ABSTRACT
Multiple Endocrine Neoplasia Type 1 (MEN1) is a rare autosomal dominantly inherited endocrine tumor predisposition syndrome, caused by mutations in the MEN1 gene. Cardinal manifestations are primary hyperparathyroidism (pHPT), pituitary adenomas (PA), and neuroendocrine tumors (NETs) of the pancreas (PanNET) and duodenum. Other manifestations are NETs of thymus, lung, and stomach, adrenal tumors, and an increased breast cancer risk in women. Malignant NETs are the most important cause of disease-related mortality, mainly NF-PanNETs, gastrinomas and thymus NETs. MEN1 can be diagnosed genetically and a clinical diagnosis in patients with negative comprehensive testing has been debated. Timely recognition of MEN1, referral for genetic testing and swift cascade screening is essential. MEN1-related pHPT (penetrance >95%) is a multiglandular disease and recurrence after initial operation is to be expected. Subtotal parathyroidectomy is the preferred initial operation. Prolactinomas are the most prevalent PA in MEN1, followed by non-functioning (NF) PAs. Treatment and treatment results do not differ from sporadic PAs. Life-time penetrance of duodenopancreatic NETs is >80%. NF-PanNETs are most frequent, followed by gastrinomas and insulinomas. Surgical resection is the mainstay of treatment, and is indicated in non-gastrinoma functional PanNETs and NF-PanNETs >2cm or with progression during follow-up. No consensus exists on the surgical treatment of MEN1-related gastrinoma. MEN1-related dpNETs are currently detected at earlier stages and more indolent small dpNETs are seen. The main challenge is to identify patients at risk for an aggressive disease course. Thymic NETs (2-8%) occur predominantly in males and have a poor prognosis. Bronchopulmonary NETs are more frequent than previously thought, occur in both sexes, and are usually indolent although cases with a deviant progressive course occur. Adrenal tumors are mostly indolent non-functioning adenomas, but adrenocortical carcinomas and pheochromocytomas do occur. Women with MEN1 have an increased (RR 2.8) risk of breast cancer, at a younger age than the general population. Given the complexity of the disease, it is strongly advised that patients, whenever possible, be followed and treated in centers of expertise.
INTRODUCTION
Multiple Endocrine Neoplasia Type 1 (MEN1) is an inherited endocrine tumor predisposition syndrome. The prevalence is estimated at 1 in 20.000 to 1 in 40.000, and is therefore considered a rare disease (1). The syndrome predisposes mutation carriers to develop several endocrine tumors (Figure 1) with a high lifetime incidence of primary hyperparathyroidism (pHPT), pituitary adenomas (PA), and neuroendocrine tumors (NETs) of the pancreas (PanNET) and duodenum (dNET) (2). These tumors are considered the cardinal manifestations of the syndrome. Besides the cardinal manifestations, patients with MEN1 are at a higher risk for developing NETs of the thymus, lung, and stomach. In addition, there is a higher risk for adrenal tumors and a higher risk for developing breast cancer in women (3, 4). Next to these endocrine manifestations, patients are at risk for developing several non-endocrine lesions of the skin and subcutaneous tumors such as lipomas.
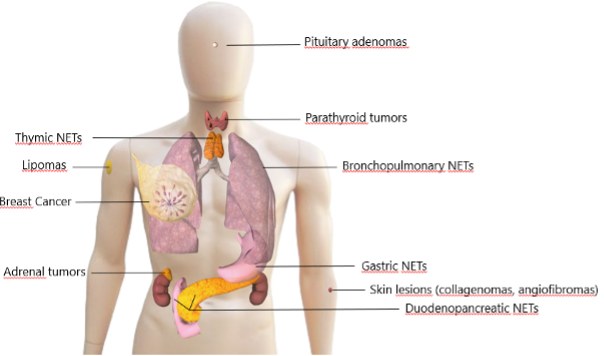
Figure1. Manifestations of MEN1
After identification of the causative MEN1 mutation an intensive lifelong surveillance program follows, if possible, starting at childhood because of the high life-time risk for developing tumors (2). This intensive follow-up is aimed at early detection of tumors to enable timely interventions in order to prevent complications and metastases of tumors and thereby preventing premature death and improving the quality of the life of patients. In 2001 the first set of clinical practice guidelines was published by Brandi et al (5). Because of paucity of scientific evidence, these guidelines were mainly based on expert opinion. Although more evidence was available, the updated clinical guidelines of Thakker, et al. which were published in 2012 were also written in the absence of robust scientific evidence (2). However, publishing the clinical practice guidelines led to more structured care of patients, which facilitated studies of the natural course of the disease and the effect of follow-up and treatment strategies. Therefore, in the last two decades large and sometimes nation-wide MEN1 cohort studies were initiated which led to new insights into the course of the disease and knowledge about more optimal follow-up and treatment. However, to date there remains a paucity of prospective data from interventional trials designed specifically for MEN1 patients.
GENETICS
The MEN1 gene (OMIM 613733 (gene) and OMIM 131100 (phenotype)), identified in 1997 (6, 7), consists of 10 exons and is localized on chromosome 11q13. Exon 1, the 5’ region upstream of exon 2 and the 3’ region of exon 10 are non-coding, so there are 9 coding exons (exons 2 through 10). The MEN1 gen encodes the protein menin, a 610 amino-acid nuclear scaffold protein that regulates gene transcription by coordinating chromatin remodeling.
In 2008, Lemos and Thakker published an overview of the 459 different germline mutations reported in the first decade since the discovery of the gene (8). In 2016, Concolino, et al. identified an additional 208 novel germline variants, of which 76 were reported as Variant of Unknown Significance (VUS) (9). Around 40% of all identified mutations are frameshift mutations, 25% nonsense, 20% missense, 10% splice-site, and the remainder 5% are rarer mutations such as in-frame deletions/insertions and partial or whole gene deletions (10). Frameshift, nonsense, and splice-site mutations, which are the majority, are predicted to be loss of function mutations that lead to truncated forms of menin (8). The variants are scattered throughout the MEN1 gene with no evident hotspots, although some mutations are found in apparently unrelated families. This is considered to be a founder effect (11). Sequence analysis of the MEN1 gene will reveal most of the variants. Since 1-2% of the variants are (partial) deletions of the MEN1 gene (8), multiplex ligation-dependent probe amplification (MLPA) or Copy Number Variation (CNV) analysis should also be included in the diagnostic DNA testing.
When a sequence variant is identified in the MEN1 gene this is classified by the genetic laboratory as either benign or likely benign (which is considered a “negative” result i.e., no disease-causing variant is found, clinically similar to no sequence variant identified), as a VUS (uncertain if the variant is disease causing) or as pathogenic or likely pathogenic (which is considered a “positive” result, a disease-causing variant is found). Especially new non-truncating (mostly missense) mutations may be difficult to classify (12).
The approximate prevalence of MEN1 has been reported as 1 in 30,000 individuals with no apparent gender bias. MEN1 follows an autosomal dominant pattern of inheritance with >95% penetrance by age 40–50 years (11). The cardinal clinical manifestations of MEN1 are primary hyperparathyroidism (pHPT), anterior pituitary adenomas (PA), and NET of the duodenum and pancreas, the so called three P’s.
If a patient is diagnosed with MEN1, he or she should be advised to undergo screening to detect manifestations and remain under lifelong surveillance in a center of expertise where care is provided by multidisciplinary teams (MDTs) comprising relevant specialists with experience in the diagnosis and treatment of patients with endocrine tumors (2).
Single center cohorts have identified certain genotypes that are associated with a more aggressive course of the disease (especially related to duodenopancreatic NETs), such as mutation in the JUND (13) or the CHES1 (14)interacting domain or nonsense/frameshift versus missense mutations (15), but since none of these associations have been independently validated, genotype cannot be used to individualize surveillance.
Evaluation of 10 Dutch families suggest genetic anticipation (decreased age of disease onset or an increased disease severity in successive generations), a known phenomenon which to date cannot be explained in autosomal dominant inherited disease genes without trinucleotide repeat expansions (“growing genes”) (16). Somatic mosaicism with subsequent germline inheritance has been described (17).
Function of the MEN1 Gene
MEN1 is considered to act as a tumor suppressor gene which is demonstrated by the identification of inactivating mutations, together with loss of heterozygosity (LOH) in MEN1-related tumors. Biochemical, proteomics, genetics and genomics approaches have identified various potential roles, which converge on the regulation of gene expression. The most consistent findings show that menin connects transcription factors including JUND (OMIM 165162), NFKB (OMIM 164011), and SMAD3 (OMIM 603109) and modulates their activities. In the nucleus, menin acts as a scaffold protein to regulate gene transcription by coordinating chromatin remodeling interacting with chromatin regulatory proteins in the MLL1 /MLL2 complex. Menin is implicated in both histone deacetylase and histone methyltransferase activity (HMT), and via the latter it regulates the expression of cyclin-dependent kinase inhibitor (CDKI) and homeobox domain genes (18, 19). While the MEN1 gene functions as a tumor suppressor gene in MEN1, it has an oncogenic role in sporadic breast cancer cells (18). Some excellent reviews on the function of the MEN1 gene can be found elsewhere (11, 18, 20).
Potential Therapeutic Opportunities (18)
Loss of menin in MEN1-associated tissues, leads to the disruption of anti-proliferative gene expression programs and to the development of endocrine tumors. Restoration of the epigenetic perturbations or correction of the function of aberrantly expressed genes in the absence of menin hold promise for molecular mechanism-based means to treat or prevent MEN1-related tumors. The fate and function of a cell are determined by its gene expression signature. As menin is a transcriptional regulator, MEN1-related tumorigenesis is likely to be the result of aberrant tumor suppressive gene expression due to the loss of menin. Restoration of the expression of menin target genes in MEN1-affected tissues could therefore have therapeutic consequences. This has been shown in a preclinical study in mice, where MEN1 replacement in pituitary tumors of Men1 (+/-) mice led to a decrease in proliferation of the pituitary tumors (21).
DIAGNOSIS AND GENETIC TESTING
Diagnosis of MEN1
Patients with MEN1 are at risk to develop different endocrine and non-endocrine manifestations. The most important of which are (with approximate lifetime prevalence in parentheses):
- Cardinal manifestations/Major criteria: pHPT (>95%), duodenopancreatic NETs (dpNETs) (>80%) and anterior pituitary tumors (50–65%)
- Minor clinical criteria: adrenal adenomas (11–35%) and bronchopulmonary, thymic, and gastric NETs (20–30%) (22, 23).
Additionally, there might be an association with meningioma (<10%). Cutaneous manifestations such as subcutaneous lipomas (but visceral, pleural, or retroperitoneal lipomas have also been described), facial angiofibromas (22-88%) and collagenomas (0-72%) are also seen (2). Women with MEN1 have a 2-3-fold elevated risk of developing breast cancer (3, 4).
Presently, MEN1 can be diagnosed genetically by identifying the germline heterozygous (likely) pathogenic variant in the MEN1 gene through DNA analysis. According to the guidelines, a diagnosis of MEN1 can also be made on familial grounds in a patient with one of the cardinal MEN1 manifestations and a first-degree family member with MEN1 (2). Additionally, a clinical diagnosis can be made in individuals with two of the three cardinal manifestations (2). However, with modern-day sensitive DNA testing, the value of the clinical criterion in patients with negative DNA testing is under debate. There is mounting evidence that patients who have clinical MEN1, but negative DNA testing have a different clinical course from patients with positive DNA testing (24, 25). The same could also be argued for patients with a familial diagnosis with negative DNA testing for the family mutation, as these may have a sporadically occurring endocrine tumor. This subject is discussed in more detail in the paragraph on genetic heterogeneity.
Patients with MEN1 suffer from high morbidity and a decreased life expectancy. In the present day and age, MEN1-related malignancy is the main MEN1-related cause of death, particularly due to duodenopancreatic and thymic NETs (26, 27). A timely and accurate diagnosis of MEN1 is paramount to improve disease outcomes. This enables early identification of tumor manifestations allowing timely treatment to reduce morbidity and improve survival (28).
It is therefore important for clinicians to consider the diagnosis of MEN1 not only in those patients meeting clinical or familial criteria, but also in patients with a suspicious family or personal medical history, but not meeting clinical or familial diagnostic criteria. In patients presenting with an endocrine tumor within the MEN1 spectrum, taking a family history of MEN1-related tumors is very important. Additionally, a young age at presentation or multifocality of tumors within a single organ may point to a diagnosis of MEN1. The combination of a major and minor criterion or two minor criteria should also raise suspicion of MEN1. In all these cases of suspected MEN1, the patient should be referred to a clinical geneticist or genetic counselor for counseling and consideration of DNA testing. For patients presenting with sporadically occurring endocrine tumors, de Laat, et al. developed and validated a prediction rule to predict the presence of an MEN1 mutation (29). In this model, recurrent pHPT, non-recurrent pHPT, dpNETs, PA, NET of the stomach, lung and thymus, a positive family history for a NET and age, predicted the risk of having MEN1. The authors developed a nonogram for clinical practice, allowing the clinician to calculate the risk of MEN1 in patients suspected of MEN1 with sporadically occurring endocrine tumors (29).
DNA Testing
According to current practice guidelines diagnostic DNA testing for MEN1 should be offered to (2):
- all patients fulfilling the diagnostic criteria of a clinical or familial MEN1 diagnosis
- all patients with a pHPT under the age of 30 or multiple (synchronous) parathyroid adenomas under the age of 40 or recurrent parathyroid adenomas
- patients with a gastrin-producing NET (irrespective the age of presentation)
- patients with multiple PanNETs (irrespective the age of presentation)
- patients with two different minor criteria
- a patient with an MEN1-related tumor with a positive family history of MEN1-related tumors
Once MEN1 is diagnosed in the proband, genetic counseling and DNA testing should be offered to family members, preferably by means of cascade screening. Data from the DutchMEN Study Group (DMSG) emphasize the importance of timely genetic testing of family members and prompt clinical screening according to MEN1 guidelines. In a study determining lag time between MEN1 diagnosis in index cases and their non-index family members, they found a median lag time of 3.5 years (range 0-30) years, in which clinically significant manifestations occurred in the non-index family members with MEN1 (30). Genetic testing in asymptomatic family members of MEN1 patients is called pre-symptomatic or predictive genetic testing and involves testing the at-risk family members for the familial MEN1 mutation. This is single-site testing, and outcome is whether the family mutation is present or absent in this particular family member.
Genetic Heterogeneity
Some studies have reported that between 5% and 10% of patient who fulfill the clinical criteria for MEN1 will not harbor mutations in the coding region or adjacent splice sites. In some of these patients a (likely) pathogenic variant can be found in the CDKN1A (OMIM 600778 ), CDKN1B (OMIM 116899 ), CDKN2B (OMIM 600431 ) or CDKN2Cgene (OMIM 603369 ). Mutations in CDKN1B are the cause of the MEN4 syndrome, the latest of the MEN syndromes and most rare, with <50 cases reported in the literature to date (31-33). Rather than being a separate phenotype, (likely) pathogenic variants in these genes are more likely to cause a MEN1 phenotype, with pHPT, PA and gastroenteropancreatic NETs as the main features, and are best met with the same guidelines for surveillance, until more is known about the phenotype of this rare syndrome.
Recent data have shown that patients with a clinical diagnosis of MEN1, in whom no (likely) pathogenic variant in the MEN1 gene can be found (genotype-negative MEN1/GN-MEN1), and who do not have another known germline mutation, have a different phenotype and clinical course compared to mutation positive patients (24, 25). Genotype-negative patients develop MEN1 manifestations at higher age, rarely develop a third main MEN1 manifestation, and have a life expectancy comparable with the general population (24, 25).
Additionally, regarding the individual manifestations, it seems that GN-MEN1 patients have less recurrent or multigland pHPT, less multifocal PanNETs, and more somatotrophinomas and less prolactinomas compared to genotype-positive patients (25). Most patients with GN-MEN1 present with the combination of pHPT and PA, followed by pHPT/dpNET and dpNET/PA (25). The apparent differences in clinical course suggest that GN-MEN1 patients do not have true MEN1, but another MEN1-like syndrome or sporadic co-incidence of two NETs (2, 33). In these patients there is usually a negative family history for MEN1-related disease. Although not specified in the current guidelines, these patients may benefit from a separate classification with alternative surveillance recommendations based on the clinical picture, as has been suggested by Pieterman, et al. (25). Important baseline considerations for an alternative surveillance are genetic counseling, comprehensive genetic testing based on the personal and family history, and baseline screening to identify any unrecognized manifestations. In these patients, there is generally no cause for surveillance of the first-degree relatives, although these decisions should be individualized and discussed in multidisciplinary teams.
GN-MEN1 patients with a positive family history of clinical MEN1 or a foregut NET and those presenting with all three main MEN1-related tumors, should be followed according to MEN1 guidelines as should their relatives. In these patients, a “false-negative result” of DNA testing should also be considered. This may either be because deletion/duplication analyses are not performed, a sequence variant exists outside of the assayed region, or polymerase chain reaction primer selection led to selective amplification of wild-type DNA (25). Additionally, somatic mosaicism or alternative mechanisms of MEN1 gene silencing could lead to inactivation of normal menin (25).
Depending on the presenting clinical picture and the family history, other hereditary syndromes causing endocrine tumors should also be considered.
If pHPT is the primary phenotype, other genes associated with hereditary pHPT are for example CDC73, CASR and RET (MEN2). Germline CDC73 (formerly HRPT2) (OMIM 607393) analysis is recommended in individuals with (suspected) Hyperparathyroidism-Jaw Tumor (HPT-JT) syndrome, familial isolated pHPT, atypical or malignant parathyroid histology, and young individuals with pHPT. These criteria would increase germline CDC73 mutation detection, enabling optimal clinical management of pHPT as well as genetic counseling and surveillance for family members at risk for developing CDC73-related disorders(34).
If PAs are the primary phenotype, mutations in the AIP gene (OMIM 605555) should be considered as these can cause (familial) pituitary adenomas. Predictors of a genetic cause of sporadic pituitary adenomas are young age of diagnosis, and also in AIP pathogenic variants there is an association with gigantism and macroadenomas (35).
PanNETs can also be seen in neurofibromatosis type 1 (NF1), Von Hippel-Lindau (VHL), and Tuberous Sclerosis Complex (TSC).
Pretest Counseling
Before DNA testing, pretest counseling is of utmost importance. The patient should be informed by genetic counseling about all aspects (medical, psychological, social, and familial implications) of the possible outcome of genetic testing. This should lead to an individual decision whether or not to opt for DNA testing. In case of DNA testing in minors the counseling should be offered to the parents, and include the minor if possible (which is obligatory over the age of 12 in the country where the authors practice (the Netherlands) to obtain informed consent for testing.
In case of diagnostic testing the patient must be informed about the possible outcomes of the DNA test (finding an (likely)pathogenic variant, finding a VUS, and not finding (likely) pathogenic variants) and the implications of these findings for the patient and family members.
In diagnostic DNA testing the MEN1 gene should be analyzed, for which Sanger sequencing can be used, or Next Generation Sequencing (NGS) techniques. To exclude deletions MLPA or CNV analysis must be performed. As mentioned earlier there is genetic heterogeneity and panel DNA diagnostics can be considered. In particular CDKN1A, CDKN1B, CDKN2B, CDKN2C, CDC73 and AIP can be added to the panel using NGS techniques, also to be completed with CNV analysis, depending on the clinical picture. In case of panel testing, the patient must be prepared for the possible findings in the different genes, differentiation of the consequences, and implications of these findings.
In case of testing for a familial (likely) pathogenic variant (so-called presymptomatic or predictive DNA testing) presence or absence of the familial mutation can be ascertained, but the differences in expression of the MEN1 syndrome, both within and between families must be emphasized.
In case of future pregnancy the possible options like invasive prenatal diagnostics and Preimplantation Genetic Diagnosis (PGD) should be discussed so the prospective parents can make an informed decision about the desired pregnancy.
Periodical Screening
The identification of an MEN1 mutation in patients and family members at risk is followed by the advice to remain under lifelong surveillance, with at least annual clinic visits including history, physical examination, biochemical screening, and radiological screening at specific intervals (2). This should preferably be carried out in centers of expertise with a dedicated multidisciplinary team well versed in management of patients with MEN1. In MEN1 there are no prophylactic treatments, so the goal of this screening & surveillance program is early detection of MEN1-related tumors to minimize morbidity by hormonal hypersecretion and to prevent malignant NETs by timely intervention. In the absence of known genotype-phenotype correlations and with a heterogeneous clinical course, even within families, the specific mutation or family history cannot solely guide the surveillance program. In the following sections screening and surveillance is discussed within each manifestation.
Screening at the Pediatric Age
The current clinical guidelines for MEN1 suggest starting clinical and biochemical screening at the age of 5 years (2), which is based on the earliest reported case of a patients with a clinical MEN1 manifestation (36). For radiologic screening in asymptomatic children, pituitary imaging is suggested from age 5 years onward (every 3 years), with abdominal imaging starting at age 10 years (every 1-3 years) and thoracic imaging starting at age 15 years (every 1-2 years) (2). However, this intensive surveillance at the pediatric age has been questioned by some groups who suggest postponing routine screening of asymptomatic patients until ages 15 or 16 years while counseling parents about typical clinical signs of MEN1 manifestations and contacting providers if they occur (37).
