4094-4100
- Fischer HR, Hackeng WH, Schopman W, Silberbusch J. Analysis of factors in hyperthyroidism, which determine the duration of suppressive treatment before recovery of thyroid stimulating hormone secretion. Clinical endocrinology 1982; 16:575-585
- Orgiazzi J. Anti-TSH receptor antibodies in clinical practice. Endocrinology and metabolism clinics of North America 2000; 29:339-355, vii
- Pekary AE, Jackson IM, Goodwin TM, Pang XP, Hein MD, Hershman JM. Increased in vitro thyrotropic activity of partially sialated human chorionic gonadotropin extracted from hydatidiform moles of patients with hyperthyroidism. The Journal of clinical endocrinology and metabolism 1993; 76:70-74
- Brenta G, Schnitman M, Fretes O, Facco E, Gurfinkel M, Damilano S, Pacenza N, Blanco A, Gonzalez E, Pisarev MA. Comparative efficacy and side effects of the treatment of euthyroid goiter with levo-thyroxine or triiodothyroacetic acid. The Journal of clinical endocrinology and metabolism 2003; 88:5287-5292
- Ladenson PW. Thyroid hormone analogues: ready for prime time. Thyroid 2011; 21:101-102
- Re RN, Kourides IA, Ridgway EC, Weintraub BD, Maloof F. The effect of glucocorticoid administration on human pituitary secretion of thyrotropin and prolactin. The Journal of clinical endocrinology and metabolism 1976; 43:338-346
- Van den Berghe G, de Zegher F, Lauwers P. Dopamine and the sick euthyroid syndrome in critical illness. Clinical endocrinology 1994; 41:731-737
- Sato T, Suzuki Y, Taketani T, Ishiguro K, Nakajima H. Age-related change in pituitary threshold for TSH release during thyroxine replacement therapy for cretinism. The Journal of clinical endocrinology and metabolism 1977; 44:553-559
- Van Sande J, Parma J, Tonacchera M, Swillens S, Dumont J, Vassart G. Somatic and germline mutations of the TSH receptor gene in thyroid diseases. The Journal of clinical endocrinology and metabolism 1995; 80:2577-2585
- Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best practice & research Clinical endocrinology & metabolism 2007; 21:277-305
- Smith PJ, Surks MI. Multiple effects of 5,5'-diphenylhydantoin on the thyroid hormone system. Endocrine reviews 1984; 5:514-524
- Bonomi M, Proverbio MC, Weber G, Chiumello G, Beck-Peccoz P, Persani L. Hyperplastic pituitary gland, high serum glycoprotein hormone alpha-subunit, and variable circulating thyrotropin (TSH) levels as hallmark of central hypothyroidism due to mutations of the TSH beta gene. The Journal of clinical endocrinology and metabolism 2001; 86:1600-1604
- Oliveira JH, Persani L, Beck-Peccoz P, Abucham J. Investigating the paradox of hypothyroidism and increased serum thyrotropin (TSH) levels in Sheehan's syndrome: characterization of TSH carbohydrate content and bioactivity. JClinEndocrinolMetab 2001; 86:1694-1699
- Bjerner J, Nustad K, Norum LF, Olsen KH, Bormer OP. Immunometric assay interference: incidence and prevention. Clinical chemistry 2002; 48:613-621
- Monchamp T, Chopra IJ, Wah DT, Butch AW. Falsely elevated thyroid hormone levels due to anti-sheep antibody interference in an automated electrochemiluminescent immunoassay. Thyroid 2007; 17:271-275
- Stockigt JR. Thyroid hormone binding and variants of transport proteins. In: DeGroot L, L. JJ, eds. Endocrinology. 6th ed. Philadelphia: Saunders; 2010:392-402.
- Sakata S, Nakamura S, Miura K. Autoantibodies against thyroid hormones or iodothyronine. Implications in diagnosis, thyroid function, treatment, and pathogenesis. Annals of internal medicine 1985; 103:579-589
- Stockigt JR, Lim CF. Medications that distort in vitro tests of thyroid function, with particular reference to estimates of serum free thyroxine. Best practice & research Clinical endocrinology & metabolism 2009; 23:753-767
- Mendel CM, Frost PH, Kunitake ST, Cavalieri RR. Mechanism of the heparin-induced increase in the concentration of free thyroxine in plasma. The Journal of clinical endocrinology and metabolism 1987; 65:1259-1264
- Jaume JC, Mendel CM, Frost PH, Greenspan FS, Laughton CW. Extremely low doses of heparin release lipase activity into the plasma and can thereby cause artifactual elevations in the serum-free thyroxine concentration as measured by equilibrium dialysis. Thyroid 1996; 6:79-83
- Spencer CA, Takeuchi M, Kazarosyan M. Current status and performance goals for serum thyrotropin (TSH) assays. Clinical chemistry 1996; 42:140-145
- Verheecke P. Free triiodothyronine concentration in serum of 1050 euthyroid children is inversely related to their age. Clinical chemistry 1997; 43:963-967
- Mariotti S, Barbesino G, Caturegli P, Bartalena L, Sansoni P, Fagnoni F, Monti D, Fagiolo U, Franceschi C, Pinchera A. Complex alteration of thyroid function in healthy centenarians. The Journal of clinical endocrinology and metabolism 1993; 77:1130-1134
- Kim B. Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid 2008; 18:141-144
