ABSTRACT
The epidemic of obesity cries out for strategies to prevent it, and for treatment when prevention fails. Yet despite the obvious need, the path to developing anti-obesity medications is strewn with failures. This review examines some historical developments in the management of obesity prior to the 20th century followed by more recent efforts to find a “cure.” Since obesity has been a human disease since paleolithic times, discussion of treatment prior to the scientific revolution in about 1500 CE will include comments on treatment of obesity in Grecian Medicine and the Treatment of Sancho “The Fat” King of Leon in 10th century northern Spain. We will then trace treatment trends through time, ending in the early 21st century. In the first half of the 20th century there were 3 main themes associated with drug treatment of obesity: use of thyroid hormones, the interlude with dinitrophenol as drug regulation was beginning to expand, and the introduction of amphetamines and Rainbow Pills. In the second half of the 20th century many new treatment approaches were explored as the scientific basis for understanding regulation of food intake expanded, particularly after the discovery of leptin in 1994. The fact that amphetamine could be addictive led to the search for compounds that acted in the central nervous system with retained the anorectic properties but were not addictive. Both anti-depressant and anticonvulsive drugs were evaluated with disappointing results. The idea of enhancing energy expenditure with thermogenic drugs was pursued but led to a dead end. Peptides, metabolic drugs and drugs that act on the gastrointestinal track are currently under exploration. This chapter makes an effort to answer the question of why it has been so difficult to develop effective medications for the management of obesity.
INTRODUCTION
“When we say that science is essentially progressive this does not mean that in his quest for truth man follows always the shortest path. Far from it, he beats about the bush, does not find what he is looking for but finds something else, retraces his steps, loses himself in various detours, and finally after many wanderings touches the goal”. Georges Sarton (1)
This brief historical review will examine some of the ‘wanderings’ that have occurred in the search for medications to use in the management of obesity (anti-obesity medications, or AOM’s). It is set against the broader long-time changes in our knowledge of how obesity develops and the result of attempts to treat it effectively. Over human history since the Cognitive Revolution some 75,000 years ago (2), knowledge has expanded logarithmically (Figure 1). There is every indication that it is continuing to expand as evidenced by the explosion of knowledge around the SARS-Covid-19 Pandemic of 2020 and its detrimental impact of people with obesity.
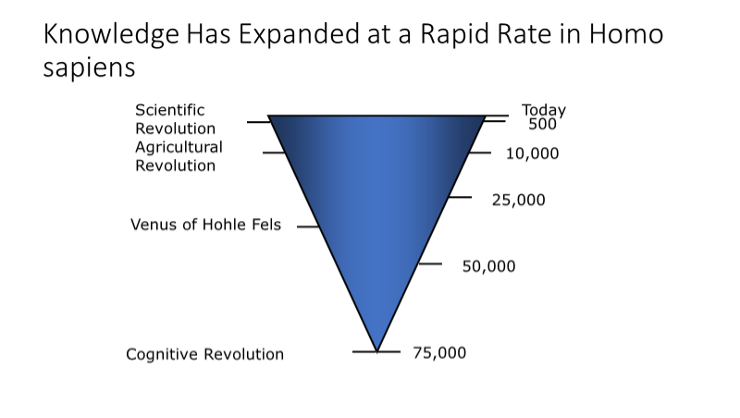
Figure 1. Expansion of Knowledge since the Cognitive Revolution
Beginning in the upper Paleolithic era between 25,000 and 35,000 years ago we find the first representations of obesity in human figurines, including the Venus of Hohle Fels (3), and mere 10,000 years later the Venus of Willendorf (4). The agricultural revolution and domestication of animals occurred about 10,000 years ago and during this period there were many other representations of the human with obesity from many parts of the world. Compared to the long 15,000 year interval between carving of the Venus of Willendorf and the Agricultural Revolution we are living in a very small slice of time. This slice might appropriately begin with the Scientific Revolution dated to 1543 CE, when Copernicus published his theory of the heliocentric nature of the planetary system, Vesalius published his anatomy on the fabric of the human body and the subsequent Industrial Revolution that began about 1750 CE (5). During the next 500 years, the oxygen theory of metabolism was articulated by Laennec (1793), the first law of thermodynamics was published by von Helmholtz (1848), the concept of cells as the basic of life was presented by Schwann and Schleiden (1839), and the first clear differentiation of types of obesity were made by Babinski (1900), Frohlich (1901), and Cushing (1912). The 20th century has brought enormous changes to health care and offers great promise for people with obesity, but the road to discovery of medications for treatment of obesity has been a tortuous one but one that finally offers promise of success.
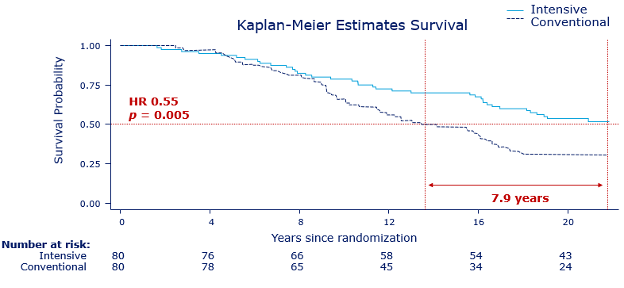
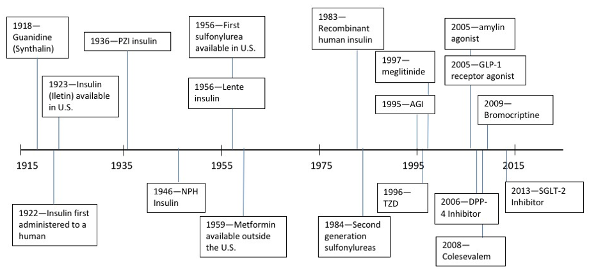
In the past 100 years, earnest efforts to deploy anti-obesity treatments were repeatedly tried and failed. The first three are the story of thyroid hormones, the story of dintrophenol, and the story of amphetamines. These 3 medications were tested in people with obesity against a background of growing governmental regulation of medicines and rapidly expanding knowledge about the causes of obesity. Although the timing for different treatments often overlap, Figure 2illustrates the gradual evolution to the present day. We begin with the colorful history of the Rainbow Pills, followed by the anorectic drugs. The year 1994 marks a major milestone – it was the year that leptin was discovered – a discovery that gave obesity a strong biological base from which further discoveries have continued (6).

Figure 2. Historical events and the development of anti-obesity medications in the past 100 years
There are three underlying principals in developing any treatment for managing obesity: First, do no harm. This is a principal that dates back at least to the time of Hippocrates 2500 years ago. The second concept is that treatments don’t work unless used. If you don’t take the medicine designed to treat obesity you can’t expect the obesity to disappear as if by magic. Third, if an individual being treated for their obesity doesn’t lose weight for one reason or another it is unreasonable to anticipate that they will get any benefits that might be associated with weight loss.
THE FAILURE OF MEDICATIONS USED TO TREAT OBESITY
Management of obesity is strewn with misadventures (Table 1) (7). Below is a selected list of failures in the search for treatments for obesity. Some of these problems occurred before marketing, but others only after the drug had been approved for marketing.
|
Table 1. Some of the Unintended Consequences that have Led to the Withdrawal of Medications Used for Management of Obesity
|
|
Year
|
Drug
|
Alleged Mechanism
|
Reason for Discontinuation
|
|
1892
|
Thyroid
|
Thermogenesis
|
Hyperthyroidism
|
|
1932
|
Dintrophenol
|
Thermogenesis
|
Disapproved due to Cataracts/Neuropathy
|
|
1937
|
Amphetamine
|
Sympathomimetic
|
Disapproved due to Addiction
|
|
1961-90
|
Human chorionic gonadotropin
|
Reduce food intake
|
Disapproved - Ineffective compared to placebo
|
|
1971
|
Aminorex
|
Sympathomimetic
|
Withdrawn after marketing due to Pulmonary hypertension
|
|
1985
|
Gelatin-based very low-calorie diet
|
Reduce food intake
|
Cardiovascular Deaths (Torsade de Points)
|
|
1991-95
|
Fluoxetine
|
Serotonin reuptake inhibitor
|
Weight regain after loss
|
|
1985-98
|
β-3 Agonists
|
Increased thermogenesis
|
Limited Effect; Increased HR
|
|
1997
|
Fenfluramine
|
Serotonergic receptor activation (? 5HT2c)
|
Withdrawn after marketing due to cardiac valvulopathy and pulmonary hypertension
|
|
1998
|
Phenylpropanolamine
|
Sympathomimetic
|
Withdrawn after marketing for Strokes
|
|
1999
|
Leptin
|
Leptin receptor agonist-reduced food intake
|
Limited Weight Loss
|
|
2003
|
Ephedrine/Caffeine & Herbal Ma Huang
|
Sympathomimetic and adrenergic blocker
|
Withdrawn after marketing for Heart attacks/stroke
|
|
2003
|
Ciliary Neurotrophic Factor
|
Acts on leptin receptor
|
Produced neutralizing antibodies
|
|
2007
|
MK-0557
|
Neuropeptide Y5 (NPY) receptor antagonist-reduced food intake.
|
Limited effectiveness
|
|
2007
|
Ecopipam
|
D2/D5 agonist-Reduce food intake
|
Suicidality
|
|
2008
|
Tesofensine
|
Triple Monoamine Reuptake Inhibitor
|
Raised blood pressure
|
|
2009
|
Melanocortin-4 Receptor Agonist
|
Reduce Food Intake
|
Limited effectiveness, priapism
|
|
2010
|
Capsinoids
|
Thermogenesis
|
Limited effectiveness
|
|
2010
|
Rimonabant
|
Endocannabinoid agonist
|
Suicidality
|
|
2011
|
Sibutramine
|
Triple Reuptake Inhibitor
|
Withdrawn after marketing for cardiovascular toxicity
|
|
2020
|
Lorcaserin
|
Serotonergic Reduce Food Intake
|
Cancer
|
TREATMENT OF OBESITY BEFORE THE SCIENTIFIC REVOLUTION, c. 1543 CE
Diet (meaning limiting total calories) and exercise have been the cornerstones of treatment for the patient with obesity since at least the time of Hippocrates 2500 years ago. Following are a few examples of where medications have been used as adjuncts, and sometimes as the main line of treatment throughout these many centuries.
Greco-Roman Medicine
The problem of obesity was well known to physicians in the Greco-Roman period from 500 BCE to 500 CE. “An Egyptian pharaoh was said to have a middle wider than the span of his slave’s outstretched arms.” Nichomachus of Smyrna “…was so huge that he couldn’t even get up from his bed” ((8); p 20). Dionysius of Heraclea was so obese that his attendants needed to stick pins into him to keep him from falling asleep on his throne. In retrospect some observers think he may have had a sleep disturbance like sleep apnea (9). The enormous size of a Roman Senator was such that he could only walk when two of his slaves carried his belly for him.
Among the earliest attempts at producing weight loss by adding compounds to the treatment regimen have been ascribed to Soranus of Ephesus, a 2nd century CE Greek physician. Much of what we know about Soranus’ work in the 2nd century CE comes from translations by Caelius Aurelianus in the 5th Century. Both describe obesity as a disease where the body keeps acquiring additional flesh beyond what is needed. They believe it is an “unsightly affliction” because there are no apparent symptoms other than the increase of flesh. The Greek model of disease involved four humors (blood, phlegm, yellow bile, and black bile) being out of balance. Treatment was needed to rebalance what was out of balance – for obesity this meant less intake and more output through purgatives and diuresis (10). Soranus’ regimen consisted of laxatives and purgatives along with exercise, heat, and massage. Indeed, laxatives have been a recurrent theme in the historic management of obesity. Induction of vomiting was also used for treating both the obese and to remove excess alcohol during Roman orgies. “Fat individuals should vomit in the middle of the day, after a running or marching exercise and before taking any food”. The emetic for this purpose was half a cup of the hyssop plant (0.15 L) ground with three liters of water to which vinegar and salt are added. Vinegar was another favorite for use in the context of humoral medicine ((8); p 18). Since obesity was considered as a “moist and cool” condition in the humoral view of health, vinegar which is dry and warm would be an appropriate balancing agent. We will see vinegar resurface in later periods.
Obesity in the Hindu Tradition
Ayurvedic Medicine originated in India more than 3,000 years ago and is derived from the Sanskrit words ayur (life) and veda (science or knowledge). In India, Ayurvedic Medicine is considered a form of medical care equal to conventional Western medicine, traditional Chinese medicine, naturopathic medicine, or homeopathic medicine. Practitioners of Ayurveda in India undergo state-recognized, institutionalized training. Two names are primarily associated with the early writings on Ayurvedic medicine, Sushruta and Charaka. Although the exact dates are unclear, they probably lived between 600 BCE and 100 CE. Sushruta, often referred to as the “Father of Indian Medicine” or the “Father of Plastic Surgery,” developed the Sushruta Samhita, one of the foundational texts of Ayurvedic Medicine. The second text, the Charaka-Samhita or “compendium of Charaka,” was composed by Charaka. Both texts remained foundational for 2 millennia and were translated into many foreign languages. They both discussed general principles of medicine, pathology, diagnoses, anatomy, therapeutics, pharmaceutics, and toxicology. The Sushruta Samhita provides more exposure to surgery. These texts are credited with very early recognition of the sugary taste of diabetic urine and that this disease often affected indolent, overweight people who ate excessively, especially sweet and fatty foods. “[O]vereating... causes illness and shortens life span. It is a contraindication to the use of compresses or mild enemas. For treatment of obesity two suggestions are made…The vigorous massage of the body with pea flour counteracts phlegm diseases and obesity...The gullet hair compress and flesh of a wolf remedy [to treat] goiters, dropsy and obesity.” (11).
Obesity in the Islamic Tradition: 10th and 11th Century
In the 7th century following the revelations from God, the prophet Muhammad began preaching his religious beliefs that gradually unified Arabia into a single entity. The Islamic religion spread rapidly across the Middle East, North Africa and into the Iberian Peninsula. Centers of cultural excellence in Baghdad in the east, and in Cordoba, on the Iberian Peninsula soon appeared. We will focus on two physician-scholars and their impact on management of obesity in the 10th and 11th century. Ibn Sinha (980-1037 CE), anglicized as Avicenna was from the Middle Eastern part of the Islamic Caliphate, and the Jewish physician Hasdai ibn Shaprut (917-970 CE) from the Western Islamic area of Cordoba on the Iberian Peninsula.
Abu Ali Ibn Sina (Avicenna) was a prolific and influential author who published the most influential medical textbook of the Middle Ages called the Canon (12, 13). The Canon contains no personal experiences and no new ideas but was rather a summary of existing knowledge and was widely used in medical schools for hundreds of years. Avicenna surpassed both Aristotle and Galen in his dialectical subtlety (14, 15), and by some estimates published more than 100 medical books, as well as books in other areas. Avicenna describes how to manage the patient with obesity in four stages: 1) Produce a rapid descent of the food from the stomach and intestines, in order to prevent completion of absorption by the mesentery (16); 2) Take food which is bulky but feebly nutritious (17); 3) Take the bath before food, often (18); and 4) Hard exercise (19).
Hasdai ibn Shaprut enters this story because he treated King Sancho I (aka Sancho the Fat). Sancho I (935-966) became king of Leon (Pamplona, Spain) in 958 A.D when the elder King Ordono III died. Sancho’s reign was short-lived because of his fatness. “The nobility of Leon thought of him as weak-willed because of his obesity” and he was deposed (20). Sancho’s grandmother, Toda Arnez, who had once ruled the kingdom, was determined to help Sancho regain his throne. First, she took him to the local physicians who were unsuccessful in helping him lose weight. Although she loathed the Muslims, who occupied the southern part of the Iberian Peninsula in the 10th century, Toda and Sancho sought the help of Hasdai ibn Shaprut (917-970 CE), a brilliant and learned Jewish physician living in Cordoba. Shaprut was physician to the Muslim Caliph, Abd al Rahman in Cordoba, a major cultural center in the 10th century. Cordoba in the 10th century occupied a place similar to Rome in the 1st century or New York City in the 20th century. It was also renowned as one of the great medical centers of the late Middle Ages. In a day when house calls were still in fashion, Shaprut went to Pamplona to evaluate Sancho, and agreed to take him as a patient, but advised that it would be a long treatment requiring Sancho to relocate to Cordoba. Because of the seriousness of the problem, both medically and politically, Sancho and his grandmother Toda moved from Pamplona to the Muslim southern Iberian city of Cordoba. The medicine that Sancho received was “Theriaca.” This mixture of ingredients is said to have originated with Mitridates VI in Grecian times. He developed it as an antidote for snake bites. It can be compounded with up to 64 or more ingredients, with principal ingredients being opium, ginger, cinnamon, myrrh, saffron, and castor oil, a well-known laxative. Over time, Sancho gradually lost weight. When he returned to Leon as a leaner man who could mount his horse, and he was able to retake his throne. From this story, it is clear that not even kings and rulers are immune to obesity or the bias against obesity that led to his overthrow. Apparently, a king may be cruel, but not fat. Another lesson is that medical treatment of obesity can be effective if properly done. For Sancho, theriac had a dramatic success and allowed him to regain his throne and his kingdom (21).
DRUGS FOR TREATMENT OF OBESITY: 1500-1900 CE
The year 1543 CE is an appropriate one for demarcating a big divide in our view of progress in the management of obesity. It was the year that the Polish monk, Copernicus, published his book showing that the planets revolved around the sun, thus destroying the long-held view that the sun rotates around the earth. Less than 50 years earlier, movable type was invented by Gutenberg in Mainz, Germany a discovery that revolutionized the transmission of knowledge. In 1492 Columbus discovered the New World, opening up new wealth, cultures and lands and products such as tobacco that affect body weight. At about the same time, the dramatic events of the Protestant Reformation begun by Martin Luther challenged the primacy of the Roman Catholic Church. In the same year as Copernicus published his book, the first scientifically accurate book on human anatomy was published by Vesalius. Heady times these!
The five centuries since the scientific revolution have seen the industrial revolution (ca 1750), the second agricultural revolution (1750-1900) and the internet revolution of the present day. This expanding base of knowledge (Figure 1) provides the intellectual basis for understanding obesity as we know it today.
Sixteenth Century
The 16th century is often labeled the “century of discovery.” It is also the century where the corpulent King Henry VIII showed what happens when excess food is abundant, and exercise too little. Christopher Columbus had just discovered the New World in 1492 and brought tobacco, tomatoes, and many other products back to Europe with him. Tobacco became a treatment for obesity. We now know that nicotine is addictive, and that smoking can both reduce food intake and stimulate energy expenditure (22). Cessation of smoking is associated with weight gain (22), making this one of the challenges of stopping smoking. In the 16th century, the use of vinegar originally described many centuries earlier was again used as a treatment for obesity. However, obesity was less common in the 16th century, and maintaining good health through diet and exercise remained the goals for healthy living.
Seventeenth Century
Obesity gradually increased in prevalence such that strategies for its management appeared in textbooks for physicians in the 17th century. There were several formulas for treating “obesitas or corpulency,” listed in the textbook by Theophile Bonet (Bonetus), a leading physician of the time. They give a flavor of the strategies beyond diet and exercise that were used by physicians to treat obesity in the 17th century. To quote from Bonet ((23); p 390):
- “Chiapinius Vitellius, Camp Master-General, a middle-aged man, grew so fat, that he was forced to sustain his belly by a swathe, which came about his neck: And observing that he was every day more unfit for the Wars than others, he voluntarily abstained from Wine, and continued to drink vinegar as long as he lived; upon which his Belly fell, and his Skin hung loose, with which he could wrap himself as with a Doublet. It was observed that he lost 87 pounds of weight. [Note: Vinegar and cleansing, or cathartic agents have a long history for treating obesity-See Grecian Medicine above] (Underlining mine).
- “Lest any great mischief should follow, we must try to subtract by medicine, what a spare diet will not; because it has been observed, that a looseness either natural, or procured by Art, does not a little good. But this must be done by degrees and slowly, since it is not safe to disturb so much matter violently, lest it should come all at once. Therefore, the best way of Purging is by Pills, of Rheubarb, Aloes each 2 drachms [1drachm = 1/8 ounce or 60 grains], Agarick 1 drachm, Cinnamon, yellow Sanders, each half a drachm. Make them up with Syrup of Chichory. They must be taken in this manner; First, 1 Scruple* must be given an hour and a half before Meal; then two or three days afterwards, take half a drachm or two scruples before Meal. Thus, purging must be often repeated at short intervals, till you think all the cacochymie is removed. *[scruple – a unit of apothecary weight equal to about 1.3 grams, or 20 grains] [Note: Purgatives also go back to Grecian Medicine]
- “A certain Goldsmith, who was extreme fat, so that he was ready to be choaked, took the following Powder in his Meat, and so he was cured; Take of Tartar two ounces, Cinnamon three ounces, Ginger one ounce, Sugar four ounces. Make a Powder.
- “Horstius found the things following to take down fat Men; especially onions, Garlick, Cresses, Leeks, Seed of Rue, and especially Vinegar of Squills: Let them purge well: Let them Sweat, and purge by Urine: Let them use violent exercise before they eat: Let them induce hunger, want of Sleep and Thirst. Let them Sweat in a Stove and continue in the Sun. Let them abstain from Drink between Dinner and Supper: for to drink between Meals makes Men fat.
- “I knew a Nobleman so fat, that he could scarce sit on Horse-back, but he was asleep; and he could scarce stir a foot. But now he is able to walk, and his body is come to itself, only by chewing of Tobacco Leaves, as he affirmed to me. For it is good for Phlegmatick and cold Bodies.
- “Let Lingua Avis, or Ash-Keyes be taken constantly about one drachm in Wine. According to Pliny it cures Hydropical persons, and makes fat people lean” (23).
Eighteenth Century
The 18th century witnessed publication of the first two English monographs dealing exclusively with obesity. In each book, the author proposed a new way of treating obesity based on his own theory of how obesity developed. The first book by Dr. Thomas Short was published in 1727 (24). From Short’s perspective, treatment of obesity required restoring the natural balance and removal of the secondary causes. If possible, one should pick a place to live where the air is not too moist or too soggy and one should not reside in flat, wet countries or in the city or the woodlands. He thought that exercise was important and that the diet should be “moderate spare and of the more detergent kind” (24).
The second book was by Dr. Malcolm Flemyng, a graduate of the medical school in Edinburgh (25). His approach to treatment of obesity was based on the results of a patient that he presented to the Royal Society in London in 1757 and subsequently published in 1760. Flemyng’s theory was that sweat, urine, and feces all contained “oil” and that effective treatment for obesity required increased loss of “oil” by one or more of these 3 routes. Thus, laxatives, diuretics and sweating were his principal approach to the treatment of obesity. To quote Flemyng:
“Now we are so happy as to be in possession of a diuretic medicine, which has that quality [increases the quantity of urine and “renders the animal oil more mixable with the watery vehicle of the blood, that otherwise it would be” (a diuretic which) in a singular degree; and is withal so safe, as that it may be taken in large quantities every day for years together, without remarkably impairing the general health: that medicine is soap” ((25); p. 19).
Flemyng believed that obesity, or corpulency as it was often called, could be prevented as well as cured by the medical use of soap. In his book he presents a physician weighing 291 pounds (20 stone 11 lbs.) who lost more than 28 lbs. (2 stone) with this treatment.
DRUG TREATMENT OF OBESITY IN MODERN TIMES: 1900-2020 CE
The progress of science from 1543 onward has had an impact on obesity that is nicely illustrated in the effects on measurement of body composition. From the time of Hippocrates 2500 years ago it has been possible to assess obesity at the whole-body level, referred to as Level I in the model of Wang et al (26). With the accurate description of human anatomy beginning with Vesalius in 1543, obesity could be viewed from the tissue and organ level (level II). In 1839 the concept of the “cell” was introduced leading to a cellular analysis of obesity or Level III in this model. As chemistry advanced in the 19th century, obesity could be viewed at the molecular level or Level IV in this model. Finally in the 20thcentury techniques for measuring body composition at the atomic level were introduced adding Level V to the model. Advancing basic science thus clearly provided a base for improving measurements of body composition and for understanding how and why obesity develops.
Thyroid Hormone: 1893-1994
THE BEGINNING
The introduction of thyroid hormone as a treatment for obesity can be dated to 1893 and represents the first drug used on a rational basis for treatment of this problem. In 1888 the Myxedema Commission published a report on a disease called myxedema, a form of severe hypothyroidism (27). They found that it resulted from failure of the thyroid gland. Patients with myxedema have a puffy type of weight gain, slowing of their thought processes and speech, and, if severe, a drop in body temperature and coma. When these patients are treated with thyroid extract, all these symptoms, including the weight gain are reversed. Proof of a cause-and-effect relationship of the thyroid gland to myxedema came when the thyroid was removed and the symptoms were corrected by treatment with thyroid hormone extract.
In 1893, less than 5 years after the Myxedema Commission Report, the use of thyroid preparations for treatment of obesity had appeared in medical literature. Thyroid extract was the major form of thyroid hormone available until thyroxine was isolated by Kendall in 1915 (28) and synthesized in 1926 by Harrington (29).
THE GENIE IS OUT OF THE B0TTLE: 1893 to 1953
Once it was clear that thyroid could increase metabolic rate, the genie was out of the bottle. In 1893 in his report on “Cases of Myxedema and Acromegalia Treated with Benefit of Sheep’s Thyroid” Dr. J.J. Putnam from the Massachusetts General Hospital included a footnote ((30); p. 130) saying:
“Dr. Barron has very kindly written to me that he has used the treatment [with thyroid] in 5 cases of ordinary corpulence. One lost twenty-eight pounds in 6 weeks, three a moderate amount, and all lost more or less. I [Putnam] am trying it in two cases but have no results to report as yet” (30).
Following in the footsteps of this this report were two others: one by Yorke-Davies in 1894 (31) and another independently by Wendelstadt (32) describing the use of thyroid substance to treat patients with obesity. Their communications, and that of Leichtenstern (33), brought a short wave of popularity for the treatment of corpulency [obesity] by thyroid preparations that, along with iodine, soon became the mainstays of patent medicines and various nostrums used for weight loss. As Foxcroft notes:
“It [iodine] was one of the secret ingredients in some of the most popular and widely advertised patent medicines against fat including: Allan’s Anti-Fat; Frank J Kellogg’s Safe Fat Reducer; Dr. Bertha C. Day’s Fort Wayne prescriptions, Marmola, Newman’s Obesity Cure, Chichester’s Corpus Lean, Rengo, Dr. Gordon’s Elegant Pill, Corpulin, Elimiton, Phy-th-rin, San-Gri-Na Trilene tablets – all these contained either fucus (bladderwrack) or thyroid extract, or Ipecac (a plant-based emetic), camphor (an appetite suppressant), potassium acetate (a diuretic) and digitalis ( a stimulant)” ((8); p. 103).
Many of these ingredients were subsequently found in “Rainbow Pills” that were popular later in the 20th century (See below).
From the beginning, use of thyroid hormone to treat obesity raised concerns (8). For example, Woods Hutchinson, a medical professor in the United States who frequently wrote for women's magazines such as Cosmopolitan, said in1894 that physicians didn't have any idea how thyroid, often prescribed with potassium (and sometimes arsenic), worked:
"Both [thyroid and arsenic] cause, in some curious manner which we do not as yet understand, such an interference with the normal metabolism of the body as to cause the burning up and elimination of considerable amounts of body fat."
Hutchinson further noted that if patients lost more than ten percent of their body weight—the "movable ten percent," he called it—the results could be injurious. "The appetite becomes impaired, the sleep broken, and the heart's action irregular." If prolonged, the drug would set up a "serious and obstinate disturbance of the nervous system, and particularly of the nerves controlling the heart, accompanied by palpitation, sweating, weakness, and intense nervousness." (8)
Also expressing concern was Professor Sajous, first President of the Endocrine Society who said:
“The fact that thyroid preparations in sufficient doses promote the rapid combustion of fats has caused them to be used extensively in this disorder…In large doses (thyroid gland) imposes hyperoxidation upon all cells…we behold gradual emaciation beginning with the adipose tissues, which are the first to succumb. Hence the use of thyroid preparations in obesity. Briefly, in all cases of obesity in which thyroid gland is rationally indicated, the feature to determine is whether directly or indirectly hypothyroidea underlies the adiposis.” (34)
Administration of thyroid hormone to obese subjects whose basal metabolism is normal seemed illogical and was contraindicated according to some physicians (35, 36). However, Evans and Strang, two highly respected physicians, did use thyroid therapy in two percent of their cases, selecting those in which the initial level of the twenty-four-hour resting metabolic rate was not greatly above “ideal” (37). Two other physicians, Lyon and Dunlop, reported on the effects of diet and thyroid treatment in 24 hospitalized patients with obesity who were prescribed a 1000 kcal/d diet. The average daily weight loss was 162 g/d. When non-toxic doses of thyroid were added, the average daily weight loss increased to 273 g/d and then fell back to 153g/d when thyroid was withdrawn (38).
Some physicians used thyroid hormone as an adjunct to a hypocaloric or sub-maintenance diet in all forms of obesity, provided that that there were no contraindications. In an outpatient study of 106 unselected patients with obesity, Bayer and Gray treated 100 with diet alone, 51 with added thyroid and 23 with added dinitrophenol (see below) (39). Diet alone produced a weight loss of 15 lbs. (6.8 kg) in 3 1/2 months, after which weight stabilized following a loss of 10 to 20 pound (4.5 to 9.1 kg). Next, 41 of these patients were given thyroid extract averaging 1½ grains (96 mg/d) per day and lost a further 11 pounds (5.0 kg). Eleven other patients were given dinitrophenol at an average dose of 165 mg/d and lost a further 12 pounds (5.4 kg). This study demonstrated the value of adding either thyroid or dinitrophenol after weight loss ceased on a sub-maintenance calorie diet. Both drugs seemed to be more effective when the metabolic rate was low. When one drug followed the other there was a small additional weight loss, which was similar at about 4 pounds (1.8 kg) with either order of transfer.
One downside to using supraphysiologic doses of thyroid hormone in the management of obesity is that it increases the catabolism of protein, and thus the loss of lean body mass, which is a proportionally larger amount of the weight loss than is the loss of fat (40).
At the time of World War II, the status of thyroid hormone was summarized in two monographs. In the first famous monograph, Obesity and Leanness by Rony, 1940 (41), the author wrote:
“When thyroid is administered to obese subjects with a normal level of basal metabolism, living on unrestricted diet, a few of the subjects lose considerable weight when the basal metabolism is increased by 10 to 20 per cent. However, administration of non-toxic doses of thyroid is not followed by appreciable loss of weight in most subjects as long as the food intake remains unrestricted. In other words, for most obese patients a sub-maintenance [hypocaloric] diet must accompany thyroid administration if consistent loss of weight is to be effected.” ((41), p 257).
Further, Rony concluded
“Therefore, in the absence of contraindications a trial with thyroid in the later stages of the sub-maintenance regime seems to be justified in most cases of obesity.” ((41), p 259).
His contraindications to use of thyroid were:
- Advanced Age. Rarely used in individuals > 50 years old and never > 60 years old.
- Hypertension
- Valvular or myocardial heart disease, arrhythmia, tachycardia, regardless of origin
- High basal metabolic rate.
- Marked vasomotor disturbances during the menopause
- Marked nervous irritability or emotional instability
- Intolerance to small doses of thyroid, manifested by rapid pulse, palpitations of the heart, tremor, insomnia or suppression of menstruation.”
In 1949, ten years after Rony’s report on thyroid hormone usage in obesity, Rynearson and Gastineau from the Mayo Clinic provided another summary (42). They noted, as documented earlier, that thyroid hormone was introduced into clinical practice in 1894 and rapidly grew in popularity because of the belief that many patients with obesity had low metabolic rates that were consistent with hypothyroidism. In addition to “replacement doses” some clinicians began using higher doses of thyroid hormone that were “calorigenic”; that is they raised metabolic rate by as much as 15-20% (43). Another rationale for the use of thyroid hormone was to restore to “normal” the metabolic rate that declined with weight loss (38, 44-47). As Rynearson and Gastineau noted, Wilder had shown that if adequate amounts of protein were provided in the diet, metabolic rate did not fall (48), removing this argument for the use of thyroid hormone. Although thyroid hormone was widely used to treat obesity in 1949, there were several, mainly academic physicians, who opposed its use including Professor Means from the Massachusetts General Hospital at Harvard Medical School (47), Professor Severinghaus from the University of Michigan (49, 50), and others (51).
Opposition to use of thyroid hormone also came from two reports that failed to find weight loss from treatment with thyroid hormone (52, 53). As Rynearson and Gastineau noted, thyroid hormone also increased heart rate, putting a load on the cardiovascular system as well as increasing protein breakdown (54, 55). Rynearson and Gastineau concluded by adding their caution about the use of thyroid hormone to treat obesity. However, caution at the academic level did not necessarily translate into caution by practitioners in the office.
TRIIODOTHYRONINE IS DISCOVERED AFTER WORLD WAR II AND THE WATER GETS MUDDIER
The identification of triiodothyronine by Gross and Pitt-Rivers in 1952 opened the door on a new chapter in the use of thyroid hormones for treatment of obesity (56). Triiodothyronine is derived from thyroxine by deiodination and works more rapidly with a shorter half-life than thyroxine. Studies of triiodothyronine were aided by the development of a radioimmunoassay that was specific for this molecule (57). During weight loss, circulating levels of T3 decline, as does energy expenditure. This raised the obvious question of whether increasing the level of this hormone back to pre-weight loss levels would reverse the lower metabolism and thus enhance weight loss and weight loss maintenance. This hypothesis was tested by Byerley & Heber (58) who showed that during a 10 day fast where serum T3 and metabolic rate both declined, restoring T3 to normal levels during the last 3 days of the fast did not raise the metabolic rate.