PRIMARY HYPERPARATHYROIDISM
Primary hyperparathyroidism (pHPT) (Figure 1) is one of the cardinal manifestations of MEN1 and has an almost complete lifetime penetrance (24, 38). It is often the first clinical manifestation of the disease and biochemical (asymptomatic) pHPT can be diagnosed several years before symptoms arise. The reported mean age of pHPT diagnosis in published MEN1 cohorts is in the fourth decade of life (39-45), with wide ranges. When interpreting these mean ages at diagnosis it is important to realize that these cohorts often span multiple decades, are made up of both index cases and family members, and contain patients who did and did not follow prospective screening programs. Recent studies reporting on MEN1 at the pediatric age show that in a screened population at least half of the pediatric patients already have primary hyperparathyroidism, although rarely seen before the age of 10 (46-49). In most cases, patients diagnosed at a pediatric age are asymptomatic and the diagnosis is made biochemically by screening (46-49). Clinical and symptomatic pHPT is usually seen in the third decade of life.
Primary hyperparathyroidism in MEN1 is a multiglandular disease, affecting all parathyroid glands, although often asymmetrically and asynchronously. Parathyroid tumors in adults with MEN1 usually represent mono- or oligoclonal proliferations that probably arise independently in each parathyroid gland (50). Tumorigenesis is initiated when the remaining normal allele of the MEN1 gene is lost (the second hit), and as this cumulative chance increases with age, normal parathyroid tissue is less often seen with increasing age (51). Supernumerary glands (that is, more than four parathyroid glands) are frequently seen in MEN1, with reported ranges between 12-30% (52). Parathyroid glands at ectopic locations are also not uncommon in MEN1, especially in the thymus.
The diagnosis of primary hyperparathyroidism can be made when there is hypercalcemia in combination with an elevated or inadequately normal parathyroid hormone (PTH). In patients with MEN1 who follow a prospective screening program from an early age, the diagnosis is often made while they are still asymptomatic. Classic objective symptoms of pHPT include polyuria and polydipsia, gastro-intestinal complaints (nausea, abdominal pain, constipation, pancreatitis), (symptomatic) urolithiasis, and decreased bone mineral density (BMD) which can lead to pathological fractures. Non-specific symptoms occurring in pHPT are fatigue, musculoskeletal complaints, neuropsychiatric symptoms such as anxiety, depression, concentration disturbance and sleep-disturbances, and a general decrease in quality of life.
The diagnosis of pHPT in patients with known MEN1 or from a known MEN1 kindred is straightforward. However, pHPT can also be the first clinical clue to an MEN1 diagnosis in a patient or family without prior MEN1 diagnosis or suspected history. The prevalence of pHPT in the general population can be up to 1% (53, 54) and among cohorts of patients with pHPT, depending on the characteristics, the incidence of MEN1 is 1-18% (2). Considering MEN1 in patients presenting with pHPT is extremely important, because the diagnosis alters the management and prognosis of pHPT, allows screening and surveillance for other endocrine tumors associated with MEN1, and allows for cascade screening within the family to identify MEN1 germline mutations carriers. Important clues to an MEN1 diagnosis in a patient presenting with pHPT are young age of onset, a family history of pHPT or other MEN1-related tumors, a personal history of other MEN1-related tumors, and multiglandular disease or persistent/recurrent pHPT (29). Recurrent pHPT is one of the strongest predictors for the presence of an MEN1 mutation (29). Compared to sporadic pHPT, patients with MEN1-related pHPT present at an earlier age, have an almost equal gender distribution compared to female predominance in sporadic pHPT, and present with lower levels of calcium and PTH (55, 56). Even though they have biochemically milder disease, BMD seems to be lower in patients with MEN1-related pHPT and renal involvement similar compared to patient with sporadic pHPT, which may reflect longer standing disease (55). MEN1-related pHPT is a multiglandular disease, as already stipulated, while sporadic pHPT is predominantly caused by single-gland adenomas (56, 57). This also affects recurrence rates which are much higher in MEN1-related pHPT (56, 57). The American Association of Endocrine Surgeons (AAES) guidelines advise genetic counseling for patients younger than 40 years with pHPT and multiglandular disease and to consider this for those with a family history or syndromic manifestations (57). The European guidelines slightly differ suggesting genetic testing for MEN1 in patients with pHPT before the age of 40, multiglandular disease, or persistent/recurrent pHPT (58).
When comparing, several studies show that patients with MEN1-related pHPT have lower BMD compared to patients with sporadic pHPT (55, 59, 60), although a Chinese study found no significant difference (61). In patients with MEN1-related pHPT, decreased BMD is frequently seen and already present at a young age (62-64). When measured the 1/3 distal radius seems most affected, so including this location in dual-energy X-ray absorptiometry (DEXA) should be considered in patients with MEN1 (62, 64). Parathyroidectomy improves BMD (59, 65), although in one small study improvement was less for patients with MEN1 compared to patients with sporadic pHPT (59). A factor contributing to the earlier and more severe bone involvement in MEN1-related pHPT may be the early-onset of the disease thereby also influencing peak bone formation. In addition, other MEN1-related diseases may also contribute to bone loss such as pituitary insufficiency caused by pituitary adenomas or their treatment, hypercortisolism (although infrequent in MEN1), and gastro-intestinal surgery (66).
Urolithiasis is also frequently seen and at a young age in patients MEN1-related pHPT (55, 62, 64). In addition, a recent study showed that patients with MEN1 age 20-59 had a higher prevalence of chronic kidney disease stage 3 compared to the general US population (67).
It is therefore important to perform Dual-energy X-ray absorptiometry (DEXA) to assess BMD as well as a renal ultrasound and 24-hour urine for calcium excretion to asses risk of urolithiasis in patients with MEN1 diagnosed with pHPT. And if initial observation is chosen, DEXA should be repeated every 2 years (68).
In patients with MEN1 and pHPT, the interplay with Zollinger-Ellison Syndrome (ZES; increased gastric acid section due to gastrinomas) is also relevant, as calcium can increase gastrin levels. In a study among 84 patients with MEN1-pHPT and ZES, successful parathyroidectomy resulted in biochemical cure of ZES without any resection of duodenal or pancreatic NETs in 20% of the patients (69). In a recent perspective paper Hackeng and colleagues propose a parathyroid-gut axis arguing that hypercalcemia may promote the gastrin-cell hyperplasia to neoplasia sequence through the calcium-sensing receptor (70). The reverse, a more severe form of pHPT among patients with MEN1-ZES has also been suggested, because in the aforementioned study of 84 patients with MEN1-pHTP and ZES, patients had a higher frequency of urolithiasis at presentation, higher serum PTH, and higher recurrences rates after initial subtotal parathyroidectomy compared to the literature (69).
Parathyroidectomy
The treatment of hyperparathyroidism in MEN1 is surgical. Intervention is aimed at achieving eucalcemia for as long as possible, while preventing permanent hypoparathyroidism and facilitating potential subsequent surgery.
The optimal timing of the initial operation is still a matter of debate, especially in (asymptomatic) children and young adults. The guidelines for the management of asymptomatic pHPT recommend surgical intervention in case of significant hypercalcemia (1 mg/dL or 0.25 mmol/L above the upper limit of normal), skeletal abnormalities (a T-score of < -2.5 at the Lumbar Spine, Total Hip, Femoral Neck or 1/3 Distal Radius or a vertebral fracture), risk of renal complications (creatinine clearance below 60 ml/min, 24-h urine calcium excretion of >400 mg/d (>10 mmol/L)), the presence of nephrolithiasis/nephrocalcinosis, or age below 50 (68). However, these guidelines are not intended for patients with MEN1 and most patients with MEN1 will meet the age-criterion regardless of other values. In patients with MEN1 surgery is indicated in case of symptoms, significant hypercalcemia, and renal or skeletal complications. In addition, concomitant gastrinoma may also provide an indication for surgical intervention of pHPT. For patients not meeting any of these criteria, there is no evidence to determine timing of surgery. Arguments have been made in favor of observation to avoid the risk of symptomatic hypoparathyroidism, multiple operations, and by allowing the disease to progress a little bit more, making the glands more easily identifiable upon intervention. However, on the other hand, data showing early bone and renal complications have made others suggest and prefer early intervention to prevent downstream disabilities (71).
For initial parathyroidectomy in patients with MEN1 there are theoretically four different strategies: focused parathyroidectomy (removing a single affected parathyroid gland), unilateral clearance (resection of all parathyroid tissue on one side, including unilateral cervical thymectomy), subtotal parathyroidectomy with concomitant cervical thymectomy, or total parathyroidectomy, cervical thymectomy and immediate auto-transplantation of parathyroid tissue (usually to the non-dominant forearm).
The initial operation recommended by most experts and guidelines is a bilateral cervical exploration, identifying all four parathyroid glands and performing a subtotal parathyroidectomy (leaving a vascularized remnant about 1.5-2 times the size of a normal gland) with concomitant cervical thymectomy (2, 57, 58, 71-73). The latter serves the dual purpose of removing any ectopic/supranumerary parathyroid glands and potentially decreases the risk of subsequent development of thymic NETs. This approach offers the best balance between persistence (persisting pHPT after operation or recurrence within 6 months after operation) and recurrence (recurrent pHPT 6 months or more after the operation preceded by a eucalcemic period) on the one hand and permanent (lasting >6 months after the operation) hypoparathyroidism on the other hand. Persistence is infrequent in subtotal (0-22%) and total parathyroidectomy (0-19%), but rates range from 0-53% in less than subtotal parathyroidectomy(58, 72). Recurrence rates are also significantly higher after less than subtotal parathyroidectomy (0-100%) compared to subtotal (0-65%) or total parathyroidectomy (0-56%) (58, 72) and occur earlier (74). Permanent hypoparathyroidism on the other hand is rarely seen after less than subtotal parathyroidectomy. When comparing subtotal with total parathyroidectomy, hypoparathyroidism is significantly more frequent after total parathyroidectomy (RR 1.61 (95%CI 1.12-2.31) (72).
Pre-operative imaging plays a limited role at initial parathyroidectomy in patients with MEN1, because the recommended initial operation always constitutes bilateral neck exploration( 58). In addition, data has shown that pre-operative imaging (consisting of neck ultrasound and sestamibi scan as first line and parathyroid computed tomography (CT) or magnetic resonance imaging (MRI) as second line) only identified 68% of the largest glands pre-operatively (75). Pre-operative imaging may have some use for identifying ectopic glands (7% of ectopic glands were identified by pre-operative imaging in one series) and for identifying concomitant thyroid abnormalities that need attention (71, 75). Similar, intra-operative PTH monitoring seems of little value during the initial parathyroidectomy (76).
Recently, several groups have advocated unilateral clearance as an initial operation, especially for young patients with MEN1 (74, 77-79). The rationale behind this approach is to provide several years of eucalcemia during acquisition of peak bone mass, while preventing hypoparathyroidism and allowing subsequent reoperations to be performed in a non-operated neck (the contra-lateral side). A prerequisite for this strategy is that pre-operative imaging concordantly shows unilateral disease. Intra-operative PTH monitoring should be used to ensure there is an adequate drop in PTH after the resection. Although persistence rates between 10-15% after unilateral clearance or single-gland excision have been reported by these groups (74, 77), others state that less than subtotal parathyroidectomy has an unacceptable failure rate (69% in one study) (80). Several remarks must be made when using retrospective studies to evaluate this strategy. The first being that intentional less than subtotal resection is a different entity from an intended subtotal or total resection in which not all glands were identified (78). Secondly, true unilateral clearance in which all parathyroid tissue on one side of the neck is removed including unilateral cervical thymectomy is a very different operative strategy from minimal invasive parathyroidectomy/single gland excision and in retrospective studies these are often lumped together under “less than subtotal” resections. Thirdly, the success of such an approach is dependent on the sensitivity of pre-operative imaging and in most retrospective studies, more sensitive imaging modalities such as 18F-fluorcholine positron emission tomography (PET)/CT have not been used. Finally, since in MEN1 inherently all parathyroids are affected, although asynchronously, such an approach may be more successful in younger patients, where there may still be normal parathyroid glands (51). Currently this approach is controversial. Therefore, prospective data are needed to determine if and when unilateral clearance can benefit patients with MEN1 at the time of their initial parathyroidectomy.
Currently, subtotal parathyroidectomy remains the initial procedure of choice, but total parathyroidectomy or unilateral clearance can be considered depending on individual circumstances. Single gland excision is generally not recommended.
After initial subtotal parathyroidectomy, the 10-year recurrence rate is approximately 50% (2). Reoperation is therefore a frequent necessity in patients with MEN1. Recurrence can be caused by parathyroid glands missed during the initial operation, parathyroid glands intentionally left in situ, growth of the remnant of a partially resected gland, supranumerary and/or ectopic glands, and hyperplasia of autotransplanted parathyroid tissue. As reoperations are more complex and have a higher risk of complications (12% not including hypoparathyroidism in one study of reoperative parathyroidectomy in MEN1 (81)), the timing of the reoperation is individualized and patients with mild biochemical recurrence are usually initially observed. When reoperation is indicated, careful examination of the operation notes and pathology reports of previous procedures, if available, is very important. In contrast to the initial surgery, pre-operative imaging is essential for surgical planning in reoperations. First-line imaging studies are neck ultrasound and Tc99m-sestamibi scan, although this may not show all enlarged glands. Second-line imaging studies are 4-dimentional CT or MRI and PET (18F-fluorocholine or 11C-methionine) (82, 83). In a small study 18F-fluorocholine PET-CT has also shown to be of added value in MEN1-related pHPT (84). If first- and second-line studies are inconclusive more invasive localization studies can be considered such as arteriography, venous sampling, and neck ultrasound with fine needle aspiration and PTH measurement (83). The exact operative strategy (bilateral or unilateral neck exploration or focused resection) is individualized based on previous operation(s) and results of preoperative imaging. If the thymus was not removed during the initial operation, its removal is recommended at reoperation (81). Intra-operative PTH monitoring is valuable for the reoperative setting in MEN1 as it can inform when the exploration can be ended (71, 81).
As a consequence of the extended initial operation necessary, as well as frequent reoperation, life-time risk of postoperative hypoparathyroidism is relatively high for patients with MEN1. Transient hypoparathyroidism, defined as lasting less than six months after parathyroidectomy, may be seen in more than 50% of patients and its absence after subtotal parathyroidectomy may even be associated with recurrence (85, 86). Rates of permanent hypoparathyroidism are dependent on the procedure performed and vary greatly between series. It is important to realize that, unless patients are truly aparathyroid, recovery of parathyroid function can occur after 6 months up to several years, and permanent hypoparathyroidism may therefore be more aptly termed “prolonged” hypoparathyroidism (86). To prevent hypoparathyroidism immediate autotransplantation is used when it is suspected that all parathyroid glands are resected or when there is concern about parathyroid tissue viability in situ (57). Cryopreservation with delayed autotransplantation can also be used as a rescue from permanent hypoparathyroidism, but is not available everywhere and its use has been under debate (57).
Non-Surgical Interventions
For those patients who require intervention, but who are not surgical candidates, cinacalcet, an allosteric agonist of the calcium receptor, can be used. It has been shown to reduce/normalize calcium and PTH in small studies in patients with MEN1, although it has no effect on bone and renal complications(87-89). Cinacalcet should be used with great caution in children, as a death from acute hypocalcemia has been reported in a 14-year-old (90). Another alternative may be ethanol ablation of enlarged parathyroid glands. A study from the Mayo Clinic reported results from 37 patients who had an average of 2.2 treatments and a mean duration of eucalcemia of 25 months. Complications were hypocalcemia in 8%, hoarseness in 5%, and cough in 1% (91).
Parathyroid Carcinoma
Parathyroid carcinoma is a very rare endocrine malignancy seen in <1% of all patients with pHPT (92). It is likewise very rare in patients with MEN1, with only 21 reported cases in the literature (based on a review published in 2020) (93). In three large series from The University of Texas MD Anderson Cancer Center, the Mayo Clinic, and The Peking Union Medical College Hospital the prevalence of parathyroid carcinomas was 2/242 (0.8%), 1/348 (0.3%) and 1/153 (0.7%) respectively and the prevalence of atypical parathyroid neoplasm was 1/242 (0.4%), 0, and 2/153 (1.3%) respectively (93-95).
Conclusion
In conclusion, pHPT in MEN1 has an almost complete penetrance and is responsible for most MEN1-related surgeries. It is a multiglandular disease and recurrence after initial operation is to be expected. End-organ damage (bone, renal) can occur early and in asymptomatic patients and should be systematically looked for. Recognizing MEN1 in a patient presenting with apparently sporadic pHPT has important consequences for both the patient and his/her family. Surgical decision making is complex both for initial and reoperations and patients with MEN1 should whenever possible be treated in centers of expertise by a high-volume endocrine surgeon. Treatment decisions are made by multidisciplinary teams in shared decision making with the patient taking into account not only medical information but also the patient’s individual situation, such as but not limited to, ability to adhere to follow-up and insurance issues.
PITUITARY ADENOMAS
In 1903, the first description of a case with MEN1 was published by Erdheim. The necropsy report of a patient with acromegaly revealed a pituitary adenoma and enlarged parathyroid glands (96). Pituitary adenomas (PAs) are one of the three cardinal features associated with MEN1 and part of the so-called ‘three Ps’ (Figure 1). PAs are in general benign lesions and do not seem to negatively affect survival in patients with MEN1(26), although cases of mortality due to PAs have been reported (27). However, they can cause significant morbidity due to mass effect on the optic chiasm or hormone secretion leading to functional symptoms or hormone deficiency.
As in other main manifestations of MEN1, loss of heterozygosity (LOH) at the MEN1 locus has been demonstrated in pituitary adenomas in patients with MEN1, confirming the role of MEN1 in the pathogenesis of these tumors (97-100). However, in contrast to PanNETs, the role of MEN1/menin in tumorigenesis of sporadic PAs seems to be limited. Although initially, before the identification of the MEN1 gene, 19-33% of sporadic PAs showed allelic loss on chromosome 11 (101, 102), subsequent studies investigating LOH, somatic mutations, and messenger mRNA expression found limited involvement of MEN1 in sporadic PAs (103-107).
As the prevalence of clinically relevant PAs is 68-98/100,000 in the general population and in general <3% of patients with a PA will have MEN1, the question is when to think of MEN1 in a patient presenting with a PA (2, 35). Obviously, MEN1 should be considered in a patient with a family history of MEN1-related tumors or presenting with other MEN1-related tumors. For patients with apparently sporadic PAs (no suspicious family history or syndromic features), a recent systematic review has shown that MEN1 mutation analysis is recommended in patients ≤ 30 years, although this was a weak recommendation based on low quality of evidence (35).
Characteristics of Pituitary Adenoma in MEN1
From the earliest descriptions of MEN1 in the 1950s PAs have been recognized as one of the main characteristics of the syndrome. However, since the original description of MEN1, the clinical picture of MEN1-related PAs has changed. In a summary of the first 85 reported cases of MEN1 (many of which were autopsy cases), Ballard found a very high prevalence of 65% of PA, with 42% being chromophobe adenomas and more than one in four being acromegaly/eosinophilic adenoma (108). With the discovery of prolactin, it was soon realized that in fact prolactinomas were the most frequently occurring PA in patients with MEN1. The discovery of the MEN1 gene in 1997 (6, 7), and more advanced genetic testing techniques such as NGS and MLPA, have allowed better identification of patients as having MEN1. This has led to the recognition that patients with a clinical diagnosis of MEN1 because they have two out of the three main MEN1-related tumors, but negative mutation analysis, have a different clinical course than mutation positive patients and arguably do not have true MEN1, but rather an MEN1-like syndrome or a co-occurrence of two sporadic tumors (24, 25). Most patients in this group have a clinical MEN1 diagnosis based on the combination PA and pHPT. As these patients may have been included in older MEN1 cohorts, before the widespread availability of genetic testing, and these patients seem to have macro-adenomas and somatotrophinomas more often, this can be one of the reasons of the changing clinical picture of MEN1-related PAs. Additionally, imaging techniques have markedly improved over the last decades and guidelines have been developed for the screening and surveillance of patients with MEN1 including regular pituitary imaging and biochemical screening using Insulin-like Growth Factor-1 (IGF-1) and prolactin (2, 5). All this has led to earlier identification of PAs in patients with MEN1 and more frequent detection of (small) non-functioning PA (NFPAs).
After the discovery of the MEN1 gene (1997), six cohorts of MEN1-PA have been published, the first two by the French multicenter Groupe d’étude des Tumeurs Endocrines (GTE) in 2002 (109) and 2008 (110), in 2015 the DutchMEN Study Group (DMSG) published the results from their national population-based database (111), which was followed by two single-center cohort from China (112) and the Mayo Clinic respectively (113). Recently, the GTE have published an update to their previous study, only including patients diagnosed since January 1st 2000 (114).
As in sporadic PAs, PAs in patients with MEN1 show a slight female predominance (52-69%) (109, 111-114). With exception of the Chinese cohort, where the mean age of diagnosis was 54 years (112), the mean/median age of diagnosis of MEN1-related PAs is in the fourth decade. Lifetime prevalence of a PA in patients with MEN1 is 49-58% (38, 111).
Although not as frequent as pHPT, PAs are often the first clinical manifestation of the MEN1 syndrome. In the Dutch cohort, in 29% of the patients with a PA, it was the first manifestation (111). In the most recent GTE cohort, 88/202 patients with a PA were the index case in their family and in 84% of these patients a PA was (one of) the first manifestation(s) (114).