- Thaler MA, Seifert-Klauss V, Luppa PB. The biomarker sex hormone-binding globulin - from established applications to emerging trends in clinical medicine. Best practice & research Clinical endocrinology & metabolism 2015; 29:749-760
- Gronhagen-Riska C, Fyhrquist F, Valimaki M, Lamberg BA. Thyroid hormones affect serum angiotensin I converting enzyme levels. Acta medica Scandinavica 1985; 217:259-264
- Stockigt JR, Stevens V, White EL, Barlow JW. "Unbound analog" radioimmunoassays for free thyroxin measure the albumin-bound hormone fraction. Clinical chemistry 1983; 29:1408-1410
- Surks MI, DeFesi CR. Normal serum free thyroid hormone concentrations in patients treated with phenytoin or carbamazepine. A paradox resolved. Jama 1996; 275:1495-1498
- Beck-Peccoz P, Romelli PB, Cattaneo MG, Faglia G, White EL, Barlow JW, Stockigt JR. Evaluation of free thyroxine methods in the presence of iodothyronine-binding autoantibodies. The Journal of clinical endocrinology and metabolism 1984; 58:736-739
- Despres N, Grant AM. Antibody interference in thyroid assays: a potential for clinical misinformation. Clinical chemistry 1998; 44:440-454
- Norden AG, Jackson RA, Norden LE, Griffin AJ, Barnes MA, Little JA. Misleading results from immunoassays of serum free thyroxine in the presence of rheumatoid factor. Clinical chemistry 1997; 43:957-962
- Beck-Peccoz P, Persani L. Variable biological activity of thyroid-stimulating hormone. EurJEndocrinol 1994; 131:331-340
- Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary tumors. Endocrine reviews 1996; 17:610-638
- Ekins R. Measurement of free hormones in blood. Endocrine reviews 1990; 11:5-46
- Grebe S. Laboratory testing in thyroid disorders. In: Luster M, Duntas L, Wartofsky L, eds. The Thyroid and Its Diseases. A Comprehensive Guide for the Clinician: Springer International Publishing AG, part of Springer Nature; 2019:129-160.
- Braverman LE. Evaluation of thyroid status in patients with thyrotoxicosis. Clinical chemistry 1996; 42:174-178
- Martino E, Bartalena L, Bogazzi F, Braverman LE. The effects of amiodarone on the thyroid. Endocrine reviews 2001; 22:240-254
- Bartalena L, Bogazzi F, Chiovato L, Hubalewska-Dydejczyk A, Links TP, Vanderpump M. 2018 European Thyroid Association (ETA) Guidelines for the Management of Amiodarone-Associated Thyroid Dysfunction. European thyroid journal 2018; 7:55-66
- Dalan R, Kon W, K LM. Phenotypic expression and challenges of a distinct form of thyrotoxicosis. Triiodothyronine-predominant Graves’ disease – aggressive, refractory and anything but banal. The Endocrinologist 2008; 18:90-94
- Takamatsu J, Hosoya T, Naito N, Yoshimura H, Kohno Y, Tarutani O, Kuma K, Sakane S, Takeda K, Mozai T. Enhanced thyroid iodine metabolism in patients with triiodothyronine-predominant Graves' disease. The Journal of clinical endocrinology and metabolism 1988; 66:147-152
- Takamatsu J, Kuma K, Mozai T. Serum triiodothyronine to thyroxine ratio: a newly recognized predictor of the outcome of hyperthyroidism due to Graves' disease. The Journal of clinical endocrinology and metabolism 1986; 62:980-983
- Sobrinho LG, Limbert ES, Santos MA. Thyroxine toxicosis in patients with iodine induced thyrotoxicosis. The Journal of clinical endocrinology and metabolism 1977; 45:25-29
- Amino N, Yabu Y, Miki T, Morimoto S, Kumahara Y, Mori H, Iwatani Y, Nishi K, Nakatani K, Miyai K. Serum ratio of triiodothyronine to thyroxine, and thyroxine-binding globulin and calcitonin concentrations in Graves' disease and destruction-induced thyrotoxicosis. The Journal of clinical endocrinology and metabolism 1981; 53:113-116
- Spencer CA, Schwarzbein D, Guttler RB, LoPresti JS, Nicoloff JT. Thyrotropin (TSH)-releasing hormone stimulation test responses employing third and fourth generation TSH assays. The Journal of clinical endocrinology and metabolism 1993; 76:494-498
- Hartoft-Nielsen ML, Lange M, Rasmussen AK, Scherer S, Zimmermann-Belsing T, Feldt-Rasmussen U. Thyrotropin-releasing hormone stimulation test in patients with pituitary pathology. HormRes 2004; 61:53-57
- Laurberg P. Persistent problems with the specificity of immunometric TSH assays. Thyroid 1993; 3:279-283
- Feldt-Rasmussen U. Serum thyroglobulin and thyroglobulin autoantibodies in thyroid diseases. Pathogenic and diagnostic aspects. Allergy 1983; 38:369-387
- Schlumberger M, Hitzel A, Toubert ME, Corone C, Troalen F, Schlageter MH, Claustrat F, Koscielny S, Taieb D, Toubeau M, Bonichon F, Borson-Chazot F, Leenhardt L, Schvartz C, Dejax C, Brenot-Rossi I, Torlontano M, Tenenbaum F, Bardet S, Bussiere F, Girard JJ, Morel O, Schneegans O, Schlienger JL, Prost A, So D, Archambeaud F, Ricard M, Benhamou E. Comparison of seven serum thyroglobulin assays in the follow-up of papillary and follicular thyroid cancer patients. The Journal of clinical endocrinology and metabolism 2007; 92:2487-2495
