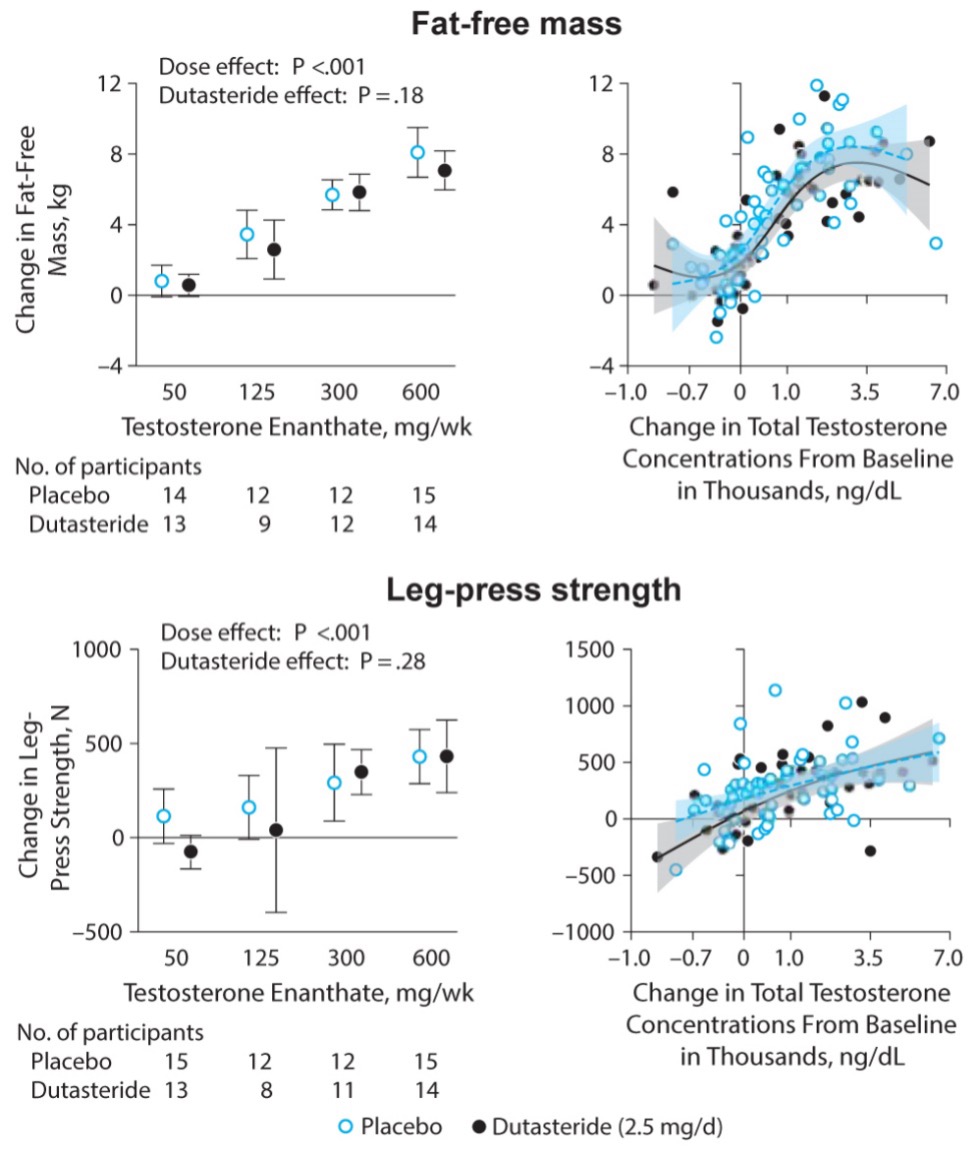
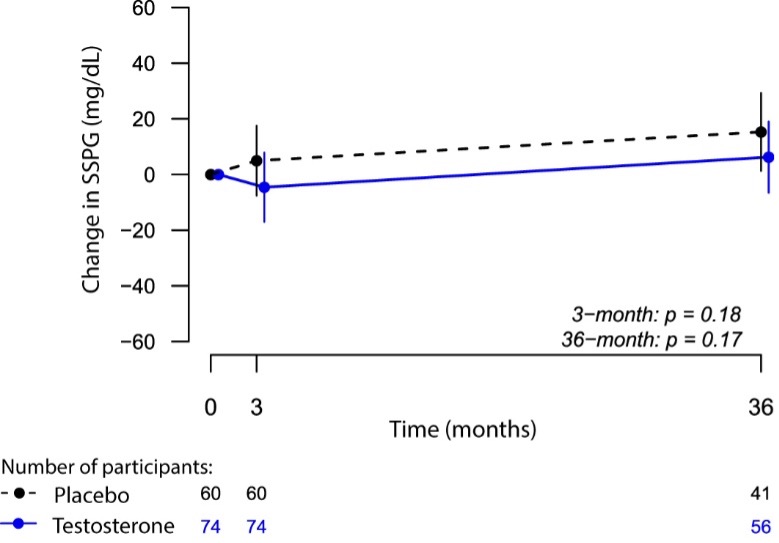
To determine whether testosterone’s effects on muscle mass and strength, sexual function, hematocrit, prostate, sebum production, and lipids are attenuated when its conversion to DHT is blocked, we administered to healthy men, 21-50 years, a long-acting GnRH-agonist to suppress endogenous testosterone. We randomized them to placebo or dutasteride (dual inhibitor of steroid 5-alpha reductase type 1 and 2) 2.5-mg daily, plus 50, 125, 300, or 600-mg testosterone enanthate weekly for 20-weeks (208). Changes in lean and fat mass, leg-press and chest-press strength, were related to testosterone dose but did not differ between placebo and dutasteride groups (208). The relation between testosterone concentrations and the changes in lean body mass, maximum voluntary muscle strength, hematocrit, and sebum production was similar between dutasteride and placebo arms (Figure 9) (208). Changes in sexual-function scores, bone markers, prostate volume, and PSA did not differ between groups (208). These data indicate that testosterone’s conversion to DHT is not essential for mediating its effects on muscle mass and strength, sexual function, hematocrit, or sebum production in men over the range of testosterone concentrations achieved in this trial (208). These data are consistent with studies that have reported that administration of steroid 5α-reductase inhibitors has little or no effect on muscle or bone mass (209-211). The isoforms of steroid 5α reductase enzyme also catalyze the 5α reduction of cortisol, progesterone, bile acids and other metabolites. In the central nervous system, 5α-reductase is the rate-limiting enzyme in the conversion of progesterone to allopregnanolone that serves as a positive allosteric modulator of gamma-aminobutyric acid (GABA) A receptors to modulate neural pathways that regulate mood, affect, and cognition (212-214). Low levels of allopregnanolone have been implicated in the pathogenesis of some forms of depressive and anxiety disorders (215). An intravenous preparation of allopregnanolone was found to be efficacious and approved for the treatment of postpartum depression (216,217) and is being investigated for the treatment of other depressive disorders. Steroid 5α-reductase enzymes are also involved in cortisol metabolism and in the pathogenesis of metabolic disease (218).
Studies of aromatase knockout mice have revealed higher fat mass and lower muscle mass in mice that are null for the P450-linked CYP19A1 aromatase gene (219). Similarly, humans with CYP19A1 mutations have decreased muscle mass and increased fat mass, and they exhibit insulin resistance (220). Data from these experiments of nature suggest that aromatization of testosterone to estradiol is important in mediating androgen effects on body composition. Finkelstein et al (221) have recently examined the relative roles of testosterone and estradiol in regulation of muscle and fat mass, and sexual function. These investigators found that testosterone’s effects on lean mass, muscle size, and strength were not significantly attenuated when its conversion to estradiol was blocked by administration of an aromatase inhibitor (221).
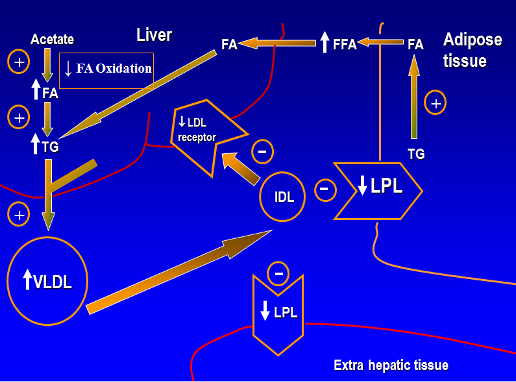
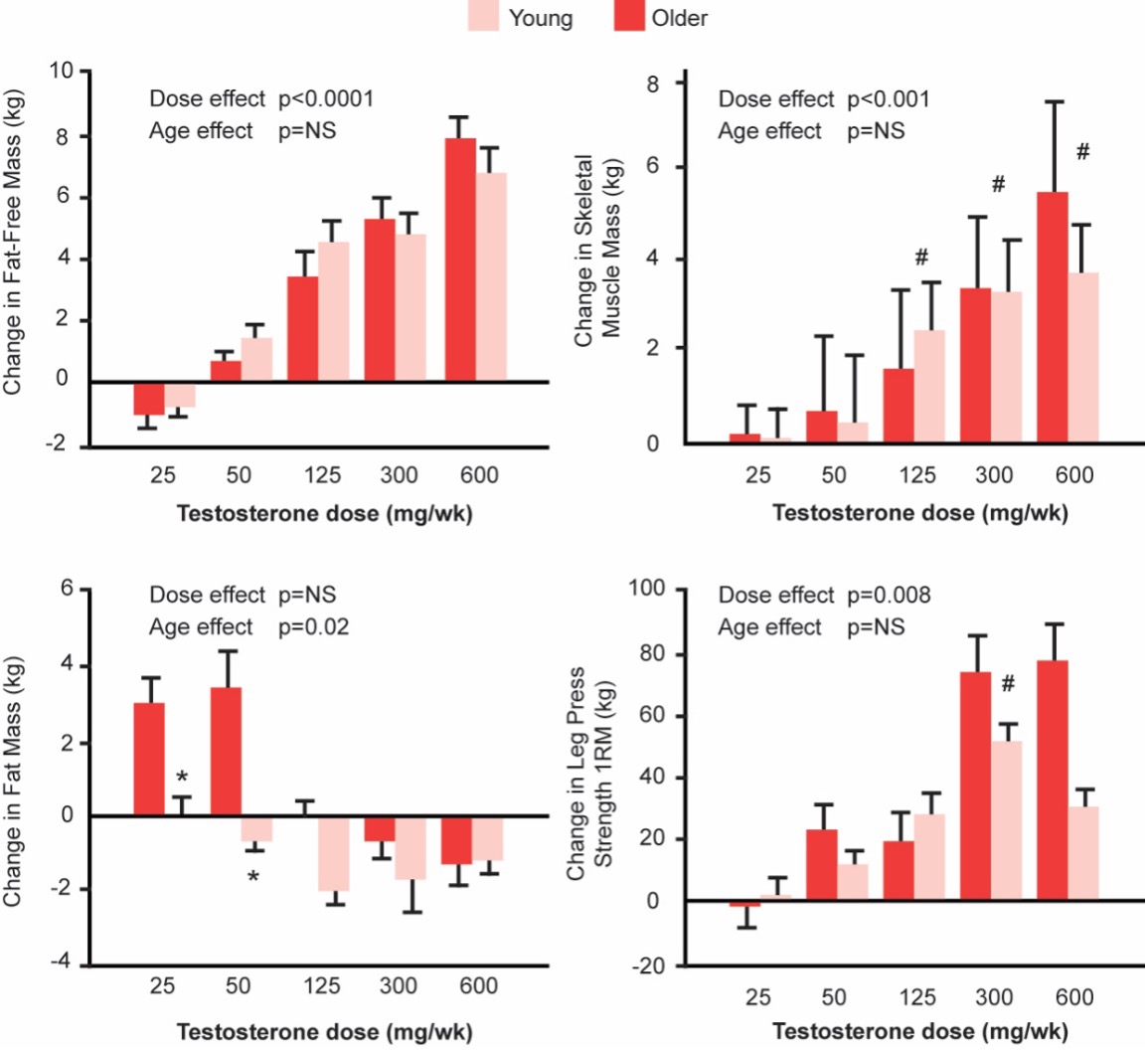
Testosterone is an important regulator of fat mass and distribution. Lowering testosterone concentrations by administration of a GnRH agonist increases fat mass, and testosterone administration in hypogonadal men decreases whole body fat mass (159,222-224). The loss of fat mass during testosterone administration occurs both in the appendices as well as the trunk and is distributed evenly between the superficial subcutaneous and deep intra-abdominal and intermuscular compartments (166,223). The effects of testosterone on whole body fat mass are related to the administered testosterone dose and the circulating testosterone concentrations (166,223).
The effects of testosterone on fat mass are mediated through its conversion to estradiol by the aromatase enzyme encoded by CYP19A1 (221). Men with inactivating mutations of CYP19A1 are characterized by increased fat mass, metabolic syndrome, hepatic steatosis, and insulin resistance (225-227). Estradiol replacement of male aromatase knockout mice reverses the adiposity and metabolic abnormalities associated with estrogen deficiency (228).
Testosterone regulates adipose tissue mass and metabolism through multiple mechanistic pathways. Androgens inhibit adipogenic differentiation of multipotent mesenchymal progenitor cells; these effects are blocked by androgen receptor blocker, bicalutamide (200,201,229). Testosterone regulates fat oxidation but does not appear to affect triglyceride secretion over short durations (230).
Testosterone, after its aromatization to estradiol, acts through the estrogen receptors in specific brain regions to regulate eating behavior, energy expenditure, and adipose tissue metabolism. The deletion of estrogen receptor α (ER-α) in specific brain regions is associated with adiposity, hyperphagia, and hypometabolism (231); estradiol acting through ER-α regulates eating behavior and energy expenditure differentially through actions on different hypothalamic neurons (231). Activation of estrogen receptor β (ER-β) by selective agonists inhibits weight gain, adiposity, increases energy expenditure and thermogenesis, and reverses hepatic steatosis in mice through direct effects on xenobiotic and bile acid receptors in the liver (232).
Sexual function in men is a complex process that includes central mechanisms for regulation of sexual desire and arousal, and local mechanisms for penile tumescence, orgasm, and ejaculation (233). Primary effects of testosterone are on sexual interest and motivation (233-238). Testosterone replacement of young, androgen deficient men improves a wide range of sexual behaviors including frequency of sexual activity, sexual daydreams, sexual thoughts, feelings of sexual desire, attentiveness to erotic stimuli, and spontaneous erections (233-241). Kwan et al (237)demonstrated that androgen-deficient men have decreased frequency of sexual thoughts and lower overall sexual activity scores; however, these men can achieve erections in response to visual erotic stimuli. Hypogonadal men have lower frequency and duration of the episodes of nocturnal penile tumescence; testosterone replacement increases both the frequency and duration of sleep-entrained, penile erections (239-241). Although both orgasm and ejaculation are believed to be androgen-independent, hypogonadal men have decreased ejaculate volume and their orgasm may be delayed.
Although hypogonadal men can achieve erections, it is possible that achievement of optimal penile rigidity might require physiologic testosterone concentrations. Testosterone regulates nitric oxide synthase activity in the cavernosal smooth muscle (242). Testosterone administration in orchiectomized rats increases penile blood flow and has trophic effects on cavernosal smooth muscle (243-245).
In male rodents, all measures of mating behavior are normalized by relatively low testosterone levels that are insufficient to maintain prostate and seminal vesicle weight (246,247). Similarly, in men, sexual function is maintained at relatively low normal levels of serum testosterone (221,238,248). Testosterone’s effects on libido are mediated through its conversion to estradiol (221).
Total and free serum testosterone levels are positively associated with sexual desire, erectile function and sexual activity in older men with unequivocally low testosterone levels and symptoms of sexual dysfunction (114). These findings suggest that low testosterone levels may contribute to impaired sexual functioning in older men.
Erectile dysfunction and androgen deficiency are two common but independently distributed, clinical disorders that sometimes co-exist in the same patient (112,113,233,249). Hypogonadism is a clinical syndrome that results from androgen deficiency (16); in contrast, erectile dysfunction is usually a manifestation of a systemic vasculopathy, often of atherosclerotic origin. Thus, androgen deficiency and erectile dysfunction have distinct pathophysiology. Eight to ten percent of middle-aged men presenting with erectile dysfunction have low testosterone levels (113,249-251).
In open-label trials, testosterone treatment has been shown to improve sexual function in young men with classical hypogonadism due to disorders of the hypothalamus, pituitary, or testes (159,252). However, previous trials evaluating the benefits of testosterone therapy in men 60 years and older with age-related decline in testosterone levels on sexual functioning have yielded inconsistent results (253), with some studies showing improvement (254,255), while others have suggested no clear benefit (23). The inconsistencies in these previous studies are due to several factors, including small sample sizes, inclusion of men who were not clearly hypogonadal or did not have sexual symptoms, inclusion of men with heterogeneous sexual disorders, variable treatment durations, and the use of outcomes assessment tools that had not been rigorously validated.
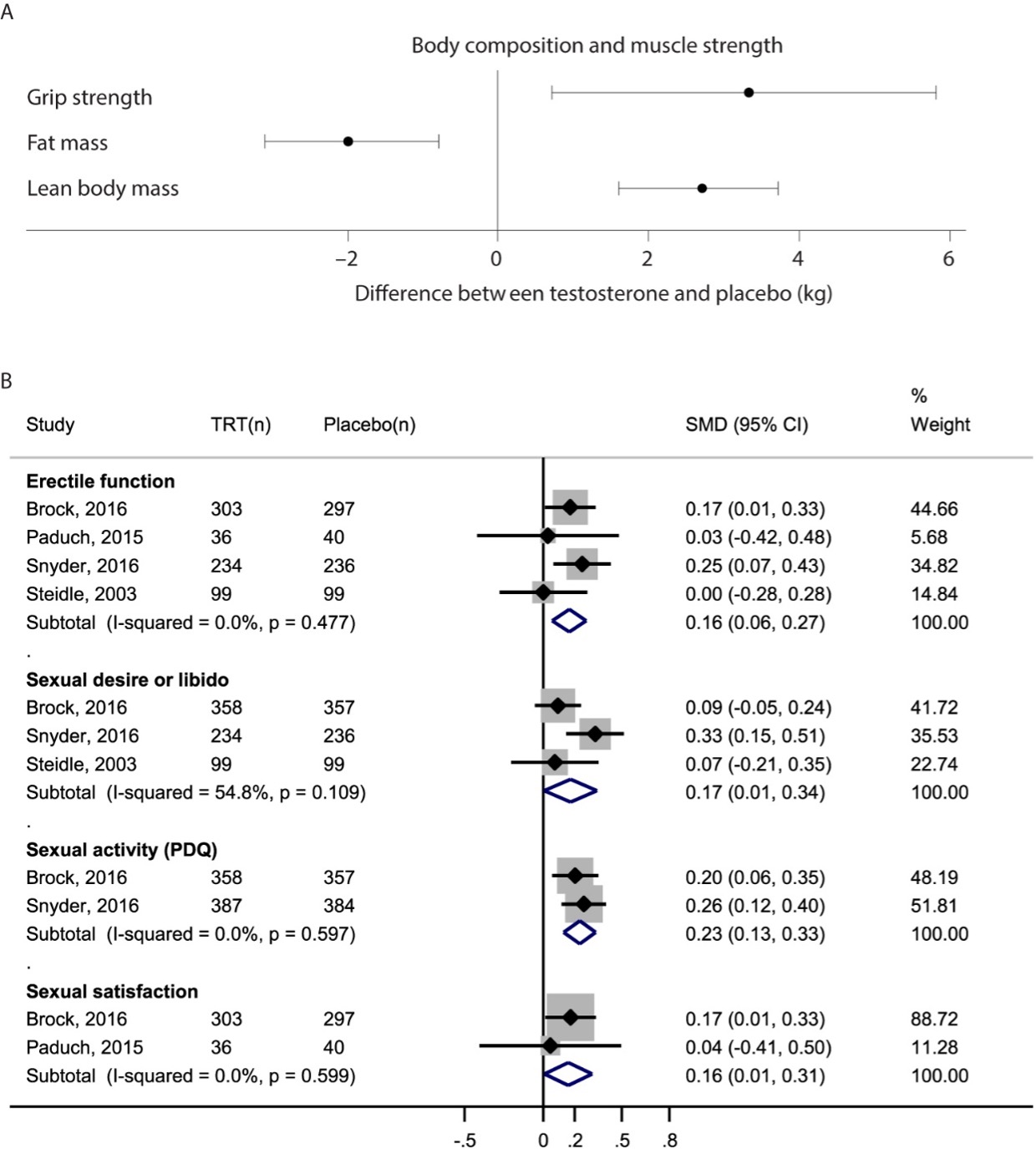
In a small number of placebo-controlled trials of testosterone that have been conducted in men with sexual symptoms and low testosterone levels (24,26,161), testosterone replacement has been associated with a small but significant increase in overall sexual activity, sexual desire, erectile function, and sexual satisfaction. A meta-analysis of these placebo-controlled trials found that testosterone replacement of hypogonadal men is associated with a small but significant increase in sexual desire [standardized mean difference (SMD): 0.17; 95% CI, 0.01, 0.34], erectile function (SMD: 0.16; 95% CI, 0.06, 0.27), and sexual satisfaction (SMD: 0.16; 95% CI, 0.01, 0.31) (256).
The Sexual Function Trial of the TTrials determined the efficacy of testosterone treatment for 1-year on sexual function in symptomatic, community-dwelling, older men ≥65 years with low testosterone levels (26). Testosterone administration for 1-year to raise testosterone concentrations into a range that is mid-normal for healthy young men was associated with significant improvements in sexual activity, desire, and erectile function (257). The treatment effects tended to wane over time, and the effect on erectile function was substantially smaller than that reported with phosphodiesterase 5 inhibitors (258). The magnitude of increase in testosterone levels was related to the improvements in sexual activity and desire, but not erectile function (257). There was no clear testosterone threshold level of effect.
Testosterone does not improve sexual function in middle-aged and older men who have normal testosterone levels and do not have any sexual symptoms (23). Testosterone replacement therapy does not improve ejaculatory function in men with ejaculatory disorder (259).
It had been speculated that testosterone administration might improve erectile response of men with ED to selective phosphodiesterase inhibitors (260-262). To determine whether the addition of testosterone to a phosphodiesterase-5-inhibitor improves erectile response, we conducted a randomized, placebo-controlled trial (263), in men, 40-to-70 years, with erectile dysfunction and low total testosterone< 11.5 nmol/L (330ng/dL) and/or free testosterone <173.5 pmol/L (50 pg/mL). All participants were initially started on sildenafil alone and the sildenafil dose was optimized based on their response during a 3 to 7-week run-in period (263). The participants were then randomized to 10-g testosterone or placebo gel for 14-weeks in combination with the optimized sildenafil dose (263). The administration of sildenafil alone was associated with substantial increases in erectile function domain (EFD) score and total and satisfactory sexual encounters (263). However, the change in EFD score in men assigned to testosterone plus sildenafil did not differ significantly from that in men assigned to placebo plus sildenafil (263). Changes in total and successful sexual encounters, quality-of-life, and marital-intimacy did not differ between testosterone and placebo groups. Even among the subsets of men with baseline testosterone <250 ng/dL or those without diabetes, there were no significant differences in EFD scores between the two arms (263). Another placebo-controlled trial of men with erectile dysfunction who were non-responders to tadalafil also did not show a greater improvement in erectile function in men assigned to the testosterone arm than in those assigned to the placebo arm (262). Thus, in randomized trials, the addition of testosterone to PDE5Is has not been shown to improve erectile function in men with erectile dysfunction (262,263).
In older hypogonadal men with low sexual desire, testosterone treatment improves sexual desire, erectile function, and overall sexual activity. Androgen deficiency is an important cause of low sexual desire disorder (233). Therefore, serum testosterone concentrations should be measured in the diagnostic evaluation of hypoactive sexual desire disorder as well as erectile dysfunction, recognizing that low sexual desire is often multifactorial; systemic illness, relationship and differentiation (the ability of individuals in a relationship to maintain their distinct identities) issues, depression, and many medications can be important antecedents or contributors to low sexual desire and sexual dysfunction.
Androgen deficiency that develops before the completion of pubertal development is associated with reduced cortical and trabecular bone mass (272,273). During the pubertal years, bone accretion, and bone length and thickness is regulated by sex steroids. During puberty, sex hormones slow long bone growth and accelerate axial growth. Prepubertal sex hormone deficiency allows continued long bone growth and slows axial growth resulting in longer limbs and a shorter trunk (eunuchoidal proportions) (274). Sex differences in bone width are also established during pubertal development. Men increase bone width by periosteal bone formation and women mostly by endocortical apposition (275). Young men with constitutional delay of puberty have lower bone mineral density (276), which does not improve spontaneously 2 years later (277). Indeed, men with hip fractures have been shown to have smaller femoral head diameters, which may potentially be related to delayed puberty (278). Therefore, individuals with sex-steroid deficiency before or during peri-pubertal years may end up with suboptimal peak bone mass and increased lifetime fracture risk. Similarly, men with acquired androgen deficiency have lower bone mineral density than age-matched controls (155).
Testosterone therapy of healthy, young, hypogonadal men is associated with significant increases in vertebral bone mineral density (156,279-283). However, bone mineral density is typically not normalized after 1-2 years of testosterone replacement therapy (156). Some hypogonadal patients included in these testosterone trials had panhypopituitarism and also suffered from growth hormone deficiency. It is possible that concomitant GH replacement might be necessary for restoration of normal bone mineral density. Excessive glucocorticoid replacement might also contribute to bone loss in these patients. In addition, some participants had experienced testosterone deficiency before the onset and completion of pubertal development; the individuals who develop androgen deficiency during the critical pubertal developmental window of bone accretion, may end up with decreased peak bone mass, and testosterone administration may not be able to restore bone mass to levels seen in eugonadal age-matched controls. Many testosterone replacement trials were less than 3 years in duration, and it is possible that a longer period of testosterone administration might be necessary to achieve maximal improvements in bone mineral density. Indeed, Behre et al (279) reported that bone mineral density in some hypogonadal men continued to increase even after many years of testosterone treatment using a scrotal transdermal patch and reached the levels expected for age-matched eugonadal controls.
The age-related decline in sex hormones is associated with age-related changes in bone mineral density and increased risk of osteoporotic fractures (131-136,284,285). Older men with hip fractures have lower testosterone levels than age-matched controls (286). Bioavailable testosterone levels have been found to be better predictors of fracture risk than total testosterone levels (287). Interestingly, a U-shaped association between endogenous testosterone concentrations and incident fractures was recently observed, with midrange plasma testosterone levels being associated with lower incidence of any fracture and with hip fracture compared to lower or higher testosterone (288). Men with osteoporosis have been found to have lower DHT levels than those without osteoporosis (289). In the Cardiovascular Health Study, in which testosterone and DHT levels were measured by liquid chromatography–tandem mass spectrometry, circulating DHT, but not testosterone, was found to be negatively associated with hip fracture risk in men (290).
In epidemiologic studies, estradiol levels are more strongly associated with bone mineral density of the spine, hip, and distal radius than total testosterone levels (132,134,135,285). Men with low bioavailable estrogen have increased risk of non-vertebral fracture which is increased further in those with low bioavailable estrogen, low bioavailable testosterone as well as high SHBG (287) suggesting a complex interplay of these hormones in fracture resilience. Mendelian randomization analysis have found that increased genetically determined estradiol levels are associated with increase in lumbar spine bone mineral density (291) and lower fracture risk (292). The CYP19A1 alleles associated with higher estradiol levels are associated with higher bone mineral density (291).
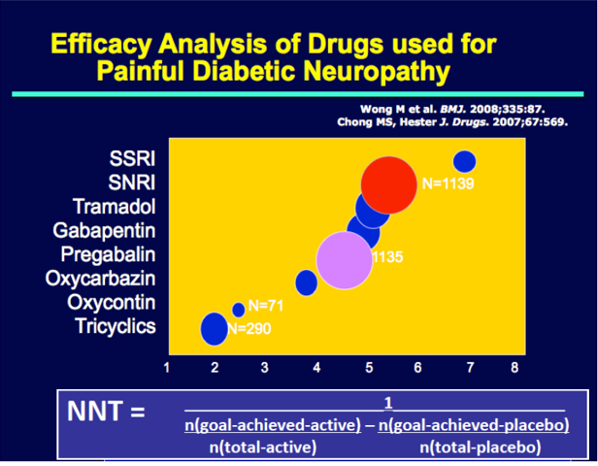
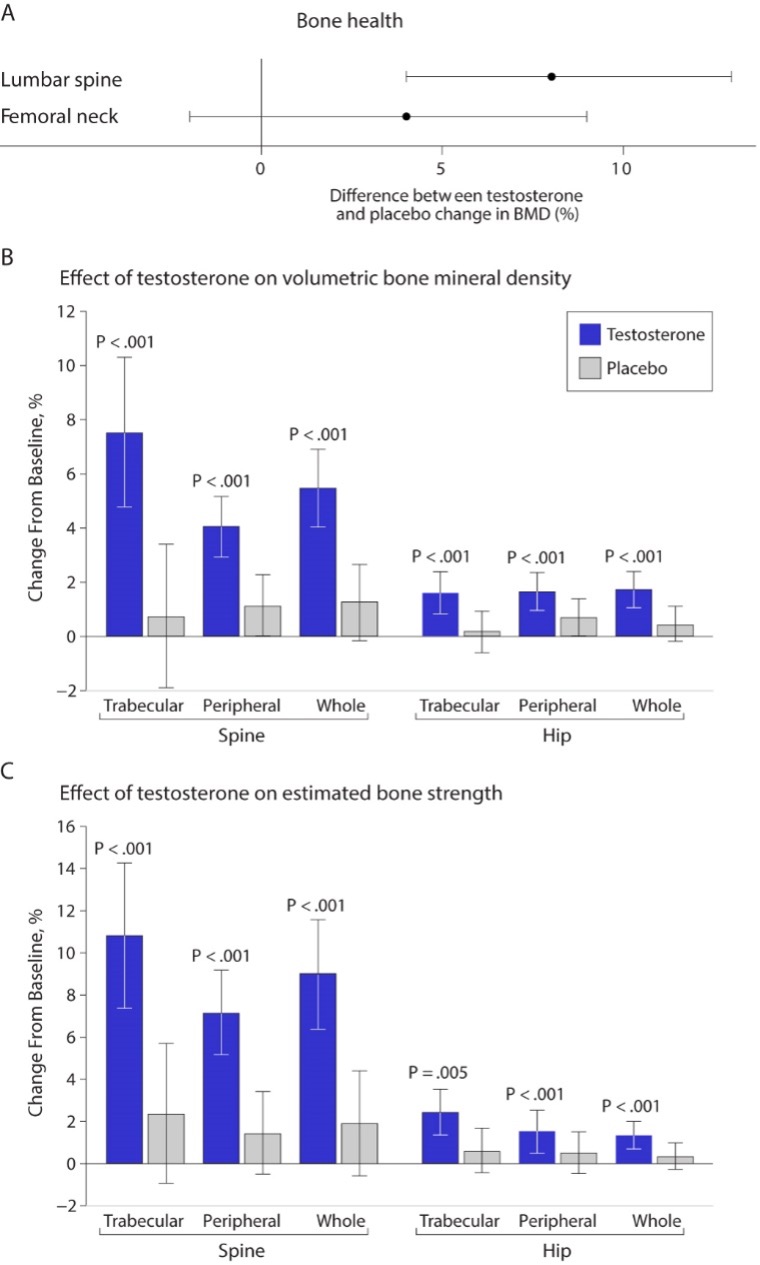
Earlier studies of testosterone replacement of relatively healthy older men that examined the effects of testosterone on bone mineral density reported inconsistent results (188,189,293,294). One study found greater increases in vertebral bone mineral density in the testosterone arm of the trial than in the placebo arm, while another study did not find any significant differences between the change in vertebral or femoral bone mineral density between testosterone and placebo groups (294). A meta-analysis of randomized trials found a significantly greater increase in lumbar bone mineral density but not in femoral bone mineral density in the testosterone arms of trials that used intramuscular testosterone than in placebo arms (Figure 10) (295); transdermal testosterone had no significant effect.
The Bone Trial of the TTrials determined the effects of testosterone replacement for 1-year in men 65 years or older with low testosterone levels on volumetric bone mineral density and bone strength using quantitative computed tomography (296). This trial found significantly greater increases in volumetric bone mineral density and estimated bone strength in the testosterone arms compared to placebo; specifically, these increases were most prominent in the spine than hip and more in trabecular than peripheral bone (Figure 10). The treatment effects on volumetric bone density and bone strength observed in the TTrials compare favorably with those reported in trials of bisphosphonates and some selective estrogen receptor modulators.
The T4Bone substudy of the T4DM trial determined the effect of 24 months of testosterone treatment on bone microarchitecture and bone mineral density of men aged 50 years or older enrolled in a community-based lifestyle program using high resolution-peripheral quantitative computed tomography (297). Compared to placebo, testosterone treatment increased cortical and total bone mineral density of the tibia and radius, as well as cortical area and thickness at both sites. Testosterone treatment also increased areal thickness at the lumbar spine (297).
Future studies are needed to determine whether these improvements from testosterone treatment are associated with reduced fracture risk in older men with low testosterone levels.