Prolactinomas are the most prevalent PA in patients with MEN1 and account for 30-80% of adenomas diagnosed in patients with clinically evident disease (42, 43, 109, 111-114). Second most prevalent are non-functioning PA comprising 36-48% in the most recent cohorts (111-114). Other functioning PAs are seen in <10%, and are in decreasing order of prevalence somatotropinomas, ACTH-producing adenomas (Cushing’s disease), and TSHomas and gonadotropinomas (the latter two being equally rare) (109, 111-114). Co-secreting tumors are seen in less than 10% (109, 111-114).
Multifocal PAs are rare in MEN1, and are found in 1.5% in the most recent GTE cohort (114) and in 4% in the 2008 GTE cohort of surgically resected MEN1-related PAs (110). In this latter cohort the prevalence of multifocal tumors was compared to that in non-MEN1 resected PAs and was found to be significantly larger. Additionally, MEN1-related resected PAs were more often plurihormonal on immunostaining (110).
Signs and symptoms in MEN1-related PAs (Table 1) are not different from those observed in sporadic PAs and are caused by size effects (chiasm compression, compression of nerves in the cavernous sinus, hypopituitarism) and effects of hormonal hypersecretion in functioning tumors.
|
Table 1. Signs and Symptoms of Pituitary Adenomas in MEN1
|
|
Related to tumor size/ growth
|
headache, visual field defects (usually bitemporal hemianopsia), diplopia, hypopituitarism
|
|
Prolactinoma
|
females: amenorrhea, galactorrhea, infertility
males: hypogonadism, impotence, lack of libido, galactorrhea (rare), infertility
|
|
Somatotrophinoma
|
Acromegaly: local overgrowth of bone (most often mandible, skull), soft tissue growth (acral enlargement, coarse facial features), hyperhidrosis, fatigue, hyperglycemia, hypertension, sleep apnea, skin tags, hypogonadism.
|
|
Corticotrophinoma
|
Cushing syndrome: central obesity, hypertension, hyperglycemia, gonadal dysfunction, moon facies, plethora, osteoporosis, proximal muscle weakness, psychological disturbance, wide purple striae, easy bruising
|
|
Thyrotropinoma
|
heat intolerance, unintentional weight loss, anxiety, tremor, palpitations, frequent bowel movements
|
|
Gonadotropinoma
|
hypogonadism, ovarian hyperstimulation in women
|
|
Pediatric specific
|
delayed or halted pubertal development, primary amenorrhea (females), accelerated linear growth, poor growth velocity, decline in school performance
|
Presently, most non-functioning PAs in MEN1 are microadenomas detected by prospective screening. These micro-adenomas show indolent behavior during follow-up. In the Dutch series after a median follow-up of 5.3 yrs, 9.7% showed minimal tumor growth which was without clinical significance in all and none progressed to macro-adenoma (111). In the Mayo Clinic cohort, in those with asymptomatic non-functioning PA (size not specified) progression to surgery was seen only in 1.7/100yr (113). In the most recent GTE cohort, after a median follow-up of 2 years (IQR 0-4), progression in Hardy classification was only seen in 1 out of 63 patients with a non-functioning micro-adenoma (2%) (114). In the Chinese cohort, of the 19 patients with non-functioning micro-adenomas, no progression to macro-adenomas was seen during a median follow-up of 3 years (112).
Prolactinomas are also mostly micro-adenoma, while 30-38% are macro-adenomas (111, 113, 114). As in sporadic PAs, GH-secreting tumors are more often macro-adenomas and ACTH-secreting tumors are generally microadenomas.
Although the youngest patient with a clinical manifestation of MEN1 described in the literature is a 5-year-old boy with gigantism and a lactosomatotroph macro-adenoma (36), PAs are rare in patients with MEN1 below the age of 10 (37, 46-49). However, pediatric cohorts show that in children and adolescents who have clinical manifestations of MEN1 up to 1/3 have PAs (37, 47-49). As in adults, most PAs the pediatric and adolescent age are prolactinomas followed by non-functioning PAs and more rarely GH or ACTH producing tumors (37, 46-49, 115). In the two largest pediatric cohorts, PAs were symptomatic in 50% of the cases and were macro-adenomas in 33-51% (47, 48).
Treatment
The treatment of MEN1-related pituitary adenomas follows the same strategy as sporadic pituitary adenomas. Management is aimed at tumor reduction, normalization of hormone secretion, and preservation of pituitary function.
Dopamine agonists are the first line of treatment for patients with prolactinoma (116), in which cabergoline has proven to be most effective at restoring normal prolactin concentrations and achieving tumor shrinkage than other dopamine agonists. With regards to adverse effects, cabergoline shows fewer side effects than bromocriptine. In case of treatment resistance, or treatment intolerance, surgery or radiotherapy are considered as second-line treatment options (116).
In Cushing’s disease(117) and acromegaly(118) surgery is the first treatment option. In addition, non-functioning PAs with mass effect or rapid progressive adenomas will also benefit from surgery.
MEN1-related functional PAs were initially considered more resistant to medical treatment than those with sporadic disease (109). However, the latest reports do not confirm this (111, 114) and treatment results seem to be in line with what is reported in sporadic PAs. The latter cohorts consist of a population with meticulous surveillance and therefore PAs are detected in an early phase (111, 114).
Pituitary Carcinoma
Pituitary carcinoma is extremely rare, and this is equally so in patients with MEN1. Although at higher risk for PA than the general population, there does not seem to be an increased risk of pituitary carcinoma. Single cases of malignant, metastatic prolactinoma (119, 120), gonadotropinoma (121), thyrotropin secreting adenoma (122), and non-functioning PA (123) have been reported.
Surveillance for Pituitary Adenoma
Current guidelines recommend examination by MRI of the pituitary gland every three years from the age of five years, and an annual blood test of IGF-I and prolactin concentrations, together with a clinical assessment (2). The young starting age – which was based on a single case-report – has been disputed, given that PAs are rarely seen before the age of 10.
The aim of surveillance imaging is to detect the PAs in an early phase before clinical symptoms become apparent. In general, surveillance leads to detection of smaller non-functioning PAs (111, 113, 114). However, early diagnosis by surveillance is not associated with smaller prolactinomas, but treatment is required less frequently and a longer safe observation period can be conducted (111). There are currently no specific recommendations for the follow-up of MEN1-related (micro-)adenomas under observation, on medical treatment or after surgical resection.
DUODENOPANCREATIC NEUROENDOCRINE TUMORS AND GASTRIC NETS
General
Duodenopancreatic neuroendocrine tumors (dpNETs) (Figure 1) are one of the cardinal features of MEN1 and highly penetrant, with a prevalence of over 80% at the age of 80 in recent cohorts (24, 38, 124). Malignant dpNETs are the most important cause of MEN1-related death (26, 125).
Duodenopancreatic NETs in MEN1 can secrete hormones that produce a clinical syndrome or be functionally silent (non-functioning, NF). Due to improved imaging techniques in the past decades, including endoscopic ultrasound (EUS) and somatostatin receptor (SSTR) imaging, non-functioning pancreatic NETs (NF-PanNETs) are now recognized as the most frequent type of dpNET in patients with MEN1. Of the functional dpNETs, gastrinomas are the most frequent, seen in approximately 30% of patients with dpNETS. In patients with MEN1, gastrinomas are almost exclusively of duodenal origin (126). Insulinomas (pancreatic in origin) are the second most common functional dpNET and occur in approximately 10-15% of patients with MEN1. More rare functional PanNETs such as glucagonomas, vipomas, somatostatinomas (127) or even rarer PanNETs secreting GHRH (128), calcitonin or PTH-related peptide (129), can also occur. Upon histological examination of the duodenum in patients with MEN1, small somatostatin-positive tumors can also be found (130) although they do not seem to give rise to the somatostatinoma syndrome.
The hallmark of duodenopancreatic involvement in MEN1 is multifocality, with the pancreas usually containing multiple NETs <5mm, called micro-adenomas, combined with one or more macroscopic PanNETs (130). These micro-adenomas already have loss of heterogeneity (LOH) of the MEN1 locus and are considered precursors to PanNETs (130). Similarly, duodenal gastrinomas in MEN1 are usually multiple and accompanied by gastrin cell hyperplasia, although LOH was demonstrated in duodenal gastrinomas, but not in gastrin cell hyperplasia (131). This multiplicity sets MEN1-related dpNETs apart from sporadic duodenal and pancreatic NETs, which are usually single tumors.
For patients with MEN1, the cumulative probability of having a dpNET increases with age, however the age of onset varies somewhat per tumor type. In a recent study from the Dutch MEN1 cohort, the modeled cumulative probability of having developed a NF-PanNET was 8.6% (95%CI 0.8-15.3%) at age 15, 12% (95% CI 5.9-17.0) at age 18, 16.1% (11.2-21.5) at age 21 and rising to 80% at age 70 (72.2-97.0)(15). Insulinomas can also occur at a young age and the prevalence of insulinoma among the larger (n>50) cohorts describing pediatric and adolescent MEN1 ranges from 6-25% (37, 46-48). Data from a recent multicenter cohort study show that half of the patients with MEN1-related insulinoma were diagnosed before the age of 30 (96 patients who underwent surgery for MEN1-related insulinoma from 46 centers in Europe and North-America between 1990-2016) (132). The onset of gastrinomas is usually later, with a reported mean age of onset around 30-35 years in the National Institutes of Health (NIH) MEN1-ZES cohort (133, 134) to 51 years in the Dutch MEN1 cohort (135). The occurrence of MEN1-related gastrinoma in childhood or adolescence is rare.
Duodenopancreatic NETs can be the first manifestation of MEN1, both in patients from known MEN1 families but also in the index case. Approximately 20-25% of all patients with gastrinoma have MEN1 (136), a rate much lower for insulinomas (approximately 5%) (2). Therefore, genetic testing for MEN1 is recommended in all patients diagnosed with gastrinoma (137). For patients presenting with a non-gastrinoma dpNET without a family history of MEN1, referral for genetic testing should be guided by the individual clinical characteristics, such as patient age, concomitant other MEN1-related tumors, multifocality of dpNETs, and family history of endocrine tumors. If a new diagnosis of MEN1 is made in a family, cascade screening and subsequent screening and lifelong surveillance of affected family members is of utmost importance, as delays may lead to preventable morbidity and mortality in non-index cases in the family (30).
Distant metastases occur in approximately 15-30% of MEN1-related dpNETs and are the most important prognostic factor for disease-related survival (125, 134, 138, 139). In the Dutch MEN1 cohort, 5- and 10-year overall survival rates were 95% and 86% for patients with dpNETs without liver metastases, compared to 65% and 50% for those with liver metastases (139). Non-functional pancreatic NETs and duodenal gastrinomas are the most frequent cause of distant metastases. Regional lymph node metastases are seen more often, but the exact reported prevalence highly depends on the type of cohort, primary dpNET, and the manner of diagnosis (i.e., surgical cohorts versus observational cohorts, surgery with or without systematic lymph node dissection, imaging with or without SSTR-PET imaging, etc.). In a recent publication from the Dutch MEN1 cohort, in 350 patients with MEN1-related NF-PanNETs without metastases at diagnosis, metastases (regional and/or distant) developed in 18%, while the cumulative probability of having any PanNET-related metastases at the age of 70 was 41.2% (95%CI 31.3-50.3) (15). Since patients with MEN1 often have multiple concomitant dpNETs and most patients with duodenal gastrinomas have concomitant NF-PanNETs, it may be difficult to determine the primary tumor for regional and distant metastases.
Unlike in MEN2, in MEN1 there is no clear genotype-phenotype correlation. Several groups however have studied the association between MEN1 germline mutation and the disease course of dpNETs in their cohorts, to see if genotype might be able to identify a subset of patients with a more aggressive clinical course. This was in part fueled by the clinical observation that in some families dpNETs seem to be more prevalent, occur at a younger age and have a higher proportion of metastatic disease.
Several associations have been reported: in the French GTE cohort mutations in the JUND interacting domain were associated with death (13), in the German Marburg cohort CHES1 loss of interaction was associated with aggressive pNETs and pNET-related mortality (14), in the Italian Florence cohort mutations in exon 8 were associated with higher risk of progression and mortality (140), in the MD Anderson cohort mutations in exon 2 were associated with a higher risk of distant metastases (141), and in the Dutch MEN1 cohort nonsense/frameshift mutations were associated with a higher cumulative probability of developing metastases in NF-PanNET (regional and/or distant) compared to missense mutations 53.9 (37.8-74.3%) vs 10% (2.6-82.7%)) (15). However, these associations up until now have not been independently validated, either because associations were not confirmed in other cohorts or validation was not performed.
In patients with MEN1, dpNETs are usually diagnosed at an early stage, especially in patients from families with MEN1 or who have had predictive genetic testing. Additionally, even in index cases, benign MEN1 manifestations may lead to the diagnosis of MEN1 and dpNETs can be diagnosed early. In the French GTE cohort and the Dutch MEN1 cohort, both spanning multiple decades, synchronous metastases were seen in 6.5 and 6.4% of patients with a dpNET respectively (125, 139). In MEN1-related dpNETs the focus of care therefore lies before the onset of metastatic disease and with a younger population than is seen in sporadic dpNETs. The goals of follow-up and treatment are to prevent metastatic disease, cure hormonal hypersecretion, and prevent complication from hormonal hypersecretion, while minimizing treatment-related complications and preserving Quality of Life. It is therefore of utmost importance that whenever possible patients with MEN1 and MEN1-related dpNETs are treated in centers of expertise with a knowledgeable and experienced multidisciplinary team.
Staging and Grading
MEN1-related dpNET are graded according to the latest WHO classification (Table 2) of digestive system tumors (2019, 5th edition) and the WHO Classification of Tumors of Endocrine Organs (2017, 4th edition) (142). Where previously dpNET grading was only covered in the Classification of Tumors of Endocrine Organs, it is now included in the classification of digestive system tumors as well (142).
.
|
Table 2. WHO Classification of Digestive Neuroendocrine Tumors
|
|
Classification
|
Ki-67 proliferation index
|
Mitotic rate (mitoses/2mm2)
|
|
Well-differentiated Neuroendocrine Tumors (NET)
|
|
NET, G1
|
<3%
|
<2
|
|
NET, G2
|
3-20%
|
2-20
|
|
NET, G3
|
>20%
|
>20
|
|
Poorly-differentiated Neuroendocrine Carcinomas (NEC)
|
|
NEC (G3)
Small-cell type
Large-cell type
|
>20%
|
>20
|
Pancreatic NETs are staged according to the AJCC UICC 8th edition Neuroendocrine tumors of the pancreas (Table 3a and b).
|
Table 3a. TNM Staging of Pancreatic Neuroendocrine Tumors (AJCC UICC 8thedition)
|
|
Primary Tumor (T)
For any T add (m) for multiple tumors e.g., T2(m).
|
|
TX
|
Tumor cannot be assessed
|
|
T1
|
Tumor limited to the pancreas*, <2 cm
|
|
T2
|
Tumor limited to the pancreas*, 2-4 cm
|
|
T3
|
Tumor limited to the pancreas*, >4 cm; or tumor invading the duodenum or CBD
|
|
T4
|
Tumor invading adjacent organs (stomach, spleen, colon, adrenal gland) or the wall of large vessels (celiac axis or the superior mesenteric artery)
|
|
Regional lymph Nodes (N)
|
|
NX
|
Regional lymph nodes cannot be assessed
|
|
N0
|
No regional lymph node involvement
|
|
N1
|
Regional lymph node involvement
|
|
Distant Metastases (M)
|
|
M0
|
No distant metastases
|
|
M1
|
Distant metastases
|
|
M1a
|
Hepatic metastases only
|
|
M1b
|
Extra-hepatic metastases only
|
|
M1c
|
Both hepatic and extra-hepatic metastases
|
* Limited to the pancreas means no invasion of adjacent organs or the wall of large vessels. Extension into peripancreatic adipose tissue is included in “limited to the pancreas”. CBD common bile duct
|
Table 3b Stage Grouping
|
|
Stage I
|
T1 N0 M0
|
|
Stage II
|
T2-3 N0 M0
|
|
Stage III
|
T4 N0 M0
Any T N1 M0
|
|
Stage IV
|
Any T Any N M1
|
Duodenal NETs are staged according to the AJCC UICC 8th edition Neuroendocrine Tumors of the duodenum and ampulla of Vater (Table 4a and b).
|
Table 4a. TNM Staging of Duodenal Neuroendocrine Tumors (AJCC UICC 8th edition)
|
|
Primary Tumor (T)
If the number of tumors is known use T (#), if unavailable or too numerous T(m) e.g., T2(3) or T2(m)
|
|
TX
|
Tumor cannot be assessed
|
|
T1
|
Tumor invades the mucosa or submucosa only and is ≤ 1 cm (duodenal)
Tumor ≤ 1 cm and confined within the sphincter of Oddi (ampullary)
|
|
T2
|
Tumor invades the muscularis propria or is >1 cm (duodenal).
Tumor invades through sphincter into duodenal submucosa or muscularis propria, or is >1 cm (ampullary).
|
|
T3
|
Tumor invades the pancreas or peripancreatic adipose tissue
|
|
T4
|
Tumor invades the visceral peritoneum (serosa) or other organs
|
|
Regional lymph Nodes (N)
|
|
NX
|
Regional lymph nodes cannot be assessed
|
|
N0
|
No regional lymph node involvement
|
|
N1
|
Regional lymph node involvement
|
|
Distant Metastases (M)
|
|
M0
|
No distant metastases
|
|
M1
|
Distant metastases
|
|
M1a
|
Hepatic metastases only
|
|
M1b
|
Extra-hepatic metastases only
|
|
M1c
|
Both hepatic and extra-hepatic metastases
|
|
Table 4b. Stage Grouping
|
|
Stage I
|
T1 N0 M0
|
|
Stage II
|
T2-3 N0 M0
|
|
Stage III
|
T4 N0 M0
Any T N1 M0
|
|
Stage IV
|
Any T Any N M1
|
Non-Functioning Pancreatic NETs
In patients with known MEN1, screening is advised for early detection of NF-PanNETs. When NF-PanNETs are diagnosed and there is no immediate indication for intervention, surveillance should be performed at regular intervals to re-evaluate indications for intervention, as well as to detect newly developing dpNETs. Current guidelines suggest to start screening for NF-PanNETs in MEN1 below the age of 10 by a combination of biochemical tests and yearly imaging (either MRI, CT or EUS) (2).
Since the publication of the guidelines, it has become clear that the diagnosis of NF-PanNETs in patients with MEN1 relies heavily on imaging, since tumors markers chromogranin A, pancreatic polypeptide, and glucagon have low accuracy for the diagnosis of NF-PanNETs as summarized in a recent systematic review (143). Additionally, since the publication of the guidelines SSTR-PET-CT has emerged as a high-sensitive diagnostic imaging tool for dpNETs and its role within the screening and surveillance of MEN1-related dpNETs has yet to be determined.
There is currently no consensus regarding the best imaging modality and interval for screening and surveillance of MEN1-related NF-PanNETs (144). In a recent systematic review on the diagnosis of NF-PanNETs in MEN1, it was concluded that for lifelong screening and surveillance CT was probably least suitable given the inferior sensitivity compared to EUS and SSTR-PET-CT in combination with the cumulative exposure to ionizing radiation, although head-to-head comparisons with MRI are not available (143). This does not mean that a CT scan cannot still be indicated and be the best imaging for specific clinical situations (for example pre-operative imaging). EUS is the most sensitive method for the diagnosis of NF-PanNETs and offers the possibility of obtaining tissue for analysis pre-operatively. However, it is also invasive, operator dependent, clinically significant PanNETs can be missed in the pancreatic tail, and for the diagnosis of NF-PanNETs in MEN1 histological confirmation is usually not necessary given the high pre-test likelihood and the typical appearance on imaging. On the other hand, tissue-based analysis prior to intervention may become more relevant in MEN1 as more novel prognostic factors are identified. MRI has the advantage of performing more homogenously throughout the pancreas and the absence of ionizing radiation although a significant proportion of NF-PanNETs >2 cm is missed. The authors of the systematic review therefore suggest alternate use of EUS and MRI (143).
Given a reported growth rate of 0.1-1.32 mm/year for small NF-PanNETs, if initial screening imaging is negative, the next imaging can be performed with an interval of two to three years, providing no clinical reason for earlier imaging. For prevalent NF-PanNETs without an indication for intervention, the interval for active surveillance imaging should be individualized according to growth rate. Initial repeat imaging should be done after 6-12 months to assess growth rate, but afterwards in small stable NF-PanNETs the interval can be extended to once every 1-2 years. The imaging modality can be either MRI or alternating with EUS. EUS alone should always be combined with an imaging method for metastases detection, since it does not offer complete abdominal imaging. The exact role of SSTR-PET-CT in the screening and surveillance of MEN1-related NF-PanNETs is still to be determined, however, based on currently available evidence, it is best employed when results may change management such as in prevalent NF-PanNETs >10 mm for early detection of metastases, or as comprehensive staging before planned interventions (143, 145).
Another dilemma is when to start radiological screening for NF-pNET in children with MEN1. As mentioned before, current guidelines advise initiating screening before the age of 10 (2). However, others have advocated postponing until the age of 16, in the absence of signs and symptoms (37). Recently, modeled data from the Dutch population-based MEN1 cohort show that the estimated age at a 1%, 2,5% and 5% risk of having developed a clinically significant NF-PanNET (≥ 20mm or documented growth of ≥1.6 mm within one year above a baseline size of ≥ 15mm) is 9.5, 13.5 and 17.8 years respectively and they conclude that there is medical indication to initiate radiological screening during the second decade of life and that starting between 13-14 years of age is justifiable (15).