- Mazzaferri EL, Kloos RT. Clinical review 128: Current approaches to primary therapy for papillary and follicular thyroid cancer. The Journal of clinical endocrinology and metabolism 2001; 86:1447-1463
- Pelttari H, Valimaki MJ, Loyttyniemi E, Schalin-Jantti C. Post-ablative serum thyroglobulin is an independent predictor of recurrence in low-risk differentiated thyroid carcinoma: a 16-year follow-up study. European journal of endocrinology 2010; 163:757-763
- Giovanella L, Feldt-Rasmussen U, Verburg FA, Grebe SK, Plebani M, Clark PM. Thyroglobulin measurement by highly sensitive assays: focus on laboratory challenges. Clinical chemistry and laboratory medicine 2015; 53:1301-1314
- Giovanella L, Clark PM, Chiovato L, Duntas L, Elisei R, Feldt-Rasmussen U, Leenhardt L, Luster M, Schalin-Jantti C, Schott M, Seregni E, Rimmele H, Smit J, Verburg FA. Thyroglobulin measurement using highly sensitive assays in patients with differentiated thyroid cancer: a clinical position paper. European journal of endocrinology 2014; 171:R33-46
- Cohen JH, 3rd, Ingbar SH, Braverman LE. Thyrotoxicosis due to ingestion of excess thyroid hormone. Endocrine reviews 1989; 10:113-124
- Pacini F, Fugazzola L, Lippi F, Ceccarelli C, Centoni R, Miccoli P, Elisei R, Pinchera A. Detection of thyroglobulin in fine needle aspirates of nonthyroidal neck masses: a clue to the diagnosis of metastatic differentiated thyroid cancer. The Journal of clinical endocrinology and metabolism 1992; 74:1401-1404
- Kim MJ, Kim EK, Kim BM, Kwak JY, Lee EJ, Park CS, Cheong WY, Nam KH. Thyroglobulin measurement in fine-needle aspirate washouts: the criteria for neck node dissection for patients with thyroid cancer. Clinical endocrinology 2009; 70:145-151
- Knudsen N, Bulow I, Jorgensen T, Perrild H, Ovesen L, Laurberg P. Serum Tg--a sensitive marker of thyroid abnormalities and iodine deficiency in epidemiological studies. The Journal of clinical endocrinology and metabolism 2001; 86:3599-3603
- Verburg FA, Luster M, Cupini C, Chiovato L, Duntas L, Elisei R, Feldt-Rasmussen U, Rimmele H, Seregni E, Smit JW, Theimer C, Giovanella L. Implications of thyroglobulin antibody positivity in patients with differentiated thyroid cancer: a clinical position statement. Thyroid 2013; 23:1211-1225
- Feldt-Rasmussen U, Giovanella L. Thyroglobulin and Tg antibodies. In: Luster M, Duntas L, Wartofsky L, eds. The Thyroid and Its Diseases. A Comprehensive Guide for the Clinician: Springer International Publishing AG, part of Springer Nature; 2019:655-672.
- Feldt-Rasmussen U, Verburg FA, Luster M, Cupini C, Chiovato L, Duntas L, Elisei R, Rimmele H, Seregni E, Smit JW, Theimer C, Giovanella L. Thyroglobulin autoantibodies as surrogate biomarkers in the management of patients with differentiated thyroid carcinoma. Current medicinal chemistry 2014; 21:3687-3692
- Feldt-Rasmussen U, Rasmussen AK. Serum thyroglobulin (Tg) in presence of thyroglobulin autoantibodies (TgAb). Clinical and methodological relevance of the interaction between Tg and TgAb in vitro and in vivo. Journal of endocrinological investigation 1985; 8:571-576
- Bliddal S, Boas M, Hilsted L, Friis-Hansen L, Juul A, Larsen T, Tabor A, Faber J, Precht DH, Feldt-Rasmussen U. Increase in thyroglobulin antibody and thyroid peroxidase antibody levels, but not preterm birth-rate, in pregnant Danish women upon iodine fortification. European journal of endocrinology 2017; 176:603-612
- Feldt-Rasmussen U. Analytical and clinical performance goals for testing autoantibodies to thyroperoxidase, thyroglobulin, and thyrotropin receptor. Clinical chemistry 1996; 42:160-163
- Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, Grimley Evans J, Hasan DM, Rodgers H, Tunbridge F, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clinical endocrinology 1995; 43:55-68
- Costagliola S, Morgenthaler NG, Hoermann R, Badenhoop K, Struck J, Freitag D, Poertl S, Weglohner W, Hollidt JM, Quadbeck B, Dumont JE, Schumm-Draeger PM, Bergmann A, Mann K, Vassart G, Usadel KH. Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves' disease. The Journal of clinical endocrinology and metabolism 1999; 84:90-97
- Schott M, Hermsen D, Broecker-Preuss M, Casati M, Mas JC, Eckstein A, Gassner D, Golla R, Graeber C, van Helden J, Inomata K, Jarausch J, Kratzsch J, Miyazaki N, Moreno MA, Murakami T, Roth HJ, Stock W, Noh JY, Scherbaum WA, Mann K. Clinical value of the first automated TSH receptor autoantibody assay for the diagnosis of Graves' disease (GD): an international multicentre trial. Clinical endocrinology 2009; 71:566-573
- Laurberg P, Nygaard B, Glinoer D, Grussendorf M, Orgiazzi J. Guidelines for TSH-receptor antibody measurements in pregnancy: results of an evidence-based symposium organized by the European Thyroid Association. European journal of endocrinology 1998; 139:584-586