Testosterone also directly stimulates osteoblastic bone formation. Androgen receptors have been demonstrated on osteoblasts and on mesenchymal stem cells (313). Testosterone stimulates cortical bone formation (314). Sclerostin is secreted by osteocytes and inhibits osteoblast differentiation. Sclerostin was found to be negatively related to total and free testosterone in men with idiopathic osteoporosis (312). Hypogonadal men have higher serum sclerostin levels than eugonadal men, and DHT directly suppresses sclerostin production in cultured human osteocytes through an AR-mediated mechanism (315). Testosterone also stimulates the production of several growth factors within the bone, including IGF-1; these growth factors may contribute to bone formation (316). Leydig cells in the testis secrete insulin-like peptide 3 (INSL3) in addition to testosterone. INSL3 has been reported to have a negative association with sclerostin in specific populations and INSL3 downregulates sclerostin protein expression in cultured osteocytes (317). Osteocalcin secreted by osteoblasts acts on Leydig cells through the GPRC6A receptor, suggesting a possible feedback mechanism for bone-testis crosstalk (318). Testosterone increases muscle mass, which may indirectly increase bone mass by increased loading. Testosterone might inhibit apoptosis of osteoblasts through non-genotropic mechanisms (319,320). In addition to its effects on bone mineral density, testosterone might reduce fall propensity because of its effects on muscle strength and reaction time.
In men androgens and estrogens both play independent roles in regulating bone resorption (301). Estradiol levels above 10 pg/ml are generally believed to be sufficient to prevent increases in bone resorption and decreases in BMD in men (311).
Testosterone replacement has been shown to increase vertebral and femoral bone mineral density, and bone strength in older men with unequivocally low testosterone levels (16). Testosterone increases bone mass by multiple mechanisms. Testosterone’s aromatization to estrogen plays an important role in regulating bone health in men. Testosterone’s effects on fracture risk have not been studied.
Several lines of evidence suggest that testosterone regulates several domains of cognition, sexually dimorphic behaviors, mood, and affect, and the neuropathology of Alzheimer’s Disease (AD). Testosterone is aromatized to estrogen in the brain, and some effects of testosterone on cognition might be mediated through its conversion to estradiol. Additionally, androgen receptors are expressed in the brain (330), and androgen effects on brain organization during development (331,332) are mediated through androgen receptor. Androgens increase neurite arborization, facilitating intercellular communication (331-334). Testosterone is metabolized in neurons as well as in glial cells to DHT, which is further converted reversibly in some cell types such as type 1 astrocytes to 5α-androstane-3α,17β-diol (335), which is a potent modulator of GABA on GABAA receptors but a weak ligand for AR and ER (336). The 3β isomer of androstanediol, 5α-androstan-3β,17β-diol, is also synthesized in the brain; this steroid is a ligand for ERβ (337). Thus, testosterone treatment may potentially expose the brain to a range of biologically active metabolites, all of which may contribute to the observed responses. Testosterone also affects serotonin, dopamine, acetylcholine (333), and calcium signaling (334). Thus, testosterone could influence cognitive function and the development and progression of AD neuropathology through multiple mechanistic pathways.
The age-related decline in serum testosterone levels has been associated with impairment in cognitive function (338). Androgens effects on cognitive function are domain-specific. For instance, observations that men outperform women in a variety of visuo-spatial skills suggest that androgens enhance visuo-spatial skills (339). In !Kung San hunter-gatherers of Southern Africa, testosterone, but not estradiol, levels correlated with better spatial ability and with worse verbal fluency (340). Women with congenital adrenal hyperplasia with high androgen levels score higher on tests of spatial cognition than their age- and gender-matched siblings (341). 46, XY rats with androgen insensitivity perform worse on tests of spatial cognition than their age-matched controls (342). Other studies have reported a complex relationship between androgen levels and spatial ability (123,343-345). Circulating levels of dihydrotestosterone, a metabolite of testosterone that is not converted to estrogen, positively correlated with verbal fluency (340). Barrett-Conner et al (122) found positive associations between total and bioavailable testosterone levels, and global cognitive functioning and mental control, but not with visuospatial skills. In the Baltimore Longitudinal Study of Aging (346), higher free testosterone index was associated with better scores on visual and verbal memory, visuospatial functioning, and visuomotor scanning. Men with low testosterone levels had lower scores on visual memory and visuospatial performance (346); however, some studies have shown no association of serum testosterone levels with domains of visual and verbal memory, and executive function in older men (347,348). In the Concord Health and Aging in Men Project, the authors found that changes in serum testosterone levels over time, rather than baseline testosterone levels, were predictive of cognitive decline (338).
A large body of preclinical and epidemiologic data shows that testosterone acts as a negative regulator of endogenous Ab amyloid accumulation in the brain, attenuates tau phosphorylation, reduces neuro-inflammation, exerts neuronal protective effect in response to injury and disease, and promotes neuronal regeneration and connectivity. However, the randomized trials data generated largely in community dwelling middle-aged and older adults without cognitive deficits or Alzheimer's Disease neuropathology have been inconclusive for reasons that are discussed below.
Testosterone acts as a negative regulator of endogenous Ab amyloid accumulation in the brain through multiple mechanisms. Surgical orchiectomy of male rats is associated with increased accumulation of Ab amyloid in the brain; the accumulation of Ab amyloid in surgically orchiectomized rats is prevented by DHT administration but not by estradiol administration (349-351). In male Brown-Norway rats, age-related decreases in testosterone and DHT are associated with increased brain levels of Ab amyloid (350). Testosterone promotes the conversion of amyloid precursor protein (APP) to soluble APP-alpha rather than A beta amyloid. Consistent with these findings, prolonged treatment of cultured cortical neurons and neuroblastoma cell lines with testosterone resulted in increased production of soluble sAPP-a and decreased production of Ab amyloid (349). This effect of testosterone on the processing of APP is mediated in part through its aromatization to estradiol (352). However, there is strong evidence of mediation through a direct androgen receptor (AR)-mediated pathway as well (350-353).
In 3XTg-AD mouse model of Alzheimer’s Disease, orchiectomy at age 3 months is associated with significantly increased accumulation of Ab amyloid in hippocampus CA1, amygdala, and subinculum at age 6 months (352,353). DHT treatment of orchiectomized mice prevents the accumulation of Ab amyloid as well as deterioration of spontaneous alternation behavior (353). DHT also reduces Tau-phosphorylation in orchiectomized triple transgenic mouse model of AD (352). Androgens upregulate the expression of neprilysin, the enzyme that catalyzes the degradation and clearance of Ab amyloid in neuronal cells (354) and decrease Ab amyloid accumulation (355).
Testosterone also attenuates AD-like tau pathology. In gonadectomized mice, testosterone as well as estradiol reduce tau phosphorylation (356,357). Androgens also reduce tau phosphorylation induced by acute heat shock and injury in male rats independent of estradiol (358).
Testosterone exerts neuroprotective effects across the lifespan in brain areas susceptible to neurodegeneration in AD (365-368). Thus, in cultured neurons, testosterone reduces neuronal apoptosis induced by oxidative stress and Abamyloid (369-371).
Testosterone promotes neuronal growth, connectivity, and functioning. Testosterone increases neurite arborization, and synapse formation facilitating intercellular communication (372-375). Testosterone also has nongenomic effects, and affects serotonin, dopamine, acetylcholine and calcium signaling (376-378). Androgen receptors are expressed in the brain, and androgen effects on organization of the brain during development are likely mediated directly through AR. Some additional effects of testosterone are mediated through its conversion to estradiol.
Testosterone also exerts protective effects against neuroinflammation. Orchiectomy as well as obesogenic diet are each associated with increased expression levels of proinflammatory cytokines TNF-alpha and IL-1beta in the cerebral cortex in middle-aged male rats (370). The castration-induced upregulation of proinflammatory cytokines TNFa and IL-1b effect is prevented by testosterone supplementation (370). Similar stimulatory effects of testosterone on the expression of proinflammatory cytokines were observed in mixed glial cell cultures in vitro (370). 5aDihydrotestosterone inhibits interleukin-1a or tumor necrosis factor a-induced proinflammatory cytokine production via androgen receptor-dependent inhibition of nuclear factor kB activation (371). Low testosterone also is associated with increased macrophage infiltration in sciatic nerve in castrated male rats (371). The mechanisms of these protective effects of testosterone on neuroinflammation are incompletely understood but appear to require both androgen receptor and estrogen receptor-mediated pathways (371).
Some but not all epidemiologic studies have found an association between low circulating testosterone levels and AD (346,379-384); the relation appears to be stronger between free testosterone levels and the risk of AD than between total testosterone and AD (346). The strength of the association between testosterone and AD is affected by apolipoprotein ε4 genotype, a genetic risk factor for AD (380); men with one or more ε4 alleles have lower testosterone levels and a higher risk of AD than men without an ε4 allele (380).
In longitudinal follow-up of male participants of the Baltimore Longitudinal Study on Aging (346), the men who were healthy at baseline and developed a clinical diagnosis of AD had significantly lower free testosterone levels than those who did not develop AD. The age-related decline in circulating free testosterone levels preceded the clinical diagnosis of AD by nearly 10 years (346).
Epidemiologic investigations of the association of circulating testosterone levels with age-related changes in cognitive function are in agreement that androgens effects on cognitive function are domain-specific. Generally, men with low testosterone levels perform less well than those with normal testosterone levels on tests of verbal fluency, visuospatial abilities, verbal memory, and executive function (121,122,385-388). Some inconsistency in findings across studies is likely related to heterogeneity of study populations, lack of standardization of cognitive assessments across studies, inaccuracy and imprecision of testosterone immunoassays, and the use of variable thresholds of testosterone levels to define "low". Some studies have suggested a curvilinear relation between testosterone levels and cognitive function; both low and high testosterone levels are associated with worse function suggesting that there may be an optimal level at which cognitive performance is optimized (386).
No adequately powered randomized placebo-controlled trials of testosterone replacement have been conducted in men with AD (389-392). The clinical trials data on the effects of testosterone on cognition have provided conflicting results; these trials were limited by their small size, inclusion of men who were not clearly hypogonadal and who did not have cognitive impairment or AD neuropathology, and use of outcomes that were not directly related to AD phenotype. Some studies have reported improvements in verbal memory and visuospatial skill while others found no effect (389,391-393).
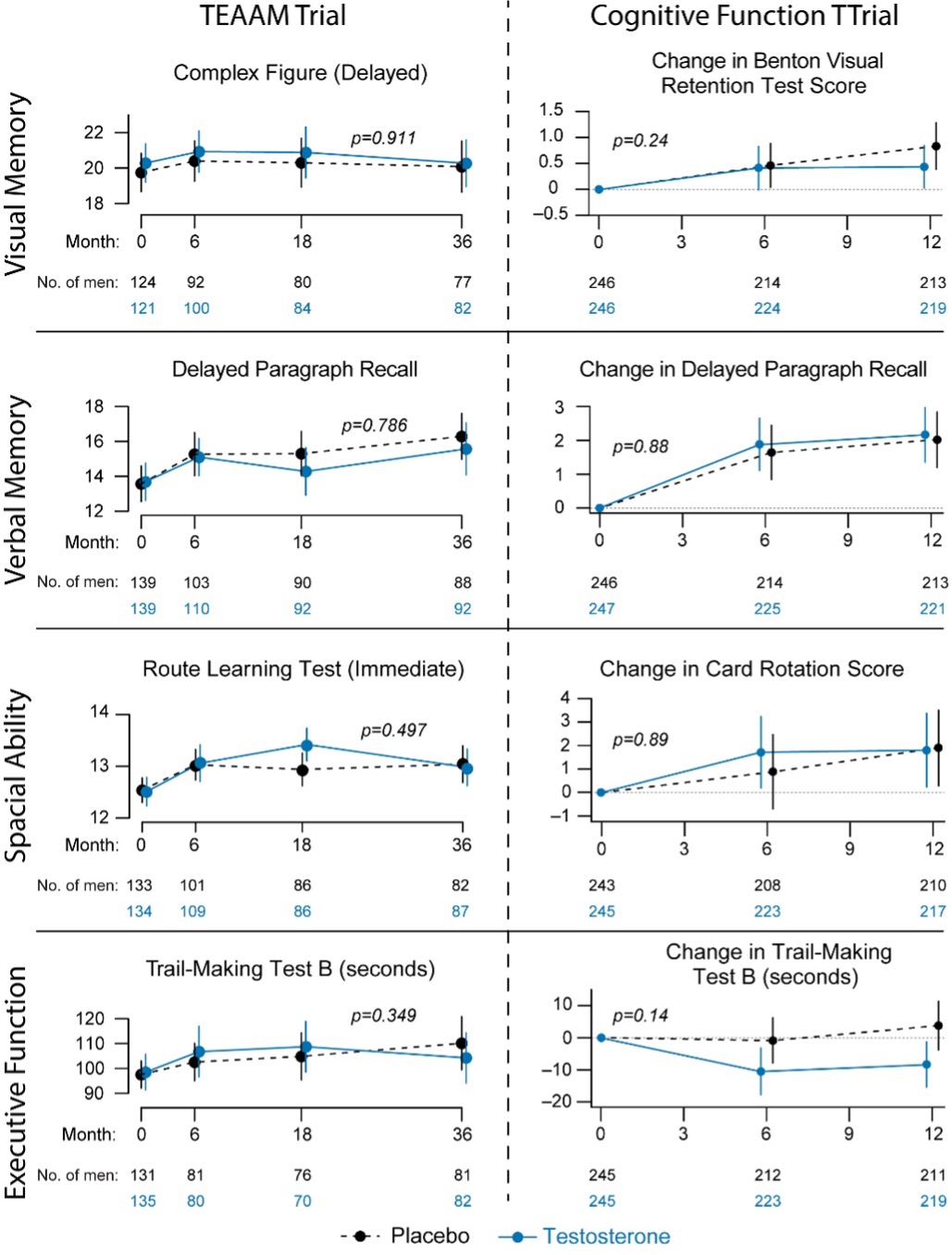
The Testosterone Trials, a set of 7 coordinated trials of community-dwelling older men with unequivocally low testosterone levels, measured using liquid chromatography tandem-mass spectrometry (LC-MS/MS), showed no significant effect on delayed paragraph recall – the primary outcome of the trial (26,390). Post hoc analysis of the TTrials data showed small but significant improvement in executive function (390). The TTrials had many attributes of good trial design - prospective allocation of participants, parallel groups, blinding, and high retention rates, but progression of AD was not a primary aim of the trial (26). The trial’s duration of one year was not long enough to evaluate effects on clinically meaningful measures of cognitive function or AD pathology. The trial did not include any measures of AD pathology, including A beta amyloid or Tau-protein or blood or CSF markers of AD. The participants in this well conducted trial were not selected prospectively based on cognitive deficits or risk of AD. A few small testosterone supplementation studies (sample size varying from 11 to 47) of 6 weeks to 6-month duration in men with cognitive impairment of AD have reported modest improvements in verbal and spatial memory but the small sample sizes, short intervention durations, variable eligibility criteria, and inclusion of men without confirmed AD, and inclusion of men with normal testosterone levels limit the interpretability of these data (391,392).
In spite of the robust preclinical data that testosterone acts as a negative regulator of endogenous Ab amyloid accumulation in the brain, attenuates tau phosphorylation, reduces neuro-inflammation, exerts neuronal protective effect in response to injury and disease, and promotes neuronal regeneration and connectivity and some epidemiologic evidence that decline in testosterone levels increases the risk of incident clinical AD, the randomized trial data on the effects of testosterone on cognition is highly equivocal. The randomized clinical trials in community dwelling older adults without cognitive deficit or AD neuropathology have not found clinically meaningful improvements in cognitive function. The inconsistency in findings cannot yet be interpreted as conclusive evidence that there is no effect. Limitations of previous studies include limited sample sizes, inclusion of men with no clear cognitive deficit or AD neuropathology, the use of a variety of neuropsychological tests that are not clinically meaningful in the context of AD or dementia; the use of differing protocols in clinical trials. The effects of testosterone therapy on clinically important outcomes in men with cognitive impairment have not been studied. The efficacy of testosterone replacement in men with cognitive impairment, such as in patients with Alzheimer’s disease, needs further investigation in larger randomized controlled trials.
Circulating testosterone concentrations have not been consistently associated with major depressive disorder in men (128,129,395-398). Rather, testosterone levels appear to be associated with a late-life low grade persistent depressive disorder (dysthymia) (128,129,395-398). Intervention trials have failed to demonstrate statistically significant or clinically meaningful improvements in patients with major depressive disorder (399). Placebo-controlled trials of testosterone in men with refractory depression also have not consistently shown a beneficial effect of testosterone (399-402). A meta-analysis of randomized trials reported modest improvements in depressive symptoms in testosterone-treated men compared to placebo-treated men (403), but there is no convincing evidence that testosterone treatment can induce remission in men with major depressive disorder (404). Two small trials in men with dysthymia have reported greater improvements in depressive symptoms in testosterone-treated men than in placebo-treated men (405,406). Adequately powered long-term randomized trials are needed to determine whether testosterone replacement therapy can induce remission in older hypogonadal men with late-onset, low grade persistent depressive disorder (dysthymia).
There is anecdotal evidence that androgens improve energy and reduced sense of fatigue (407). Testosterone administration increases hemoglobin and red cell mass, stimulates 2, 3 DPG concentrations thereby shifting the oxygen – hemoglobin dissociation curve favorably to improve greater oxygen delivery, and induces muscle capillarity (322,408,409). Additionally, testosterone stimulates mitochondrial biogenesis and mitochondrial quality (410). All of these adaptations would be expected to improve net oxygen delivery to the muscle, improve aerobic performance, and reduce fatigability. The effects of testosterone on fatigue and vitality have been studied in some randomized trials. Endogenous levels of total and free testosterone are not significantly associated with vitality in older hypogonadal men with sexual dysfunction, diminished vitality, and/or mobility limitation (114). In the Vitality Trial of the TTrials, testosterone treatment for 1-year did not improve vitality in older men with low vitality measured using the Functional Assessment of Chronic Illness Therapy (FACIT)-scale but men receiving testosterone did report a small but statistically significant improvement in mood. These findings are consistent with other randomized controlled studies (23,171,190), showing no clear benefit on fatigue and health-related quality of life with testosterone therapy.
Supraphysiologic doses of androgenic steroids such as those abused by athletes and recreational bodybuilders have been associated with aggressive responses to provocative situations (411), increased scores on Young’s manic scale, and with affective and psychotic disorders in some individuals (412); these adverse effects have not been reported with physiologic testosterone replacement.
By improving some aspects of physical and sexual function, testosterone supplementation might be expected to improve health-related quality of life. However, only a few small trials have evaluated the effects of testosterone on health-related quality of life. A systematic review of a small number of randomized trials has not revealed a significant improvement in composite health-related quality of life scores, but testosterone therapy improves scores on the physical component of MOS SF-36 (16,159).
Short-term testosterone administration in healthy, young, androgen-deficient men with classical hypogonadism is associated with a low frequency of relatively mild adverse effects such as acne, oiliness of skin, and breast tenderness. However, the long-term risks of testosterone supplementation in older men are largely unknown. There are several unique considerations in older men that may increase their risks of testosterone administration. Serum total and free testosterone concentrations are higher in older men than young men at any dose of testosterone therapy, due to decreased testosterone clearance in older men (61). Older men exhibit greater increments in hemoglobin and hematocrit in response to testosterone administration than young men (321), adjusting for testosterone dose. Altered responsiveness of older men to testosterone administration might make them susceptible to a higher frequency of adverse events, such as erythrocytosis, or to unique adverse events not observed in young hypogonadal men. The baseline prevalence of disorders such as prostate cancer, benign prostatic hypertrophy, and cardiovascular disease that might be exacerbated by testosterone administration is high in older men; therefore, small changes in risk in either direction could have enormous public health impact. Furthermore, the clustering of co-morbid conditions in the frail elderly might render these men more susceptible to the adverse effects of testosterone therapy than healthy young hypogonadal men.
The contraindications for testosterone administration include history of prostate or breast cancer (16). Benign prostatic hypertrophy by itself is not a contraindication, unless it is associated with severe symptoms, as indicated by IPSS symptom score of greater than 21. Testosterone should not be given without prior evaluation and treatment to men with baseline hematocrit greater than 50%, severe untreated sleep apnea, or congestive heart failure with Class III or IV symptoms (16). Testosterone suppresses spermatogenesis and should not be prescribed to men who are considering having a child in the near future.
The risks of testosterone administration include acne, oiliness of skin, erythrocytosis, induction or exacerbation of sleep apnea, leg edema, transient breast tenderness or enlargement, and reversible suppression of baseline spermatogenesis (16) (Table 2). Abnormalities of liver enzymes, hepatic neoplasms, and peliosis hepatis that have been reported previously with orally administered, 17-alpha alkylated androgens, have not been observed with replacement doses of transdermal or injectable testosterone formulations. The two major areas of concern and uncertainty are the effects of long-term testosterone administration on prostate cancer and major adverse cardiovascular events.
Adapted with permission from the Endocrine Society Guideline for Testosterone Therapy in Men with Hypogonadism in: Bhasin et al J Clin Endocrinol Metab 2018;103(5):1715-1744.
The long-term consequences of testosterone supplementation on the risk of heart disease remain unknown and have been the subject of debate (145,413-417). Some known effects of testosterone such as increase in hematocrit, suppression of plasma HDL cholesterol, and salt and water retention, might be expected to increase cardiovascular risk. Some other effects such as testosterone’s vasodilator effect on coronary arteries resulting in increased coronary blood flow, reduction of whole body and abdominal fat mass, and improved brachial reactivity might be perceived as beneficial. Testosterone’s effects on coagulation are complex; testosterone administration is associated with stimulation of both anti-coagulant and pro-coagulant proteins.
Cross-sectional studies of middle-aged men found a positive relationship between serum testosterone levels and plasma HDL-cholesterol concentrations (415,418-421). Lower testosterone levels in men are associated with higher levels of dense LDL particles (418), triglycerides (421,422) and prothrombotic factors (423).
The effects of androgen supplementation on plasma lipids depend on the dose, the route of administration (oral or parenteral), the type of androgen (aromatizable or not), and the subject population (whether young or old, and hypogonadal or not). Supraphysiological doses of testosterone and non-aromatizable androgens frequently employed by bodybuilders undoubtedly decrease plasma HDL-cholesterol levels (424-427). However, administration of replacement doses of testosterone in older men has been associated with only a modest decrease or no change in plasma HDL-cholesterol (16,22,23,25,184,186-189,428-430), and without a significant effect on cholesterol efflux capacity from macrophages (431), suggesting preserved HDL function.
Cross-sectional studies have found a positive association between circulating testosterone concentrations and tissue plasminogen activator activity (432), and a negative relationship between testosterone and plasminogen activator inhibitor-1 activity, fibrinogen, and some other prothrombotic factors (432), suggesting an antithrombotic effect of testosterone. However, testosterone increases hematocrit (433), as well as neutrophil, monocyte and platelet counts (434). In men, higher neutrophil counts – even within the normal range - are associated with cardiovascular disease (435). Similarly, higher monocyte counts within the normal range have been suggested as a risk-factor for coronary artery plaque formation and cardiovascular mortality (436). Additionally, testosterone administration increases thromboxane A2 receptor density on human platelets, increasing platelet aggregability ex vivo (437,438). Observational studies have not found a consistent relationship between testosterone treatment and the risk of venous thromboembolism (439-443), although one study reported a small increase in VTE risk in the first few months after starting testosterone treatment (439).
Cross-sectional studies have reported conflicting findings on the association of endogenous testosterone levels and inflammatory markers (444-449). Intervention trials of testosterone generally have not found a significant effect of testosterone on inflammatory markers (430,450). Even supraphysiological doses of testosterone have been found not to affect C-reactive protein (451). Similarly, a prospective cohort study did not find meaningful changes in inflammatory markers in men with prostate cancer receiving androgen deprivation therapy (452).
Whether variation of testosterone within the normal range is associated with risk of coronary artery disease remains controversial. Of the 30 cross-sectional studies reviewed by Alexandersen (145), 18 reported lower testosterone levels in men with coronary heart disease, 11 found similar testosterone levels in controls and men with coronary artery disease and 1 found higher levels of DHEAS. Prospective studies have failed to reveal an association of total testosterone levels and coronary artery disease (146-150,453-455). The common carotid artery intimal media thickness, a marker of generalized atherosclerosis, is negatively associated with circulating testosterone levels (150).
In some animal models, orchiectomy accelerates and testosterone administration retards atherogenesis progression (466). The protective effect of testosterone on aortic atherogenesis in this preclinical model is mediated through its conversion to estradiol by the CYP19A1 in the blood vessel wall (466).
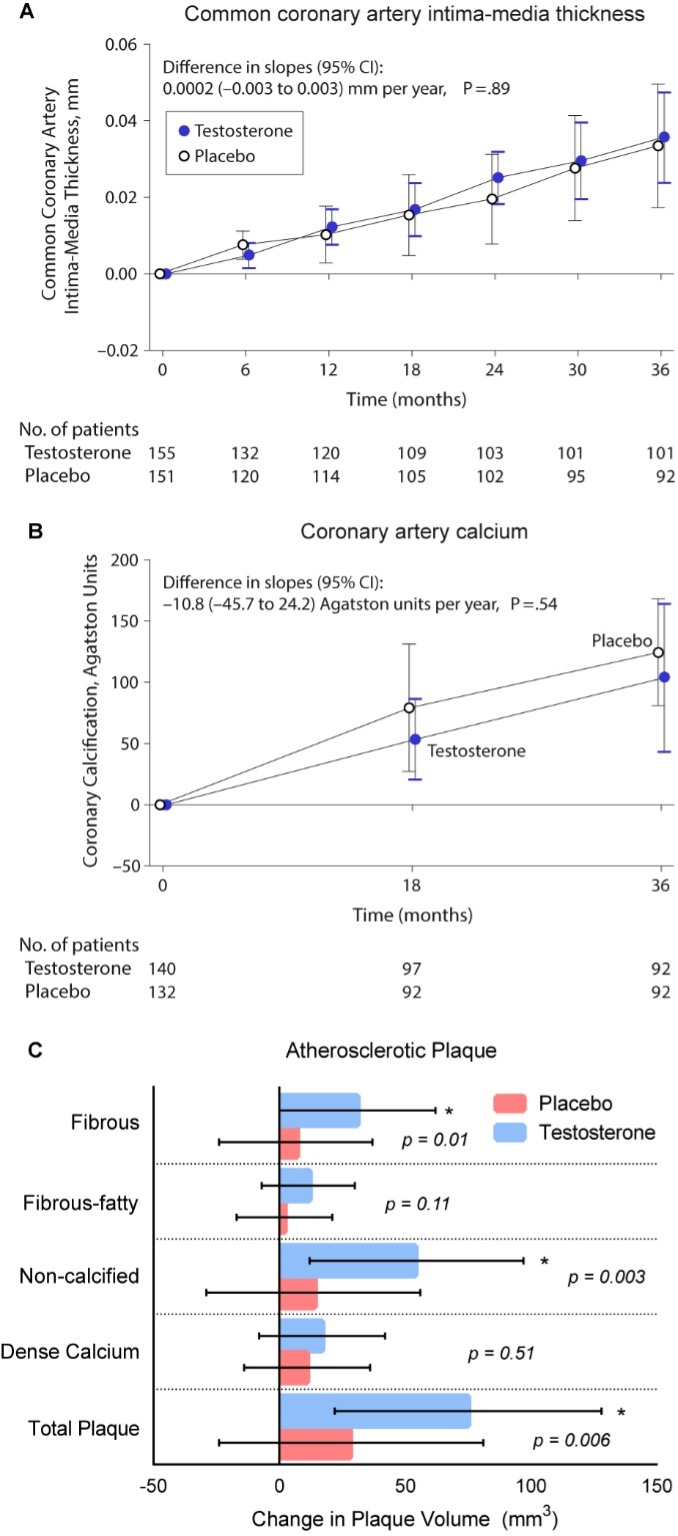
Two large placebo-controlled trials have evaluated the effects of testosterone treatment on atherogenesis progression in middle-aged and older men. The Testosterone’s Effects on Atherosclerosis Progression in Aging Men (TEAAM) Trial determined the effects of testosterone therapy on progression of subclinical atherosclerosis in the common carotid artery using sonographic measurement of common carotid artery intima-media thickness (CCA-IMT) and the coronary artery calcium scores measured using MDCT. The participants in the TEAAM Trial were 308 men, 60 years and older, with total testosterone between 100 and 400 ng/dL or free testosterone below 50 pg/mL (23). Men were randomized to receive either 75 mg of transdermal testosterone gel or placebo gel daily and received for 3 years. Neither the progression of CCA-IMT nor coronary artery calcium scores differed between the men randomized to the testosterone and placebo groups (Figure 12) (23).
In the cardiovascular trial of the TTrials, 138 men with serum total testosterone below 275 ng/dL received either testosterone gel or placebo gel for one year and were evaluated by coronary computed tomographic angiography for progression of non-calcified and calcified coronary artery plaque volume, as well as coronary artery calcium score (467). Consistent with the findings of the TEAAM Trial, the changes in coronary artery calcium scores did not differ between the testosterone and placebo groups over one year of intervention. However, the increase in non-calcified plaque volume (primary endpoint) was significantly greater in men assigned to the testosterone arm than in those assigned to placebo arm (Figure 12) (467); there were baseline differences in non-calcified plaque volume between the two groups. The clinical implications of these findings to cardiovascular risk remain to be established.
Cross-sectional studies have also linked low androgen levels in men to an increased risk of atrial fibrillation (482-484), and normalization of testosterone levels with testosterone replacement is associated with a decreased incidence of atrial fibrillation compared with untreated hypogonadal men (485). These findings need corroboration in randomized trials.
To-date, no randomized trials have been large enough or of sufficiently long duration to determine the effects of testosterone treatment on MACE (416). The frequency of MACE reported in randomized testosterone trials has been low—even lower than that expected for the age and comorbid conditions of the participants (18,486,487). A randomized trial of testosterone in older men (The TOM Trial) with mobility limitation was stopped early due to a higher frequency of cardiovascular-related events in men assigned to testosterone than in those assigned to placebo (22), heightening concern about the cardiovascular safety of testosterone in frail older men. In contrast to many other testosterone trials in older men, which recruited relatively healthy older men, the participants in the TOM trial had a high prevalence of chronic conditions, such as heart disease, diabetes mellitus, obesity, hypertension, and hyperlipidaemia (22). Men, 75 years of age or older, and men with high on-treatment testosterone levels seemed to be at the greatest risk of cardiovascular-related events. In secondary analyses, these events were found to be associated with changes in serum free testosterone and estradiol levels (488). The dose of testosterone used in the TOM trial was higher than that used in some previous trials, but not dissimilar from or lower than that used in some other trials. The cardiovascular events were small in number and of variable clinical significance. The TOM trial was not designed for cardiovascular events; therefore, the cardiovascular events were not a pre-specified endpoint, and were not collected in a standardized manner, nor adjudicated prospectively. Additionally, many of the cardiovascular events were not MACE.
The higher cardiovascular adverse event incidence in testosterone-treated older men observed in the TOM trial was not reproduced in two larger trials of longer duration published more recently; in the TEAAM trial, the incidence of major adverse cardiac events throughout the 3 years of intervention was similar between groups (23). Similarly, in the TTrials, the number of MACE (myocardial infarction, stroke or death related to cardiovascular disease) during the one year of treatment was similar in the two groups, with seven men in each group experiencing an event (26). The number of MACE in the TEAAM and Ttrials were too few to permit strong inferences on the effects of testosterone treatment on MACE.
The Hormonal Regulators of Muscle and Metabolism in Aging (HORMA) trial reported a significantly greater increase in blood pressure in men treated with testosterone than in those treated with placebo (489). Testosterone administration causes salt and water retention (490), which can induce edema and worsen pre-existing heart failure.
The long-term effects of testosterone replacement therapy on MACE remain unknown. The FDA conducted an extensive review and concluded “the studies...have significant limitations that weaken their evidentiary value for confirming a causal relationship between testosterone and adverse cardiovascular outcomes”. Nevertheless, the FDA directed the pharmaceutical companies to add in the drug label information about a possible increased risk of cardiovascular events with testosterone use. An independent review conducted by the European Medicines Agency also found no consistent evidence of an increased risk of coronary heart disease associated with testosterone treatment of hypogonadal men. Long-term randomized trials of the effects of testosterone replacement on MACE are needed and are particularly important because even small changes in incidence rates could have significant public health impact.
A large randomized, placebo-controlled trial to study the effects of testosterone replacement therapy on the incidence of major adverse cardiovascular events in men 45 to 80 years of age with low testosterone levels and one or more symptoms of testosterone deficiency, who are at increased risk for cardiovascular events is currently underway (The TRAVERSE Trial, NCT03518034). The intervention duration is up to 5 years in this trial of over 6,000 men. The efficacy outcomes include adjudicated clinical fractures, remission of low-grade persistent depressive disorder (dysthymia), progression from pre-diabetes to diabetes, correction of anemia, and overall sexual activity, sexual desire, and erectile function. This randomized, placebo-controlled trial offers an historical opportunity to advance our understanding of the cardiovascular safety and long-term efficacy of testosterone replacement in middle-aged and older hypogonadal men.
Testosterone levels are lower in men with type 2 diabetes mellitus compared with controls (505-510). Low total testosterone levels have been associated with lower insulin sensitivity (505,511) and increased risk of type 2 diabetes mellitus and metabolic syndrome in community dwelling men both cross-sectionally and longitudinally (508-510,512-520). However, the association of free testosterone and type 2 diabetes mellitus has been inconsistent; some studies have reported a weak relationship (509,510,512) while others have failed to find any relationship (508,514). Circulating sex hormone binding globulin (SHBG) and some SHBG polymorphisms also have been associated negatively with the risk of type 2 diabetes (508-510,512-516,521-524). For instance, individuals with the rs6257and rs179994 variant alleles of the SHBG single nucleotide polymorphism (SNP) have lower plasma SHBG levels and a higher risk of type 2 diabetes (521-524). Similarly, individuals with the rs6259 variant have higher SHBG levels and lower type 2 diabetes risk (524). We performed longitudinal analyses of men participating in the Massachusetts Male Aging Study (525), a population-based study of men aged 40-70 years (Figure 13) to evaluate whether SHBG is an independent predictor of T2DM (526). After adjustment for age, body mass index, hypertension, smoking, alcohol intake and physical activity, the hazard ratio for incident type 2 diabetes was 2.0 for each one SD decrease in SHBG and 1.29 for each one SD decrease in total testosterone (525). Free testosterone was not significantly associated with type 2 diabetes. The strong association of T2DM risk with SHBG persisted even after additional adjustment for free testosterone. The association of SHBG polymorphisms with type 2 diabetes suggests a potential role of SHBG in the pathogenesis of type 2 diabetes. In a Mendelian randomization analysis of the UK Biobank data, genetically determined free testosterone levels were associated with the risk of type 2 diabetes mellitus in a sexually dimorphic manner after adjusting for SHBG levels; men with low genetically determined free testosterone levels had increased risk of type 2 diabetes while women with low genetically determined free testosterone levels had reduced risk of type 2 diabetes (527).