It is important to remember that each screening and surveillance schedule should be tailored to the needs of the individual patient in his or her unique circumstances, should be based on well-informed shared decisions making between providers and patients (and parents if applicable), with multidisciplinary team input when necessary.
The only curative treatment for NF-PanNETs in MEN1 is surgical resection, and the goal of surgical intervention in NF-PanNETs is to prevent metastases and thereby NF-PanNET-related mortality, while preserving as much pancreatic tissue as possible and limiting treatment-related morbidity and mortality. Although theoretically, total duodenopancreatectomy would prevent metastatic disease altogether, short-term morbidity associated with this complex major surgery is high and the subsequent life-long brittle diabetes that follows rarely justifies such major intervention when balanced against the risk of distant metastases and PanNET-related death.
Since the risk of future metastases and disease-related death must be balanced against short- and long-term treatment-related morbidity and mortality, information regarding prognosis in MEN1-related NF-PanNETs is of vital importance to make well-informed decisions regarding timing and extent of intervention. However, presently there is a paucity of prognostic factors on which to base these decisions (146). The most important factor to date is tumor size, with the risk of (distant) metastases increasing with increased size. Recent data from retrospective cohort studies have shown that small (<2cm) NF-PanNETs generally have an indolent course, that surgical resection of small NF-PanNETs does not seem to offer benefit over active surveillance, and that the risk of metastases and disease-related death is low, albeit not zero (124, 146-150). Most small NF-PanNETs are stable during follow-up, but there is a subset with progression in size (150). Generally, size progression is also considered to be a prognostic factor. An important tissue-based prognostic factor is tumor grade, with grade 2 tumors being more often associated with metastases (146). Grade 3 NF-PanNETs or NECs are rarely seen in patients with MEN1, but are associated with a worse prognosis. More recently, advancements in molecular techniques have identified several potential prognostic biomarkers for NF-PanNETs, mostly in sporadic NF-PanNETs, but limited data in MEN1-related NF-PanNETs is also available. Mutations in alpha-thalassemia/mental retardation X-linked (ATRX) and death domain-associated protein (DAXX), which lead to the alternative lengthening of telomeres (ALT) phenotype have been found to be associated with decreased disease-free survival and higher rates of distant metastases (146, 151). Mutations in DAXX and ATRX result in loss of nuclear expressions of their proteins by immunohistochemistry (IHC) and ALT can be identified in tissue-samples by telomere-specific fluorescence in situ hybridization (FISH). Next to DAXX/ATRX and ALT, the differential expression of transcription factors aristaless-related homeobox gene (ARX) and pancreatic and duodenal homeobox 1 (PDX1) as assessed by IHC was also found to be associated with risk of metastases (152, 153). In patients with MEN1-related NF-PanNETs, one study showed that liver metastases were only seen in ARX+ or ARX-/PDX1- tumors and that ALT positivity was only seen in ARX+ or ARX-/PDX1- tumors and significantly correlated with relapse rate (152). However, since the publication of these data, a large international cohort of 1322 NETs (not including MEN1-related NETs), was evaluated by immunolabelling for ARX/PDX1, ATRX/DAXX and by telomere-specific FISH for ALT and it was found that ATRX/DAXX and ALT, but not ARX/PDX1 were independent negative prognostic factors (151).
A recent study by Fahrmann, et al. identified a 3-marker polyamine signature that distinguished patients with metastatic dpNETs from controls and which yield an AUC of 0.84 (95% CI: 0.62-1.00) with 66.7% sensitivity at 95% specificity for distinguishing cases form controls in an independent test set (154). These results form the basis for prospective testing of plasma polyamines as a prognostic factor for MEN1-related dpNETs.
Further validation of these molecular markers in MEN1, may also change the role of pre-intervention EUS-guided aspiration or biopsy.
So, when to intervene in MEN1-related NF-PanNETs? Presently, these decisions are mostly based on tumor size and growth, with the current guidelines suggesting considering surgical resection in NF-PanNETs >10 mm or those that show significant growth during follow-up (doubling of tumor size over a 3–6-month interval and exceeding 10 mm) (2). Given the emerging evidence that most NF-PanNETs <20 mm are indolent in nature as described above, a more recent consensus statement states that surgical resection is indicated for NF-PanNETs >20 mm and those that progress during surveillance (155). Additionally, the presence of suspicious lymph nodes, or a higher grade on EUS-guided aspiration may guide intervention decisions. In all cases these decisions should be made in multidisciplinary teams and in shared decision making with the patient. The extent of resection depends on multiple patient-, tumor- and MEN1-related factors and should be individualized.
Gastrinoma
Gastrinomas, NETs secreting gastrin, cause the Zollinger-Ellison Syndrome (ZES). ZES is a syndrome characterized by tumor-related hypergastrinemia leading to gastric acid hypersecretion.
Sign and symptoms of ZES/Gastrinoma are gastro-esophageal reflux disease (GERD), (proton-pump inhibitor (PPI) responsive) diarrhea, abdominal pain, nausea/vomiting, weight loss, and peptic ulcer disease. Complications may arise from the peptic ulcer disease including upper gastro-intestinal bleeding, strictures, and bowel perforation.
Before the introduction of PPIs, complications from gastric acid hypersecretion were an important cause of death in patients with MEN1 (134). With the arrival of proton pump inhibitors, gastric acid hypersecretion can be effectively treated, although higher dosages are needed than for the treatment of non-ZES hyperacidity.
The diagnosis of gastrinoma in MEN1 is challenging at present. The gold standard for the diagnosis is the demonstration of inappropriate fasting hypergastrinemia without the use of antisecretory drugs. The diagnosis is established if the fasting serum gastrin (FSG) is more than tenfold the upper limit of normal with a gastric pH of less than two (after ruling out retained antrum) (156, 157). When gastric pH is low and FSG is <10-fold upper limit of normal, additional testing is needed to establish the diagnosis, such as a secretin provocative test or measuring basal acid output (156, 157). The latter situation occurs in 60% of ZES, and this might even be higher in MEN1, given the early detection through prospective screening programs. Due to unreliable gastrin assays, the limited availability of secretin and therewith the loss of expertise in performing the secretin provocative test, the wide-spread use of PPIs and the risk associated with cessation of PPI for proper testing, the diagnosis of gastrinoma is challenging (157, 158). Additionally, the value of non-biochemical tests in the diagnosis of gastrinoma (such as SSTR-PET-CT and EUS/ esophagogastroduodenoscopy (EGD)-guided cytology/biopsy) might also need re-evaluation (157). Recently experts have therefore suggested possible new criteria for diagnosis of ZES in patients with fasting hypergastrinemia with and without PPI use (157).
Patients with MEN1 are screened for the presence of a gastrinoma by at least annually assessing clinical symptoms and fasting serum gastrin (2). If a diagnosis of gastrinoma is established or suspected, EGD should be performed to assess the presence of complications of gastric acid hypersecretion, type II gastric NETs, and possibly to identify duodenal gastrinomas. Duodenal gastrinomas in MEN1 are small, but despite their small size 70-80% are metastatic to the regional lymph nodes at the time of diagnosis (159). However, these regional lymph node metastases do not seem to have a negative impact on overall survival (159). In MEN1, attributing locoregional lymph nodes to the correct primary dpNET is important for adequate treatment planning and prognostic inferences. It is also challenging however, given that most patients with MEN1 and duodenal gastrinoma(s) also have concomitant PanNETs, and the duodenal gastrinoma(s) may not be visible on imaging due to their small size. Hackeng, et al. studied 137 microscopic and macroscopic dpNETs and 36 matched metastases (lymph node and distant) in 10 patients with MEN1 to unravel the relationship between the multiple primary dpNETs in MEN1 and the metastases (160). They found that most patients had a single NET of origin for their metastases, but multiple metastatic primaries were also seen. In addition, and very important for MEN1-related gastrinomas, in 6 patients with MEN1 and hypergastrinemia, periduodenopancreatic lymph node metastases clustered with minute duodenal gastrinomas and not with larger pancreatic NETs. So a duodenal origin for periduodenopancreatic lymph node metastases in patients with MEN1 and hypergastrinemia should always be considered (160).
Although its role still needs to be delineated, in MEN1-gastrinoma next to EUS/EGD, SSTR-PET-CT may be very useful for staging to visualize duodenal gastrinomas, lymph node metastases, and concomitant PanNETs.
Presently, as in other MEN1-related dpNETs, surgery is the only potentially curative treatment. And although MEN1-related ZES has been historically been considered a surgically incurable disease, more recent small studies have shown, that when the correct target organ is addressed, namely the duodenum and not the pancreas, biochemical cure can be achieved after partial pancreaticoduodenectomy(PD), combined with regional lymph node dissection (159). However, this must be balanced against the risk of peri-operative and long-term complications and loss of quality of life. Overall survival is generally good in duodenum-preserving operations as well, but persistence or recurrence of ZES occurs in 6-100% (159). Most often, hyperacidity can be adequately controlled with PPIs, making the main goal of surgical resection the prevention of distant metastases and disease-related death. The majority of MEN1-ZES patients with associated small PanNETs have an indolent disease course with excellent overall survival even without surgical intervention (161). Still, in retrospective studies around a quarter of the patients develop liver metastases and around 15% shows aggressive growth (138), and presently there are no good markers to predict which patients with MEN1-ZES will have a more aggressive disease course. In a study from the NIH age at ZES diagnosis (≤33), FSG levels ≥10,000 pg/mL, pancreatic tumors >3 cm, presence of liver/bone metastases and presence of gastric carcinoids were associated with aggressive tumor growth (138). In a study from the DutchMEN Study Group, overall survival rates of MEN1-gastrinoma were 83% and 65% at 5 and 10 years respectively, which was significantly worse than age- and gender-matched patients without gastrinoma. FSG ≥ 20x upper limit of normal, PanNETs≥ 2cm, synchronous liver metastases, EGD suspicious for gastric NETs, and multiple concurrent NETs were associated with decreased overall survival (135). A recent study from the French GTE, ZES was independently associated with a higher risk of distant metastases, but did not significantly seem to be associated with decreased overall survival (125).
So presently, if surgical intervention should be performed, when surgical intervention should be performed and how (to what extent) surgical intervention should be performed for gastrinoma in MEN1 are all controversial topics. Treatment of patients in centers of expertise with a highly dedicated multidisciplinary team and experienced surgeons is therefore very important. Treatment decisions for MEN1-ZES should be made after MDT discussion in shared decision making with the patient.
As stands to reason, treatment of MEN1-ZES patients with (high-dose) PPI is mandatory and patients should not cease this treatment without consultation with their provider. If specific testing without PPI is needed this needs to be performed under close supervision in centers of expertise.
As mentioned in the section on parathyroid tumors, the interplay between pHPT and ZES in MEN1 is important to realize as hypercalcemia can increase gastrin levels. Additionally, a recent paper on the Tasmanian MEN1 cohort showed an association between H. pylori seropositivity and hypergastrinemia and severe ZES-range hypergastrinemia. Further work is needed to fully elucidate this relationship, but testing for H. Pylori and eradication if positive might be consider in patients with MEN1-ZES (162).
Insulinoma
As already stated, MEN1-related insulinomas occur at a young age and are the most frequent functional PanNET at the pediatric age. Early recognition of signs and symptoms of insulinoma is of extreme importance in both children and adults. Signs and symptoms may be erroneously attributed to epilepsy or behavioral or neurological disorders, especially if insulinoma is the presenting manifestation of MEN1 in an index case. In children this can lead to decline in school performance and in children and adults alike episodes of hypoglycemia can lead to accidents or irrational behavior.
As insulinomas secrete insulin inappropriately and lead to hypoglycemia the signs and symptoms are those of hypoglycemia; both adrenergic symptoms (such as fast heartbeat, jitteriness/shakiness, sweating and pale skin) as well as neuroglycopenic symptoms (such as mental status changes and irritability). Symptoms are relieved with food (glucose) intake. They usually occur during fasting, before meals or after exercise, but can occasionally occur at other times. In patients fulfilling Whipple’s triad (symptoms and/ or signs consistent with hypoglycemia, a low plasma glucose concentration, and resolution of symptoms/signs after plasma glucose concentration is raised) diagnosis can be established by a supervised fast (163). In patients with MEN1, screening for insulinoma is advised from the age of five by careful history taking and measurement of fasting insulin and – more importantly – glucose (2). However, almost all patients with MEN1 and insulinoma are symptomatic, therefore the history is probably the most important element.
When the diagnosis of insulinoma is made, localization in MEN1 can be challenging, if there are multiple PanNETs. These usually are concomitant NF-PanNETs, since in surgical series multiple insulin-positive PanNETs in patients operated for insulinoma were seen in 8-40% (132, 164-166).
For MEN1-related insulinoma, especially if conventional imaging shows multiple PanNETs and correctly identifying the insulinoma(s) among them would change surgical strategy, 86Ga-Exendin-4 PET-CT is very promising. Although there is limited data in MEN1 patients, a recent meta-analysis showed a positive predictive value (PPV) of 94%, with a negative predictive value (NPV) of 67%; In MEN1 PPV was 95% with NPV 96%, although based on a limited number of patients (167, 168).
Surgical resection is the treatment of choice for MEN1-related insulinomas and is associated with a high cure rate. In a retrospective cohort study of 40 European and 6 North-American institutes 92 patients with MEN1-related insulinomas who underwent surgical resection were followed for a median of 8 years after surgery (132). Overall, after different surgical procedures, only 1 patient had persistence of hypoglycemia and six had recurrent hypoglycemia, four due to new primaries and 2 due to development of liver metastases, leading to a 10-year hypoglycemia-free survival of 91% (95% CI 80-96). For those with unifocal insulinoma based on pre- and intra-operative assessment (n=63), 1/46 (2.2%) undergoing pancreas resection had persistent disease, while among those who underwent enucleation 1/17 (6%) had recurrence of hypoglycemia based on a new primary insulinoma. For those with multifocal insulinoma (n=33), of whom 30 underwent pancreatic resection, mostly distal pancreatic resection, and three had multiple enucleations, 15% had recurrent hypoglycemia (9% based on new primaries and 6% based on liver metastases) (132).
Therefore, given the better outcomes of pancreatic function over the long-term and young age of the patients, if surgically feasible, enucleation seems the better option for solitary insulinomas in MEN1, provided of course that concomitant functional and non-functional tumors do not make a different strategy necessary (132).
Among MEN1-related dpNETs, insulinomas have the best oncological prognosis (26, 125, 169). Data from the international MEN1 Insulinoma Study Group and the DutchMEN Study Group show that for surgically resected insulinomas 10-yr liver-metastases free survival was 87% (72-91%) (169). Malignant insulinoma is rare, both in sporadic and MEN1-related insulinoma. In the two largest MEN1-insulinoma cohorts synchronous liver metastases were seen in 3.8-8.1% and metachronous liver metastases in 0-2.2% after a median follow-up of 8-9 years (132, 165).
Rare Functional dpNETS
Functional dpNETs besides gastrinomas and insulinomas, are rare in MEN1 and are seen in <1% of patients with dpNETs(2). These include PanNETs producing vaso-active intestinal peptide (VIPoma), somatostatin, glucagon and other (ectopic) hormones such as growth hormone releasing hormone (GHRH), calcitonin, or PTH-related peptide (PTHrP). A rare functional tumor is considered if there are elevated hormone levels in conjunction with a fitting clinical syndrome. Without a clinical syndrome, tumors are not considered functional but merely hypersecreting. This is relevant as for example glucagon can be elevated in patients with MEN1 and PanNETs without the patient having the glucagonoma syndrome. VIPomas lead to watery diarrhea, hypokalemia, achlorhydria and dehydration, the somatostatinoma syndrome consists of diabetes mellitus, diarrhea, steatorrhea and cholelithiasis, while glucagonomas give rise to necrolytic migratory erythema, diabetes mellitus, and weight loss. Tumors producing GHRH, calcitonin, and PTHrP lead to acromegaly, diarrhea and hypercalcemia, respectively. In these rare functional dpNETs without synchronous distant metastases surgery is generally indicated.
Although for most non-metastatic functional dpNETs in MEN1 surgery is indicated, there may be a (temporary) need to control the hormonal syndrome medically. As such gastrinomas are treated with high-dose PPI, insulinomas with diazoxide or frequent feedings and in all cases somatostatin analogues might be considered if needed to control the functional syndrome.
Local resection of sporadic small dNETs is increasingly considered as an alternative to surgery (170), and current European Neuroendocrine Tumor Society (ENETS) guidelines recommend endoscopic management for dNETs ≤ 10mm in size, confined to the submucosal layer and without lymph node and distant metastases (171). However, since in MEN1 dNETs are usually multiple, may grow beyond the submucosa and, in case of gastrinomas, are associated with lymph nodes in up to 80% of the cases, this is not generally recommended (159).
Similarly, for PanNETs EUS-guided intervention using ethanol or radio-frequent ablation has been reported in around 80 and 70 cases respectively in the literature, with only a handful procedures performed in patients with MEN1 (172). Whether or not interventional EUS may play a role in treatment of MEN1-related PanNETs is therefore unclear at the present time.
There is much interest in chemoprevention in small NF-PanNETs using somatostatin analogues (SSA). SSA have proven anti-proliferative effect in advanced PanNETs (173, 174) and the question has been raised if SSA may be used to prevent progression and metastases of small NF-PanNETs in patients with MEN1. In mouse models of Men1PanNET lanreotide and pasireotide showed the ability to decrease tumor proliferation. In a retrospective non-controlled study of 20 patients with small NF-PanNETs who received long-acting octreotide for 12-75 months 10% had an objective tumor response, 80% stable disease, and 10% showed progression (175). In another small (n=8) prospective series patients with small NF-PanNETs were treated with SSA for up to 72 months, with stable disease in all, however again without a control group (176). In a recent observational cohort study lanreotide was compared with standard of care active surveillance in n=42 patients with pNETs <2 cm (N=23 lanreotide vs n=19 active surveillance) during a median follow-up of 6 years (177). The study showed improved RECIST-defined progression-free survival (PFS) in the lanreotide group. In both groups however, one patient developed distant (liver) metastases (177). Limitations include sample size, non-experimental and therefore non-randomized design, and non-blinded outcome evaluation. In addition, improved RECIST PFS is not yet known to predict longer overall survival for MEN1 patients with small NF-PanNETs. Ideally this is further evaluated in a randomized double-blind trial. The most important challenge in the design of such a study however, is the definition of appropriate surrogate endpoint for distant metastases and overall survival (144).
The treatment of stage IV dpNET in patients with MEN1 is similar to that of patients with sporadic dpNETs(178). There is very limited evidence regarding MEN1-specific outcome data, and from the limited evidence available there seems to be no difference with sporadic NETs. In landmark studies leading to approval of lanreotide (173), everolimus (179), sunitinib (180) and peptide receptor radionuclide therapy (PPRT) (181), patients with MEN1 were either excluded, only single cases included or MEN1-status was not mentioned (182). With the advancing molecular understanding of MEN1-related NETs, MEN1-specific targeted therapies might be possible in the future, which in turn might benefit the almost 50% MEN1-mutated sporadic PanNETs (182).
Gastric NETs in MEN1 (Type II Gastric NETs)
NETs of the stomach (Figure 1), formerly called gastric carcinoids or carcinoids of the stomach, are classified into three different types(171):
- Type I, associated with atrophic gastritis
- Type II, associated with MEN1/ZES
- Type III, without associated conditions
Gastric NETs are graded according to the latest WHO classification of digestive system tumors (2019, 5th edition) as described above for dpNETs (Table 2) (142). TNM staging is shown in Table 5a and b.
|
Table 5a. TNM Staging of Neuroendocrine Tumors of the Stomach (AJCC UICC 8th edition)
|
|
Primary Tumor (T)
For any T, add (m) for multiple tumors, for multiple tumors with different Ts, use the highest (e.g., if three tumors’ sizes 0.5, 0.5, and 1.5 cm, T stage should be T2(m).
|
|
TX
|
Tumor cannot be assessed
|
|
T0
|
No evidence of primary tumor
|
|
T1
|
Invades lamina propria or submucosa and ≤ 1 cm
|
|
T2
|
Invades muscularis propria or >1cm
|
|
T3
|
Invades through the muscularis propria into subserosal tissue without penetration of overlying serosa
|
|
T4
|
Invades visceral peritoneum (serosal) or other organs or adjacent structures
|
|
Regional lymph Nodes (N)
|
|
NX
|
Regional lymph nodes cannot be assessed
|
|
N0
|
No regional lymph node involvement
|
|
N1
|
Regional lymph node involvement
|
|
Distant Metastases (M)
|
|
M0
|
No distant metastases
|
|
M1
|
Distant metastases
|
|
M1a
|
Hepatic metastases only
|
|
M1b
|
Extra-hepatic metastases only
|
|
M1c
|
Both hepatic and extra-hepatic metastases
|
|
Table 5b. Stage Grouping
|
|
Stage I
|
T1 N0 M0
|
|
Stage II
|
T2-3 N0 M0
|
|
Stage III
|
T4 N0 M0
Any T N1 M0
|
|
Stage IV
|
Any T Any N M1
|
Gastric NETs are tumors of the gastric entero-chromaffin like (ECL) cells, which develop in MEN1 due to the trophic effect of gastrin on ECL-cells combined with the predisposing germline MEN1 mutation. Both components seem to be necessary for the development of gastric NETs in patients with MEN1. Gastric NETs are rarely seen in sporadic ZES patients and loss of heterogeneity was demonstrated in 75% of MEN1-related gastric NETs (183, 184), while in patients with MEN1 gastric NETs occur almost exclusively in patients with gastrinoma and regression of gastric NETs has been reported after normalization of hypergastrinemia (185, 186). ECL-cell hyperplasia is considered a precursor lesion for gastric NETs (187).