- Kahaly GJ, Bartalena L, Hegedüs L, Leenhardt L, Poppe K, Pearce SH. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. European thyroid journal 2018:167–186
- Hesarghatta Shyamasunder A, Abraham P. Measuring TSH receptor antibody to influence treatment choices in Graves' disease. Clinical endocrinology 2017; 86:652-657
- Massart C, Gibassier J, d'Herbomez M. Clinical value of M22-based assays for TSH-receptor antibody (TRAb) in the follow-up of antithyroid drug treated Graves' disease: comparison with the second generation human TRAb assay. Clinica chimica acta; international journal of clinical chemistry 2009; 407:62-66
- Barbesino G, Tomer Y. Clinical review: Clinical utility of TSH receptor antibodies. The Journal of clinical endocrinology and metabolism 2013; 98:2247-2255
- Dumitrescu AM, Korwutthikulrangsri M, Refetoff S. Impaired sensitivity to thyroid hormone: defects of transport, metabolism, and action. In: Braverman LE, Cooper DS, Kopp P, eds. Werner and Ingbar's the thyroid: a fundamental and clinical text. 11th ed. Philadelphia: Lippincott, Williams & Wilkins; 2020:868-907.
- Burmeister LA. Reverse T3 does not reliably differentiate hypothyroid sick syndrome from euthyroid sick syndrome. Thyroid 1995; 5:435-441
- Roti E, Uberti ED. Iodine excess and hyperthyroidism. Thyroid 2001; 11:493-500
- Weber C, Scholz GH, Lamesch P, Paschke R. Thyroidectomy in iodine induced thyrotoxic storm. Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association 1999; 107:468-472
- O'Connell R, Parkin L, Manning P, Bell D, Herbison P, Holmes J. A cluster of thyrotoxicosis associated with consumption of a soy milk product. Australian and New Zealand journal of public health 2005; 29:511-512
- Vejbjerg P, Knudsen N, Perrild H, Laurberg P, Andersen S, Rasmussen LB, Ovesen L, Jorgensen T. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281-1286
- Carle A, Andersen SL, Boelaert K, Laurberg P. MANAGEMENT OF ENDOCRINE DISEASE: Subclinical thyrotoxicosis: prevalence, causes and choice of therapy. European journal of endocrinology 2017; 176:R325-r337
- Karmisholt J, Andersen S, Laurberg P. Interval between tests and thyroxine estimation method influence outcome of monitoring of subclinical hypothyroidism. The Journal of clinical endocrinology and metabolism 2008; 93:1634-1640
- Hollander CS, Mitsuma T, Nihei N, Shenkman L, Burday SZ, Blum M. Clinical and laboratory observations in cases of triiodothyronine toxicosis confirmed by radioimmunoassay. Lancet 1972; 1:609-611
- Sterling K, Refetoff S, Selenkow HA. T3 thyrotoxicosis. Thyrotoxicosis due to elevated serum triiodothyronine levels. Jama 1970; 213:571-575
- Lum SM, Kaptein EM, Nicoloff JT. Influence of nonthyroidal illnesses on serum thyroid hormone indices in hyperthyroidism. The Western journal of medicine 1983; 138:670-675
- Inada M, Sterling K. Thyroxine transport in thyrotoxicosis and hypothyroidism. The Journal of clinical investigation 1967; 46:1442-1450
- Nauman JA, Nauman A, Werner SC. Total and free triiodothyronine in human serum. The Journal of clinical investigation 1967; 46:1346-1355
- Homsanit M, Sriussadaporn S, Vannasaeng S, Peerapatdit T, Nitiyanant W, Vichayanrat A. Efficacy of single daily dosage of methimazole vs. propylthiouracil in the induction of euthyroidism. Clinical endocrinology 2001; 54:385-390
- Davies PH, Franklyn JA, Daykin J, Sheppard MC. The significance of TSH values measured in a sensitive assay in the follow-up of hyperthyroid patients treated with radioiodine. The Journal of clinical endocrinology and metabolism 1992; 74:1189-1194
- Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, Rivkees SA, Samuels M, Sosa JA, Stan MN, Walter MA. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid 2016; 26:1343-1421
- Biondi B, Bartalena L, Cooper DS, Hegedus L, Laurberg P, Kahaly GJ. The 2015 European Thyroid Association Guidelines on Diagnosis and Treatment of Endogenous Subclinical Hyperthyroidism. European thyroid journal 2015; 4:149-163
- Persani L, Brabant G, Dattani M, Bonomi M, Feldt-Rasmussen U, Fliers E, Gruters A, Maiter D, Schoenmakers N, van Trotsenburg ASP. 2018 European Thyroid Association (ETA) Guidelines on the Diagnosis and Management of Central Hypothyroidism. European thyroid journal 2018; 7:225-237
- Ross DS, Daniels GH, Gouveia D. The use and limitations of a chemiluminescent thyrotropin assay as a single thyroid function test in an out-patient endocrine clinic. The Journal of clinical endocrinology and metabolism 1990; 71:764-769
- Fish LH, Schwartz HL, Cavanaugh J, Steffes MW, Bantle JP, Oppenheimer JH. Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. The New England journal of medicine 1987; 316:764-770
- Shimon I, Cohen O, Lubetsky A, Olchovsky D. Thyrotropin suppression by thyroid hormone replacement is correlated with thyroxine level normalization in central hypothyroidism. Thyroid 2002; 12:823-827