There is no evidence that testosterone causes prostate cancer (539). A retrospective analysis of the Registry of Hypogonadism in Men (RHYME) (540) and several meta-analyses of randomized controlled trials (486,541,542) did not find an increased risk of prostate cancer in men receiving testosterone. Also, there is no consistent relationship between endogenous serum testosterone levels and the risk of prostate cancer (16,18,123,343,542-545). A meta-analyses of prospective cohort studies did not find a significant association between endogenous total testosterone levels and prostate cancer (542). Conversely, an analysis of 20 prospective studies found that men in the lowest tenth of free testosterone concentration had a lower risk of prostate cancer (OR=0.77, 95%CI= 0.69 to 0.86; p<0.001) compared with men with higher concentrations (545). Similarly, in the male participants in the UK Biobank followed for a mean of 6.9 years, higher genetically determined free testosterone was associated with a higher risk of prostate cancer, while total testosterone was not associated with prostate cancer risk (Figure 15) (546). The men with Klinefelter Syndrome have lower risk of prostate cancer than the general population. Taken together, these data suggest that life-long exposure to testosterone treatment in hypogonadal men could potentially increase the risk of prostate cancer.
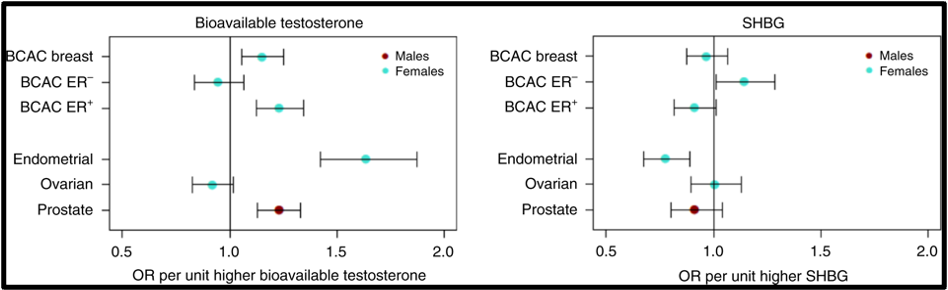
Figure15. Mendelian Randomization: Genetically Determined Bio-Testosterone Associated with Increased Prostate Cancer Risk. Legend: UK Biobank Study: Genetic determinants of bioavailable testosterone were positively associated with risk of prostate cancer in men and ER+ breast cancer and endometrial cancer in women. Reproduced with permission from: Ruth KS, et al. Nat Med. 2020;26(2):252‐258.
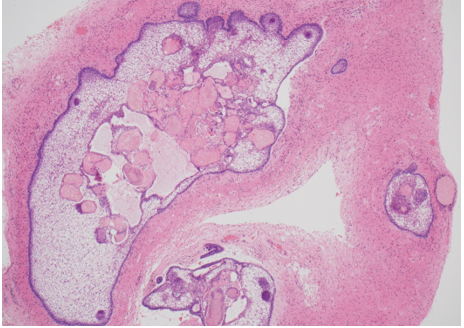
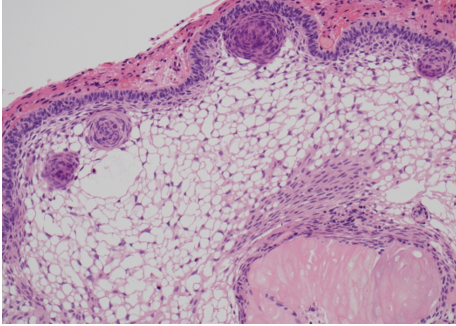
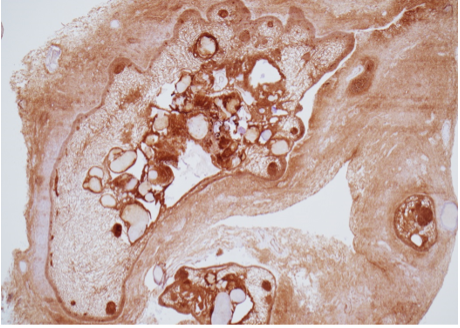
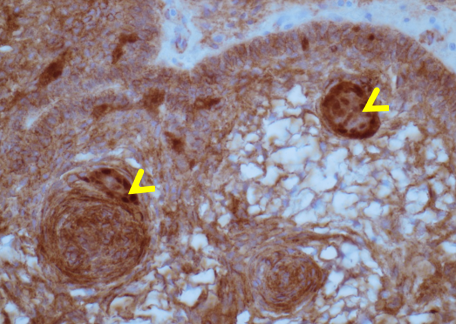
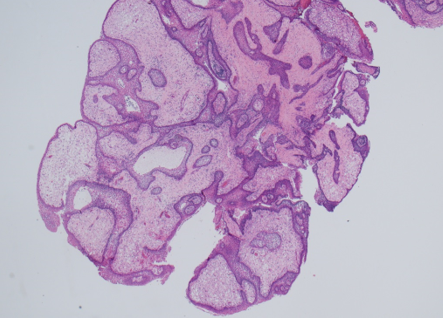
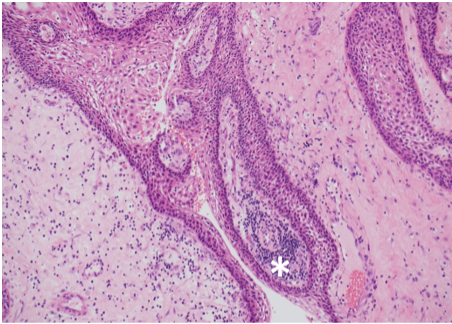
Prostate cancer is an androgen–dependent tumor, and testosterone treatment is known to promote the growth of metastatic prostate cancer (544,547). Testosterone administration has been historically contraindicated in men with history of prostate cancer (16,543). The prevalence of subclinical, microscopic foci of prostate cancer in older men is high (548-555). There is concern that testosterone administration might make these subclinical foci of cancer grow and become clinically overt. In addition, older men with low testosterone levels may have prostate cancer (556,557). Morgentaler et al (556,557) reported a high prevalence of biopsy-detectable prostate cancer in men with low total or free testosterone levels despite normal PSA levels and normal digital rectal examinations. However, this study did not have a control group, and we do not know whether sextant biopsies of age-matched controls with normal testosterone levels would yield a similarly high incidence of biopsy-detectable cancer. Therefore, this study should not be interpreted to conclude that there is a higher prevalence of prostate cancer in older men with low testosterone levels, or that low testosterone levels are an indication for performing prostate biopsy.
Effects of Testosterone Therapy on Prostate Events
None of the testosterone trials in middle-aged or older men had sufficient power or intervention duration to detect meaningful differences in the incidence of prostate cancer between testosterone and placebo-treated men. Testosterone treatment of hypogonadal men increases PSA levels (16,558), which may lead to urological referral for prostate biopsy. A systematic review of randomized testosterone trials in middle-aged and older men found (413) that men treated with testosterone in clinical trials were at significantly higher risk for undergoing prostate biopsy than placebo-treated men (413). Because of the high prevalence of subclinical prostate cancer in older men, the higher number of prostate biopsies in testosterone-treated men could lead to increased detection rates of subclinical prostate cancer in comparison with placebo-treated men. Thus, testosterone therapy of middle-aged and older men is associated with a higher risk of prostate biopsy and a bias towards detection of a higher number of prostate events (18,413).
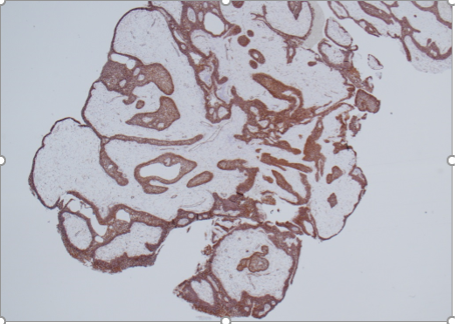
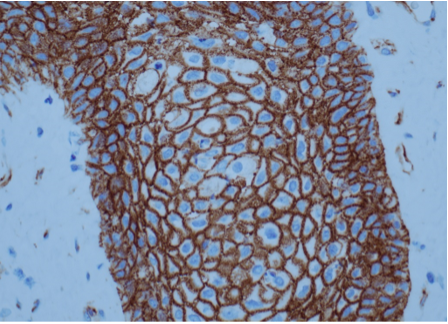
Administration of exogenous testosterone or suppression of circulating levels of testosterone by administration of a GnRH antagonist is not associated with proportionate changes in intra-prostatic testosterone or DHT concentrations. For instance, in a randomized controlled trial, Marks et al (559) measured intraprostatic testosterone and DHT levels in older men treated with placebo or testosterone. Surprisingly, intraprostatic DHT concentrations were not significantly higher in testosterone-treated men than in placebo-treated men (559). Similarly, the expression levels of androgen-dependent genes in the prostate were not significantly altered by testosterone administration (559). In separate studies, lowering of circulating testosterone levels by administration of a GnRH antagonist was not associated with changes in intraprostatic androgen concentrations (560,561).
Effects of Testosterone Replacement on Serum PSA Levels
Serum PSA levels are lower in androgen–deficient men and are restored to normal following testosterone replacement (16,558,562-570). Lowering of serum testosterone concentrations by withdrawal of androgen therapy in young, hypogonadal men is associated with a decrease in serum PSA levels. Similarly, treatment of men with benign prostatic hyperplasia with a 5-alpha reductase inhibitor, finasteride, is associated with a significant lowering of serum and prostatic PSA levels (570,571). However, serum PSA levels do not increase progressively in healthy hypogonadal men with replacement doses of testosterone. The increase in PSA levels during testosterone replacement might trigger evaluation and biopsy in some patients (16,543).
More intensive PSA screening and follow-up of men receiving testosterone replacement might lead to an increased number of prostate biopsies and the detection of subclinical prostate cancers that would have otherwise remained undetected (16,543). Serum PSA levels tend to fluctuate when measured repeatedly in the same individual over time (572-574). There is considerable test-retest variability in PSA measurements (572-574). Some of this variability is due to the inherent assay variability, and a significant portion of this variability is due to unknown factors. Fluctuations are larger in men with high mean PSA levels. Variability can be even greater if measurements are performed in different laboratories that use dissimilar assay methodology (572-574).
An important issue is what increment in PSA level should warrant a prostate biopsy in older men receiving testosterone replacement. To address this issue, we conducted a systematic review of published studies of testosterone replacement in hypogonadal men (543). This review indicated that the weighted effect size of the change in PSA after testosterone replacement in young, hypogonadal men is 0.68 standard deviation units (95% confidence interval 0.55 to 0.82). This means that the effect of testosterone replacement therapy is to increase PSA levels by an average 0.68 standard deviations over baseline. Because the average standard deviation was 0.47 in this systematic analysis, the standard deviation score of 0.68 translates into an average increase in serum PSA levels of about 0.30 ng/ml in young hypogonadal men (543). The average change in serum PSA levels after testosterone replacement in studies of older men was 0.43 ng/mL (543). The data from the Proscar Long-Term Efficacy and Safety Study (PLESS) demonstrated that the 90% confidence interval for the change in PSA values measured 3 to 6 months apart is 1.4 ng/mL (570). Therefore, a change in PSA of >1.4 ng/ml between any two values measured 3 to 6 month apart in the same patient is unusual (16,543). In the TTrials, 2.4% of men receiving testosterone had increases above 1.4 ng/mL at 3 months, and 4.7% at 12 months (26).
Carter et al, based on the analysis of PSA data from the Baltimore Longitudinal Study of Aging, reported that PSA velocity, defined as the annual rate of change of PSA, is different in men who develop prostate cancer than in those who do not (575-577). Thus, PSA velocity greater than 0.7 ng/ml/year was unusual in men without prostate cancer whose baseline PSA was between 4 and 10 ng/ml (575-577). However, most men being considered for testosterone replacement will have baseline PSA less than 4 ng/ml. In a subsequent analysis, the same group reported that the PSA velocity in men with baseline PSA between 2 and 4 ng/ml was 0.2 ng/ml/year (577). Because test-to-retest variability in PSA measurement is far greater than this threshold, it is likely that the use of this threshold of 0.2 ng/ml/year to select men for prostate biopsy would lead to many unnecessary biopsies.
In eugonadal, young men, administration of supraphysiological doses of testosterone does not further increase serum PSA levels (166,169,578). These data are consistent with dose response studies in young men that demonstrate that maximal serum concentrations of PSA are achieved at testosterone levels that are at the lower end of the normal male range; higher testosterone concentrations are not associated with higher PSA levels (166,169).
In summary, these data suggest that the administration of replacement doses of testosterone to androgen-deficient men can be expected to produce a modest increment in serum PSA levels. Increments in PSA levels after testosterone supplementation in androgen-deficient men are generally less than 0.5 ng/mL and increments in excess of 1.4 ng/mL over a 3–6-month period are unusual. Nevertheless, administration of testosterone to men with baseline PSA levels between 2.6 and 4.0 ng/mL will cause PSA levels to exceed 4.0 ng/mL in some men. Increments in PSA levels above 4 ng/mL will trigger a urological consultation and many of these men will be asked to undergo prostate biopsies. However, considering the controversy over prostate cancer screening and monitoring, the decision to monitor PSA levels during testosterone treatment and the decision to refer a patient for consideration of prostate biopsy should be made only after informing him of the risks and benefits of prostate cancer screening and monitoring and engaging the patient in a shared decision-making process.
Monitoring PSA Levels in Older Men Receiving Testosterone Replacement (Tables 3 and 4)
Older men considering testosterone supplementation should undergo evaluation of risk factors for prostate cancer; the Endocrine Society guideline suggest a baseline PSA measurement and a digital prostate examination (16). Prostate cancer screening has some risks; therefore, initiation of prostate monitoring should be a shared decision, made only after a discussion of the risks and benefits of prostate cancer monitoring. Men with history of prostate cancer, should not be given androgen supplementation and those with palpable abnormalities of the prostate or PSA levels greater than 3 ng/ml should undergo urological evaluation. After initiation of testosterone replacement therapy, PSA levels should be repeated at 3 months and annually thereafter (16). Although measurements of free PSA and PSA density have been proposed to enhance the specificity of PSA measurement, long term data, especially from studies of testosterone replacement in older men, are lacking. Considering the interassay variability and the longitudinal change in PSA previously discussed, an Endocrine Society Expert Panel recently suggested that men receiving testosterone replacement should be referred to urological consultation if: 1) PSA increases more than 1.4 ng/mL in the first 12 months of treatment; 2) a PSA above 4 ng/mL is confirmed; or 3) a prostatic abnormality is detected on digital rectal examination (16). After 12 months of treatment, prostate monitoring should follow standard guidelines for prostate cancer screening taking into account the age and race of the patient (16).
|
Table 3. Recommendations for Monitoring of Men Receiving Testosterone Therapy
|
|
A. Explain the potential benefits and risks of monitoring for prostate cancer and engage the patient in shared decision making regarding the prostate monitoring plan.
|
|
B. Evaluate the patient at 3–12 months after treatment initiation and then annually to assess whether symptoms have responded to treatment and whether the patient is suffering from any adverse effects
|
|
C. Monitor testosterone concentrations 3–6 months after initiation of therapy:
· --Therapy should aim to raise testosterone into the mid-normal range.
· --Injectable testosterone enanthate or cypionate: measure testosterone midway between injections. If midinterval T is >600 ng/dL (24.5 nmol/L) or <350 ng/dL (14.1 nmol/L), adjust dose or frequency.
· --Transdermal gels: assess testosterone 2–8 h following the gel application, after the patient has been on treatment for at least 1 week; adjust dose to achieve testosterone in the mid-normal range.
· --Transdermal patches: assess testosterone 3–12 h after application; adjust dose to achieve concentration in the mid-normal range.
· --Buccal T bioadhesive tablet: assess concentrations immediately before or after application of fresh system.
· --Testosterone pellets: measure concentrations at the end of the dosing interval. Adjust the number of pellets and/or the dosing interval to maintain serum T concentrations in the mid-normal range.
· --Oral T undecanoate: monitor serum T concentrations 3–5h after ingestion with a fat-containing meal.
· --Injectable testosterone undecanoate: measure serum T levels at the end of the dosing interval just prior to the next injection and aim to achieve nadir levels in low-mid range.
|
|
D. Check hematocrit at baseline, 3–6 months after starting treatment, and then annually. If hematocrit is >54%, stop therapy until hematocrit decreases to a safe level; evaluate the patient for hypoxia and sleep apnea; reinitiate therapy with a reduced dose.
|
|
E. Measure BMD of lumbar spine and/or femoral neck after 1–2 year of testosterone therapy in hypogonadal men with osteoporosis, consistent with regional standard of care.
|
|
F. For men 55–69 years of age and for men 40–69 years of age who are at increased risk for prostate cancer who choose prostate monitoring, perform digital rectal examination and check PSA level before initiating treatment; check PSA and perform digital rectal examination 3–12 months after initiating testosterone treatment, and then in accordance with guidelines for prostate cancer screening depending on the age and race of the patient.
|
|
G. Obtain urological consultation if there is:
· An increase in serum PSA concentration.1.4 ng/mL within 12 months of initiating testosterone treatment
· A confirmed PSA > 4 ng/mL at any time
· Detection of a prostatic abnormality on digital rectal examination
· Substantial worsening of lower urinary tract symptoms
|
Adapted with permission from the Endocrine Society Guideline for Testosterone Therapy in Men with Hypogonadism in: Bhasin et al J Clin Endocrinol Metab 2018;103(5):1715-1744.
|
Table 4. Indications for Urological Consultation in Men Receiving Testosterone Replacement
|
|
1. 1) An increase in serum or plasma PSA concentration >1.4 ng/mL within any 12-month period after initiating testosterone treatment
|
|
2. 2) A PSA >4.0 ng/mL
|
|
3. 3) Detection of a prostatic abnormality on digital rectal examination
|
|
4. 4) An AUA/IPSS prostate symptom score of >19
|
Adapted with permission from the Endocrine Society Guideline for Testosterone Therapy in Men with Hypogonadism in: Bhasin et al J Clin Endocrinol Metab 2018;103(5):1715-1744.
Testosterone and Benign Prostatic Hypertrophy
Testosterone replacement can be administered safely to men with benign prostatic hypertrophy who have mild to moderate symptom scores. The severity of symptoms associated with benign prostatic hypertrophy can be assessed by using either the International Prostate Symptom Score (IPSS) or the American Urological Association (AUA) Symptom questionnaires. Androgen deficiency is associated with decreased prostate volume and androgen replacement increases prostate volume compared to age–matched controls (559,562,566,567). Meta-analyses of testosterone trials have not found statistically significant difference in lower urinary tract symptoms scores in hypogonadal men receiving testosterone replacement compared to placebo (Figure 16) (256,579). However, in patients with pre–existing, severe symptoms of benign prostatic hypertrophy, even small increases in prostate volume during testosterone administration may exacerbate obstructive symptoms. In these men, testosterone should either not be administered or administered with careful monitoring of obstructive symptoms.
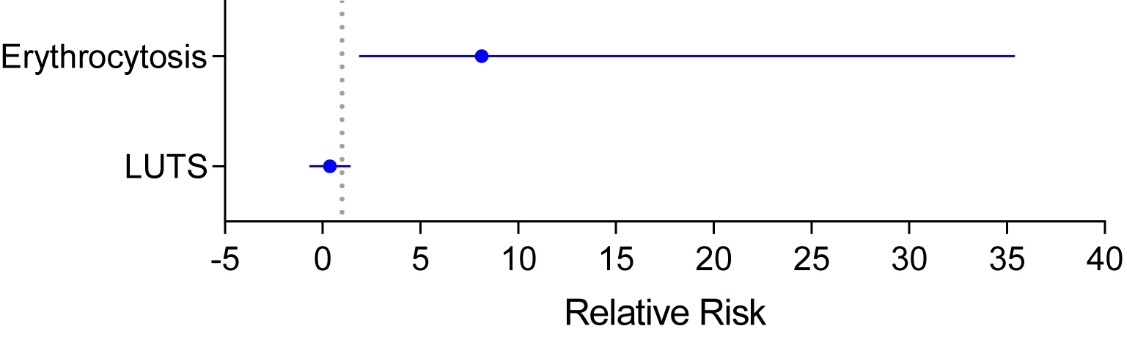
Figure 16. Adverse events associated with testosterone therapy in randomized trials. The relative risk and 95% CI for development of erythrocytosis (RR= 8.14; 95%CI= 1.87 to 35.40) and lower urinary tract symptoms (LUTS; RR= 0.38; 95%CI= -0.67 to 1.43) in randomized testosterone trials derived from meta-analyses published by Ponce et al., 2018 are shown. The figure was adapted with permission from Ponce et al. J Clin Endocrinol Metab. 2018;103(5):1745-54.
ERYTHROCYTOSIS
Testosterone replacement is associated with increased red cell mass and hemoglobin levels (Figure 16) (256,329,580-585). Therefore, testosterone replacement should not be administered to men with baseline hematocrit of 52% or greater without appropriate evaluation and treatment of erythrocytosis (16) (Table 3). Administration of testosterone to androgen–deficient young men is typically associated with a small increase in hemoglobin levels. Clinically significant erythrocytosis is uncommon in young hypogonadal men during testosterone replacement therapy, but can occur in men with sleep apnea, significant smoking history, or chronic obstructive lung disease. Testosterone administration in older men is associated with greater increments in hemoglobin than observed in young, hypogonadal men (321). The magnitude of hemoglobin increase during testosterone therapy appears related to the testosterone dose, the increase in testosterone concentrations during testosterone therapy, and age (321). Testosterone replacement by means of a transdermal system has been reported to produce a lesser increase in hemoglobin levels than that associated with intramuscular testosterone enanthate and cypionate presumably because of the substantially higher testosterone dose and average circulating testosterone levels achieved with testosterone esters (586).
Testosterone increases hemoglobin and hematocrit by multiple mechanisms (322,408,409,587). Testosterone administration stimulates iron-dependent erythropoiesis by suppressing hepcidin transcription and increasing iron availability for erythropoiesis (322,408,409,587). Additionally, testosterone stimulates erythropoiesis by a direct effect on bone marrow hematopoietic progenitors and increasing the numbers of myeloid progenitors. Testosterone also stimulates erythropoietin and alters the set-point of the relationship between erythropoietin and hemoglobin (322). Testosterone supplementation can correct anemia in older men with unexplained anemia of aging and anemia of inflammation (322,329,409). Suppression of testosterone secretion in men receiving androgen deprivation therapy reduces hematocrit and hemoglobin levels by slowing erythropoiesis independently of changes in erythropoietin levels (588).
Monitoring Hematocrit During Testosterone Replacement Therapy (Table 3)
Hematocrit levels should be measured at baseline and 3 months after institution of testosterone replacement or after increase in dosage, and every 12 months thereafter. It is not clear what absolute hematocrit level or magnitude of change in hematocrit warrants discontinuation of testosterone administration. Plasma viscosity increases disproportionately as hematocrit rises above 50%. Hematocrit levels above 54% may be associated with increased risk of neuro-occlusive events. Therefore, testosterone dose should be withheld if hematocrit rises above 54%; once hematocrit falls to a safe level, testosterone therapy may be re-initiated at a reduced dose or with a different formulation (16).
SLEEP APNEA
Circulating testosterone concentrations are related to sleep rhythm and are generally higher during sleep than during waking hours (589-592). Testosterone secretory peaks coincide with the onset of rapid-eye movement sleep. Aging is associated with decreased sleep efficiency, reduced numbers of REM sleep episodes, and altered REM sleep latency, which may contribute to lower circulating testosterone concentrations (590-594). The degree of sleep-disordered breathing increases with age and is associated with reduced overnight plasma bioavailable testosterone. Thus, changes in sleep efficiency and architecture are associated with alterations in testosterone levels in older men (590-594). Sleep apnea and disordered sleep are often associated with low testosterone levels (595), particularly in patients with more severe cases of OSA (i.e. severe hypoxemia) (596). Some potential mechanisms by which OSA may decrease endogenous testosterone levels include disruption of pulsatile luteinizing hormone secretion from restricted sleep and/or recurrent nocturnal hypoxia (597,598), which is further exacerbated by obesity. OSA treatment with continuous positive airway pressure has been demonstrated to increase serum testosterone levels (599).
Testosterone can induce or exacerbate sleep apnea in some individuals, particularly those with obesity or chronic obstructive lung disease (589-594,600). This appears to be due to direct effects of testosterone on laryngeal muscles. Testosterone administration depresses hypercapnic ventilator drive and induces apnea in primate infants (594). Short-term administration of high doses of testosterone shortens sleep duration and worsens sleep apnea in older men (601). The frequency of sleep apnea in randomized testosterone trials in older men has been very low (16,486) and no randomized trial has reported an increased incidence of OSA or OSA worsening in men randomized to the testosterone arm compared to the placebo arm.
Testosterone should not be given to men with severe untreated OSA without evaluation and treatment of sleep apnea. Several screening instruments can be used to detect sleep apnea. A history of loud snoring, and daytime somnolence, in an obese individual with hypertension increases the likelihood of having sleep apnea; such patients should be referred for a sleep study.
BREAST ENLARGEMENT AND TENDERNESS
Testosterone administration can induce breast tenderness; however, gynecomastia is an uncommon complication of testosterone replacement therapy. Even with administration of supraphysiological doses of testosterone enanthate, less than 4% of men in a contraceptive trial developed detectable breast enlargement (580). Breast cancer is listed as a contraindication for testosterone replacement therapy primarily because of concern that increased estrogen levels during testosterone treatment might exacerbate breast cancer growth. There are, however, few case reports of breast cancer occurring as a complication of testosterone treatment. Men with Klinefelter’s syndrome have a higher risk of breast cancer than the general population (602).
An Individualized, Patient-Centric Approach to Shared Decision Making in the Evaluation and Treatment of Older Men with Low Testosterone Levels
Recent large randomized clinical trials, especially the TTrials, have substantially expanded our understanding of the efficacy and short-term safety of testosterone in older men with low testosterone levels. However, none of the trials has been long enough or large enough to determine the effects of testosterone treatment on major adverse cardiovascular events and prostate cancer risk. Furthermore, the long-term efficacy of testosterone treatment in improving hard outcomes – physical disability, fractures, falls, progression to dementia, progression from prediabetes to diabetes, remission of late-life low grade persistent depressive disorder (dysthymia) - remains to be established. Adherence with testosterone treatment is poor and in one survey, nearly 50% of men prescribed testosterone, discontinued treatment within 3 months. Population level screening of all older men for androgen deficiency is not justified (16) because of the lack of agreement on a case definition, the paucity of data on the performance characteristics of the screening instruments (e.g., the ADAM questionnaire (603), the Aging Male Symptoms questionnaire (604), and the MMAS questionnaire (605)) and the lack of clarity on the public health impact of the androgen deficiency syndrome in the general population.
Recognizing the lack of evidence of the long-term safety and efficacy of testosterone therapy in older men with symptomatic androgen deficiency, the expert panel of the Endocrine Society recommended against testosterone therapy of all men 65 years or older with low testosterone levels (16). Instead, the panel suggested that “in men >65 years who have symptoms or conditions suggestive of testosterone deficiency (such as low libido or unexplained anemia) and consistently and unequivocally low morning testosterone, clinicians offer testosterone therapy on an individualized basis after explicit discussion of the potential risks and benefits” (16).
The decision to offer testosterone treatment to older men with low testosterone levels should be guided by an individualized assessment of potential benefits and risks (Figure 17) (606). Determine whether the patient has clear evidence of testosterone deficiency recognizing the imprecision and inaccuracy of many available immunoassays and the substantial overlap in the symptoms of hypogonadism and aging per se. Perform a careful general health evaluation to identify conditions, such as prostate cancer, erythrocytosis, heart failure, or a hypercoagulable state that could increase the risk of harm. Weigh the burden of patient's symptoms and conditions associated with testosterone deficiency against the potential benefits and the uncertainty of long-term harm. Evaluate prostate cancer risk recognizing that prostate cancer screening and monitoring has some risks. Weigh the bother and distress associated with symptoms of testosterone deficiency and patient's values and risk tolerance against the uncertainty of benefits and long-term risks, and the burden, cost, and risks of treatment and monitoring. The participation of the patient who is well informed of the potential benefits and risks in the shared decision to initiate testosterone treatment can enable a more thoughtful treatment plan and increased adherence with the treatment and monitoring (606).
Figure 17. An evidence-based, individualized approach to testosterone therapy in older men with testosterone deficiency. The decision to offer testosterone treatment to older men with low testosterone levels should be guided by an individualized assessment of potential benefits and risks. Testosterone deficiency needs to be evaluated using reliable assays for the measurement of total and free testosterone levels. Patients should also be evaluated for conditions that are likely to respond to testosterone replacement therapy (TRT) as well as conditions that could be adversely impacted, such as prostate cancer, erythrocytosis, heart failure, or a hypercoagulable state. It is important to consider each patient’s burden of symptoms, individual preferences, and risk tolerance against the uncertainty of long-term benefits and risks, the burden and risks of monitoring, and the cost. Reproduced with permission from Bhasin S. 2021. J Clin Invest. 2021;131(4):e146607.
Testosterone therapy can be instituted using any of the available approved formulations based on considerations of pharmacokinetics, patient convenience and preference, cost, and formulation-specific adverse effects (16). The men receiving testosterone therapy should be monitored using a standardized monitoring plan to facilitate early detection of adverse events and to minimize the risk of unnecessary prostate biopsies (Table 2), as recommended by the Endocrine Society expert panel (Table 3) (16).
CHANGES IN THE SPERMATOGENIC COMPARTMENT OF THE TESTIS
Women are more fertile below the age of 40, and fecundity decrease after age 35 and fertility ceases at the inception of menopause, around age 50. Increasing age in women confers greater risk for infertility, spontaneous abortion, and genetic and chromosomal defects among offspring. In contrast, there is no critical age at which sperm production or function, and fertility cease in men (607-614). Although serum testosterone concentrations decrease below the normal range in a significant minority of older men, men over the age of 60 years commonly father children; the oldest father on record was 94-years old (607,609). Even though many older men are fertile, the overall fertility and fecundity decline with aging. The interpretability of data on the effects of aging on male fertility is limited by the small size of the studies and the low overall event rates.
Paternal age is associated with an increased risk of germ line mutations in FGFR2, FGFR3, and RET genes and inherited autosomal dominant diseases, such as Apert's syndrome, achondroplasia, and Costello Syndrome, respectively, in the offspring of older men (614-623). These monogenic disorders have been referred to as paternal age effect (PAE) disorders. Approximately one third of babies with diseases due to new autosomal dominant mutations are fathered by men aged 40 years or older (624).
Some other disorders such as schizophrenia, autism, and bipolar disorder have also been linked to paternal age (Figure 18) (615,616,622,623). The rate of de novo mutations increases with paternal age (622), which may contribute to the increase risk of neurodevelopmental diseases such as schizophrenia and autism (622).
Figure 18. Impact of paternal age on incidence of schizophrenia and early-onset bipolar disorder. Increasing paternal age at conception increases the relative risk of having an offspring with schizophrenia (panel A; figure adapted from Malaspina et al. Arch Gen Psychiatry. 2001 Apr;58(4):361-7.) and the odds ratio of having a child with early-onset bipolar disorder (compared to fathers aged 20 to 24 years; panel B; data derived from Frans et al. Arch Gen Psychiatry. 2008 Sep;65(9):1034-40)
The accumulation of these de novo germ line mutations with increasing paternal age has been explained by the “selfish spermatogonial selection" hypothesis (618,619). According to this hypothesis, the somatic mutations in male germ cells that enhance the proliferation of germ cells could lead to within-testis expansion of mutant clonal lines (620,621), thus favoring the propagation of germ cells carrying these pathogenic mutations, and increasing the risk of mutations in the offspring of older fathers (620,621). Interestingly, the risk of autism has also been associated with the age of the father as well as the grandparent (623). These concerns have prompted the American Society of Reproductive Medicine to state in their guidelines that semen donors should be younger than 40 years of age so that potential hazards related to aging are diminished (610).
Some cardiac defects have also been attributed to aberrant genetic input from older men. For instance, a case-control study of 4,110 individuals with congenital heart defects born between 1952 and 1973 in British Columbia, found a general pattern of increasing risk with increasing paternal age among cases relative to controls for ventricular septal defects, atrial septal defects, and patent ductus arteriosus (617,624). The risk of schizophrenia has also been reported to increase with paternal age (618) and possible loci affecting this risk have been identified (625). In addition, a modest proportion of preeclampsia, normally associated with increased maternal risk factors including age, might be attributable to an increase in paternal age although no gene loci have been identified (626). These observations need further corroboration.