According to Garrow in 1974, the results of weight studies with triiodothyronine vary from modest enthusiasm to outright condemnation (59). In one study (60), T3 was given in more or less physiological levels of 105 mcg/d to 29 patients with obesity. Weight loss averaged 8.6 kg (19 lbs) over 17 weeks. However, after one-year weight loss was only 3.6 kg, leading the authors to conclude that T3 in these doses was not a valuable tool for treating obesity. In a second study, Hollingsworth et al (61) treated 17 patients who weighed between 110 and 179 kg with an 800 kcal/d diet along with a placebo or triiodothyronine 225 mcg/d, about triple the maintenance dose. Weight loss over 6 months in the patients treated with T3 was 21.9 kg compared to 13.3 kg in the placebo-treated patients. In still another early study, Drenick and Fisler used triiodothyronine or thyroxine to help patients who had lost weight in the hospital maintain their weight loss (62). These 21 men had lost 42.3 kg in the hospital and were able to maintain most of it, although with troublesome side effects of T3. After reviewing these three studies using triiodothyronine, Garrow concludes:
“Thyroid preparations are not a satisfactory substitute for a low energy diet, but in a severely obese person who has become adapted to a low energy diet after several months, and hence has ceased to lose weight, thyroid hormones may provide the only practical line of treatment. It should be emphasized that this line of treatment should only be used if it is certain that the patient is in fact on a low energy diet, and usually this means supervision in hospital for a period of at least 2-3 weeks with appropriate measurements of metabolic rate.” ((59); p 171)
Against this pessimistic view of triiodothyronine, Garrow relates his personal experience with individuals who are “resistant” to weight loss even under observation. He studied two patients living on the United Kingdom’s Medical Research Council’s metabolic ward at the Northwick Park Hospital who claimed to have “refractory” obesity. The first was a 54-year-old woman who had a low metabolic rate, but normal thyroid function tests and who lost only 1 kg/week while eating an 800 kcal/d diet while under observation. Dr. Garrow treated her with 100 mcg/d of triiodothyronine along with a selective beta-blocker to prevent tachycardia. On this treatment she lost 21 kg in 6 weeks with remission of her angina pectoris. A similar patient with significant osteoarthritis also benefited from triiodothyronine. Garrow summarizes these studies by saying:
“…there are patients who do not lose much weight on an 800 kcal diet under close supervision, and this is due to a low metabolic rate, although there is no clinical or biochemical evidence of hypothyroidism. In such cases it seems justifiable, if the obesity is disabling, to increase the metabolic rate with thyroid preparations, providing that this process is properly controlled” ((59); p 279-281).
It was about this time, in 1981, that 3 cases of death were reported in individuals who were taking high doses of thyroxine (T4) (63), an observation that raised a red flag. However, the debate about use of thyroid hormones to treat obesity has continued into the 21st Century. In a review in 2002, Krotkiewski concluded the following on the use of triiodothyronine, thyroxine, or thyroid extract to ameliorate the metabolic effects of a very low calorie diet on serum T3 or metabolic rate (64):
“Thus, it seems reasonable to recommend small doses of T3 as an adjunct to dietary treatment of obesity in the following groups of patients:
- In patients receiving beta-adrenergic receptor blockers, showing verified resistance to dietary therapy.
- In overweight patients on T4 replacement therapy after successful treatment of hyperthyroidism.
- In overweight patients on habitual food intake receiving T4 replacement therapy (previously hypothyroid).
- In patients showing `dietary treatment-resistant' weight increase while stopping cigarette smoking.
- In patients eating a very-low calorie diet (VLCD) and/or a low-calorie diet (LCD) showing low T3, parallel to slowed rate of body weight loss despite continued calorie restriction.
- In patients with abdominal obesity and metabolic syndrome, resistant to dietary treatment or showing inadequate improvement in associated metabolic aberrations.
- In patients showing, before or during dietary treatment, signs and symptoms of sub-clinical hypothyroidism.
Triiodothyronine acts through one of 2 receptors, the T3 receptor alpha (TRα) and T3 receptor beta (TRβ). The TRα receptor appears to mediate the effects on heart rate, whereas the TRβ receptor mediates the effect on cholesterol and on metabolic rate (65). Activation of the TRβ receptor may provide a strategy for lowering lipoprotein cholesterol and for increasing metabolic rate in animals and human beings.
Thus, the status of thyroid hormone in management of the patient with obesity is still open for further investigations with the last word yet to be written.
Dinitrophenol: 1918-1938
Dinitrophenol has had more than one life. First, as an explosive during World War I. Then, as a weight loss agent. Finally, as a drug for potential use in neurological diseases (66). I will focus on its use as a weight loss drug.
DINITROPHENOL: INITIAL BENEFITS AND TOXICITIES
During the rapid growth of the chemical industry in Germany in the late 19th and early 20th centuries, many compounds were made for dyeing cloth. A major offshoot of this development was the introduction of dyes to stain histological samples for study of tissue structure under the microscope. Another outgrowth was a supply of chemicals to synthetic organic chemists and pharmacologists. Paul Ehrlich (1854-1915) was a pioneer with these dyes and can be called “The Father of Pharmacology.” Among his many contributions to biomedical science is his concept of the “magic bullet” – the idea that a chemical molecule could act like a key in a lock to provide a way to target chemicals to treat disease-causing processes within cells. One of the fruits of his labor was arsphenamine, or salvarsan; also called “606,” for the number of different molecules that were tried before he found his “magic bullet” for the treatment of syphilis (5).
Another product of the chemical industry that had a direct impact on obesity was the synthesis of 2,4-dinitrophenol. French factory workers preparing this chemical in munitions factories during World War I were noted to lose weight. As Perkins put it in his lengthy report on Munitions Intoxications in France: “Workers claim that they have grown thin to a notable extent after several months of work with DNP” ((67); p 2341).
This observation was picked up by Tainter and his colleagues at Stanford University in 1931. They initially conducted animal experiments with DNP before using it in patients (68, 69). During studies for drug safety, they noted that the therapeutic index of dinitrophenol, that is the relation of therapeutic to toxic effects, was razor thin, so they proceeded carefully with their clinical studies. They treated 20 men and 150 women with obesity using doses of dinitrophenol up to 0.3 g daily along with a moderately restricted diet. Of these individuals, 71 had been treated previously with dietary measures, thyroid hormone, or both. The average total dose of drug was 26 kg per patient and an average treatment duration of 88 days. Weight loss averaged 17.1 lb. (7.8 kg) or 1.4 lb. (0.64 kg) per week and only 5 patients failed to lose weight. The largest weight loss was 82 lb. (37.3 kg) which occurred over 198 days. Mild toxicity occurred in 28 individuals, and consisted of skin rashes, pruritus, and peripheral neuritis. There was no evidence of changes in red or white blood cells and no change in blood pressure. In his review in 2007, Colman noted that, by 1934, Tainter estimated that as many as 100,000 Americans had used dinitrophenol emphasizing the “desire” of Americans to become slimmer (70).
In 1934 Tainter and his colleagues wrote: “It can now be said that dinitrophenol is of definite value as a drug for treating obesity” (71). He also reported three deaths, skin rashes and the yellow skin color associated with DNP as well as rashes and peripheral neuritis in some patients – a finding indicating the drug should be discontinued (72). However, it wasn’t long until other problems surfaced including cataracts (73) as well as neuropathy. By one estimate, 2,500 people lost their sight using dinitrophenol. Reports of deaths also continued to be reported.
Other clinicians did not have the same enthusiasm as Tainter. In 1935, McGavack from San Francisco reviewed the use of dinitrophenol by some 290 individuals and reached several conclusions (74). First, the loss of weight using diet and dinitrophenol was not strikingly greater than with diet alone. Second, many people had distressing symptoms from its use. Third, there were significant toxic effects of the DNP on body function and tissues. Finally, the reported deaths from dinitrophenol when used in accepted therapeutic dosages made it hard to justify for widespread use in treating overweight, which he called the relatively benign condition. This was supported by two other studies that compared the effects of DNP to other ways of managing the patient with obesity. Bayer and Gray (39) reported on 100 individuals who were treated diet alone (920 kcals/d), or with diet and the addition of either thyroid extract (up to 3 grains/d) or dinitrophenol up to 300 mg/d. On diet alone, weight stabilized in 72 of these patients after 4 months and an average 15 lb. (6.8kg) weight loss. Thyroid extract induced an extra weight loss averaging 11 lb. (5.0 kg) in 41 patients during 90 days of treatment with thyroid extract, compared to dinitrophenol, which produced an average loss of 12 lbs. (5.5 kg) in 13 patients over 50 days. Dinitrophenol seemed to be most effective when the BMR was normal. In a second report Strang and Evans observed greater weight losses in the cases receiving dinitrophenol than in those treated by diet alone but felt that the difference was hardly striking enough to offset the discomfort and possible damage of treatment with DNP (75).
With this information as a background in 1935, the Council on Pharmacy and Chemistry of the American Medical Association concluded that the dinitrophenol was too hazardous to include in the AMA publication of New and Non-official Remedies (76). At the time that dinitrophenol was being touted for obesity in 1935, the US FDA was limited in its power to regulate drugs based on The Food and Drugs Act, which was passed in 1906 and brought the FDA into existence. It wasn’t until this law was updated as the Food, Drug, and Cosmetic Act of 1938 that the FDA had the authority needed to act against drugs like dinitrophenol. In 1938, they turned their attention on the Isabella Laboratories which was selling capsules containing 1.5 g of dinitrophenol as Formula 281. When the hazards became clear, the FDA moved to ban DNP as too dangerous for use in humans (70).
THE DINITROPHENOL RENAISSANCE: 1980 –
Although it had been banned in 1938, dinitrophenol made a come-back in 1981 (77). In this year, a Texas physician named Dr. Bachynsky began to process industrial DNP and put it into tablets which he dispensed and marketed under the trade name ‘Mitcal’. He advertised that ‘Mitcal’ produced weight loss by a mechanism he called intracellular hyperthermia i.e., uncoupling of oxidative phosphorylation. In subsequent court proceedings it was alleged that over 14,000 people were treated with Mitcal by Dr. Bachynsky. Individuals using Mitcal started reporting adverse effects, such as fever, shortness of breath and sweating, to the US Food and Drugs Administration in late 1982. Additionally in 1984, there was a fatality associated with an intentional overdose of ‘Mitcal’. Dr. Bachynsky was convicted in 1986 of drug law violations, fined and prohibited from dispensing DNP to any patients. However, Dr. Bachynsky continued to use DNP for a variety of different ‘medicinal claims.’ He was eventually jailed for fraud in 2008 in the USA for developing DNP for use in Europe as a cancer treatment again called intracellular hyperthermia therapy.
Outside of the medical community, DNP is still marketed on the Internet without regulation, primarily to body builders who are attempting to lose weight. To address this growing use, the Food Standards Agency in 2011 issued a warning that this product is “not fit for human consumption” given its short- and long-term effects. Nonetheless, gym enthusiasts continue to obtain and use DNP, even though there were sporadic reports of deaths (78). To obtain insight into the reasons for this continued use of DNP, Ainsworth et al interviewed 14 users who reported that the internet was the main source of their information about DNP (79). The authors found that these individuals valued “self-control” and their own judgment in minimizing potential risks. This is, of course, against the background of continuing deaths associated with use of DNP. In their review, Grundlingh et al plotted the deaths associated with DNP by decade (77). In the 1930s there were 8 deaths, which declined to only one per decade in the 1940s and 1950s during the time of stringent regulation. Subsequently, after 4 decades with no deaths, thirteen deaths were recorded in the first decade of the 21st century.
In addition, DNP at doses that do not affect weight is being explored for its potential use in neurodegenerative disease where mitochondrial dysfunction is often observed. DNP has been shown to induce neurotrophic growth factors involved in neuronal health, cognition, and learning. It is now being investigated through approved FDA channels with an Investigational New Drug Application for a group of neurodegenerative diseases such as Huntington’s disease, multiple sclerosis, and Duchenne’s muscular dystrophy (80).
In summary, DNP has had many lives. As an explosive during World War I, as a treatment for obesity during the 1930’s (first discredited and then resurfacing in the 1980s), as a weight control agent for body-builders using the internet to obtain supplies, and most recently as a potential agent for treatment of neurodegenerative diseases.
Amphetamine: 1932-1968
SYNTHESIS AND INITIAL TESTING
The rise, fall, and return to restricted medical use of amphetamine is the story of the first psychoactive mood-altering prescription drug (81). Amphetamine might be described as a Janus drug – that is it has two faces – one the pharmacological side to constrict blood vessels, suppress food intake, and either stimulate the central nervous system or calm it in individuals with attention deficit disorder. The other face is the potential for abuse, first recognized in the late 1930s and brought into stark focus by “street-use” of methamphetamine.
Amphetamine is a sympathomimetic drug originally labeled in 1910 (82). The name amphetamine comes from its chemical structure – α-methyl-β-phenethylamine. It was first synthesized in 1887 by the Romanian chemist Edeleanu, who was working in Germany. The stimulant properties of his compound, phenylisopropylamine as he called it, were unknown until it was independently resynthesized and tested clinically by Gordon Alles. Alles, a chemist in California, was searching for an alternative decongestant to compete with ephedrine in the treatment of allergy and asthma. In 1929 Alles synthesized what he called beta-phenyl-isopropylamine (amphetamine). To test the clinical effects of his discovery, Alles became a “human guinea” when he was injected with 50 mg, a relatively large dose of his product. Within seven minutes he noted that his nose was dry and clear when he sniffed. His blood pressure climbed dramatically and by 17 minutes he noted heart palpitations. He also had a “feeling of well-being.” That evening at a dinner party he grew chatty, considering himself unusually witty. He recorded that he had a “Rather sleepless night. Mind seemed to run from one subject to another” (83). In 1932 Alles patented his compound and its medical uses (81).
In 1933 Smith, Kline and French (SKF), an American pharmaceutical company began selling Benzedrine, a decongestant that they had patented that was identical to the one Alles had patented (81).
1935 was another key year in the history of amphetamine. Alles thought that the stimulant properties of amphetamine might help people suffering with narcolepsy, a disorder of sleep. He supplied amphetamine to Myron Prinzmetal, who had worked with Alles as a medical student at the University of California, San Francisco (UCSF), to test in patients with narcolepsy. Prinzmetal et al published their affirmation of this effect in 1935 (84) and their findings were soon replicated by Ullrich et al in 1936 (85). Both groups showed that the drug did reduce sleep time in people with narcolepsy, and this became an indication for amphetamine use. Of interest to this paper, these authors also noted that the patients who were treated for narcolepsy also lost weight.
To explore amphetamine as an agent that might improve mood, SKF teamed up with Dr. Abraham Myerson, a highly respected professor of psychiatry working at both Tufts and Harvard who operated a well-funded laboratory. His books for the public had given him public visibility. In addition to his work on amphetamine for psychiatric problems, Myerson picked up on the reported weight loss during treatment for narcolepsy. Together with Mark Falcon-Lesses, Myerson designed a clinical trial to assess the effects of amphetamine for weight loss (86). A group of 16 women and 1 man were treated for 6 to 23 weeks with Benzedrine sulfate in doses of 10 to 30 mg/d. Patients weighed between 145 and 316 lbs. (66 to 144 kg) when the trial began and lost an average of 1.46 lbs/week (0.66 kg/week) during treatment. There were no untoward side effects, and the results were published in the prestigious New England Journal of Medicine in 1938 (86). As Rasmussen summarized the situation in 1938: ”Despite the study by Lesses and Myerson, SKF did not market Benzedrine Sulfate to treat obesity, although the firm did follow-up earlier observations by Nathanson and others that Benzedrine resulted in weight loss. For instance, SKF sponsored testing of the drug’s impact on metabolic rate and trials evaluating its effects on appetite. Initially, SKF thought that marketing of Benzedrine for weight loss might interfere with its development as a respectable psychiatric drug. On the other hand, SKF might have realized that amphetamine was “selling itself” for weight loss since many smaller companies were profiting from the weight loss market, even though they were violating the patents owned by SKF. However, by the end of World War II amphetamine and methamphetamine were extensively used by both the Allied and Axis forces because they seemed to enhance performance and SKF would move to capture the weight-loss market” (87).
In a review of 1946, Bett described the setting for amphetamine (Benzedrine) and obesity this way: “The value of Benzedrine sulphate as an ‘adjunct' to diet and other measures in the treatment of obesity may thus be summarized: it decreases appetite and helps the patient to follow a diet, possibly also by producing a sense of well-being. Increasing activity, it promotes proper balance between energy output and caloric intake. It educates the patient to new eating habits, so that weight loss is maintained after stopping the drug. It has no appreciable effect on basal metabolic rate.” (88).
AMPHETAMINE AND ADDICTION
A warning about the risk of addiction with the use of amphetamine was sounded in an editorial in the Journal of the American Medical Association (JAMA) in 1938 (89). The observation that the use of Benzedrine over long periods is "certainly not without danger, particularly to the circulatory system," prompted this comment from Lesses : (90)"…the drugs to which human beings become addicted are the narcotics. There is no evidence in the entire literature of medicine that stimulants become habit forming." This, in retrospect, was clearly inaccurate. Even though in Myerson’s clinical experience with Benzedrine for more than two years in a very large number of cases he had not seen "a single case of addiction in the sense that a person, otherwise, now feels it necessary to take the drug habitually and in ascending doses to produce the desired effect." In 1937 abuse of the drug was reported among Midwestern college students, particularly at the University of Minnesota. Moreover Benzedrine Sulfate tablets were taking on the identity as “pep pills” or “pepper-uppers” in the popular imagination as Myerson recognized two years later (91). Based on the growing evidence of abuse, amphetamine became a strictly regulated prescription drug, but its illegal use in the popular culture has been difficult to control and addiction to methamphetamine has become a major modern problem.
AMPHETAMINE REVIVAL FOR TREATING ATTENTION DEFICIT/HYPERACTIVITY DISORDER
Racemic amphetamine can be used to treat Attention Deficit Hyperactivity Disorder (ADHD) and binge-eating disorder. Lisdexamfetamine, a combination of lysine and d-amphetamine, which is hydrolyzed once inside the red blood cell has become a drug of choice for this diagnosis (92). Although recreational use of amphetamine produces serious risk of addiction, this is unlikely to occur when amphetamine-derivatives are used at therapeutic doses for long-term treatment of attention deficit disorder. In fact, lifetime stimulant therapy for ADHD that begins during childhood reduces the risk of developing substance abuse as an adult.
Rainbow Pills: 1940-1968
Rainbow Pills are a continuation of the amphetamine story (93). The Miriam Webster Dictionary defines Rainbow Pills as “any of a combination of pills (as of amphetamines, laxatives, and thyroid hormones) typically of different colors that were formerly taken to curb appetite and promote weight loss” (“Rainbow pill.” The Merriam-Webster.com Medical Dictionary, Merriam-Webster Inc., https://www.merriam-webster.com/medical/rainbow%20pill. Accessed 5 January 2020).
I have picked the start date of 1940 for the beginning of the Rainbow Pill period since it was the date for the first of 27 meetings sponsored by Western Research Laboratories held to introduce weight loss specialists to the use of their Rainbow Pills. Their 27th Annual Symposium on Obesity was held in Denver on April 13-15, 1967, and I was in attendance. Amphetamine was one of the many-colored pills included in what came to be called the "rainbow diet pill" regimen. In addition to amphetamine, Rainbow Pills included thyroid hormone, laxatives, diuretics, and digitalis (Table 2).
|
Table 2. Ingredients in One or More of The Rainbow Pills
|
|
Ingredients for Weight Loss
|
Ingredients to Mask Side Effects
|
|
d-Amphetamine
|
Cardiac glycosides (digitalis)
|
|
Diuretics
|
Barbiturates
|
|
Thyroid hormones
|
Corticosteroids
|
|
Laxatives
|
Potassium
|
|
Phenolphthalein
|
Belladonna
|
|
Herbal ingredients
|
Glandular extracts
|
Adapted from (93).
Digitalis was included since it can cause “nausea,” with weight loss as a consequence. The downside of digitalis is that it has a narrow window of tolerance between its therapeutic effect and toxicity. Diuretics were included to increase fluid (and weight) loss even though this is not fat loss. As we noted earlier, “diuresis” was one strategy used for weight loss dating from Greco-Roman times. Rainbow Pills often contained a barbiturate to suppress some of the side effects of amphetamine stimulation. To reduce middleman costs, the distribution of these pills to patients was directly from the physicians’ own offices rather than through a pharmacy.
The route to regulate Rainbow Pills was a prolonged one. Following the Pure Food and Drug Act of 1906 and the formation of the U.S. Food and Drug Administration (FDA), enforcement powers of the FDA were significantly enhanced with passage of the Food, Drug and Cosmetic Act of 1938. In 1962, the Kefauver-Harris amendmentrequired drug manufacturers to provide proof of both the safety and effectiveness of their drugs prior to approval for sale to the public. It also required drug advertising to disclose accurate information about side effects, and stopped cheap generic drugs being marketed as expensive drugs under new trade names and calling them new "breakthrough" medications.
In 1967-1968, a number of deaths attributed to use of Rainbow Pills triggered a Senate investigation and the gradual implementation of greater restrictions on drug marketing for obesity management. At the beginning of my research career into the causes and treatment of obesity, the U.S. Senate Select Committee held their first hearings on the misuse of anti-obesity medications (94). As a young Assistant Professor, I was invited to give a talk at the 28th Annual Western Research Laboratories Symposium on Obesity being held in the elegant Brown Palace hotel in downtown Denver, Colorado. The title of my talk was “Some New Thoughts on the Treatment of Obesity: Growth Hormone; Thyroid Hormone and Appetite Control.” Other topics on the program included “Abnormal Thyroid Transport Mechanisms in Obesity” by Dr. Irving B. Perlstein; “Exercise and Fitness Programs for the Overweight” by Captain McHargue from the US Air Force Academy; “Is the sound medical management of obesity successful? (A preliminary study of 6,000 cases histories) by Wilmer Asher; “Digitalis and The ‘Normal’ Heart, Some Controversial Aspects” by Richard Bloomfield; and finally “Obesity and its effects on Man-Hours of Work Loss” by Dr. Raymond E. Dietz. The invitees included physicians and their staff involved in delivering weight loss programs around the country. At the Saturday morning breakfast, a prominent member of the group sat down next to me. He had been summoned to testify before Senator Hart at the Rainbow Pill Hearings in the U.S. Senate (94). He said, slamming his fist on the table, that “Senator Hart had accused him of making more than $1 million dollars a year.” He went on to say, “I hardly ever made more than $750,000 per year”! This was 1968, and I was a young faculty member making a mere $12,000 per year. I listened in astonishment! I was undeterred from my academic career by the possibilities of making large amounts of money from treating patients with obesity, and my table mate continued to operate his financially rewarding clinics for many more years.
RAINBOW PILLS RESURFACE
After the rainbow diet pills were banned in the US in the late 1960s, they largely disappeared from the market during the 1970s and 1980s. They reappeared in Brazil in South America and in Spain in the 1980s. The re-entry of Rainbow Pills into the United States market was facilitated by passage of the Dietary Supplement Health and Education Act and Supplement Act (DSHEA Act) of 1994 which states that dietary supplements do not require premarket review by the FDA. More about the DSHEA Act later. Thus, federal regulators are powerless to stop the pills from hitting store shelves, and thanks to the Internet, the distribution network of rainbow pills is larger than ever and they can purchased in many forms (93).
Benzocaine
Benzocaine is a topical anesthetic that in lozenge form can numb the nerve endings in the mouth, which might, in turn, reduce the pleasure of eating. To test this, 40 women with obesity received one of four treatments: 1) a chewing gum containing 96 mg/day of benzocaine alone, 2) phenylpropanolamine alone at a dose of 75 mg/day, 3) the combination of these two agents; and 4) a placebo gum and pill. At the end of 8 weeks, those treated with phenylpropanolamine lost twice as much weight as the placebo-treated group, whereas those receiving benzocaine lost essentially no weight (95, 96). Interestingly, weight loss in the group receiving the combination of phenylpropanolamine and benzocaine was equal to that of the placebo group. Clearly local anesthesia in the mouth using benzocaine is of no value in the treatment of obesity.
Anorectic Sympathomimetic Drugs: 1950-1997
Amphetamine is the grandfather of this class of drugs. Amphetamine reduces food intake, and through activation of the sympathetic nervous system, raises blood pressure and has neurophysiological effects. Efforts by chemists to separate these physiological effects occupied much of the period between the end of World War II with approval of the final sympathomimetic anorectic drugs, fenfluramine and chlorpentermine, in 1973.
The anorectic sympathomimetic drugs can be divided into three groups. The first is a non-prescription sympathomimetic appetite suppressant, phenylpropanolamine that is no longer approved in the US for management of obesity. The second group is comprised of sympathomimetic drugs approved before 1973. The third group is comprised of drugs approved after 1973, until the removal of fenfluramine and dexfenfluramine from the market in 1997.
PHENYLPROPANOLAMINE FOR WEIGHT LOSS: 1938-2000
Phenylpropanolamine was synthesized as early as 1910 (82). Like other sympathomimetic drugs, it can raise blood pressure and constrict small blood vessels, thus leading to its use as a nasal decongestant. Phenylpropanolamine was first used commercially during the 1930s as an intravenous treatment for postoperative hypotension (97). The drug was patented in 1938, and in 1939 it was noted to suppress appetite. From the 1960s until it was removed from the US market in 2000, it was sold without prescription as a decongestant since the US Food and Drug Administration (FDA) recognized it as safe under the standard of Generally Recognized As Safe (GRAS). Phenylpropanolamine was given GRAS status as an appetite suppressant based on an analysis of weight loss in published studies (98-100). The weight loss was modest – only an additional 0.5 kg per week. This was similar to the results reported by Scoville for other anorectic drugs (101). Other studies performed after 1985 reported slower weight loss, averaging only 0.21 kg/week more than in the placebo-treated subjects. In addition, weight loss began slowing after the first 4 weeks, and at the end of these studies, phenylpropanolamine subjects had lost only 0.14 kg/week more than those receiving placebo. There is only one controlled trial of phenylpropanolamine that lasted up to 20 weeks (100). After 6 weeks, the phenylpropanolamine-treated group had lost 2.4 kg (0.43 kg/week) compared with 1.1 kg (0.18 kg/week) in the placebo group. Twenty-four subjects continued in an optional extension to 20 weeks and lost 5.1 kg (6.5%) compared with 12 placebo-treated subjects who only lost 0.4 kg (0.5%) of their initial body weight (P < 0.05).
For many years phenylpropanolamine was sold over-the-counter under the trade name Dexatrim, which was owned by Thompson Medical Company. When the patent protection for phenylpropanolamine ended in the 1980s, Ciba-Geigy began a marketing campaign for Acutrim (Oros TM), an over-the-counter product containing phenylpropanolamine. In a randomized 14-week randomized, clinical trial, participants who took Acutrim lost 8.0% of their body weight compared to 5.4% in those receiving the placebo (102).
Concerns about detrimental effects of phenylpropanolamine on blood pressure and the cardiovascular system had been a recurring issue. To examine this possibility critically, a double-blind, multicenter clinical trial funded in part by Thompson Medical, examined the effects of phenylpropanolamine on the changes in blood pressure in 881 individuals (103). In this parallel arm study, one group received a placebo three times a day, a second group received 75 mg of a sustained-release form of phenylpropanolamine once daily followed by two placebo capsules, and a third group received 25 mg of immediate-release phenylpropanolamine three times a day. Thirty percent of the 811 participants were above their ideal body weight. Blood pressure increased significantly in the first 6 hours following the 25 mg dose, and even more in those receiving the sustained-release preparation. However, the authors did not consider this to be clinically important (103). Baseline body weight and diastolic blood pressure were significant and independent determinants of the pressor effect of phenylpropanolamine.
This study, however, did not allay concerns about effects of phenylpropanolamine on blood pressure and the risk of stroke. To provide more perspective, an epidemiological study was designed to compare individuals using phenylpropanolamine who had had a hemorrhagic stroke with a control group without a history of stroke. This study found a significant association between hemorrhagic stroke and the use of phenylpropanolamine (104). As a result of this study and two years after Thomson Medical sold its interest in Dexatrim to Chattem Pharmaceuticals, the FDA took steps on Nov 6, 2000 to remove phenylpropanolamine from all drug products in the United States and requested that all drug companies discontinue marketing products containing phenylpropanolamine. This was one of the first examples of off-target effects derailing the clinical use of a drug in the management of obesity.
ANORECTICS: 1959-1973
Anorectic drugs refer to medications derived from the chemical backbone of amphetamine, which act as sympathomimetics, specifically mimicking the effects of the neurotransmitter norepinephrine. After attention was drawn to the risk of dependence and addiction from amphetamine, organic chemists began the search for drugs that retained the appetite suppressant properties but that did not have the abuse potential of amphetamine (105). The structural features needed to retain anorectic activity were defined. They included a chain between the amino group and the phenyl ring restricted to two carbon atoms; the binding of the amino group to a secondary carbon atom; and substitutions at other positions, which may lead to reduction in anorectic activity.
A large number of molecules were synthesized and tested for their effects on food intake and risk of habituation (106)and some of the marketed one are shown in Table 3.
|
Table 3. Drugs Evaluated by the Food and Drug Administration in 1973
|
|
Generic Name
|
Proprietary Names
|
Year Approved
|
DEA Schedule
|
Current Status
|
|
d,l-amphetamine
|
Benzedrine & Many others
|
1936
|
II
|
Not approved for obesity
|
|
d-amphetamine
|
Dexedrine & Many others
|
Before 1952
|
II
|
Not approved for obesity
|
|
Methamphetamine
|
Desoxyn & Many others
|
1947
|
II
|
Not approved for obesity
|
|
Phenmetrazine
|
Preludin
|
1959
|
II
|
Not Marketed
|
|
Benzphetamine
|
Didrex
|
1962
|
III
|
Available by prescription
|
|
Phendimetrazine
|
Plegine
|
1960
|
III
|
Available by prescription
|
|
Chlorphentermine
|
Pre-Sate
|
1962
|
III
|
Not Marketed
|
|
Phentermine
|
Ionamin, Wilpo
|
1959
|
IV
|
Most widely used drug for obesity
|
|
Mazindol
|
Sanorex; Mazanor
|
1980
|
III
|
Not Marketed
|
|
Diethylpropion
|
Tenuate; Tepanil
|
1959
|
IV
|
Available by prescription
|
Data from (100, 107)
At the request of the Bureau of Drugs at the FDA, a group of “Consultants on Anorectic Drugs” was empaneled. They issued their report in 1973, the same year that the NIH sponsored a major conference on obesity (107). In his letter to the Director of the Bureau of Drugs at the FDA, Dr. Thaddeus Prout, Chairman of this review group summarized the view of the consultants on current anorectic drugs (108). Their conclusions are summarized in abbreviated form as follows:
- “Adult obese subjects instructed in dietary management and treated with anorectic drugs on the average tend to lose more weight than those treated with placebo in relatively short-term trials.”
- “The amount of weight loss associated with the use of an “anorectic” drug varies from trial to trial.” [Note: In an aside this germane comment was made: “Dr. Scoville noted in answer to a question that there were no statistically significant overall differences comparing two anorectic drugs.” (p 498).]
- “The magnitude of increased weight loss of drug treated patients over placebo treated patients was only a fraction of a pound a week.”
- “…the total impact of drug-induced weight loss over that of diet alone must be considered clinically trivial. The limited usefulness of these agents must be measured against any possible risk factors inherent in their use.”.
- “The amphetamines, including methamphetamine have been widely abused in numerous populations. It is thus in the best interest of the public health to limit the use of amphetamines as far as is compatible with adequate therapy.”
- “Evidence presented for newer “anorectic” congeners of the amphetamine family and non-amphetamine drugs do not set them apart as having higher benefit or lower risks than older available drugs. The risk potential of fenfluramine may be an exception to this general statement.”
- “There is no evidence in the data reviewed which showed that combination of an ‘anorectic’ agent with other drugs increase the benefits or reduce the risks of the ‘anorectic’ agent.”
- “There is no clinical data which support the parenteral use of these drugs in the treatment of obesity.”
Based on these findings, The FDA Consultant Panel also made several recommendations summarized below:
- “That all “anorectics” reviewed (dl-amphetamine, d-amphetamine, methamphetamine, benzphetamine, phentermine, chlorphentermine, chlortermine, phenmetrazine, phendimetrazine, fenfluramine, mazindol, diethylpropion), with the exception of fenfluramine, be placed on Schedule II on the basis of abuse potential.”
- That combinations of “anorectics” with other drugs be evaluated in accordance with the policy of the FDA on combination drugs”.
- That amphetamines prepared for, or in a form suitable for, parenteral use not be approved for use in the treatment of obesity.