- Ferretti E, Persani L, Jaffrain-Rea ML, Giambona S, Tamburrano G, Beck-Peccoz P. Evaluation of the adequacy of levothyroxine replacement therapy in patients with central hypothyroidism. JClinEndocrinolMetab 1999; 84:924-929
- Ain KB, Pucino F, Shiver TM, Banks SM. Thyroid hormone levels affected by time of blood sampling in thyroxine-treated patients. Thyroid 1993; 3:81-85
- Centanni M, Gargano L, Canettieri G, Viceconti N, Franchi A, Delle Fave G, Annibale B. Thyroxine in goiter, Helicobacter pylori infection, and chronic gastritis. The New England journal of medicine 2006; 354:1787-1795
- Ianiro G, Mangiola F, Di Rienzo TA, Bibbo S, Franceschi F, Greco AV, Gasbarrini A. Levothyroxine absorption in health and disease, and new therapeutic perspectives. European review for medical and pharmacological sciences 2014; 18:451-456
- Mersebach H, Rasmussen AK, Kirkegaard L, Feldt-Rasmussen U. Intestinal adsorption of levothyroxine by antacids and laxatives: case stories and in vitro experiments. Pharmacology & toxicology 1999; 84:107-109
- Singh N, Weisler SL, Hershman JM. The acute effect of calcium carbonate on the intestinal absorption of levothyroxine. Thyroid 2001; 11:967-971
- Benvenga S, Bartolone L, Pappalardo MA, Russo A, Lapa D, Giorgianni G, Saraceno G, Trimarchi F. Altered intestinal absorption of L-thyroxine caused by coffee. Thyroid 2008; 18:293-301
- Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, Pacini F, Randolph GW, Sawka AM, Schlumberger M, Schuff KG, Sherman SI, Sosa JA, Steward DL, Tuttle RM, Wartofsky L. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016; 26:1-133
- Bartalena L, Pinchera A. Levothyroxine suppressive therapy: harmful and useless or harmless and useful? Journal of endocrinological investigation 1994; 17:675-677
- Leite V. The Importance of the 2015 American Thyroid Association Guidelines for Adults with Thyroid Nodules and Differentiated Thyroid Cancer in Minimising Overdiagnosis and Overtreatment of Thyroid Carcinoma. European endocrinology 2018; 14:13-14
- Surks MI, Schadlow AR, Oppenheimer JH. A new radioimmunoassay for plasma L-triiodothyronine: measurements in thyroid disease and in patients maintained on hormonal replacement. The Journal of clinical investigation 1972; 51:3104-3113
- Larsen PR. Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones. The New England journal of medicine 1982; 306:23-32
- Roti E, Minelli R, Gardini E, Braverman LE. The use and misuse of thyroid hormone. Endocrine reviews 1993; 14:401-423
- Wiersinga WM, Duntas L, Fadeyev V, Nygaard B, Vanderpump MP. 2012 ETA Guidelines: The Use of L-T4 + L-T3 in the Treatment of Hypothyroidism. European thyroid journal 2012; 1:55-71
- Kim BW, Bianco AC. For some, L-thyroxine replacement might not be enough: a genetic rationale. The Journal of clinical endocrinology and metabolism 2009; 94:1521-1523
- Panicker V, Saravanan P, Vaidya B, Evans J, Hattersley AT, Frayling TM, Dayan CM. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. The Journal of clinical endocrinology and metabolism 2009; 94:1623-1629
- De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. The Journal of clinical endocrinology and metabolism 1999; 84:151-164
- Medici BB, la Cour JL, Michaelsson LF, Faber JO, Nygaard B. Neither Baseline nor Changes in Serum Triiodothyronine during Levothyroxine/Liothyronine Combination Therapy Predict a Positive Response to This Treatment Modality in Hypothyroid Patients with Persistent Symptoms. European thyroid journal 2017; 6:89-93
- Michaelsson LF, Medici BB, la Cour JL, Selmer C, Roder M, Perrild H, Knudsen N, Faber J, Nygaard B. Treating Hypothyroidism with Thyroxine/Triiodothyronine Combination Therapy in Denmark: Following Guidelines or Following Trends? European thyroid journal 2015; 4:174-180
- Van den Berghe G. Novel insights into the neuroendocrinology of critical illness. European journal of endocrinology 2000; 143:1-13
- Mebis L, Debaveye Y, Visser TJ, Van den Berghe G. Changes within the thyroid axis during the course of critical illness. Endocrinology and metabolism clinics of North America 2006; 35:807-821, x
- Van den Berghe G, Wouters P, Weekers F, Mohan S, Baxter RC, Veldhuis JD, Bowers CY, Bouillon R. Reactivation of pituitary hormone release and metabolic improvement by infusion of growth hormone-releasing peptide and thyrotropin-releasing hormone in patients with protracted critical illness. The Journal of clinical endocrinology and metabolism 1999; 84:1311-1323
- Felicetta JV, Green WL, Haas LB, Kenny MA, Sherrard DJ, Brunzell JD. Thyroid function and lipids in patients with chronic renal disease treated by hemodialysis: with comments on the "free thyroxine index". Metabolism: clinical and experimental 1979; 28:756-763
- Feldt-Rasmussen U. Thyroid function tests and the effects og drugs. In: Wass J, Arlt W, Semple R, eds. Oxford Textbook of Endocrinology and Diabetes. 3rd ed: Oxford University Press; 2021:346-352.