Although there is a positive association between paternal age and incidence of aneuploidy, it has been difficult to dissociate the effect of paternal age from the confounding influence of the advanced maternal age. After accounting for various confounders, there does not appear to be a major independent effect of increased paternal age on the incidence of autosomal aneuploidies (608,609,615,616,627,628). The existence of a paternal age effect on Down syndrome is controversial. Earlier studies from the 1960s and 1970s found no correlation between Down syndrome and paternal age (e.g.,(629)). However, a study in New York from 1983 to 1997 found a significant greater numbers of mothers and fathers 35 years of age and older, respectively, among parents of patients with Down’s syndrome (630). Among the cases of Down syndrome evaluated, paternal age had a significant effect only when the mothers were 35 years of age or older, and was the highest when both the Mother and Father were older than 40 years in which case the risk of Down Syndrome was 6 times that observed among couples younger than 35 years of age (630).
Changes in Fertility of Older Men
A review of studies examining fertility at different ages demonstrated significant age-related differences in fertility rates in men; men older than 50 have lower pregnancy rates, increased time to pregnancy, and subfecundity compared to younger men (608,609,615,631,632). Some changes in fertility rates might be related to age-related decrease in sexual activity. A literature review found no significant change in sperm concentration with aging when comparing men under the age of 30 to those greater than 50 years (613). However, in general, semen volume, sperm motility, and the number of morphologically normal sperm decrease with advancing age (Table 5; (45,608-614,622,627,628,631,633)). A number of these studies, however, did not control for important confounding variables. Of the 21 studies in which sperm densities were compared among men of different age groups (613), only four studies adjusted for the duration of abstinence, well known to affect sperm concentration. In addition, there is significant heterogeneity in the populations studied; most of the studies examined data from semen of sperm donors while others examined men from infertility clinics. Sperm donors might represent a healthier group of men than the general population; conversely men in infertility clinics might be more likely to have abnormalities of sperm number or function. Even studies that have controlled for abstinence as well as alcohol and tobacco use have shown an age-related decrease in semen volume. In one study of men whose partners had bilateral tubal obstruction or absence of both tubes and who were treated by conventional IVF, the odds ratio of failure to conceive was higher for men 40 years of age or older (634).
|
Table 5: Changes in Semen Quality and Fertility in Men with Age
|
|
Parameter
|
Age comparison
|
Change
|
|
Semen volume
|
30 versus ³50 years
|
3-22% decrease
|
|
Sperm concentration
|
Varying
|
None
|
|
Abnormal sperm morphology
|
£30 versus ³ 50 years
|
4-18% increase
|
|
Time to pregnancy
|
<30-35 versus >30-50 years
|
5-20% increase
|
|
Pregnancy rates
|
<30 versus > 50 years
|
23-38% decrease
|
|
Subfecundity
|
Varying
|
11-250% increase
|
From a Literature Review by Kidd et al., 2001 (501)
CHANGES IN THE GERM CELL COMPARTMENT
In a comparison of younger men (21-25 years) with older men (>50) referred for andrological evaluation, the ejaculate volume, progressive sperm motility, and sperm morphology were lower in older men than younger men after adjustment for duration of sexual abstinence (635). The older men also had a higher frequency of sperm tail defects, suggesting epididymal dysfunction (636). In addition, the fructose content was significantly lower in older men suggesting a defect in the seminal vesicle contribution to semen. There were no significant differences in sperm concentration and testicular size between the young and older men in this study.
Necropsies on adult men of different ages have revealed that the testicular volume is lower only in men in the 8th decade of life (637). A recent study examined testicular germ cells obtained by orchiectomy from 36 older men with advanced prostate cancer and by testicular biopsy from 21 younger men with obstructive azoospermia, as controls (638). The ratios of primary spermatocytes, round spermatids, and elongated spermatids to Sertoli cells were significantly decreased in the testes of older men, but the ratio of spermatogonia to Sertoli cell number remained unchanged (638,639). Older men are characterized by lower rates of germ cell apoptosis and cell proliferation compared with younger men, suggesting that germ cell proliferation and apoptosis diminish with aging (639).
Other studies evaluating the fidelity of the germ cell compartment are cross-sectional and depend on analyses of sperm number and semen quality; large-scale chromosomal analyses in healthy community dwelling men are scarce as most data are derived from fertility clinics. A review of studies examining semen quality at different ages demonstrated significant age-related decrease in semen volume and sperm morphology. The change in sperm morphology has been hypothesized to be due to an increase in aneuploidy with age. Härkönen et al (628) found that sperm morphology was directly associated with the number of chromosomes in sperm and that men with higher aneuploidy rates for chromosomes 13, 18, 21, X and Y had lower sperm motility and sperm concentrations. Despite the changes in sperm morphology and motility from older men, in vitro fertilizing capacity of the sperm is well preserved (45,634).
There are several difficulties in interpreting these data on age-related changes in sperm density and function. The normal range for sperm concentration in men is wide where sperm concentration above 15 million/ml (total sperm per ejaculate > 39 million) is considered normal. Thus, even though average sperm concentrations decline with aging, they are still are in the normal range (45,632,638). Furthermore, normal sperm counts do not always correlate with normal sperm function.
Studies in flies demonstrate more germ cells during larval than adult stages suggesting age-related quiescence of the germ line (640). Significant age-related decreases in germ cells and spermatogenesis also have been reported in rodents and primates (641-645). The Brown Norway rat has been studied as a model of aging of the human male reproductive system because in this rodent model, serum testosterone levels decrease with aging, as they do in humans (642-644). Along with changes in hypothalamic-pituitary hormones, alterations in sperm counts, sperm maturation, Sertoli cell number, and progeny outcomes have been observed in this rodent model (Table 6) (626,642-651). Analysis of ribosomal DNA from germ cells of the male brown Norway rat has revealed hypermethylation of ribosomal DNA (645,652). Alterations in ribosomes have been theorized to promote aging of cells by multiplying errors in protein synthesis which initially might elude gross morphological analysis but eventually might lead to germ cell degeneration (652). Further assessment of spermatogonial stem cell populations is needed. In many animal models of life span extension, there is a trade-off between longer life and fecundity, although there are some exceptions (653).
|
Table 6. Changes in the Reproductive Axis in the Brown-Norway Rat
|
|
Parameter
|
Change
|
Reference
|
|
GnRH
|
¯
|
530, 531
|
|
FSH
|
|
530, 531
|
|
LH
|
®
|
530, 531
|
|
Testosterone
|
¯
|
530-532, 534
|
|
Germ Cells
|
¯
|
535
|
|
Sertoli Cells
|
¯
|
531, 537
|
|
Spermatogenesis
|
¯
|
531, 537
|
|
Seminiferous Tubules
|
¯
|
531, 537
|
|
Seminiferous Tubule Function
|
altered
|
534, 537
|
|
Epididymal function
|
¯
|
538
|
|
Sperm morphology
|
altered
|
538
|
|
Sperm motility
|
¯
|
538
|
|
Sperm Count
|
¯
|
532
|
CHANGES IN SUPPORTIVE CELLS AND ACCESSORY GLANDS
Since Sertoli and Leydig cells are crucial to spermatogenesis, changes in these cells could affect sperm number and function. Age-related changes in the supporting structures for sperm maturation have been described in the Brown Norway rat. These changes include reductions in the numbers of Leydig and Sertoli cells (643-645). Changes in the supporting cells and structures for sperm maturation have been invoked to explain the age-related decrease in sperm number and fecundity in rats. In stallions, the numbers of Sertoli cells decreases with aging but individual Sertoli cells display a remarkable capacity to accommodate greater numbers of developing germ cells (654).
In men, Sertoli cell number has been reported to be lower in men aged 50 to 85 years than in men aged 20 to 48 years (655). The apoptotic rate of primary spermatocytes in aged men was also significantly elevated compared with that of younger men, resulting in a decrease of the number of primary spermatocytes per Sertoli cell (639), leading the authors to suggest that there might be a failure of the Sertoli cells to support spermatogenesis in older men.
Sertoli cells produce inhibin, which regulates gonadotropin expression from the pituitary. Inhibin B has been identified as the physiologically important form of inhibin in men and as a valuable serum marker of Sertoli cell function and spermatogenesis. Higher gonadotropins and lower inhibin levels in older men suggest a decline in Sertoli cell function (655); however changes in circulating inhibin B levels with advancing age have been inconsistent (70,655-657). Overall, these data suggest a possible decline in Sertoli cell number and function in older.
Aging is accompanied by a progressive, albeit variable, decline of Leydig cell function with a decrease of mean serum free (or bioavailable) testosterone levels in the population between age 25 and 75 years (658). Total Leydig cell volume and the absolute number of Leydig cells decline with advancing age, although total testis weight does not change substantially with age (658-662). In one study, age accounted for more than a third of the variation in Leydig cell number, and explained more than half the variation in daily sperm production (661). This might in part be explained by a fusion of Leydig cells resulting in fewer but multinucleated Leydig cells with age (662). The functionality of the multinucleated cells is not known.
REFERENCES
1. Croft DP, Brent LJ, Franks DW, Cant MA. The evolution of prolonged life after reproduction. Trends Ecol Evol 2015; 30:407-416
2. Bhasin S, Kerr C, Oktay K, Racowsky C. The Implications of Reproductive Aging for the Health, Vitality and Economic Welfare of Human Societies. J Clin Endocrinol Metab 2019;
3. Stone L. Declining fertility in America. American Enterprise Institute. 2018; https://www.aei.org/wp-content/uploads/2018/12/Declining-Fertility-in-America.pdf. Accessed June 24, 2021.
4. Schmidt L, Sobotka T, Bentzen JG, Nyboe Andersen A, Reproduction E, Society Task F. Demographic and medical consequences of the postponement of parenthood. Hum Reprod Update 2012; 18:29-43
5. CDC. Birth Data. 2019; https://www.cdc.gov/nchs/nvss/births.htm. Accessed June 24, 2021.
6. Live Births and Birth Rates, by Year. https://www.infoplease.com/us/population/live-births-and-birth-rates-year. Accessed June 24, 2021.
7. Mathews TJ, Hamilton BE. Mean age of mothers is on the rise: United States, 2000–2014. NCHS data brief, no 232. Hyattsville, MD: NCHS; 2016.
8. Menken J, Trussell J, Larsen U. Age and infertility. Science 1986; 233:1389-1394
9. Goetzinger KR, Shanks AL, Odibo AO, Macones GA, Cahill AG. Advanced Maternal Age and the Risk of Major Congenital Anomalies. Am J Perinatol 2017; 34:217-222
10. American Society for Reproductive M. Guidelines for sperm donation. Fertil Steril 2004; 82 Suppl 1:S9-12
11. Brandt JS, Cruz Ithier MA, Rosen T, Ashkinadze E. Advanced paternal age, infertility, and reproductive risks: A review of the literature. Prenat Diagn 2019; 39:81-87
12. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing Group for the Women's Health Initiative I. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA 2002; 288:321-333
13. Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998; 280:605-613
14. Harman SM, Naftolin F, Brinton EA, Judelson DR. Is the estrogen controversy over? Deconstructing the Women's Health Initiative study: a critical evaluation of the evidence. Ann N Y Acad Sci 2005; 1052:43-56
15. Barrett-Connor E. Hormones and heart disease in women: the timing hypothesis. Am J Epidemiol 2007; 166:506-510
16. Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2018; 103:1715-1744
17. Petak SM, Nankin HR, Spark RF, Swerdloff RS, Rodriguez-Rigau LJ, American Association of Clinical E. American Association of Clinical Endocrinologists Medical Guidelines for clinical practice for the evaluation and treatment of hypogonadism in adult male patients--2002 update. Endocr Pract 2002; 8:440-456
18. Spitzer M, Huang G, Basaria S, Travison TG, Bhasin S. Risks and benefits of testosterone therapy in older men. Nat Rev Endocrinol 2013; 9:414-424
19. Wang C, Nieschlag E, Swerdloff R, Behre HM, Hellstrom WJ, Gooren LJ, Kaufman JM, Legros JJ, Lunenfeld B, Morales A, Morley JE, Schulman C, Thompson IM, Weidner W, Wu FC, International Society of A, International Society for the Study of Aging M, European Association of U, European Academy of A, American Society of A. Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. J Androl 2009; 30:1-9
20. Matsumoto AM. Andropause: clinical implications of the decline in serum testosterone levels with aging in men. J Gerontol A Biol Sci Med Sci 2002; 57:M76-99
21. Bhasin S, Buckwalter JG. Testosterone supplementation in older men: a rational idea whose time has not yet come. J Androl 2001; 22:718-731
22. Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette AM, Eder R, Tennstedt S, Ulloor J, Zhang A, Choong K, Lakshman KM, Mazer NA, Miciek R, Krasnoff J, Elmi A, Knapp PE, Brooks B, Appleman E, Aggarwal S, Bhasin G, Hede-Brierley L, Bhatia A, Collins L, LeBrasseur N, Fiore LD, Bhasin S. Adverse events associated with testosterone administration. N Engl J Med 2010; 363:109-122
23. Basaria S, Harman SM, Travison TG, Hodis H, Tsitouras P, Budoff M, Pencina KM, Vita J, Dzekov C, Mazer NA, Coviello AD, Knapp PE, Hally K, Pinjic E, Yan M, Storer TW, Bhasin S. Effects of Testosterone Administration for 3 Years on Subclinical Atherosclerosis Progression in Older Men With Low or Low-Normal Testosterone Levels: A Randomized Clinical Trial. JAMA 2015; 314:570-581
24. Brock G, Heiselman D, Maggi M, Kim SW, Rodriguez Vallejo JM, Behre HM, McGettigan J, Dowsett SA, Hayes RP, Knorr J, Ni X, Kinchen K. Effect of Testosterone Solution 2% on Testosterone Concentration, Sex Drive and Energy in Hypogonadal Men: Results of a Placebo Controlled Study. J Urol 2016; 195:699-705
25. Emmelot-Vonk MH, Verhaar HJ, Nakhai Pour HR, Aleman A, Lock TM, Bosch JL, Grobbee DE, van der Schouw YT. Effect of testosterone supplementation on functional mobility, cognition, and other parameters in older men: a randomized controlled trial. JAMA 2008; 299:39-52
26. Snyder PJ, Bhasin S, Cunningham GR, Matsumoto AM, Stephens-Shields AJ, Cauley JA, Gill TM, Barrett-Connor E, Swerdloff RS, Wang C, Ensrud KE, Lewis CE, Farrar JT, Cella D, Rosen RC, Pahor M, Crandall JP, Molitch ME, Cifelli D, Dougar D, Fluharty L, Resnick SM, Storer TW, Anton S, Basaria S, Diem SJ, Hou X, Mohler ER, 3rd, Parsons JK, Wenger NK, Zeldow B, Landis JR, Ellenberg SS, Testosterone Trials I. Effects of Testosterone Treatment in Older Men. N Engl J Med 2016; 374:611-624
27. Snyder PJ, Bhasin S, Cunningham GR, Matsumoto AM, Stephens-Shields AJ, Cauley JA, Gill TM, Barrett-Connor E, Swerdloff RS, Wang C, Ensrud KE, Lewis CE, Farrar JT, Cella D, Rosen RC, Pahor M, Crandall JP, Molitch ME, Resnick SM, Budoff M, Mohler ER, 3rd, Wenger NK, Cohen HJ, Schrier S, Keaveny TM, Kopperdahl D, Lee D, Cifelli D, Ellenberg SS. Lessons From the Testosterone Trials. Endocr Rev 2018; 39:369-386
28. Srinivas-Shankar U, Roberts SA, Connolly MJ, O'Connell MD, Adams JE, Oldham JA, Wu FC. Effects of testosterone on muscle strength, physical function, body composition, and quality of life in intermediate-frail and frail elderly men: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab 2010; 95:639-650
29. Shi Z, Araujo AB, Martin S, O'Loughlin P, Wittert GA. Longitudinal changes in testosterone over five years in community-dwelling men. J Clin Endocrinol Metab 2013; 98:3289-3297
30. Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710-717
31. Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR, Baltimore Longitudinal Study of A. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. Baltimore Longitudinal Study of Aging. J Clin Endocrinol Metab 2001; 86:724-731
32. Feldman HA, Longcope C, Derby CA, Johannes CB, Araujo AB, Coviello AD, Bremner WJ, McKinlay JB. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab 2002; 87:589-598
33. Morley JE, Kaiser FE, Perry HM, 3rd, Patrick P, Morley PM, Stauber PM, Vellas B, Baumgartner RN, Garry PJ. Longitudinal changes in testosterone, luteinizing hormone, and follicle-stimulating hormone in healthy older men. Metabolism 1997; 46:410-413
34. Zmuda JM, Cauley JA, Kriska A, Glynn NW, Gutai JP, Kuller LH. Longitudinal relation between endogenous testosterone and cardiovascular disease risk factors in middle-aged men. A 13-year follow-up of former Multiple Risk Factor Intervention Trial participants. Am J Epidemiol 1997; 146:609-617
35. Gray A, Feldman HA, McKinlay JB, Longcope C. Age, disease, and changing sex hormone levels in middle-aged men: results of the Massachusetts Male Aging Study. J Clin Endocrinol Metab 1991; 73:1016-1025
36. Simon D, Preziosi P, Barrett-Connor E, Roger M, Saint-Paul M, Nahoul K, Papoz L. The influence of aging on plasma sex hormones in men: the Telecom Study. Am J Epidemiol 1992; 135:783-791
37. Ferrini RL, Barrett-Connor E. Sex hormones and age: a cross-sectional study of testosterone and estradiol and their bioavailable fractions in community-dwelling men. Am J Epidemiol 1998; 147:750-754
38. Dai WS, Kuller LH, LaPorte RE, Gutai JP, Falvo-Gerard L, Caggiula A. The epidemiology of plasma testosterone levels in middle-aged men. Am J Epidemiol 1981; 114:804-816
39. Pollanen P, Harkonen K, Makinen J, Irjala K, Saad F, Hubler D, Oettel M, Koskenuo M, Huhtaniemi I. Facts and fictions of andropause: lessons from the Turku male Ageing Study. Paper presented at: 1st International Congress on Male Health 2001
40. Wu FCW. 2003 Subclinical hypogonadism. Endocrine Society Meeting; 2003; Philadelphia, PA.
41. Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278-1281
42. Harman SM, Tsitouras PD. Reproductive hormones in aging men. I. Measurement of sex steroids, basal luteinizing hormone, and Leydig cell response to human chorionic gonadotropin. J Clin Endocrinol Metab 1980; 51:35-40
43. Harman SM, Tsitouras PD, Costa PT, Blackman MR. Reproductive hormones in aging men. II. Basal pituitary gonadotropins and gonadotropin responses to luteinizing hormone-releasing hormone. J Clin Endocrinol Metab 1982; 54:547-551
44. Murono EP, Nankin HR, Lin T, Osterman J. The aging Leydig cell: VI. Response of testosterone precursors to gonadotrophin in men. Acta Endocrinol (Copenh) 1982; 100:455-461
45. Nieschlag E, Lammers U, Freischem CW, Langer K, Wickings EJ. Reproductive functions in young fathers and grandfathers. J Clin Endocrinol Metab 1982; 55:676-681
46. Tenover JS, Matsumoto AM, Plymate SR, Bremner WJ. The effects of aging in normal men on bioavailable testosterone and luteinizing hormone secretion: response to clomiphene citrate. J Clin Endocrinol Metab 1987; 65:1118-1126
47. Wu FC, Tajar A, Beynon JM, Pye SR, Silman AJ, Finn JD, O'Neill TW, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Kula K, Lean ME, Pendleton N, Punab M, Boonen S, Vanderschueren D, Labrie F, Huhtaniemi IT, Group E. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123-135
48. Bhasin S, Pencina M, Jasuja GK, Travison TG, Coviello A, Orwoll E, Wang PY, Nielson C, Wu F, Tajar A, Labrie F, Vesper H, Zhang A, Ulloor J, Singh R, D'Agostino R, Vasan RS. Reference ranges for testosterone in men generated using liquid chromatography tandem mass spectrometry in a community-based sample of healthy nonobese young men in the Framingham Heart Study and applied to three geographically distinct cohorts. J Clin Endocrinol Metab 2011; 96:2430-2439
49. In: Liverman CT, Blazer DG, eds. Testosterone and Aging: Clinical Research Directions. Washington (DC)2004.
50. Orwoll E, Lambert LC, Marshall LM, Phipps K, Blank J, Barrett-Connor E, Cauley J, Ensrud K, Cummings S. Testosterone and estradiol among older men. J Clin Endocrinol Metab 2006; 91:1336-1344
51. Zumoff B, Strain GW, Kream J, O'Connor J, Rosenfeld RS, Levin J, Fukushima DK. Age variation of the 24-hour mean plasma concentrations of androgens, estrogens, and gonadotropins in normal adult men. J Clin Endocrinol Metab 1982; 54:534-538
52. Pirke KM, Doerr P. Age related changes and interrelationships between plasma testosterone, oestradiol and testosterone-binding globulin in normal adult males. Acta Endocrinol (Copenh) 1973; 74:792-800
53. Mohr BA, Bhasin S, Link CL, O'Donnell AB, McKinlay JB. The effect of changes in adiposity on testosterone levels in older men: longitudinal results from the Massachusetts Male Aging Study. Eur J Endocrinol 2006; 155:443-452
54. Wu FC, Tajar A, Pye SR, Silman AJ, Finn JD, O'Neill TW, Bartfai G, Casanueva F, Forti G, Giwercman A, Huhtaniemi IT, Kula K, Punab M, Boonen S, Vanderschueren D, European Male Aging Study G. Hypothalamic-pituitary-testicular axis disruptions in older men are differentially linked to age and modifiable risk factors: the European Male Aging Study. J Clin Endocrinol Metab 2008; 93:2737-2745
55. Sartorius G, Spasevska S, Idan A, Turner L, Forbes E, Zamojska A, Allan CA, Ly LP, Conway AJ, McLachlan RI, Handelsman DJ. Serum testosterone, dihydrotestosterone and estradiol concentrations in older men self-reporting very good health: the healthy man study. Clin Endocrinol (Oxf) 2012; 77:755-763
56. Camacho EM, Huhtaniemi IT, O'Neill TW, Finn JD, Pye SR, Lee DM, Tajar A, Bartfai G, Boonen S, Casanueva FF, Forti G, Giwercman A, Han TS, Kula K, Keevil B, Lean ME, Pendleton N, Punab M, Vanderschueren D, Wu FC, Group E. Age-associated changes in hypothalamic-pituitary-testicular function in middle-aged and older men are modified by weight change and lifestyle factors: longitudinal results from the European Male Ageing Study. Eur J Endocrinol 2013; 168:445-455
57. Tajar A, Forti G, O'Neill TW, Lee DM, Silman AJ, Finn JD, Bartfai G, Boonen S, Casanueva FF, Giwercman A, Han TS, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Vanderschueren D, Huhtaniemi IT, Wu FC, Group E. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 2010; 95:1810-1818
58. McKinlay JB, Longcope C, Gray A. The questionable physiologic and epidemiologic basis for a male climacteric syndrome: preliminary results from the Massachusetts Male Aging Study. Maturitas 1989; 11:103-115
59. Tajar A, Huhtaniemi IT, O'Neill TW, Finn JD, Pye SR, Lee DM, Bartfai G, Boonen S, Casanueva FF, Forti G, Giwercman A, Han TS, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Vanderschueren D, Wu FC, Group E. Characteristics of androgen deficiency in late-onset hypogonadism: results from the European Male Aging Study (EMAS). J Clin Endocrinol Metab 2012; 97:1508-1516
60. Ohlsson C, Wallaschofski H, Lunetta KL, Stolk L, Perry JR, Koster A, Petersen AK, Eriksson J, Lehtimaki T, Huhtaniemi IT, Hammond GL, Maggio M, Coviello AD, Group ES, Ferrucci L, Heier M, Hofman A, Holliday KL, Jansson JO, Kahonen M, Karasik D, Karlsson MK, Kiel DP, Liu Y, Ljunggren O, Lorentzon M, Lyytikainen LP, Meitinger T, Mellstrom D, Melzer D, Miljkovic I, Nauck M, Nilsson M, Penninx B, Pye SR, Vasan RS, Reincke M, Rivadeneira F, Tajar A, Teumer A, Uitterlinden AG, Ulloor J, Viikari J, Volker U, Volzke H, Wichmann HE, Wu TS, Zhuang WV, Ziv E, Wu FC, Raitakari O, Eriksson A, Bidlingmaier M, Harris TB, Murray A, de Jong FH, Murabito JM, Bhasin S, Vandenput L, Haring R. Genetic determinants of serum testosterone concentrations in men. PLoS Genet 2011; 7:e1002313
61. Coviello AD, Lakshman K, Mazer NA, Bhasin S. Differences in the apparent metabolic clearance rate of testosterone in young and older men with gonadotropin suppression receiving graded doses of testosterone. J Clin Endocrinol Metab 2006; 91:4669-4675
62. Ishimaru T, Pages L, Horton R. Altered metabolism of androgens in elderly men with benign prostatic hyperplasia. J Clin Endocrinol Metab 1977; 45:695-701
63. Mulligan T, Iranmanesh A, Johnson ML, Straume M, Veldhuis JD. Aging alters feed-forward and feedback linkages between LH and testosterone in healthy men. Am J Physiol 1997; 273:R1407-1413
64. Winters SJ, Sherins RJ, Troen P. The gonadotropin-suppressive activity of androgen is increased in elderly men. Metabolism 1984; 33:1052-1059
65. Urban RJ, Veldhuis JD, Blizzard RM, Dufau ML. Attenuated release of biologically active luteinizing hormone in healthy aging men. J Clin Invest 1988; 81:1020-1029
66. Zwart AD, Urban RJ, Odell WD, Veldhuis JD. Contrasts in the gonadotropin-releasing hormone dose-response relationships for luteinizing hormone, follicle-stimulating hormone and alpha-subunit release in young versus older men: appraisal with high-specificity immunoradiometric assay and deconvolution analysis. Eur J Endocrinol 1996; 135:399-406
67. Gruenewald DA, Naai MA, Marck BT, Matsumoto AM. Age-related decrease in hypothalamic gonadotropin-releasing hormone (GnRH) gene expression, but not pituitary responsiveness to GnRH, in the male Brown Norway rat. J Androl 2000; 21:72-84
68. Bonavera JJ, Swerdloff RS, Sinha Hakim AP, Lue YH, Wang C. Aging results in attenuated gonadotropin releasing hormone-luteinizing hormone axis responsiveness to glutamate receptor agonist N-methyl-D-aspartate. J Neuroendocrinol 1998; 10:93-99
69. Deslypere JP, Kaufman JM, Vermeulen T, Vogelaers D, Vandalem JL, Vermeulen A. Influence of age on pulsatile luteinizing hormone release and responsiveness of the gonadotrophs to sex hormone feedback in men. J Clin Endocrinol Metab 1987; 64:68-73
70. Mahmoud AM, Goemaere S, De Bacquer D, Comhaire FH, Kaufman JM. Serum inhibin B levels in community-dwelling elderly men. Clin Endocrinol (Oxf) 2000; 53:141-147
71. Pincus SM, Mulligan T, Iranmanesh A, Gheorghiu S, Godschalk M, Veldhuis JD. Older males secrete luteinizing hormone and testosterone more irregularly, and jointly more asynchronously, than younger males. Proc Natl Acad Sci U S A 1996; 93:14100-14105
72. Keenan DM, Veldhuis JD. Disruption of the hypothalamic luteinizing hormone pulsing mechanism in aging men. Am J Physiol Regul Integr Comp Physiol 2001; 281:R1917-1924
73. Eendebak R, Ahern T, Swiecicka A, Pye SR, O'Neill TW, Bartfai G, Casanueva FF, Maggi M, Forti G, Giwercman A, Han TS, Slowikowska-Hilczer J, Lean MEJ, Punab M, Pendleton N, Keevil BG, Vanderschueren D, Rutter MK, Tampubolon G, Goodacre R, Huhtaniemi IT, Wu FCW, Group E. Elevated luteinizing hormone despite normal testosterone levels in older men-natural history, risk factors and clinical features. Clin Endocrinol (Oxf) 2018; 88:479-490
74. Morales A, Lunenfeld B, International Society for the Study of the Aging M. Investigation, treatment and monitoring of late-onset hypogonadism in males. Official recommendations of ISSAM. International Society for the Study of the Aging Male. Aging Male 2002; 5:74-86
75. Society E. Proceedings of the Andropause Consensus Panel. 2002
76. Bhasin S, Tenover JS. Age-associated sarcopenia--issues in the use of testosterone as an anabolic agent in older men. J Clin Endocrinol Metab 1997; 82:1659-1660
77. Perry HM, 3rd, Miller DK, Patrick P, Morley JE. Testosterone and leptin in older African-American men: relationship to age, strength, function, and season. Metabolism 2000; 49:1085-1091
78. Melton LJ, 3rd, Khosla S, Riggs BL. Epidemiology of sarcopenia. Mayo Clin Proc 2000; 75 Suppl:S10-12; discussion S12-13
79. Baumgartner RN, Koehler KM, Gallagher D, Romero L, Heymsfield SB, Ross RR, Garry PJ, Lindeman RD. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147:755-763
80. Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ. Predictors of skeletal muscle mass in elderly men and women. Mech Ageing Dev 1999; 107:123-136