- “The single-entity oral “anorectic” preparations including the amphetamine be permitted to be labeled for restricted use in obesity provide that they are used in association with a specific weight reduction program and that the clinically trivial contribution of these drugs to the overall weight reduction is properly emphasized. To carry out the latter recommendation, a statement such as that made in the conclusions drawn from this review must be included in all labeling and promotional products. This statement should include the following points: Studies of the effect of “anorectic” drugs in the treatment of obesity when compared with the effects of patients treated in a similar manner without the use of the drugs demonstrated that the magnitude of weight loss of drug treated patients over no-drug treated patients was only a fraction of a pound a week. The rate of weight loss was greatest in the first weeks of study for both the drug and the non-drug treated subjects and tended to decrease in succeeding weeks. The natural history of obesity is measured in years whereas the studies offered for review are restricted to a few weeks duration. Thus, the total impact of ‘drug induced” weight loss over that of diet alone must be considered clinically trivial. The limited usefulness of these agents must be measured against any possible risk factors such as nervousness, insomnia, and drug habituation that might be inherent in their use. Moreover, these agents can only be recommended for short term use in the treatment of obesity in a carefully monitored and specified weight reduction program under the care of a physician” (108).
- That future approval of all ‘anorectic’ drugs prepared for future use be based on demonstration of efficacy as measured by statistical superiority of the drug over placebo in trials using FDA recommended protocols. These protocols should include provisions, among others, for the testing of a specific target populations, specification of a minimum duration trial to assure clinical relevance of the study, and give consideration to the handling of patient dropout.
- Further, that appropriate summary data derived from efficacy studies be presented in labeling and in all promotional material to indicate the degree of weight loss that was found.For this purpose guidelines note in (4) above should be supplemented by the addition of the specific facts found for the specific drug under consideration.
Shortly after this letter, the US FDA issued new Guidelines on Labeling for Single-Entity Amphetamine Products. The FDA also approved d,l-fenfluramine, chlortermine, and mazindol. These were the last drugs in this class to be approved. Chlortermine is no longer marketed and d,l-fenfluramine and d-fenfluramine have been removed from the market as described below. All the derivatives of amphetamine have been tarred with the same brush – that of risk for addiction. Since then, for better or for worse and whether deserved or not, as amphetamine fell from grace, a similar pall fell over the entire class of derived compounds. As we will see, one of these derivatives, fenfluramine, had no demonstrated abuse potential at all and yet was still regulated by the U.S. Government as though it did.
AMINOREX- ANORECTIC WITHDRAWN FOR TOXICITY
Aminorex demonstrates the law of unintended consequences as applied to drug development for obesity management. This drug is an “amphetamine-like” drug that was developed by McNeil pharmaceuticals in the United States and licensed in 1965 for sale in Germany, Austria, and Switzerland under the trade names of Menocil or Apiquel (109). Soon after its launch, Berne, Switzerland noted an increase in the diagnosis of pulmonary hypertension. This was eventually linked to the use of aminorex, which appears to have been responsible for a 5 to 20-fold increase in incidence of primary pulmonary hypertension (PPH) in Switzerland. In Berne, up to 20% of those taking aminorex who were admitted to the hospital died, with up to 50% more dying over the next 10 years. A retrospective analysis in deaths in Austria, Switzerland, and Germany during the late 1960s showed that this drug was responsible for most of the cases of PPH between 1968 and 1972. Experimental studies subsequently confirmed that aminorex can produce pulmonary hypertension in animals (110-112).
This outbreak of PPH was the canary in the coal mine signaling a potential relationship between some β-phenethylamines and PPH, a form of plexogenic arteriopathy (113). Primary pulmonary hypertension (PPH) is a rare disease that occurs with a frequency of about 1–2 per million persons per year. Pulmonary artery systolic pressure is directly related to body mass index. The risk of PPH appears to have a genetic basis which was brought to the fore by 3 anorectic drugs, aminorex, fenfluramine, and chlorphentermine which modulate serotonin activity (113). A retrospective case-control study including 95 cases from several European centers and 355 matched controls estimated that the use of appetite suppressant medications may have increased the odds ratio by 10 to 23-fold for an incidence to 28–43 cases per million per year (114). Kramer and Lane reexamined the data from the aminorex cases to provide a comparison with fenfluramine (113). They estimated the odds ratio for developing primary pulmonary hypertension after exposure to aminorex was 97.8 (95% CI 78.9 –121.3) and that nearly 80% of the cases of this disease in the affected countries could be attributed to aminorex. Using the French and Belgian cases in the dexfenfluramine study (114), the authors estimated the odds ratio for developing primary pulmonary hypertension after exposure to dexfenfluramine to be 3.7 (95% CI 5 1.9 –7.2) for 3 months or more exposure and 7.0 (95% CI 5 2.8 –17.6) for exposures lasting more than 12 months (111). Dexfenfluramine, in contrast to aminorex, was estimated to increase the background rate by 20% or less. Such unanticipated “off-target” toxicity and the capacity for widespread involvement on a population basis given the high prevalence of obesity as a chronic disease has influenced the US FDA’s cautionary approach to initial approval and recommendation for swift removal in after-market surveillance for all drugs in this category.
PHENTERAMINE- A SURVIVOR
Phentermine is one of the derivatives of α-methyl-β-phenethylamine backbone of amphetamine that was approved for marketing by the U.S. FDA in 1959. Unlike other members of this group, which have had problems with toxicity or lack of use, phentermine accounts for over 75% of the anti-obesity medications used in 2019 (115). Phentermine, like many other sympathomimetic amines, acts on the trace amine-associated receptor 1 (TAAR1) where it facilitates the efflux of norepinephrine, and to a lesser extent the efflux of dopamine and serotonin (116, 117)
At the time of phentermine’s approval in 1959, most clinical trials for weight loss drugs lasted 12 weeks or less. However, several studies have evaluated the longer-term use of phentermine (118-120). In one older study (118), a group of patients received a placebo, the second group received phentermine resin 30 mg/day, while the third group was treated with phentermine resin alternating with placebo at 1-month intervals. The two groups given phentermine in either intermittent or continuous form lost similar amount of weight which averaged 20.5% of their initial body weight compared to only 6% in the placebo group. The group treated intermittently showed the effect of stopping and starting an active drug to placebo relative to continuous treatment. These authors concluded that intermittent phentermine was preferable because it was cheaper, gave equivalent weight loss, and reduced exposure to medication; although this runs counter to current concepts of chronic disease management in which continuous use is recommended. In another older study (120), patients with osteoarthritis were treated with phentermine-resin, 30mg/day for 6 months or with placebo. The group treated with phentermine lost 12.6% of their body weight, compared to 9.2% in the placebo group. In a third older study (119), 59 subjects were treated for 14 weeks with either a placebo or phentermine 30 mg/day. The group receiving phentermine lost 8.7% of their body weight compared with only 2.0% in the placebo-treated group. Two more recent studies have evaluated the effects of phentermine in children (121) and in adults (122). In a retrospective study of children treated with lifestyle or lifestyle plus phentermine, the group receiving phentermine lost significantly more weight at 1, 3 and 6 months with no significant changes in blood pressure (121). A study in adults compared phentermine alone, canagliflozin alone, or phentermine plus canagliflozin against placebo in a double-blind RCT. Over 6 months, the placebo-treated group lost 1.1% vs 4.6% in the group receiving phentermine alone and 8.1% in the combined treatment group (122). Despite the significant weight loss, blood pressure was not different from placebo and heart rate was significantly increased in the group treated with phentermine supporting the concerns about the effect of phentermine on the cardiovascular system.
As with other sympathomimetic drugs, concerns have been raised about an increase in blood pressure and abuse in those taking phentermine. Subsequent studies have demonstrated that when used appropriately (e.g., those without contraindications such as uncontrolled hypertension or hyperthyroidism, active vascular disease, arrhythmias, or glaucoma), phentermine does not result in behaviors associated with substance abuse or withdrawal symptoms (123, 124), or raise blood pressure on average. In fact, observational studies and a small number of prospective randomized, controlled studies have demonstrated either neutral effects or reductions in blood pressure during long-term phentermine use and no increased risk for adverse cardiac events (125-128). A recent summary of reported after market adverse events of the FDA approved combination tablet Qsymia, containing both phentermine and topiramate, has failed to link use of this drug with cardiac valvulopathy, myopathy, or pulmonary hypertension (129). Phentermine is the most widely prescribed anti-obesity drug used for management of obesity today because it is a generic drug and thus low in cost and appears to have a track record of safety (115). While it is a “survivor” in terms of continued use as an anti-obesity medication, its reputation often suffers amongst providers and the public from “guilt by association” with fenfluramine use as described below.
Fenfluramine/Dexfenfluramine: 1973-1997 – Doomed by its Toxicity
Fenfluramine has structural similarities to amphetamine. However, to the surprise of most workers in the field it had a very different mechanism of action. Fenfluramine was developed in France in the 1960s and marketed there in 1964 (130). Instead of being a sympathomimetic drug and mimicking the effects of norepinephrine, as other anorectic drugs do, fenfluramine acted to block the re-update of serotonin at its nerve endings. In addition, one of the metabolites of fenfluramine, norfenfluramine, acted as a serotonin agonist (131, 132). Treatment of animals with most sympathomimetic drugs reduces brain norepinephrine; treatment with fenfluramine does not. Rather, fenfluramine reduces brain serotonin. The discovery that fenfluramine acted on the serotonergic system to reduce food intake led to a whole new area of research into drugs for the management of obesity.
Following clinical trials, fenfluramine was approved by the US FDA in 1973 for management of obesity (130). It was given a Schedule IV designation by the Drug Enforcement Agency because of its chemical similarity to other sympathomimetic anorectic drugs with abuse potential. However, fenfluramine behaves differently and many who took the drug complained of a dysphoria, and it was not used to replace amphetamine-drug abuse in substitution trials.
The anorectic effects of fenfluramine reside almost entirely in the dextro-isomer of this compound, named dexfenfluramine. When this was recognized, The International Dexfenfluramine (or INDEX) trial was designed to establish the safety and efficacy of dexfenfluramine (133). With this data in hand, dexfenfluramine was approved by the US FDA for management of obesity in the United Stated in 1994.
The first set-back for fenfluramine occurred in 1981 when the first 2 cases of pulmonary hypertension were reported (134), similar to the earlier reports of pulmonary hypertension in patients treated with aminorex. Between 1980 and 1993 there had been a total of 12 cases of pulmonary hypertension associated with fenfluramine (135). This data was emerging as a series of publications showed dramatic weight loss efficacy by combining fenfluramine with phentermine (commonly referred to a “Fen-Phen”), two anti-obesity medications with differing mechanisms of actions (136). The weight loss of 8.4 kg with the combination was similar to the individual agents, but there were fewer side effects when they were used together. This was followed by a single center NIH-funded trial of fenfluramine and phentermine lasting 4 years (137). By the end of the first 32 weeks, those receiving the combination of fenfluramine and phentermine, had lost 15.9% of their weight compared to 4.9% in the placebo group. The results supported and amplified the pilot data from this group. Patients with obesity treated with the combination of phentermine and fenfluramine lost more weight and, in many cases, were able to maintain the lower weight for more than two years during this study.
When the dramatic weight loss with Fen-Phen began to circulate widely in the 1990’s, fenfluramine and phentermine use exploded in popularity across the country. Patients and doctors alike were thrilled with the results and for the first time it appeared that Americans with obesity were winning the battle of expanding waistlines. Medical offices dispensing Fen-Phen opened up all over the country. In 1994 prescriptions for fenfluramine were mentioned less than 100,000 times among drug prescriptions. However, in 1995 when Fen-Phen hit the market, mentions of fenfluramine rose by 5-fold to over 500,000 prescriptions before peaking at nearly 4 million prescriptions (138).
The second set-back for fenfluramine and, by association, for phentermine occurred in July 1997 when the first case-series of valvular heart disease in patients taking Fen-Phen appeared in the prestigious New England Journal of Medicine (139). Despite the fact that the total participants in that study numbered only 24, the report included only women, pre-existing valvular heart disease could not be ruled out, and no control group was included to account for the known associations between obesity and both valvopathy and pulmonary hypertension, urgent meetings by the U.S. Food and Drug Administration with academic groups around the country assembled enough information to convince the FDA that up to 30% of the patients treated with fenfluramine (alone and in combination with phentermine) might develop valvular heart disease (140). On September 15, 1997, fenfluramine and dexfenfluramine were removed from the market worldwide. The Fen-Phen success had been shattered by the off-target consequences foretold by aminorex decades earlier and added another sad ending to a therapy that offered such promise in the management of the patient with obesity.
Sibutramine: 1988-2010
Sibutramine is a triple amine reuptake inhibitor affecting norepinephrine, serotonin, and dopamine. Its use as a weight loss medication was an outgrowth of a clinical program searching for effective drugs to treat depression conducted by Boots Pharmaceuticals in the United Kingdom (141). The effects of sibutramine on weight loss caught the attention of the company in 1988, which then commissioned a short clinical trial. During this eight-week study with two doses of sibutramine, Weintraub and his colleagues demonstrated a doubling of weight loss over that achieved with placebo with the low dose and a nearly four-fold increase with the high dose over the weight loss compared to the placebo group (142). With this promising background a full-scale clinical program was designed. There was a clear dose-response to the drug in a six-month clinical trial (143). In a two-year trial, sibutramine reduced body weight by nearly 12% at 6 months. In contrast, the weight loss in the placebo-group gradually faded and at two years they were only 3% lighter than at baseline. These strong clinical results were marred by an increase in blood pressure. Following the phase 3 clinical trials, approval for marketing was requested from the US FDA and the Committee for Proprietary Medicinal Products (CPMP) in Europe. Because of the increased blood pressure observed in some patients, the CPMP would only approve the drug if the company would conduct a long-term study to evaluate potential cardiovascular outcomes. This trial was begun, and the drug was approved for marketing by Knoll Pharmaceuticals, a Division of BASF in Germany who had purchased it from Boots Pharmaceutical and who subsequently sold it Abbott Laboratories in the US.
The Sibutramine Outcomes (SCOUT) trial was the result of this requirement (144). It began enrolling participants in January 2003 with a completion date of November 2005. By this time a total of 10,742 individuals with obesity and either a high risk of cardiovascular disease or diabetes had been randomized to treatment for up to six years with either sibutramine or placebo to determine whether use of sibutramine along with a weight management program would impact the risk for cardiovascular complications in this selected population. Despite reductions in blood pressure from baseline in both the sibutramine and placebo groups, to the dismay of the investigators sibutramine produced a significantly greater increase in CVD events (HR: 1.16, 95% CI 1.03-1.31, P = 0.02) as estimated from a composite end-point of nonfatal myocardial infarction (MI), nonfatal cerebrovascular accidents (CVA), cardiac arrest, and CV death (144). In absolute terms, cardiovascular events occurred in 11.4% of those assigned to treatment with sibutramine versus 10.0% for those treated with placebo.
Following this report, the U.S. FDA convened their Endocrine Advisory Panel to ask the question of whether this drug should continue to be marketed. The Panel recommended that sibutramine should be removed, although not everyone agreed with this conclusion. In a dissenting article, "Sibutramine: gone, but not forgotten", Dr. David Haslam (chairman of the National Obesity Forum) wrote that the SCOUT study was flawed as it only covered high-risk patients and did not consider patients with obesity who do not have cardiovascular complications or similar contraindications(145). The trial also had two other issues. First, patients with CVD who did not lose weight when taking the drug nonetheless continued treatment for up to 6 years, something that no responsible physician would do. Second, the intent-to-treat analysis of the results included patients who lost weight as well as those who did not lose weight. If your hypothesis is that the benefits in reducing the risk of cardiovascular disease and diabetes result from weight loss, then including patients who do not lose weight unduly biases the conclusions. It is, so to speak, like shooting yourself in the foot. A re-analysis of the data using only those who lost weight while taking sibutramine showed a lower rate of CVD events in those treated with sibutramine (146). Despite this re-analysis, the European Medicines Agency recommended suspension of marketing authorization for sibutramine, and on January 21, 2010, based on the results of the SCOUT trial, it was removed from the market in Europe. Abbott Laboratories in the US followed suit and announced on October 8, 2010 that it was terminating marketing of sibutramine in the US market under pressure from the FDA.
Lessons Learned:
- The first lesson from the SCOUT trial is that using intent-to-treat statistical methods, which retains individuals who do not lose weight, the drug produced harm.
- The second lesson is that when individuals who successfully lose weight while taking the drug are analyzed, instead of all comers, there is an overall statistical benefit – that is those who lose weight benefit from the weight loss
- The third lesson is that since individuals with obesity are generally at increased risk for detrimental cardiovascular outcomes, requiring documentation that new anti-obesity drugs do not have detrimental CVD effects became the rule.
- Despite its withdrawal from the market in 2010, sibutramine can still be found in products for sale on-line. The U.S. FDA found it in 69 of 72 products that were tested. These products appear to come from overseas, mainly China, and may pose health hazards (https://www.fda.gov/drugs/questions-answers/questions-and-answers-about-fdas-initiative-against-contaminated-weight-loss-products. Content Current as of 02/28/2018. Accessed March 9, 2021)
Tesofensine
Tesofensine, like sibutramine, is a triple monoamine reuptake inhibitor, which means it blocks the reuptake of serotonin, norepinephrine, and dopamine. The initial clinical data on the potential effectiveness of this drug for weight loss came from studies in neurological diseases including Parkinson’s disease and Alzheimer’s disease where treatment with tesofensine produced significant weight loss (147). This was followed by a randomized double-blind clinical trial in 203 patients who were randomly assigned to placebo or doses of 0.25, 0.5 or 1 mg/d of tesofensine (148). After 24 weeks the placebo-treated group lost 2.0% of their body weight compared with 6.5% in those given 0.25 mg/d of tesofensine, 11.2% in the 0.5 mg/d dose and 12.6% in the 1.0 mg/d dose. Although the clinical profile was relatively free of untoward side effects, there was, as with sibutramine, a small increase in heart rate and blood pressure. The cardiovascular risks identified in the SCOUT trial of sibutramine may also apply to tesofensine and provide a potential hurdle for approval of this drug.
Although no further data has appeared regarding tesofensine as a single anti-obesity agent since 2013, the combination of tesofensine (up to 0.5 mg/d) and metoprolol (50 mg/d) (Tesomet) was granted FDA approval in 2021 as an orphan drug in the treatment of both Prader-Willi syndrome and hypothalamic obesity based on results of a recent phase 2A trial (149). The combined use with a beta-blocker mitigated changes to pulse rate and BP and may be standard in development of other anti-obesity medications from this class of drugs.
Lorcaserin- Potential Cancer Risk
Lorcaserin is the most recent casualty among drugs used in the management of patients with obesity. It is a selective serotonin (5-HT2C) receptor agonist (thereby avoiding potential for adverse effects on cardiac valves and pulmonary artery pressures induced by other serotonin receptors—see fenfluramine above), which reduced food intake in experimental animals and humans. Following completion of Phase 3 studies (150), an application for approval of Lorcaserin for marketing was submitted to the US FDA. An FDA Advisory Panel met on September 16, 2010, but rather than approving the drug, they voted 9-5 against approval based on two concerns, one about efficacy of the drug and the other about its safety. They were particularly concerned about the findings of tumors in experimental animals treated with lorcaserin. Following this Advisory Committee, the FDA decided, on 23 October 2010, not to approve lorcaserin for marketing. Arena, the manufacturer of lorcaserin conducted a new round of studies and on May 10, 2012, another FDA Advisory Panel was convened. This time they voted to recommend lorcaserin for marketing. A trial combining lorcaserin, a serotonergic drug, with phentermine, enhanced weight loss somewhat but did not mimic the dramatic results with Fen-Phen (151).
Even though lorcaserin had been engineered to be selective for the 5-HT2C receptor rather than the 5-HT2B receptor, which is located on heart values and mediates the valvulopathy associated with fenfluramine and other serotonin medications, a post-marketing cardiovascular outcomes trial was commissioned. This trial, called CAMELLIA-TIMI, enrolled 12,000 patients who were at high risk for cardiovascular events. After a median of 3.3 years of treatment with Lorcaserin or placebo, the authors concluded that: “In a high-risk population of overweight or obese patients, lorcaserin facilitated sustained weight loss without a higher rate of major cardiovascular events than that in the placebo”, termed non-inferiority (152). In the original publication of this trial, the overall incidence of cancer was slightly greater but not statistically different between the lorcaserin (7.7%) and placebo (7.1%) groups over the 3.3 years of follow-up. However, during post marketing monitoring, the FDA found that the point estimates for cancer rate ratios were consistently greater than 1.0 in the lorcaserin group compared to placebo, and finally became significant 2.5 years after study completion (153). The main cancers were skin, prostate, GI, and respiratory. Based on this significant increase in cancer risk, the FDA requested Eisai, the manufacturer of Lorcaserin (Belviq), to withdraw the drug on Feb 13, 2020 and the company complied.
Although there were signals in the initial submission that might have been a warning for this risk, it was not until well after completion of the trial and several years into general use that a potentially latent severe adverse event became apparent.
Histamine and Histamine Antagonists
Histamine is a naturally occurring monoamine produced by decarboxylation of the amino acid, histidine. Histamine is located in neurons in the brain, in mast cells scattered around the body, and in the gastrointestinal track. There are four histamine receptors that mediate its effects, mainly located in the brain, the gut, and the immune system. The H-1 receptors in the brain are involved in wakefulness, appetite regulation, and endocrine function. In the periphery, H-1 receptors are in smooth muscles and endothelium where they are involved in bronchoconstriction, vasodilatation, and the sensation of itching. The H-2 receptors are in the stomach where they mediate gastric acid secretion, as well as in the brain, although their function in the brain is less clear. Injection of histamine into the brains of animals reduced food intake (154). Conversely, blocking histamine receptors with alpha-fluormethylhistidine, which irreversibly inhibits histidine decarboxylase, increases food intake. Similarly blocking histamine breakdown with metoprine, an inhibitor of histamine N-methyltransferase, also suppresses food intake in animals (155). Another piece of evidence suggesting a role for histamine receptors in food intake comes from data showing antipsychotic drugs that produce weight gain bind to histamine H-1 receptors with higher affinity than with any other monoamine receptor (156). Finally, the use of H-1 containing products to relive allergy is associated with weight gain (157).
The H-3 histamine receptor differs from the other two by being an autoreceptor which inhibits the action of other histamine receptors. There have been several studies examining histamine-related drugs as potential candidates for management of obesity. Betahistine is a weak, orally active agonist of the histamine H-1 but a potent antagonist of the histamine H-3 auto-receptor, which raises the ambient levels of histamine and has been approved to treat vertigo. In a randomized, placebo-controlled 12-week clinical trial, 281 people with obesity were given 8, 16 or 24 mg/d of betahistine or placebo twice daily. The overall weight loss was not significantly different between treatment groups, but the subgroup of white women under age 50 lost -4.24±3.87 kg compared to -1.65±2.96 kg (P<0.005) (158).
The histamine H-2 receptor antagonist, cimetidine was reported to reduce body weight in experimental animals (159)and in patients with type 2 diabetes (160), prompting one of the authors to conduct a trial in patients with obesity (161). In this small randomized clinical trial by a single author, a general practitioner from Norway, 60 participants (five females) received 200 mg of cimetidine or a matching placebo 3 times a day before meals along with a high fiber diet. The treatment group lost 9.5 kg, which was significantly more weight than the 2.2 kg lost by placebo-treated controls. The editors of the British Medical Journal who were reviewing this paper received another manuscript from Rasmussen and colleagues in Denmark attempting to replicate the Norwegian Study (162). These investigators also included 60 patients (nine female) in the same age range as in the Norwegian study. In the first eight weeks participants were randomized to either cimetidine or placebo. In this trial weight loss was almost identical with the cimetidine-treated group losing 5.7 kg compared to 5.9 kg in the placebo-group. When this second paper was received by the British Medical Journal, the editor was concerned by the contradictory results of the two group and possibility of scientific misconduct. Dr. Richard Smith, editor of BMJ, sent both papers to Dr. John Garrow, MD, PhD, one of the leading British scientists in obesity research (163). In his published response Garrow noted that a weight loss of 9.5 kg in 8 weeks is a remarkably good result in this period, so good in fact that it might not be a measured outcome, but rather a manufactured one. There was also no attrition in either group, which is again remarkable. The similarity of weight loss from week to week and the fact that both the heavier and lighter patients lost the same amount of weight was also surprising and unexpected. Garrow was “baffled” at why there should be such a large difference in weight loss between these two studies (7.4 kg in the Norwegian Study and 0.2 kg in the Danish Study) both having been conducted in nearby Nordic nations. The editor of BMJ, Dr. Richard Smith was also concerned as he noted in a paper about scientific misconduct that he published after he left his role as editor of the British Medical Journal (164). The case was referred to the University of Oslo, and to the faculty in Tromsö where Dr. Stoa-Birketvedt was a general practitioner. No evidence of misconduct was identified after reviewing the records and she was exonerated. As Dr. Florholmen, one of Stoa-Birketvedt’s collaborators on several studies and Dr. Leeds note in a comment on the Scientific Misconduct, “…scientific journals have an equally strong moral obligation to make a clear statement when a suspect investigator has been found not guilty” (165). This was not done in this case, but Stoa-Berketvedt and her colleagues went on to report a 42-month follow-up of her initial study. Twenty-two of the original patients continued in a cimetidine intervention group and received cimetidine for eight weeks, twice a year along with behavioral strategies and a diet. The 33 subjects in the non-intervention group received neither the cimetidine nor the behavior/diet treatment. After 42 months, body weight was stable in those in the cimetidine group compared to a gain of 7.5 kg in the other group (166).
Ranitidine, another histamine H-2 antagonist was tested for its potential to slow weight gain in patients receiving anti-psychotic drugs. In a meta-analysis, patients receiving ranitidine along with their anti-psychotic medication, gained less weight than their controls (167), suggesting a role for the H2 receptor in modulating energy balance under selected circumstances.
Fluoxetine for Weight Loss
Fluoxetine is a selective serotonin reuptake inhibitor (SSRI) approved by the U.S. FDA to treat depression and several other conditions including obsessive–compulsive disorder (OCD), bulimia nervosa, panic disorder, and premenstrual dysphoric disorder. Fluoxetine was discovered by Eli Lilly & Company in 1972 and it began widespread use in 1986 (168). The World Health Organization's List of Essential Medicines includes fluoxetine, and in 2016 there were more than 23 million prescriptions for this drug, making it the 29th most prescribed medication in the United States. Fluoxetine at a dose of 60 mg per day (3 times the usual dose for treatment of depression) produced dose-related weight loss in overweight patients, which initiated studies into whether this drug, at this higher dose, could be developed as an anti-obesity medication. Initial studies up to 6 months in duration were promising, but with longer duration of treatment two unexpected outcomes occurred. First, there were substantial differences in weight loss between studies. In a meta-analysis of 6 studies, one study reported a loss of –14.5 kg and another reported a weight gain of + 0.40 kg (169). Another meta-analysis showed no significant weight loss difference between fluoxetine and placebo (170). The mean difference in weight loss at 12 months of -0.33 kg (95%CI –1.49 to 0.82 kg) favoring fluoxetine.
The second problem for fluoxetine was that after 16 to 24 weeks of treatment, patients began to regain weight, even while continuing the drug. Darga et al. were the first to note the rebound in weight with continued treatment (171). After a weight loss of 11.7% during 29 weeks of treatment, weight began to rebound and was only 7.8% at 12 months which was not significantly different from placebo. In a review of 6 studies with 719 patients randomized to fluoxetine and 722 randomized to placebo, weight loss at 6 months in the placebo group was 2.2 kg versus 4.8 kg in the fluoxetine-treated group (172). At one year, however the weight losses were no longer statistically different at 1.8 kg in the placebo group and 2.4 kg in the fluoxetine-treated group. Regaining 50% of the lost weight during the second six months of treatment on fluoxetine terminated clinical trials by Eli Lilly of fluoxetine as a weight loss agent. Similar results were true for another SSRI, sertraline (172, 173).
This weight “rebound” after six months of treatment with these SSRI’s was part of the reason that the US Food and Drug Administration moved to require trials lasting up to a year for new weight loss medications (172, 174).
Bupropion as a Single Agent for Weight Loss
Bupropion (WellbutrinR; Others) was first synthesized in 1969 by Nariman Mehta and patented by Burroughs-Wellcome Company in 1974. It is a norepinephrine- and dopamine-reuptake inhibitor approved by the US Food and Drug Administration for the treatment of depression and as an aid in smoking cessation. It was the 28th most prescribed medication in 2016 and the fourth most prescribed antidepressant in the U.S. with more than 23 million prescriptions. Two multi-center clinical trials have examined the effect of bupropion, as a single agent, on weight loss in patients with obesity, in patients with depressive symptoms, and in patients with uncomplicated obesity. In one study, 421 overweight patients with depressive symptom were randomized with 213 receiving the 400 mg/d dose of bupropion and 209 receiving the placebo for 24 weeks. The 121 subjects in the bupropion group who completed the trial lost 6.0 ± 0.5 % of initial body weight compared to 2.8 ± 0.5 % in the 108 subjects in the placebo group (p<0.001) (175). Another study in patients with uncomplicated overweight randomized 327 subjects in equal proportions to bupropion 300 mg/d, bupropion 400 mg/d, or placebo (176). Of the 69% who remained in the study after 24 weeks, weight losses were 5 ± 1% in the placebo group, 7.2 ± 1% in the 300 mg/d dose, and 10.1 ± 1% in the group receiving 400 mg/d (p<0.001). At 24 weeks, the placebo group was further randomized to either the 300mg or 400mg group and the trial was extended to week 48. In this trial, non-depressed subjects responded better to bupropion than those with depressive symptoms.
Even though the combination of bupropion and naltrexone is currently approved for management of complicated overweight and obesity, bupropion alone is a reasonable choice for people with depression and problems with weight control.
Topiramate as a Single Agent for Weight Loss
Topiramate is a substituted fructose molecule that was discovered by Bruce E. Maryanoff and Joseph F. Gardocki working at the McNeil Pharmaceuticals Laboratories in 1979. It was approved by the US FDA in 1996 to treat epilepsy with generalized or focal seizures and subsequently to prevent migraine headaches. It has also been used to treat alcohol dependence. The drug acts on several systems. It is a carbonic anhydrase inhibitor that can produce a mild metabolic acidosis and altered taste for carbonated beverages. It acts as a voltage-gated modular of sodium and calcium channels. It also activates both the GABA-A receptors and AMPA/kainite receptors.
In the 1990s, Johnson & Johnson, the parent company of McNeil Pharmaceuticals, noted that topiramate use was associated with significant weight loss and initiated trials to evaluate its effectiveness as a stand-alone medication for overweight and obesity. The magnitude of the weight loss at the higher doses of 192 and 256 mg/d rivals that of the Fen/Phen combination discussed earlier (177-179). Reaching a weight-loss plateau at the two highest doses did not occur until nearly one year of treatment. After the initial studies, further clinical development was abandoned because of the negative effects of this drug on mental function, including memory loss and difficulties with concentration and attention. For patients with these side effects, the clinical efficacy for weight loss did not counterbalance the side effects and its development as a single agent was suspended.
Zonisamide as a Single Agent for Weight Loss
Zonisamide is chemically a sulfonamide that is approved by the US FDA for use in Parkinson’s Disease and as an antiepileptic drug. It has serotonergic and dopaminergic activity, as well as being an inhibitor of carbonic anhydrase (like topiramate) and is an inhibitor of sodium and calcium channels. Weight loss was noted in the clinical trials for the treatment of epilepsy. To evaluate its potential use in management of obesity, Gadde et al conducted in a 16-week randomized controlled trial in 60 obese subjects (180) who were prescribed a calorie-restricted diet and a placebo or doses of zonisamide 100 mg/d and increasing to 400 mg/d. The zonisamide group lost 6.6% of initial body weight at 16 weeks compared to 1% in the placebo group. In a second randomized placebo-controlled trial, 225 patients with obesity received either placebo or zonisamide 200 mg/d or 400 mg/d. Neurological, psychiatric, and gastrointestinal adverse events occurred in the zonisamide-treated groups, but weight loss in the placebo-treated group was 4.0 kg after 1 year, 4.4 kg in the group receiving zonisamide 200 mg/d and 7.3 kg in the group receiving zonisamide 400 mg/d (181). Similar to topiramate, for patients with these side effects, the clinical efficacy does not outrank them enough to use as an anti-obesity medication.
Naltrexone as a Single Agent for Weight Loss
Both exogenous and endogenous opioids can stimulate food intake (182), effects that can be blocked by antagonists to opioid receptors. Naloxone, a short-term antagonist to opioid receptors, reduces fat intake in experimental animals (183). This suggested that naloxone might be a potential treatment for obesity. To test this idea Atkinson et al, in1982, gave single doses of naloxone to both lean individuals and individuals with obesity and found a reduction of food intake only in those with obesity (184). The problem for naloxone as a treatment for obesity is its short duration of action.
Naltrexone in contrast, is a longer-lasting derivative that has been tested in several clinical trials (185-190). These trials were disappointing, and naltrexone has not been tried at the higher doses that might be required to produce a reduction in weight because of its potential for hepatic toxicity. In 1992 de Zwaan and Mitchell concluded in a review of naloxone and naltrexone (191) that “studies do not justify the routine use of naloxone and naltrexone in patients with Prader-Willi syndrome, obesity, bulimia nervosa, or anorexia nervosa because of their unprofitable risk/benefit ratios, although further work, particularly focused on some of the newer antagonists, should be undertaken.” Limited duration of action, limited efficacy, and toxicity at higher doses (192) were the downfall of opioid receptor antagonists as single anti-obesity agents.
Ecopipam and Dopamine D1/D5 Receptor Modulators for Weight Loss
Dopamine in the dorsal striatum of the brain is involved with the behavioral restraint of eating and the emotional components regulating eating behavior in human beings (193). This suggests that blockade of dopamine receptors might be a strategy for management patients with obesity. At least five dopamine receptors have been identified (194). The D1 and D5 receptors are very similar to each other, as are D2, D3, and D4 receptors. Agonists to the D1/D5 receptors reduce the duration of feeding primarily by decreasing the frequency of eating. On the other hand, D2 agonists reduce the rate of eating. A role for D1-like receptors in modulation of food intake has been established in animal models (195), suggesting that such a drug might have clinical use.