- John-Kalarickal J, Pearlman G, Carlson HE. New medications which decrease levothyroxine absorption. Thyroid 2007; 17:763-765
- Liwanpo L, Hershman JM. Conditions and drugs interfering with thyroxine absorption. Best practice & research Clinical endocrinology & metabolism 2009; 23:781-792
- Siraj ES, Gupta MK, Reddy SS. Raloxifene causing malabsorption of levothyroxine. Archives of internal medicine 2003; 163:1367-1370
- Stevenson HP, Archbold GP, Johnston P, Young IS, Sheridan B. Misleading serum free thyroxine results during low molecular weight heparin treatment. Clinical chemistry 1998; 44:1002-1007
- Hawkins RC. Furosemide interference in newer free thyroxine assays. Clinical chemistry 1998; 44:2550-2551
- Sapin R, Schlienger JL, Gasser F, Noel E, Lioure B, Grunenberger F, Goichot B, Grucker D. Intermethod discordant free thyroxine measurements in bone marrow-transplanted patients. Clinical chemistry 2000; 46:418-422
- Franklyn JA, Black EG, Betteridge J, Sheppard MC. Comparison of second and third generation methods for measurement of serum thyrotropin in patients with overt hyperthyroidism, patients receiving thyroxine therapy, and those with nonthyroidal illness. The Journal of clinical endocrinology and metabolism 1994; 78:1368-1371
- Haymart MR, Esfandiari NH, Stang MT, Sosa JA. Controversies in the Management of Low-Risk Differentiated Thyroid Cancer. Endocrine reviews 2017; 38:351-378
- Guimaraes V, DeGroot LJ. Moderate hypothyroidism in preparation for whole body 131I scintiscans and thyroglobulin testing. Thyroid 1996; 6:69-73
- Ladenson PW, Braverman LE, Mazzaferri EL, Brucker-Davis F, Cooper DS, Garber JR, Wondisford FE, Davies TF, DeGroot LJ, Daniels GH, Ross DS, Weintraub BD. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma. The New England journal of medicine 1997; 337:888-896
- Maxon HR, 3rd, Smith HS. Radioiodine-131 in the diagnosis and treatment of metastatic well differentiated thyroid cancer. Endocrinology and metabolism clinics of North America 1990; 19:685-718
- Cailleux AF, Baudin E, Travagli JP, Ricard M, Schlumberger M. Is diagnostic iodine-131 scanning useful after total thyroid ablation for differentiated thyroid cancer? The Journal of clinical endocrinology and metabolism 2000; 85:175-178
- Wartofsky L. Management of low-risk well-differentiated thyroid cancer based only on thyroglobulin measurement after recombinant human thyrotropin. Thyroid 2002; 12:583-590
- Giovanella L, Avram AM, Clerc J, Hindie E, Taieb D, Verburg FA. Postoperative serum thyroglobulin and neck ultrasound to drive decisions about iodine-131 therapy in patients with differentiated thyroid carcinoma: an evidence-based strategy? European journal of nuclear medicine and molecular imaging 2018;
- Pacini F, Molinaro E, Castagna MG, Agate L, Elisei R, Ceccarelli C, Lippi F, Taddei D, Grasso L, Pinchera A. Recombinant human thyrotropin-stimulated serum thyroglobulin combined with neck ultrasonography has the highest sensitivity in monitoring differentiated thyroid carcinoma. The Journal of clinical endocrinology and metabolism 2003; 88:3668-3673
- Brassard M, Borget I, Edet-Sanson A, Giraudet AL, Mundler O, Toubeau M, Bonichon F, Borson-Chazot F, Leenhardt L, Schvartz C, Dejax C, Brenot-Rossi I, Toubert ME, Torlontano M, Benhamou E, Schlumberger M. Long-term follow-up of patients with papillary and follicular thyroid cancer: a prospective study on 715 patients. The Journal of clinical endocrinology and metabolism 2011; 96:1352-1359
- Sunny SS, Hephzibah J, Mathew D, Bondu JD, Shanthly N, Oommen R. Stimulated Serum Thyroglobulin Levels versus Unstimulated Serum Thyroglobulin in the Follow-up of Patients with Papillary Thyroid Carcinoma. World journal of nuclear medicine 2018; 17:41-45
- Spencer CA, Lopresti JS. Measuring thyroglobulin and thyroglobulin autoantibody in patients with differentiated thyroid cancer. Nature clinical practice Endocrinology & metabolism 2008; 4:223-233
- Spencer CA. Clinical review: Clinical utility of thyroglobulin antibody (TgAb) measurements for patients with differentiated thyroid cancers (DTC). The Journal of clinical endocrinology and metabolism 2011; 96:3615-3627
- Chopra IJ, Solomon DH, Huang TS. Serum thyrotropin in hospitalized psychiatric patients: evidence for hyperthyrotropinemia as measured by an ultrasensitive thyrotropin assay. Metabolism: clinical and experimental 1990; 39:538-543
- De Groot L, Abalovich M, Alexander EK, Amino N, Barbour L, Cobin RH, Eastman CJ, Lazarus JH, Luton D, Mandel SJ, Mestman J, Rovet J, Sullivan S. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. The Journal of clinical endocrinology and metabolism 2012; 97:2543-2565