81. Workshop on sarcopenia: muscle atrophy in old age. Airlie, Virginia, September 19-21, 1994. Proceedings. J Gerontol A Biol Sci Med Sci 1995; 50 Spec No:1-161
82. Dutta C, Hadley EC. The significance of sarcopenia in old age. J Gerontol A Biol Sci Med Sci 1995; 50 Spec No:1-4
83. Kohrt WM, Malley MT, Dalsky GP, Holloszy JO. Body composition of healthy sedentary and trained, young and older men and women. Med Sci Sports Exerc 1992; 24:832-837
84. American Medical Association white paper on elderly health. Report of the Council on Scientific Affairs. Arch Intern Med 1990; 150:2459-2472
85. Borkan GA, Hults DE, Gerzof SG, Robbins AH, Silbert CK. Age changes in body composition revealed by computed tomography. J Gerontol 1983; 38:673-677
86. Borkan GA, Norris AH. Fat redistribution and the changing body dimensions of the adult male. Hum Biol 1977; 49:495-513
87. Cohn SH, Vartsky D, Yasumura S, Sawitsky A, Zanzi I, Vaswani A, Ellis KJ. Compartmental body composition based on total-body nitrogen, potassium, and calcium. Am J Physiol 1980; 239:E524-530
88. Novak LP. Aging, total body potassium, fat-free mass, and cell mass in males and females between ages 18 and 85 years. J Gerontol 1972; 27:438-443
89. Schoeller DA. Changes in total body water with age. Am J Clin Nutr 1989; 50:1176-1181; discussion 1231-1175
90. Evans WJ. Effects of exercise on body composition and functional capacity of the elderly. J Gerontol A Biol Sci Med Sci 1995; 50 Spec No:147-150
91. Fleg JL, Lakatta EG. Role of muscle loss in the age-associated reduction in VO2 max. J Appl Physiol (1985) 1988; 65:1147-1151
92. Proctor DN, Balagopal P, Nair KS. Age-related sarcopenia in humans is associated with reduced synthetic rates of specific muscle proteins. J Nutr 1998; 128:351S-355S
93. Lexell J, Henriksson-Larsen K, Winblad B, Sjostrom M. Distribution of different fiber types in human skeletal muscles: effects of aging studied in whole muscle cross sections. Muscle Nerve 1983; 6:588-595
94. Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci 1995; 50 Spec No:11-16
95. Frontera WR, Hughes VA, Lutz KJ, Evans WJ. A cross-sectional study of muscle strength and mass in 45- to 78-yr-old men and women. J Appl Physiol (1985) 1991; 71:644-650
96. Harris T. Muscle mass and strength: relation to function in population studies. J Nutr 1997; 127:1004S-1006S
97. Skelton DA, Greig CA, Davies JM, Young A. Strength, power and related functional ability of healthy people aged 65-89 years. Age Ageing 1994; 23:371-377
98. Roy TA, Blackman MR, Harman SM, Tobin JD, Schrager M, Metter EJ. Interrelationships of serum testosterone and free testosterone index with FFM and strength in aging men. Am J Physiol Endocrinol Metab 2002; 283:E284-294
99. O'Donnell AB, Travison TG, Harris SS, Tenover JL, McKinlay JB. Testosterone, dehydroepiandrosterone, and physical performance in older men: results from the Massachusetts Male Aging Study. J Clin Endocrinol Metab 2006; 91:425-431
100. Mohr BA, Bhasin S, Kupelian V, Araujo AB, O'Donnell AB, McKinlay JB. Testosterone, sex hormone-binding globulin, and frailty in older men. J Am Geriatr Soc 2007; 55:548-555
101. van den Beld AW, de Jong FH, Grobbee DE, Pols HA, Lamberts SW. Measures of bioavailable serum testosterone and estradiol and their relationships with muscle strength, bone density, and body composition in elderly men. J Clin Endocrinol Metab 2000; 85:3276-3282
102. Schaap LA, Pluijm SM, Smit JH, van Schoor NM, Visser M, Gooren LJ, Lips P. The association of sex hormone levels with poor mobility, low muscle strength and incidence of falls among older men and women. Clin Endocrinol (Oxf) 2005; 63:152-160
103. Orwoll E, Lambert LC, Marshall LM, Blank J, Barrett-Connor E, Cauley J, Ensrud K, Cummings SR, Osteoporotic Fractures in Men Study G. Endogenous testosterone levels, physical performance, and fall risk in older men. Arch Intern Med 2006; 166:2124-2131
104. Krasnoff JB, Basaria S, Pencina MJ, Jasuja GK, Vasan RS, Ulloor J, Zhang A, Coviello A, Kelly-Hayes M, D'Agostino RB, Wolf PA, Bhasin S, Murabito JM. Free testosterone levels are associated with mobility limitation and physical performance in community-dwelling men: the Framingham Offspring Study. J Clin Endocrinol Metab 2010; 95:2790-2799
105. Hyde Z, Flicker L, Almeida OP, Hankey GJ, McCaul KA, Chubb SA, Yeap BB. Low free testosterone predicts frailty in older men: the health in men study. J Clin Endocrinol Metab 2010; 95:3165-3172
106. Travison TG, Nguyen AH, Naganathan V, Stanaway FF, Blyth FM, Cumming RG, Le Couteur DG, Sambrook PN, Handelsman DJ. Changes in reproductive hormone concentrations predict the prevalence and progression of the frailty syndrome in older men: the concord health and ageing in men project. J Clin Endocrinol Metab 2011; 96:2464-2474
107. Cawthon PM, Ensrud KE, Laughlin GA, Cauley JA, Dam TT, Barrett-Connor E, Fink HA, Hoffman AR, Lau E, Lane NE, Stefanick ML, Cummings SR, Orwoll ES, Osteoporotic Fractures in Men Research G. Sex hormones and frailty in older men: the osteoporotic fractures in men (MrOS) study. J Clin Endocrinol Metab 2009; 94:3806-3815
108. Travison TG, Morley JE, Araujo AB, O'Donnell AB, McKinlay JB. The relationship between libido and testosterone levels in aging men. J Clin Endocrinol Metab 2006; 91:2509-2513
109. Zitzmann M, Faber S, Nieschlag E. Association of specific symptoms and metabolic risks with serum testosterone in older men. J Clin Endocrinol Metab 2006; 91:4335-4343
110. Marberger M, Roehrborn CG, Marks LS, Wilson T, Rittmaster RS. Relationship among serum testosterone, sexual function, and response to treatment in men receiving dutasteride for benign prostatic hyperplasia. J Clin Endocrinol Metab 2006; 91:1323-1328
111. Araujo AB, Esche GR, Kupelian V, O'Donnell AB, Travison TG, Williams RE, Clark RV, McKinlay JB. Prevalence of symptomatic androgen deficiency in men. J Clin Endocrinol Metab 2007; 92:4241-4247
112. Kaiser FE, Viosca SP, Morley JE, Mooradian AD, Davis SS, Korenman SG. Impotence and aging: clinical and hormonal factors. J Am Geriatr Soc 1988; 36:511-519
113. Korenman SG, Morley JE, Mooradian AD, Davis SS, Kaiser FE, Silver AJ, Viosca SP, Garza D. Secondary hypogonadism in older men: its relation to impotence. J Clin Endocrinol Metab 1990; 71:963-969
114. Cunningham GR, Stephens-Shields AJ, Rosen RC, Wang C, Ellenberg SS, Matsumoto AM, Bhasin S, Molitch ME, Farrar JT, Cella D, Barrett-Connor E, Cauley JA, Cifelli D, Crandall JP, Ensrud KE, Fluharty L, Gill TM, Lewis CE, Pahor M, Resnick SM, Storer TW, Swerdloff RS, Anton S, Basaria S, Diem S, Tabatabaie V, Hou X, Snyder PJ. Association of sex hormones with sexual function, vitality, and physical function of symptomatic older men with low testosterone levels at baseline in the testosterone trials. J Clin Endocrinol Metab 2015; 100:1146-1155
115. O'Connor DB, Lee DM, Corona G, Forti G, Tajar A, O'Neill TW, Pendleton N, Bartfai G, Boonen S, Casanueva FF, Finn JD, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Labrie F, Lean ME, Punab M, Silman AJ, Vanderschueren D, Wu FC, European Male Ageing Study G. The relationships between sex hormones and sexual function in middle-aged and older European men. J Clin Endocrinol Metab 2011; 96:E1577-1587
116. Dodge HH, Du Y, Saxton JA, Ganguli M. Cognitive domains and trajectories of functional independence in nondemented elderly persons. J Gerontol A Biol Sci Med Sci 2006; 61:1330-1337
117. Liu-Ambrose T, Pang MY, Eng JJ. Executive function is independently associated with performances of balance and mobility in community-dwelling older adults after mild stroke: implications for falls prevention. Cerebrovasc Dis 2007; 23:203-210
118. Janowsky JS, Oviatt SK, Orwoll ES. Testosterone influences spatial cognition in older men. Behav Neurosci 1994; 108:325-332
119. Janowsky JS. The role of androgens in cognition and brain aging in men. Neuroscience 2006; 138:1015-1020
120. Fales CL, Knowlton BJ, Holyoak KJ, Geschwind DH, Swerdloff RS, Gonzalo IG. Working memory and relational reasoning in Klinefelter syndrome. J Int Neuropsychol Soc 2003; 9:839-846
121. Alexander GM, Swerdloff RS, Wang C, Davidson T, McDonald V, Steiner B, Hines M. Androgen-behavior correlations in hypogonadal men and eugonadal men. II. Cognitive abilities. Horm Behav 1998; 33:85-94
122. Barrett-Connor E, Goodman-Gruen D, Patay B. Endogenous sex hormones and cognitive function in older men. J Clin Endocrinol Metab 1999; 84:3681-3685
123. Gouchie C, Kimura D. The relationship between testosterone levels and cognitive ability patterns. Psychoneuroendocrinology 1991; 16:323-334
124. Shute VJ, Pellegrino JW, Hubert L, Reynolds RW. The relationship between androgen levels and human spatial abilities. Bulletin of the Psychonomic Society 2013; 21:465-468
125. Margolese HC. The male menopause and mood: testosterone decline and depression in the aging male--is there a link? J Geriatr Psychiatry Neurol 2000; 13:93-101
126. Barrett-Connor E, Von Muhlen DG, Kritz-Silverstein D. Bioavailable testosterone and depressed mood in older men: the Rancho Bernardo Study. J Clin Endocrinol Metab 1999; 84:573-577
127. van Honk J, Tuiten A, Verbaten R, van den Hout M, Koppeschaar H, Thijssen J, de Haan E. Correlations among salivary testosterone, mood, and selective attention to threat in humans. Horm Behav 1999; 36:17-24
128. Seidman SN, Araujo AB, Roose SP, McKinlay JB. Testosterone level, androgen receptor polymorphism, and depressive symptoms in middle-aged men. Biol Psychiatry 2001; 50:371-376
129. Seidman SN, Araujo AB, Roose SP, Devanand DP, Xie S, Cooper TB, McKinlay JB. Low testosterone levels in elderly men with dysthymic disorder. Am J Psychiatry 2002; 159:456-459
130. Markianos M, Tripodianakis J, Sarantidis D, Hatzimanolis J. Plasma testosterone and dehydroepiandrosterone sulfate in male and female patients with dysthymic disorder. J Affect Disord 2007; 101:255-258
131. Fink HA, Ewing SK, Ensrud KE, Barrett-Connor E, Taylor BC, Cauley JA, Orwoll ES. Association of testosterone and estradiol deficiency with osteoporosis and rapid bone loss in older men. J Clin Endocrinol Metab 2006; 91:3908-3915
132. Greendale GA, Edelstein S, Barrett-Connor E. Endogenous sex steroids and bone mineral density in older women and men: the Rancho Bernardo Study. J Bone Miner Res 1997; 12:1833-1843
133. Amin S, Zhang Y, Sawin CT, Evans SR, Hannan MT, Kiel DP, Wilson PW, Felson DT. Association of hypogonadism and estradiol levels with bone mineral density in elderly men from the Framingham study. Ann Intern Med 2000; 133:951-963
134. Khosla S, Melton LJ, 3rd, Atkinson EJ, O'Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab 1998; 83:2266-2274
135. Center JR, Nguyen TV, Sambrook PN, Eisman JA. Hormonal and biochemical parameters in the determination of osteoporosis in elderly men. J Clin Endocrinol Metab 1999; 84:3626-3635
136. Mellstrom D, Johnell O, Ljunggren O, Eriksson AL, Lorentzon M, Mallmin H, Holmberg A, Redlund-Johnell I, Orwoll E, Ohlsson C. Free testosterone is an independent predictor of BMD and prevalent fractures in elderly men: MrOS Sweden. J Bone Miner Res 2006; 21:529-535
137. Shores MM, Moceri VM, Gruenewald DA, Brodkin KI, Matsumoto AM, Kivlahan DR. Low testosterone is associated with decreased function and increased mortality risk: a preliminary study of men in a geriatric rehabilitation unit. J Am Geriatr Soc 2004; 52:2077-2081
138. Shores MM, Matsumoto AM, Sloan KL, Kivlahan DR. Low serum testosterone and mortality in male veterans. Arch Intern Med 2006; 166:1660-1665
139. Araujo AB, Kupelian V, Page ST, Handelsman DJ, Bremner WJ, McKinlay JB. Sex steroids and all-cause and cause-specific mortality in men. Arch Intern Med 2007; 167:1252-1260
140. Laughlin GA, Barrett-Connor E, Bergstrom J. Low serum testosterone and mortality in older men. J Clin Endocrinol Metab 2008; 93:68-75
141. Yeap BB, Marriott RJ, Antonio L, Chan YX, Raj S, Dwivedi G, Reid CM, Anawalt BD, Bhasin S, Dobs AS, Hankey GJ, Matsumoto AM, Norman PE, O'Neill TW, Ohlsson C, Orwoll ES, Vanderschueren D, Wittert GA, Wu FCW, Murray K. Serum Testosterone is Inversely and Sex Hormone-binding Globulin is Directly Associated with All-cause Mortality in Men. J Clin Endocrinol Metab 2021; 106:e625-e637
142. Araujo AB, Dixon JM, Suarez EA, Murad MH, Guey LT, Wittert GA. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab 2011; 96:3007-3019
143. Corona G, Rastrelli G, Monami M, Guay A, Buvat J, Sforza A, Forti G, Mannucci E, Maggi M. Hypogonadism as a risk factor for cardiovascular mortality in men: a meta-analytic study. Eur J Endocrinol 2011; 165:687-701
144. Litman HJ, Bhasin S, O'Leary MP, Link CL, McKinlay JB, Investigators BS. An investigation of the relationship between sex-steroid levels and urological symptoms: results from the Boston Area Community Health survey. BJU Int 2007; 100:321-326
145. Alexandersen P, Haarbo J, Christiansen C. The relationship of natural androgens to coronary heart disease in males: a review. Atherosclerosis 1996; 125:1-13
146. Barrett-Connor E, Khaw KT. Endogenous sex hormones and cardiovascular disease in men. A prospective population-based study. Circulation 1988; 78:539-545
147. Cauley JA, Gutai JP, Kuller LH, Dai WS. Usefulness of sex steroid hormone levels in predicting coronary artery disease in men. Am J Cardiol 1987; 60:771-777
148. Contoreggi CS, Blackman MR, Andres R, Muller DC, Lakatta EG, Fleg JL, Harman SM. Plasma levels of estradiol, testosterone, and DHEAS do not predict risk of coronary artery disease in men. J Androl 1990; 11:460-470
149. Yarnell JW, Beswick AD, Sweetnam PM, Riad-Fahmy D. Endogenous sex hormones and ischemic heart disease in men. The Caerphilly prospective study. Arterioscler Thromb 1993; 13:517-520
150. Hak AE, Witteman JC, de Jong FH, Geerlings MI, Hofman A, Pols HA. Low levels of endogenous androgens increase the risk of atherosclerosis in elderly men: the Rotterdam study. J Clin Endocrinol Metab 2002; 87:3632-3639
151. Bassey EJ, Fiatarone MA, O'Neill EF, Kelly M, Evans WJ, Lipsitz LA. Leg extensor power and functional performance in very old men and women. Clin Sci (Lond) 1992; 82:321-327
152. Rantanen T, Avela J. Leg extension power and walking speed in very old people living independently. J Gerontol A Biol Sci Med Sci 1997; 52:M225-231
153. Whipple RH, Wolfson LI, Amerman PM. The relationship of knee and ankle weakness to falls in nursing home residents: an isokinetic study. J Am Geriatr Soc 1987; 35:13-20
154. Wolfson L, Judge J, Whipple R, King M. Strength is a major factor in balance, gait, and the occurrence of falls. J Gerontol A Biol Sci Med Sci 1995; 50 Spec No:64-67
155. Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT. Drug insight: Testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nat Clin Pract Endocrinol Metab 2006; 2:146-159
156. Katznelson L, Finkelstein JS, Schoenfeld DA, Rosenthal DI, Anderson EJ, Klibanski A. Increase in bone density and lean body mass during testosterone administration in men with acquired hypogonadism. J Clin Endocrinol Metab 1996; 81:4358-4365
157. Bhasin S, Storer TW, Berman N, Yarasheski KE, Clevenger B, Phillips J, Lee WP, Bunnell TJ, Casaburi R. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab 1997; 82:407-413
158. Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men--a clinical research center study. J Clin Endocrinol Metab 1996; 81:3469-3475
159. Snyder PJ, Peachey H, Berlin JA, Hannoush P, Haddad G, Dlewati A, Santanna J, Loh L, Lenrow DA, Holmes JH, Kapoor SC, Atkinson LE, Strom BL. Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab 2000; 85:2670-2677
160. Wang C, Swerdloff RS, Iranmanesh A, Dobs A, Snyder PJ, Cunningham G, Matsumoto AM, Weber T, Berman N, Testosterone Gel Study G. Transdermal testosterone gel improves sexual function, mood, muscle strength, and body composition parameters in hypogonadal men. J Clin Endocrinol Metab 2000; 85:2839-2853
161. Steidle C, Schwartz S, Jacoby K, Sebree T, Smith T, Bachand R, North American AATGSG. AA2500 testosterone gel normalizes androgen levels in aging males with improvements in body composition and sexual function. J Clin Endocrinol Metab 2003; 88:2673-2681
162. Bhasin S, Ellenberg SS, Storer TW, Basaria S, Pahor M, Stephens-Shields AJ, Cauley JA, Ensrud KE, Farrar JT, Cella D, Matsumoto AM, Cunningham GR, Swerdloff RS, Wang C, Lewis CE, Molitch ME, Barrett-Connor E, Crandall JP, Hou X, Preston P, Cifelli D, Snyder PJ, Gill TM. Effect of testosterone replacement on measures of mobility in older men with mobility limitation and low testosterone concentrations: secondary analyses of the Testosterone Trials. Lancet Diabetes Endocrinol 2018; 6:879-890
163. Storer TW, Basaria S, Traustadottir T, Harman SM, Pencina K, Li Z, Travison TG, Miciek R, Tsitouras P, Hally K, Huang G, Bhasin S. Effects of Testosterone Supplementation for 3 Years on Muscle Performance and Physical Function in Older Men. J Clin Endocrinol Metab 2017; 102:583-593
164. Storer TW, Bhasin S, Travison TG, Pencina K, Miciek R, McKinnon J, Basaria S. Testosterone Attenuates Age-Related Fall in Aerobic Function in Mobility Limited Older Men With Low Testosterone. J Clin Endocrinol Metab 2016; 101:2562-2569
165. Traustadottir T, Harman SM, Tsitouras P, Pencina KM, Li Z, Travison TG, Eder R, Miciek R, McKinnon J, Woodbury E, Basaria S, Bhasin S, Storer TW. Long-Term Testosterone Supplementation in Older Men Attenuates Age-Related Decline in Aerobic Capacity. J Clin Endocrinol Metab 2018; 103:2861-2869
166. Bhasin S, Woodhouse L, Casaburi R, Singh AB, Bhasin D, Berman N, Chen X, Yarasheski KE, Magliano L, Dzekov C, Dzekov J, Bross R, Phillips J, Sinha-Hikim I, Shen R, Storer TW. Testosterone dose-response relationships in healthy young men. Am J Physiol Endocrinol Metab 2001; 281:E1172-1181
167. Storer TW, Magliano L, Woodhouse L, Lee ML, Dzekov C, Dzekov J, Casaburi R, Bhasin S. Testosterone dose-dependently increases maximal voluntary strength and leg power, but does not affect fatigability or specific tension. J Clin Endocrinol Metab 2003; 88:1478-1485
168. Woodhouse LJ, Reisz-Porszasz S, Javanbakht M, Storer TW, Lee M, Zerounian H, Bhasin S. Development of models to predict anabolic response to testosterone administration in healthy young men. Am J Physiol Endocrinol Metab 2003; 284:E1009-1017
169. Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med 1996; 335:1-7
170. Blackman MR, Sorkin JD, Munzer T, Bellantoni MF, Busby-Whitehead J, Stevens TE, Jayme J, O'Connor KG, Christmas C, Tobin JD, Stewart KJ, Cottrell E, St Clair C, Pabst KM, Harman SM. Growth hormone and sex steroid administration in healthy aged women and men: a randomized controlled trial. JAMA 2002; 288:2282-2292
171. Bhasin S, Apovian CM, Travison TG, Pencina K, Moore LL, Huang G, Campbell WW, Li Z, Howland AS, Chen R, Knapp PE, Singer MR, Shah M, Secinaro K, Eder RV, Hally K, Schram H, Bearup R, Beleva YM, McCarthy AC, Woodbury E, McKinnon J, Fleck G, Storer TW, Basaria S. Effect of Protein Intake on Lean Body Mass in Functionally Limited Older Men: A Randomized Clinical Trial. JAMA Intern Med 2018; 178:530-541
172. Bhasin S, Apovian CM, Travison TG, Pencina K, Huang G, Moore LL, Campbell WW, Howland A, Chen R, Singer MR, Shah M, Eder R, Schram H, Bearup R, Beleva YM, McCarthy AC, Li Z, Woodbury E, McKinnon J, Storer TW, Basaria S. Design of a randomized trial to determine the optimum protein intake to preserve lean body mass and to optimize response to a promyogenic anabolic agent in older men with physical functional limitation. Contemp Clin Trials 2017; 58:86-93
173. Urban RJ, Bodenburg YH, Gilkison C, Foxworth J, Coggan AR, Wolfe RR, Ferrando A. Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am J Physiol 1995; 269:E820-826
174. Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab 2002; 282:E601-607
175. Kong A, Edmonds P. Testosterone therapy in HIV wasting syndrome: systematic review and meta-analysis. Lancet Infect Dis 2002; 2:692-699
176. Johns K, Beddall MJ, Corrin RC. Anabolic steroids for the treatment of weight loss in HIV-infected individuals. Cochrane Database Syst Rev 2005:CD005483
177. Bhasin S, Storer TW, Javanbakht M, Berman N, Yarasheski KE, Phillips J, Dike M, Sinha-Hikim I, Shen R, Hays RD, Beall G. Testosterone replacement and resistance exercise in HIV-infected men with weight loss and low testosterone levels. JAMA 2000; 283:763-770
178. Grinspoon S, Corcoran C, Askari H, Schoenfeld D, Wolf L, Burrows B, Walsh M, Hayden D, Parlman K, Anderson E, Basgoz N, Klibanski A. Effects of androgen administration in men with the AIDS wasting syndrome. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 1998; 129:18-26
179. Storer TW, Woodhouse LJ, Sattler F, Singh AB, Schroeder ET, Beck K, Padero M, Mac P, Yarasheski KE, Geurts P, Willemsen A, Harms MK, Bhasin S. A randomized, placebo-controlled trial of nandrolone decanoate in human immunodeficiency virus-infected men with mild to moderate weight loss with recombinant human growth hormone as active reference treatment. J Clin Endocrinol Metab 2005; 90:4474-4482
180. Grinspoon S, Corcoran C, Parlman K, Costello M, Rosenthal D, Anderson E, Stanley T, Schoenfeld D, Burrows B, Hayden D, Basgoz N, Klibanski A. Effects of testosterone and progressive resistance training in eugonadal men with AIDS wasting. A randomized, controlled trial. Ann Intern Med 2000; 133:348-355
181. Bhasin S, Storer TW, Asbel-Sethi N, Kilbourne A, Hays R, Sinha-Hikim I, Shen R, Arver S, Beall G. Effects of testosterone replacement with a nongenital, transdermal system, Androderm, in human immunodeficiency virus-infected men with low testosterone levels. J Clin Endocrinol Metab 1998; 83:3155-3162
182. Grinspoon S, Corcoran C, Stanley T, Baaj A, Basgoz N, Klibanski A. Effects of hypogonadism and testosterone administration on depression indices in HIV-infected men. J Clin Endocrinol Metab 2000; 85:60-65
183. Knapp PE, Storer TW, Herbst KL, Singh AB, Dzekov C, Dzekov J, LaValley M, Zhang A, Ulloor J, Bhasin S. Effects of a supraphysiological dose of testosterone on physical function, muscle performance, mood, and fatigue in men with HIV-associated weight loss. Am J Physiol Endocrinol Metab 2008; 294:E1135-1143
184. Sih R, Morley JE, Kaiser FE, Perry HM, 3rd, Patrick P, Ross C. Testosterone replacement in older hypogonadal men: a 12-month randomized controlled trial. J Clin Endocrinol Metab 1997; 82:1661-1667
185. Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Lenrow DA, Holmes JH, Dlewati A, Santanna J, Rosen CJ, Strom BL. Effect of testosterone treatment on body composition and muscle strength in men over 65 years of age. J Clin Endocrinol Metab 1999; 84:2647-2653
186. Page ST, Amory JK, Bowman FD, Anawalt BD, Matsumoto AM, Bremner WJ, Tenover JL. Exogenous testosterone (T) alone or with finasteride increases physical performance, grip strength, and lean body mass in older men with low serum T. J Clin Endocrinol Metab 2005; 90:1502-1510
187. Schroeder ET, Singh A, Bhasin S, Storer TW, Azen C, Davidson T, Martinez C, Sinha-Hikim I, Jaque SV, Terk M, Sattler FR. Effects of an oral androgen on muscle and metabolism in older, community-dwelling men. Am J Physiol Endocrinol Metab 2003; 284:E120-128
188. Kenny AM, Prestwood KM, Gruman CA, Marcello KM, Raisz LG. Effects of transdermal testosterone on bone and muscle in older men with low bioavailable testosterone levels. J Gerontol A Biol Sci Med Sci 2001; 56:M266-272
189. Nair KS, Rizza RA, O'Brien P, Dhatariya K, Short KR, Nehra A, Vittone JL, Klee GG, Basu A, Basu R, Cobelli C, Toffolo G, Dalla Man C, Tindall DJ, Melton LJ, 3rd, Smith GE, Khosla S, Jensen MD. DHEA in elderly women and DHEA or testosterone in elderly men. N Engl J Med 2006; 355:1647-1659
190. Travison TG, Basaria S, Storer TW, Jette AM, Miciek R, Farwell WR, Choong K, Lakshman K, Mazer NA, Coviello AD, Knapp PE, Ulloor J, Zhang A, Brooks B, Nguyen AH, Eder R, LeBrasseur N, Elmi A, Appleman E, Hede-Brierley L, Bhasin G, Bhatia A, Lazzari A, Davis S, Ni P, Collins L, Bhasin S. Clinical meaningfulness of the changes in muscle performance and physical function associated with testosterone administration in older men with mobility limitation. J Gerontol A Biol Sci Med Sci 2011; 66:1090-1099
191. Kenny AM, Kleppinger A, Annis K, Rathier M, Browner B, Judge JO, McGee D. Effects of transdermal testosterone on bone and muscle in older men with low bioavailable testosterone levels, low bone mass, and physical frailty. J Am Geriatr Soc 2010; 58:1134-1143
192. Snyder PJ, Ellenberg SS, Cunningham GR, Matsumoto AM, Bhasin S, Barrett-Connor E, Gill TM, Farrar JT, Cella D, Rosen RC, Resnick SM, Swerdloff RS, Cauley JA, Cifelli D, Fluharty L, Pahor M, Ensrud KE, Lewis CE, Molitch ME, Crandall JP, Wang C, Budoff MJ, Wenger NK, Mohler ERR, Bild DE, Cook NL, Keaveny TM, Kopperdahl DL, Lee D, Schwartz AV, Storer TW, Ershler WB, Roy CN, Raffel LJ, Romashkan S, Hadley E. The Testosterone Trials: Seven coordinated trials of testosterone treatment in elderly men. Clin Trials 2014; 11:362-375
193. de Labra C, Guimaraes-Pinheiro C, Maseda A, Lorenzo T, Millan-Calenti JC. Effects of physical exercise interventions in frail older adults: a systematic review of randomized controlled trials. BMC Geriatr 2015; 15:154
194. Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J 2005; 19:422-424
195. Houston DK, Nicklas BJ, Ding J, Harris TB, Tylavsky FA, Newman AB, Lee JS, Sahyoun NR, Visser M, Kritchevsky SB, Health ABCS. Dietary protein intake is associated with lean mass change in older, community-dwelling adults: the Health, Aging, and Body Composition (Health ABC) Study. Am J Clin Nutr 2008; 87:150-155
196. Huang G, Pencina K, Li Z, Apovian CM, Travison TG, Storer TW, Gagliano-Juca T, Basaria S, Bhasin S. Effect of Protein Intake on Visceral Abdominal Fat and Metabolic Biomarkers in Older Men With Functional Limitations: Results From a Randomized Clinical Trial. J Gerontol A Biol Sci Med Sci 2021; 76:1084-1089
197. Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. Am J Physiol Endocrinol Metab 2002; 283:E154-164
198. Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone-induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. Am J Physiol Endocrinol Metab 2003; 285:E197-205
199. Bhasin S, Taylor WE, Singh R, Artaza J, Sinha-Hikim I, Jasuja R, Choi H, Gonzalez-Cadavid NF. The mechanisms of androgen effects on body composition: mesenchymal pluripotent cell as the target of androgen action. J Gerontol A Biol Sci Med Sci 2003; 58:M1103-1110
200. Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway. Endocrinology 2003; 144:5081-5088
201. Gupta V, Bhasin S, Guo W, Singh R, Miki R, Chauhan P, Choong K, Tchkonia T, Lebrasseur NK, Flanagan JN, Hamilton JA, Viereck JC, Narula NS, Kirkland JL, Jasuja R. Effects of dihydrotestosterone on differentiation and proliferation of human mesenchymal stem cells and preadipocytes. Mol Cell Endocrinol 2008; 296:32-40
202. Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez-Cadavid NF, Bhasin S. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells: nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology 2006; 147:141-154
203. Singh R, Bhasin S, Braga M, Artaza JN, Pervin S, Taylor WE, Krishnan V, Sinha SK, Rajavashisth TB, Jasuja R. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/ beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology 2009; 150:1259-1268
204. Jasuja R, Costello JC, Singh R, Gupta V, Spina CS, Toraldo G, Jang H, Li H, Serra C, Guo W, Chauhan P, Narula NS, Guarneri T, Ergun A, Travison TG, Collins JJ, Bhasin S. Combined administration of testosterone plus an ornithine decarboxylase inhibitor as a selective prostate-sparing anabolic therapy. Aging Cell 2014; 13:303-310
205. Bartsch W, Krieg M, Voigt KD. Quantification of endogenous testosterone, 5 alpha-dihydrotestosterone and 5 alpha-androstane-3 alpha, 17 beta-diol in subcellular fractions of the prostate, bulbocavernosus/levator ani muscle, skeletal muscle and heart muscle of the rat. J Steroid Biochem 1980; 13:259-264
206. Wilson JD. The role of 5alpha-reduction in steroid hormone physiology. Reprod Fertil Dev 2001; 13:673-678
207. Andriole GL, Kirby R. Safety and tolerability of the dual 5alpha-reductase inhibitor dutasteride in the treatment of benign prostatic hyperplasia. Eur Urol 2003; 44:82-88