Ecopipam is a selective dopamine D1/D5 antagonist that was evaluated in four randomized, double-blind, multicenter trials that compared ecopipam (n=1667) to placebo (n=1118) in individuals with obesity, with or without type 2 diabetes (196). In dose-ranging Phase 2 studies, subjects received 10, 30, or 100 mg daily for 12 weeks. In a 52-week Phase 3 study, individuals received a single 50 mg or 100 mg daily dose of ecopipam combined with a weight loss program. Primary efficacy variables were the proportion of subjects losing more than 5% of their weight from baseline at 12 weeks (Phase 2) or the distribution of percentage weight loss from baseline at 52 weeks (Phase 3). After 12 weeks in the Phase 2 study, 26% of subjects administered 100 mg/d of ecopipam lost more than 5% of their weight vs. 6% of placebo-treated subjects (P<0.01). In the Phase 3 studies, ecopipam 100 mg produced a 3.1% to 4.3% greater weight loss than placebo at 52 weeks. More subjects administered ecopipam vs. placebo achieved a 5% to 10% or >10% weight loss in two phase 3 studies in the patients without diabetes.
These clinical trials were stopped prematurely because of unexpected psychiatric adverse events which occurred in 31% of those receiving ecopipam compared to 15% in those receiving placebo. These events included depression, anxiety, and suicidal ideation. Although ecopipam was effective for achieving and maintaining modest weight loss in subjects with obesity, the adverse effects on mood terminated its development as a drug for the management of weight loss (196). As is apparent in several non-selective centrally-acting medications, off target effects in the brain (in this case depression and suicidal ideation) can derail development or proceeding to approval.
Dopamine D2 Agonists as Single Weight Loss Agents
Prolactinomas are tumors of the pituitary gland that produce prolactin, and they are associated with weight gain. These tumors can be treated medically with dopamine D2 agonists such as cabergoline or bromocriptine. Bromocriptine, a specific D2 agonist, has been reported to decrease body weight in patients with obesity (197), but in a systematic analysis of this drug and cabergoline in the treatment of prolactinomas, there was no significant effect on body weight, although there was a significant reduction in hemoglobin A1c in patients with diabetes (198). A form of this drug (bromocriptine) is available for the treatment of diabetes (199), but it does not affect body weight.
Cannabinoid Receptor Antagonists
The rimonabant story is one of a “near-miss” with promising beginnings but a sad, and possibly inappropriate, ending. Tetrahydrocannabinol is the active ingredient of Marihuana-Cannabis and was shown to stimulate food intake in 1984. There are two endogenous cannabinoid receptors CB-1 (470 amino acids in length) and CB-2 (360 amino acids in length) that were identified six years later. The CB-1 receptor has almost all the amino acids that comprise the CB-2 receptor with additional amino acids at both ends. CB-1 receptors are distributed throughout the brain in the areas related to feeding. They are also found on fat cells and in the gastrointestinal track. The CB-2 receptors are primarily on immune cells. There are two well-characterized endogenous endocannabinoids called anandamide and 2-arachidonyl glycerol. When injected into the brain, these endocannabinoids increase food intake. The rewarding properties of cannabinoid agonists are mediated through the mesolimbic dopaminergic system (200-203).
Rimonabant was the first specific antagonist of the CB-1 receptor. It inhibits intake of sweet foods by marmosets and reduces intake of high-fat foods in rats. Whereas intake of standard rat chow is not affected by rimonabant. In addition to inhibiting the intake of highly palatable foods, pair-feeding experiments in diet-induced obese rats showed that the rimonabant-treated animals lost 21% of their body weight compared with 14% in the pair-fed controls (202-205).
With this background and the safety of the drug, a clinical program testing rimonabant as a potential weight loss drug was initiated and four phase 3 trials of rimonabant for the treatment of patients with obesity have been published. The first trial, called Rimonabant in Obesity (RIO)-Europe (206), was intended to be conducted only in Europe, but slow recruitment led to inclusion of 276 subjects from the United States. In this 2-year trial, a total of 1507 patients with a BMI > 30 kg/m2 without comorbidities or a BMI >27 kg/m2 with hypertension or dyslipidemia were stratified on whether they lost more or less than 2 kg during a run-in period. They were then randomized in a ratio of 1:2:2 to receive placebo, 5 mg/day or 20 mg/day of rimonabant. A diet with 600 kcal/day below energy requirements was prescribed. There was a 4-week run-in period followed by 52 weeks of drug treatment. Of those who started, 61% (920) completed the 1st year. From baseline at the end of the run-in period, those in the placebo group who completed the trial lost 2.3 kg, those on the low-dose of rimonabant lost 3.6 kg, and those receiving the high-dose group lost 8.6 kg. After this and other extensive trials, Rimonabant was approved in Europe, but was not approved by the FDA because of concerns about suicidality. As this concern grew, marketing of rimonabant was suspended in Europe on Oct 23, 2008 just over 2 years after its approval because of these psychiatric problems.
Against the problem of depression and suicidal ideation, were benefits on blood pressure, lipid profile, and the cardiovascular risk profile. At the time of its removal from the market, several trials were on-going. In the SERENADE Trial (207), rimonabant significantly improved control and body weight in patients with diabetes. In the ARPEGGIO trial, also in patients with diabetes, rimonabant proved to be beneficial for control of diabetes and for the loss of weight (208). Progression of arterial thickening over 30 months was not slowed by rimonabant, compared to placebo (209). This and several other studies suggest that a 5% loss of body weight over a 30-month period with rimonabant is not sufficient to modify the progression of atherosclerosis in the carotid artery in obese patients with metabolic syndrome. In another cardiovascular outcome trial 18 months of treatment failed to show an effect for rimonabant on disease progression with the primary end point of percent atheroma volume (PAV) but, tantalizingly, there was a favorable effect on the secondary end point of total atheroma volume (TAV) (210). Ultimately, the CRESCENDO trial to examine cardiovascular outcomes in a total 18,695 patients randomized to rimonabant or placebo was stopped prematurely due to concerns about increased suicide rates in individuals receiving rimonabant (211). This led to termination of all the rimonabant studies along with studies of other cannabinoid antagonists. Whether the benefits would have outweighed the risks is something we will never know.
In a recurring theme for centrally acting agents, specificity for both receptor type and brain location are important in avoiding off-target behavioral effects that can doom an anti-obesity medication.
The two cannabinoid receptors are also widely distributed and include the liver and fat cells, which raises the possibility that drugs acting only on the peripheral CB-1 receptors might be effective treatments for obesity. Experimental data in animals shows that this is indeed possible, and we will have to await further developments in this area (212).
Yohimbine
Yohimbine is an alpa-2 adrenergic antagonist that modulates lipolytic response to epinephrine. It is an indole alkaloid derived from the bark of the Pausinystalia johimbe tree in Africa as well as from an unrelated tree in South America (Aspidosperma quebracho-blanco). Yohimbine is a plant extract sold as a dietary supplement that has had a number of claims for beneficial responses, including as an aphrodisiac, weight control, and erectile dysfunction, but data supporting any of these claims is scant or absent (213).
Drugs That Increase Energy Expenditure
As unwanted weight gain leading to overweight and obesity results from a prolonged positive imbalance between energy intake and energy expenditure, it would be ideal for an anti-obesity medicine (or combination of medicines) to impact both. Energy expenditure can be partitioned into several components (214). The first is basal or resting energy expenditure, which is the energy needed to maintain body temperature, brain activity, cellular gradients for sodium and potassium, cardiac function, respiration, and other “basal” functions. This so-called “basal” or “resting” energy expenditure accounts for about 2/3 of total daily energy expenditure in most people and can be modified with exposure to cold or heat. The second component is the energy needed to move the body (activity), which is also the most variable component since it depends on the amount and intensity of the activity performed. Some very active people can increase total energy expenditure by 50% or more above basal levels, whereas in others with low levels of physical activity, this will account for less than 25% of daily energy use. The final component of energy expenditure is that involved with the ingestion, digestion, and absorption of food. This is the so-called “thermic effect of food,” or in earlier times, “specific dynamic action.”
The regulatory model of body weight would point to development of anti-obesity drugs that increase energy expenditure through promotion of negative energy balance. Thyroid hormone is the prototypical thermogenic agent and has already been discussed in detail. Other drugs in several categories are considered below based on their mechanisms of action (215). The first are agents that are sympathomimetic amines with ephedrine at the top of the list. Next are the drugs acting on the adrenergic receptors that affect thermogenesis. The third group are the catechins, which occur naturally in tea. Finally, we discuss capsaicin that produces the feelings of “heat” on the tongue when eating hot peppers.
EPHEDRA AND EPHEDRINE
The story of ephedra and ephedrine, one of its active components, is mixed with good science, commercial exploitation, and governmental inadequacies. There were several key players in this story. The first is Arne Astrup, MD a professor of Medicine in Copenhagen who was a key figure in demonstrating the efficacy of using the combination of ephedrine and caffeine in the management of obesity. The second are two US Senators, Senator Orin Hatch representing the State of Utah and Senator Tom Harkin representing the State of Iowa. Finally, there is Michael Ellis, CEO of Metabolife, the largest purveyor of ephedra compounds and who is also representative of the entrepreneurs who marked their products directly to the consumer.
Ephedrine with and without caffeine
Ephedrine has been used to treat asthma for more than 75 years. As a sympathomimetic amine, it also stimulates thermogenesis in animals and human beings (216). In 1971 Dr. Eriksen, a Danish general practitioner in the town of Elsinore, Denmark noted that his asthmatic patients receiving ephedrine, caffeine, and phenobarbital often lost weight. Word of his “discovery” spread and by 1977 over 70,000 people were taking this compound called, appropriately, the ‘Elsinore Pill’ (217). One side effect of the Elsinore Pill was skin rashes, which were thought to be related to the phenobarbital. To understand the Elsinore pill better, Malchow-Moller et al. conducted a trial comparing ephedrine and caffeine without phenobarbital against diethylpropion, a known anorectic drug (217). Over the 12 weeks of treatment both active medications produced a similar weight loss of just over 8 kg, which was much more than placebo.
Following-up on this lead, Astrup and his colleagues explored ephedrine alone and in combination with caffeine as a potential treatment for obesity (218). The duration of the thermogenic effect of ephedrine is amplified by theophylline or caffeine, which slows the degradation of cyclic-AMP. Caffeine and other xanthines appear to prevent the body from becoming resistant to the effects of ephedrine by inhibiting phosphodiesterase and the adenosine receptor (219). The response to ephedrine was dose-related and persisted during chronic treatment. The mechanism by which ephedrine affects body weight is, in part, due to its effect upon thermogenesis, but it also clearly reduces food intake.
Several clinical trials with ephedrine and ephedrine plus caffeine have been reported (220). In a 6-month trial, Astrup et al. compared placebo, caffeine 600 mg/day, ephedrine 60 mg/day, and the combination of caffeine 600 mg/day with ephedrine 60 mg/day in 180 subjects with obesity (221). Withdrawals were equal in all groups with 141 subjects completing the trial. Weight loss was significantly greater in the group receiving the combination of caffeine and ephedrine (16.6 kg vs. 13.2 kg, P = 0.0015). Weight loss in the caffeine-only group and the ephedrine-only group was not different from that in the placebo group. Side effects of tremor, insomnia, and dizziness were transient and by eight weeks were no different than the placebo group. Blood pressure fell equally in all four groups. After six months the medication was stopped for two weeks to assess withdrawal symptoms. One hundred twenty-seven subjects then entered a 6-month open-label study of caffeine 600 mg/day with ephedrine 60 mg/day (221). The 99 subjects who completed the study lost an additional 1.1 kg, and no clinically significant withdrawal symptoms were observed.
The combination of caffeine and ephedrine, like the combination of phentermine and fenfluramine produced 16% weight loss during the initial six months that was maintained over the ensuing six months. In a 15-wk trial, patients with obesity lost significantly more weight when receiving ephedrine 20 mg and caffeine 200 mg daily compared to patients receiving dexfenfluramine 30 mg a day (222). Based on these and other data, the Danish Health Authority approved the use of ephedrine and caffeine as a combination drug for the management of obesity. After problems with ephedra, the plant-based source of ephedrine surfaced in this United States in the 1990s, marketing authority was withdrawn in Denmark.
Ephedra: An Herbal Medication
Ephedra has its historical roots going back several thousand years in Chinese medicine. Ephedrine is one of the principal alkaloids in the ephedra plant and was originally isolated by Nagai in 1885 and then rediscovered by Chen and Schmidt in the 1920s (223, 224). During the middle of the 20th century, ephedrine was a major drug used to treat asthma, only falling out of favor when newer and better drugs, such as amphetamine, were developed.
Ephedra sinica, also known by its Chinese name Ma huang, has been used medicinally to treat asthma, hay fever, and common colds. It is a source of six principal components, including ephedrine and pseudoephedrine, which are sympathomimetic amines that provide the stimulant and decongestant properties of this plant. Dietary supplements containing ephedra alkaloids were widely marketed over the counter for treatment of obesity in the 1990s. This direct marketing to the public without having to go through the Food and Drug Administration to prove their safety and efficacy was made possible when the US Congress passed the Dietary Supplements and Health Education Act in 1994 (DSHEA) (225). This law effectively put dietary supplements out of the reach of regulation by the US Food and Drug Administration. The work described above on ephedrine by Astrup and his colleagues in the 1980s and 1990s provided the rationale for the mass marketing of ephedra to the public in the United States since there were no regulatory barriers in the way. Under the DSHEA act, products could be marketed if a health claim was not raised in the marketing materials. However, weight loss was an obvious market for these products. Metabolife became one of the leading distributors of ephedra products and amassed sales that surpassed a billion dollars a year. Although they were not regulated by the FDA, they needed to protect themselves from US Federal Trade Commission, which could go after them for false advertising. Based on the work of Astrup et al (221), Metabolife commissioned a study of their product compared to placebo, hopeful that the outcome would support their claims. Indeed, a scientific paper by Boozer C, et. al. did just that (226).
However, complaints that ephedra alkaloids, such as those marketed by Metabolife, were unsafe began to reach the US Food and Drug Administration, including serious side effects and deaths. Unknown to the FDA at the time, Metabolife had received over 14,000 complaints of adverse events associated with its product that were not initially made public. In response to accumulating evidence of adverse effects and deaths related to ephedra, in 1997 the US Food and Drug Administration (FDA) reviewed the data and then finally banned the sale of supplements containing ephedra in 2004. This ban was challenged in court by the manufacturers of ephedra products, but it was ultimately upheld by the US Court of Appeals of the Tenth Circuit in 2006. Documentation of adverse events that could be attributed to unregulated ephedra supplements published in a report in JAMA (227). These effects include severe skin reactions, irritability, nervousness, dizziness, trembling, insomnia, headache, dehydration, profuse perspiration, itchy scalp and skin, vomiting, and hyperthermia. Cardiovascular effects including irregular heartbeat, heart attack, stroke and death were the most serious.
Michael Ellis, co-founder of Metabolife, was sentenced to six months in federal prison in 2008 for his failure to report adverse effects from his company's products to the US FDA. In the midst of this, the Dietary Supplement Industry created a public relations group called the Ephedra Education Council, to oppose the changes to the marketing of products containing ephedra, and commissioned a scientific review by a private consulting firm, which, as might be expected, reported that ephedra was safe. The Ephedra Education Council also attempted to block publication of a study confirming wide discrepancies between the labeled potency of supplements and the actual amount of ephedra in the product. Both Senators Orrin Hatch (R. Utah) and Tom Harkin (D. Iowa), authors of the Dietary Supplements Health and Education Act, questioned the scientific basis for the US FDA's proposed labeling changes and suggested that the number of problems reported were insufficient to warrant regulatory action. At the time, Senator Hatch's son was working for a firm hired to lobby Congress and the US FDA on behalf of ephedra manufacturers—a potential conflict of interest.
BETA-AGONISTS
Broadly, adipose tissue is divided into “white” fat, “beige” fat and “brown” fat. Beige and brown adipocytes are progressively enriched with iron-containing mitochondria, resulting in progressively darker brownish hues. Animal studies reported that heat production by brown fat in mammals could be increased by diet. The thermogenic response to a meal was mediated, in part, by release of norepinephrine from the rich supply of sympathetic nerves that supply brown adipose tissue and could thus be blocked by severing the sympathetic nerves to this tissue. The adrenergic receptor mediating this response was different from the previously defined beta-1 and beta-2 adrenergic receptors and was subsequently named the beta-3 receptor (228). Activation of the beta-3 receptor increased the activity of a mitochondrial uncoupling protein, allowing for free passage of proteins across the inner membrane, thereby short-circuiting the production of high energy phosphate bonds in ATP during metabolism and “wasting” the energy instead as heat. Stock and Rothwell (229) showed that brown adipose tissue plays a key role in dissipating additional calories rather than storing them as fat. In the animal that has overeaten a highly palatable diet, there is a thermogenic response to norepinephrine that is blocked by propranolol, a β-adrenergic blocking drug, confirming the importance of β-adrenergic receptors in this response.
Cloning of the beta-3 adrenergic receptor by Strosberg (230) opened up the way to develop drugs that could increase energy expenditure (231, 232). The first generation of beta-3 agonists were developed against the rodent receptor and highlight the problems associated with cross-species differences. A few of these compounds were tested in human beings, and the data are summarized in Table 4. Four drugs demonstrated an increase in energy expenditure and an increase in heart rate (BRL 26830A; BRL35135; Ro16–8714; Ro40–2148). The only first generation atypical β-adrenergic agonist to have a long-term clinical trial is BRL 26830A (R*,R*)-(6)-methyl-4-[2-[(2-hydroxy-2-phenylethyl)amino]propyl]-benzoate, (E)-2-butenedioate (2:1) (220, 233). This compound stimulates oxygen consumption even in the leptin deficient genetically obese (ob/ob) mouse and causes weight loss and a decrease in body fat, while at the same time conserving or increasing lean body mass (231). In human studies, it increases oxygen consumption for up to six weeks (234).
In an 18-wk double-blind randomized trial in 40 patients with obesity prescribed
an 800-kcal, low-fat, high-fiber diet, Connacher et al (235) reported that the treated group lost a mean ± SD of 15.4 ± 6.6 kg over 18 weeks compared with 10.0 ± 5.9 kg in the placebo group (P=0.02). The metabolic rate decreased less in the experimental group (5.35 to 5.11 kJ/min) than in the placebo-treated group (5.22 to 4.98 kJ/min) (P = 0.08). An increase in tremor, probably due to activation of β-2-adrenergic receptors, was the principal drawback (236). There were no effects on hunger or satiety (237).
A second drug from Beecham Research Laboratories (BRL-35135) was structurally similar to the previous drug (BRL 26830A) and also produced an increase in RMR and an improvement in glucose tolerance. The mild tremor noted with the previous drug was also seen with this compound (238). Another compound from the Imperial Chemical Industries (ICI-D7114) developed against the rodent receptor was nearly devoid of thermogenic activity (239). A pair of compounds developed by Hoffmann LaRoche (Nutley, NJ) (Ro 16–8714 and Ro 40–2148) were thermogenic in lean and obese subjects (240) but caused significant increases in heart rate. The final compound from the rodent receptor assays (CL-316, -243, or BTA-243) was not thermogenic but in a short-term clinical trial improved glucose disposal (241).
The question then became would the thermogenic responses be sufficient to produce weight loss without untoward side effects. Several compounds have reached clinical evaluation, but the therapeutic response with the most specific drugs developed against the human beta-3 receptor have been disappointing too (242, 243). TAK-677 is a novel beta-3 agonist that was tested over four weeks in 65 healthy younger men and women with obesity and a BMI of 33.9 ± 2.1 (mean ± SD). Subjects were randomized to 3 groups and after 28 days of treatment with placebo, 0.2 mg or 1 mg in two divided doses daily, the 24-hour energy expenditure in a whole room calorimeter fell by 39 kcal/d in the placebo group and rose by 13 kcal/d with the higher dose of TAK-677. Heart rate increased significantly by 8 beats/min in the higher dose group. This drug was not developed further because of the concerns for potential CV toxicity (242). The recent identification of brown adipose tissue by positron emission tomography in human beings may reawaken interest in the idea of enhancing thermogenesis.
Studies on the beta-3 adrenergic receptor demonstrate several things:
First, drugs developed based on biology of other species may not work in humans.
Second, predicting the in vivo effects of agonist drugs from in vitro data can be hazardous.
And third, the action on other (off target) receptors, the heart in this case, may vitiate the targeted biology, in this case energy expenditure (244).
Despite these disappointing results, thermogenic drugs for the treatment of obesity are still under investigation, including peroxisome proliferator-activated receptor (PPAR)-α and -γ agonists and retinoids. Although much of this research is still in the early stages, the β-3 adrenergic receptor agonist (ARA) mirabegron, approved for the treatment of overactive bladder, has been studied in humans. Preclinical results suggest that β -3 ARAs could also improve obesity-associated metabolic diseases by increasing thermogenesis in BAT, lipolysis in white adipose tissue, or insulin sensitivity. A small study of 14 women receiving 100 mg/d of mirabegron XR for 4 weeks demonstrated increased activity in BAT as well as improved insulin sensitivity (245).
|
Table 4. Clinical Studies with B-3 Adrenergic Agonists
|
|
Author (Ref No)
|
Year
|
Subjects
|
Length
|
Diet
|
Energy
Expend
|
Heart Rate
|
Weight Loss
|
Comments
|
|
|
|
|
|
|
|
Placebo
|
Drug
|
Placebo
|
Drug
|
|
|
Beecham Research Laboratories (BRL) 26836A
|
|
|
Zed (246)
|
1985
|
Obese (14F/2M)
|
6 wk
|
Restricted
|
|
--
|
--
|
-6.56 kg
-7.3%
|
-9.34 kg
-10.1%
|
Abstract; RCT; Initial weight 92.7 kg drug grp; 89.8 kg placebo grp.
|
|
Abraham (247)
|
1987
|
Obese
|
6 wk
|
Restricted
|
|
--
|
--
|
-232 g/d
|
-291 g/d
|
Abstract; Protein loss 17.1 g/d placebo; 8.9 g/d for BRL
|
|
Smith (248)
|
1987
|
Lean (12M)
|
1 d
|
Ad-lib
|
|
|
|
+0.3 kg
|
-0.2 kg
|
RCT; fasting insulin decreased
|
|
Chapman (249)
|
1988
|
Obese (43F#)
|
6 wk
|
1000 kcal/d
|
|
No Change
|
No Change
|
|
|
RCT; 50 mg/d; Tremor noted
|
|
Connacher (235)
|
1988
|
Obese
(32F/8M)
|
18 wk
|
800 kcal/d
|
REE up 11.6%
|
No Change
|
No Change
|
10.0 kg
|
15.4 kg
|
RCT; 200 mg/d 2 week then 400 mg/d for 16 wks.
|
|
Connacher (236)
|
1990
|
18 (Sex Not given)
|
Single dose
|
Ad-lib
|
|
|
|
|
|
Tremor increased
|
|
Connacher (233)
|
1992
|
Obese (12F)
|
3 wk
|
Ad-lib
|
No Δ VO2or RQ
|
No Change
|
No Change
|
|
|
100 mg tid improved insulin sensitivity and down-regulated adrenergic receptors.
|
|
Beecham Research Laboratories (BRL) 35135
|
|
|
Mitchell (238)
|
1989
|
Obese
(6F/4M)
|
10d
|
Wt Maintaining
|
|
|
|
No Change
|
No Change
|
AUC for glucose and insulin reduced Glucose Thermogenesis unchanged
|
|
Smith (250)
|
1990
|
T2D
|
|
|
|
|
|
|
|
Mild Tremor
|
|
Cawthorne (234)
|
1992
|
Lean M
Obese (6F/4M)
|
Single
10d
|
Wt Maintaining
|
Up 16% vs 4% placebo
|
|
|
|
|
Thermogenic response to glucose increased from 10.1% to 16.0% by BRL
|
|
Imperial Chemical Industries (ICI) D7114
|
|
|
Toubro (251)
|
1993
|
Lean
|
14d
|
|
No Change
|
|
|
|
|
Abstract- No effect on glucose disposal
|
|
Goldberg (239)
|
1995
|
Lean (16M)
|
14d
|
|
No Change in SEE
|
|
|
|
No Change
|
RCT Parallel arm
|
|
Hoffmann-LaRoche Ro 16-8714 (Ro 40-2148)
|
|
|
Henny (240)
|
1987
|
Lean M
|
Acute
|
|
REE Up with dose
|
|
Up by 2% 8% & 49%
|
|
|
3 doses; REE, HR and SBP increased with dose
|
|
Henny (252)
|
1988
|
Lean 6M
Obese 6
|
Acute
|
|
REE up in both groups
|
|
HR, SBP & FFA Up
|
|
|
Single dose comparison L vs Ob. Both up’d REE, HR, SBP & FFA
|
|
Jequier (228)
|
1992
|
|
|
|
|
|
|
|
|
Compared acute Epi, Dopamine and Ro16-8714.
|
|
Haesler (253)
|
1994
|
Obese 12F
|
14d
|
|
REE Not increased
|
|
|
|
|
Ro-2148. Parallel Group; 400 mg bid. Glucose thermogenesis up.
|
|
Lederle Laboratories CL 316,243 (BIA-243)
|
|
|
Wheeldon (254)
|
1994
|
Lean (8M)
|
Acute
|
|
|
|
|
|
|
Salbutamol & CL35135 with nadolol and Bispropol; Sal & CL has chronotropic and inotropic effects
|
|
Weyer (241)
|
1998
|
Lean (14M)
|
56d
|
Ad-lib
|
24h EE not changed
|
|
|
No Change
|
No Change
|
Parallel arm; 4 placebo, 10 CL; Insulin action increased
|
|
Lederle Laboratories L-796568
|
|
|
Van Baak (243)
|
2002
|
OW-OB (12M)
|
Acute
|
Ad-Lib
|
Increased 8% high dose
|
No Change
|
No Change
|
|
|
2 Center cross-over study; placebo, 250 & 1000 mg doses; SBP, glycerol & FFA increased; No change Temp, leptin, NE, DBP
|
|
Larsen (255)
|
2002
|
OW-OB (20M)
|
28d
|
Ad-lib
|
24h EE not changed
|
|
|
No Change
|
No Change
|
Parallel arm, 2 center RCT; RQ unchanged; GTT not changed; TG decreased
|
|
Takeda (TAK) 647
|
|
|
Redman (242)
|
2007
|
Obese
|
28d
|
Isocaloric
|
|
|
|
|
|
RCT; placebo & 2 doses
|
F=Female; M=Male; #= Post-menopausal; REE = Resting energy expenditure; RQ=respiratory quotient; tid=three times a day; bid=twice daily; RCT=randomized control trial; wk=week; d=day; Δ=change
CAPSINOIDS
Capsinoids are non-pungent compounds with molecular structures similar to capsaicin, which comes from red papers and gives them that “hot” taste. For this reason, capsinoids were thought to have thermogenic properties that could be harnessed in the management of obesity. This concept was tested in a clinical trial including 13 healthy subjects who received four doses of the capsinoids (1, 3, 6 and 12 mg) and placebo using a crossover, randomized, double-blind design. To test the thermogenic effect, resting metabolic rate was measured by indirect calorimetry for 45 min before and 120 min after ingesting each of the capsinoids or placebo. There were no significant effects of any dose on energy expenditure compared to placebo, and there was no effect on blood pressure or axillary temperature (256). Further development of these compounds was halted due to clinical ineffectiveness.
The possibility that peptides might provide the basis for developing drugs to control food intake began well before 1994, but I have selected 1994 as the beginning of this period because it was the year that leptin was discovered—a discovery that made obesity a scientifically respectable field (6). Leptin became foundational for our current understanding of the physiological regulation of body weight and helped to unravel the genetic and molecular basis for the neurocircuitry implicated in the pathophysiologic changes that lead to unwanted weight gain and obesity. Following the discovery of leptin and advancement of the weight regulation model, obesity wasn’t “your fault” anymore. In this section we will discuss several peptide drugs, including cholecystokinin, human growth hormone, inhibitors of insulin release, human chorionic gonadotrophin, leptin, and ciliary neurotrophic factor.
CHOLECYSTOKININ
Cholecystokinin (CCK), also called pancreozymin, is a gastrointestinal peptide produced in and secreted from enteroendocrine “I” cells in the duodenum. CCK, along with gastrin, which is secreted from the “G” cells in the stomach, share five C-terminal amino acids. The cholecystokinin group of peptides are produced as a 150-amino acid precursor called pre-pro-cholecystokinin with several forms identified by the number of amino acids they contain, e.g., CCK58, CCK33, CCK22 and CCK8 with 58, 33, 22 or 8 amino acids respectively. Most CCK peptides have a sulfate group attached to a tyrosine located seven residues from the C-terminal end of the molecule. This modification is crucial for the ability of CCK to activate the cholecystokinin-1 receptor as the non-sulfated CCK peptides are inactive. When injected parenterally, CCK-8 produces a dose-related reduction in food intake in lean experimental animals and human beings with obesity (257-259). There are two CCK receptors: CCK1 (CCKA) and CCK2 (CCKB). The former is located primarily in the gastrointestinal track and the latter in the brain. Peptide analogs of CCK provide one avenue for developing drugs that could reduce food intake (260, 261). Another approach to manipulating CCK is with benzodiazepines that act as CCK agonists (262). A third approach is to use antagonists to proteolytic degradation of CCK and CCK-releasing factors in the GI track.
CCK releases digestive enzymes from the pancreas and stimulates the release of bile from the gall bladder. Fatty acids and certain amino acids are the major stimulators of CCK release. CCK suppresses hunger by decreasing the rate of gastric emptying. It also stimulates the vagus nerve, an effect which is blocked by capsaicin. Vagotomy prevents the reduction in food intake produced by the peripheral injection of CCK, suggesting that afferent messages generated in the gastroduodenal/hepatic circuit are relayed to the brain by the vagus nerve (220). These vagal messages initiated by CCK activate several neuronal complexes in the brain including the nucleus of the tractus solitarius (NTS), the lateral parabrachial nucleus, and the central nucleus of the amygdala, as assessed by expression of the early gene product c-fos (263). The production of early satiety by CCK does not require an intact medial hypothalamus because it occurs in human beings with hypothalamic injury and obesity (264). In human studies CCK reduces food intake by 6–63% (average 27%) in lean subjects and 13–33% (average 21%) in individuals with obesity, with a small number of studies reporting GI side effects (220). However, animal studies suggest that suppressed food intake returns to baseline quickly (a true form of tolerance) and any initial weight loss is not sustained. Currently, no drugs appear to be in development using this pathway, suggesting limited effectiveness or potential side effects.
HUMAN GROWTH HORMONE
Individuals who are obese secrete less human growth hormone (hGH) (265). This is important in relation to unwanted weight gain because this hormone enhances lipolysis (266), increases metabolic rate (267), and leads to changes in fat patterning in children with hypopituitarism treated with growth hormone (268). Early studies with hGH showed that it would lead to reduced protein loss in people with obesity (269). In adults with growth hormone deficiency, treatment with hGH reduces body fat (270), whereas effective treatment of patients with acromegaly who secrete excessive amounts of growth hormone reduces circulating growth hormone levels and is associated with an increase in body fat (271).
Most trials of human growth hormone in patients with obesity have been short term, lasting from 21 days to five weeks (272), but a few have lasted up to 39 weeks (273). Most of the subjects were women. Metabolic rate was increased in those patients when measured, and respiratory exchange rate was reduced, indicating that more fat was being burned (oxidized). Insulin-like growth factor I, the liver-derived hormone released by hGH, increased FFA levels, and the rate of fat loss relative to protein loss (272). However, a number of side effects were also noted (273) that reduce the enthusiasm for long-term treatment with hGH. In a long-term, randomized placebo-controlled trial lasting nine months, 15 men were treated with hGH daily and 15 with placebo injection (274). In these men, GH enhanced fat loss with a larger percentage of this loss coming from visceral fat than from the subcutaneous fat. In a review in 2008, Rixhon et al noted that treatment with growth hormone affects fat distribution, may or may not improve metabolic profile, but does not lead to much actual weight loss (275).
Human growth hormone is thus important for growth and maintaining a healthy body composition (increased lean mass, less visceral fat), but its value is obesity is limited by the fact that it does not produce weight loss.
MANIPULATION OF INSULIN SECRETION BY DIAZOXIDE OR OCTREOTIDE
Treatment with peripheral insulin in patients with type 1 diabetes with the goal of near normalization of glucose control is well known to produce weight gain (276). One hypothesis for the development of obesity is that dietary carbohydrate (glucose) stimulates insulin release, which in turn promotes obesity (277), although this hypothesis is thought by many to be overly simplistic (278). Even though insulin has been clearly established to be an important mediator of normal weight regulation through its central actions and that insulin resistance or deficiency leads to hyperphagia and weight gain (279, 280), a few studies with experimental animals and in small numbers of patients with obesity following hypothalamic injury have implicated hyperinsulinemia as a potential driver of weight gain, and that lowering insulin can reduce food intake and body fat (281, 282). There are at least two ways of reducing insulin secretion pharmacologically: the administration of diazoxide or the use of octreotide. Diazoxide lowers insulin secretion from the pancreatic beta cell and increases glucose release from the liver. Octreotide is an octapeptide that mimics the naturally occurring somatostatin, also called growth hormone inhibiting hormone. Its effects are mediated by SSTR2 and SSTR5 receptors, which inhibit the release of several hormones, including growth hormone, insulin and glucagon, and thus is non-specific in its effect.