- Subclinical hypothyroidism in the infertile female population: a guideline. Fertility and sterility 2015; 104:545-553
- Patton PE, Samuels MH, Trinidad R, Caughey AB. Controversies in the management of hypothyroidism during pregnancy. Obstetrical & gynecological survey 2014; 69:346-358
- Vila L, Velasco I, Gonzalez S, Morales F, Sanchez E, Torrejon S, Soldevila B, Stagnaro-Green A, Puig-Domingo M. Controversies in endocrinology: On the need for universal thyroid screening in pregnant women. European journal of endocrinology 2014; 170:R17-30
- Korevaar TI. Evidence-Based Tightrope Walking: The 2017 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease During Pregnancy and the Postpartum. Thyroid 2017; 27:309-311
- Negro R, Stagnaro-Green A. Clinical aspects of hyperthyroidism, hypothyroidism, and thyroid screening in pregnancy. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists 2014; 20:597-607
- Maraka S, Singh Ospina NM, Mastorakos G, O'Keeffe DT. Subclinical Hypothyroidism in Women Planning Conception and During Pregnancy: Who Should Be Treated and How? Journal of the Endocrine Society 2018; 2:533-546
- Feldt-Rasmussen U, Bliddal S, Rasmussen AK, Boas M, Hilsted L, Main K. Challenges in interpretation of thyroid function tests in pregnant women with autoimmune thyroid disease. Journal of thyroid research 2011; 2011:598712
- Ball R, Freedman DB, Holmes JC, Midgley JE, Sheehan CP. Low-normal concentrations of free thyroxin in serum in late pregnancy: physiological fact, not technical artefact. Clinical chemistry 1989; 35:1891-1896
- Roti E, Gardini E, Minelli R, Bianconi L, Flisi M. Thyroid function evaluation by different commercially available free thyroid hormone measurement kits in term pregnant women and their newborns. Journal of endocrinological investigation 1991; 14:1-9
- Sapin R, d'Herbomez M. Free thyroxine measured by equilibrium dialysis and nine immunoassays in sera with various serum thyroxine-binding capacities. Clinical chemistry 2003; 49:1531-1535
- d'Herbomez M, Forzy G, Gasser F, Massart C, Beaudonnet A, Sapin R. Clinical evaluation of nine free thyroxine assays: persistent problems in particular populations. Clinical chemistry and laboratory medicine 2003; 41:942-947
- Lee RH, Spencer CA, Mestman JH, Miller EA, Petrovic I, Braverman LE, Goodwin TM. Free T4 immunoassays are flawed during pregnancy. American journal of obstetrics and gynecology 2009; 200:260.e261-266
- Feldt-Rasmussen U. Laboratory measurement of thyroid-related hormones, proteins, and autoantibodies in serum. In: Braverman LE, Cooper DS, Kopp P, eds. Werner and Ingbar's the thyroid: a fundamental and clinical text. 11th ed. Philadelphia: Lippincott, Williams & Wilkins; 2020:868-907.
- Bliddal S, Feldt-Rasmussen U, Boas M, Faber J, Juul A, Larsen T, Precht DH. Gestational age-specific reference ranges from different laboratories misclassify pregnant women's thyroid status: comparison of two longitudinal prospective cohort studies. European journal of endocrinology 2014; 170:329-339
- Welsh KJ, Soldin SJ. DIAGNOSIS OF ENDOCRINE DISEASE: How reliable are free thyroid and total T3 hormone assays? EurJEndocrinol 2016; 175:R255-R263
- Boas M, Forman JL, Juul A, Feldt-Rasmussen U, Skakkebaek NE, Hilsted L, Chellakooty M, Larsen T, Larsen JF, Petersen JH, Main KM. Narrow intra-individual variation of maternal thyroid function in pregnancy based on a longitudinal study on 132 women. European journal of endocrinology 2009; 161:903-910
- Hallengren B, Lantz M, Andreasson B, Grennert L. Pregnant women on thyroxine substitution are often dysregulated in early pregnancy. Thyroid 2009; 19:391-394
- Alexander EK, Marqusee E, Lawrence J, Jarolim P, Fischer GA, Larsen PR. Timing and magnitude of increases in levothyroxine requirements during pregnancy in women with hypothyroidism. The New England journal of medicine 2004; 351:241-249
- Chan S, Boelaert K. Optimal management of hypothyroidism, hypothyroxinaemia and euthyroid TPO antibody positivity preconception and in pregnancy. Clinical endocrinology 2015; 82:313-326
- Vaidya B, Anthony S, Bilous M, Shields B, Drury J, Hutchison S, Bilous R. Detection of thyroid dysfunction in early pregnancy: Universal screening or targeted high-risk case finding? The Journal of clinical endocrinology and metabolism 2007; 92:203-207
- Horacek J, Spitalnikova S, Dlabalova B, Malirova E, Vizda J, Svilias I, Cepkova J, Mc Grath C, Maly J. Universal screening detects two-times more thyroid disorders in early pregnancy than targeted high-risk case finding. European journal of endocrinology 2010; 163:645-650