In the NIH-ZES cohort, 57 patients were extensively studied for gastric ECL-cell changes. All of the patients were found to have proliferative ECL-cell changes, with advanced changes in 53% and gastric NETs in 23% (188). More recently, data on ECL-cell changes in patients with MEN1 was reported from the Marburg MEN1 database (185). They reported on 38 MEN1 patients who underwent regular screening including EGD, regardless of gastrinoma status. Sixteen of these patients had a gastrinoma diagnosis, 13 of whom had biochemical ZES at the time of first EGD. They found that ECL-changes and gastric NETs were exclusively seen in patients with MEN1-gastrinoma albeit in a lower percentage than in the NIH. They found ECL hyperplasia in 62.5% of patients with a gastrinoma diagnosis, versus 0% in those without gastrinoma. No advanced ECL-cell changes were seen and gastric NETs were found in 12.5% of patients with a gastrinoma diagnosis (185). These differences might also reflect practice changes with earlier gastrinoma diagnosis due to screening and surveillance and more frequent surgical treatment of gastrinoma in the Marburg cohort compared to the NIH cohort.
In the NIH cohort, higher levels of FSG as well as longer duration of ZES were associated with a higher risk of advanced ECL-cells changes and gastric NETs (188). Higher levels of FSG as risk factor could not be confirmed in the Marburg cohort, however numbers of MEN1-ZES were small (185). As mentioned above, in the section on gastrinoma, the presence of gastric NETs in patients with MEN1-ZES was associated with a more aggressive disease course (138) and decreased overall survival (135).
Therefore, in the current MEN1 Clinical Practice Guidelines, EGD with biopsy, is recommend every 3 years in patients with MEN1 and hypergastrinemia. Although treatment of MEN1-related gastric NETs is not well established (2, 171), the guideline suggest that lesions <10 mm may remain under endoscopic surveillance, while larger tumors require endoscopic resection or local resection, which is analogue to the treatment of type I gastric NETs (2, 171).
The prognosis of MEN1-related gastric NETs is generally good, with metastases (regional and distant) reported in 10-30% and disease-related death <10% (171). Nevertheless, aggressive symptomatic and metastatic cases leading to mortality have been reported (189, 190).
Conclusion
In conclusion, dpNETs are highly prevalent in patients with MEN1 reaching a more than 80% penetrance at the age of 80. NF-PanNETs are most frequently seen, followed by gastrinomas and insulinomas. Most MEN1-related dpNETs are diagnosed at an early stage and NF-PanNETs <2 cm generally have an indolent course. However, distant metastatic dpNETs (mostly NF-PanNETs and gastrinoma) are the most important cause of MEN1-related mortality. Treatment goals for MEN1-related dpNETs are therefore to prevent metastatic disease, cure hormonal hypersecretion, and prevent complication from hormonal hypersecretion, while minimizing treatment-related complications and preserving Quality of Life. Surgical resection is the mainstay for treatment, and is indicated in non-gastrinoma functional PanNETs and NF-PanNETs >2cm or with progression during follow-up. No consensus exists on the surgical treatment of MEN1-related gastrinoma. With increasing awareness of MEN1, increasingly refined and defined screening and surveillance programs, and increasing sensitive imaging modalities, MEN1-related dpNETs are detected at earlier stage and more indolent small dpNETs are seen. The main challenge at this point is therefore identifying those patients who are at risk for a more aggressive disease course and distant metastases to be able to offer those patients close follow-up schedules and earlier and more aggressive treatment, while limiting treatment-related morbidity in patients with low risk. Novel prognostic indicators are therefore needed, ideally blood-based, so minimal invasive assessment is possible. Other future directions are the investigation of chemoprevention in small NF-PanNETs.
Gastric NETs in MEN1 are almost exclusively seen in patients with gastrinoma and usually have an indolent course. Screening with EGD should be performed in all patients with MEN1-ZES.
THORACIC NEUROENDOCRINE TUMORS
General
Thoracic NETs occurring as part of the MEN1 syndrome are thymic (thNET) and bronchopulmonary NETs (bpNET) (Figure 1), although recently it was suggested that thymomas may also be part of the MEN1-related tumor spectrum (191). These tumors are not considered main disease-defining manifestations. As in other MEN1-related tumors, loss of heterogeneity (LOH) at the MEN1 locus was demonstrated in bpNETs in patients with MEN1 (97). This in contrast to thymic NETs, where until recently no LOH at the MEN1 locus was found. However, in a recent publication by the NIH, LOH was seen at the MEN1 locus in both MEN1-related thymic NETs (8 out of 12) and two (out of two) thymomas in patients with MEN1 (191). MEN1 is also the most frequently mutated gene in sporadic well-differentiated bpNETs, this is not described in thymus NET (192). However, approximately 25% of patients with thNET have germline mutations in MEN1, therefore it is very important to consider the diagnosis of MEN1 in patients presenting with a sporadic thNET (193). Thymic and bronchopulmonary NETs in MEN1 generally develop in adults. In pediatric and adolescent series (age up to 21, 31 in one series), there is only one reported case of thNET (diagnosed at age 16) and two cases of bpNET (diagnosed at age 15 and 20 respectively) (37, 46-48).
Staging and Grading
bpNET and thNET are classified according to the WHO classification (Table 6). TNM staging for thymicNETs is shown in Table 7a and b. TNM staging of bronchopulmonary NETs follows the same classification as bronchogenic lung carcinomas (Table 8a and b).
|
Table 6. WHO Classification of Bronchopulmonary and Thymic Neuroendocrine Tumors
|
|
Classification
|
Mitotic Rate and Necrosis
|
|
Well-differentiated
|
|
Typical Carcinoid, NET G1
|
Mitotic rate <2 and absence of necrosis
|
|
Atypical Carcinoid, NET G2
|
Mitotic rate 2-10 and/or presence of necrosis
|
|
Poorly differentiated
|
|
Neuro-endocrine carcinomas
Small-cell type
Large-cell type
|
Mitotic rate >10
|
|
Table 7a. Staging of Thymic Neuroendocrine Tumors (AJCC UICC 8th edition)
|
|
Primary Tumor (T)
|
|
TX
|
Tumor cannot be assessed
|
|
T0
|
No evidence of primary tumor
|
|
T1
T1a
T1b
|
Tumor encapsulated or extending into the mediastinal fat; may involve the mediastinal pleura
Tumor with no mediastinal pleura involvement
Tumor with direct invasion of mediastinal pleura
|
|
T2
|
Tumor with direct invasion of the pericardium (either partial or full thickness)
|
|
T3
|
Tumor with direct invasion into any of the following: Lung, brachiocephalic vein, superior vena cava, phrenic nerve, chest wall, or extrapericardial pulmonary artery or veins
|
|
T4
|
Tumor with invasion into any of the following: Aorta (ascending, arch, or descending), arch vessels, intrapericardial pulmonary artery, myocardium, trachea, esophagus
|
|
Regional lymph Nodes (N)
|
|
NX
|
Regional lymph nodes cannot be assessed
|
|
N0
|
No regional lymph node involvement
|
|
N1
|
Metastasis in anterior (perithymic) lymph nodes
|
|
N2
|
Metastasis in deep intrathoracic or cervical lymph nodes
|
|
Distant Metastases (M)
|
|
M0
|
No pleural, pericardial, or distant metastasis
|
|
M1
|
Pleural, pericardial, or distant metastasis
|
|
M1a
|
Separate pleural or pericardial nodule(s)
|
|
M1b
|
Pulmonary intraparenchymal nodule or distant organ metastasis
|
|
Table 7b. Stage Grouping
|
|
Stage I
|
T1 N0 M0
|
|
Stage II
|
T2 N0 M0
|
|
Stage IIIa
|
T3 N0 M0
|
|
Stage IIIb
|
T4 N0 M0
|
|
Stage IVa
|
Any T N1 M0
Any T N0-1 M1a
|
|
Stage IVb
|
Any T N2 M0-M1a
|
|
|
Any T Any N M1b
|
|
Table 8a. TNM Staging of Bronchopulmonary Neuroendocrine Tumors (AJCC UICC 8th edition)
|
|
Primary Tumor (T)
If the number of tumors is known used T (#), if unavailable or too numerous T(m) (e.g., T2a(2) or T2a(m)).
|
|
TX
|
Primary tumor cannot be assessed or tumor proven by presence of malignant cells in sputum or bronchial washings but not visualized by imaging or bronchoscopy
|
|
T0
|
No evidence of primary tumor
|
|
Tis
|
Tumor in situ
|
|
T1
T1a(mi)
T1a
T1b
T1c
|
Tumor ≤3 cm in greatest dimension surrounded by lung or visceral pleura without bronchoscopic evidence of invasion more proximal than the lobar bronchus (i.e., not in the main bronchus)*
Minimally invasive adenocarcinoma
Tumor ≤1 cm in greatest dimension
Tumor >1 cm but ≤2 cm in greatest dimension
Tumor >2 cm but ≤3 cm in greatest dimension
|
|
T2
T2a
T2b
|
Tumor >3 cm but ≤5 cm or tumor with any of the following features:
Involves main bronchus regardless of distance from the carina but without involvement of the carina
Invades visceral pleura
Associated with atelectasis or obstructive pneumonitis that extends to the hilar region, involving part or all of the lung
Tumor >3 cm but ≤4 cm in greatest dimension
Tumor >4 cm but ≤5 cm in greatest dimension
|
|
T3
|
Tumor >5 cm but ≤7 cm in greatest dimension or associated with separate tumor nodule(s) in the same lobe as the primary tumor or directly invades any of the following structures: chest wall (including the parietal pleura and superior sulcus tumors), phrenic nerve, parietal pericardium
|
|
T4
|
Tumor >7 cm in greatest dimension or associated with separate tumor nodule(s) in a different ipsilateral lobe than that of the primary tumor or invades any of the following structures: diaphragm, mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, esophagus, vertebral body, and carina
|
|
Regional lymph Nodes (N)
|
|
NX
|
Regional lymph nodes cannot be assessed
|
|
N0
|
No regional lymph node involvement
|
|
N1
|
Metastasis in ipsilateral peribronchial and/or ipsilateral hilar lymph nodes and intrapulmonary nodes, including involvement by direct extension
|
|
N2
|
Metastasis in ipsilateral mediastinal and/or subcarinal lymph node(s)
|
|
N3
|
Metastasis in contralateral mediastinal, contralateral hilar, ipsilateral or contralateral scalene, or supraclavicular lymph node(s)
|
|
Distant Metastases (M)
|
|
M0
|
No distant metastasis
|
|
M1
|
Distant metastases
|
|
M1a
|
Separate tumor nodule(s) in a contralateral lobe; tumor with pleural or pericardial nodule(s) or malignant pleural or pericardial effusion
|
|
M1b
|
Single extrathoracic metastasis
|
|
M1c
|
Multiple extrathoracic metastases in one or more organs
|
|
Table 8b. Stage Grouping
|
|
Stage 0
|
Tis N0 M0
|
|
Stage IA
|
T1 N0 M0
|
|
Stage IB
|
T2a N0 M0
|
|
Stage IIA
|
T2b N0 M0
|
|
Stage IIB
|
T1a-c N1 M0
T2 N1 M0
T3 N0 M0
|
|
Stage IIIA
|
T1a-c N2 M0
T2 N2 M0
T3 N1 M0
T4 N0-1 M0
|
|
Stage IIIB
|
T1a-c N3 M0
T2 N3 M0
T3 N2 M0
T4 N2 M0
|
|
Stage IIIC
|
T3-4 N3 M0
|
|
Stage IVA
|
Any T Any N M1a-b
|
|
Stage IVb
|
Any T Any N M1c
|
Thymic NET
ThNETs develop in 2.0 - 8.2% of MEN1 patients, with a median age at diagnosis of 43 years (range 16–72 years) (194-201). There is a strong male predominance (male to female ratio 4:1) in MEN1-related thNET, which is more pronounced in American and European cohorts compared to Asian series (194). Although one of the earliest studies on MEN1-related thNET suggested a higher prevalence of truncating MEN1 mutations in patients with thNET (202), no clear genotype-phenotype relationship has been described in later cohorts (196, 197, 199, 200). Furthermore, familial clustering of thNET within MEN1 families has been reported in a number of studies (193, 197, 199) but others could not find comparable results (196, 200). The suggested link between smoking and the occurrence of thNET in MEN1 remains controversial as well, as the portion of (heavy) smokers varied significantly among studies (193-196, 199, 201).
With the exception of a small subset of ACTH-producing tumors, most thNETs are functionally silent. As a result, the majority of patients only experiences symptoms when the tumor has reached an advanced stage, underlining the importance of periodic thoracic imaging for a timely detection.
MEN1-related thNET are characterized by their aggressive nature, illustrated by their frequent presentation with metastatic disease (53.5% of patients), usually located in lymph nodes, bones and lungs (194). Despite the low prevalence of thNETs among patients with MEN1, they are responsible for 19% of MEN1-related deaths (134). The poor prognosis of MEN1-related thNET has also been illustrated in a meta-analysis of 99 MEN-1 thNETs: median survival was 8.4 years, and the 10-year survival rate was 33%. An older age at diagnosis, a tumor diameter >5 cm and the presence of metastasis were associated with worse outcome (194).
Total (thoracic) thymectomy, including excision of the tumor, the entire thymus and perithymic fat, is the recommended treatment of choice (2, 203). Additional radiotherapy and chemotherapy may be used in patients with unresectable or metastatic disease. Data from the earlier mentioned meta-analysis suggested that adjunctive therapy after surgery tended to result in a better survival compared to surgery alone (after adjusting for gender, age at diagnosis, tumor size and smoking), but this effect did not reach statistical significance (HR 0.557, 95%CI: 0.110–2.817) (194). Prophylactic cervical thymectomy, generally performed during parathyroid surgery for primary hyperparathyroidism, may decrease the chance of the occurrence of thNET. However, several cases of thNET have been reported in patients after this procedure, indicating that surveillance imaging is still required in these patients (197, 202).
Bronchopulmonary NET
Histopathologically proven bpNETs occur in 4.7-6.6% of MEN1 patients, but a much higher proportion of MEN1 patients may be diagnosed with lesions radiologically suspect of bpNET (22.9%, 26.0% and 29.3% in the Dutch, Tasman and German cohort respectively) (195, 204-207). BpNETs are diagnosed at a median age of ± 45 years and the reported age at bpNET diagnosis ranges between 20 and 69 years. Although the earliest report suggested a female predominance among MEN1 patients with bpNET (205), later studies could not find a relationship between the occurrence of bpNET and sex (200, 204, 206, 207). Likewise, genotype (the type of mutation) or smoking status does not seem to influence the development of bpNET in MEN1 patients (204, 206, 207).
Only a minority of patients experience symptoms (dyspnea, cough, hemoptysis), which explains the high rate of bpNET (77–100%) diagnosed through periodic thoracic imaging surveillance (195, 206, 207). Growth analysis of lung lesions highly suspect of bpNET have demonstrated their overall indolent course, illustrated by a tumor doubling time of ±12 years at long-term follow-up in a Dutch national cohort study (204). However, a very small number of lesions showed sudden aggressive tumor growth. Unfortunately, no prognostic factors for tumor growth have been identified to date.
The vast majority of MEN1-related bpNETs are well-differentiated NETs (typical and atypical carcinoids); only five cases of poorly differentiated neuroendocrine carcinomas have been identified in MEN1 patients until now, all in the French Groupe d’étude des Tumeurs Endocrines (GTE) cohort (206). Considering the large cohort size (n=1023 MEN1 patients), long-term follow-up, high frequency of smokers and lack of molecular analyses confirming a causal relationship with the MEN1 syndrome, a sporadic coincidental occurrence of neuroendocrine carcinomas in MEN1 patients might also be a possible explanation for the manifestation of these carcinomas in this particular study. The overall benign histopathological characteristics of MEN1-related bpNET may explain their usually good prognosis: large cohort studies have shown that bpNETs do not significantly affect survival in MEN1 patients (204, 206), although a few (eight) aggressive cases with fatal outcome have been described (206, 207). A recent comparison between patients with MEN1-related and sporadic bpNET with comparable histopathological features showed a significantly higher disease-specific mortality in sporadic bpNET, however this has not yet been confirmed in other cohorts (208).
Data from the largest cohort of histologically proven bpNETs in MEN1 patients (n=51) suggested that patients with distant metastasis at diagnosis and non-operated patients had a significantly worse survival (206). Additionally, females, patients with a typical carcinoid (compared to atypical carcinoid) and those without lymph node involvement tended to have a better survival (p=0.07, p=0.08 and p=0.08, respectively (206). However, the most recent Dutch cohort study could not find any prognostic factors (204).
Surgical resection is considered the first treatment of choice, and should be done as lung-sparing as possible, including considering endobronchial resection if feasible (2, 203). Given their usually indolent course, watch-and-wait policy may be considered in patients with small, non-central, slow-growing lesions (203). Parallel to treatment regimens in sporadic bpNET, additional radiotherapy and/or chemotherapy could be used in case of persistent or metastasized disease, although data on the effect of these regimens in aggressive MEN1-related bpNET is very limited.
Surveillance
Current clinical guidelines recommend thoracic imaging (CT or MRI) every 1-2 years for detection of thymic and bronchopulmonary NETs (2). The optimal imaging modality still remains to be elucidated, although CT scans are used in the far majority of cases. A direct comparison between MRI and CT scans among MEN1 patients is lacking, and the role of nuclear imaging in screening programs for thoracic NETs has to be determined yet (209-212). Furthermore, the frequency of periodic surveillance is subject of debate; on one hand, the overall indolent course of bpNET and rareness of thNET might argue for less frequent screening – thereby diminishing radiation exposure, physical and psychological distress for patients, and health care costs –, but the lack of predictors for (sudden) aggressive tumor growth in bpNET and the aggressive nature of thNET plead for frequent thoracic imaging in order to enable timely intervention if necessary. Therefore, treating physicians should inform their patients about the benefits and disadvantages of a strict surveillance program, in order to come to a personalized surveillance strategy based on shared decision-making.
ADRENAL TUMORS
Adrenal involvement (Figure 1) is frequently seen in patients with MEN1 and considered to be part of the syndrome though not one of the cardinal manifestations. Mice with heterozygous inactivation of the Men1 gene develop adrenocortical lesions to a greater proportion than Men1 wild-type controls (213, 214) and the adrenal tumors show loss of heterogeneity (LOH) and loss of menin staining. In humans with MEN1, LOH is rarely seen in benign adrenocortical tumors (215-217). It has been hypothesized that the development of adrenal tumors in MEN1 might be related to PanNETs and hyperinsulinemia, because in some cohorts an association was seen between the occurrence of PanNETs, hyperinsulinemia, and adrenal lesions. However, in the largest series to date, no difference was found in the prevalence of main MEN1 manifestations between those with and without adrenal lesions (217).
In retrospective cohorts on adrenal involvement in MEN1 the reported prevalence greatly differs from 20-73% (216-225). Prevalence in part differs by the way adrenal lesions are defined (i.e. also including hyperplasia) and the manner of diagnosis, with prevalence being the highest (73%) in an EUS study (n=49) including all adrenal lesions from ‘plump’ adrenals to adenomas (221). In the series (n=27) with the second-highest prevalence (63%) all CT scans were re-read with the purpose of classifying adrenal lesions and every adrenocortical lesion >5 mm was considered a nodule (225). The largest series to date from the French GTE (n=715) has the lowest prevalence of 20.4% (217). Adrenal lesions are rarely the reason for an MEN1 diagnosis or the first manifestation of the disease, and are most frequently diagnosed asymptomatically by screening/surveillance imaging during follow-up or at the time of initial comprehensive imaging after the diagnosis of MEN1 is made (217, 223). Mean age of diagnosis is usually in the fifth decade, but ranges vary widely (217, 223, 224).
The French GTE series compared MEN1-related adrenal lesions to a cohort of sporadic incidentalomas (n=144) and found that adrenal lesions in patients with MEN1 were diagnosed at a younger age and were similar in size and in prevalence of bilateral lesions (217).
Benign Adrenocortical Tumors
Most MEN1-related adrenal lesions are benign adrenocortical lesions and include hyperplasia, (macro)nodular hyperplasia, and adenomas. Bilateral lesions are frequently seen, but again prevalence reported varies widely from 12.5% to >50% in different series( 216-219, 221-224). Most adrenocortical lesions in MEN1 are non-functioning and generally stable over the course of follow-up (216-224). In a minority of the cases ACTH-independent hypercortisolism, autonomous cortisol secretion, or primary hyperaldosteronism are seen (216-219, 221-224). Interestingly, in the French series, when comparing MEN1-related adrenal lesions with adrenal incidentalomas, functional tumors were more common (15% vs 6.9%), especially primary hyperaldosteronism and ACTH-independent hypercortisolism (217). Pheochromocytomas on the other hand were more common among sporadic incidentalomas.