The hypothesis that inhibiting insulin release could treat obesity was tested by Lustig et al. who treated patients with hypothalamic obesity using octreotide. In a randomized clinical trial of 18 patients, Lustig and colleagues demonstrated that octreotide could indeed suppress insulin levels, and at the same time resulted in stabilized body weight and BMI in this difficult to manage population with obesity (283). In a follow-up study, Lustig et al treated 172 (28M/144F) patients with evidence of insulin hypersecretion for six months with one of three doses of octreotide or placebo (284). The higher doses produced small but statistically significant weight loss, which appeared to be greater in those with higher insulin levels. The data were mixed, but those with the greatest decrease in insulin did show the largest reductions in weight (283, 284). A third study tested octreotide in 20 women with polycystic ovary syndrome who received a low-calorie diet for one month followed by 6 months of diet and either octreotide or placebo. There was no difference in weight loss, but there were improvements in hyperandrogenism and the insulin-IGF-I system (285). Because of its non-specific inhibition of hormonal secretion, poor efficacy in weight loss, and intolerable side effects in many patients (nausea, vomiting, and induction of type 2 diabetes due to inhibition of insulin secretion), octreotide has not been pursued further as a clinical therapy for weight management and cannot truly be considered a test of the “insulin as obesogenic hormone” hypothesis.
On the other hand, diazoxide has more specificity as an inhibitor of insulin secretion and has also been tested in patients with obesity. In a meta-analysis of seven studies, four with diazoxide and three with octreotide, Huang et al reported that suppression of insulin secretion in patients with obesity led to reduced body weight and fat mass and unchanged lean mass at the cost of slightly increased blood glucose (286). While this concept merits further development, like many complex physiological systems, insulin has several recognized roles in governing fat mass. In addition to its capacity to enhance peripheral fat storage, preclinical models have demonstrated that insulin acts as both a satiety factor and feed-back hormonal signal reflecting adipose tissue stores that inhibits appetite and enhances energy expenditure (279, 280). These functions of insulin, which have strong support in animal studies, plus the increased risk for causing type 2 diabetes when insulin secretion is pharmacologically suppressed, has dampened enthusiasm for this line of investigation.
HUMAN CHORIONIC GONADOTROPIN: 1954-2011
The story of the use of human Chorionic Gonadotropin (HCG) in the treatment of obesity involves several groups of actors. The first was Dr. Simeons who described the HCG protocols in 1954 and 1956. The second were the clinics that have used this hormone as a treatment for obesity. The third were the investigators who conducted clinical trials to test the effectiveness of HCG treatment plan. And the final actors were the US Government and its regulatory apparatus.
Human chorionic gonadotropin (HCG) is a peptide that comes from the placenta after implantation of a fetus. Its role is to maintain hormonal activity during part of pregnancy. Concentrations of HCG in the blood and urine provide the basis for pregnancy tests. The biological function of HCG and its chemical composition is similar to that of luteinizing hormone produced in the pituitary. HCG maintains hormonal secretion from the corpus luteum during pregnancy. Some early experimental studies with HCG suggested it may have metabolic effects on adipose tissue, but these remain to be confirmed (287, 288).
The clinical use of human chorionic gonadotropin (HCG) in the treatment of obesity can be dated to a paper published in the prestigious medical journal The Lancet in 1954. A British Endocrinologist, A.T. Simeon’s (289, 290) described studies in India on pregnant women and on “fat boys” with pituitary problems (Fröhlich’s Syndrome) who were eating a calorie-deficient diet. He noted that when treated with HCG, both groups lost weight and had a reduction in appetite.His clinical method subsequently appeared in a small booklet called Pounds and Inches that outlined his program in detail (291). This program involved a 500 total kcal per day diet along with daily injections of human chorionic gonadotrophin (hCG) lasting between 20 and 40 days.
There is little doubt that adhering to a 500 kcal/d diet will produce significant weight loss in a short period of time. The question was, however, whether the injections of HCG contributed pharmacologically to the effect or whether they were simply a placebo. In an early study, Carne compared 20 patients prescribed the 500 kcal/d diet half of whom received injections of saline and half HCG (292). They also compared a second group eating the same diet with half receiving shots of HCG and the other half receiving no injection. The authors concluded that patients receiving injections lost more weight than those not receiving injections, but that injections of HCG were not better than injections of saline. Other studies subsequently published came to the same conclusion: treatment with HCG was not significantly different from treatment with placebo. A young physician, Frank Greenway, MD working with me carried out one of these studies (293). We randomly assigned patients to receive “placebo injections” or injections of HCG. All patients were instructed in a 500 kcal/d diet. As expected on such a severe caloric restriction, all patients rapidly lost weight, but there was no difference in weight loss between those receiving HCG and those receiving placebo injections in this study.
Based on this and other studies, the US FDA ordered the following disclaimer in advertisements for HCG clinics.
“These weight reduction treatments include the injection of HCG, a drug which has not been approved by the Food and Drug Administration as safe and effective in the treatment of obesity or weight control. There is no substantial evidence that HCG increases weight loss beyond that resulting from caloric restriction, that it causes a more attractive or "normal" distribution of fat, or that it decreases the hunger and discomfort associated with calorie-restrictive diets.”
Despite this negative clinical trial data, the popularity of treatment with HCG was so great that it became the basis for a group of clinics dispensing it along with the 500 kcal/d diet. Ten years after the FDA made its pronouncement, there is still no evidence that HCG increases weight loss. To investigate the use of this treatment, a Los Angeles Times reporter, Robert Steinbrook, wrote a story titled “Doctors Use of Obesity Treatment Debunked by FDA” (294). For this story, Steinbrook “randomly telephoned 40 medical weight-loss programs that advertised in Los Angeles yellow pages [and] found 19 that offered HCG shots, including the Lindora Medical Clinic, one of Southern California’s largest chains of weight-loss clinics.” In his article he said: “At Lindora medical clinics, the patient information booklet states that HCG works by “mobilizing your abnormal fat into the blood stream as nutrients. This permits you to accept a very low caloric intake so that you can lose weight rapidly and feel well and not be overly hungry.” Steinbrook went on to say: “The FDA’s warning directly contradicts those assertions.” In a telephone interview with Dr. Marshall Stamper, Lindora’s founder and owner Stamper said that “Most of the patients that come through the clinic use [HCG].” However, as Steinbrook noted in his story, “HCG shots have been condemned by the American Medical Association, the California Medical Association, and the Federal Trade Commission on the basis of studies in the Journal of the American Medical Association and elsewhere in the mid-1970s that concluded the shots were no more effective than salt-water placebo injections or diet alone.” In his phone interview Dr. Stamper went on to say “They [patients] prefer it [HCG] to other methods,” “adding that he uses the shots himself on occasion for arthritis of his fingers, another condition for which the FDA has not approved HCG. We tell the patients point-blank that it doesn’t cause weight loss, but that they will do better on it. I feel the patients have definitely benefited. I feel it goes beyond the placebo effect,” Stamper said.
In 1995, a full ten years after Steinbrook’s story, HCG was still in use. A meta-analysis of studies with HCG published in 1995 (295) found that studies of HCG were generally of poor quality and the authors of this review concluded that "there is no scientific evidence that HCG is effective in the treatment of obesity; it does not bring about weight-loss or fat-redistribution, nor does it reduce hunger or induce a feeling of well-being.” (295)
Nonetheless, interest in the "HCG diet" continues into the 21st century as told in the story by Kevin Mark Trudeau published April 2007 and called The Weight Loss Cure "They" Don't Want You to Know About. The book describes a weight loss plan using HCG similar to the one published by Simeons in 1954 using human chorionic gonadotrophin (296).
Wikipedia describes Kevin Mark Trudeau as:
“….an American fraudster, author, salesman, and pool enthusiast, known for promotion of his books and resulting legal cases involving the Federal Trade commission (FTC). His ubiquitous late-night infomercials, which promoted unsubstantiated health, diet, and financial advice, earned him a fortune but eventually resulted in civil and criminal penalties for fraud, larceny, and contempt of court. In the early 1990s, Trudeau was convicted of larceny and credit card fraud. In 1998, he was accused of grossly misrepresenting the contents of his book, The Weight-Loss Cure “They” Don’t Want You to Know About. [The book uses the Simeon’s approach]. In a 2004 settlement, Trudeau agreed to pay a $500,000 fine and cease marketing of all products except his books, which are protected under the First Amendment. However, in 2011, he was fined $37.6 million for violating the 2004 settlement, and ordered to post a $2 million bond before engaging in any future infomercial advertising. In 2013, facing further prosecution for violations of the 2011 agreement and non-payment of the $37-million judgment, Trudeau filed for bankruptcy protection. His claims of insolvency were challenged by FTC lawyers, who maintained that he was hiding money in shell companies, and cited examples of continued lavish spending, such as $359 for a haircut. In November 2013, Trudeau was convicted of criminal contempt, and was sentenced to 10 years in federal prison. He will be eligible for release in 2022. Infomercials starring Trudeau and promoting his books — under the auspices of a private California corporation of undisclosed ownership — continue to air regularly on United States television stations.” (297)
Finally, on December 6, 2011, the United States Food and Drug Administration (FDA) prohibited the sale of over-the-counter diet products like HCG and indicated that they were fraudulent. Even later, on November 15, 2016, the American Medical Association (AMA) passed a policy statement saying that "The use of human chorionic gonadotropin (HCG) for weight loss is inappropriate." There is no scientific evidence that HCG is effective in the treatment of obesity. The authors stated “…the use of HCG should be regarded as an inappropriate therapy for weight reduction…” Yet, to this day, the internet still contains information about the HCG Diet, HCG Diet Cookbooks, and products claiming to contain HCG.
LEPTIN
The ‘obese’ mouse is a recessively inherited form of obesity that provided a major breakthrough in understanding the basic biology of food intake and regulation of body weight. This animal is obese because it lacks leptin, a hormone produced almost entirely in adipose tissue that was discovered in 1994 by positional cloning of the “ob” gene from the ‘obese’ mouse (6). This discovery revolutionized the field of obesity research by making it clear that obesity, in at least some cases, was a biochemical problem and not a problem of “will power.”
The principal site of action for leptin is in the brain, where activation of leptin receptors reduces food intake and increases energy expenditure, in part by increasing activity of the sympathetic nervous system. Several variants of the leptin receptor have been identified and cloned. Leptin inhibits hunger by reducing the production and release of neuropeptide Y (NPY) and agouti-related protein (AGRP), both potent stimulators of hunger, and by promoting satiety through synthesis of alpha melanocyte stimulating hormone (α-MSH), a hormone that suppresses food intake(298-300). Genetic deficiency in the leptin receptor, like the absence of leptin itself, is associated with a marked increase in food intake and massive obesity in both animals and humans (301, 302). Since leptin secretion is proportional to adipocyte cell size and number, as one might anticipate, circulating levels of leptin are highly correlated with the amount of body fat. In the cases of leptin deficient human beings and animals, leptin replacement cures their obesity by restoring normal appetite regulation and food intake and increasing the activity of the sympathetic nervous system (303).
Excitement following the discovery of leptin led to the hope that it might also cure “simple” (or polygenic) obesity once and for all. To test this, a randomized, double-blind clinical trial with leptin was performed. The results were a great disappointment when published in 1999 (304). There were 54 lean individuals with a mean weight of 72 kg and 73 subjects with obesity weighing on average 90 kg who were assigned to treatment with daily injections of leptin (r-met Hu Leptin) at doses of 0.01 mg/kg, 0.03 mg/kg, 0.1 mg/kg, or 0.3 mg/kg or placebo. The lean individuals maintained a eucaloric diet whereas the patients with obesity ate diets reduced by 500kcal/d below maintenance energy needs. All participants were treated for four weeks, and the subjects with obesity for an additional 20 weeks. There was a significant dose-related weight loss in the lean subjects at 4 weeks and in the individuals with obesity at 24 weeks. At the end of four weeks, 60 of the 70 patients with obesity who remained in the study elected to continue for an additional 20 weeks. Among the subjects who completed the study (n = 53 lean at 4 weeks and n = 47 obese at the end of 20 weeks), the authors found a significant dose-response effect for weight loss from baseline at 4 weeks and from baseline in the obese subjects treated for 24 weeks, but the differences were relatively small (weight changes for obese subjects at 24 weeks were 21.7 kg for placebo; 20.7 kg at 0.01 mg/kg; 21.4 kg at 0.03 mg/kg; 22.4 kg at 0.10 mg/kg; and 27.1 kg at 0.3 mg/kg). Reactions at the site of injection were the most common adverse event, but only two subjects withdrew for this reason. Glycemic control was unchanged during the study.
In a second human study, daily injections of leptin at doses of 0, 0.01 mg/kg, 0.05 mg/kg, 0.1 mg/kg and 0.3 mg/kg daily were relatively ineffective in the treatment of obesity (305). This limited response to leptin in subjects with obesity and higher levels of circulating leptin, contrasted sharply to the dramatic weight loss when leptin was used in the rare individuals with leptin deficiency (303), in whom leptin was much more effective. These studies showed that human leptin could produce weight loss in human beings with obesity, but the weight loss was disappointingly small. This fact, along with the side effects and route of injection, as well as the probability that antibodies against native leptin would develop led to discontinuation of this drug for commercial uses in obesity. On the other hand, these findings opened the door to exploration of “downstream” neuronal pathways from the leptin receptor, where it is now thought that the basis for leptin resistance resides.
Although leptin failed as an agent for treatment of the patient with obesity, it did find a role in ameliorating many of the symptoms of lipodystrophy (306-309). During 4 months of therapy with leptin in patients with lipodystrophy, triglyceride levels decreased by 60%, liver volume was reduced by an average of 28%, and resting metabolic rate decreased significantly (306).
Leptin treatment has been shown to ameliorate many of the adaptive effects associated with calorie restriction, including attenuating the decreased 24-hour energy expenditure, and thyroid hormone levels (310, 311). Leptin also reversed many of the changes in the brain as assessed by functional magnetic resonance (fMRI) associated with hunger in subjects stabilized at 10% below their usual weight (312). Leptin has been shown to help restore normal gonadal pulsatility and menses in women with leptin deficiency (313) and hypothalamic amenorrhea, typically due to extreme exercise or anorexia nervosa (314). While considered a failure as a single agent, the possibility that leptin may resurface in combination with some other peptide or oral agent remains open for the future.
CILIARY NEUROTROPHIC FACTOR
The receptor subunits for leptin share sequence similarities with the hypothalamic receptor for ciliary neurotrophic factor (CNTF), a neurocytokine that promotes neurotransmitter synthesis and neurite outgrowth in selected populations of brain cells (315). Leptin and CNTF both produce similar patterns of activation in brain cells. Treatment with CNTF in either ob/ob mice, which lack circulating leptin protein, or in db/db mice, which lack the leptin receptor, reduced the adiposity, hyperphagia, and hyperinsulinemia. CNTF was similarly effective in mice with diet-induced obesity. These findings suggest that this system may be a valuable target for drugs to manage obesity. Further support for this idea came from a published trial showing that CNTF produced weight loss in patients with amyotrophic lateral sclerosis (316).
A modified version of CNTF with a 15 amino-acid truncation of the C-terminal end and two amino acid substitutions was developed and tested in human beings with obesity (317). Phase 3 clinical trials for this modified ciliary neurotrophic factor were conducted in 2003 and demonstrated a small positive effect in some, but not all patients. A major problem was that nearly 70% of the participants developed antibodies against the recombinant CNTF after approximately three months of treatment. In the minority of subjects who did not develop the antibodies, weight loss averaged 12.5 pounds (5.7 kg) in one year, versus 4.5 pounds (2.0 kg) for the placebo-treated group. Development of neutralizing antibodies to peptide agents such as leptin and CNTF are pitfalls to this class of drugs (peptides) as they blunt the therapeutic responses.
Neuropeptide Y
Regulation of food intake in the brain involves a series of pathways located at the base of the brain in the arcuate and adjacent nuclei (298). This network involves secretion of peptides that increase food intake, including neuropeptide Y (NPY) and agouti-related peptide (AGRP) as well as the release of peptides that inhibit food intake, including α-MSH (derived from cleavage of pro-opiomelanocortin or POMC) and cocaine-amphetamine related peptide (CART). The effects of NPY are transmitted through 5 widely distributed receptors, Y-1, Y-2, Y-4, Y-5 and Y-6. Activation of NPY receptors Y-1 or Y-5 dramatically increases food intake in animals, and continued stimulation of the NPY receptor produces obesity, making an antagonist to these NPY receptors an attractive target for an anti-obesity drug.
Seeing this opportunity, Merck and Company developed MK-0557 which is a highly selective NPY-5 receptor antagonist with 98% receptor occupancy at 1.25 mg dose vs 3% for placebo using positron emission tomography. In the initial 12-week dose-ranging study weight losses were 0.7, 1.6, 2.2, 2.1 & 2.3 kg for placebo, 0.2, 1, 5 & 25 mg dose respectively (p=0.041) (318). Despite these small weight losses, a subsequent trial lasting 52-weeks was undertaken to evaluate it use in the prevention of weight regain. The placebo group had -1.1kg weight loss compared -2.2 kg in the group treated with 1 mg/d (319). This small, but statistically significant difference was not substantial enough for further drug development. While this study demonstrated that the NPY-5 receptor was involved in modulating food intake and body weight in human beings, the magnitude of weight loss was too small for further commercial development of this drug.
Melanocortin-4 Receptor Agonists
The melanocortin-4 receptor (MC4R) plays a central role in the regulation of food intake (320, 321). Individuals with genetic defects in this receptor are obese and disabling this receptor in experimental animals produces obesity (322). This receptor thus appeared to be a good target for pharmaceutical intervention prompting Merck and Company to pursue it (323). Their target drug, MK-0493, went into clinical trials as a selective and novel melanocortin-4 receptor agonist. However single doses of 200 mg and 500 mg were disappointing, producing only a marginally significant effect on 24-hr food intake compared to placebo. Nonetheless in two subsequent weight loss trials (one following stepped titration), MK-0493 produced a small, but not statistically significant weight loss relative to baseline. These clinical studies suggest that an MC4R agonist alone may not be an easy clinical target. While this limited clinical response to this agonist led to a termination of the clinical development program at Merck, recent reports led the FDA to approve another agonist to the MC4 receptor, setmelanotide, which was shown to be effective in the treatment for 3 rare genetic forms of obesity representing neuro signaling defects upstream from the MC4R: leptin deficiency, dysfunction in the PCSK1 (encoding for prohormone convertase 1/3), and deficiency of POMC (324, 325).
Testosterone and Anabolic Steroids
Testosterone is the principal product of the male testis and is responsible for changes which occur in males during puberty, including masculinization, voice changes, and changes in distribution of fat and muscle. Testosterone can also be produced by the adrenal gland, the ovary, and by conversion of androstenedione in peripheral tissues with 25% of testosterone in women coming from the ovary, 25% from the adrenal gland, and 50% from peripheral conversion. Levels of testosterone are associated with masculine dominance and these levels decline slowly after their peak in mid-life.
Reports of disordered reproductive function in men with obesity appeared in the mid-1970s as abstracts. In one study of 10 men with massive obesity, total serum testosterone and free testosterone correlated negatively with body weight but did not correlate significantly with sex-hormone-binding-globulin (326). The effects of obesity are probably due to reduced release of gonadotrophins from the pituitary since there was a normal response of testosterone to injections of human chorionic gonadotropin (HCG) acting directly on the testis (326). When clomiphene, a drug which enhances the release of the pituitary gonadotrophins was given, it also produced an increased release of testosterone by enhancing release of gonadotrophins from the pituitary (326). In a recent review, low total testosterone and low serum sex hormone-binding globulin were noted to be highly prevalent in men with obesity (327). However, it was only those with low levels of free testosterone along with signs and/or symptoms of hypogonadism that should be considered androgen deficient and thus merit treatment (327). These alterations are reversible upon weight loss as summarized in pooled data from 9 analyses in which Grossmann showed a linear relationship between weight loss and the increase in total testosterone (328, 329). Whether low testosterone is a biomarker or a true risk factor for metabolic disturbances remains unclear (330).
Testosterone levels are positively related to the amount of visceral fat in women and negatively correlated with visceral fat in men (331, 332). This inverse relationship between testosterone and visceral fat suggested that visceral fat might be reduced by treatment with testosterone. This hypothesis was tested using men with low-normal levels of circulating testosterone (≤ 0.20 nmol/liter) and a BMI greater than 25 kg/m2 (333). In the first study, testosterone was given orally twice daily for 8 months in doses of 80 mg. The 11 men who received testosterone had a significant decrease in the amount of visceral fat as measured by computed tomography compared to the 12 men who received placebo. There were no changes in body weight, subcutaneous fat, or lean body mass, but insulin sensitivity was improved.
In a second study, 31 men were randomly allocated to three groups receiving either placebo, testosterone, or dihydrotestosterone (334). In this study, testosterone and dihydrotestosterone were given as a gel applied to the arms daily for 9 months. The placebo group received only the gel. The testosterone-treated group had a significant decrease in waist circumference and visceral fat. On the other hand, the group treated with dihydrotestosterone had an increase in visceral fat compared to placebo. Insulin sensitivity was also improved by treatment with testosterone.
These intriguing results with testosterone, led to two additional studies examining the effects of anabolic steroids in both men and women (335, 336). The idea was to emphasize the effects on muscle and fat and reduce the masculinizing effects of testosterone. The first study lasted 9-months and included 30 healthy men who were overweight with mean BMI values between 33.8 –34.5 kg/m2 and testosterone values between 2 and 5 ng/ml (336). During the first 3 months when oxandrolone, an oral anabolic steroid was given, there was a significantly greater decrease in subcutaneous fat and a greater fall in visceral fat than in the groups treated with placebo or testosterone enanthate injected every 2 wk. There was also a significant drop in HDL cholesterol levels, which is a known side effect of orally administered anabolic steroids. The anabolic steroid group was thus changed from an oral to an injectable drug, nandrolone decanoate. Surprisingly, the group treated with bi-weekly injections of testosterone did not replicate the earlier data with testosterone published by the Swedish investigators (333, 334). Moreover, the biweekly injections of nandrolone failed to maintain the difference seen during daily oral treatment with oxandrolone. This suggests that frequent, if not daily, administration of steroid may be needed to obtain the effects on visceral fat.
In a second trial 30 post-menopausal women with obesity were treated for 9 months with the anabolic steroid nandrolone (335). During this time, they lost more fat and gained more lean mass than the comparison groups. Treatment with the anabolic steroid also produced a gain in visceral fat and a relatively greater loss of SC abdominal fat. The conclusion from the four studies is that visceral and total body fat can be manipulated separately, and that testosterone plays an important role in differential fat distribution in both men and women.
In older, obese hypogonadal men, adding testosterone for 6 months to lifestyle therapy does not further improve overall physical function. However, testosterone may attenuate the weight loss–induced reduction in muscle mass and hip bone mineral density (BMD) and may further improve aerobic capacity (337).
In summary, testosterone in men is related to their level of body fat and exogenous testosterone can influence both fat and muscle mass. Oral administration of testosterone or other steroids may have significant effects on circulating levels of lipids. The effects of obesity on testosterone may be related to changes in the central mechanisms that modulate the feedback of testosterone and luteinizing hormone levels, in which case weight loss benefits testosterone levels rather than vice versa. Ongoing studies are being conducted to determine if testosterone supplementation and the benefits on body composition are outweighed by increased risk for adverse cardiovascular events.
Dehydroepiandrosterone
Dehydroepiandrosterone [DHEA (3β-Hydroxyandrost-5-en-17-one)] and its sulfated derivate, DHEA-S are the most abundant steroid products of the human adrenal gland. At high levels of BMI, in the range of 40–60 kg/m2, there is a significant negative correlation of DHEA with BMI. Interestingly, the levels of DHEA and testosterone decline with age in both men and in women. Despite the high circulating levels of this steroid, no major function has been identified for it, although it can act as a weak androgen, a weak estrogen, and has effects in the central nervous system.
In contrast to human beings, most other animals have low levels of DHEA (338). When mice, rats, cats, and dogs are given DHEA in their diet they lost weight. This suggests that DHEA might be useful in treating patients with obesity (339-342). As a result, several clinical trials of DHEA have been completed in human beings. In a 28-day study with 10 volunteers of normal weight there was no effect on body weight or on insulin sensitivity as assessed by the euglycemic-hyperinsulinemic clamp even when the volunteers were given 1,600 mg/d of DHEA, a dose that is near the limit where hepatic toxicity becomes a risk (343). Another clinical study showed that DHEA had no effect on energy expenditure, body composition, or protein turnover (344). A third 28-day study in men with obesity also showed no improvement in body fat or insulin sensitivity (342). In women with obesity, there was likewise no change in body fat, but there was a decrease in insulin sensitivity (345). In one provocative trial, etiocholanedione, a metabolite of DHEA was tested in a 20 week double-blind cross-over study (346). Weight loss during treatment with etiocholandione was 2.8 kg compared to a weight loss of 0.21 kg (P<0.05) in the placebo group. This study has not been followed up and these compounds appear not to be under further study. All in all, a review of the subject by Clore (339) concluded that “a significant role for DHEA in the pharmacologic treatment of human obesity is unlikely.”
Conjugated Linoleic Acid
Dietary supplements containing ‘conjugated’ linoleic acid (CLA) have been widely promoted as weight loss agents. In this context, the word “conjugated” refers to the position of the double-bond between carbons 9-11 or 10-12 in the fatty acid chain of linoleic acid. In a meta-analysis of 13 randomized, controlled trials lasting up to 6 months, Larsen et al reported that there was little evidence that conjugated linoleic acid produced weight loss in human beings (347). One report suggests it may lower body fat without having an effect on body weight (348). Liver toxicity is one concern with the use of CLA with the trans-10, cis-12 isomeric form. One review said that this CLA isomer may adversely affect health by producing lipodystrophy and insulin resistance and by decreasing milk fat production in lactating women (347). Ultimately, Dwyer et al concluded that there is little evidence of benefit and there is a potential for harm when using CLA for weight reduction (349).
Hydroxycitrate
Hydroxycitric acid is a derivative of citric acid and is a component of some tropical plants including Garcia cambogia. One form of hydroxycitrate is a potent inhibitor of a final step in the tricarboxylic acid cycle (Krebs Cycle) involving the conversion of citrate to acetyl CoA and oxaloacetate. Studies in animals suggested that hydroxycitrate modulated lipid metabolism and thus might be useful in treating obesity (350). Garcinia cambogia is a significant plant source of hydroxycitrate. Two clinical trials with Garcinia cambogia have been published, but the results are not promising. In one randomized clinical trial there was no significant effect on body weight or fat mass (351). A more recent report suggests that either alone or with Green Tea Garcinia cambogia may cause liver damage (352) Clinically trivial differences in weight loss and potential liver toxicity made this drug a bad bet for weight loss (353).
Drugs Acting on the Gastrointestinal Track
DRUGS THAT MODIFY DIGESTION OR ABSORPTION OF NUTRIENTS
Reducing absorption of ingested nutrients is one potential strategy for lowering body weight. Drugs affecting metabolism and absorption of fat and carbohydrate have both been investigated to test this hypothesis. Orlistat is a drug approved around the world that uses this strategy by blocking intestinal lipases, thus leaving some “undigested” triglyceride in the GI tract. Because it is currently marketed, it will not be discussed further. Cetilostat is a second drug that blocks intestinal lipases. It has been tested in clinical trials and is approved for clinical use in Japan. Two other drugs, cholestyramine and neomycin have also been tested, but abandoned.
Cetilstat A Blocker of Fat Digestion
Cetilistat, (ATL-962), like orlistat, is a gastrointestinal lipase inhibitor. A 5-day trial of cetilistat in 90 normal volunteers showed a three to seven-fold increase in fecal fat loss that was dose-dependent, but only 11% of subjects had more than one oily stool (354). This data suggested that cetilistat might have fewer gastrointestinal side-effects than orlistat.A subsequent 12-week study with 372 patients compared placebo with doses of 60, 120 or 240 mg of cetilistat given three times a day (355). Weight loss ranged between 3.5 and 4 kg compared with a 2 kg loss in the placebo-treated group. In a second phase II study, doses of 40, 80 and 120 mg three times a day were given to patients with type 2 diabetes (356) with a dose-related reduction in body weight and in hemoglobin A1c. Side-effects have reduced the enthusiasm for this drug. However, it proved effective in vitro against the SARS-COVID-19 virus in a drug screen, which may give it a new life (357).
Cholestyramine a Binder of Sterols and Fatty Acids
Cholestyramine is a sequestrant that binds bile acids in the gastrointestinal track to prevent their reabsorption. As a strong ion exchange resin it can exchange its chloride anions with anionic bile acids and bind them strongly in the resin matrix, subsequently excreting the cholestyramine and adherent fat in the stool (358). Although originally developed for lowering cholesterol (Questran, Questran Light, Cholybar, and Olestyr), the idea that this drug could be used to enhance fat loss in the stools of patients with obesity as a strategy for weight loss was tested in two clinical studies. In the first, two patients with marked obesity received 36 g/d of cholestyramine for 9 days along with a 1200 kcal/d diet with 80% of the calories coming from fat. Although there was a statistically significant increase in fecal fat loss, it was disappointingly (287). A second study was conducted by Campbell et al in six patients with obesity using 30 g/d of cholestyramine (359). These results were also disappointing. Finally, cholestyramine was tried in an out-patient obesity clinic, where patients with obesity failed to lose significant amounts of weight (Bray Unpublished data). Thus, increasing fecal loss of fatty acids by binding them to resins produced clinically insignificant weight loss.
Neomycin as an Inhibitor of Fat Absorption
Neomycin is an antibiotic found in many topical medications, including creams, ointments, and eye drops. It was discovered in 1949 and approved for medical use in 1952. Like cholestyramine, it increases the loss of fat in the stools(358). In doses of 6-12 g/d it consistently produces steatorrhea (360), and in doses of 3 g/d it impaired fat absorption in 5 of 10 patients (361). Its use in obesity was limited by the fact that it alters the intestinal mucosa and is not safe in a chronic disease such as obesity (362), making it unsuitable for long-term use.
Olestra (Sucrose Polyester): An Indigestible Fat Replacement
Sucrose polyester (Olestra) is formed from sucrose by adding five or more fatty acids to a molecule of sucrose to produce a compound that has the physical characteristics of triglyceride, but which cannot be digested by pancreatic lipase. Short-term studies replacing dietary triglycerides with olestra showed two patterns of adaptation. Substitution of fat with sucrose polyester (olestra) in a single breakfast meal was followed by energy compensation over the next 24–36 hours in healthy young males (363). In contrast, substitution of dietary fat with sucrose polyester (olestra) in the noon or evening meal to lower digestible fat from 40% of intake to 30% of intake, was not followed by energy or nutrient compensation over the next 24 hours (364). Further lowering of digestible fat intake from 30% of intake to nearly 20% of energy intake over three meals in healthy subjects led them to feel less satiated at the end of the study and they compensated for nearly 75% of the energy deficit over the next day (365). In longer term studies lasting either 2 or 10 weeks, substitution of sucrose polyester (olestra) for one-third of the dietary fat in a diet with 40% fat reduced energy intake by about 15% (366).
In each of the short-term experiments there was only partial compensation for the energy deficit, suggesting that when the energy density of the diet is changed surreptitiously, individuals continued to eat for the same mass of food, even though it provided less metabolizable energy. Weight loss in the two-week experiment was 1.5 kg and in the 10-week experiment it was more than 5 kg, a weight loss that was significantly greater than in the control group. In a third study lasting nine months, 45 healthy men who were overweight were randomly assigned to one of three diets: a control diet with 33% of energy from fat, a fat-reduced diet containing 25% of energy from fat, and a diet where one-third of the dietary fat was replaced by olestra to achieve a diet containing 25% metabolizable energy from fat (367). Among the 36 men who completed the 9-month study, the sub-group receiving the sucrose polyester (olestra) as a replacement for regular dietary fat lost 6.27 ±1.66kg of body weight and 5.85 ±1.34 kg of body fat over the 9-month trial. This contrasted with the sub-group eating the control 33% fat diet who lost only half as much at 3.8 ± 1.34 kg of body weight and 3.45 ±1.0 kg of body fat. The sub-group eating the 25% low fat diet lost even less body weight (1.79 ± 0.81 kg) and body fat (1.68±0.75 kg). After 9 months it was clear that the men assigned to the fat-substituted diet had lost significantly more weight and fat than the men in the other two groups. In addition, the men eating the two regular fat diets (33% fat and 25% fat) asked for significantly more food than the men assigned to the olestra fat-substituted group, indicating higher levels of hunger (P < 0.05). In contrast, the men eating the olestra substituted fat-reduced diet asked for almost no extra foods suggesting that they were more satiated. The authors concluded that replacement of dietary fat with olestra reduces body weight and total body fat when compared with a 25% fat diet or a control diet containing 33% fat (367). Subjects eating the olestra-containing diet had substantial decreases in serum carotene, lycopene, lutein and zeaxanthin, which occurred by 12 weeks. Replacement with a multi-vitamin was sufficient to overcome these changes (368). Weight loss and the loss of fat also impacted cardiovascular risk factors with a significant effect of diet mainly on total cholesterol, low-density lipoprotein cholesterol, and triglyceride levels, all of which decreased in the olestra-substituted group but not the other two groups by 9 months (369). After adjustment for the percentage of fat loss during the trial, only the changes in triglycerides remained statistically significant (369).