- Negro R, Formoso G, Mangieri T, Pezzarossa A, Dazzi D, Hassan H. Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroid disease: effects on obstetrical complications. The Journal of clinical endocrinology and metabolism 2006; 91:2587-2591
- Thangaratinam S, Tan A, Knox E, Kilby MD, Franklyn J, Coomarasamy A. Association between thyroid autoantibodies and miscarriage and preterm birth: meta-analysis of evidence. Bmj 2011; 342:d2616
- Lazarus JH. The continuing saga of postpartum thyroiditis. The Journal of clinical endocrinology and metabolism 2011; 96:614-616
- Stuckey BG, Yeap D, Turner SR. Thyroxine replacement during super-ovulation for in vitro fertilization: a potential gap in management? Fertility and sterility 2010; 93:2414.e2411-2413
- Casey BM, Dashe JS, Wells CE, McIntire DD, Leveno KJ, Cunningham FG. Subclinical hyperthyroidism and pregnancy outcomes. Obstetrics and gynecology 2006; 107:337-341
- Khan I, Okosieme O, Lazarus J. Antithyroid drug therapy in pregnancy: a review of guideline recommendations. Expert review of endocrinology & metabolism 2017; 12:269-278
- Andersen SL, Olsen J, Wu CS, Laurberg P. Birth defects after early pregnancy use of antithyroid drugs: a Danish nationwide study. The Journal of clinical endocrinology and metabolism 2013; 98:4373-4381
- Yang J, Yao LP, Dong MJ, Xu Q, Zhang J, Weng WW, Chen F. Clinical Characteristics and Outcomes of Propylthiouracil-Induced Antineutrophil Cytoplasmic Antibody-Associated Vasculitis in Patients with Graves' Disease: A Median 38-Month Retrospective Cohort Study from a Single Institution in China. Thyroid 2017; 27:1469-1474
- Akmal A, Kung J. Propylthiouracil, and methimazole, and carbimazole-related hepatotoxicity. Expert opinion on drug safety 2014; 13:1397-1406
- Luton D, Le Gac I, Vuillard E, Castanet M, Guibourdenche J, Noel M, Toubert ME, Leger J, Boissinot C, Schlageter MH, Garel C, Tebeka B, Oury JF, Czernichow P, Polak M. Management of Graves' disease during pregnancy: the key role of fetal thyroid gland monitoring. The Journal of clinical endocrinology and metabolism 2005; 90:6093-6098
- Bliddal S, Rasmussen AK, Sundberg K, Feldt-Rasmussen U. Careful assessment of maternal thyroid function can prevent cases of fetal goitrous hypothyroidism. Fetal diagnosis and therapy 2013; 34:66-67
- Stagnaro-Green A. Approach to the patient with postpartum thyroiditis. The Journal of clinical endocrinology and metabolism 2012; 97:334-342
- Feldt-Rasmussen U, Hoier-Madsen M, Rasmussen NG, Hegedus L, Hornnes P. Anti-thyroid peroxidase antibodies during pregnancy and postpartum. Relation to postpartum thyroiditis. Autoimmunity 1990; 6:211-214
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: Where do we stand in 2013? Thyroid 2013; 23:523-528
- Bech K, Hoier-Madsen M, Feldt-Rasmussen U, Jensen BM, Molsted-Pedersen L, Kuhl C. Thyroid function and autoimmune manifestations in insulin-dependent diabetes mellitus during and after pregnancy. Acta endocrinologica 1991; 124:534-539
- Pearce EN. Thyroid disorders during pregnancy and postpartum. Best practice & research Clinical obstetrics & gynaecology 2015; 29:700-706
- Azizi F. The occurrence of permanent thyroid failure in patients with subclinical postpartum thyroiditis. European journal of endocrinology 2005; 153:367-371
- Stagnaro-Green A, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Negro R. High rate of persistent hypothyroidism in a large-scale prospective study of postpartum thyroiditis in southern Italy. The Journal of clinical endocrinology and metabolism 2011; 96:652-657
- Stuckey BG, Kent GN, Allen JR, Ward LC, Brown SJ, Walsh JP. Low urinary iodine postpartum is associated with hypothyroid postpartum thyroid dysfunction and predicts long-term hypothyroidism. Clinical endocrinology 2011; 74:631-635
- Bergink V, Pop VJM, Nielsen PR, Agerbo E, Munk-Olsen T, Liu X. Comorbidity of autoimmune thyroid disorders and psychiatric disorders during the postpartum period: a Danish nationwide register-based cohort study. Psychological medicine 2018; 48:1291-1298
- Covinsky M, Laterza O, Pfeifer JD, Farkas-Szallasi T, Scott MG. An IgM lambda antibody to Escherichia coli produces false-positive results in multiple immunometric assays. Clinical chemistry 2000; 46:1157-1161
- Kailajarvi M, Takala T, Gronroos P, Tryding N, Viikari J, Irjala K, Forsstrom J. Reminders of drug effects on laboratory test results. Clinical chemistry 2000; 46:1395-1400
- Ismail AA, Walker PL, Cawood ML, Barth JH. Interference in immunoassay is an underestimated problem. Annals of clinical biochemistry 2002; 39:366-373
- Kricka LJ. Interferences in immunoassay--still a threat. Clinical chemistry 2000; 46:1037-1038