208. Bhasin S, Travison TG, Storer TW, Lakshman K, Kaushik M, Mazer NA, Ngyuen AH, Davda MN, Jara H, Aakil A, Anderson S, Knapp PE, Hanka S, Mohammed N, Daou P, Miciek R, Ulloor J, Zhang A, Brooks B, Orwoll K, Hede-Brierley L, Eder R, Elmi A, Bhasin G, Collins L, Singh R, Basaria S. Effect of testosterone supplementation with and without a dual 5alpha-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial. JAMA 2012; 307:931-939
209. Borst SE, Conover CF, Carter CS, Gregory CM, Marzetti E, Leeuwenburgh C, Vandenborne K, Wronski TJ. Anabolic effects of testosterone are preserved during inhibition of 5alpha-reductase. Am J Physiol Endocrinol Metab 2007; 293:E507-514
210. Matsumoto AM, Tenover L, McClung M, Mobley D, Geller J, Sullivan M, Grayhack J, Wessells H, Kadmon D, Flanagan M, Zhang GK, Schmidt J, Taylor AM, Lee M, Waldstreicher J, Pless Study G. The long-term effect of specific type II 5alpha-reductase inhibition with finasteride on bone mineral density in men: results of a 4-year placebo controlled trial. J Urol 2002; 167:2105-2108
211. Amory JK, Anawalt BD, Matsumoto AM, Page ST, Bremner WJ, Wang C, Swerdloff RS, Clark RV. The effect of 5alpha-reductase inhibition with dutasteride and finasteride on bone mineral density, serum lipoproteins, hemoglobin, prostate specific antigen and sexual function in healthy young men. J Urol 2008; 179:2333-2338
212. Agis-Balboa RC, Pinna G, Zhubi A, Maloku E, Veldic M, Costa E, Guidotti A. Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc Natl Acad Sci U S A 2006; 103:14602-14607
213. Agis-Balboa RC, Guidotti A, Pinna G. 5alpha-reductase type I expression is downregulated in the prefrontal cortex/Brodmann's area 9 (BA9) of depressed patients. Psychopharmacology (Berl) 2014; 231:3569-3580
214. Rasmusson AM, King MW, Valovski I, Gregor K, Scioli-Salter E, Pineles SL, Hamouda M, Nillni YI, Anderson GM, Pinna G. Relationships between cerebrospinal fluid GABAergic neurosteroid levels and symptom severity in men with PTSD. Psychoneuroendocrinology 2019; 102:95-104
215. Stoffel-Wagner B. Neurosteroid metabolism in the human brain. Eur J Endocrinol 2001; 145:669-679
216. Kanes S, Colquhoun H, Gunduz-Bruce H, Raines S, Arnold R, Schacterle A, Doherty J, Epperson CN, Deligiannidis KM, Riesenberg R, Hoffmann E, Rubinow D, Jonas J, Paul S, Meltzer-Brody S. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet 2017; 390:480-489
217. Frieder A, Fersh M, Hainline R, Deligiannidis KM. Pharmacotherapy of Postpartum Depression: Current Approaches and Novel Drug Development. CNS Drugs 2019; 33:265-282
218. Nasiri M, Nikolaou N, Parajes S, Krone NP, Valsamakis G, Mastorakos G, Hughes B, Taylor A, Bujalska IJ, Gathercole LL, Tomlinson JW. 5alpha-Reductase Type 2 Regulates Glucocorticoid Action and Metabolic Phenotype in Human Hepatocytes. Endocrinology 2015; 156:2863-2871
219. Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A 2000; 97:12735-12740
220. Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, Korach KS, Simpson ER. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med 1997; 337:91-95
221. Finkelstein JS, Lee H, Burnett-Bowie SA, Pallais JC, Yu EW, Borges LF, Jones BF, Barry CV, Wulczyn KE, Thomas BJ, Leder BZ. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med 2013; 369:1011-1022
222. Mauras N, Hayes V, Welch S, Rini A, Helgeson K, Dokler M, Veldhuis JD, Urban RJ. Testosterone deficiency in young men: marked alterations in whole body protein kinetics, strength, and adiposity. J Clin Endocrinol Metab 1998; 83:1886-1892
223. Woodhouse LJ, Gupta N, Bhasin M, Singh AB, Ross R, Phillips J, Bhasin S. Dose-dependent effects of testosterone on regional adipose tissue distribution in healthy young men. J Clin Endocrinol Metab 2004; 89:718-726
224. Wang C, Cunningham G, Dobs A, Iranmanesh A, Matsumoto AM, Snyder PJ, Weber T, Berman N, Hull L, Swerdloff RS. Long-term testosterone gel (AndroGel) treatment maintains beneficial effects on sexual function and mood, lean and fat mass, and bone mineral density in hypogonadal men. J Clin Endocrinol Metab 2004; 89:2085-2098
225. Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab 2004; 89:61-70
226. Maffei L, Rochira V, Zirilli L, Antunez P, Aranda C, Fabre B, Simone ML, Pignatti E, Simpson ER, Houssami S, Clyne CD, Carani C. A novel compound heterozygous mutation of the aromatase gene in an adult man: reinforced evidence on the relationship between congenital oestrogen deficiency, adiposity and the metabolic syndrome. Clin Endocrinol (Oxf) 2007; 67:218-224
227. Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 1995; 80:3689-3698
228. Hewitt KN, Pratis K, Jones ME, Simpson ER. Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology 2004; 145:1842-1848
229. Chazenbalk G, Singh P, Irge D, Shah A, Abbott DH, Dumesic DA. Androgens inhibit adipogenesis during human adipose stem cell commitment to preadipocyte formation. Steroids 2013; 78:920-926
230. Host C, Gormsen LC, Christensen B, Jessen N, Hougaard DM, Christiansen JS, Pedersen SB, Jensen MD, Nielsen S, Gravholt CH. Independent effects of testosterone on lipid oxidation and VLDL-TG production: a randomized, double-blind, placebo-controlled, crossover study. Diabetes 2013; 62:1409-1416
231. Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, Zhang X, Zou F, Gent LM, Hahner LD, Khan SA, Elias CF, Elmquist JK, Clegg DJ. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 2011; 14:453-465
232. Ponnusamy S, Tran QT, Thiyagarajan T, Miller DD, Bridges D, Narayanan R. An estrogen receptor beta-selective agonist inhibits non-alcoholic steatohepatitis in preclinical models by regulating bile acid and xenobiotic receptors. Exp Biol Med (Maywood) 2017; 242:606-616
233. Bhasin S, Enzlin P, Coviello A, Basson R. Sexual dysfunction in men and women with endocrine disorders. Lancet 2007; 369:597-611
234. Arver S, Dobs AS, Meikle AW, Allen RP, Sanders SW, Mazer NA. Improvement of sexual function in testosterone deficient men treated for 1 year with a permeation enhanced testosterone transdermal system. J Urol 1996; 155:1604-1608
235. Bancroft J, Wu FC. Changes in erectile responsiveness during androgen replacement therapy. Arch Sex Behav 1983; 12:59-66
236. Davidson JM, Camargo CA, Smith ER. Effects of androgen on sexual behavior in hypogonadal men. J Clin Endocrinol Metab 1979; 48:955-958
237. Kwan M, Greenleaf WJ, Mann J, Crapo L, Davidson JM. The nature of androgen action on male sexuality: a combined laboratory-self-report study on hypogonadal men. J Clin Endocrinol Metab 1983; 57:557-562
238. Bagatell CJ, Heiman JR, Rivier JE, Bremner WJ. Effects of endogenous testosterone and estradiol on sexual behavior in normal young men. J Clin Endocrinol Metab 1994; 78:711-716
239. Carani C, Granata AR, Bancroft J, Marrama P. The effects of testosterone replacement on nocturnal penile tumescence and rigidity and erectile response to visual erotic stimuli in hypogonadal men. Psychoneuroendocrinology 1995; 20:743-753
240. Cunningham GR, Hirshkowitz M, Korenman SG, Karacan I. Testosterone replacement therapy and sleep-related erections in hypogonadal men. J Clin Endocrinol Metab 1990; 70:792-797
241. Carani C, Bancroft J, Granata A, Del Rio G, Marrama P. Testosterone and erectile function, nocturnal penile tumescence and rigidity, and erectile response to visual erotic stimuli in hypogonadal and eugonadal men. Psychoneuroendocrinology 1992; 17:647-654
242. Lugg J, Ng C, Rajfer J, Gonzalez-Cadavid N. Cavernosal nerve stimulation in the rat reverses castration-induced decrease in penile NOS activity. Am J Physiol 1996; 271:E354-361
243. Shabsigh R. The effects of testosterone on the cavernous tissue and erectile function. World J Urol 1997; 15:21-26
244. Mills TM, Lewis RW, Stopper VS. Androgenic maintenance of inflow and veno-occlusion during erection in the rat. Biol Reprod 1998; 59:1413-1418
245. Mills TM, Dai Y, Stopper VS, Lewis RW. Androgenic maintenance of the erectile response in the rat. Steroids 1999; 64:605-609
246. Bhasin S, Fielder T, Peacock N, Sod-Moriah UA, Swerdloff RS. Dissociating antifertility effects of GnRH-antagonist from its adverse effects on mating behavior in male rats. Am J Physiol 1988; 254:E84-91
247. Fielder TJ, Peacock NR, McGivern RF, Swerdloff RS, Bhasin S. Testosterone dose-dependency of sexual and nonsexual behaviors in the gonadotropin-releasing hormone antagonist-treated male rat. J Androl 1989; 10:167-173
248. Buena F, Swerdloff RS, Steiner BS, Lutchmansingh P, Peterson MA, Pandian MR, Galmarini M, Bhasin S. Sexual function does not change when serum testosterone levels are pharmacologically varied within the normal male range. Fertil Steril 1993; 59:1118-1123
249. Morley JE, Korenman SG, Mooradian AD, Kaiser FE. Sexual dysfunction in the elderly male. J Am Geriatr Soc 1987; 35:1014-1022
250. Buvat J, Lemaire A. Endocrine screening in 1,022 men with erectile dysfunction: clinical significance and cost-effective strategy. J Urol 1997; 158:1764-1767
251. Citron JT, Ettinger B, Rubinoff H, Ettinger VM, Minkoff J, Hom F, Kan P, Alloo R. Prevalence of hypothalamic-pituitary imaging abnormalities in impotent men with secondary hypogonadism. J Urol 1996; 155:529-533
252. Burris AS, Banks SM, Carter CS, Davidson JM, Sherins RJ. A long-term, prospective study of the physiologic and behavioral effects of hormone replacement in untreated hypogonadal men. J Androl 1992; 13:297-304
253. Cunningham GR, Toma SM. Clinical review: Why is androgen replacement in males controversial? J Clin Endocrinol Metab 2011; 96:38-52
254. Allan CA, Forbes EA, Strauss BJ, McLachlan RI. Testosterone therapy increases sexual desire in ageing men with low-normal testosterone levels and symptoms of androgen deficiency. Int J Impot Res 2008; 20:396-401
255. Cavallini G, Caracciolo S, Vitali G, Modenini F, Biagiotti G. Carnitine versus androgen administration in the treatment of sexual dysfunction, depressed mood, and fatigue associated with male aging. Urology 2004; 63:641-646
256. Ponce OJ, Spencer-Bonilla G, Alvarez-Villalobos N, Serrano V, Singh-Ospina N, Rodriguez-Gutierrez R, Salcido-Montenegro A, Benkhadra R, Prokop LJ, Bhasin S, Brito JP. The efficacy and adverse events of testosterone replacement therapy in hypogonadal men: A systematic review and meta-analysis of randomized, placebo-controlled trials. J Clin Endocrinol Metab 2018;
257. Cunningham GR, Stephens-Shields AJ, Rosen RC, Wang C, Bhasin S, Matsumoto AM, Parsons JK, Gill TM, Molitch ME, Farrar JT, Cella D, Barrett-Connor E, Cauley JA, Cifelli D, Crandall JP, Ensrud KE, Gallagher L, Zeldow B, Lewis CE, Pahor M, Swerdloff RS, Hou X, Anton S, Basaria S, Diem SJ, Tabatabaie V, Ellenberg SS, Snyder PJ. Testosterone Treatment and Sexual Function in Older Men With Low Testosterone Levels. J Clin Endocrinol Metab 2016; 101:3096-3104
258. Muller A, Smith L, Parker M, Mulhall JP. Analysis of the efficacy and safety of sildenafil citrate in the geriatric population. BJU Int 2007; 100:117-121
259. Paduch DA, Polzer PK, Ni X, Basaria S. Testosterone Replacement in Androgen-Deficient Men With Ejaculatory Dysfunction: A Randomized Controlled Trial. J Clin Endocrinol Metab 2015; 100:2956-2962
260. Shabsigh R, Kaufman JM, Steidle C, Padma-Nathan H. Randomized study of testosterone gel as adjunctive therapy to sildenafil in hypogonadal men with erectile dysfunction who do not respond to sildenafil alone. J Urol 2004; 172:658-663
261. Aversa A, Isidori AM, Spera G, Lenzi A, Fabbri A. Androgens improve cavernous vasodilation and response to sildenafil in patients with erectile dysfunction. Clin Endocrinol (Oxf) 2003; 58:632-638
262. Buvat J, Montorsi F, Maggi M, Porst H, Kaipia A, Colson MH, Cuzin B, Moncada I, Martin-Morales A, Yassin A, Meuleman E, Eardley I, Dean JD, Shabsigh R. Hypogonadal men nonresponders to the PDE5 inhibitor tadalafil benefit from normalization of testosterone levels with a 1% hydroalcoholic testosterone gel in the treatment of erectile dysfunction (TADTEST study). J Sex Med 2011; 8:284-293
263. Spitzer M, Basaria S, Travison TG, Davda MN, Paley A, Cohen B, Mazer NA, Knapp PE, Hanka S, Lakshman KM, Ulloor J, Zhang A, Orwoll K, Eder R, Collins L, Mohammed N, Rosen RC, DeRogatis L, Bhasin S. Effect of testosterone replacement on response to sildenafil citrate in men with erectile dysfunction: a parallel, randomized trial. Ann Intern Med 2012; 157:681-691
264. Stepan JJ, Lachman M, Zverina J, Pacovsky V, Baylink DJ. Castrated men exhibit bone loss: effect of calcitonin treatment on biochemical indices of bone remodeling. J Clin Endocrinol Metab 1989; 69:523-527
265. Goldray D, Weisman Y, Jaccard N, Merdler C, Chen J, Matzkin H. Decreased bone density in elderly men treated with the gonadotropin-releasing hormone agonist decapeptyl (D-Trp6-GnRH). J Clin Endocrinol Metab 1993; 76:288-290
266. Daniell HW. Osteoporosis after orchiectomy for prostate cancer. J Urol 1997; 157:439-444
267. Eriksson S, Eriksson A, Stege R, Carlstrom K. Bone mineral density in patients with prostatic cancer treated with orchidectomy and with estrogens. Calcif Tissue Int 1995; 57:97-99
268. Smith MR, Lee WC, Brandman J, Wang Q, Botteman M, Pashos CL. Gonadotropin-releasing hormone agonists and fracture risk: a claims-based cohort study of men with nonmetastatic prostate cancer. J Clin Oncol 2005; 23:7897-7903
269. Lopez AM, Pena MA, Hernandez R, Val F, Martin B, Riancho JA. Fracture risk in patients with prostate cancer on androgen deprivation therapy. Osteoporos Int 2005; 16:707-711
270. Shahinian VB, Kuo YF, Freeman JL, Goodwin JS. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 2005; 352:154-164
271. Vanderschueren D, Van Herck E, Schot P, Rush E, Einhorn T, Geusens P, Bouillon R. The aged male rat as a model for human osteoporosis: evaluation by nondestructive measurements and biomechanical testing. Calcif Tissue Int 1993; 53:342-347
272. Finkelstein JS, Klibanski A, Neer RM, Greenspan SL, Rosenthal DI, Crowley WF, Jr. Osteoporosis in men with idiopathic hypogonadotropic hypogonadism. Ann Intern Med 1987; 106:354-361
273. Bonjour JP, Theintz G, Buchs B, Slosman D, Rizzoli R. Critical years and stages of puberty for spinal and femoral bone mass accumulation during adolescence. J Clin Endocrinol Metab 1991; 73:555-563
274. Bird RJ, Hurren BJ. Anatomical and clinical aspects of Klinefelter's syndrome. Clin Anat 2016; 29:606-619
275. Schoenau E, Neu CM, Mokov E, Wassmer G, Manz F. Influence of puberty on muscle area and cortical bone area of the forearm in boys and girls. J Clin Endocrinol Metab 2000; 85:1095-1098
276. Finkelstein JS, Neer RM, Biller BM, Crawford JD, Klibanski A. Osteopenia in men with a history of delayed puberty. N Engl J Med 1992; 326:600-604
277. Finkelstein JS, Klibanski A, Neer RM. A longitudinal evaluation of bone mineral density in adult men with histories of delayed puberty. J Clin Endocrinol Metab 1996; 81:1152-1155
278. Seeman E, Duan Y, Fong C, Edmonds J. Fracture site-specific deficits in bone size and volumetric density in men with spine or hip fractures. J Bone Miner Res 2001; 16:120-127
279. Behre HM, Kliesch S, Leifke E, Link TM, Nieschlag E. Long-term effect of testosterone therapy on bone mineral density in hypogonadal men. J Clin Endocrinol Metab 1997; 82:2386-2390
280. Leifke E, Korner HC, Link TM, Behre HM, Peters PE, Nieschlag E. Effects of testosterone replacement therapy on cortical and trabecular bone mineral density, vertebral body area and paraspinal muscle area in hypogonadal men. Eur J Endocrinol 1998; 138:51-58
281. Guo CY, Jones TH, Eastell R. Treatment of isolated hypogonadotropic hypogonadism effect on bone mineral density and bone turnover. J Clin Endocrinol Metab 1997; 82:658-665
282. Aminorroaya A, Kelleher S, Conway AJ, Ly LP, Handelsman DJ. Adequacy of androgen replacement influences bone density response to testosterone in androgen-deficient men. Eur J Endocrinol 2005; 152:881-886
283. Wang C, Swerdloff RS, Iranmanesh A, Dobs A, Snyder PJ, Cunningham G, Matsumoto AM, Weber T, Berman N. Effects of transdermal testosterone gel on bone turnover markers and bone mineral density in hypogonadal men. Clin Endocrinol (Oxf) 2001; 54:739-750
284. Boonen S, Vanderschueren D, Geusens P, Bouillon R. Age-associated endocrine deficiencies as potential determinants of femoral neck (type II) osteoporotic fracture occurrence in elderly men. Int J Androl 1997; 20:134-143
285. Khosla S, Melton LJ, 3rd, Robb RA, Camp JJ, Atkinson EJ, Oberg AL, Rouleau PA, Riggs BL. Relationship of volumetric BMD and structural parameters at different skeletal sites to sex steroid levels in men. J Bone Miner Res 2005; 20:730-740
286. Stanley HL, Schmitt BP, Poses RM, Deiss WP. Does hypogonadism contribute to the occurrence of a minimal trauma hip fracture in elderly men? J Am Geriatr Soc 1991; 39:766-771
287. LeBlanc ES, Nielson CM, Marshall LM, Lapidus JA, Barrett-Connor E, Ensrud KE, Hoffman AR, Laughlin G, Ohlsson C, Orwoll ES, Osteoporotic Fractures in Men Study G. The effects of serum testosterone, estradiol, and sex hormone binding globulin levels on fracture risk in older men. J Clin Endocrinol Metab 2009; 94:3337-3346
288. Yeap BB, Alfonso H, Chubb SAP, Center JR, Beilin J, Hankey GJ, Almeida OP, Golledge J, Norman PE, Flicker L. U-Shaped Association of Plasma Testosterone, and no Association of Plasma Estradiol, with Incidence of Fractures in Men. J Clin Endocrinol Metab 2020; 105
289. Ilangovan R, Sittadjody S, Balaganesh M, Sivakumar R, Ravi Sankar B, Balasubramanian K, Srinivasan S, Subramanian C, Thompson DM, Queimado L, Srinivasan N. Dihydrotestosterone is a determinant of calcaneal bone mineral density in men. J Steroid Biochem Mol Biol 2009; 117:132-138
290. Rosenberg EA, Buzkova P, Fink HA, Robbins JA, Shores MM, Matsumoto AM, Mukamal KJ. Testosterone, dihydrotestosterone, bone density, and hip fracture risk among older men: The Cardiovascular Health Study. Metabolism 2021; 114:154399
291. Eriksson AL, Perry JRB, Coviello AD, Delgado GE, Ferrucci L, Hoffman AR, Huhtaniemi IT, Ikram MA, Karlsson MK, Kleber ME, Laughlin GA, Liu Y, Lorentzon M, Lunetta KL, Mellstrom D, Murabito JM, Murray A, Nethander M, Nielson CM, Prokopenko I, Pye SR, Raffel LJ, Rivadeneira F, Srikanth P, Stolk L, Teumer A, Travison TG, Uitterlinden AG, Vaidya D, Vanderschueren D, Zmuda JM, Marz W, Orwoll ES, Ouyang P, Vandenput L, Wu FCW, de Jong FH, Bhasin S, Kiel DP, Ohlsson C. Genetic Determinants of Circulating Estrogen Levels and Evidence of a Causal Effect of Estradiol on Bone Density in Men. J Clin Endocrinol Metab 2018; 103:991-1004
292. Nethander M, Vandenput L, Eriksson AL, Windahl S, Funck-Brentano T, Ohlsson C. Evidence of a Causal Effect of Estradiol on Fracture Risk in Men. J Clin Endocrinol Metab 2019; 104:433-442
293. Amory JK, Watts NB, Easley KA, Sutton PR, Anawalt BD, Matsumoto AM, Bremner WJ, Tenover JL. Exogenous testosterone or testosterone with finasteride increases bone mineral density in older men with low serum testosterone. J Clin Endocrinol Metab 2004; 89:503-510
294. Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Holmes JH, Dlewati A, Staley J, Santanna J, Kapoor SC, Attie MF, Haddad JG, Jr., Strom BL. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab 1999; 84:1966-1972
295. Tracz MJ, Sideras K, Bolona ER, Haddad RM, Kennedy CC, Uraga MV, Caples SM, Erwin PJ, Montori VM. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab 2006; 91:2011-2016
296. Snyder PJ, Kopperdahl DL, Stephens-Shields AJ, Ellenberg SS, Cauley JA, Ensrud KE, Lewis CE, Barrett-Connor E, Schwartz AV, Lee DC, Bhasin S, Cunningham GR, Gill TM, Matsumoto AM, Swerdloff RS, Basaria S, Diem SJ, Wang C, Hou X, Cifelli D, Dougar D, Zeldow B, Bauer DC, Keaveny TM. Effect of Testosterone Treatment on Volumetric Bone Density and Strength in Older Men With Low Testosterone: A Controlled Clinical Trial. JAMA Intern Med 2017; 177:471-479
297. Ng Tang Fui M, Hoermann R, Bracken K, Handelsman DJ, Inder WJ, Stuckey BGA, Yeap BB, Ghasem-Zadeh A, McLachlan R, Robledo KP, Jesudason D, Zajac JD, Wittert GA, Grossmann M. Effect of Testosterone treatment on bone microarchitecture and bone mineral density in men: a two-year RCT. J Clin Endocrinol Metab 2021;
298. Anderson FH, Francis RM, Peaston RT, Wastell HJ. Androgen supplementation in eugonadal men with osteoporosis: effects of six months' treatment on markers of bone formation and resorption. J Bone Miner Res 1997; 12:472-478
299. Leder BZ, LeBlanc KM, Schoenfeld DA, Eastell R, Finkelstein JS. Differential effects of androgens and estrogens on bone turnover in normal men. J Clin Endocrinol Metab 2003; 88:204-210
300. Falahati-Nini A, Riggs BL, Atkinson EJ, O'Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest 2000; 106:1553-1560
301. Riggs BL, Khosla S, Melton LJ, 3rd. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 2002; 23:279-302
302. Sims NA, Clement-Lacroix P, Minet D, Fraslon-Vanhulle C, Gaillard-Kelly M, Resche-Rigon M, Baron R. A functional androgen receptor is not sufficient to allow estradiol to protect bone after gonadectomy in estradiol receptor-deficient mice. J Clin Invest 2003; 111:1319-1327
303. Syed FA, Modder UI, Fraser DG, Spelsberg TC, Rosen CJ, Krust A, Chambon P, Jameson JL, Khosla S. Skeletal effects of estrogen are mediated by opposing actions of classical and nonclassical estrogen receptor pathways. J Bone Miner Res 2005; 20:1992-2001
304. Vidal O, Lindberg MK, Hollberg K, Baylink DJ, Andersson G, Lubahn DB, Mohan S, Gustafsson JA, Ohlsson C. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc Natl Acad Sci U S A 2000; 97:5474-5479
305. Lindberg MK, Alatalo SL, Halleen JM, Mohan S, Gustafsson JA, Ohlsson C. Estrogen receptor specificity in the regulation of the skeleton in female mice. J Endocrinol 2001; 171:229-236
306. Vandenput L, Ederveen AG, Erben RG, Stahr K, Swinnen JV, Van Herck E, Verstuyf A, Boonen S, Bouillon R, Vanderschueren D. Testosterone prevents orchidectomy-induced bone loss in estrogen receptor-alpha knockout mice. Biochem Biophys Res Commun 2001; 285:70-76
307. Vandenput L, Swinnen JV, Boonen S, Van Herck E, Erben RG, Bouillon R, Vanderschueren D. Role of the androgen receptor in skeletal homeostasis: the androgen-resistant testicular feminized male mouse model. J Bone Miner Res 2004; 19:1462-1470
308. Venken K, De Gendt K, Boonen S, Ophoff J, Bouillon R, Swinnen JV, Verhoeven G, Vanderschueren D. Relative impact of androgen and estrogen receptor activation in the effects of androgens on trabecular and cortical bone in growing male mice: a study in the androgen receptor knockout mouse model. J Bone Miner Res 2006; 21:576-585
309. Ren J, Wang XH, Wang GC, Wu JH. 17beta estradiol regulation of connexin 43-based gap junction and mechanosensitivity through classical estrogen receptor pathway in osteocyte-like MLO-Y4 cells. Bone 2013; 53:587-596
310. Dias JP, Melvin D, Simonsick EM, Carlson O, Shardell MD, Ferrucci L, Chia CW, Basaria S, Egan JM. Effects of aromatase inhibition vs. testosterone in older men with low testosterone: randomized-controlled trial. Andrology 2016; 4:33-40
311. Finkelstein JS, Lee H, Leder BZ, Burnett-Bowie SA, Goldstein DW, Hahn CW, Hirsch SC, Linker A, Perros N, Servais AB, Taylor AP, Webb ML, Youngner JM, Yu EW. Gonadal steroid-dependent effects on bone turnover and bone mineral density in men. J Clin Invest 2016; 126:1114-1125
312. Huber DM, Bendixen AC, Pathrose P, Srivastava S, Dienger KM, Shevde NK, Pike JW. Androgens suppress osteoclast formation induced by RANKL and macrophage-colony stimulating factor. Endocrinology 2001; 142:3800-3808
313. Colvard DS, Eriksen EF, Keeting PE, Wilson EM, Lubahn DB, French FS, Riggs BL, Spelsberg TC. Identification of androgen receptors in normal human osteoblast-like cells. Proc Natl Acad Sci U S A 1989; 86:854-857
314. Kasperk CH, Wergedal JE, Farley JR, Linkhart TA, Turner RT, Baylink DJ. Androgens directly stimulate proliferation of bone cells in vitro. Endocrinology 1989; 124:1576-1578
315. Lapauw B, Vandewalle S, Taes Y, Goemaere S, Zmierczak H, Collette J, Kaufman JM. Serum sclerostin levels in men with idiopathic osteoporosis. Eur J Endocrinol 2013; 168:615-620
316. Zhang XZ, Kalu DN, Erbas B, Hopper JL, Seeman E. The effects of gonadectomy on bone size, mass, and volumetric density in growing rats are gender-, site-, and growth hormone-specific. J Bone Miner Res 1999; 14:802-809
317. Di Nisio A, De Toni L, Rocca MS, Ghezzi M, Selice R, Taglialavoro G, Ferlin A, Foresta C. Negative Association Between Sclerostin and INSL3 in Isolated Human Osteocytes and in Klinefelter Syndrome: New Hints for Testis-Bone Crosstalk. J Clin Endocrinol Metab 2018; 103:2033-2041
318. Zoch ML, Clemens TL, Riddle RC. New insights into the biology of osteocalcin. Bone 2016; 82:42-49
319. Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 2001; 104:719-730
320. Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 2002; 298:843-846
321. Coviello AD, Kaplan B, Lakshman KM, Chen T, Singh AB, Bhasin S. Effects of graded doses of testosterone on erythropoiesis in healthy young and older men. J Clin Endocrinol Metab 2008; 93:914-919
322. Bachman E, Travison TG, Basaria S, Davda MN, Guo W, Li M, Connor Westfall J, Bae H, Gordeuk V, Bhasin S. Testosterone induces erythrocytosis via increased erythropoietin and suppressed hepcidin: evidence for a new erythropoietin/hemoglobin set point. J Gerontol A Biol Sci Med Sci 2014; 69:725-735
323. Wu JY, Scadden DT, Kronenberg HM. Role of the osteoblast lineage in the bone marrow hematopoietic niches. J Bone Miner Res 2009; 24:759-764
324. Valderrabano RJ, Wu JY. Bone and blood interactions in human health and disease. Bone 2019; 119:65-70
325. Valderrabano RJ, Lui LY, Lee J, Cummings SR, Orwoll ES, Hoffman AR, Wu JY, Osteoporotic Fractures in Men Study Research G. Bone Density Loss Is Associated With Blood Cell Counts. J Bone Miner Res 2017; 32:212-220
326. Valderrabano RJ, Lee J, Lui LY, Hoffman AR, Cummings SR, Orwoll ES, Wu JY, Osteoporotic Fractures in Men Study Research G. Older Men With Anemia Have Increased Fracture Risk Independent of Bone Mineral Density. J Clin Endocrinol Metab 2017; 102:2199-2206
327. Valderrabano RJ, Buzkova P, Chang PY, Zakai NA, Fink HA, Robbins JA, Wu JY, Lee JS, Cardiovascular Health Study g. Associations of hemoglobin and change in hemoglobin with risk of incident hip fracture in older men and women: the cardiovascular health study. Osteoporos Int 2021; 32:1669-1677
328. Ferrucci L, Maggio M, Bandinelli S, Basaria S, Lauretani F, Ble A, Valenti G, Ershler WB, Guralnik JM, Longo DL. Low testosterone levels and the risk of anemia in older men and women. Arch Intern Med 2006; 166:1380-1388
329. Roy CN, Snyder PJ, Stephens-Shields AJ, Artz AS, Bhasin S, Cohen HJ, Farrar JT, Gill TM, Zeldow B, Cella D, Barrett-Connor E, Cauley JA, Crandall JP, Cunningham GR, Ensrud KE, Lewis CE, Matsumoto AM, Molitch ME, Pahor M, Swerdloff RS, Cifelli D, Hou X, Resnick SM, Walston JD, Anton S, Basaria S, Diem SJ, Wang C, Schrier SL, Ellenberg SS. Association of Testosterone Levels With Anemia in Older Men: A Controlled Clinical Trial. JAMA Intern Med 2017; 177:480-490
330. Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol 1990; 294:76-95
331. Lu S, Simon NG, Wang Y, Hu S. Neural androgen receptor regulation: effects of androgen and antiandrogen. J Neurobiol 1999; 41:505-512
332. Lustig RH. Sex hormone modulation of neural development in vitro. Horm Behav 1994; 28:383-395
333. Rubinow DR, Schmidt PJ. Androgens, brain, and behavior. Am J Psychiatry 1996; 153:974-984
334. Tobin VA, Millar RP, Canny BJ. Testosterone acts directly at the pituitary to regulate gonadotropin-releasing hormone-induced calcium signals in male rat gonadotropes. Endocrinology 1997; 138:3314-3319
335. Melcangi RC, Celotti F, Castano P, Martini L. Differential localization of the 5 alpha-reductase and the 3 alpha-hydroxysteroid dehydrogenase in neuronal and glial cultures. Endocrinology 1993; 132:1252-1259
336. Reddy DS. Testosterone modulation of seizure susceptibility is mediated by neurosteroids 3alpha-androstanediol and 17beta-estradiol. Neuroscience 2004; 129:195-207
337. Handa RJ, Sharma D, Uht R. A role for the androgen metabolite, 5alpha androstane 3beta, 17beta diol (3beta-diol) in the regulation of the hypothalamo-pituitary-adrenal axis. Front Endocrinol (Lausanne) 2011; 2:65
338. Hsu B, Cumming RG, Waite LM, Blyth FM, Naganathan V, Le Couteur DG, Seibel MJ, Handelsman DJ. Longitudinal Relationships between Reproductive Hormones and Cognitive Decline in Older Men: The Concord Health and Ageing in Men Project. J Clin Endocrinol Metab 2015; 100:2223-2230