Olestra was approved by the FDA as a food additive in 1996 and soon appeared in chips sold under the brand name “WOW.” Because of perceived or real side-effects, the use of olestra containing products declined and in 2002 Proctor-Gamble, the originator of olestra, sold its manufacturing facility in Cincinnati. Some products with olestra may still be available. In the end, while this food product produced beneficial effects, it never caught on with the public.
Fat emulsion: OlibraTM
Food and its digested products act on the intestine at various levels to secrete hormones that modulate food intake. One of these signals is the entry of fats and fatty acids into the ileum which activates both neural and humoral factors that inhibit gastric emptying, prolong gastric transit time, and can produce satiety. This mechanism is referred to as the “ileal brake.” Longer chain fatty acids (> 12 carbons) and free fatty acids, rather than triglycerides, are the most effective stimuli for the ileal brake. OlibraTM is a fat emulsion composed of fractionated palm and oat oil in the proportion of 95:5. The palm oil is emulsified by hydrophilic galactolipids derived from oat oil (Lipid Technologies Provider AB, Karishamn, Sweden). This product increases satiety and reduces food intake in some studies (370), but not in others (371). To test the clinical effectiveness of this fat emulsion, a phase-2 randomized double-blind controlled trial of 12 weeks duration was conducted in 74 men and women with obesity (372). In this study, “Olibra” had no significant effect on food intake, appetite or ratings of satiety and did not differentially affect, body weight, or body composition.
Amylase Blockers of Carbohydrate Digestion
Amylase is an enzyme found in the saliva and intestine that hydrolyzes dietary starch into simple sugars. Salivary α-amylase is encoded by a gene called AMY1. In epidemiological studies, low AMY1 gene expression was associated with obesity (373). Moreover, low serum concentrations of amylase have been associated with elevated BMI (374). In a weight loss study, individuals who were overweight or obese and carried the AMY1-AMY2 (rs11185098) genotype, which is associated with higher amylase activity, had greater weight loss during the 2-year POUNDS Lost dietary intervention study (375), suggesting that medications modifying starch digestion might be of potential value in the management of obesity.
To test the effect of blocking starch digestion on body weight, a clinical trial with 59 people was designed to give some participants an amylase-starch (glucosidase) inhibitor called BAY e 4609 (Acarbose) or a placebo (376). Although insulin and glucose tolerance improved, the drug did not produce weight loss. A somewhat different conclusion was reached by William-Olsson et al (377) who treated 24 women with acarbose after they had already lost weight and found that it helped prevent weight regain. In a third study, Wolever et al. reported that after one-year, 354 individuals with type 2 diabetes who received acarbose lost 0.46 kg compared to a weight gain of 0.33 kg among those in the placebo group. Although statistically significant, this result was clinically small (378). In this case, despite the relation of the AMY-1 gene expression to weight change, α-amylase blockers produce either no effect or a clinically trivial effect on body weight.
Enterostatin A Peptide That Affects Fat Intake
Enterostatin is a penta-peptide produced by trypsin cleavage of pancreatic procolipase in the intestine. Procolipase is secreted from the pancreas in response to dietary fat. Enterostatin, is highly conserved across a number of species (379, 380) and selectively reduces fat intake in the rat by nearly 50% (381). The dose-response curve for enterostatin is U-shaped with an optimal inhibitory effect on feeding at 1 nmol. Higher and lower doses are less effective, and at high doses enterostatin actually stimulates food intake. Chronic infusion of enterostatin reduced body weight in animals (382), but parenteral enterostatin did not reduce food intake in human beings (383). In the end, limited effectiveness terminated development of this product.
SUMMARY AND PERSPECTIVE
This chapter provides a historical perspective on earnest but often misguided approaches advocated by providers in the treatment of obesity, as well as the scientific challenges and adverse patient outcomes that have plagued development of effective weight loss drugs. Besides the typical toxicities associated with any drug development program, including rashes, gastrointestinal distress, hepatic metabolism, and the development of antibodies against some peptides, a number of other specific toxicities have been particularly vexing. One example is off-target effects. This can occur when receptor activation (or inhibition) impacts more than one intra-cellular signaling system, such as when changes in blood pressure occur in response to sibutramine and tesofensine. Off-target effects can also occur when receptor specificity is low and more than one receptor within a drug class are affected. For example, drugs that either raise systemic levels of serotonin, or lack specificity for the serotonin receptor such as fenfluramine, will activate both the 5HT2c and 5HT2b receptors. Activation of the 5HT2c receptors lowered food intake whereas simultaneous activation of the 5HT2b receptor produces cardiac valvulopathy. Or an endocannaboid receptor antagonist may inhibit food intake and cause weight loss but may also affect mood centers and increase risk for suicidality. Finally, drugs that modulate brain neurotransmitters can result in different behaviors depending on where the drug is acting. For example, modulators of GABA activity, like topiramate, can simultaneously impact appetite and alter mood. Or dual or triple amine uptake inhibitors that can, on the one hand, reduce schizophrenic symptoms while increasing hunger signals and food intake. Food intake can be a pleasurable experience through activation of hedonic/pleasure circuitry involving dopamine, which has similarities to many addictive substances, including amphetamine. Many anorectic drugs have had to thread a small therapeutic window between improved appetite control and similar adverse behavioral effects.
The patient is equally as important in the management of obesity as the medication. Beside the biological effects alluded to above, patients often have unrealistic expectations about how much weight loss drugs can produce. When the treatment doesn’t meet their expectations, they can become frustrated and discontinue therapy even when health benefits from even modest weight loss occur. The second reality that often, and mistakenly, leads patients and providers to stop anti-obesity medication use is the weight plateau that usually occurs after 3-4 months (or up to one year in the case of GLP-1 receptor agonists) of continuous treatment. Ultimately a new weight plateau is to be expected since the body weight set point will still defend its fat stores. This does not indicate tolerance to the drug or that it stopped working, but more the limit of the therapeutic effectiveness expected for that drug. This is similar to the management of any chronic disease. An example would be initiating an angiotensin converting enzyme (ACE) inhibitor for hypertension. It is not expected that this one drug will result in normalization of BP in every patient or that, in taking the ACE inhibitor, the patient will be able to set and achieve a target blood pressure of their choosing. Instead, the medication is started and after appropriate follow-up, the physiological response is assessed and if deemed inadequate, adjustments are made in dose or combination therapy is initiated. Similar examples can be made for first line therapies for type 2 diabetes (metformin) and hyperlipidemia (statins).
Besides off-target effects and inter-individual variability in weight loss response, another barrier to effective obesity management is the cost of medications, especially with a chronic disease as highly prevalent as obesity. Until a medication or combination of medications is developed that can meet the patient’s weight loss goals and produce a large enough weight loss to make it cost-effective, challenges will remain in effectively treating patients with obesity.
REFERENCES
- Sarton G. The History of Science and the New Humanism. Colver lectures,. 1931, New York: H. Holt and company. 178 pages.
- Harari YN. Sapiens: A Brief History of Humankind. First U.S. edition. ed. 2015, New York: Harper. 443 pages.
- Conard NJ. A female figurine from the basal Aurignacian of Hohle Fels Cave in southwestern Germany. Nature.2009;459(7244):248-52. DOI: 10.1038/nature07995. PMID: 19444215.
- Weber GW, Lukeneder A, Harzhauser M, Mitteroecker P, Wurm L, Hollaus LM, Kainz S, Haack F, Antl-Weiser W, Kern A. The microstructure and the origin of the Venus from Willendorf. Sci Rep. 2022;12(1):2926. DOI: 10.1038/s41598-022-06799-z. PMID: 35228605.
- Bray GA. The Battle of the Bulge : A History of Obesity Research. 2007, Pittsburgh, Pa.: Dorrance Pub. Co. 847 pages.
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425-32. DOI: 10.1038/372425a0. PMID: 7984236.
- Bray GA. Why do we need drugs to treat the patient with obesity? Obesity (Silver Spring). 2013;21(5):893-9. DOI: 10.1002/oby.20394. PMID: 23520198.
- Foxcroft L. Calories & Corsets: A History of Dieting Over 2,000 Years. 2011, London: Profile Books. 232 pages.
- Kryger MH. Sleep apnea. From the needles of Dionysius to continuous positive airway pressure. Arch Intern Med. 1983;143(12):2301-3. DOI: 10.1001/archinte.143.12.2301. PMID: 6360064.
- Papavramidou N, Christopoulou-Aletra H. Management of obesity in the writings of Soranus of Ephesus and Caelius Aurelianus. Obes Surg. 2008;18(6):763-5. DOI: 10.1007/s11695-007-9362-1. PMID: 18386109.
- Dorje G, Parfionovich Y. Tibetan Medical Paintings: Illustrations to the Blue Beryl Treatise of Sangye Gyamtso (1653-1705). 1992, New York: H.N. Abrams, Inc.
- Talbott JH. A Biographical History of Medicine: Excerpts and Essays on the Men and Their Work. 1970, New York,: Grune & Stratton. 1211 pages.
- Sarton G. Sarton on the History of Science. Essays by George Sarton. 1962, Cambridge,: Harvard University Press. 383 pages.
- Ullmann M. Islamic medicine. Islamic surveys. 1978, Edinburgh: Edinburgh University Press. 138 pages.
- Campbell D. Arabian Medicine and its Influence on the Middle Ages. Trübner's oriental series. 1926, London,: K. Paul, Trench, Trubner & co., ltd.
- Castiglioni A. A History of Medicine. 1941, New York,: A. A. Knopf. 1013 pages.
- Sigerist HE. A History of Medicine. Vol. 1. Primitive and Archaic Medicine. Vol. 2. Early Greek, Hindu, and Persian Medicine. Publication / Historical Library, Yale Medical Library. 1961, New York,: Oxford University Press.
- Major RH. A History of Medicine. 1954, Springfield, IL: Charles C Thomas.
- Gruner OC. A Treatise on the Canon of Medicine of Avicenna, Incorporating a Translation of the First Book. 1930, London,: Luzac & Co. 612 pages.
- Hopkins KD, Lehmann ED. Successful medical treatment of obesity in 10th century Spain. Lancet.1995;346(8972):452. DOI: 10.1016/s0140-6736(95)92830-8. PMID: 7623606.
- Irisarri Ád. El viaje de la reina: De cómo la intrépida reina Toda de Navarra realiza un viaje de Pamplona a Córdoba en el año mil. 6* ed ed. Novela histórica. 1998, Barcelona: Emecé Editores. 348 pages.
- Chao AM, Wadden TA, Ashare RL, Loughead J, Schmidt HD. Tobacco Smoking, Eating Behaviors, and Body Weight: A Review. Curr Addict Rep. 2019;6:191-199. DOI: 10.1007/s40429-019-00253-3. PMID: 33224710.
- Bonet T. A guide to the Practical Physician : Shewing from the most approved authors, both ancient and modern, the truest and safest way of curing all diseases, internal and external, whether by medicine, surgery, or diet. 1684, London: Thomas Flesher. 788 pages.
- Short T. A Discourse Concerning the Causes and Effects of Corpulency : Together with the Method for its Prevention and Cure. 1727, London: Oxford Arms in Warwick Lane. 80 pages.
- Flemyng M. A Discourse on the Nature, Causes, and Cure of Corpulency. Illustrated by a Remarkable Case, Read Before the Royal Society, November 1757. And Now First Published, by Malcolm Flemyng, M.D. 1760: L. Davis and C. Reymers.
- Wang ZM, Pierson RN, Jr., Heymsfield SB. The five-level model: a new approach to organizing body-composition research. Am J Clin Nutr. 1992;56(1):19-28. DOI: 10.1093/ajcn/56.1.19. PMID: 1609756.
- Report of a Committee of the Clinical Society of London Nominated December 14, 1883, to Investigate the Subject of Myxœdema. Trans Clinical Society of London. 1888;21(suppl):1-215.
- Kendall EC. A Method for the Decomposition of the Proteins of the Thyroid, with a Description of Certain Constituents. Journal of Biological Chemistry. 1915;20(4):501-509. DOI: 10.1016/s0021-9258(18)88214-x.
- Harington CR. Chemistry of Thyroxine: Constitution and Synthesis of Desiodo-Thyroxine. Biochem J.1926;20(2):300-13. DOI: 10.1042/bj0200300. PMID: 16743659.
- Putnam JJ. Cases of Myxedema and Acromegalia Treated with Benefit of Sheep’s Thyroid: Recent Observations respecting the pathology of the cachexias following disease of the thyroid: clinical relationships of Grave’s Disease and Acromegalia. The American Journal of the Medical Sciences. 1893;106(2):125-148.
- Yorke-Davies NE. Thyroid Tabloids in Obesity. Bmj. 1894;2(1749):42-43. DOI: 10.1136/bmj.2.1749.42.
- Wendelstadt. Ueber Entfettungscuren mit Schilddrüsenfütterung. Dtsch Med Wochenschr. 1894;20(50):934-935.
- Leichtenstern O. Ueber Myxödem und über Entfettungscuren mit Schilddrüsenfütterung1). Dtsch Med Wochenschr. 1894;20(50):932-933.
- Sajous CEdM. The Internal Secretions and the Principles of Medicine. 7th ed. 1916, Philadelphia: F.A. Davis.
- Strang JM, McClugage HB, Evans FA. Further studies in the dietary correction of obesity. Am J Clin Sci.1930;179:687-694.
- Short JJ. The Increased Metabolism of Obesity. Journal of the American Medical Association.1936;106(21):1776-1779. DOI: 10.1001/jama.1936.02770210002002.
- Evans FA. The Treatment of Obesity with Low Caloric Diets. Journal of the American Medical Association.1931;97(15):1063-1069. DOI: 10.1001/jama.1931.02730150019007.
- Lyon DM, Dunlop DM. The Treatment of Obesity: A Comparison of the Effects of Diet and of Thyroid Extract. QJM: An International Journal of Medicine. 1932;1(2):331-352. DOI: 10.1093/oxfordjournals.qjmed.a066590.
- Bayer LM, Gray H. Obesity treatment by diet, thyroid, and dinitrophenol: Result on 106 outpatients. American Journal of the Medical Sciences. 1935;189(1):86-90.
- Bray GA. Thyroid Hormones in the Treatment of Obesity. in Obesity in Perspective. (G.A. Bray, Ed.), Section IV, Vol 2, Part 2, Chapter. 55, Washington, D.C., U.S. Govt Printing Office, 1975. Washington DC: U.S. Govt. Print. Off. Pages 449-456.
- Rony HR. Obesity and Leanness. 2nd ed. 1940, Philadelphia: Lea & Febiger. 300 pages.
- Rynearson EH, Gastineau CF. Obesity. 1st ed. 1949, Springfield, IL: C. C. Thomas. 134 pages.
- Bram I. Therapeutic hyperthryroidism, in International clinics; a quarterly of clinical lectures. 1937, J.B. Lippincott.: Philadelphia. p. 48-61.
- Benedict FG, Miles WR, Roth P, Smith HM. Human Vitality and Efficiency Under Prolonged Restricted Diet. 1919, Washington,: Carnegie institution of Washington. 701 pages.
- Brown EG, Ohlson MA. Weight reduction of obese women of college age; clinical results and basal metabolism. J Am Diet Assoc. 1946;22:849-57. PMID: 20998120.
- Cutting WC. The Treatment of Obesity1. The Journal of Clinical Endocrinology & Metabolism. 1943;3(2):85-88. DOI: 10.1210/jcem-3-2-85.
- Means JH. Therapeutics of the Thyroid. Journal of the American Medical Association. 1935;105(1):24-28. DOI: 10.1001/jama.1935.92760270002009.
- Wilder RM. The treatment of obesity, in International clinics; a quarterly of clinical lectures. 1933, J.B. Lippincott.: Philadelphia. p. 24-28.
- Sevringhaus EL. Diagnostic and therapeutic problems of obesity. J. Mich. Med. Soc. 1943;42:530-536.
- Sevringhaus EL. Endocrine Therapy in General Practice. 5th ed. 1945, Chicago: Year Book Publishers, Inc. 223 pages.
- Barborka CJ. Obesity. Medical Clinics of North America. 1937;21(1):23-39. DOI: 10.1016/s0025-7125(16)37221-2.
- Bronstein IP, Halpern LJ, Brown AW. Obesity in children. The Journal of Pediatrics. 1942;21(4):485-496. DOI: 10.1016/s0022-3476(42)80242-5.
- Grulee CG. The American Journal of Diseases of Children. AMA Am J Dis Child. 1951;81(3):374-93. DOI: 10.1001/archpedi.1951.02040030384005. PMID: 14810171.
- Mason EH. The Treatment of Obesity. Can Med Assoc J. 1924;14(11):1052-6. PMID: 20315168.
- Lyon DM, Dunlop DM, Stewart CP. Respiratory quotient in obese subjects. Biochem J. 1932;26(4):1107-17. DOI: 10.1042/bj0261107. PMID: 16744913.
- Gross J, Pitt-Rivers R. The identification of 3:5:3'-L-triiodothyronine in human plasma. Lancet.1952;1(6705):439-41. DOI: 10.1016/s0140-6736(52)91952-1. PMID: 14898765.
- Chopra IJ, Solomon DH, Beall GN. Radioimmunoassay for measurement of triiodothyronine in human serum. J Clin Invest. 1971;50(10):2033-41. DOI: 10.1172/JCI106696. PMID: 4107265.
- Byerley LO, Heber D. Metabolic effects of triiodothyronine replacement during fasting in obese subjects. J Clin Endocrinol Metab. 1996;81(3):968-76. DOI: 10.1210/jcem.81.3.8772559. PMID: 8772559.
- Garrow JS. Energy balance and obesity in man. 2d ed. 1978, Amsterdam; New York New York: Elsevier/North-Holland Biomedical Press. xii, 243 p.
- Goodman NG. Triiodothyronine and placebo in the treatment of obesity. A study of fifty-five patients. Med Ann Dist Columbia. 1969;38(12):658-62 passim. PMID: 4902411.
- Hollingsworth DR, Amatruda TT, Jr., Scheig R. Quantitative and qualitative effects of L-triiodothyronine in massive obesity. Metabolism. 1970;19(11):934-45. DOI: 10.1016/0026-0495(70)90040-5. PMID: 4920912.
- Drenick EJ, Fisler JL. Prevention of recurrent weight gain with large doses of synthetic thyroid hormones. Curr Ther Res Clin Exp. 1970;12(9):570-6. PMID: 4989686.
- Bhasin S, Wallace W, Lawrence JB, Lesch M. Sudden death associated with thyroid hormone abuse. Am J Med. 1981;71(5):887-90. DOI: 10.1016/0002-9343(81)90392-2. PMID: 7304660.
- Krotkiewski M. Thyroid hormones in the pathogenesis and treatment of obesity. Eur J Pharmacol. 2002;440(2-3):85-98. DOI: 10.1016/s0014-2999(02)01420-6. PMID: 12007527.
- Grover GJ, Mellstrom K, Ye L, Malm J, Li YL, Bladh LG, Sleph PG, Smith MA, George R, Vennstrom B, Mookhtiar K, Horvath R, Speelman J, Egan D, Baxter JD. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci U S A. 2003;100(17):10067-72. DOI: 10.1073/pnas.1633737100. PMID: 12888625.
- Bleasdale EE, Thrower SN, Petroczi A. Would You Use It With a Seal of Approval? Important Attributes of 2,4-Dinitrophenol (2,4-DNP) as a Hypothetical Pharmaceutical Product. Front Psychiatry. 2018;9:124. DOI: 10.3389/fpsyt.2018.00124. PMID: 29731723.
- Perkins RG. A Study of the Munitions Intoxications in France. Public Health Reports (1896-1970).1919;34(43):2335-2374. DOI: 10.2307/4575357.
- Cutting WC, Tainter ML. Actions of dinitrophenol. Proceedings of the Society for Experimental Biology and Medicine. 1932;29:1268-1269.
- Cutting WC, Mehrtens HG, Tainter ML. Actions and Uses of Dinitrophenol. Journal of the American Medical Association. 1933;101(3):193-195. DOI: 10.1001/jama.1933.02740280013006.
- Colman E. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul Toxicol Pharmacol.2007;48(2):115-7. DOI: 10.1016/j.yrtph.2007.03.006. PMID: 17475379.
- Tainter ML, Cutting WC, Stockton AB. Use of Dinitrophenol in Nutritional Disorders : A Critical Survey of Clinical Results. Am J Public Health Nations Health. 1934;24(10):1045-53. DOI: 10.2105/ajph.24.10.1045. PMID: 18014064.
- Tainter ML. Dinitrophenol in the Treatment of Obesity. Journal of the American Medical Association.1935;105(5):332-337. DOI: 10.1001/jama.1935.02760310006002.
- Horner WD. Cataract Following Di-Nitrophenol Treatment for Obesity. Archives of Ophthalmology.1936;16(3):447-461. DOI: 10.1001/archopht.1936.00840210121009.
- McGavack TH. Dinitrophenol and Reducing. Cal West Med. 1936;44(2):77-8. PMID: 18743565.
- Strang JM. An Evaluation of Dinitrophenol as an Aid in Weight Reduction. JAMA: The Journal of the American Medical Association. 1935;104(22):1957-1963. DOI: 10.1001/jama.1935.02760220003002.
- Council on Pharmacy and chemistry. Journal of the American Medical Association. 1935;105(1):31-33. DOI: 10.1001/jama.1935.02760270033012.
- Grundlingh J, Dargan PI, El-Zanfaly M, Wood DM. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol. 2011;7(3):205-12. DOI: 10.1007/s13181-011-0162-6. PMID: 21739343.
- Holborow A, Purnell RM, Wong JF. Beware the yellow slimming pill: fatal 2,4-dinitrophenol overdose. BMJ Case Rep. 2016;2016. DOI: 10.1136/bcr-2016-214689. PMID: 27045052.
- Ainsworth NP, Vargo EJ, Petroczi A. Being in control? A thematic content analysis of 14 in-depth interviews with 2,4-dinitrophenol users. Int J Drug Policy. 2018;52:106-114. DOI: 10.1016/j.drugpo.2017.12.012. PMID: 29331928.
- Geisler JG. 2,4 Dinitrophenol as Medicine. Cells. 2019;8(3). DOI: 10.3390/cells8030280. PMID: 30909602.
- Rasmussen N. America's first amphetamine epidemic 1929-1971: a quantitative and qualitative retrospective with implications for the present. Am J Public Health. 2008;98(6):974-85. DOI: 10.2105/AJPH.2007.110593. PMID: 18445805.
- Barger G, Dale HH. Chemical structure and sympathomimetic action of amines. J Physiol. 1910;41(1-2):19-59. DOI: 10.1113/jphysiol.1910.sp001392. PMID: 16993040.
- Hicks J. Fast Times: The Life, Death, and Rebirth of Amphetamine, in Science History Institute. 2012.
- Prinzmetal M. The Use of Benzedrine for the Treatment of Narcolepsy. Journal of the American Medical Association. 1935;105(25):2051-2054. DOI: 10.1001/jama.1935.02760510023006.
- Ulrich H, Trapp CE, Vidgoff B. The Treatment of Narcolepsy with Benzedrine Sulphate. Annals of Internal Medicine. 1936;9(9):1213-1221. DOI: 10.7326/0003-4819-9-9-1213.
- Lesses MF, Myerson A. Human Autonomic Pharmacology. New England Journal of Medicine. 1938;218(3):119-124. DOI: 10.1056/nejm193801202180307.
- Rasmussen N. Making the first anti-depressant: amphetamine in American medicine, 1929-1950. J Hist Med Allied Sci. 2006;61(3):288-323. DOI: 10.1093/jhmas/jrj039. PMID: 16492800.
- Bett WR. Benzedrine sulphate in clinical medicine; a survey of the literature. Postgrad Med J.1946;22(250):205-18. DOI: 10.1136/pgmj.22.250.205. PMID: 20997404.
- Benzedrine Sulfate—a Warning. Journal of the American Medical Association. 1938;110(12):901-902. DOI: 10.1001/jama.1938.02790120043013.
- Lesses MF. Benzedrine Sulfate. Journal of the American Medical Association. 1938;110(18):1507-1508. DOI: 10.1001/jama.1938.02790180095025.
- Myerson A. The Rationale for Amphetamine (Benzedrine) Sulphate Therapy. Am J Clin Sci. 1940;199(5):729-737.
- Heal DJ, Smith SL, Gosden J, Nutt DJ. Amphetamine, past and present--a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-96. DOI: 10.1177/0269881113482532. PMID: 23539642.
- Cohen PA, Goday A, Swann JP. The return of rainbow diet pills. Am J Public Health. 2012;102(9):1676-86. DOI: 10.2105/AJPH.2012.300655. PMID: 22813089.
- United States Congress Senate Committee on the Judiciary Subcommittee on Antitrust and Monopoly. Diet pill industry hearings before the Subcommittee on Antitrust and Monopoly of the Committee on the Judiciary, United States Senate, Ninetieth Congress, second session, pursuent to S. Res. 26. 1968, Washington, D.C.: U.S. G.P.O. 731 pages.
- Greenway F. A double-blind clinical evaluation of the anorectic activity of phenylpropanolamine versus placebo. Clin Ther. 1989;11(5):584-9. PMID: 2680083.
- Greenway FL. Clinical studies with phenylpropanolamine: a metaanalysis. Am J Clin Nutr. 1992;55(1 Suppl):203S-205S. DOI: 10.1093/ajcn/55.1.203s. PMID: 1530830.
- Morgan JP, Kagan DV, Brody JS. Phenylpropanolamine : risks, benefits, and controversies. Clinical pharmacology and therapeutics series. 1985, New York: Praeger. xxi, 426 p.
- Hoebel BG, Cooper J, Kamin MC, Willard D. Appetite suppression by phenylpropanolamine in humans. Obesity and Bariatric Medicine. 1975;4:l92-l99.
- Weintraub M. Phenylpropanolamine as an anorexiant agent in weight control: a review of published and unpublished studies, in Morgan JP, Kagan DV, Brody is, eds. Phenylpropanolamine. Risks, benefits, and controversies. New York: Praeger Scientific. 1985. p. 53-79.
- Schteingart DE. Effectiveness of phenylpropanolamine in the management of moderate obesity. Int J Obes Relat Metab Disord. 1992;16(7):487-93. PMID: 1323545.
- Scoville BA. Review of amphetamine-like drugs by the Food and Drug Administration: clinical data and value judgments, in Obesity in perspective : a conference. 1975, U.S. Govt. Print. Off.: Washington. p. 441–443.
- Weintraub M, Ginsberg G, Stein EC, Sundaresan PR, Schuster B, O'Connor P, Byrne LM. Phenylpropanolamine OROS (Acutrim) vs. placebo in combination with caloric restriction and physician-managed behavior modification. Clin Pharmacol Ther. 1986;39(5):501-9. DOI: 10.1038/clpt.1986.87. PMID: 3516509.
- Blackburn GL, Morgan JP, Lavin PT, Noble R, Funderburk FR, Istfan N. Determinants of the pressor effect of phenylpropanolamine in healthy subjects. JAMA. 1989;261(22):3267-72. PMID: 2716162.
- Kernan WN, Viscoli CM, Brass LM, Broderick JP, Brott T, Feldmann E, Morgenstern LB, Wilterdink JL, Horwitz RI. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med. 2000;343(25):1826-32. DOI: 10.1056/NEJM200012213432501. PMID: 11117973.
- Samuel PD, Burland WL. Drug Treatment of Obesity, in Obesity in perspective : a conference. 1975, U.S. Govt. Print. Off.: Washington. p. 419-428.
- Heil GC, Ross ST. Chemical Agents Affecting Appetite, in Obesity in perspective : a conference. 1975, U.S. Govt. Print. Off.: Washington. p. 409-418.
- Bray GA. Treatment of obesity with drugs and invasive procedures, in Obesity in America. 1979, DHEW Publication No. (NIH) 79-359: Washington DC. p. 179-205.
- Final Report to the Director, Bureau of Drugs, by the Chairman, Consultants on Anorectic Drugs, in Obesity in perspective : a conference. 1975, U.S. Govt. Print. Off.: Washington. p. 409-418.
- Maier J, Mayer FP, Brandt SD, Sitte HH. DARK Classics in Chemical Neuroscience: Aminorex Analogues. ACS Chem Neurosci. 2018;9(10):2484-2502. DOI: 10.1021/acschemneuro.8b00415. PMID: 30269490.
- Colman E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann Intern Med. 2005;143(5):380-5. DOI: 10.7326/0003-4819-143-5-200509060-00013. PMID: 16144896.
- Kramer MS, Lane DA. Aminorex, dexfenfluramine, and primary pulmonary hypertension. J Clin Epidemiol.1998;51(4):361-4. DOI: 10.1016/s0895-4356(97)00289-8. PMID: 9539893.
- McQuillan BM, Picard MH, Leavitt M, Weyman AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation. 2001;104(23):2797-802. DOI: 10.1161/hc4801.100076. PMID: 11733397.
- Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation. 1999;100(8):869-75. DOI: 10.1161/01.cir.100.8.869. PMID: 10458725.
- Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, Simonneau G, Begaud B. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609-16. DOI: 10.1056/NEJM199608293350901. PMID: 8692238.
- Saxon DR, Iwamoto SJ, Mettenbrink CJ, McCormick E, Arterburn D, Daley MF, Oshiro CE, Koebnick C, Horberg M, Young DR, Bessesen DH. Antiobesity Medication Use in 2.2 Million Adults Across Eight Large Health Care Organizations: 2009-2015. Obesity (Silver Spring). 2019;27(12):1975-1981. DOI: 10.1002/oby.22581. PMID: 31603630.
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39(1):32-41. DOI: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. PMID: 11071707.
- Underhill SM, Colt MS, Amara SG. Amphetamine Stimulates Endocytosis of the Norepinephrine and Neuronal Glutamate Transporters in Cultured Locus Coeruleus Neurons. Neurochem Res. 2020;45(6):1410-1419. DOI: 10.1007/s11064-019-02939-6. PMID: 31912366.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J. 1968;1(5588):352-4. DOI: 10.1136/bmj.1.5588.352. PMID: 15508204.
- Langlois KJ, Forbes JA, Bell GW, Grant GF, Jr. A double-blind clinical evaluation of the safety and efficacy of phentermine hydrochloride (Fastin) in the treatment of exogenous obesity. Curr Ther Res Clin Exp.1974;16(4):289-96. PMID: 4208343.
- Willims RA, Foulsham BM. Weight reduction in osteoarthritis using phentermine. Practitioner.1981;225(1352):231-2. PMID: 7022428.
- Ryder JR, Kaizer A, Rudser KD, Gross A, Kelly AS, Fox CK. Effect of phentermine on weight reduction in a pediatric weight management clinic. Int J Obes (Lond). 2017;41(1):90-93. DOI: 10.1038/ijo.2016.185. PMID: 27773937.
- Hollander P, Bays HE, Rosenstock J, Frustaci ME, Fung A, Vercruysse F, Erondu N. Coadministration of Canagliflozin and Phentermine for Weight Management in Overweight and Obese Individuals Without Diabetes: A Randomized Clinical Trial. Diabetes Care. 2017;40(5):632-639. DOI: 10.2337/dc16-2427. PMID: 28289041.
- Hendricks EJ, Srisurapanont M, Schmidt SL, Haggard M, Souter S, Mitchell CL, De Marco DG, Hendricks MJ, Istratiy Y, Greenway FL. Addiction potential of phentermine prescribed during long-term treatment of obesity. Int J Obes (Lond). 2014;38(2):292-8. DOI: 10.1038/ijo.2013.74. PMID: 23736363.
- Hendricks EJ, Greenway FL. A study of abrupt phentermine cessation in patients in a weight management program. Am J Ther. 2011;18(4):292-9. DOI: 10.1097/MJT.0b013e3181d070d7. PMID: 20592662.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring).2011;19(12):2351-60. DOI: 10.1038/oby.2011.94. PMID: 21527891.
- Hendricks EJ, Rothman RB. Phentermine therapy for obesity does not elevate blood pressure. Diabetes Obes Metab. 2011;13(10):963-4. DOI: 10.1111/j.1463-1326.2011.01435.x. PMID: 21896124.
- Garvey WT, Ryan DH, Look M, Gadde KM, Allison DB, Peterson CA, Schwiers M, Day WW, Bowden CH. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr.2012;95(2):297-308. DOI: 10.3945/ajcn.111.024927. PMID: 22158731.
- Ritchey ME, Harding A, Hunter S, Peterson C, Sager PT, Kowey PR, Nguyen L, Thomas S, Cainzos-Achirica M, Rothman KJ, Andrews EB, Anthony MS. Cardiovascular Safety During and After Use of Phentermine and Topiramate. J Clin Endocrinol Metab. 2019;104(2):513-522. DOI: 10.1210/jc.2018-01010. PMID: 30247575.
- Gorelik E, Gorelik B, Masarwa R, Perlman A, Hirsh-Raccah B, Matok I. The cardiovascular safety of antiobesity drugs-analysis of signals in the FDA Adverse Event Report System Database. Int J Obes (Lond).2020;44(5):1021-1027. DOI: 10.1038/s41366-020-0544-4. PMID: 32152496.
- Burland WL. A review of experience with fenfluramine, in Obesity in Perspective. Fogarty International Center Series of Preventive Medicine (GA Bray, Ed.). Section IV, Vol 2, Part 2, Capter 55. 1976, U.S. Govt. Print. Off.: Washington DC. p. 429-440 (DHEW Publicatioin #75-708).