Adrenocortical Carcinoma
Adrenocortical carcinoma (ACC) is a rare occurrence among patients with MEN1, with a 2019 review of literature identifying 19 published cases (226). In the Swedish cohort, one patient had an ACC and in the tumor LOH at the MEN1 locus was seen (216, 222). Three papers reporting from the same German institutions reported two, four and one case of ACC respectively, but some of these might represent the same patients (220, 224, 227). In the large French series, eight patients with 10 ACCs were reported, which was 5% of patients with an adrenal lesion, but 13.8% of those with an adrenal tumor (>10mm) (217). ACC prevalence was also significantly higher than in the sporadic adrenal incidentaloma cohort (217). There are also several case reports describing patients with MEN1-related ACC (226, 228-233). It is important to mention that there are several cases reported were the ACC developed from an initially observed relatively small adrenocortical tumor, although when reported these lesions did not have Houndsfield Units (HU) ≤ 10. In the French cohort one patient had two nodules (8 and 13 mm, 38 HU) in one adrenal, which grew to 29 and 30 mm after 5 years, which turned out to be ACCs. Another patient had a 25 mm calcified lesion (40 HU) which grew to 40 mm in 4 years(217). One case report describes a female patient with a 2 cm left adrenal tumor which grew to 3 cm in 7 years (advised to undergo surgery but refused) and then to 4 cm in 4 years, after which the tumor was removed and turned out to be an ACC (233). In other cases more rapidly progressing tumors are described (220, 231). When ACCs are functioning, they are mostly cortisol producing or sex-steroid producing. In sporadic ACC,MEN1 is considered one of the driver genes (234).
Pheochromocytoma
Pheochromocytoma is one of the hallmark conditions of Multiple Endocrine Neoplasia type 2 (MEN2), caused by germline mutations in the RET oncogene. In MEN1, the occurrence of pheochromocytoma is rare, with a 2020 case report and review of literature describing 20 published cases (235). The authors identified LOH at the MEN1 locus in the resected pheochromocytoma of the patient they report (235), and in another published series, two resected pheochromocytomas from patients with MEN1 were examined and LOH at the MEN1 locus was found in both, with one having absent menin staining and one weak menin staining (236).
Screening, Treatment and Follow-up
In patients with MEN1, minimal recommended screening for adrenal lesions as per the current guidelines, is abdominal imaging with CT or MRI every 3 years for those without adrenal lesions (2). Since abdominal imaging is also performed to screen for and/or surveille pancreatic lesions this can often be combined. However, care should be taken that the adrenals are properly imaged in the right phase to not only judge their size but also, should a lesion be present, to judge its characteristics. The preponderance to develop adrenal lesions should be mentioned in the clinical information to the radiologist and the images should be read by a radiologist experienced in adrenal imaging.
If an adrenal lesion is identified hormonal screening is recommended if patients are symptomatic or lesions are >1 cm (2). The current guidelines recommend that screening be focused on hyperaldosteronism and hypercortisolism (2), but given that pheochromocytomas do occur in MEN1 as described above and the consequence of missing the diagnosis can be serious, it is prudent to screen for the presence of a pheochromocytoma by either plasma free (nor)metanephrines or urinary fractionated (nor)metanephrines.
Indications for surgical resection parallel those of adrenal incidentalomas, being clinically significant hormone excess and/or concerns about malignancy either due to atypical characteristics on imaging, size (>4 cm), or significant growth over a 6-month period, which in the adrenal incidentaloma guidelines is suggested as increase in 20% (in addition to at least 5mm increase in actual size) (2, 237). Given the reports of ACCs in MEN1 arising from initially small lesions, some authors recommend to use a size cut-off of 3 cm in MEN1 (220).
If there is no indication for surgery, surveillance imaging is indicated, initially after 6 months. In the absence of a surgical indication at this point, frequency of further imaging follow-up should be determined individually and discussed by the multidisciplinary team. Unlike in sporadic adrenal incidentalomas, surveillance cannot be ended, given that multiple adrenal lesions can arise. There may be indications during follow-up to repeat initially negative hormonal screening, such as the development of symptoms or a new adrenal lesion.
CUTANEOUS LESIONS
Facial angiofibromas and collagenomas are the main skin lesions in MEN1 (Figure 1) (238, 239). Frequencies of 64% for angiofibromas and 62% for collagenomas have been described. Multiple angiofibromas and collagenomas are present in 77–81% of the MEN1 patients (238). Primarily angiofibromas are seen in patients with MEN1 (238, 240). An odds ratio of 6.6 (95% CI, 1.09–40.43) for cutaneous lesions in MEN1 in 29 patients with MEN1 in comparison with their non- affected family members is described (240).
These findings are further supported by the allelic loss of the MEN1 gene in six angiofibromas, three collagenomas, and one lipoma, suggesting that loss of function of the wild-type MEN1 gene product plays a role in the development of these skin lesions in patients with MEN1(241). Melanomas and other skin lesions are also described in the MEN1 population, but not with significant prevalences.
Lipomas (Figure 1) are reported in 17-34% of patients with MEN1 (238-240). Loss of heterozygosity of the MEN1 gene is described in MEN1-related lipomas (97, 241, 242) and may also play a role in sporadic lipomas (242). Menin seems to be an important factor for adipogenesis and contributes to lipoma development (243, 244).
A case of a novel MEN1 gene mutation with a recurrent sarcoma addresses the need for cautiousness of (atypical) skin lesions in patients with MEN1 (245).
BREAST CANCER AND MEN1
A higher incidence of breast cancer (Figure 1) was found in four independent MEN1 cohorts in the Netherlands, France, Tasmania and the United States. In the Dutch cohort a relative risk of 2.83 was found, which was significantly higher than in the general Dutch population (3). The median age for breast cancer was 45 years, which is approximately 15 years younger than the general Dutch population. The increased risk for breast cancer for MEN1 carriers was not associated with other breast cancer risk factors or a familial breast cancer risk. Considering the younger age of breast cancer occurrence and an earlier age of breast cancer, surveillance should be considered. Breast cancer surveillance from the age of 40 is initiated in the Dutch MEN1 cohort (4). After the latter publication, several cases of early breast cancer in MEN1 patients were reported (246-249).
These epidemiological findings are supported by basic research. Loss-of-function Men1 mouse models have shown an increased incidence of both in situ and invasive mammary cancer (250). Menin, the tumor suppressor protein encoded by MEN1, is co-localized with the estrogen receptor (ER) alfa in breast cancer cells. In this manner, menin functions as a direct activator of Erα (251). In sporadic ER-positive breast cancer, menin seems to have a proliferative role, which is in contrast with breast cancer in MEN1 carriers, in whom LOH of the MEN1 gene could be found (3, 252). Recent studies showed that reduced menin staining is associated with ER-negative breast cancer and in ER-positive breast cancer with larger tumors, higher grade tumor, and luminal subtypes tumors. Providing further evidence that there is an important role of menin in ERα regulation and the breast cancer formation (253).
PSYCHOSOCIAL ASPECTS
Recently there has been more interest in the psychosocial wellbeing of patients with MEN1 (254-259).The first study was published in 2003, which showed that psychosocial outcomes such as anxiety, depression, intrusion, and avoidance are not altered by the hospital or home setting. A higher burden of disease led to more depression. Compared to the population-based norm values, patients with MEN1 scored lower for General Health and Social Functioning according to the SF-36 (260).
Postoperatively, quality of life (QOL) scores did not differ after pancreaticoduodenal surgery in MEN1 patients in comparison with the general population. Financial difficulties caused by the treatment were significantly worse in MEN1 patients (261). Financial burden seems to be associated with having MEN1. The degree of financial burden has a linear relationship with worse health-related QOL. Patients were three-times more likely to be unemployed in comparison with the US population (256).
The largest QOL-related study showed that employment status was the most consistent predictor for QOL. This is in line with the former studies. The health-related QOL according to the SF-36 was significantly lower for patients with MEN1 on all subscales except for the physical functioning scale. Patients who are aware of their PA and PanNET have worse QOL scores in comparison with patients who are not aware of having these tumors(259). The degree of fear of disease recurrence is high in patients with MEN1. This fear is negatively associated with health-related QOL and is higher in patients who consider themselves at high risk for developing a MEN1-related tumor. More MEN1-related manifestations lead to more fear of disease occurrence (258). In comparison with other chronic diseases MEN1 scores worse regarding anxiety, depression and fatigue (257).
QOL did not overly differ from the general population in the Italian cohort (255) and patients were more optimistic than in the Swedish cohort (260). This could be due to cultural differences, population selection, and awareness of the disease and its implications.
The high response rate in the MEN1 population illustrates the motivation of patients to participate in research and care about their wellbeing (259, 260).
CONCLUSION
In conclusion, in the past decades there have been great advances in the understanding of the natural course of MEN1-related tumors, which has had direct consequences on clinical care. In the coming decade one of the main research objectives will be the identification of individual predictors of disease course which can guide personalized treatment and surveillance. Increasing international collaborations will enable prospective studies. Given the complexity of the disease, it is strongly advised that patients, whenever possible, be followed and treated in centers of expertise. If this is not feasible, consultation with a center of expertise should be considered.
REFERENCES
- Lloyd, R.V., et al., WHO Classification of Tumours of Endocrine Organs. 4th ed. 2017, Lyon: IARC.
- Thakker, R.V., et al., Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). The Journal of clinical endocrinology & metabolism, 2012. 97(9): p. 2990-3011.
- Dreijerink, K.M.a., et al., Breast-Cancer Predisposition in Multiple Endocrine Neoplasia Type 1. New England Journal of Medicine, 2014. 371(6): p. 583-584.
- van Leeuwaarde, R.S., et al., MEN1-Dependent Breast Cancer: Indication for Early Screening? Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab, 2017. 102(6): p. 2083-2090.
- Brandi, M.L., et al., Guidelines for diagnosis and therapy of MEN type 1 and type 2. The Journal of clinical endocrinology & metabolism, 2001. 86(12): p. 5658-71.
- Chandrasekharappa, S.C., et al., Positional Cloning of the Gene for Multiple Endocrine Neoplasia-Type 1 Science, 1997. 276: p. 404-407.
- Lemmens, I., et al., Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Human molecular genetics, 1997. 6: p. 1177-83.
- Lemos, M.C. and R.V. Thakker, Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Human mutation, 2008. 29(1): p. 22-32.
- Concolino, P., A. Costella, and E. Capoluongo, Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet, 2016. 209(1-2): p. 36-41.
- Kooblall, K.G., et al., Multiple Endocrine Neoplasia Type 1 (MEN1) 5'UTR Deletion, in MEN1 Family, Decreases Menin Expression. J Bone Miner Res, 2021. 36(1): p. 100-109.
- Agarwal, S.K., The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer, 2017. 24(10): p. T119-t134.
- Romanet, P., et al., Proposition of adjustments to the ACMG-AMP framework for the interpretation of MEN1 missense variants. Hum Mutat, 2019. 40(6): p. 661-674.
- Thevenon, J., et al., Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: a Groupe d'etude des Tumeurs Endocrines (GTE) cohort study. Human molecular genetics, 2013. 22: p. 1940-1948.
- Bartsch, D.K., et al., Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab, 2014. 99(11): p. E2387-91.
- Klein Haneveld, M.J., et al., Initiating pancreatic neuroendocrine tumour (pNET) screening in young MEN1 patients: results from the DutchMEN Study Group. J Clin Endocrinol Metab, 2021.
- van den Broek, M.F.M., et al., Clues For Genetic Anticipation In Multiple Endocrine Neoplasia Type 1. J Clin Endocrinol Metab, 2020. 105(7).
- Beijers, H., et al., Germline and somatic mosaicism in a family with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur J Endocrinol, 2019. 180(2): p. K15-k19.
- Dreijerink, K.M.A., H.T.M. Timmers, and M. Brown, Twenty years of menin: emerging opportunities for restoration of transcriptional regulation in MEN1. Endocr Relat Cancer, 2017. 24(10): p. T135-t145.
- Canaff, L., et al., Impaired transforming growth factor-β (TGF-β) transcriptional activity and cell proliferation control of a menin in-frame deletion mutant associated with multiple endocrine neoplasia type 1 (MEN1). The Journal of biological chemistry, 2012. 287: p. 8584-97.
- Iyer, S. and S.K. Agarwal, Epigenetic regulation in the tumorigenesis of MEN1-associated endocrine cell types. J Mol Endocrinol, 2018. 61(1): p. R13-r24.
- Walls, G.V., et al., MEN1 gene replacement therapy reduces proliferation rates in a mouse model of pituitary adenomas. Cancer research, 2012. 72: p. 5060-8.
- Pieterman, C.R., et al., Care for patients with multiple endocrine neoplasia type 1: the current evidence base. Fam Cancer, 2011. 10(1): p. 157-71.
- van Leeuwaarde, R.S., et al., The future: medical advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer, 2017. 24(10): p. T179-t193.
- de Laat, J.M., et al., MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med, 2016. 14(1): p. 182.
- Pieterman, C.R.C., et al., Understanding the clinical course of genotype-negative MEN1 patients can inform management strategies. Surgery, 2020.
- Goudet, P., et al., Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World journal of surgery, 2010. 34(2): p. 249-55.
- Geerdink, E.A., R.B. Van der Luijt, and C.J. Lips, Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? European journal of endocrinology, 2003. 149(6): p. 577-82.
- de Laat, J.M., R.S. van Leeuwaarde, and G.D. Valk, The Importance of an Early and Accurate MEN1 Diagnosis. Front Endocrinol (Lausanne), 2018. 9: p. 533.
- de Laat, J.M., et al., Predicting the risk of multiple endocrine neoplasia type 1 for patients with commonly occurring endocrine tumors. European journal of endocrinology, 2012. 167(2): p. 181-7.
- van Leeuwaarde, R.S., et al., Impact of delay in diagnosis in outcomes in MEN1: results from the Dutch MEN1 study group. J Clin Endocrinol Metab, 2016: p. jc20153766.
- Alrezk, R., F. Hannah-Shmouni, and C.A. Stratakis, MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer, 2017. 24(10): p. T195-t208.
- Frederiksen, A., et al., Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J Clin Endocrinol Metab, 2019. 104(9): p. 3637-3646.
- Georgitsi, M., et al., Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. The Journal of clinical endocrinology and metabolism, 2007. 92: p. 3321-3325.
- van der Tuin, K., et al., CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J Clin Endocrinol Metab, 2017. 102(12): p. 4534-4540.
- van den Broek, M.F.M., et al., Clinical Relevance of Genetic Analysis in Patients With Pituitary Adenomas: A Systematic Review. Front Endocrinol (Lausanne), 2019. 10: p. 837.
- Stratakis, C.A., et al., Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab, 2000. 85(12): p. 4776-80.
- Manoharan, J., et al., Is Routine Screening of Young Asymptomatic MEN1 Patients Necessary? World J Surg, 2017. 41(8): p. 2026-2032.
- Romanet, P., et al., UMD-MEN1 Database: An Overview of the 370 MEN1 Variants Present in 1676 Patients From the French Population. J Clin Endocrinol Metab, 2019. 104(3): p. 753-764.
- Goudet, P., et al., Hyperparathyroidism in multiple endocrine neoplasia type I: surgical trends and results of a 256-patient series from Groupe D'etude des Néoplasies Endocriniennes Multiples Study Group. World J Surg, 2001. 25(7): p. 886-90.
- Pieterman, C.R., et al., Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clinical endocrinology, 2009. 70(4): p. 575-81.
- Marini, F., F. Giusti, and M.L. Brandi, Multiple endocrine neoplasia type 1: extensive analysis of a large database of Florentine patients. Orphanet J Rare Dis, 2018. 13(1): p. 205.
- Giusti, F., et al., Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine, 2017. 58(2): p. 349-359.
- Sakurai, A., et al., Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clinical endocrinology, 2012. 76(4): p. 533-9.
- Soczomski, P., et al., Multiple endocrine neoplasia type 1 in Poland: a two-centre experience. Endokrynol Pol, 2019. 70(5): p. 385-391.
- Waldmann, J., et al., Screening of patients with multiple endocrine neoplasia type 1 (MEN-1): a critical analysis of its value. World journal of surgery, 2009. 33(6): p. 1208-18.
- Herath, M., et al., Paediatric and young adult manifestations and outcomes of multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf), 2019. 91(5): p. 633-638.
- Goudet, P., et al., MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'etude des Tumeurs Endocrines. J Clin Endocrinol Metab, 2015. 100(4): p. 1568-77.
- Shariq, O.A., et al., Multiple endocrine neoplasia type 1 in children and adolescents: Clinical features and treatment outcomes. Surgery, 2021.
- Vannucci, L., et al., MEN1 in children and adolescents: Data from patients of a regional referral center for hereditary endocrine tumors. Endocrine, 2018. 59(2): p. 438-448.
- Marx, S.J. and D.M. Lourenço, Jr., Questions and Controversies About Parathyroid Pathophysiology in Children With Multiple Endocrine Neoplasia Type 1. Front Endocrinol (Lausanne), 2018. 9: p. 359.
- Doherty, G.M., T.C. Lairmore, and M.K. DeBenedetti, Multiple endocrine neoplasia type 1 parathyroid adenoma development over time. World J Surg, 2004. 28(11): p. 1139-42.
- d'Alessandro, A.F., et al., Supernumerary parathyroid glands in hyperparathyroidism associated with multiple endocrine neoplasia type 1. Rev Assoc Med Bras (1992), 2012. 58(3): p. 323-7.
- Yeh, M.W., et al., Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab, 2013. 98(3): p. 1122-9.
- Press, D.M., et al., The prevalence of undiagnosed and unrecognized primary hyperparathyroidism: a population-based analysis from the electronic medical record. Surgery, 2013. 154(6): p. 1232-7; discussion 1237-8.
- Eller-Vainicher, C., et al., Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity. J Bone Miner Res, 2009. 24(8): p. 1404-10.
- Twigt, B.A., et al., Differences between sporadic and MEN related primary hyperparathyroidism; clinical expression, preoperative workup, operative strategy and follow-up. Orphanet J Rare Dis, 2013. 8: p. 50.
- Wilhelm, S.M., et al., The American Association of Endocrine Surgeons Guidelines for Definitive Management of Primary Hyperparathyroidism. JAMA Surg, 2016. 151(10): p. 959-968.
- Iacobone, M., et al., Hereditary hyperparathyroidism--a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg, 2015. 400(8): p. 867-86.
- Silva, A.M., et al., Operative intervention for primary hyperparathyroidism offers greater bone recovery in patients with sporadic disease than in those with multiple endocrine neoplasia type 1-related hyperparathyroidism. Surgery, 2017. 161(1): p. 107-115.
- Wang, W., et al., Impaired geometry, volumetric density, and microstructure of cortical and trabecular bone assessed by HR-pQCT in both sporadic and MEN1-related primary hyperparathyroidism. Osteoporos Int, 2020. 31(1): p. 165-173.
- Kong, J., et al., Clinical and Genetic Analysis of Multiple Endocrine Neoplasia Type 1-Related Primary Hyperparathyroidism in Chinese. PLoS One, 2016. 11(11): p. e0166634.
- Lourenço, D.M., Jr., et al., Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type 1-associated primary hyperparathyroidism. J Bone Miner Res, 2010. 25(11): p. 2382-91.
- Burgess, J.R., et al., Osteoporosis in multiple endocrine neoplasia type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Arch Surg, 1999. 134(10): p. 1119-23.
- Lourenco, D.M., Jr., et al., Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. European journal of endocrinology, 2008. 159(3): p. 259-74.
- Coutinho, F.L., et al., Bone mineral density analysis in patients with primary hyperparathyroidism associated with multiple endocrine neoplasia type 1 after total parathyroidectomy. Clin Endocrinol (Oxf), 2010. 72(4): p. 462-8.
- Kann, P.H., et al., Peripheral bone mineral density in correlation to disease-related predisposing conditions in patients with multiple endocrine neoplasia type 1. J Endocrinol Invest, 2012. 35(6): p. 573-9.
- Green, P., et al., High prevalence of chronic kidney disease in patients with multiple endocrine neoplasia type 1 and improved kidney function after parathyroidectomy. Surgery, 2019. 165(1): p. 124-128.
- Bilezikian, J.P., et al., Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab, 2014. 99(10): p. 3561-9.
- Norton, J.A., et al., Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg, 2008. 247(3): p. 501-10.
- Hackeng, W.M., et al., A Parathyroid-Gut Axis: Hypercalcemia and the Pathogenesis of Gastrinoma in Multiple Endocrine Neoplasia 1. Mol Cancer Res, 2021. 19(6): p. 946-949.
- Nobecourt, P.F., et al., Intraoperative Decision-Making and Technical Aspects of Parathyroidectomy in Young Patients With MEN1 Related Hyperparathyroidism. Front Endocrinol (Lausanne), 2018. 9: p. 618.
- Nastos, C., et al., Optimal extent of initial parathyroid resection in patients with multiple endocrine neoplasia syndrome type 1: A meta-analysis. Surgery, 2021. 169(2): p. 302-310.
- Schreinemakers, J.M., et al., The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg, 2011. 35(9): p. 1993-2005.
- Manoharan, J., et al., Single gland excision for MEN1-associated primary hyperparathyroidism. Clin Endocrinol (Oxf), 2020. 92(1): p. 63-70.
- Nilubol, N., et al., Preoperative localizing studies for initial parathyroidectomy in MEN1 syndrome: is there any benefit? World J Surg, 2012. 36(6): p. 1368-74.