339. Maccoby EE, Jacklin CN. The Psychology of Sex Differences. Stanford, CA: Stanford University Press; 1974.
340. Christiansen K. Sex hormone-related variations of cognitive performance in !Kung San hunter-gatherers of Namibia. Neuropsychobiology 1993; 27:97-107
341. Nass R, Baker S. Androgen effects on cognition: congenital adrenal hyperplasia. Psychoneuroendocrinology 1991; 16:189-201
342. Jones BA, Watson NV. Spatial memory performance in androgen insensitive male rats. Physiol Behav 2005; 85:135-141
343. Silverman I, Kastuk D, Choi J, Phillips K. Testosterone levels and spatial ability in men. Psychoneuroendocrinology 1999; 24:813-822
344. Hooven CK, Chabris CF, Ellison PT, Kosslyn SM. The relationship of male testosterone to components of mental rotation. Neuropsychologia 2004; 42:782-790
345. Moffat SD, Hampson E. A curvilinear relationship between testosterone and spatial cognition in humans: possible influence of hand preference. Psychoneuroendocrinology 1996; 21:323-337
346. Moffat SD, Zonderman AB, Metter EJ, Blackman MR, Harman SM, Resnick SM. Longitudinal assessment of serum free testosterone concentration predicts memory performance and cognitive status in elderly men. J Clin Endocrinol Metab 2002; 87:5001-5007
347. LeBlanc ES, Wang PY, Janowsky JS, Neiss MB, Fink HA, Yaffe K, Marshall LM, Lapidus JA, Stefanick ML, Orwoll ES, Osteoporotic Fractures in Men Research G. Association between sex steroids and cognition in elderly men. Clin Endocrinol (Oxf) 2010; 72:393-403
348. Lessov-Schlaggar CN, Reed T, Swan GE, Krasnow RE, DeCarli C, Marcus R, Holloway L, Wolf PA, Carmelli D. Association of sex steroid hormones with brain morphology and cognition in healthy elderly men. Neurology 2005; 65:1591-1596
349. Gouras GK, Xu H, Gross RS, Greenfield JP, Hai B, Wang R, Greengard P. Testosterone reduces neuronal secretion of Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci U S A 2000; 97:1202-1205
350. Ramsden M, Nyborg AC, Murphy MP, Chang L, Stanczyk FZ, Golde TE, Pike CJ. Androgens modulate beta-amyloid levels in male rat brain. J Neurochem 2003; 87:1052-1055
351. Pike CJ, Nguyen TV, Ramsden M, Yao M, Murphy MP, Rosario ER. Androgen cell signaling pathways involved in neuroprotective actions. Horm Behav 2008; 53:693-705
352. Rosario ER, Carroll J, Pike CJ. Testosterone regulation of Alzheimer-like neuropathology in male 3xTg-AD mice involves both estrogen and androgen pathways. Brain Res 2010; 1359:281-290
353. Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci 2006; 26:13384-13389
354. Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci 2008; 28:8430-8441
355. Yao M, Nguyen TV, Rosario ER, Ramsden M, Pike CJ. Androgens regulate neprilysin expression: role in reducing beta-amyloid levels. J Neurochem 2008; 105:2477-2488
356. Papasozomenos S, Shanavas A. Testosterone prevents the heat shock-induced overactivation of glycogen synthase kinase-3 beta but not of cyclin-dependent kinase 5 and c-Jun NH2-terminal kinase and concomitantly abolishes hyperphosphorylation of tau: implications for Alzheimer's disease. Proc Natl Acad Sci U S A 2002; 99:1140-1145
357. Papasozomenos SC. The heat shock-induced hyperphosphorylation of tau is estrogen-independent and prevented by androgens: implications for Alzheimer disease. Proc Natl Acad Sci U S A 1997; 94:6612-6617
358. Liu XA, Zhu LQ, Zhang Q, Shi HR, Wang SH, Wang Q, Wang JZ. Estradiol attenuates tau hyperphosphorylation induced by upregulation of protein kinase-A. Neurochem Res 2008; 33:1811-1820
359. Jones KJ, Brown TJ, Damaser M. Neuroprotective effects of gonadal steroids on regenerating peripheral motoneurons. Brain Res Brain Res Rev 2001; 37:372-382
360. Jones KJ, Coers S, Storer PD, Tanzer L, Kinderman NB. Androgenic regulation of the central glia response following nerve damage. J Neurobiol 1999; 40:560-573
361. Hammond J, Le Q, Goodyer C, Gelfand M, Trifiro M, LeBlanc A. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J Neurochem 2001; 77:1319-1326
362. Kujawa KA, Emeric E, Jones KJ. Testosterone differentially regulates the regenerative properties of injured hamster facial motoneurons. J Neurosci 1991; 11:3898-3906
363. Kujawa KA, Jacob JM, Jones KJ. Testosterone regulation of the regenerative properties of injured rat sciatic motor neurons. J Neurosci Res 1993; 35:268-273
364. Tetzlaff JE, Huppenbauer CB, Tanzer L, Alexander TD, Jones KJ. Motoneuron injury and repair: New perspectives on gonadal steroids as neurotherapeutics. J Mol Neurosci 2006; 28:53-64
365. Rhodes ME, Frye CA. Androgens in the hippocampus can alter, and be altered by, ictal activity. Pharmacol Biochem Behav 2004; 78:483-493
366. Nunez JL, Jurgens HA, Juraska JM. Androgens reduce cell death in the developing rat visual cortex. Brain Res Dev Brain Res 2000; 125:83-88
367. Hamson DK, Wainwright SR, Taylor JR, Jones BA, Watson NV, Galea LA. Androgens increase survival of adult-born neurons in the dentate gyrus by an androgen receptor-dependent mechanism in male rats. Endocrinology 2013; 154:3294-3304
368. Ramsden M, Shin TM, Pike CJ. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience 2003; 122:573-578
369. Nguyen TV, Yao M, Pike CJ. Androgens activate mitogen-activated protein kinase signaling: role in neuroprotection. J Neurochem 2005; 94:1639-1651
370. Jayaraman A, Lent-Schochet D, Pike CJ. Diet-induced obesity and low testosterone increase neuroinflammation and impair neural function. J Neuroinflammation 2014; 11:162
371. Xu J, Itoh Y, Hayashi H, Takii T, Miyazawa K, Onozaki K. Dihydrotestosterone inhibits interleukin-1alpha or tumor necrosis factor alpha-induced proinflammatory cytokine production via androgen receptor-dependent inhibition of nuclear factor-kappaB activation in rheumatoid fibroblast-like synovial cell line. Biol Pharm Bull 2011; 34:1724-1730
372. Roof RL. The dentate gyrus is sexually dimorphic in prepubescent rats: testosterone plays a significant role. Brain Res 1993; 610:148-151
373. Scharfman HE, MacLusky NJ. Differential regulation of BDNF, synaptic plasticity and sprouting in the hippocampal mossy fiber pathway of male and female rats. Neuropharmacology 2014; 76 Pt C:696-708
374. Parducz A, Garcia-Segura LM. Sexual differences in the synaptic connectivity in the rat dentate gyrus. Neurosci Lett 1993; 161:53-56
375. Meyer G, Ferres-Torres R, Mas M. The effects of puberty and castration on hippocampal dendritic spines of mice. A Golgi study. Brain Res 1978; 155:108-112
376. Naghdi N, Asadollahi A. Genomic and nongenomic effects of intrahippocampal microinjection of testosterone on long-term memory in male adult rats. Behav Brain Res 2004; 153:1-6
377. Naghdi N, Majlessi N, Bozorgmehr T. The effect of intrahippocampal injection of testosterone enanthate (an androgen receptor agonist) and anisomycin (protein synthesis inhibitor) on spatial learning and memory in adult, male rats. Behav Brain Res 2005; 156:263-268
378. Flores-Ramos M, Alcauter S, Lopez-Titla M, Bernal-Santamaria N, Calva-Coraza E, Edden RAE. Testosterone is related to GABA+ levels in the posterior-cingulate in unmedicated depressed women during reproductive life. J Affect Disord 2019; 242:143-149
379. Hogervorst E, Combrinck M, Smith AD. Testosterone and gonadotropin levels in men with dementia. Neuro Endocrinol Lett 2003; 24:203-208
380. Hogervorst E, Lehmann DJ, Warden DR, McBroom J, Smith AD. Apolipoprotein E epsilon4 and testosterone interact in the risk of Alzheimer's disease in men. Int J Geriatr Psychiatry 2002; 17:938-940
381. Hogervorst E, Williams J, Budge M, Barnetson L, Combrinck M, Smith AD. Serum total testosterone is lower in men with Alzheimer's disease. Neuro Endocrinol Lett 2001; 22:163-168
382. Moffat SD, Zonderman AB, Metter EJ, Kawas C, Blackman MR, Harman SM, Resnick SM. Free testosterone and risk for Alzheimer disease in older men. Neurology 2004; 62:188-193
383. Paoletti AM, Congia S, Lello S, Tedde D, Orru M, Pistis M, Pilloni M, Zedda P, Loddo A, Melis GB. Low androgenization index in elderly women and elderly men with Alzheimer's disease. Neurology 2004; 62:301-303
384. Rosario ER, Chang L, Stanczyk FZ, Pike CJ. Age-related testosterone depletion and the development of Alzheimer disease. JAMA 2004; 292:1431-1432
385. Hier DB, Crowley WF, Jr. Spatial ability in androgen-deficient men. N Engl J Med 1982; 306:1202-1205
386. Hogervorst E, Matthews FE, Brayne C. Are optimal levels of testosterone associated with better cognitive function in healthy older women and men? Biochim Biophys Acta 2010; 1800:1145-1152
387. Matousek RH, Sherwin BB. Sex steroid hormones and cognitive functioning in healthy, older men. Horm Behav 2010; 57:352-359
388. Muller M, Aleman A, Grobbee DE, de Haan EH, van der Schouw YT. Endogenous sex hormone levels and cognitive function in aging men: is there an optimal level? Neurology 2005; 64:866-871
389. Hua JT, Hildreth KL, Pelak VS. Effects of Testosterone Therapy on Cognitive Function in Aging: A Systematic Review. Cogn Behav Neurol 2016; 29:122-138
390. Resnick SM, Matsumoto AM, Stephens-Shields AJ, Ellenberg SS, Gill TM, Shumaker SA, Pleasants DD, Barrett-Connor E, Bhasin S, Cauley JA, Cella D, Crandall JP, Cunningham GR, Ensrud KE, Farrar JT, Lewis CE, Molitch ME, Pahor M, Swerdloff RS, Cifelli D, Anton S, Basaria S, Diem SJ, Wang C, Hou X, Snyder PJ. Testosterone Treatment and Cognitive Function in Older Men With Low Testosterone and Age-Associated Memory Impairment. JAMA 2017; 317:717-727
391. Lu PH, Masterman DA, Mulnard R, Cotman C, Miller B, Yaffe K, Reback E, Porter V, Swerdloff R, Cummings JL. Effects of testosterone on cognition and mood in male patients with mild Alzheimer disease and healthy elderly men. Arch Neurol 2006; 63:177-185
392. Fukai S, Akishita M, Yamada S, Toba K, Ouchi Y. Effects of testosterone in older men with mild-to-moderate cognitive impairment. J Am Geriatr Soc 2010; 58:1419-1421
393. Cherrier MM, Craft S, Matsumoto AH. Cognitive changes associated with supplementation of testosterone or dihydrotestosterone in mildly hypogonadal men: a preliminary report. J Androl 2003; 24:568-576
394. Huang G, Wharton W, Bhasin S, Harman SM, Pencina KM, Tsitouras P, Li Z, Hally KA, Asthana S, Storer TW, Basaria S. Effects of long-term testosterone administration on cognition in older men with low or low-to-normal testosterone concentrations: a prespecified secondary analysis of data from the randomised, double-blind, placebo-controlled TEAAM trial. Lancet Diabetes Endocrinol 2016; 4:657-665
395. Devanand DP, Nobler MS, Singer T, Kiersky JE, Turret N, Roose SP, Sackeim HA. Is dysthymia a different disorder in the elderly? Am J Psychiatry 1994; 151:1592-1599
396. Shores MM, Sloan KL, Matsumoto AM, Moceri VM, Felker B, Kivlahan DR. Increased incidence of diagnosed depressive illness in hypogonadal older men. Arch Gen Psychiatry 2004; 61:162-167
397. Schweiger U, Deuschle M, Weber B, Korner A, Lammers CH, Schmider J, Gotthardt U, Heuser I. Testosterone, gonadotropin, and cortisol secretion in male patients with major depression. Psychosom Med 1999; 61:292-296
398. Bhasin S, Seidman SN. Testosterone Treatment of Depressive Disorders in Men – Too Much Smoke, not Enough High Quality Evidence. JAMA Psychiatry 2018; in press
399. Seidman SN, Spatz E, Rizzo C, Roose SP. Testosterone replacement therapy for hypogonadal men with major depressive disorder: a randomized, placebo-controlled clinical trial. J Clin Psychiatry 2001; 62:406-412
400. Wang C, Alexander G, Berman N, Salehian B, Davidson T, McDonald V, Steiner B, Hull L, Callegari C, Swerdloff RS. Testosterone replacement therapy improves mood in hypogonadal men--a clinical research center study. J Clin Endocrinol Metab 1996; 81:3578-3583
401. Pope HG, Jr., Cohane GH, Kanayama G, Siegel AJ, Hudson JI. Testosterone gel supplementation for men with refractory depression: a randomized, placebo-controlled trial. Am J Psychiatry 2003; 160:105-111
402. Seidman SN, Miyazaki M, Roose SP. Intramuscular testosterone supplementation to selective serotonin reuptake inhibitor in treatment-resistant depressed men: randomized placebo-controlled clinical trial. J Clin Psychopharmacol 2005; 25:584-588
403. Walther A, Breidenstein J, Miller R. Association of Testosterone Treatment With Alleviation of Depressive Symptoms in Men: A Systematic Review and Meta-analysis. JAMA Psychiatry 2019; 76:31-40
404. Bhasin S, Seidman S. Testosterone Treatment of Depressive Disorders in Men: Too Much Smoke, Not Enough High-Quality Evidence. JAMA Psychiatry 2019; 76:9-10
405. Shores MM, Kivlahan DR, Sadak TI, Li EJ, Matsumoto AM. A randomized, double-blind, placebo-controlled study of testosterone treatment in hypogonadal older men with subthreshold depression (dysthymia or minor depression). J Clin Psychiatry 2009; 70:1009-1016
406. Seidman SN, Orr G, Raviv G, Levi R, Roose SP, Kravitz E, Amiaz R, Weiser M. Effects of testosterone replacement in middle-aged men with dysthymia: a randomized, placebo-controlled clinical trial. J Clin Psychopharmacol 2009; 29:216-221
407. Rabkin JG, Ferrando SJ, Wagner GJ, Rabkin R. DHEA treatment for HIV+ patients: effects on mood, androgenic and anabolic parameters. Psychoneuroendocrinology 2000; 25:53-68
408. Guo W, Bachman E, Li M, Roy CN, Blusztajn J, Wong S, Chan SY, Serra C, Jasuja R, Travison TG, Muckenthaler MU, Nemeth E, Bhasin S. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 2013; 12:280-291
409. Guo W, Li M, Bhasin S. Testosterone supplementation improves anemia in aging male mice. J Gerontol A Biol Sci Med Sci 2014; 69:505-513
410. Guo W, Wong S, Li M, Liang W, Liesa M, Serra C, Jasuja R, Bartke A, Kirkland JL, Shirihai O, Bhasin S. Testosterone plus low-intensity physical training in late life improves functional performance, skeletal muscle mitochondrial biogenesis, and mitochondrial quality control in male mice. PLoS One 2012; 7:e51180
411. Kouri EM, Lukas SE, Pope HG, Jr., Oliva PS. Increased aggressive responding in male volunteers following the administration of gradually increasing doses of testosterone cypionate. Drug Alcohol Depend 1995; 40:73-79
412. Pope HG, Jr., Katz DL. Affective and psychotic symptoms associated with anabolic steroid use. Am J Psychiatry 1988; 145:487-490
413. Calof OM, Singh AB, Lee ML, Kenny AM, Urban RJ, Tenover JL, Bhasin S. Adverse events associated with testosterone replacement in middle-aged and older men: a meta-analysis of randomized, placebo-controlled trials. J Gerontol A Biol Sci Med Sci 2005; 60:1451-1457
414. Rolf C, Nieschlag E. Potential adverse effects of long-term testosterone therapy. Baillieres Clin Endocrinol Metab 1998; 12:521-534
415. Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev 2003; 24:313-340
416. Gagliano-Juca T, Basaria S. Trials of testosterone replacement reporting cardiovascular adverse events. Asian J Androl 2018; 20:131-137
417. Gagliano-Juca T, Basaria S. Testosterone replacement therapy and cardiovascular risk. Nat Rev Cardiol 2019; 16:555-574
418. Haffner SM, Laakso M, Miettinen H, Mykkanen L, Karhapaa P, Rainwater DL. Low levels of sex hormone-binding globulin and testosterone are associated with smaller, denser low density lipoprotein in normoglycemic men. J Clin Endocrinol Metab 1996; 81:3697-3701
419. Khaw KT, Barrett-Connor E. Endogenous sex hormones, high density lipoprotein cholesterol, and other lipoprotein fractions in men. Arterioscler Thromb 1991; 11:489-494
420. Page ST, Mohr BA, Link CL, O'Donnell AB, Bremner WJ, McKinlay JB. Higher testosterone levels are associated with increased high-density lipoprotein cholesterol in men with cardiovascular disease: results from the Massachusetts Male Aging Study. Asian J Androl 2008; 10:193-200
421. Zhang N, Zhang H, Zhang X, Zhang B, Wang F, Wang C, Zhao M, Yu C, Gao L, Zhao J, Guan Q. The relationship between endogenous testosterone and lipid profile in middle-aged and elderly Chinese men. Eur J Endocrinol 2014; 170:487-494
422. Haffner SM, Mykkanen L, Valdez RA, Katz MS. Relationship of sex hormones to lipids and lipoproteins in nondiabetic men. J Clin Endocrinol Metab 1993; 77:1610-1615
423. De Pergola G, De Mitrio V, Sciaraffia M, Pannacciulli N, Minenna A, Giorgino F, Petronelli M, Laudadio E, Giorgino R. Lower androgenicity is associated with higher plasma levels of prothrombotic factors irrespective of age, obesity, body fat distribution, and related metabolic parameters in men. Metabolism 1997; 46:1287-1293
424. Crist DM, Peake GT, Stackpole PJ. Lipemic and lipoproteinemic effects of natural and synthetic androgens in humans. Clin Exp Pharmacol Physiol 1986; 13:513-518
425. Herbst KL, Amory JK, Brunzell JD, Chansky HA, Bremner WJ. Testosterone administration to men increases hepatic lipase activity and decreases HDL and LDL size in 3 wk. Am J Physiol Endocrinol Metab 2003; 284:E1112-1118
426. Hurley BF, Seals DR, Hagberg JM, Goldberg AC, Ostrove SM, Holloszy JO, Wiest WG, Goldberg AP. High-density-lipoprotein cholesterol in bodybuilders v powerlifters. Negative effects of androgen use. JAMA 1984; 252:507-513
427. Thompson PD, Cullinane EM, Sady SP, Chenevert C, Saritelli AL, Sady MA, Herbert PN. Contrasting effects of testosterone and stanozolol on serum lipoprotein levels. JAMA 1989; 261:1165-1168
428. Snyder PJ, Peachey H, Berlin JA, Rader D, Usher D, Loh L, Hannoush P, Dlewati A, Holmes JH, Santanna J, Strom BL. Effect of transdermal testosterone treatment on serum lipid and apolipoprotein levels in men more than 65 years of age. Am J Med 2001; 111:255-260
429. Whitsel EA, Boyko EJ, Matsumoto AM, Anawalt BD, Siscovick DS. Intramuscular testosterone esters and plasma lipids in hypogonadal men: a meta-analysis. Am J Med 2001; 111:261-269
430. Mohler ER, 3rd, Ellenberg SS, Lewis CE, Wenger NK, Budoff MJ, Lewis MR, Barrett-Connor E, Swerdloff RS, Stephens-Shields A, Bhasin S, Cauley JA, Crandall JP, Cunningham GR, Ensrud KE, Gill TM, Matsumoto AM, Molitch ME, Pahor M, Preston PE, Hou X, Cifelli D, Snyder PJ. The Effect of Testosterone on Cardiovascular Biomarkers in the Testosterone Trials. J Clin Endocrinol Metab 2018; 103:681-688
431. Rubinow KB, Vaisar T, Tang C, Matsumoto AM, Heinecke JW, Page ST. Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity. J Lipid Res 2012; 53:1376-1383
432. Glueck CJ, Glueck HI, Stroop D, Speirs J, Hamer T, Tracy T. Endogenous testosterone, fibrinolysis, and coronary heart disease risk in hyperlipidemic men. J Lab Clin Med 1993; 122:412-420
433. Eugster M, Reinhart WH. The influence of the haematocrit on primary haemostasis in vitro. Thromb Haemost 2005; 94:1213-1218
434. Gagliano-Juca T, Pencina KM, Guo W, Li Z, Huang G, Basaria S, Bhasin S. Differential effects of testosterone on circulating neutrophils, monocytes, and platelets in men: Findings from two trials. Andrology 2020; 8:1324-1331
435. Welsh C, Welsh P, Mark PB, Celis-Morales CA, Lewsey J, Gray SR, Lyall DM, Iliodromiti S, Gill JMR, Pell J, Jhund PS, Sattar N. Association of Total and Differential Leukocyte Counts With Cardiovascular Disease and Mortality in the UK Biobank. Arterioscler Thromb Vasc Biol 2018; 38:1415-1423
436. Choi SH, Kim JH, Lim S, Lim JY, Kim KW, Park KS, Jang HC. Monocyte count as a predictor of cardiovascular mortality in older Korean people. Age Ageing 2017; 46:433-438
437. Ajayi AA, Mathur R, Halushka PV. Testosterone increases human platelet thromboxane A2 receptor density and aggregation responses. Circulation 1995; 91:2742-2747
438. Ajayi AA, Halushka PV. Castration reduces platelet thromboxane A2 receptor density and aggregability. QJM 2005; 98:349-356
439. Martinez C, Suissa S, Rietbrock S, Katholing A, Freedman B, Cohen AT, Handelsman DJ. Testosterone treatment and risk of venous thromboembolism: population based case-control study. BMJ 2016; 355:i5968
440. Ramasamy R, Scovell J, Mederos M, Ren R, Jain L, Lipshultz L. Association Between Testosterone Supplementation Therapy and Thrombotic Events in Elderly Men. Urology 2015; 86:283-285
441. Sharma R, Oni OA, Chen G, Sharma M, Dawn B, Sharma R, Parashara D, Savin VJ, Barua RS, Gupta K. Association Between Testosterone Replacement Therapy and the Incidence of DVT and Pulmonary Embolism: A Retrospective Cohort Study of the Veterans Administration Database. Chest 2016; 150:563-571
442. Shores MM. Testosterone treatment and cardiovascular events in prescription database studies. Asian J Androl 2018; 20:138-144
443. Schoenfeld MJ, Shortridge E, Cui Z, Muram D. Medication adherence and treatment patterns for hypogonadal patients treated with topical testosterone therapy: a retrospective medical claims analysis. J Sex Med 2013; 10:1401-1409
444. Zhang Y, Gao Y, Tan A, Yang X, Zhang H, Zhang S, Wu C, Lu Z, Wang M, Liao M, Qin X, Li L, Hu Y, Mo Z. Endogenous sex hormones and C-reactive protein in healthy Chinese men. Clin Endocrinol (Oxf) 2013; 78:60-66
445. Kaplan SA, Johnson-Levonas AO, Lin J, Shah AK, Meehan AG. Elevated high sensitivity C-reactive protein levels in aging men with low testosterone. Aging Male 2010; 13:108-112
446. Pastuszak AW, Kohn TP, Estis J, Lipshultz LI. Low Plasma Testosterone Is Associated With Elevated Cardiovascular Disease Biomarkers. J Sex Med 2017; 14:1095-1103
447. Tsilidis KK, Rohrmann S, McGlynn KA, Nyante SJ, Lopez DS, Bradwin G, Feinleib M, Joshu CE, Kanarek N, Nelson WG, Selvin E, Platz EA. Association between endogenous sex steroid hormones and inflammatory biomarkers in US men. Andrology 2013; 1:919-928
448. Maggio M, Basaria S, Ble A, Lauretani F, Bandinelli S, Ceda GP, Valenti G, Ling SM, Ferrucci L. Correlation between testosterone and the inflammatory marker soluble interleukin-6 receptor in older men. J Clin Endocrinol Metab 2006; 91:345-347
449. Haring R, Baumeister SE, Volzke H, Dorr M, Kocher T, Nauck M, Wallaschofski H. Prospective inverse associations of sex hormone concentrations in men with biomarkers of inflammation and oxidative stress. J Androl 2012; 33:944-950
450. Ng MK, Liu PY, Williams AJ, Nakhla S, Ly LP, Handelsman DJ, Celermajer DS. Prospective study of effect of androgens on serum inflammatory markers in men. Arterioscler Thromb Vasc Biol 2002; 22:1136-1141
451. Singh AB, Hsia S, Alaupovic P, Sinha-Hikim I, Woodhouse L, Buchanan TA, Shen R, Bross R, Berman N, Bhasin S. The effects of varying doses of T on insulin sensitivity, plasma lipids, apolipoproteins, and C-reactive protein in healthy young men. J Clin Endocrinol Metab 2002; 87:136-143
452. Gagliano-Juca T, Burak MF, Pencina KM, Li Z, Edwards RR, Travison TG, Basaria S. Metabolic Changes in Androgen Deprived Non-Diabetic Men with Prostate Cancer are not mediated by Cytokines or aP2. J Clin Endocrinol Metab 2018;
453. Phillips GB, Pinkernell BH, Jing TY. The association of hypotestosteronemia with coronary artery disease in men. Arterioscler Thromb 1994; 14:701-706
454. Tibblin G, Adlerberth A, Lindstedt G, Bjorntorp P. The pituitary-gonadal axis and health in elderly men: a study of men born in 1913. Diabetes 1996; 45:1605-1609
455. Arnlov J, Pencina MJ, Amin S, Nam BH, Benjamin EJ, Murabito JM, Wang TJ, Knapp PE, D'Agostino RB, Sr., Bhasin S, Vasan RS. Endogenous sex hormones and cardiovascular disease incidence in men. Ann Intern Med 2006; 145:176-184
456. Wu SZ, Weng XZ. Therapeutic effects of an androgenic preparation on myocardial ischemia and cardiac function in 62 elderly male coronary heart disease patients. Chin Med J (Engl) 1993; 106:415-418
457. Jaffe MD. Effect of testosterone cypionate on postexercise ST segment depression. Br Heart J 1977; 39:1217-1222
458. Ong PJ, Patrizi G, Chong WC, Webb CM, Hayward CS, Collins P. Testosterone enhances flow-mediated brachial artery reactivity in men with coronary artery disease. Am J Cardiol 2000; 85:269-272
459. Rosano GM, Leonardo F, Pagnotta P, Pelliccia F, Panina G, Cerquetani E, della Monica PL, Bonfigli B, Volpe M, Chierchia SL. Acute anti-ischemic effect of testosterone in men with coronary artery disease. Circulation 1999; 99:1666-1670
460. Webb CM, Adamson DL, de Zeigler D, Collins P. Effect of acute testosterone on myocardial ischemia in men with coronary artery disease. Am J Cardiol 1999; 83:437-439, A439
461. English KM, Steeds RP, Jones TH, Diver MJ, Channer KS. Low-dose transdermal testosterone therapy improves angina threshold in men with chronic stable angina: A randomized, double-blind, placebo-controlled study. Circulation 2000; 102:1906-1911
462. Thompson PD, Ahlberg AW, Moyna NM, Duncan B, Ferraro-Borgida M, White CM, McGill CC, Heller GV. Effect of intravenous testosterone on myocardial ischemia in men with coronary artery disease. Am Heart J 2002; 143:249-256
463. Yue P, Chatterjee K, Beale C, Poole-Wilson PA, Collins P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation 1995; 91:1154-1160
464. Yu J, Akishita M, Eto M, Ogawa S, Son BK, Kato S, Ouchi Y, Okabe T. Androgen receptor-dependent activation of endothelial nitric oxide synthase in vascular endothelial cells: role of phosphatidylinositol 3-kinase/akt pathway. Endocrinology 2010; 151:1822-1828
465. Scragg JL, Jones RD, Channer KS, Jones TH, Peers C. Testosterone is a potent inhibitor of L-type Ca(2+) channels. Biochem Biophys Res Commun 2004; 318:503-506
466. Nathan L, Shi W, Dinh H, Mukherjee TK, Wang X, Lusis AJ, Chaudhuri G. Testosterone inhibits early atherogenesis by conversion to estradiol: critical role of aromatase. Proc Natl Acad Sci U S A 2001; 98:3589-3593
467. Budoff MJ, Ellenberg SS, Lewis CE, Mohler ER, 3rd, Wenger NK, Bhasin S, Barrett-Connor E, Swerdloff RS, Stephens-Shields A, Cauley JA, Crandall JP, Cunningham GR, Ensrud KE, Gill TM, Matsumoto AM, Molitch ME, Nakanishi R, Nezarat N, Matsumoto S, Hou X, Basaria S, Diem SJ, Wang C, Cifelli D, Snyder PJ. Testosterone Treatment and Coronary Artery Plaque Volume in Older Men With Low Testosterone. JAMA 2017; 317:708-716
468. Salem JE, Alexandre J, Bachelot A, Funck-Brentano C. Influence of steroid hormones on ventricular repolarization. Pharmacol Ther 2016; 167:38-47
469. Ridley JM, Shuba YM, James AF, Hancox JC. Modulation by testosterone of an endogenous hERG potassium channel current. J Physiol Pharmacol 2008; 59:395-407
470. Er F, Michels G, Brandt MC, Khan I, Haase H, Eicks M, Lindner M, Hoppe UC. Impact of testosterone on cardiac L-type calcium channels and Ca2+ sparks: acute actions antagonize chronic effects. Cell Calcium 2007; 41:467-477
471. Vicente J, Johannesen L, Galeotti L, Strauss DG. Mechanisms of sex and age differences in ventricular repolarization in humans. Am Heart J 2014; 168:749-756
472. Zhang Y, Ouyang P, Post WS, Dalal D, Vaidya D, Blasco-Colmenares E, Soliman EZ, Tomaselli GF, Guallar E. Sex-steroid hormones and electrocardiographic QT-interval duration: findings from the third National Health and Nutrition Examination Survey and the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol 2011; 174:403-411
473. Gagliano-Juca T, Icli TB, Pencina KM, Li Z, Tapper J, Huang G, Travison TG, Tsitouras P, Harman SM, Storer TW, Bhasin S, Basaria S. Effects of Testosterone Replacement on Electrocardiographic Parameters in Men: Findings From Two Randomized Trials. J Clin Endocrinol Metab 2017; 102:1478-1485
474. Schwartz JB, Volterrani M, Caminiti G, Marazzi G, Fini M, Rosano GM, Iellamo F. Effects of testosterone on the Q-T interval in older men and older women with chronic heart failure. Int J Androl 2011; 34:e415-421
475. Gagliano-Juca T, Travison TG, Kantoff PW, Nguyen PL, Taplin ME, Kibel AS, Huang G, Bearup R, Schram H, Manley R, Beleva YM, Edwards RR, Basaria S. Androgen Deprivation Therapy Is Associated With Prolongation of QTc Interval in Men With Prostate Cancer. J Endocr Soc 2018; 2:485-496
476. Zhang Y, Post WS, Blasco-Colmenares E, Dalal D, Tomaselli GF, Guallar E. Electrocardiographic QT interval and mortality: a meta-analysis. Epidemiology 2011; 22:660-670
477. Noseworthy PA, Peloso GM, Hwang SJ, Larson MG, Levy D, O'Donnell CJ, Newton-Cheh C. QT interval and long-term mortality risk in the Framingham Heart Study. Ann Noninvasive Electrocardiol 2012; 17:340-348
478. Nielsen JB, Graff C, Rasmussen PV, Pietersen A, Lind B, Olesen MS, Struijk JJ, Haunso S, Svendsen JH, Kober L, Gerds TA, Holst AG. Risk prediction of cardiovascular death based on the QTc interval: evaluating age and gender differences in a large primary care population. Eur Heart J 2014; 35:1335-1344
479. Keating NL, O'Malley AJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. J Clin Oncol 2006; 24:4448-4456
480. Haque R, UlcickasYood M, Xu X, Cassidy-Bushrow AE, Tsai H-T, Keating NL, Van Den Eeden SK, Potosky AL. Cardiovascular disease risk and androgen deprivation therapy in patients with localised prostate cancer: a prospective cohort study. British Journal of Cancer 2017;
481. Salem JE, Waintraub X, Courtillot C, Shaffer CM, Gandjbakhch E, Maupain C, Moslehi JJ, Badilini F, Haroche J, Gougis P, Fressart V, Glazer AM, Hidden-Lucet F, Touraine P, Lebrun-Vignes B, Roden DM, Bachelot A, Funck-Brentano C. Hypogonadism as a Reversible Cause of Torsades de Pointes in Men. Circulation 2018; 138:110-113
482. Magnani JW, Moser CB, Murabito JM, Sullivan LM, Wang N, Ellinor PT, Vasan RS, Benjamin EJ, Coviello AD. Association of sex hormones, aging, and atrial fibrillation in men: the Framingham Heart Study. Circ Arrhythm Electrophysiol 2014; 7:307-312
483. Rosenberg MA, Shores MM, Matsumoto AM, Buzkova P, Lange LA, Kronmal RA, Heckbert SR, Mukamal KJ. Serum androgens and risk of atrial fibrillation in older men: The Cardiovascular Health Study. Clin Cardiol 2018; 41:830-836
484. Zeller T, Schnabel RB, Appelbaum S, Ojeda F, Berisha F, Schulte-Steinberg B, Brueckmann BE, Kuulasmaa K, Jousilahti P, Blankenberg S, Palosaari T, Salomaa V, Karakas M. Low testosterone levels are predictive for incident atrial fibrillation and ischaemic stroke in men, but protective in women - results from the FINRISK study. Eur J Prev Cardiol 2018; 25:1133-1139
485. Sharma R, Oni OA, Gupta K, Sharma M, Sharma R, Singh V, Parashara D, Kamalakar S, Dawn B, Chen G, Ambrose JA, Barua RS. Normalization of Testosterone Levels After Testosterone Replacement Therapy Is Associated With Decreased Incidence of Atrial Fibrillation. J Am Heart Assoc 2017; 6
486. Fernandez-Balsells MM, Murad MH, Lane M, Lampropulos JF, Albuquerque F, Mullan RJ, Agrwal N, Elamin MB, Gallegos-Orozco JF, Wang AT, Erwin PJ, Bhasin S, Montori VM. Clinical review 1: Adverse effects of testosterone therapy in adult men: a systematic review and meta-analysis. J Clin Endocrinol Metab 2010; 95:2560-2575
487. Xu L, Freeman G, Cowling BJ, Schooling CM. Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials. BMC Med 2013; 11:108
488. Basaria S, Davda MN, Travison TG, Ulloor J, Singh R, Bhasin S. Risk factors associated with cardiovascular events during testosterone administration in older men with mobility limitation. J Gerontol A Biol Sci Med Sci 2013; 68:153-160
489. Sattler FR, Castaneda-Sceppa C, Binder EF, Schroeder ET, Wang Y, Bhasin S, Kawakubo M, Stewart Y, Yarasheski KE, Ulloor J, Colletti P, Roubenoff R, Azen SP. Testosterone and growth hormone improve body composition and muscle performance in older men. J Clin Endocrinol Metab 2009; 94:1991-2001
490. Johannsson G, Gibney J, Wolthers T, Leung KC, Ho KK. Independent and combined effects of testosterone and growth hormone on extracellular water in hypopituitary men. J Clin Endocrinol Metab 2005; 90:3989-3994
491. Alexander GC, Iyer G, Lucas E, Lin D, Singh S. Cardiovascular Risks of Exogenous Testosterone Use Among Men: A Systematic Review and Meta-Analysis. Am J Med 2017; 130:293-305
492. Albert SG, Morley JE. Testosterone therapy, association with age, initiation and mode of therapy with cardiovascular events: a systematic review. Clin Endocrinol (Oxf) 2016; 85:436-443
493. Corona G, Maseroli E, Rastrelli G, Isidori AM, Sforza A, Mannucci E, Maggi M. Cardiovascular risk associated with testosterone-boosting medications: a systematic review and meta-analysis. Expert Opin Drug Saf 2014; 13:1327-1351
494. Corona G, Rastrelli G, Di Pasquale G, Sforza A, Mannucci E, Maggi M. Testosterone and Cardiovascular Risk: Meta-Analysis of Interventional Studies. J Sex Med 2018; 15:820-838
495. Vigen R, O'Donnell CI, Baron AE, Grunwald GK, Maddox TM, Bradley SM, Barqawi A, Woning G, Wierman ME, Plomondon ME, Rumsfeld JS, Ho PM. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA 2013; 310:1829-1836
496. Shores MM, Smith NL, Forsberg CW, Anawalt BD, Matsumoto AM. Testosterone treatment and mortality in men with low testosterone levels. J Clin Endocrinol Metab 2012; 97:2050-2058
497. Finkle WD, Greenland S, Ridgeway GK, Adams JL, Frasco MA, Cook MB, Fraumeni JF, Jr., Hoover RN. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PLoS One 2014; 9:e85805
498. Baillargeon J, Urban RJ, Kuo YF, Ottenbacher KJ, Raji MA, Du F, Lin YL, Goodwin JS. Risk of Myocardial Infarction in Older Men Receiving Testosterone Therapy. Ann Pharmacother 2014; 48:1138-1144