- Garattini S, Bizzi A, Codegoni AM, Caccia S, Mennini T. Progress report on the anorexia induced by drugs believed to mimic some of the effects of serotonin on the central nervous system. Am J Clin Nutr. 1992;55(1 Suppl):160S-166S. DOI: 10.1093/ajcn/55.1.160s. PMID: 1728827.
- van Galen KA, Ter Horst KW, Serlie MJ. Serotonin, food intake, and obesity. Obes Rev. 2021;22(7):e13210. DOI: 10.1111/obr.13210. PMID: 33559362.
- Guy-Grand B, Apfelbaum M, Crepaldi G, Gries A, Lefebvre P, Turner P. International trial of long-term dexfenfluramine in obesity. Lancet. 1989;2(8672):1142-5. DOI: 10.1016/s0140-6736(89)91499-2. PMID: 2572857.
- Douglas JG, Munro JF, Kitchin AH, Muir AL, Proudfoot AT. Pulmonary hypertension and fenfluramine. Br Med J (Clin Res Ed). 1981;283(6296):881-3. DOI: 10.1136/bmj.283.6296.881. PMID: 6793158.
- Brenot F, Herve P, Petitpretz P, Parent F, Duroux P, Simonneau G. Primary pulmonary hypertension and fenfluramine use. Br Heart J. 1993;70(6):537-41. DOI: 10.1136/hrt.70.6.537. PMID: 8280518.
- Weintraub M, Hasday JD, Mushlin AI, Lockwood DH. A double-blind clinical trial in weight control. Use of fenfluramine and phentermine alone and in combination. Arch Intern Med. 1984;144(6):1143-8. PMID: 6375610.
- Weintraub M, Sundaresan PR, Madan M, Schuster B, Balder A, Lasagna L, Cox C. Long-term weight control study. I (weeks 0 to 34). The enhancement of behavior modification, caloric restriction, and exercise by fenfluramine plus phentermine versus placebo. Clin Pharmacol Ther. 1992;51(5):586-94. DOI: 10.1038/clpt.1992.69. PMID: 1587072.
- Stafford RS, Radley DC. National trends in antiobesity medication use. Arch Intern Med. 2003;163(9):1046-50. DOI: 10.1001/archinte.163.9.1046. PMID: 12742801.
- Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337(9):581-8. DOI: 10.1056/NEJM199708283370901. PMID: 9271479.
- Ryan DH, Bray GA, Helmcke F, Sander G, Volaufova J, Greenway F, Subramaniam P, Glancy DL. Serial echocardiographic and clinical evaluation of valvular regurgitation before, during, and after treatment with fenfluramine or dexfenfluramine and mazindol or phentermine. Obes Res. 1999;7(4):313-22. DOI: 10.1002/j.1550-8528.1999.tb00414.x. PMID: 10440587.
- King DJ, Devaney N. Clinical pharmacology of sibutramine hydrochloride (BTS 54524), a new antidepressant, in healthy volunteers. Br J Clin Pharmacol. 1988;26(5):607-11. DOI: 10.1111/j.1365-2125.1988.tb05303.x. PMID: 3207566.
- Weintraub M, Rubio A, Golik A, Byrne L, Scheinbaum ML. Sibutramine in weight control: a dose-ranging, efficacy study. Clin Pharmacol Ther. 1991;50(3):330-7. DOI: 10.1038/clpt.1991.144. PMID: 1914367.
- Bray GA, Blackburn GL, Ferguson JM, Greenway FL, Jain AK, Mendel CM, Mendels J, Ryan DH, Schwartz SL, Scheinbaum ML, Seaton TB. Sibutramine produces dose-related weight loss. Obes Res. 1999;7(2):189-98. DOI: 10.1002/j.1550-8528.1999.tb00701.x. PMID: 10102256.
- James WP, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, Torp-Pedersen C, Sharma AM, Shepherd GM, Rode RA, Renz CL, Investigators S. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010;363(10):905-17. DOI: 10.1056/NEJMoa1003114. PMID: 20818901.
- Haslam D. Sibutramine: gone, but not forgotten. Practical Diabetes International. 2010;27(3):96-97. DOI: 10.1002/pdi.1453.
- Caterson ID, Finer N, Coutinho W, Van Gaal LF, Maggioni AP, Torp-Pedersen C, Sharma AM, Legler UF, Shepherd GM, Rode RA, Perdok RJ, Renz CL, James WP, Investigators S. Maintained intentional weight loss reduces cardiovascular outcomes: results from the Sibutramine Cardiovascular OUTcomes (SCOUT) trial. Diabetes Obes Metab. 2012;14(6):523-30. DOI: 10.1111/j.1463-1326.2011.01554.x. PMID: 22192338.
- Astrup A, Meier DH, Mikkelsen BO, Villumsen JS, Larsen TM. Weight loss produced by tesofensine in patients with Parkinson's or Alzheimer's disease. Obesity (Silver Spring). 2008;16(6):1363-9. DOI: 10.1038/oby.2008.56. PMID: 18356831.
- Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9653):1906-1913. DOI: 10.1016/S0140-6736(08)61525-1. PMID: 18950853.
- Huynh KD, Klose MC, Krogsgaard K, Drejer J, Byberg S, Madsbad S, Magkos F, Aharaz A, Edsberg B, Tfelt-Hansen J, Astrup A, Feldt-Rasmussen U. Weight Loss, Improved Body Composition and Fat Distribution by Tesomet in Acquired Hypothalamic Obesity. Journal of the Endocrine Society. 2021;5(Supplement_1):A64-A65. DOI: 10.1210/jendso/bvab048.130.
- Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, Bays H, Shanahan WR. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-56. DOI: 10.1056/NEJMoa0909809. PMID: 20647200.
- Smith SR, Garvey WT, Greenway FL, Zhou S, Fain R, Pilson R, Fujioka K, Aronne LJ. Coadministration of lorcaserin and phentermine for weight management: A 12-week, randomized, pilot safety study. Obesity (Silver Spring). 2017;25(5):857-865. DOI: 10.1002/oby.21811. PMID: 28440045.
- Bohula EA, Wiviott SD, McGuire DK, Inzucchi SE, Kuder J, Im K, Fanola CL, Qamar A, Brown C, Budaj A, Garcia-Castillo A, Gupta M, Leiter LA, Weissman NJ, White HD, Patel T, Francis B, Miao W, Perdomo C, Dhadda S, Bonaca MP, Ruff CT, Keech AC, Smith SR, Sabatine MS, Scirica BM, Committee C-TS, Investigators. Cardiovascular Safety of Lorcaserin in Overweight or Obese Patients. N Engl J Med.2018;379(12):1107-1117. DOI: 10.1056/NEJMoa1808721. PMID: 30145941.
- Sharretts J, Galescu O, Gomatam S, Andraca-Carrera E, Hampp C, Yanoff L. Cancer Risk Associated with Lorcaserin - The FDA's Review of the CAMELLIA-TIMI 61 Trial. N Engl J Med. 2020;383(11):1000-1002. DOI: 10.1056/NEJMp2003873. PMID: 32905671.
- Itowi N, Nagai K, Nakagawa H, Watanabe T, Wada H. Changes in the feeding behavior of rats elicited by histamine infusion. Physiol Behav. 1988;44(2):221-6. DOI: 10.1016/0031-9384(88)90142-4. PMID: 3237828.
- Lecklin A, Tuomisto L, MacDonald E. Metoprine, an inhibitor of histamine N-methyltransferase but not catechol-O-methyltransferase, suppresses feeding in sated and in food deprived rats. Methods Find Exp Clin Pharmacol.1995;17(1):47-52. PMID: 7542717.
- Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28(3):519-26. DOI: 10.1038/sj.npp.1300027. PMID: 12629531.
- Ratliff JC, Barber JA, Palmese LB, Reutenauer EL, Tek C. Association of prescription H1 antihistamine use with obesity: results from the National Health and Nutrition Examination Survey. Obesity (Silver Spring).2010;18(12):2398-400. DOI: 10.1038/oby.2010.176. PMID: 20706200.
- Barak N, Greenway FL, Fujioka K, Aronne LJ, Kushner RF. Effect of histaminergic manipulation on weight in obese adults: a randomized placebo controlled trial. Int J Obes (Lond). 2008;32(10):1559-65. DOI: 10.1038/ijo.2008.135. PMID: 18698316.
- Stoa-Birketvedt G, Lovhaug N, Vonen B, Florholmen J. H2-receptor antagonist reduces food intake and weight gain in rats by non-gastric acid secretory mechanisms. Acta Physiol Scand. 1997;161(4):489-94. DOI: 10.1046/j.1365-201X.1997.00249.x. PMID: 9429656.
- Stoa-Birketvedt G, Paus PN, Ganss R, Ingebretsen OC, Florholmen J. Cimetidine reduces weight and improves metabolic control in overweight patients with type 2 diabetes. Int J Obes Relat Metab Disord. 1998;22(11):1041-5. DOI: 10.1038/sj.ijo.0800721. PMID: 9822940.
- Stoa-Birketvedt G. Effect of cimetidine suspension on appetite and weight in overweight subjects. BMJ.1993;306(6885):1091-3. DOI: 10.1136/bmj.306.6885.1091. PMID: 8388285.
- Rasmussen MH, Andersen T, Breum L, Gotzsche PC, Hilsted J. Cimetidine suspension as adjuvant to energy restricted diet in treating obesity. BMJ. 1993;306(6885):1093-6. DOI: 10.1136/bmj.306.6885.1093. PMID: 8388286.
- Garrow J. Does cimetidine cause weight loss? BMJ. 1993;306(6885):1084. DOI: 10.1136/bmj.306.6885.1084. PMID: 8495153.
- Smith R. Research misconduct: the poisoning of the well. J R Soc Med. 2006;99(5):232-7. DOI: 10.1258/jrsm.99.5.232. PMID: 16672756.
- Florholmen J, Leeds AR. Research misconduct: Dr Grethe Stoa Birketvedt not guilty of scientific misconduct. J R Soc Med. 2010;103(6):214. DOI: 10.1258/jrsm.2010.10k023. PMID: 20513898.
- Birketvedt GS, Thom E, Bernersen B, Florholmen J. Combination of diet, exercise and intermittent treatment of cimetidine on body weight and maintenance of weight loss. A 42 months follow-up study. Med Sci Monit.2000;6(4):699-703. PMID: 11208394.
- Gu XJ, Chen R, Sun CH, Zheng W, Yang XH, Wang SB, Ungvari GS, Ng CH, Golenkov A, Lok GKI, Li L, Chow IHI, Wang F, Xiang YT. Effect of adjunctive ranitidine for antipsychotic-induced weight gain: A systematic review of randomized placebo-controlled trials. J Int Med Res. 2018;46(1):22-32. DOI: 10.1177/0300060517716783. PMID: 28718688.
- Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764-74. DOI: 10.1038/nrd1821. PMID: 16121130.
- Li Z, Maglione M, Tu W, Mojica W, Arterburn D, Shugarman LR, Hilton L, Suttorp M, Solomon V, Shekelle PG, Morton SC. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med. 2005;142(7):532-46. DOI: 10.7326/0003-4819-142-7-200504050-00012. PMID: 15809465.
- Avenell A, Brown TJ, McGee MA, Campbell MK, Grant AM, Broom J, Jung RT, Smith WC. What interventions should we add to weight reducing diets in adults with obesity? A systematic review of randomized controlled trials of adding drug therapy, exercise, behaviour therapy or combinations of these interventions. J Hum Nutr Diet. 2004;17(4):293-316. DOI: 10.1111/j.1365-277X.2004.00530.x. PMID: 15250841.
- Darga LL, Carroll-Michals L, Botsford SJ, Lucas CP. Fluoxetine's effect on weight loss in obese subjects. Am J Clin Nutr. 1991;54(2):321-5. DOI: 10.1093/ajcn/54.2.321. PMID: 1858696.
- Goldstein DJ, Rampey AH, Jr., Roback PJ, Wilson MG, Hamilton SH, Sayler ME, Tollefson GD. Efficacy and safety of long-term fluoxetine treatment of obesity--maximizing success. Obes Res. 1995;3 Suppl 4:481S-490S. DOI: 10.1002/j.1550-8528.1995.tb00216.x. PMID: 8697047.
- Wadden TA, Bartlett SJ, Foster GD, Greenstein RA, Wingate BJ, Stunkard AJ, Letizia KA. Sertraline and relapse prevention training following treatment by very-low-calorie diet: a controlled clinical trial. Obes Res.1995;3(6):549-57. DOI: 10.1002/j.1550-8528.1995.tb00189.x. PMID: 8653531.
- Sayler ME, Goldstein DJ, Roback PJ, Atkinson RL. Evaluating success of weight loss programs, with an application to fluoxetine weight reduction clinical trial data. Int J Obes Relat Metab Disord. 1994;18(11):742-51. PMID: 7866474.
- Jain AK, Kaplan RA, Gadde KM, Wadden TA, Allison DB, Brewer ER, Leadbetter RA, Richard N, Haight B, Jamerson BD, Buaron KS, Metz A. Bupropion SR vs. placebo for weight loss in obese patients with depressive symptoms. Obes Res. 2002;10(10):1049-56. DOI: 10.1038/oby.2002.142. PMID: 12376586.
- Anderson JW, Greenway FL, Fujioka K, Gadde KM, McKenney J, O'Neil PM. Bupropion SR enhances weight loss: a 48-week double-blind, placebo- controlled trial. Obes Res. 2002;10(7):633-41. DOI: 10.1038/oby.2002.86. PMID: 12105285.
- Bray GA, Hollander P, Klein S, Kushner R, Levy B, Fitchet M, Perry BH. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res. 2003;11(6):722-33. DOI: 10.1038/oby.2003.102. PMID: 12805393.
- Wilding J, Van Gaal L, Rissanen A, Vercruysse F, Fitchet M, Group O-S. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int J Obes Relat Metab Disord. 2004;28(11):1399-410. DOI: 10.1038/sj.ijo.0802783. PMID: 15486569.
- Toplak H, Hamann A, Moore R, Masson E, Gorska M, Vercruysse F, Sun X, Fitchet M. Efficacy and safety of topiramate in combination with metformin in the treatment of obese subjects with type 2 diabetes: a randomized, double-blind, placebo-controlled study. Int J Obes (Lond). 2007;31(1):138-46. DOI: 10.1038/sj.ijo.0803382. PMID: 16703004.
- Gadde KM, Franciscy DM, Wagner HR, 2nd, Krishnan KR. Zonisamide for weight loss in obese adults: a randomized controlled trial. JAMA. 2003;289(14):1820-5. DOI: 10.1001/jama.289.14.1820. PMID: 12684361.
- Gadde KM, Kopping MF, Wagner HR, 2nd, Yonish GM, Allison DB, Bray GA. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-64. DOI: 10.1001/2013.jamainternmed.99. PMID: 23147455.
- Nogueiras R, Romero-Pico A, Vazquez MJ, Novelle MG, Lopez M, Dieguez C. The opioid system and food intake: homeostatic and hedonic mechanisms. Obes Facts. 2012;5(2):196-207. DOI: 10.1159/000338163. PMID: 22647302.
- Holtzman SG. Suppression of appetitive behavior in the rat by naloxone: lack of effect of prior morphine dependence. Life Sci. 1979;24(3):219-26. DOI: 10.1016/0024-3205(79)90222-4. PMID: 570626.
- Atkinson RL. Naloxone decreases food intake in obese humans. J Clin Endocrinol Metab. 1982;55(1):196-8. DOI: 10.1210/jcem-55-1-196. PMID: 7042740.
- Atkinson RL, Berke LK, Drake CR, Bibbs ML, Williams FL, Kaiser DL. Effects of long-term therapy with naltrexone on body weight in obesity. Clin Pharmacol Ther. 1985;38(4):419-22. DOI: 10.1038/clpt.1985.197. PMID: 4042525.
- Hatsukami DK, Mitchell JE, Morley JE, Morgan SF, Levine AS. Effect of naltrexone on mood and cognitive functioning among overweight men. Biol Psychiatry. 1986;21(3):293-300. DOI: 10.1016/0006-3223(86)90050-8. PMID: 3947710.
- Maggio CA, Presta E, Bracco EF, Vasselli JR, Kissileff HR, Pfohl DN, Hashim SA. Naltrexone and human eating behavior: a dose-ranging inpatient trial in moderately obese men. Brain Res Bull. 1985;14(6):657-61. DOI: 10.1016/0361-9230(85)90115-7. PMID: 3896411.
- Malcolm R, O'Neil PM, Sexauer JD, Riddle FE, Currey HS, Counts C. A controlled trial of naltrexone in obese humans. Int J Obes. 1985;9(5):347-53. PMID: 3908352.
- Mitchell JE, Morley JE, Levine AS, Hatsukami D, Gannon M, Pfohl D. High-dose naltrexone therapy and dietary counseling for obesity. Biol Psychiatry. 1987;22(1):35-42. DOI: 10.1016/0006-3223(87)90127-2. PMID: 3790639.
- Novi RF, Lamberto M, Visconti P, Maurino M, Ardizzone A, Mantovan M, Meluzzi A, Francesetti G, Molinatti GM. [The role of opioid antagonists in the treatment of obesity. Results of a clinical trial with naltrexone]. Minerva Endocrinol. 1990;15(2):121-3. PMID: 2098653.
- de Zwaan M, Mitchell JE. Opiate antagonists and eating behavior in humans: a review. J Clin Pharmacol.1992;32(12):1060-72. PMID: 1487543.
- Pfohl DN, Allen JI, Atkinson RL, Knopman DS, Malcolm RJ, Mitchell JE, Morley JE. Naltrexone hydrochloride (Trexan): a review of serum transaminase elevations at high dosage. NIDA Res Monogr. 1986;67:66-72. PMID: 3092099.
- Volkow ND, Wang GJ, Maynard L, Jayne M, Fowler JS, Zhu W, Logan J, Gatley SJ, Ding YS, Wong C, Pappas N. Brain dopamine is associated with eating behaviors in humans. Int J Eat Disord. 2003;33(2):136-42. DOI: 10.1002/eat.10118. PMID: 12616579.
- Terry P, Gilbert DB, Cooper SJ. Dopamine receptor subtype agonists and feeding behavior. Obes Res. 1995;3 Suppl 4:515S-523S. DOI: 10.1002/j.1550-8528.1995.tb00221.x. PMID: 8697052.
- Hoebel BG, Hernandez L, Schwartz DH, Mark GP, Hunter GA. Microdialysis studies of brain norepinephrine, serotonin, and dopamine release during ingestive behavior. Theoretical and clinical implications. Ann N Y Acad Sci. 1989;575:171-91; discussion 192-3. DOI: 10.1111/j.1749-6632.1989.tb53242.x. PMID: 2699187.
- Astrup A, Greenway FL, Ling W, Pedicone L, Lachowicz J, Strader CD, Kwan R, Ecopipam Obesity Study G. Randomized controlled trials of the D1/D5 antagonist ecopipam for weight loss in obese subjects. Obesity (Silver Spring). 2007;15(7):1717-31. DOI: 10.1038/oby.2007.205. PMID: 17636090.
- Cincotta AH, Meier AH. Bromocriptine (Ergoset) reduces body weight and improves glucose tolerance in obese subjects. Diabetes Care. 1996;19(6):667-70. DOI: 10.2337/diacare.19.6.667. PMID: 8725871.
- Andersen IB, Andreassen M, Krogh J. The effect of dopamine agonists on metabolic variables in adults with type 2 diabetes: A systematic review with meta analysis and trial sequential analysis of randomized clinical trials. Diabetes Obes Metab. 2021;23(1):58-67. DOI: 10.1111/dom.14183. PMID: 32869474.
- Keche Y. Bromocriptine mesylate: Food and Drug Administration approved new approach in therapy of non-insulin dependant diabetes mellitus with poor glycemic control. J Pharm Bioallied Sci. 2010;2(2):148-50. DOI: 10.4103/0975-7406.67000. PMID: 21814451.
- Di Marzo V, Deutsch DG. Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol Dis.1998;5(6 Pt B):386-404. DOI: 10.1006/nbdi.1998.0214. PMID: 9974173.
- Gelfand EV, Cannon CP. Rimonabant: a selective blocker of the cannabinoid CB1 receptors for the management of obesity, smoking cessation and cardiometabolic risk factors. Expert Opin Investig Drugs.2006;15(3):307-15. DOI: 10.1517/13543784.15.3.307. PMID: 16503766.
- Kirkham TC. Endocannabinoids in the regulation of appetite and body weight. Behav Pharmacol. 2005;16(5-6):297-313. DOI: 10.1097/00008877-200509000-00004. PMID: 16148436.
- Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27(1):73-100. DOI: 10.1210/er.2005-0009. PMID: 16306385.
- Bensaid M, Gary-Bobo M, Esclangon A, Maffrand JP, Le Fur G, Oury-Donat F, Soubrie P. The cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol Pharmacol. 2003;63(4):908-14. DOI: 10.1124/mol.63.4.908. PMID: 12644592.
- Juan-Pico P, Fuentes E, Bermudez-Silva FJ, Javier Diaz-Molina F, Ripoll C, Rodriguez de Fonseca F, Nadal A. Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium.2006;39(2):155-62. DOI: 10.1016/j.ceca.2005.10.005. PMID: 16321437.
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S, Group RI-ES. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365(9468):1389-97. DOI: 10.1016/S0140-6736(05)66374-X. PMID: 15836887.
- Rosenstock J, Hollander P, Chevalier S, Iranmanesh A, Group SS. SERENADE: the Study Evaluating Rimonabant Efficacy in Drug-naive Diabetic Patients: effects of monotherapy with rimonabant, the first selective CB1 receptor antagonist, on glycemic control, body weight, and lipid profile in drug-naive type 2 diabetes. Diabetes Care. 2008;31(11):2169-76. DOI: 10.2337/dc08-0386. PMID: 18678611.
- Hollander PA, Amod A, Litwak LE, Chaudhari U, Group AS. Effect of rimonabant on glycemic control in insulin-treated type 2 diabetes: the ARPEGGIO trial. Diabetes Care. 2010;33(3):605-7. DOI: 10.2337/dc09-0455. PMID: 20009090.
- O'Leary DH, Reuwer AQ, Nissen SE, Despres JP, Deanfield JE, Brown MW, Zhou R, Zabbatino SM, Job B, Kastelein JJ, Visseren FL, investigators A. Effect of rimonabant on carotid intima-media thickness (CIMT) progression in patients with abdominal obesity and metabolic syndrome: the AUDITOR Trial. Heart.2011;97(14):1143-50. DOI: 10.1136/hrt.2011.223446. PMID: 21610270.
- Nissen SE, Nicholls SJ, Wolski K, Rodes-Cabau J, Cannon CP, Deanfield JE, Despres JP, Kastelein JJ, Steinhubl SR, Kapadia S, Yasin M, Ruzyllo W, Gaudin C, Job B, Hu B, Bhatt DL, Lincoff AM, Tuzcu EM, Investigators S. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA. 2008;299(13):1547-60. DOI: 10.1001/jama.299.13.1547. PMID: 18387931.
- Topol EJ, Bousser MG, Fox KA, Creager MA, Despres JP, Easton JD, Hamm CW, Montalescot G, Steg PG, Pearson TA, Cohen E, Gaudin C, Job B, Murphy JH, Bhatt DL, Investigators C. Rimonabant for prevention of cardiovascular events (CRESCENDO): a randomised, multicentre, placebo-controlled trial. Lancet.2010;376(9740):517-23. DOI: 10.1016/S0140-6736(10)60935-X. PMID: 20709233.
- Lu D, Dopart R, Kendall DA. Controlled downregulation of the cannabinoid CB1 receptor provides a promising approach for the treatment of obesity and obesity-derived type 2 diabetes. Cell Stress Chaperones.2016;21(1):1-7. DOI: 10.1007/s12192-015-0653-5. PMID: 26498013.
- Berlin I, Crespo-Laumonnier B, Turpin G, Puech AJ. The alpha-2 adrenoceptor antagonist yohimbine does not facilitate weight loss but blocks adrenaline induced platelet aggregation in obese subjects. Therapie.1989;44(4):301. PMID: 2595649.
- Heymsfield SB, Smith B, Dahle J, Kennedy S, Fearnbach N, Thomas DM, Bosy-Westphal A, Muller MJ. Resting Energy Expenditure: From Cellular to Whole-Body Level, a Mechanistic Historical Perspective. Obesity (Silver Spring). 2021;29(3):500-511. DOI: 10.1002/oby.23090. PMID: 33624441.
- Jimenez-Munoz CM, Lopez M, Albericio F, Makowski K. Targeting Energy Expenditure-Drugs for Obesity Treatment. Pharmaceuticals (Basel). 2021;14(5). DOI: 10.3390/ph14050435. PMID: 34066399.
- Astrup A, Lundsgaard C, Madsen J, Christensen NJ. Enhanced thermogenic responsiveness during chronic ephedrine treatment in man. Am J Clin Nutr. 1985;42(1):83-94. DOI: 10.1093/ajcn/42.1.83. PMID: 4014068.
- Malchow-Moller A, Larsen S, Hey H, Stokholm KH, Juhl E, Quaade F. Ephedrine as an anorectic: the story of the 'Elsinore pill'. Int J Obes. 1981;5(2):183-7. PMID: 7228474.
- Astrup A, Toubro S, Cannon S, Hein P, Madsen J. Thermogenic synergism between ephedrine and caffeine in healthy volunteers: a double-blind, placebo-controlled study. Metabolism. 1991;40(3):323-9. DOI: 10.1016/0026-0495(91)90117-f. PMID: 2000046.
- Dulloo AG. Ephedrine, xanthines and prostaglandin-inhibitors: actions and interactions in the stimulation of thermogenesis. Int J Obes Relat Metab Disord. 1993;17 Suppl 1:S35-40. PMID: 8384178.
- Bray GA, Greenway FL. Current and potential drugs for treatment of obesity. Endocr Rev. 1999;20(6):805-75. DOI: 10.1210/edrv.20.6.0383. PMID: 10605627.
- Astrup A, Breum L, Toubro S, Hein P, Quaade F. The effect and safety of an ephedrine/caffeine compound compared to ephedrine, caffeine and placebo in obese subjects on an energy restricted diet. A double blind trial. Int J Obes Relat Metab Disord. 1992;16(4):269-77. PMID: 1318281.
- Breum L, Pedersen JK, Ahlstrom F, Frimodt-Moller J. Comparison of an ephedrine/caffeine combination and dexfenfluramine in the treatment of obesity. A double-blind multi-centre trial in general practice. Int J Obes Relat Metab Disord. 1994;18(2):99-103. PMID: 8148931.
- Lee MR. The history of Ephedra (ma-huang). J R Coll Physicians Edinb. 2011;41(1):78-84. DOI: 10.4997/JRCPE.2011.116. PMID: 21365072.
- Elhadef K, Smaoui S, Fourati M, Ben Hlima H, Chakchouk Mtibaa A, Sellem I, Ennouri K, Mellouli L. A Review on Worldwide Ephedra History and Story: From Fossils to Natural Products Mass Spectroscopy Characterization and Biopharmacotherapy Potential. Evid Based Complement Alternat Med.2020;2020:1540638. DOI: 10.1155/2020/1540638. PMID: 32419789.
- Bass IS, Young AL. Dietary Supplement Health and Education Act : A Legislative History and Analysis. 1996, Washington D.C.: Food and Drug Law Institute. 319 pages.
- Boozer CN, Daly PA, Homel P, Solomon JL, Blanchard D, Nasser JA, Strauss R, Meredith T. Herbal ephedra/caffeine for weight loss: a 6-month randomized safety and efficacy trial. Int J Obes Relat Metab Disord.2002;26(5):593-604. DOI: 10.1038/sj.ijo.0802023. PMID: 12032741.
- Shekelle PG, Hardy ML, Morton SC, Maglione M, Mojica WA, Suttorp MJ, Rhodes SL, Jungvig L, Gagne J. Efficacy and safety of ephedra and ephedrine for weight loss and athletic performance: a meta-analysis. JAMA.2003;289(12):1537-45. DOI: 10.1001/jama.289.12.1537. PMID: 12672771.
- Jequier E, Munger R, Felber JP. Thermogenic effects of various beta-adrenoceptor agonists in humans: their potential usefulness in the treatment of obesity. Am J Clin Nutr. 1992;55(1 Suppl):249S-251S. DOI: 10.1093/ajcn/55.1.249s. PMID: 1345888.
- Stock MJ, Rothwell NJ. The role of brown fat in diet-induced thermogenesis. Int J Vitam Nutr Res.1986;56(2):205-10. PMID: 3460973.
- Strosberg AD. Structure and function of the beta 3-adrenergic receptor. Annu Rev Pharmacol Toxicol.1997;37:421-50. DOI: 10.1146/annurev.pharmtox.37.1.421. PMID: 9131260.
- Yen TT. Antiobesity and antidiabetic beta-agonists: lessons learned and questions to be answered. Obes Res.1994;2(5):472-80. DOI: 10.1002/j.1550-8528.1994.tb00095.x. PMID: 16353599.
- Arch JR, Wilson S. Prospects for beta 3-adrenoceptor agonists in the treatment of obesity and diabetes. Int J Obes Relat Metab Disord. 1996;20(3):191-9. PMID: 8653138.
- Connacher AA, Bennet WM, Jung RT, Rennie MJ. Metabolic effects of three weeks administration of the beta-adrenoceptor agonist BRL 26830A. Int J Obes Relat Metab Disord. 1992;16(9):685-94. PMID: 1356939.
- Cawthorne MA, Sennitt MV, Arch JR, Smith SA. BRL 35135, a potent and selective atypical beta-adrenoceptor agonist. Am J Clin Nutr. 1992;55(1 Suppl):252S-257S. DOI: 10.1093/ajcn/55.1.252s. PMID: 1345889.
- Connacher AA, Jung RT, Mitchell PE. Weight loss in obese subjects on a restricted diet given BRL 26830A, a new atypical beta adrenoceptor agonist. Br Med J (Clin Res Ed). 1988;296(6631):1217-20. DOI: 10.1136/bmj.296.6631.1217. PMID: 2898268.
- Connacher AA, Lakie M, Powers N, Elton RA, Walsh EG, Jung RT. Tremor and the anti-obesity drug BRL 26830A. Br J Clin Pharmacol. 1990;30(4):613-5. DOI: 10.1111/j.1365-2125.1990.tb03821.x. PMID: 1981320.
- MacLachlan M, Connacher AA, Jung RT. Psychological aspects of dietary weight loss and medication with the atypical beta agonist BRL 26830A in obese subjects. Int J Obes. 1991;15(1):27-35. PMID: 1672679.
- Mitchell TH, Ellis RD, Smith SA, Robb G, Cawthorne MA. Effects of BRL 35135, a beta-adrenoceptor agonist with novel selectivity, on glucose tolerance and insulin sensitivity in obese subjects. Int J Obes. 1989;13(6):757-66. PMID: 2695481.
- Goldberg GR, Prentice AM, Murgatroyd PR, Haines W, Tuersley MD. Effects on metabolic rate and fuel selection of a selective beta-3 agonist (ICI D7114) in healthy lean men. Int J Obes Relat Metab Disord.1995;19(9):625-31. PMID: 8574272.
- Henny C, Schutz Y, Buckert A, Meylan M, Jequier E, Felber JP. Thermogenic effect of the new beta-adrenoreceptor agonist Ro 16-8714 in healthy male volunteers. Int J Obes. 1987;11(5):473-83. PMID: 2892807.
- Weyer C, Tataranni PA, Snitker S, Danforth E, Jr., Ravussin E. Increase in insulin action and fat oxidation after treatment with CL 316,243, a highly selective beta3-adrenoceptor agonist in humans. Diabetes.1998;47(10):1555-61. DOI: 10.2337/diabetes.47.10.1555. PMID: 9753292.
- Redman LM, de Jonge L, Fang X, Gamlin B, Recker D, Greenway FL, Smith SR, Ravussin E. Lack of an effect of a novel beta3-adrenoceptor agonist, TAK-677, on energy metabolism in obese individuals: a double-blind, placebo-controlled randomized study. J Clin Endocrinol Metab. 2007;92(2):527-31. DOI: 10.1210/jc.2006-1740. PMID: 17118998.
- van Baak MA, Hul GB, Toubro S, Astrup A, Gottesdiener KM, DeSmet M, Saris WH. Acute effect of L-796568, a novel beta 3-adrenergic receptor agonist, on energy expenditure in obese men. Clin Pharmacol Ther.2002;71(4):272-9. DOI: 10.1067/mcp.2002.122527. PMID: 11956510.
- Arch JR. The discovery of drugs for obesity, the metabolic effects of leptin and variable receptor pharmacology: perspectives from beta3-adrenoceptor agonists. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(2):225-40. DOI: 10.1007/s00210-008-0271-1. PMID: 18612674.
- O'Mara AE, Johnson JW, Linderman JD, Brychta RJ, McGehee S, Fletcher LA, Fink YA, Kapuria D, Cassimatis TM, Kelsey N, Cero C, Sater ZA, Piccinini F, Baskin AS, Leitner BP, Cai H, Millo CM, Dieckmann W, Walter M, Javitt NB, Rotman Y, Walter PJ, Ader M, Bergman RN, Herscovitch P, Chen KY, Cypess AM. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest.2020;130(5):2209-2219. DOI: 10.1172/JCI131126. PMID: 31961826.
- Zed CA, Harris GS, Harrison PJ, Robb GH. Anti-obesity activity of a novel β-adrenoceptor agonist (BRL 26830A) in diet restricted obese subjects. Int J Obes. 1985;9:231 (abstract).