- Nilubol, N., et al., Utility of intraoperative parathyroid hormone monitoring in patients with multiple endocrine neoplasia type 1-associated primary hyperparathyroidism undergoing initial parathyroidectomy. World J Surg, 2013. 37(8): p. 1966-72.
- Kluijfhout, W.P., et al., Unilateral Clearance for Primary Hyperparathyroidism in Selected Patients with Multiple Endocrine Neoplasia Type 1. World J Surg, 2016. 40(12): p. 2964-2969.
- Montenegro, F.L.M., et al., Could the Less-Than Subtotal Parathyroidectomy Be an Option for Treating Young Patients With Multiple Endocrine Neoplasia Type 1-Related Hyperparathyroidism? Front Endocrinol (Lausanne), 2019. 10: p. 123.
- Choi, H.R., et al., Benefit of diverse surgical approach on short-term outcomes of MEN1-related hyperparathyroidism. Sci Rep, 2020. 10(1): p. 10634.
- Nilubol, N., et al., Limited Parathyroidectomy in Multiple Endocrine Neoplasia Type 1-Associated Primary Hyperparathyroidism: A Setup for Failure. Ann Surg Oncol, 2016. 23(2): p. 416-23.
- Keutgen, X.M., et al., Reoperative Surgery in Patients with Multiple Endocrine Neoplasia Type 1 Associated Primary Hyperparathyroidism. Ann Surg Oncol, 2016. 23(Suppl 5): p. 701-707.
- Bioletto, F., et al., Comparison of the diagnostic accuracy of 18F-Fluorocholine PET and 11C-Methionine PET for parathyroid localization in primary hyperparathyroidism: a systematic review and meta-analysis. Eur J Endocrinol, 2021. 185(1): p. 109-120.
- Hindié, E., et al., Primary Hyperparathyroidism: Defining the Appropriate Preoperative Imaging Algorithm. J Nucl Med, 2021. 62(Suppl 2): p. 3s-12s.
- Gauthé, M., et al., (18)F-fluorocholine PET/CT in MEN1 Patients with Primary Hyperparathyroidism. World J Surg, 2020. 44(11): p. 3761-3769.
- Fyrsten, E., et al., Long-Term Surveillance of Treated Hyperparathyroidism for Multiple Endocrine Neoplasia Type 1: Recurrence or Hypoparathyroidism? World J Surg, 2016. 40(3): p. 615-21.
- Pieterman, C.R., et al., Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg, 2012. 255(6): p. 1171-8.
- Filopanti, M., et al., MEN1-related hyperparathyroidism: response to cinacalcet and its relationship with the calcium-sensing receptor gene variant Arg990Gly. Eur J Endocrinol, 2012. 167(2): p. 157-64.
- Giusti, F., et al., Cinacalcet therapy in patients affected by primary hyperparathyroidism associated to Multiple Endocrine Neoplasia Syndrome type 1 (MEN1). Endocrine, 2016. 52(3): p. 495-506.
- Moyes, V.J., et al., Clinical Use of Cinacalcet in MEN1 Hyperparathyroidism. Int J Endocrinol, 2010. 2010: p. 906163.
- Geslot, A., et al., New therapies for patients with multiple endocrine neoplasia type 1. Ann Endocrinol (Paris), 2021. 82(2): p. 112-120.
- Singh Ospina, N., et al., Safety and efficacy of percutaneous parathyroid ethanol ablation in patients with recurrent primary hyperparathyroidism and multiple endocrine neoplasia type 1. J Clin Endocrinol Metab, 2015. 100(1): p. E87-90.
- Williams, M.D., et al., Pathology data set for reporting parathyroid carcinoma and atypical parathyroid neoplasm: recommendations from the International Collaboration on Cancer Reporting. Hum Pathol, 2021. 110: p. 73-82.
- Song, A., et al., Prevalence of Parathyroid Carcinoma and Atypical Parathyroid Neoplasms in 153 Patients With Multiple Endocrine Neoplasia Type 1: Case Series and Literature Review. Front Endocrinol (Lausanne), 2020. 11: p. 557050.
- Christakis, I., et al., Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. Int J Surg, 2016. 31: p. 10-6.
- Singh Ospina, N., et al., Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol (Oxf), 2016. 84(2): p. 244-249.
- Erdheim, J., Zur normalen und pathologischen Histologie der Glandula thyreoidea, parathyreoidea, und Hypophysis. Beitr Pathol Anat, 1903. 33: p. 158-236.
- Dong, Q., et al., Loss of heterozygosity at 11q13: analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. The Journal of clinical endocrinology & metabolism, 1997. 82(5): p. 1416-20.
- Thakker, R.V., The molecular genetics of the multiple endocrine neoplasia syndromes. Clinical Endocrinology, 1993. 38(1): p. 1-14.
- Weil, R.J., et al., 11Q13 Allelic Loss in Pituitary Tumors in Patients With Multiple Endocrine Neoplasia Syndrome Type 1. Clinical Cancer Research, 1998. 4(7): p. 1673-1678.
- Yoshimoto, K., et al., Allele Loss on Chromosome 11 in a Pituitary Tumor from a Patient with Multiple Endocrine Neoplasia Type 1. 1991. p. 886-889.
- Thakker, R.V., et al., Association of somatotrophinomas with loss of alleles on chromosome 11 and with gsp mutations. J Clin Invest, 1993. 91(6): p. 2815-21.
- Boggild, M.D., et al., Molecular genetic studies of sporadic pituitary tumors. J Clin Endocrinol Metab, 1994. 78(2): p. 387-92.
- Asa, S.L., K. Somers, and S. Ezzat, The MEN-1 gene is rarely down-regulated in pituitary adenomas. J Clin Endocrinol Metab, 1998. 83(9): p. 3210-2.
- Farrell, W.E., et al., Sequence analysis and transcript expression of the MEN1 gene in sporadic pituitary tumours. Br J Cancer, 1999. 80(1-2): p. 44-50.
- Prezant, T.R., J. Levine, and S. Melmed, Molecular characterization of the men1 tumor suppressor gene in sporadic pituitary tumors. J Clin Endocrinol Metab, 1998. 83(4): p. 1388-91.
- Tanaka, C., et al., Analysis of loss of heterozygosity on chromosome 11 and infrequent inactivation of the MEN1 gene in sporadic pituitary adenomas. J Clin Endocrinol Metab, 1998. 83(8): p. 2631-4.
- Zhuang, Z., et al., Mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res, 1997. 57(24): p. 5446-51.
- Ballard, H.S., B. Fame, and R.J. Hartsock, Familial multiple endocrine adenoma-peptic ulcer complex. Medicine (Baltimore), 1964. 43(0025-7974 (Print)): p. 481-516.
- Verges, B., et al., Pituitary disease in MEN type 1 (MEN1): Data from the France-Belgium MEN1 multicenter study. Journal of Clinical Endocrinology & Metabolism, 2002. 87(2): p. 457-465.
- Trouillas, J., et al., Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol, 2008. 32(4): p. 534-43.
- de Laat, J.M., et al., Long-term natural course of pituitary tumors in patients with MEN1: results from the Dutch MEN1 study group (DMSG). J Clin.Endocrinol.Metab, 2015(1945-7197 (Electronic)): p. JC20152015-JC20152015.
- Wu, Y., et al., Pituitary adenomas in patients with multiple endocrine neoplasia type 1: a single-center experience in China. Pituitary, 2019. 22(2): p. 113-123.
- Cohen-Cohen, S., et al., Pituitary adenomas in the setting of multiple endocrine neoplasia type 1: A single-institution experience. Journal of Neurosurgery, 2021. 134(4): p. 1132-1138.
- Bras, M.L., et al., Pituitary adenoma in patients with multiple endocrine neoplasia type 1 – a cohort study. 2021: p. 1-34.
- Makri, A., et al., Children with MEN1 gene mutations may present first (and at a young age) with Cushing disease. Clin Endocrinol (Oxf), 2018. 89(4): p. 437-443.
- Melmed, S., et al., Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab, 2011. 96(2): p. 273-88.
- Nieman, L.K., et al., The diagnosis of Cushing's syndrome: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism, 2008. 93(5): p. 1526-1540.
- Katznelson, L., et al., Acromegaly: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism, 2014. 99(11): p. 3933-3951.
- Incandela, F., et al., Malignancy course of pituitary adenoma in MEN1 syndrome: Clinical-Neuroradiological signs. Eur J Radiol Open, 2020. 7: p. 100242.
- Philippon, M., et al., Long-term control of a MEN1 prolactin secreting pituitary carcinoma after temozolomide treatment. Ann Endocrinol (Paris), 2012. 73(3): p. 225-9.
- Benito, M., et al., Gonadotroph tumor associated with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab, 2005. 90(1): p. 570-4.
- Scheithauer, B.W., et al., Multiple endocrine neoplasia type 1-associated thyrotropin-producing pituitary carcinoma: report of a probable de novo example. Hum Pathol, 2009. 40(2): p. 270-8.
- Morokuma, H., et al., A case of nonfunctioning pituitary carcinoma that responded to temozolomide treatment. Case Rep Endocrinol, 2012. 2012: p. 645914.
- Triponez, F., et al., Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Annals of surgery, 2006. 243(2): p. 265-72.
- Vinault, S., et al., Metastatic Potential and Survival of Duodenal and Pancreatic Tumors in Multiple Endocrine Neoplasia Type 1: A GTE and AFCE Cohort Study (Groupe d'etude des Tumeurs Endocrines and Association Francophone de Chirurgie Endocrinienne). Ann Surg, 2018.
- Pipeleers-Marichal, M., et al., Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. The New England journal of medicine, 1990. 322(11): p. 723-7.
- Levy-Bohbot, N., et al., Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterologie clinique et biologique, 2004. 28(11): p. 1075-81.
- Borson-Chazot, F., et al., Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Ann Endocrinol (Paris), 2012. 73(6): p. 497-502.
- Milanesi, A., et al., Concurrent primary hyperparathyroidism and humoral hypercalcemia of malignancy in a patient with multiple endocrine neoplasia type 1. Pancreas, 2011. 40(4): p. 634-7.
- Klöppel, G., et al., Hyperplasia to neoplasia sequence of duodenal and pancreatic neuroendocrine diseases and pseudohyperplasia of the PP-cells in the pancreas. Endocr Pathol, 2014. 25(2): p. 181-5.
- Anlauf, M., et al., Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut, 2007. 56(5): p. 637-44.
- van Beek, D.J., et al., Surgery for multiple endocrine neoplasia type 1-related insulinoma: long-term outcomes in a large international cohort. Br J Surg, 2020. 107(11): p. 1489-1499.
- Gibril, F., et al., Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine, 2004. 83(1): p. 43-83.
- Ito, T., et al., Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine, 2013. 92(3): p. 135-81.
- van Beek, D.J., et al., Prognostic factors and survival in MEN1 patients with gastrinomas: Results from the DutchMEN study group (DMSG). J Surg Oncol, 2019. 120(6): p. 966-975.
- Jensen, R.T. and T. Ito, Gastrinoma, in Endotext, K.R. Feingold, et al., Editors. 2000, MDText.com, Inc. Copyright © 2000-2021, MDText.com, Inc.: South Dartmouth (MA).
- Hampel, H., et al., A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med, 2015. 17(1): p. 70-87.
- Gibril, F., et al., Prospective study of the natural history of gastrinoma in patients with MEN1: definition of an aggressive and a nonaggressive form. The Journal of clinical endocrinology & metabolism, 2001. 86(11): p. 5282-93.
- Conemans, E.B., et al., Prognostic factors for survival of MEN1 patients with duodenopancreatic tumors metastatic to the liver: results from the DMSG. Endocr Pract, 2017. 23(6): p. 641-648.
- Giudici, F., et al., Natural History of MEN1 GEP-NET: Single-Center Experience After a Long Follow-Up. World J Surg, 2017. 41(9): p. 2312-2323.
- Christakis, I., et al., Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: A 23-year experience at a single institution. Surgery, 2018. 163(1): p. 212-217.
- Nagtegaal, I.D., et al., The 2019 WHO classification of tumours of the digestive system. Histopathology, 2020. 76(2): p. 182-188.
- van Treijen, M.J.C., et al., Diagnosing Nonfunctional Pancreatic NETs in MEN1: The Evidence Base. J Endocr Soc, 2018. 2(9): p. 1067-1088.
- Pieterman, C.R.C., et al., HEREDITARY ENDOCRINE TUMOURS: CURRENT STATE-OF-THE-ART AND RESEARCH OPPORTUNITIES: MEN1-related pancreatic NETs: identification of unmet clinical needs and future directives. Endocr Relat Cancer, 2020. 27(8): p. T9-t25.
- Cuthbertson, D.J., et al., The Impact of (68)Gallium DOTA PET/CT in Managing Patients With Sporadic and Familial Pancreatic Neuroendocrine Tumours. Front Endocrinol (Lausanne), 2021. 12: p. 654975.
- Sadowski, S.M., et al., Prognostic factors for the outcome of nonfunctioning pancreatic neuroendocrine tumors in MEN1: a systematic review of literature. Endocr Relat Cancer, 2020. 27(6): p. R145-r161.
- Partelli, S., et al., Active Surveillance versus Surgery of Nonfunctioning Pancreatic Neuroendocrine Neoplasms </=2 cm in MEN1 Patients. Neuroendocrinology, 2016. 103(6): p. 779-786.
- Nell, S., et al., Management of MEN1 Related Nonfunctioning Pancreatic NETs: A Shifting Paradigm: Results From the DutchMEN1 Study Group. Ann Surg, 2018. 267(6): p. 1155-1160.
- Triponez, F., et al., Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World journal of surgery, 2006. 30(5): p. 654-62; discussion 663-4.
- Pieterman, C.R.C., et al., Long-Term Natural Course of Small Nonfunctional Pancreatic Neuroendocrine Tumors in MEN1-Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab, 2017. 102(10): p. 3795-3805.
- Hackeng, W.M., et al., Non-functional pancreatic neuroendocrine tumours: ATRX/DAXX and alternative lengthening of telomeres (ALT) are prognostically independent from ARX/PDX1 expression and tumour size. Gut, 2021.
- Cejas, P., et al., Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med, 2019. 25(8): p. 1260-1265.
- Chan, C.S., et al., ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun, 2018. 9(1): p. 4158.
- Fahrmann, J.F., et al., A Blood-Based Polyamine Signature Associated With Disease Progression in Patients With Multiple Endocrine Neoplasia Type 1-Related Duodenopancreatic Neuroendocrine Tumors. Journal of the Endocrine Society, 2021. 5(Supplement_1): p. A1009-A1009.
- Niederle, B., et al., Multiple Endocrine Neoplasia Type 1 (MEN1) and the Pancreas - Diagnosis and Treatment of Functioning and Non-Functioning Pancreatic and Duodenal Neuroendocrine Neoplasia within the MEN1 Syndrome - An International Consensus Statement. Neuroendocrinology, 2020.
- Falconi, M., et al., ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology, 2016. 103(2): p. 153-71.
- Metz, D.C., et al., Diagnosis of Zollinger-Ellison syndrome in the era of PPIs, faulty gastrin assays, sensitive imaging and limited access to acid secretory testing. Int J Endocr Oncol, 2017. 4(4): p. 167-185.
- Singh Ospina, N., et al., Assessing for Multiple Endocrine Neoplasia Type 1 in Patients Evaluated for Zollinger-Ellison Syndrome-Clues to a Safer Diagnostic Process. Am J Med, 2017. 130(5): p. 603-605.
- Albers, M.B., J. Manoharan, and D.K. Bartsch, Contemporary surgical management of the Zollinger-Ellison syndrome in multiple endocrine neoplasia type 1. Best Pract Res Clin Endocrinol Metab, 2019. 33(5): p. 101318.
- Hackeng, W.M., et al., Metastatic Patterns of Duodenopancreatic Neuroendocrine Tumors in Patients With Multiple Endocrine Neoplasia Type 1. Am J Surg Pathol, 2021.
- Norton, J.A., et al., Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. Annals of surgery, 2001. 234(4): p. 495-505; discussion 505-6.
- Endall, R., et al., The Relationship of Gastrinoma in MEN 1 to Helicobacter pylori infection. J Clin Endocrinol Metab, 2020. 105(3).
- Cryer, P.E., et al., Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2009. 94(3): p. 709-28.
- Bartsch, D.K., et al., Enucleation and limited pancreatic resection provide long-term cure for insulinoma in multiple endocrine neoplasia type 1. Neuroendocrinology, 2013. 98(4): p. 290-8.
- Vezzosi, D., et al., Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d'étude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol, 2015. 172(3): p. 309-19.
- Tonelli, F., et al., Operation for insulinomas in multiple endocrine neoplasia type 1: When pancreatoduodenectomy is appropriate. Surgery, 2017. 161(3): p. 727-734.
- Antwi, K., et al., 68Ga-Exendin-4 PET/CT Detects Insulinomas in Patients With Endogenous Hyperinsulinemic Hypoglycemia in MEN-1. J Clin Endocrinol Metab, 2019. 104(12): p. 5843-5852.
- Shah, R., et al., Exendin-4-based imaging in insulinoma localization: Systematic review and meta-analysis. Clin Endocrinol (Oxf), 2021. 95(2): p. 354-364.
- van Beek, D.J., et al., Prognosis after surgery for multiple endocrine neoplasia type 1-related pancreatic neuroendocrine tumors: Functionality matters. Surgery, 2021. 169(4): p. 963-973.
- Dasari, B.V.M., et al., Outcomes of Surgical and Endoscopic Resection of Duodenal Neuroendocrine Tumours (NETs): a Systematic Review of the Literature. J Gastrointest Surg, 2018. 22(9): p. 1652-1658.
- Delle Fave, G., et al., ENETS Consensus Guidelines Update for Gastroduodenal Neuroendocrine Neoplasms. Neuroendocrinology, 2016. 103(2): p. 119-24.
- Rimbaş, M., et al., Interventional endoscopic ultrasound for pancreatic neuroendocrine neoplasms. Dig Endosc, 2020. 32(7): p. 1031-1041.
- Caplin, M.E., et al., Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med, 2014. 371(3): p. 224-33.
- Caplin, M.E., et al., Anti-tumour effects of lanreotide for pancreatic and intestinal neuroendocrine tumours: the CLARINET open-label extension study. Endocr Relat Cancer, 2016. 23(3): p. 191-9.
- Ramundo, V., et al., Impact of long-acting octreotide in patients with early-stage MEN1-related duodeno-pancreatic neuroendocrine tumours. Clinical endocrinology, 2014. 80(6): p. 850-5.
- Cioppi, F., et al., The LARO-MEN1 study: a longitudinal clinical experience with octreotide Long-Acting Release in patients with Multiple Endocrine Neoplasia type 1 Syndrome. Clin Cases Miner Bone Metab, 2017. 14(2): p. 123-130.
- Faggiano, A., et al., Lanreotide Therapy vs Active Surveillance in MEN1-Related Pancreatic Neuroendocrine Tumors < 2 Centimeters. J Clin Endocrinol Metab, 2020. 105(1).
- Pavel, M., et al., ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology, 2016. 103(2): p. 172-85.
- Yao, J.C., et al., Everolimus for advanced pancreatic neuroendocrine tumors. The New England journal of medicine, 2011. 364(6): p. 514-23.
- Raymond, E., et al., Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. The New England journal of medicine, 2011. 364(6): p. 501-13.
- Strosberg, J., et al., Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med, 2017. 376(2): p. 125-135.
- Frost, M., K.E. Lines, and R.V. Thakker, Current and emerging therapies for PNETs in patients with or without MEN1. Nat Rev Endocrinol, 2018. 14(4): p. 216-227.
- Debelenko, L.V., et al., The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology, 1997. 113(3): p. 773-81.
- Lee, L., et al., Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger-Ellison Syndrome. Int J Mol Sci, 2019. 20(20).
- Manoharan, J., et al., Gastric enterochromaffin-like cell changes in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf), 2021. 95(3): p. 439-446.
- Richards, M.L., et al., Regression of type II gastric carcinoids in multiple endocrine neoplasia type 1 patients with Zollinger-Ellison syndrome after surgical excision of all gastrinomas. World J Surg, 2004. 28(7): p. 652-8.
- Kloppel, G., M. Anlauf, and A. Perren, Endocrine precursor lesions of gastroenteropancreatic neuroendocrine tumors. Endocrine pathology, 2007. 18: p. 150-155.
- Berna, M.J., et al., A prospective study of gastric carcinoids and enterochromaffin-like cell changes in multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: identification of risk factors. J Clin Endocrinol Metab, 2008. 93(5): p. 1582-91.
- Bordi, C., et al., Aggressive forms of gastric neuroendocrine tumors in multiple endocrine neoplasia type I. Am J Surg Pathol, 1997. 21(9): p. 1075-82.
- Norton, J.A., et al., Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger-Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgical treatment. Surgery, 2004. 136(6): p. 1267-74.
- Mandl, A., et al., Two distinct classes of thymic tumors in patients with MEN1 show LOH at the MEN1 locus. Endocr Relat Cancer, 2021. 28(11): p. L15-l19.
- Volante, M., et al., Molecular Pathology of Well-Differentiated Pulmonary and Thymic Neuroendocrine Tumors: What Do Pathologists Need to Know? Endocr Pathol, 2021. 32(1): p. 154-168.