499. Khaw KT, Barrett-Connor E. Lower endogenous androgens predict central adiposity in men. Ann Epidemiol 1992; 2:675-682
500. Seidell JC, Bjorntorp P, Sjostrom L, Kvist H, Sannerstedt R. Visceral fat accumulation in men is positively associated with insulin, glucose, and C-peptide levels, but negatively with testosterone levels. Metabolism 1990; 39:897-901
501. Marin P, Oden B, Bjorntorp P. Assimilation and mobilization of triglycerides in subcutaneous abdominal and femoral adipose tissue in vivo in men: effects of androgens. J Clin Endocrinol Metab 1995; 80:239-243
502. Holmang A, Bjorntorp P. The effects of testosterone on insulin sensitivity in male rats. Acta Physiol Scand 1992; 146:505-510
503. Hobbs CJ, Jones RE, Plymate SR. Nandrolone, a 19-nortestosterone, enhances insulin-independent glucose uptake in normal men. J Clin Endocrinol Metab 1996; 81:1582-1585
504. Ramirez ME, McMurry MP, Wiebke GA, Felten KJ, Ren K, Meikle AW, Iverius PH. Evidence for sex steroid inhibition of lipoprotein lipase in men: comparison of abdominal and femoral adipose tissue. Metabolism 1997; 46:179-185
505. Pitteloud N, Mootha VK, Dwyer AA, Hardin M, Lee H, Eriksson KF, Tripathy D, Yialamas M, Groop L, Elahi D, Hayes FJ. Relationship between testosterone levels, insulin sensitivity, and mitochondrial function in men. Diabetes Care 2005; 28:1636-1642
506. Endre T, Mattiasson I, Berglund G, Hulthen UL. Low testosterone and insulin resistance in hypertension-prone men. J Hum Hypertens 1996; 10:755-761
507. Defay R, Papoz L, Barny S, Bonnot-Lours S, Caces E, Simon D. Hormonal status and NIDDM in the European and Melanesian populations of New Caledonia: a case-control study. The CALedonia DIAbetes Mellitus (CALDIA) Study Group. Int J Obes Relat Metab Disord 1998; 22:927-934
508. Oh JY, Barrett-Connor E, Wedick NM, Wingard DL, Rancho Bernardo S. Endogenous sex hormones and the development of type 2 diabetes in older men and women: the Rancho Bernardo study. Diabetes Care 2002; 25:55-60
509. Haffner SM, Shaten J, Stern MP, Smith GD, Kuller L. Low levels of sex hormone-binding globulin and testosterone predict the development of non-insulin-dependent diabetes mellitus in men. MRFIT Research Group. Multiple Risk Factor Intervention Trial. Am J Epidemiol 1996; 143:889-897
510. Stellato RK, Feldman HA, Hamdy O, Horton ES, McKinlay JB. Testosterone, sex hormone-binding globulin, and the development of type 2 diabetes in middle-aged men: prospective results from the Massachusetts male aging study. Diabetes Care 2000; 23:490-494
511. Dhindsa S, Ghanim H, Batra M, Kuhadiya ND, Abuaysheh S, Sandhu S, Green K, Makdissi A, Hejna J, Chaudhuri A, Punyanitya M, Dandona P. Insulin Resistance and Inflammation in Hypogonadotropic Hypogonadism and Their Reduction After Testosterone Replacement in Men With Type 2 Diabetes. Diabetes Care 2016; 39:82-91
512. Selvin E, Feinleib M, Zhang L, Rohrmann S, Rifai N, Nelson WG, Dobs A, Basaria S, Golden SH, Platz EA. Androgens and diabetes in men: results from the Third National Health and Nutrition Examination Survey (NHANES III). Diabetes Care 2007; 30:234-238
513. Svartberg J, Jenssen T, Sundsfjord J, Jorde R. The associations of endogenous testosterone and sex hormone-binding globulin with glycosylated hemoglobin levels, in community dwelling men. The Tromso Study. Diabetes Metab 2004; 30:29-34
514. Laaksonen DE, Niskanen L, Punnonen K, Nyyssonen K, Tuomainen TP, Valkonen VP, Salonen R, Salonen JT. Testosterone and sex hormone-binding globulin predict the metabolic syndrome and diabetes in middle-aged men. Diabetes Care 2004; 27:1036-1041
515. Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 2006; 295:1288-1299
516. Kapoor D, Aldred H, Clark S, Channer KS, Jones TH. Clinical and biochemical assessment of hypogonadism in men with type 2 diabetes: correlations with bioavailable testosterone and visceral adiposity. Diabetes Care 2007; 30:911-917
517. Yeap BB, Chubb SA, Hyde Z, Jamrozik K, Hankey GJ, Flicker L, Norman PE. Lower serum testosterone is independently associated with insulin resistance in non-diabetic older men: the Health In Men Study. Eur J Endocrinol 2009; 161:591-598
518. Vikan T, Schirmer H, Njolstad I, Svartberg J. Low testosterone and sex hormone-binding globulin levels and high estradiol levels are independent predictors of type 2 diabetes in men. Eur J Endocrinol 2010; 162:747-754
519. Muller M, Grobbee DE, den Tonkelaar I, Lamberts SW, van der Schouw YT. Endogenous sex hormones and metabolic syndrome in aging men. J Clin Endocrinol Metab 2005; 90:2618-2623
520. Chin KY, Ima-Nirwana S, Mohamed IN, Aminuddin A, Ngah WZ. Total testosterone and sex hormone-binding globulin are significantly associated with metabolic syndrome in middle-aged and elderly men. Exp Clin Endocrinol Diabetes 2013; 121:407-412
521. Svartberg J, Schirmer H, Wilsgaard T, Mathiesen EB, Njolstad I, Lochen ML, Jorde R. Single-nucleotide polymorphism, rs1799941 in the Sex Hormone-Binding Globulin (SHBG) gene, related to both serum testosterone and SHBG levels and the risk of myocardial infarction, type 2 diabetes, cancer and mortality in men: the Tromso Study. Andrology 2014; 2:212-218
522. Perry JR, Weedon MN, Langenberg C, Jackson AU, Lyssenko V, Sparso T, Thorleifsson G, Grallert H, Ferrucci L, Maggio M, Paolisso G, Walker M, Palmer CN, Payne F, Young E, Herder C, Narisu N, Morken MA, Bonnycastle LL, Owen KR, Shields B, Knight B, Bennett A, Groves CJ, Ruokonen A, Jarvelin MR, Pearson E, Pascoe L, Ferrannini E, Bornstein SR, Stringham HM, Scott LJ, Kuusisto J, Nilsson P, Neptin M, Gjesing AP, Pisinger C, Lauritzen T, Sandbaek A, Sampson M, Magic, Zeggini E, Lindgren CM, Steinthorsdottir V, Thorsteinsdottir U, Hansen T, Schwarz P, Illig T, Laakso M, Stefansson K, Morris AD, Groop L, Pedersen O, Boehnke M, Barroso I, Wareham NJ, Hattersley AT, McCarthy MI, Frayling TM. Genetic evidence that raised sex hormone binding globulin (SHBG) levels reduce the risk of type 2 diabetes. Hum Mol Genet 2010; 19:535-544
523. Saltiki K, Stamatelopoulos K, Voidonikola P, Lazaros L, Mantzou E, Georgiou I, Anastasiou E, Papamichael C, Alevizaki M. Association of the SHBG gene promoter polymorphism with early markers of atherosclerosis in apparently healthy women. Atherosclerosis 2011; 219:205-210
524. Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, Buring JE, Gaziano JM, Liu S. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med 2009; 361:1152-1163
525. Lakshman KM, Bhasin S, Araujo AB. Sex hormone-binding globulin as an independent predictor of incident type 2 diabetes mellitus in men. J Gerontol A Biol Sci Med Sci 2010; 65:503-509
526. Goldman AL, Bhasin S, Wu FCW, Krishna M, Matsumoto AM, Jasuja R. A Reappraisal of Testosterone's Binding in Circulation: Physiological and Clinical Implications. Endocr Rev 2017; 38:302-324
527. Ruth KS, Day FR, Tyrrell J, Thompson DJ, Wood AR, Mahajan A, Beaumont RN, Wittemans L, Martin S, Busch AS, Erzurumluoglu AM, Hollis B, O'Mara TA, Endometrial Cancer Association C, McCarthy MI, Langenberg C, Easton DF, Wareham NJ, Burgess S, Murray A, Ong KK, Frayling TM, Perry JRB. Using human genetics to understand the disease impacts of testosterone in men and women. Nat Med 2020; 26:252-258
528. Yialamas MA, Dwyer AA, Hanley E, Lee H, Pitteloud N, Hayes FJ. Acute sex steroid withdrawal reduces insulin sensitivity in healthy men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2007; 92:4254-4259
529. Basaria S, Muller DC, Carducci MA, Egan J, Dobs AS. Relation between duration of androgen deprivation therapy and degree of insulin resistance in men with prostate cancer. Arch Intern Med 2007; 167:612-613
530. Nguyen PL, Jarolim P, Basaria S, Zuflacht JP, Milian J, Kadivar S, Graham PL, Hyatt A, Kantoff PW, Beckman JA. Androgen deprivation therapy reversibly increases endothelium-dependent vasodilation in men with prostate cancer. J Am Heart Assoc 2015; 4
531. Keating NL, O'Malley AJ, Freedland SJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst 2010; 102:39-46
532. Jones TH, Arver S, Behre HM, Buvat J, Meuleman E, Moncada I, Morales AM, Volterrani M, Yellowlees A, Howell JD, Channer KS, Investigators T. Testosterone replacement in hypogonadal men with type 2 diabetes and/or metabolic syndrome (the TIMES2 study). Diabetes Care 2011; 34:828-837
533. Corona G, Monami M, Rastrelli G, Aversa A, Sforza A, Lenzi A, Forti G, Mannucci E, Maggi M. Type 2 diabetes mellitus and testosterone: a meta-analysis study. Int J Androl 2011; 34:528-540
534. Basu R, Dalla Man C, Campioni M, Basu A, Nair KS, Jensen MD, Khosla S, Klee G, Toffolo G, Cobelli C, Rizza RA. Effect of 2 years of testosterone replacement on insulin secretion, insulin action, glucose effectiveness, hepatic insulin clearance, and postprandial glucose turnover in elderly men. Diabetes Care 2007; 30:1972-1978
535. Gianatti EJ, Dupuis P, Hoermann R, Strauss BJ, Wentworth JM, Zajac JD, Grossmann M. Effect of testosterone treatment on glucose metabolism in men with type 2 diabetes: a randomized controlled trial. Diabetes Care 2014; 37:2098-2107
536. Grossmann M, Hoermann R, Wittert G, Yeap BB. Effects of testosterone treatment on glucose metabolism and symptoms in men with type 2 diabetes and the metabolic syndrome: a systematic review and meta-analysis of randomized controlled clinical trials. Clin Endocrinol (Oxf) 2015; 83:344-351
537. Huang G, Pencina KM, Li Z, Basaria S, Bhasin S, Travison TG, Storer TW, Harman SM, Tsitouras P. Long-Term Testosterone Administration on Insulin Sensitivity in Older Men With Low or Low-Normal Testosterone Levels. J Clin Endocrinol Metab 2018; 103:1678-1685
538. Wittert G, Bracken K, Robledo KP, Grossmann M, Yeap BB, Handelsman DJ, Stuckey B, Conway A, Inder W, McLachlan R, Allan C, Jesudason D, Fui MNT, Hague W, Jenkins A, Daniel M, Gebski V, Keech A. Testosterone treatment to prevent or revert type 2 diabetes in men enrolled in a lifestyle programme (T4DM): a randomised, double-blind, placebo-controlled, 2-year, phase 3b trial. Lancet Diabetes Endocrinol 2021; 9:32-45
539. Warburton D, Hobaugh C, Wang G, Lin H, Wang R. Testosterone replacement therapy and the risk of prostate cancer. Asian J Androl 2015; 17:878-881; discussion 880
540. Debruyne FM, Behre HM, Roehrborn CG, Maggi M, Wu FC, Schroder FH, Jones TH, Porst H, Hackett G, Wheaton OA, Martin-Morales A, Meuleman E, Cunningham GR, Divan HA, Rosen RC, Investigators R. Testosterone treatment is not associated with increased risk of prostate cancer or worsening of lower urinary tract symptoms: prostate health outcomes in the Registry of Hypogonadism in Men. BJU Int 2017; 119:216-224
541. Cui Y, Zong H, Yan H, Zhang Y. The effect of testosterone replacement therapy on prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis 2014; 17:132-143
542. Boyle P, Koechlin A, Bota M, d'Onofrio A, Zaridze DG, Perrin P, Fitzpatrick J, Burnett AL, Boniol M. Endogenous and exogenous testosterone and the risk of prostate cancer and increased prostate-specific antigen (PSA) level: a meta-analysis. BJU Int 2016; 118:731-741
543. Bhasin S, Singh AB, Mac RP, Carter B, Lee MI, Cunningham GR. Managing the risks of prostate disease during testosterone replacement therapy in older men: recommendations for a standardized monitoring plan. J Androl 2003; 24:299-311
544. Fowler JE, Jr., Whitmore WF, Jr. The response of metastatic adenocarcinoma of the prostate to exogenous testosterone. J Urol 1981; 126:372-375
545. Watts EL, Appleby PN, Perez-Cornago A, Bueno-de-Mesquita HB, Chan JM, Chen C, Cohn BA, Cook MB, Flicker L, Freedman ND, Giles GG, Giovannucci E, Gislefoss RE, Hankey GJ, Kaaks R, Knekt P, Kolonel LN, Kubo T, Le Marchand L, Luben RN, Luostarinen T, Mannisto S, Metter EJ, Mikami K, Milne RL, Ozasa K, Platz EA, Quiros JR, Rissanen H, Sawada N, Stampfer M, Stanczyk FZ, Stattin P, Tamakoshi A, Tangen CM, Thompson IM, Tsilidis KK, Tsugane S, Ursin G, Vatten L, Weiss NS, Yeap BB, Allen NE, Key TJ, Travis RC. Low Free Testosterone and Prostate Cancer Risk: A Collaborative Analysis of 20 Prospective Studies. Eur Urol 2018;
546. Watts EL, Fensom GK, Smith Byrne K, Perez-Cornago A, Allen NE, Knuppel A, Gunter MJ, Holmes MV, Martin RM, Murphy N, Tsilidis KK, Yeap BB, Key TJ, Travis RC. Circulating insulin-like growth factor-I, total and free testosterone concentrations and prostate cancer risk in 200 000 men in UK Biobank. Int J Cancer 2021; 148:2274-2288
547. Manni A, Bartholomew M, Caplan R, Boucher A, Santen R, Lipton A, Harvey H, Simmonds M, White-Hershey D, Gordon R, et al. Androgen priming and chemotherapy in advanced prostate cancer: evaluation of determinants of clinical outcome. J Clin Oncol 1988; 6:1456-1466
548. Babaian RJ, Johnston DA, Naccarato W, Ayala A, Bhadkamkar VA, Fritsche HH, Jr. The incidence of prostate cancer in a screening population with a serum prostate specific antigen between 2.5 and 4.0 ng/ml: relation to biopsy strategy. J Urol 2001; 165:757-760
549. Gatling RR. Prostate carcinoma: an autopsy evaluation of the influence of age, tumor grade, and therapy on tumor biology. South Med J 1990; 83:782-784
550. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68:7-30
551. Guileyardo JM, Johnson WD, Welsh RA, Akazaki K, Correa P. Prevalence of latent prostate carcinoma in two U.S. populations. J Natl Cancer Inst 1980; 65:311-316
552. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87-108
553. Cook MB, Rosenberg PS, McCarty FA, Wu M, King J, Eheman C, Anderson WF. Racial disparities in prostate cancer incidence rates by census division in the United States, 1999-2008. Prostate 2015; 75:758-763
554. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017; 67:7-30
555. Holmang S, Marin P, Lindstedt G, Hedelin H. Effect of long-term oral testosterone undecanoate treatment on prostate volume and serum prostate-specific antigen concentration in eugonadal middle-aged men. Prostate 1993; 23:99-106
556. Morgentaler A, Bruning CO, 3rd, DeWolf WC. Occult prostate cancer in men with low serum testosterone levels. JAMA 1996; 276:1904-1906
557. Hoffman MA, DeWolf WC, Morgentaler A. Is low serum free testosterone a marker for high grade prostate cancer? J Urol 2000; 163:824-827
558. Cunningham GR, Ellenberg SS, Bhasin S, Matsumoto AM, Parsons JK, Preston P, Cauley JA, Gill TM, Swerdloff RS, Wang C, Ensrud KE, Lewis CE, Pahor M, Crandall JP, Molitch ME, Cifelli D, Basaria S, Diem SJ, Stephens-Shields AJ, Hou X, Snyder PJ. Prostate-Specific Antigen Levels During Testosterone Treatment of Hypogonadal Older Men: Data from a Controlled Trial. J Clin Endocrinol Metab 2019; 104:6238-6246
559. Marks LS, Mazer NA, Mostaghel E, Hess DL, Dorey FJ, Epstein JI, Veltri RW, Makarov DV, Partin AW, Bostwick DG, Macairan ML, Nelson PS. Effect of testosterone replacement therapy on prostate tissue in men with late-onset hypogonadism: a randomized controlled trial. JAMA 2006; 296:2351-2361
560. Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, Nelson PS, Matsumoto AM, Bremner WJ. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab 2006; 91:3850-3856
561. Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME, Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res 2007; 67:5033-5041
562. Behre HM, Bohmeyer J, Nieschlag E. Prostate volume in testosterone-treated and untreated hypogonadal men in comparison to age-matched normal controls. Clin Endocrinol (Oxf) 1994; 40:341-349
563. Cooper CS, Perry PJ, Sparks AE, MacIndoe JH, Yates WR, Williams RD. Effect of exogenous testosterone on prostate volume, serum and semen prostate specific antigen levels in healthy young men. J Urol 1998; 159:441-443
564. Douglas TH, Connelly RR, McLeod DG, Erickson SJ, Barren R, 3rd, Murphy GP. Effect of exogenous testosterone replacement on prostate-specific antigen and prostate-specific membrane antigen levels in hypogonadal men. J Surg Oncol 1995; 59:246-250
565. Guay AT, Perez JB, Fitaihi WA, Vereb M. Testosterone treatment in hypogonadal men: prostate-specific antigen level and risk of prostate cancer. Endocr Pract 2000; 6:132-138
566. Meikle AW, Arver S, Dobs AS, Adolfsson J, Sanders SW, Middleton RG, Stephenson RA, Hoover DR, Rajaram L, Mazer NA. Prostate size in hypogonadal men treated with a nonscrotal permeation-enhanced testosterone transdermal system. Urology 1997; 49:191-196
567. Sasagawa I, Nakada T, Kazama T, Satomi S, Terada T, Katayama T. Volume change of the prostate and seminal vesicles in male hypogonadism after androgen replacement therapy. Int Urol Nephrol 1990; 22:279-284
568. Shibasaki T, Sasagawa I, Suzuki Y, Yazawa H, Ichiyanagi O, Matsuki S, Miura M, Nakada T. Effect of testosterone replacement therapy on serum PSA in patients with Klinefelter syndrome. Arch Androl 2001; 47:173-176
569. Svetec DA, Canby ED, Thompson IM, Sabanegh ES, Jr. The effect of parenteral testosterone replacement on prostate specific antigen in hypogonadal men with erectile dysfunction. J Urol 1997; 158:1775-1777
570. Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, McConnell JD, Andriole GL, Geller J, Bracken BR, Tenover JS, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med 1992; 327:1185-1191
571. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA, Jr. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349:215-224
572. Manseck A, Pilarsky C, Froschermaier S, Menschikowski M, Wirth MP. Diagnostic significance of prostate-specific antigen velocity at intermediate PSA serum levels in relation to the standard deviation of different test systems. Urol Int 1998; 60:25-27
573. Riehmann M, Rhodes PR, Cook TD, Grose GS, Bruskewitz RC. Analysis of variation in prostate-specific antigen values. Urology 1993; 42:390-397
574. Wenzl TB, Riehmann M, Cook TD, Bruskewitz RC. Variation in PSA values in patients without malignancy. Urology 1998; 52:932
575. Carter HB, Pearson JD. PSA velocity for the diagnosis of early prostate cancer. A new concept. Urol Clin North Am 1993; 20:665-670
576. Carter HB, Pearson JD, Waclawiw Z, Metter EJ, Chan DW, Guess HA, Walsh PC. Prostate-specific antigen variability in men without prostate cancer: effect of sampling interval on prostate-specific antigen velocity. Urology 1995; 45:591-596
577. Fang J, Metter EJ, Landis P, Carter HB. PSA velocity for assessing prostate cancer risk in men with PSA levels between 2.0 and 4.0 ng/ml. Urology 2002; 59:889-893; discussion 893-884
578. Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab 2005; 90:678-688
579. Kathrins M, Doersch K, Nimeh T, Canto A, Niederberger C, Seftel A. The Relationship Between Testosterone-Replacement Therapy and Lower Urinary Tract Symptoms: A Systematic Review. Urology 2016; 88:22-32
580. Wu FC, Farley TM, Peregoudov A, Waites GM. Effects of testosterone enanthate in normal men: experience from a multicenter contraceptive efficacy study. World Health Organization Task Force on Methods for the Regulation of Male Fertility. Fertil Steril 1996; 65:626-636
581. Carter HB, Pearson JD, Metter EJ, Chan DW, Andres R, Fozard JL, Rosner W, Walsh PC. Longitudinal evaluation of serum androgen levels in men with and without prostate cancer. Prostate 1995; 27:25-31
582. Fried W, Marver D, Lange RD, Gurney CW. Studies on the erythropoietic stimulating factor in the plasma of mice after receiving testosterone. J Lab Clin Med 1966; 68:947-951
583. Rencricca NJ, Solomon J, Fimian WJ, Jr., Howard D, Rizzoli V, Stohlman F, Jr. The effect of testosterone on erythropoiesis. Scand J Haematol 1969; 6:431-436
584. Naets JP, Wittek M. The mechanism of action of androgens on erythropoiesis. Ann N Y Acad Sci 1968; 149:366-376
585. Dhindsa S, Ghanim H, Batra M, Kuhadiya ND, Abuaysheh S, Green K, Makdissi A, Chaudhuri A, Dandona P. Effect of testosterone on hepcidin, ferroportin, ferritin and iron binding capacity in patients with hypogonadotropic hypogonadism and type 2 diabetes. Clin Endocrinol (Oxf) 2016; 85:772-780
586. Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab 1999; 84:3469-3478
587. Bachman E, Feng R, Travison T, Li M, Olbina G, Ostland V, Ulloor J, Zhang A, Basaria S, Ganz T, Westerman M, Bhasin S. Testosterone suppresses hepcidin in men: a potential mechanism for testosterone-induced erythrocytosis. J Clin Endocrinol Metab 2010; 95:4743-4747
588. Gagliano-Juca T, Pencina KM, Ganz T, Travison TG, Kantoff PW, Nguyen PL, Taplin ME, Kibel AS, Li Z, Huang G, Edwards RR, Nemeth E, Basaria S. Mechanisms Responsible for Reduced Erythropoiesis during Androgen Deprivation Therapy in Men with Prostate Cancer. Am J Physiol Endocrinol Metab 2018;
589. Cistulli PA, Grunstein RR, Sullivan CE. Effect of testosterone administration on upper airway collapsibility during sleep. Am J Respir Crit Care Med 1994; 149:530-532
590. Emery MJ, Hlastala MP, Matsumoto AM. Depression of hypercapnic ventilatory drive by testosterone in the sleeping infant primate. J Appl Physiol (1985) 1994; 76:1786-1793
591. Schiavi RC, White D, Mandeli J. Pituitary-gonadal function during sleep in healthy aging men. Psychoneuroendocrinology 1992; 17:599-609
592. Hoyos CM, Killick R, Yee BJ, Grunstein RR, Liu PY. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf) 2012; 77:599-607
593. Matsumoto AM, Sandblom RE, Schoene RB, Lee KA, Giblin EC, Pierson DJ, Bremner WJ. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713-721
594. Schneider BK, Pickett CK, Zwillich CW, Weil JV, McDermott MT, Santen RJ, Varano LA, White DP. Influence of testosterone on breathing during sleep. J Appl Physiol (1985) 1986; 61:618-623
595. Kirbas G, Abakay A, Topcu F, Kaplan A, Unlu M, Peker Y. Obstructive sleep apnoea, cigarette smoking and serum testosterone levels in a male sleep clinic cohort. J Int Med Res 2007; 35:38-45
596. Luboshitzky R, Aviv A, Hefetz A, Herer P, Shen-Orr Z, Lavie L, Lavie P. Decreased pituitary-gonadal secretion in men with obstructive sleep apnea. J Clin Endocrinol Metab 2002; 87:3394-3398
597. Gambineri A, Pelusi C, Pasquali R. Testosterone levels in obese male patients with obstructive sleep apnea syndrome: relation to oxygen desaturation, body weight, fat distribution and the metabolic parameters. J Endocrinol Invest 2003; 26:493-498
598. Leproult R, Van Cauter E. Effect of 1 week of sleep restriction on testosterone levels in young healthy men. JAMA 2011; 305:2173-2174
599. Luboshitzky R, Lavie L, Shen-Orr Z, Lavie P. Pituitary-gonadal function in men with obstructive sleep apnea. The effect of continuous positive airways pressure treatment. Neuro Endocrinol Lett 2003; 24:463-467
600. Hoyos CM, Yee BJ, Phillips CL, Machan EA, Grunstein RR, Liu PY. Body compositional and cardiometabolic effects of testosterone therapy in obese men with severe obstructive sleep apnoea: a randomised placebo-controlled trial. Eur J Endocrinol 2012; 167:531-541
601. Liu PY, Yee B, Wishart SM, Jimenez M, Jung DG, Grunstein RR, Handelsman DJ. The short-term effects of high-dose testosterone on sleep, breathing, and function in older men. J Clin Endocrinol Metab 2003; 88:3605-3613
602. Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA, Group UKCC. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst 2005; 97:1204-1210
603. Morley JE, Charlton E, Patrick P, Kaiser FE, Cadeau P, McCready D, Perry HM, 3rd. Validation of a screening questionnaire for androgen deficiency in aging males. Metabolism 2000; 49:1239-1242
604. T'Sjoen G, Goemaere S, De Meyere M, Kaufman JM. Perception of males' aging symptoms, health and well-being in elderly community-dwelling men is not related to circulating androgen levels. Psychoneuroendocrinology 2004; 29:201-214
605. Smith KW, Feldman HA, McKinlay JB. Construction and field validation of a self-administered screener for testosterone deficiency (hypogonadism) in ageing men. Clin Endocrinol (Oxf) 2000; 53:703-711
606. Bhasin S. Testosterone replacement in aging men: an evidence-based patient-centric perspective. J Clin Invest 2021; 131
607. Seymour FI, Duffy C, Korner A. A Case of Authenticated Fertility in a Man, Aged 94. Journal of the American Medical Association 1935; 105:1423
608. Plas E, Berger P, Hermann M, Pfluger H. Effects of aging on male fertility? Exp Gerontol 2000; 35:543-551
609. Rolf C, Nieschlag E. Reproductive functions, fertility and genetic risks of ageing men. Exp Clin Endocrinol Diabetes 2001; 109:68-74
610. New guidelines for the use of semen donor insemination: 1990. The American Fertility Society. Fertil Steril 1990; 53:1S-13S
611. Practice Committee of American Society for Reproductive M, Practice Committee of Society for Assisted Reproductive T. 2008 Guidelines for gamete and embryo donation: a Practice Committee report. Fertil Steril 2008; 90:S30-44
612. Silber SJ. Effect of Age on Male Fertility. Seminars in Reproductive Medicine 1991; 9:241-248
613. Kidd SA, Eskenazi B, Wyrobek AJ. Effects of male age on semen quality and fertility: a review of the literature. Fertil Steril 2001; 75:237-248
614. Sloter E, Nath J, Eskenazi B, Wyrobek AJ. Effects of male age on the frequencies of germinal and heritable chromosomal abnormalities in humans and rodents. Fertil Steril 2004; 81:925-943
615. Wiener-Megnazi Z, Auslender R, Dirnfeld M. Advanced paternal age and reproductive outcome. Asian J Androl 2012; 14:69-76
616. Fonseka KG, Griffin DK. Is there a paternal age effect for aneuploidy? Cytogenet Genome Res 2011; 133:280-291
617. Lian ZH, Zack MM, Erickson JD. Paternal age and the occurrence of birth defects. Am J Hum Genet 1986; 39:648-660
618. Brown AS, Schaefer CA, Wyatt RJ, Begg MD, Goetz R, Bresnahan MA, Harkavy-Friedman J, Gorman JM, Malaspina D, Susser ES. Paternal age and risk of schizophrenia in adult offspring. Am J Psychiatry 2002; 159:1528-1533
619. Goriely A, McGrath JJ, Hultman CM, Wilkie AO, Malaspina D. "Selfish spermatogonial selection": a novel mechanism for the association between advanced paternal age and neurodevelopmental disorders. Am J Psychiatry 2013; 170:599-608
620. Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 2012; 90:175-200
621. Lim J, Maher GJ, Turner GD, Dudka-Ruszkowska W, Taylor S, Rajpert-De Meyts E, Goriely A, Wilkie AO. Selfish spermatogonial selection: evidence from an immunohistochemical screen in testes of elderly men. PLoS One 2012; 7:e42382
622. Wyrobek AJ, Eskenazi B, Young S, Arnheim N, Tiemann-Boege I, Jabs EW, Glaser RL, Pearson FS, Evenson D. Advancing age has differential effects on DNA damage, chromatin integrity, gene mutations, and aneuploidies in sperm. Proc Natl Acad Sci U S A 2006; 103:9601-9606
623. Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature 2012; 488:471-475
624. Olshan AF, Schnitzer PG, Baird PA. Paternal age and the risk of congenital heart defects. Teratology 1994; 50:80-84
625. Frans EM, Sandin S, Reichenberg A, Langstrom N, Lichtenstein P, McGrath JJ, Hultman CM. Autism risk across generations: a population-based study of advancing grandpaternal and paternal age. JAMA Psychiatry 2013; 70:516-521
626. Harlap S, Paltiel O, Deutsch L, Knaanie A, Masalha S, Tiram E, Caplan LS, Malaspina D, Friedlander Y. Paternal age and preeclampsia. Epidemiology 2002; 13:660-667
627. Kimura M, Itoh N, Takagi S, Sasao T, Takahashi A, Masumori N, Tsukamoto T. Balance of apoptosis and proliferation of germ cells related to spermatogenesis in aged men. J Androl 2003; 24:185-191
628. Luetjens CM, Rolf C, Gassner P, Werny JE, Nieschlag E. Sperm aneuploidy rates in younger and older men. Hum Reprod 2002; 17:1826-1832
629. Fisch H, Hyun G, Golden R, Hensle TW, Olsson CA, Liberson GL. The influence of paternal age on down syndrome. J Urol 2003; 169:2275-2278
630. Stene E, Stene J, Stengel-Rutkowski S. A reanalysis of the New York State prenatal diagnosis data on Down's syndrome and paternal age effects. Hum Genet 1987; 77:299-302
631. Sloter ED, Marchetti F, Eskenazi B, Weldon RH, Nath J, Cabreros D, Wyrobek AJ. Frequency of human sperm carrying structural aberrations of chromosome 1 increases with advancing age. Fertil Steril 2007; 87:1077-1086
632. Hellstrom WJ, Overstreet JW, Sikka SC, Denne J, Ahuja S, Hoover AM, Sides GD, Cordell WH, Harrison LM, Whitaker JS. Semen and sperm reference ranges for men 45 years of age and older. J Androl 2006; 27:421-428
633. Eskenazi B, Wyrobek AJ, Sloter E, Kidd SA, Moore L, Young S, Moore D. The association of age and semen quality in healthy men. Hum Reprod 2003; 18:447-454
634. Gallardo E, Simon C, Levy M, Guanes PP, Remohi J, Pellicer A. Effect of age on sperm fertility potential: oocyte donation as a model. Fertil Steril 1996; 66:260-264
635. Jung A, Schuppe HC, Schill WB. Comparison of semen quality in older and younger men attending an andrology clinic. Andrologia 2002; 34:116-122
636. Haidl G, Badura B, Hinsch KD, Ghyczy M, Gareiss J, Schill WB. Disturbances of sperm flagella due to failure of epididymal maturation and their possible relationship to phospholipids. Hum Reprod 1993; 8:1070-1073
637. Handelsman DJ, Staraj S. Testicular size: the effects of aging, malnutrition, and illness. J Androl 1985; 6:144-151
638. Handelsman DJ. Male reproductive ageing: human fertility, androgens and hormone dependent disease. Novartis Found Symp 2002; 242:66-77; discussion 77-81
639. Johnson L, Zane RS, Petty CS, Neaves WB. Quantification of the human Sertoli cell population: its distribution, relation to germ cell numbers, and age-related decline. Biol Reprod 1984; 31:785-795
640. Tulina N, Matunis E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 2001; 294:2546-2549
641. Black A, Lane MA. Nonhuman primate models of skeletal and reproductive aging. Gerontology 2002; 48:72-80
642. Gruenewald DA, Naai MA, Hess DL, Matsumoto AM. The Brown Norway rat as a model of male reproductive aging: evidence for both primary and secondary testicular failure. J Gerontol 1994; 49:B42-50
643. Wang C, Leung A, Sinha-Hikim AP. Reproductive aging in the male brown-Norway rat: a model for the human. Endocrinology 1993; 133:2773-2781
644. Taylor GT, Weiss J, Pitha J. Epididymal sperm profiles in young adult, middle-aged, and testosterone-supplemented old rats. Gamete Res 1988; 19:401-409
645. Oakes CC, Smiraglia DJ, Plass C, Trasler JM, Robaire B. Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proc Natl Acad Sci U S A 2003; 100:1775-1780
646. Zirkin BR, Santulli R, Strandberg JD, Wright WW, Ewing LL. Testicular steroidogenesis in the aging brown Norway rat. J Androl 1993; 14:118-123
647. Wright WW, Fiore C, Zirkin BR. The effect of aging on the seminiferous epithelium of the brown Norway rat. J Androl 1993; 14:110-117
648. Le Magueresse B, Jegou B. In vitro effects of germ cells on the secretory activity of Sertoli cells recovered from rats of different ages. Endocrinology 1988; 122:1672-1680
649. Humphreys PN. The histology of the testis in aging and senile rats. Exp Gerontol 1977; 12:27-34
650. Syntin P, Robaire B. Sperm structural and motility changes during aging in the Brown Norway rat. J Androl 2001; 22:235-244
651. Tramer F, Rocco F, Micali F, Sandri G, Panfili E. Antioxidant systems in rat epididymal spermatozoa. Biol Reprod 1998; 59:753-758
652. Edelmann P, Gallant J. On the translational error theory of aging. Proc Natl Acad Sci U S A 1977; 74:3396-3398
653. Marden JH, Rogina B, Montooth KL, Helfand SL. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc Natl Acad Sci U S A 2003; 100:3369-3373
654. Jones LS, Berndtson WE. A quantitative study of Sertoli cell and germ cell populations as related to sexual development and aging in the stallion. Biol Reprod 1986; 35:138-148
655. Tenover JS, McLachlan RI, Dahl KD, Burger HG, de Kretser DM, Bremner WJ. Decreased serum inhibin levels in normal elderly men: evidence for a decline in Sertoli cell function with aging. J Clin Endocrinol Metab 1988; 67:455-459
656. Bohring C, Krause W. Serum levels of inhibin B in men of different age groups. Aging Male 2003; 6:73-78
657. Haji M, Tanaka S, Nishi Y, Yanase T, Takayanagi R, Hasegawa Y, Sasamoto S, Nawata H. Sertoli cell function declines earlier than Leydig cell function in aging Japanese men. Maturitas 1994; 18:143-153
658. Kaler LW, Neaves WB. Attrition of the human Leydig cell population with advancing age. Anat Rec 1978; 192:513-518
659. Neaves WB, Johnson L, Petty CS. Age-related change in numbers of other interstitial cells in testes of adult men: evidence bearing on the fate of Leydig cells lost with increasing age. Biol Reprod 1985; 33:259-269
660. Paniagua R, Amat P, Nistal M, Martin A. Ultrastructure of Leydig cells in human ageing testes. J Anat 1986; 146:173-183
661. Neaves WB, Johnson L, Porter JC, Parker CR, Jr., Petty CS. Leydig cell numbers, daily sperm production, and serum gonadotropin levels in aging men. J Clin Endocrinol Metab 1984; 59:756-763
662. Nistal M, Santamaria L, Paniagua R, Regadera J, Codesal J. Multinucleate Leydig cells in normal human testes. Andrologia 1986; 18:268-272