- Teh, B.T., et al., Clinicopathologic Studies of Thymic Carcinoids in Multiple Endocrine Neoplasia Type 1. Medicine (Baltimore), 1997. 76(1): p. 21-29.
- Ye, L., et al., Clinical features and prognosis of thymic neuroendocrine tumours associated with multiple endocrine neoplasia type 1: A single-centre study, systematic review and meta-analysis. Clinical Endocrinology, 2017. 87(6): p. 706-716.
- Singh Ospina, N., et al., Thymic and Bronchial Carcinoid Tumors in Multiple Endocrine Neoplasia Type 1: The Mayo Clinic Experience from 1977 to 2013. Hormones and Cancer, 2015. 6(5-6): p. 247-253.
- Sakurai, A., et al., Thymic neuroendocrine tumour in multiple endocrine neoplasia type 1: Female patients are not rare exceptions. Clinical Endocrinology, 2013. 78(2): p. 248-254.
- Goudet, P., et al., Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: A comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines). World Journal of Surgery, 2009. 33(6): p. 1197-1207.
- Gibril, F., et al., Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. Journal of Clinical Endocrinology and Metabolism, 2003. 88(3): p. 1066-1081.
- Ferolla, P., et al., Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: The Italian series. Journal of Clinical Endocrinology and Metabolism, 2005. 90(5): p. 2603-2609.
- de Laat, J.M., et al., Natural course and survival of neuroendocrine tumors of thymus and lung in MEN1 patients. Journal of Clinical Endocrinology and Metabolism, 2014. 99(9): p. 3325-3333.
- Christakis, I., et al., Clinical Features, Treatments, and Outcomes of Patients with Thymic Carcinoids and Multiple Endocrine Neoplasia Type 1 Syndrome at MD Anderson Cancer Center. Hormones and Cancer, 2016. 7(4): p. 279-287.
- Lim, L.C., et al., Thymic carcinoid in multiple endocrine neoplasia 1: Genotype-phenotype correlation and prevention. Journal of Internal Medicine, 2006. 259(4): p. 428-432.
- Sadowski, S.M., et al., The future: surgical advances in MEN1 therapeutic approaches and management strategies. Endocrine-related cancer, 2017. 24(10): p. T243-T260.
- van den Broek, M.F.M., et al., The Management of Neuroendocrine Tumors of the Lung in MEN1: Results From the Dutch MEN1 Study Group. The Journal of clinical endocrinology and metabolism, 2021. 106(2): p. e1014-e1027.
- Sachithanandan, N., R.A. Harle, and J.R. Burgess, Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer, 2005. 103(3): p. 509-515.
- Lecomte, P., et al., Histologically Proven Bronchial Neuroendocrine Tumors in MEN1: A GTE 51-Case Cohort Study. World Journal of Surgery, 2018. 42(1): p. 143-152.
- Bartsch, D.K., et al., Bronchopulmonary Neuroendocrine Neoplasms and Their Precursor Lesions in Multiple Endocrine Neoplasia Type 1. Neuroendocrinology, 2016. 103(3-4): p. 240-247.
- van den Broek, M.F.M., et al., Well-Differentiated Bronchopulmonary Neuroendocrine Tumors: More Than One Entity. J Thorac Oncol, 2021. 16(11): p. 1810-1820.
- Sadowski, S.M., et al., Results of 68Gallium-DOTATATE PET/CT Scanning in Patients with Multiple Endocrine Neoplasia Type 1. Journal of the American College of Surgeons, 2015. 221(2): p. 509-517.
- Lastoria, S., et al., Role of 68Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1). Endocrine, 2016. 52(3): p. 488-494.
- Froeling, V., et al., Impact of Ga-68 DOTATOC PET/CT on the diagnosis and treatment of patients with multiple endocrine neoplasia. Annals of Nuclear Medicine, 2012. 26(9): p. 738-743.
- Albers, M.B., et al., Limited Value of Ga-68-DOTATOC-PET-CT in Routine Screening of Patients with Multiple Endocrine Neoplasia Type 1. World journal of surgery, 2017. 41(6): p. 1521-1527.
- Harding, B., et al., Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia. Endocr Relat Cancer, 2009. 16(4): p. 1313-27.
- Loffler, K.a., et al., Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. International journal of cancer. Journal international du cancer, 2007. 120: p. 259-67.
- Ludwig, L., et al., Loss of wild-type MEN1 gene expression in multiple endocrine neoplasia type 1-associated parathyroid adenoma. Endocr J, 1999. 46(4): p. 539-44.
- Skogseid, B., et al., Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab, 1992. 75(1): p. 76-81.
- Gatta-Cherifi, B., et al., Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol, 2012. 166(2): p. 269-79.
- Barzon, L., et al., Multiple endocrine neoplasia type 1 and adrenal lesions. J Urol, 2001. 166(1): p. 24-7.
- Burgess, J.R., et al., Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg, 1996. 131(7): p. 699-702.
- Langer, P., et al., Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg, 2002. 26(8): p. 891-6.
- Schaefer, S., et al., Natural course of small adrenal lesions in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Eur J Endocrinol, 2008. 158(5): p. 699-704.
- Skogseid, B., et al., Adrenal lesion in multiple endocrine neoplasia type 1. Surgery, 1995. 118(6): p. 1077-82.
- Ventura, M., M. Melo, and F. Carrilho, Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab, 2019. 63(5): p. 516-523.
- Waldmann, J., et al., Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg, 2007. 392(4): p. 437-43.
- Whitley, S.A., et al., The appearance of the adrenal glands on computed tomography in multiple endocrine neoplasia type 1. Eur J Endocrinol, 2008. 159(6): p. 819-24.
- Wang, W., et al., Adrenocortical carcinoma in patients with MEN1: a kindred report and review of the literature. Endocr Connect, 2019. 8(3): p. 230-238.
- Dotzenrath, C., et al., Malignant endocrine tumors in patients with MEN 1 disease. Surgery, 2001. 129(1): p. 91-5.
- Febrero, B., A. Ríos, and J.M. Rodríguez, Aggressive adrenal carcinoma in a young patient with Multiple Endocrine Neoplasia 1 syndrome. Med Clin (Barc), 2017. 149(10): p. 463.
- Griniatsos, J.E., et al., Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol, 2011. 9: p. 6.
- Haase, M., et al., A new mutation in the menin gene causes the multiple endocrine neoplasia type 1 syndrome with adrenocortical carcinoma. Endocrine, 2011. 39(2): p. 153-9.
- Harada, K., et al., A novel case of myxoid variant of adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1. Endocr J, 2019. 66(8): p. 739-744.
- Kharb, S., et al., Hidden diagnosis of multiple endocrine neoplasia-1 unraveled during workup of virilization caused by adrenocortical carcinoma. Indian J Endocrinol Metab, 2013. 17(3): p. 514-8.
- Ohara, N., et al., Lung adenocarcinoma and adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1. Respir Med Case Rep, 2017. 20: p. 77-81.
- Assié, G., et al., Integrated genomic characterization of adrenocortical carcinoma. Nat Genet, 2014. 46(6): p. 607-12.
- Tepede, A.A., et al., 18F-FDOPA PET/CT accurately identifies MEN1-associated pheochromocytoma. Endocrinol Diabetes Metab Case Rep, 2020. 2020.
- Dénes, J., et al., Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab, 2015. 100(3): p. E531-41.
- Fassnacht, M., et al., Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol, 2016. 175(2): p. G1-g34.
- Asgharian, B., et al., Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: Prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. Journal of Clinical Endocrinology and Metabolism, 2004. 89(11): p. 5328-5336.
- Darling, T.N., et al., Multiple Facial Angiofibromas and Collagenomas in Patients With Multiple Endocrine Neoplasia Type 1. Arch Dermatol., 1997. 7(133): p. 853-857.
- Vidal, A., et al., Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. Journal of the European Academy of Dermatology and Venereology, 2008. 22(7): p. 835-838.
- Pack, S., et al., Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. Journal of Investigative Dermatology, 1998. 110(4): p. 438-440.
- Vortmeyer, A.O., et al., Multiple endocrine neoplasia 1 gene alterations in MEN1-associated and sporadic lipomas. Journal of the National Cancer Institute, 1998. 90(5): p. 398-399.
- Dreijerink, K.M.A., et al., The Multiple Endocrine Neoplasia Type 1 (MEN1) Tumor Suppressor Regulates Peroxisome Proliferator-Activated Receptor γ-Dependent Adipocyte Differentiation. Molecular and Cellular Biology, 2009. 29(18): p. 5060-5069.
- Parekh, V.I., et al., Consequence of Menin Deficiency in Mouse Adipocytes Derived by in Vitro Differentiation. International Journal of Endocrinology, 2015. 2015.
- Radman, M. and T. Milicevic, A novel mutation of the MEN1 gene in a patient with multiple endocrine neoplasia type 1 and recurrent fibromyxoid sarcoma- A case report. BMC Medical Genetics, 2020. 21(1): p. 1-4.
- Cheah, S.K., et al., Breast cancer in multiple endocrine neoplasia type 1 (MEN1). Endocrinology, Diabetes & Metabolism Case Reports, 2021. 2021(May): p. 1-7.
- Jeong, Y.J., H.K. Oh, and J.G. Bong, Multiple endocrine neoplasia type 1 associated with breast cancer: A case report and review of the literature. Oncology letters, 2014. 8(1): p. 230-234.
- Kim, S.H. and J.H. Park, Adrenal incidentaloma, breast cancer and unrecognized multiple endocrine neoplasia type 1. Acta Endocrinologica, 2019. 15(4): p. 513-517.
- Maria, B.C., et al., Incidental finding of MEN-1 syndrome during staging and follow-up of breast carcinoma. BMJ Case Reports, 2020. 13(12): p. 1-6.
- Seigne, C., et al., High incidence of mammary intraepithelial neoplasia development in Men1-disrupted murine mammary glands. The Journal of pathology, 2013. 229(4): p. 546-58.
- Imachi, H., et al., Menin, a product of the MENI gene, binds to estrogen receptor to enhance its activity in breast cancer cells: possibility of a novel predictive factor for tamoxifen resistance. Breast cancer research and treatment, 2010. 122(2): p. 395-407.
- Dreijerink, K.M., et al., Article Enhancer-Mediated Oncogenic Function of the Menin Tumor Suppressor in Breast Cancer. Cell Reports, 2017. In Press(1).
- Teinturier, R., et al., Reduced menin expression leads to decreased ERα expression and is correlated with the occurrence of human luminal B-like and ER-negative breast cancer subtypes. Breast Cancer Research and Treatment, 2021(0123456789).
- Goswami, S., et al., Disease and treatment factors associated with lower quality of life scores in adults with multiple endocrine neoplasia type I. Surgery, 2017: p. 1-8.
- Marini, F., et al., Management impact: effects on quality of life and prognosis in MEN1. Endocrine-Related Cancer, 2017. 24(10): p. T227-T242.
- Peipert, B.J., et al., Financial burden is associated with worse health-related quality of life in adults with multiple endocrine neoplasia type 1. Surgery, 2017: p. 1-8.
- Peipert, B.J., et al., Health-related quality of life in MEN1 patients compared with other chronic conditions and the united states general population. Surgery (United States), 2017. 163: p. 205-211.
- van Leeuwaarde, R.S., et al., High fear of disease occurrence is associated with low quality of life in patients with Multiple Endocrine Neoplasia type 1 (MEN1): Results from the Dutch MEN1 Study Group. The Journal of Clinical Endocrinology & Metabolism, 2018. 103(June): p. 2354-2361.
- van Leeuwaarde, R.S., et al., Health-related quality of life in patients with Multiple Endocrine Neoplasia type 1. Neuroendocrinology, 2020: p. 1-8.
- Berglund, G., et al., Quality of life in patients with multiple endocrine neoplasia type 1 ( MEN1 ). Familial Cancer, 2003. 1(2): p. 27-33.
- You, Y.N., et al., Pancreatoduodenal surgery in patients with multiple endocrine neoplasia type 1: Operative outcomes, long-term function, and quality of life. Surgery, 2007. 142(6): p. 829-836.
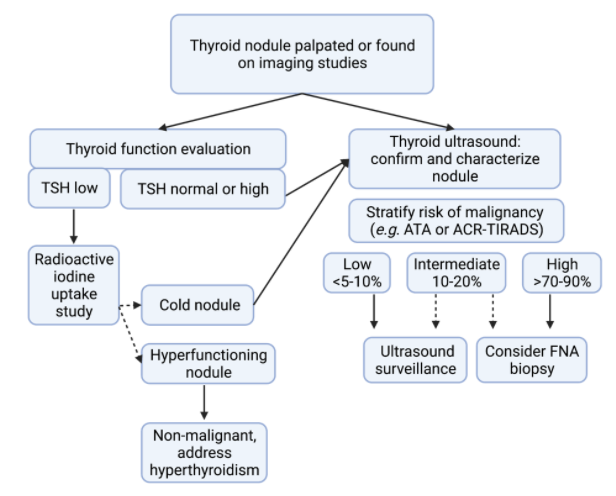
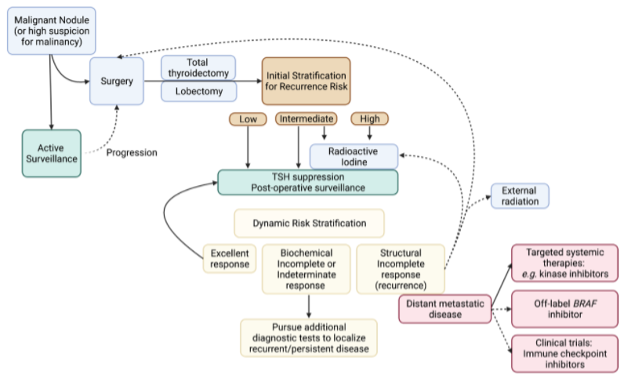






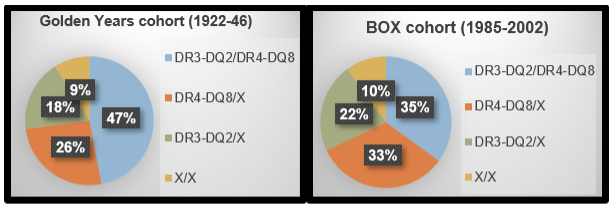








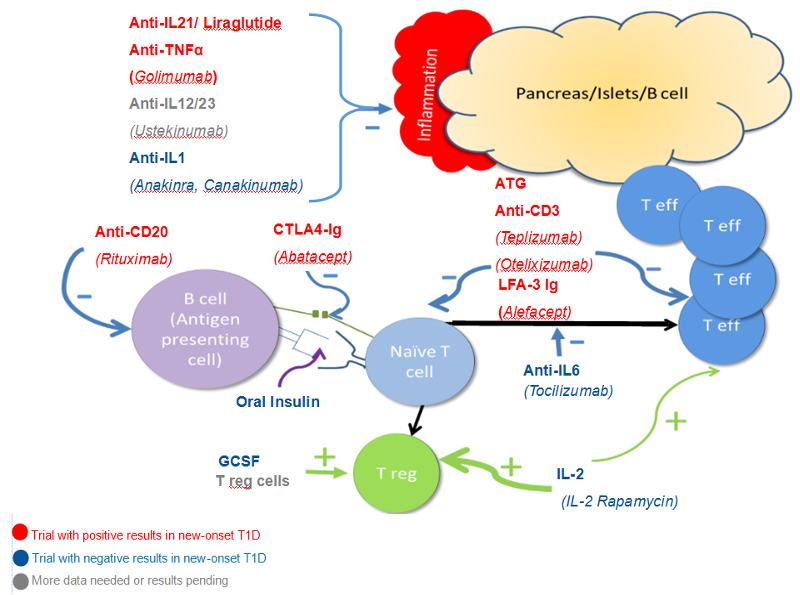



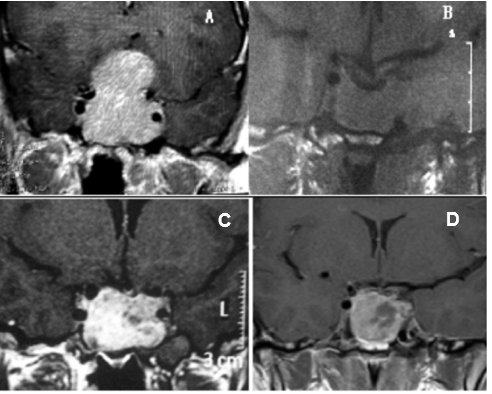
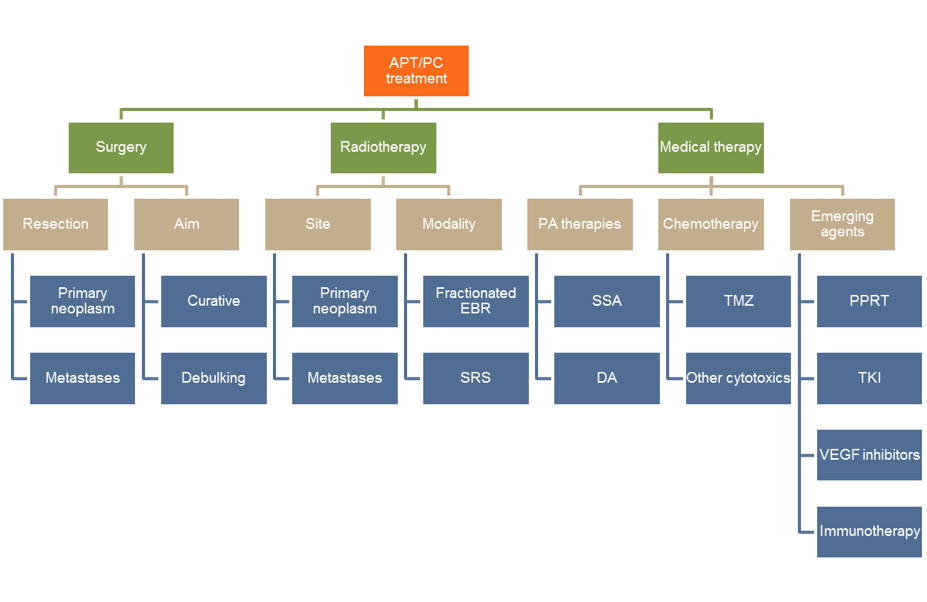
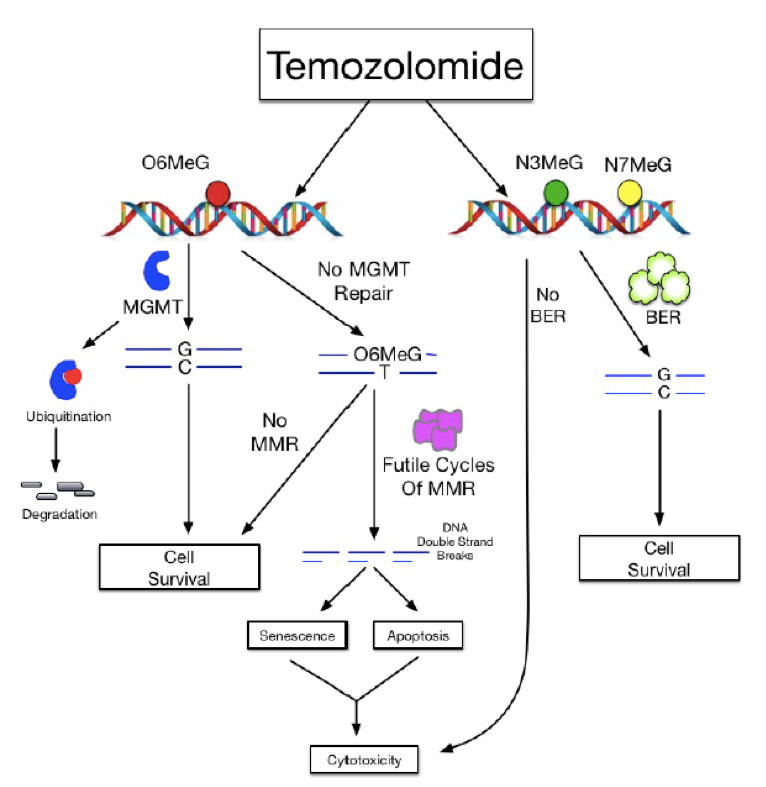
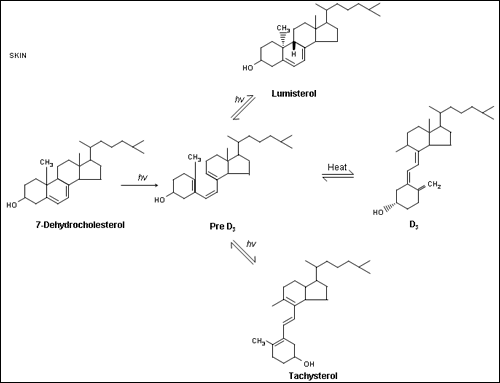
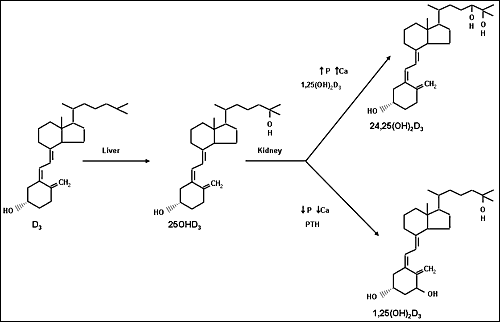
{kind=link}