- Abraham R, Zed C, Mitchell T, Parr J, Wynn V. The effect of a novel β-agonist BRL26830A on weight and protein loss in obese patients. Int J Obes. 1987;11:306A (Abstract).
- Smith SA, Cawthorne MA, Fay LC, McCullough DA, Mitchell TH. Effect of a novel β-adrenoceptor agonist on insulin sensitivity in lean healthy male volunteers. Diabetes. 1987;36:15A (Abstract).
- Chapman BJ, Farquahar DL, Galloway SM, Simpson GK, Munro JF. The effects of a new beta-adrenoceptor agonist BRL 26830A in refractory obesity. Int J Obes. 1988;12(2):119-23. PMID: 2898457.
- Smith SA, Sennitt MV, Cawthorne MA. BRL 35135: an orally active antihyperglycemic agent with weight reducing effects, in New Antidiabetic Drugs, C.J. Bailey and P.R. Flatt, Editors. 1990, Smith-Gordon and Company Limited London. p. 177–189.
- Toubro S, Astrup A. Randomised comparison of diets for maintaining obese subjects' weight after major weight loss: ad lib, low fat, high carbohydrate diet v fixed energy intake. BMJ. 1997;314(7073):29-34. DOI: 10.1136/bmj.314.7073.29. PMID: 9001476.
- Henny C, Buckert A, Schutz Y, Jequier E, Felber JP. Comparison of thermogenic activity induced by the new sympathomimetic Ro 16-8714 between normal and obese subjects. Int J Obes. 1988;12(3):227-36. PMID: 2899062.
- Haesler E, Golay A, Guzelhan C, Schutz Y, Hartmann D, Jequier E, Felber JP. Effect of a novel beta-adrenoceptor agonist (Ro 40-2148) on resting energy expenditure in obese women. Int J Obes Relat Metab Disord. 1994;18(5):313-22. PMID: 7914795.
- Wheeldon NM, McDevitt DG, McFarlane LC, Lipworth BJ. Beta-adrenoceptor subtypes mediating the metabolic effects of BRL 35135 in man. Clin Sci (Lond). 1994;86(3):331-7. DOI: 10.1042/cs0860331. PMID: 7908864.
- Larsen TM, Toubro S, van Baak MA, Gottesdiener KM, Larson P, Saris WH, Astrup A. Effect of a 28-d treatment with L-796568, a novel beta(3)-adrenergic receptor agonist, on energy expenditure and body composition in obese men. Am J Clin Nutr. 2002;76(4):780-8. DOI: 10.1093/ajcn/76.4.780. PMID: 12324291.
- Galgani JE, Ryan DH, Ravussin E. Effect of capsinoids on energy metabolism in human subjects. Br J Nutr.2010;103(1):38-42. DOI: 10.1017/S0007114509991358. PMID: 19671203.
- Corwin RL, Gibbs J, Smith GP. Increased food intake after type A but not type B cholecystokinin receptor blockade. Physiol Behav. 1991;50(1):255-8. DOI: 10.1016/0031-9384(91)90529-w. PMID: 1946726.
- Greenway FL, Bray GA. Cholecystokinin and satiety. Life Sci. 1977;21(6):769-72. DOI: 10.1016/0024-3205(77)90403-9. PMID: 916798.
- Kissileff HR, Gordon RJ, Thornton JC, Laferrere B, Albu J, Pi-Sunyer X, Geliebter A. Combined effects of cholecystokinin-8 and gastric distension on food intake in humans. Am J Physiol Regul Integr Comp Physiol.2019;317(1):R39-R48. DOI: 10.1152/ajpregu.00339.2018. PMID: 30916576.
- Moran TH, Sawyer TK, Seeb DH, Ameglio PJ, Lombard MA, McHugh PR. Potent and sustained satiety actions of a cholecystokinin octapeptide analogue. Am J Clin Nutr. 1992;55(1 Suppl):286S-290S. DOI: 10.1093/ajcn/55.1.286s. PMID: 1728841.
- Asin KE, Bednarz L, Nikkel AL, Gore PA, Jr., Nadzan AM. A-71623, a selective CCK-A receptor agonist, suppresses food intake in the mouse, dog, and monkey. Pharmacol Biochem Behav. 1992;42(4):699-704. DOI: 10.1016/0091-3057(92)90017-a. PMID: 1513850.
- Henke BR, Willson TM, Sugg EE, Croom DK, Dougherty RW, Jr., Queen KL, Birkemo LS, Ervin GN, Grizzle MK, Johnson MF, James MK. 3-(1H-indazol-3-ylmethyl)-1,5-benzodiazepines: CCK-A agonists that demonstrate oral activity as satiety agents. J Med Chem. 1996;39(14):2655-8. DOI: 10.1021/jm960249k. PMID: 8709093.
- Hamamura M, Leng G, Emson PC, Kiyama H. Electrical activation and c-fos mRNA expression in rat neurosecretory neurones after systemic administration of cholecystokinin. J Physiol. 1991;444:51-63. DOI: 10.1113/jphysiol.1991.sp018865. PMID: 1822561.
- Boosalis MG, Gemayel N, Lee A, Bray GA, Laine L, Cohen H. Cholecystokinin and satiety: effect of hypothalamic obesity and gastric bubble insertion. Am J Physiol. 1992;262(2 Pt 2):R241-4. DOI: 10.1152/ajpregu.1992.262.2.R241. PMID: 1539732.
- Veldhuis JD, Liem AY, South S, Weltman A, Weltman J, Clemmons DA, Abbott R, Mulligan T, Johnson ML, Pincus S, et al. Differential impact of age, sex steroid hormones, and obesity on basal versus pulsatile growth hormone secretion in men as assessed in an ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab.1995;80(11):3209-22. DOI: 10.1210/jcem.80.11.7593428. PMID: 7593428.
- Gertner JM. Effects of growth hormone on body fat in adults. Horm Res. 1993;40(1-3):10-5. DOI: 10.1159/000183761. PMID: 8300043.
- Bray GA. Calorigenic effect of human growth hormone in obesity. J Clin Endocrinol Metab. 1969;29(1):119-22. DOI: 10.1210/jcem-29-1-119. PMID: 5762314.
- Gertner JM. Growth hormone actions on fat distribution and metabolism. Horm Res. 1992;38 Suppl 2:41-3. DOI: 10.1159/000182592. PMID: 1292980.
- Bray GA, Raben MS, Londono J, Gallagher TF, Jr. Effects of triiodothyronine, growth hormone and anabolic steroids on nitrogen excretion and oxygen consumption of obese patients. J Clin Endocrinol Metab.1971;33(2):293-300. DOI: 10.1210/jcem-33-2-293. PMID: 4935638.
- Bengtsson BA, Eden S, Lonn L, Kvist H, Stokland A, Lindstedt G, Bosaeus I, Tolli J, Sjostrom L, Isaksson OG. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J Clin Endocrinol Metab.1993;76(2):309-17. DOI: 10.1210/jcem.76.2.8432773. PMID: 8432773.
- Brummer RJ, Lonn L, Kvist H, Grangard U, Bengtsson BA, Sjostrom L. Adipose tissue and muscle volume determination by computed tomography in acromegaly, before and 1 year after adenomectomy. Eur J Clin Invest. 1993;23(4):199-205. DOI: 10.1111/j.1365-2362.1993.tb00762.x. PMID: 8500511.
- Snyder DK, Clemmons DR, Underwood LE. Treatment of obese, diet-restricted subjects with growth hormone for 11 weeks: effects on anabolism, lipolysis, and body composition. J Clin Endocrinol Metab. 1988;67(1):54-61. DOI: 10.1210/jcem-67-1-54. PMID: 3379136.
- Thompson JL, Butterfield GE, Gylfadottir UK, Yesavage J, Marcus R, Hintz RL, Pearman A, Hoffman AR. Effects of human growth hormone, insulin-like growth factor I, and diet and exercise on body composition of obese postmenopausal women. J Clin Endocrinol Metab. 1998;83(5):1477-84. DOI: 10.1210/jcem.83.5.4826. PMID: 9589642.
- Johannsson G, Marin P, Lonn L, Ottosson M, Stenlof K, Bjorntorp P, Sjostrom L, Bengtsson BA. Growth hormone treatment of abdominally obese men reduces abdominal fat mass, improves glucose and lipoprotein metabolism, and reduces diastolic blood pressure. J Clin Endocrinol Metab. 1997;82(3):727-34. DOI: 10.1210/jcem.82.3.3809. PMID: 9062473.
- Rixhon M, Tichomirowa MA, Tamagno G, Daly AF, Beckers A. Current and future perspectives on recombinant growth hormone for the treatment of obesity. Expert Rev Endocrinol Metab. 2008;3(1):75-90. DOI: 10.1586/17446651.3.1.75. PMID: 30743787.
- Diabetes C, Complications Trial Research G, Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, Rand L, Siebert C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-86. DOI: 10.1056/NEJM199309303291401. PMID: 8366922.
- Ludwig DS, Ebbeling CB. The Carbohydrate-Insulin Model of Obesity: Beyond "Calories In, Calories Out". JAMA Intern Med. 2018;178(8):1098-1103. DOI: 10.1001/jamainternmed.2018.2933. PMID: 29971406.
- Hall KD. A review of the carbohydrate-insulin model of obesity. Eur J Clin Nutr. 2017;71(3):323-326. DOI: 10.1038/ejcn.2016.260. PMID: 28074888.
- Woods SC, Porte D, Jr., Bobbioni E, Ionescu E, Sauter JF, Rohner-Jeanrenaud F, Jeanrenaud B. Insulin: its relationship to the central nervous system and to the control of food intake and body weight. Am J Clin Nutr.1985;42(5 Suppl):1063-71. DOI: 10.1093/ajcn/42.5.1063. PMID: 3904396.
- Vogt MC, Bruning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism - from embryo to old age. Trends Endocrinol Metab. 2013;24(2):76-84. DOI: 10.1016/j.tem.2012.11.004. PMID: 23265947.
- Bray GA, Gallagher TF, Jr. Manifestations of hypothalamic obesity in man: a comprehensive investigation of eight patients and a reveiw of the literature. Medicine (Baltimore). 1975;54(4):301-30. DOI: 10.1097/00005792-197507000-00002. PMID: 1152672.
- Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59(3):719-809. DOI: 10.1152/physrev.1979.59.3.719. PMID: 379887.
- Lustig RH, Hinds PS, Ringwald-Smith K, Christensen RK, Kaste SC, Schreiber RE, Rai SN, Lensing SY, Wu S, Xiong X. Octreotide therapy of pediatric hypothalamic obesity: a double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2003;88(6):2586-92. DOI: 10.1210/jc.2002-030003. PMID: 12788859.
- Lustig RH, Greenway F, Velasquez-Mieyer P, Heimburger D, Schumacher D, Smith D, Smith W, Soler N, Warsi G, Berg W, Maloney J, Benedetto J, Zhu W, Hohneker J. A multicenter, randomized, double-blind, placebo-controlled, dose-finding trial of a long-acting formulation of octreotide in promoting weight loss in obese adults with insulin hypersecretion. Int J Obes (Lond). 2006;30(2):331-41. DOI: 10.1038/sj.ijo.0803074. PMID: 16158082.
- Gambineri A, Patton L, De Iasio R, Cantelli B, Cognini GE, Filicori M, Barreca A, Diamanti-Kandarakis E, Pagotto U, Pasquali R. Efficacy of octreotide-LAR in dieting women with abdominal obesity and polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90(7):3854-62. DOI: 10.1210/jc.2004-2490. PMID: 15827099.
- Huang Z, Wang W, Huang L, Guo L, Chen C. Suppression of Insulin Secretion in the Treatment of Obesity: A Systematic Review and Meta-Analysis. Obesity (Silver Spring). 2020;28(11):2098-2106. DOI: 10.1002/oby.22955. PMID: 33150747.
- Bray GA. Drug therapy for the obese patient, in The Obese Patient. Major Problems in Internal Medicine, Vol 9. 1976, W.B. Saunders Company: Philadelphia. p. 353-410.
- Fleigelman R, Fried GH. Metabolic effects of human chorionic gonadotropin (HCG) in rats. Proc Soc Exp Biol Med. 1970;135(2):317-9. DOI: 10.3181/00379727-135-35043. PMID: 5479992.
- Simeons AT. Chorionic gonadotropin in geriatrics. J Am Geriatr Soc. 1956;4(1):36-40. DOI: 10.1111/j.1532-5415.1956.tb01142.x. PMID: 13286062.
- Simeons AT. The action of chorionic gonadotrophin in the obese. Lancet. 1954;267(6845):946-7. DOI: 10.1016/s0140-6736(54)92556-8. PMID: 13213083.
- Simeons ATW. Pounds and Inches: A New Approach to Obesity. 6th revised edition ed. 2010, Roma: Arti grafichi Scalia: Popular Publishing.
- Carne S. The action of chorionic gonadotrophin in the obese. Lancet. 1961;2(7215):1282-4. DOI: 10.1016/s0140-6736(61)91142-4. PMID: 13876697.
- Greenway FL, Bray GA. Human chorionic gonadotropin (HCG) in the treatment of obesity: a critical assessment of the Simeons method. West J Med. 1977;127(6):461-3. PMID: 595585.
- Steinbrook R. Doctors Use of Obesity Treatment Debunked by FDA, in Los Angeles Times Sunday. 1986: Los Angeles.
- Lijesen GK, Theeuwen I, Assendelft WJ, Van Der Wal G. The effect of human chorionic gonadotropin (HCG) in the treatment of obesity by means of the Simeons therapy: a criteria-based meta-analysis. Br J Clin Pharmacol.1995;40(3):237-43. DOI: 10.1111/j.1365-2125.1995.tb05779.x. PMID: 8527285.
- Trudeau K. The Weight Loss Cure "They" Don't Want You to Know About. 2008, Elk Grove Village, IL: Alliance Publishing. 255 pages.
- Kevin Mark Trudeau. July 10, 2021]; Available from: https://en.wikipedia.org/wiki/Kevin_Trudeau.
- Schwartz MW, Seeley RJ, Zeltser LM, Drewnowski A, Ravussin E, Redman LM, Leibel RL. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr Rev. 2017;38(4):267-296. DOI: 10.1210/er.2017-00111. PMID: 28898979.
- Schwartz MW, Porte D, Jr. Diabetes, obesity, and the brain. Science. 2005;307(5708):375-9. DOI: 10.1126/science.1104344. PMID: 15662002.
- Flier JS. Clinical review 94: What's in a name? In search of leptin's physiologic role. J Clin Endocrinol Metab.1998;83(5):1407-13. DOI: 10.1210/jcem.83.5.4779. PMID: 9589630.
- Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O'Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903-8. DOI: 10.1038/43185. PMID: 9202122.
- Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398-401. DOI: 10.1038/32911. PMID: 9537324.
- Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med.1999;341(12):879-84. DOI: 10.1056/NEJM199909163411204. PMID: 10486419.
- Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568-75. DOI: 10.1001/jama.282.16.1568. PMID: 10546697.
- Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, Farid AR, Smith FJ, Campfield LA. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab.2000;85(11):4003-9. DOI: 10.1210/jcem.85.11.6955. PMID: 11095423.
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570-8. DOI: 10.1056/NEJMoa012437. PMID: 11856796.
- Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109(10):1345-50. DOI: 10.1172/JCI15001. PMID: 12021250.
- Park JY, Chong AY, Cochran EK, Kleiner DE, Haller MJ, Schatz DA, Gorden P. Type 1 diabetes associated with acquired generalized lipodystrophy and insulin resistance: the effect of long-term leptin therapy. J Clin Endocrinol Metab. 2008;93(1):26-31. DOI: 10.1210/jc.2007-1856. PMID: 17940115.
- Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53(1):27-35. DOI: 10.1007/s00125-009-1502-9. PMID: 19727665.
- Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, Gallagher D, Mayer L, Murphy E, Leibel RL. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115(12):3579-86. DOI: 10.1172/JCI25977. PMID: 16322796.
- Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002;87(5):2391-4. DOI: 10.1210/jcem.87.5.8628. PMID: 11994393.
- Rosenbaum M, Sy M, Pavlovich K, Leibel RL, Hirsch J. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J Clin Invest. 2008;118(7):2583-91. DOI: 10.1172/JCI35055. PMID: 18568078.
- Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110(8):1093-103. DOI: 10.1172/JCI15693. PMID: 12393845.
- Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351(10):987-97. DOI: 10.1056/NEJMoa040388. PMID: 15342807.
- Gloaguen I, Costa P, Demartis A, Lazzaro D, Di Marco A, Graziani R, Paonessa G, Chen F, Rosenblum CI, Van der Ploeg LH, Cortese R, Ciliberto G, Laufer R. Ciliary neurotrophic factor corrects obesity and diabetes associated with leptin deficiency and resistance. Proc Natl Acad Sci U S A. 1997;94(12):6456-61. DOI: 10.1073/pnas.94.12.6456. PMID: 9177239.
- A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTF Treatment Study Group. Neurology. 1996;46(5):1244-9. DOI: 10.1212/wnl.46.5.1244. PMID: 8628460.
- Ettinger MP, Littlejohn TW, Schwartz SL, Weiss SR, McIlwain HH, Heymsfield SB, Bray GA, Roberts WG, Heyman ER, Stambler N, Heshka S, Vicary C, Guler HP. Recombinant variant of ciliary neurotrophic factor for weight loss in obese adults: a randomized, dose-ranging study. JAMA. 2003;289(14):1826-32. DOI: 10.1001/jama.289.14.1826. PMID: 12684362.
- Erondu N, Gantz I, Musser B, Suryawanshi S, Mallick M, Addy C, Cote J, Bray G, Fujioka K, Bays H, Hollander P, Sanabria-Bohorquez SM, Eng W, Langstrom B, Hargreaves RJ, Burns HD, Kanatani A, Fukami T, MacNeil DJ, Gottesdiener KM, Amatruda JM, Kaufman KD, Heymsfield SB. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metab. 2006;4(4):275-82. DOI: 10.1016/j.cmet.2006.08.002. PMID: 17011500.
- Erondu N, Wadden T, Gantz I, Musser B, Nguyen AM, Bays H, Bray G, O'Neil PM, Basdevant A, Kaufman KD, Heymsfield SB, Amatruda JM. Effect of NPY5R antagonist MK-0557 on weight regain after very-low-calorie diet-induced weight loss. Obesity (Silver Spring). 2007;15(4):895-905. DOI: 10.1038/oby.2007.620. PMID: 17426325.
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257(5074):1248-51.
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480-4.
- Ayers KL, Glicksberg BS, Garfield AS, Longerich S, White JA, Yang P, Du L, Chittenden TW, Gulcher JR, Roy S, Fiedorek F, Gottesdiener K, Cohen S, North KE, Schadt EE, Li SD, Chen R, Van der Ploeg LHT. Melanocortin 4 Receptor Pathway Dysfunction in Obesity: Patient Stratification Aimed at MC4R Agonist Treatment. J Clin Endocrinol Metab. 2018;103(7):2601-2612. DOI: 10.1210/jc.2018-00258. PMID: 29726959.
- Krishna R, Gumbiner B, Stevens C, Musser B, Mallick M, Suryawanshi S, Maganti L, Zhu H, Han TH, Scherer L, Simpson B, Cosgrove D, Gottesdiener K, Amatruda J, Rolls BJ, Blundell J, Bray GA, Fujioka K, Heymsfield SB, Wagner JA, Herman GA. Potent and selective agonism of the melanocortin receptor 4 with MK-0493 does not induce weight loss in obese human subjects: energy intake predicts lack of weight loss efficacy. Clin Pharmacol Ther. 2009;86(6):659-66. DOI: 10.1038/clpt.2009.167. PMID: 19741604.
- Collet TH, Dubern B, Mokrosinski J, Connors H, Keogh JM, Mendes de Oliveira E, Henning E, Poitou-Bernert C, Oppert JM, Tounian P, Marchelli F, Alili R, Le Beyec J, Pepin D, Lacorte JM, Gottesdiener A, Bounds R, Sharma S, Folster C, Henderson B, O'Rahilly S, Stoner E, Gottesdiener K, Panaro BL, Cone RD, Clement K, Farooqi IS, Van der Ploeg LHT. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab. 2017;6(10):1321-1329. DOI: 10.1016/j.molmet.2017.06.015. PMID: 29031731.
- Clement K, van den Akker E, Argente J, Bahm A, Chung WK, Connors H, De Waele K, Farooqi IS, Gonneau-Lejeune J, Gordon G, Kohlsdorf K, Poitou C, Puder L, Swain J, Stewart M, Yuan G, Wabitsch M, Kuhnen P, Setmelanotide P, Investigators LPT. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8(12):960-970. DOI: 10.1016/S2213-8587(20)30364-8. PMID: 33137293.
- Glass AR, Swerdloff RS, Bray GA, Dahms WT, Atkinson RL. Low serum testosterone and sex-hormone-binding-globulin in massively obese men. J Clin Endocrinol Metab. 1977;45(6):1211-9. DOI: 10.1210/jcem-45-6-1211. PMID: 338622.
- Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(5):1715-1744. DOI: 10.1210/jc.2018-00229. PMID: 29562364.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab. 2011;96(8):2341-53. DOI: 10.1210/jc.2011-0118. PMID: 21646372.
- Kelly DM, Jones TH. Testosterone and obesity. Obes Rev. 2015;16(7):581-606. DOI: 10.1111/obr.12282. PMID: 25982085.
- Lapauw B, Kaufman JM. MANAGEMENT OF ENDOCRINE DISEASE: Rationale and current evidence for testosterone therapy in the management of obesity and its complications. Eur J Endocrinol. 2020;183(6):R167-R183. DOI: 10.1530/EJE-20-0394. PMID: 33105105.
- Evans DJ, Hoffmann RG, Kalkhoff RK, Kissebah AH. Relationship of androgenic activity to body fat topography, fat cell morphology, and metabolic aberrations in premenopausal women. J Clin Endocrinol Metab.1983;57(2):304-10. DOI: 10.1210/jcem-57-2-304. PMID: 6345569.
- Seidell JC, Bjorntorp P, Sjostrom L, Kvist H, Sannerstedt R. Visceral fat accumulation in men is positively associated with insulin, glucose, and C-peptide levels, but negatively with testosterone levels. Metabolism.1990;39(9):897-901. DOI: 10.1016/0026-0495(90)90297-p. PMID: 2202881.
- Marin P, Holmang S, Jonsson L, Sjostrom L, Kvist H, Holm G, Lindstedt G, Bjorntorp P. The effects of testosterone treatment on body composition and metabolism in middle-aged obese men. Int J Obes Relat Metab Disord. 1992;16(12):991-7. PMID: 1335979.
- Marin P, Holmang S, Gustafsson C, Jonsson L, Kvist H, Elander A, Eldh J, Sjostrom L, Holm G, Bjorntorp P. Androgen treatment of abdominally obese men. Obes Res. 1993;1(4):245-51. DOI: 10.1002/j.1550-8528.1993.tb00618.x. PMID: 16350577.
- Lovejoy JC, Bray GA, Bourgeois MO, Macchiavelli R, Rood JC, Greeson C, Partington C. Exogenous androgens influence body composition and regional body fat distribution in obese postmenopausal women--a clinical research center study. J Clin Endocrinol Metab. 1996;81(6):2198-203. DOI: 10.1210/jcem.81.6.8964851. PMID: 8964851.
- Lovejoy JC, Bray GA, Greeson CS, Klemperer M, Morris J, Partington C, Tulley R. Oral anabolic steroid treatment, but not parenteral androgen treatment, decreases abdominal fat in obese, older men. Int J Obes Relat Metab Disord. 1995;19(9):614-24. PMID: 8574271.
- Barnouin Y, Armamento-Villareal R, Celli A, Jiang B, Paudyal A, Nambi V, Bryant MS, Marcelli M, Garcia JM, Qualls C, Villareal DT. Testosterone Replacement Therapy Added to Intensive Lifestyle Intervention in Older Men With Obesity and Hypogonadism. J Clin Endocrinol Metab. 2021;106(3):e1096-e1110. DOI: 10.1210/clinem/dgaa917. PMID: 33351921.
- Gabai G, Mongillo P, Giaretta E, Marinelli L. Do Dehydroepiandrosterone (DHEA) and Its Sulfate (DHEAS) Play a Role in the Stress Response in Domestic Animals? Front Vet Sci. 2020;7:588835. DOI: 10.3389/fvets.2020.588835. PMID: 33195624.
- Clore JN. Dehydroepiandrosterone and body fat. Obes Res. 1995;3 Suppl 4:613S-616S. DOI: 10.1002/j.1550-8528.1995.tb00234.x. PMID: 8697065.
- Lardy H, Kneer N, Wei Y, Partridge B, Marwah P. Ergosteroids. II: Biologically active metabolites and synthetic derivatives of dehydroepiandrosterone. Steroids. 1998;63(3):158-65. DOI: 10.1016/s0039-128x(97)00159-1. PMID: 9558717.
- Svec F, Porter JR. The actions of exogenous dehydroepiandrosterone in experimental animals and humans. Proc Soc Exp Biol Med. 1998;218(3):174-91. DOI: 10.3181/00379727-218-44285. PMID: 9648935.
- Usiskin KS, Butterworth S, Clore JN, Arad Y, Ginsberg HN, Blackard WG, Nestler JE. Lack of effect of dehydroepiandrosterone in obese men. Int J Obes. 1990;14(5):457-63. PMID: 2143499.
- Nestler JE, Barlascini CO, Clore JN, Blackard WG. Dehydroepiandrosterone reduces serum low density lipoprotein levels and body fat but does not alter insulin sensitivity in normal men. J Clin Endocrinol Metab.1988;66(1):57-61. DOI: 10.1210/jcem-66-1-57. PMID: 2961787.
- Welle S, Jozefowicz R, Statt M. Failure of dehydroepiandrosterone to influence energy and protein metabolism in humans. J Clin Endocrinol Metab. 1990;71(5):1259-64. DOI: 10.1210/jcem-71-5-1259. PMID: 2146282.
- Mortola JF, Yen SS. The effects of oral dehydroepiandrosterone on endocrine-metabolic parameters in postmenopausal women. J Clin Endocrinol Metab. 1990;71(3):696-704. DOI: 10.1210/jcem-71-3-696. PMID: 2144295.
- Zumoff B, Strain GW, Heymsfield SB, Lichtman S. A randomized double-blind crossover study of the antiobesity effects of etiocholanedione. Obes Res. 1994;2(1):13-8. DOI: 10.1002/j.1550-8528.1994.tb00038.x. PMID: 16353603.
- Larsen TM, Toubro S, Astrup A. Efficacy and safety of dietary supplements containing CLA for the treatment of obesity: evidence from animal and human studies. J Lipid Res. 2003;44(12):2234-41. DOI: 10.1194/jlr.R300011-JLR200. PMID: 12923219.
- Riserus U, Smedman A, Basu S, Vessby B. Metabolic effects of conjugated linoleic acid in humans: the Swedish experience. Am J Clin Nutr. 2004;79(6 Suppl):1146S-1148S. PMID: 15159248.
- Dwyer JT, Allison DB, Coates PM. Dietary supplements in weight reduction. J Am Diet Assoc. 2005;105(5 Suppl 1):S80-6. DOI: 10.1016/j.jada.2005.02.028. PMID: 15867902.
- Sullivan AC, Triscari J, Hamilton JG, Miller ON. Effect of (-)-hydroxycitrate upon the accumulation of lipid in the rat. II. Appetite. Lipids. 1974;9(2):129-34. DOI: 10.1007/BF02532137. PMID: 4815800.
- Heymsfield SB, Allison DB, Vasselli JR, Pietrobelli A, Greenfield D, Nunez C. Garcinia cambogia (hydroxycitric acid) as a potential antiobesity agent: a randomized controlled trial. JAMA. 1998;280(18):1596-600. DOI: 10.1001/jama.280.18.1596. PMID: 9820262.
- Vuppalanchi R, Bonkovsky HL, Ahmad J, Barnhart H, Durazo F, Fontana RJ, Gu J, Khan I, Kleiner DE, Koh C, Rockey DC, Phillips EJ, Li YJ, Serrano J, Stolz A, Tillmann HL, Seeff LB, Hoofnagle JH, Navarro VJ, Drug-Induced Liver Injury N. Garcinia cambogia, Either Alone or in Combination With Green Tea, Causes Moderate to Severe Liver Injury. Clin Gastroenterol Hepatol. 2022;20(6):e1416-e1425. DOI: 10.1016/j.cgh.2021.08.015. PMID: 34400337.
- Onakpoya I, Hung SK, Perry R, Wider B, Ernst E. The Use of Garcinia Extract (Hydroxycitric Acid) as a Weight loss Supplement: A Systematic Review and Meta-Analysis of Randomised Clinical Trials. J Obes.2011;2011:509038. DOI: 10.1155/2011/509038. PMID: 21197150.
- Bryson A, de la Motte S, Dunk C. Reduction of dietary fat absorption by the novel gastrointestinal lipase inhibitor cetilistat in healthy volunteers. Br J Clin Pharmacol. 2009;67(3):309-15. DOI: 10.1111/j.1365-2125.2008.03311.x. PMID: 19220279.
- Kopelman P, Bryson A, Hickling R, Rissanen A, Rossner S, Toubro S, Valensi P. Cetilistat (ATL-962), a novel lipase inhibitor: a 12-week randomized, placebo-controlled study of weight reduction in obese patients. Int J Obes (Lond). 2007;31(3):494-9. DOI: 10.1038/sj.ijo.0803446. PMID: 16953261.
- Kopelman P, Groot Gde H, Rissanen A, Rossner S, Toubro S, Palmer R, Hallam R, Bryson A, Hickling RI. Weight loss, HbA1c reduction, and tolerability of cetilistat in a randomized, placebo-controlled phase 2 trial in obese diabetics: comparison with orlistat (Xenical). Obesity (Silver Spring). 2010;18(1):108-15. DOI: 10.1038/oby.2009.155. PMID: 19461584.
- Yuan S, Chan JFW, Chik KKH, Chan CCY, Tsang JOL, Liang R, Cao J, Tang K, Chen LL, Wen K, Cai JP, Ye ZW, Lu G, Chu H, Jin DY, Yuen KY. Discovery of the FDA-approved drugs bexarotene, cetilistat, diiodohydroxyquinoline, and abiraterone as potential COVID-19 treatments with a robust two-tier screening system. Pharmacol Res. 2020;159:104960. DOI: 10.1016/j.phrs.2020.104960. PMID: 32473310.
- Goldsmith GA, Hamilton JG, Miller ON. Lowering of serum lipid concentrations: mechanisms used by unsaturated fats, nicotinic acid, and neomycin: excretion of sterols and bile acids. Arch Intern Med.1960;105:512-7. PMID: 13848368.
- Campbell UD, Juhl E, Quaade F. [Obesity therapy with cholestyramine]. Nord Med. 1970;84(51):1628-9. PMID: 5490740.
- Faloon WW, Paes IC, Woolfolk D, Nankin H, Wallace K, Haro EN. Effect of neomycin and kanamycin upon intestinal absortion. Ann N Y Acad Sci. 1966;132(2):879-87. DOI: 10.1111/j.1749-6632.1966.tb43008.x. PMID: 5227780.
- Hvidt S, Kjeldsen K. Malabsorption Induced by Small Doses of Neomycin Sulphate. JAMA: The Journal of the American Medical Association. 1963;186(3):229-229. DOI: 10.1001/jama.1963.03710030149127.
- Dobbins WO, 3rd, Herrero BA, Mansbach CM. Morphologic alterations associated with neomycin induced malabsorption. Am J Med Sci. 1968;255:63-77. DOI: 10.1097/00000441-196801000-00011. PMID: 5635294.
- Rolls BJ, Pirraglia PA, Jones MB, Peters JC. Effects of olestra, a noncaloric fat substitute, on daily energy and fat intakes in lean men. Am J Clin Nutr. 1992;56(1):84-92. DOI: 10.1093/ajcn/56.1.84. PMID: 1609767.
- Cotton JR, Burley VJ, Weststrate JA, Blundell JE. Fat substitution and food intake: effect of replacing fat with sucrose polyester at lunch or evening meals. Br J Nutr. 1996;75(4):545-56. DOI: 10.1079/bjn19960158. PMID: 8672407.
- Cotton JR, Weststrate JA, Blundell JE. Replacement of dietary fat with sucrose polyester: effects on energy intake and appetite control in nonobese males. Am J Clin Nutr. 1996;63(6):891-6. DOI: 10.1093/ajcn/63.6.891. PMID: 8644683.
- Roy HJ, Most MM, Sparti A, Lovejoy JC, Volaufova J, Peters JC, Bray GA. Effect on body weight of replacing dietary fat with olestra for two or ten weeks in healthy men and women. J Am Coll Nutr. 2002;21(3):259-67. DOI: 10.1080/07315724.2002.10719219. PMID: 12074254.
- Bray GA, Lovejoy JC, Most-Windhauser M, Smith SR, Volaufova J, Denkins Y, de Jonge L, Rood J, Lefevre M, Eldridge AL, Peters JC. A 9-mo randomized clinical trial comparing fat-substituted and fat-reduced diets in healthy obese men: the Ole Study. Am J Clin Nutr. 2002;76(5):928-34. DOI: 10.1093/ajcn/76.5.928. PMID: 12399262.
- Tulley RT, Vaidyanathan J, Wilson JB, Rood JC, Lovejoy JC, Most MM, Volaufova J, Peters JC, Bray GA. Daily intake of multivitamins during long-term intake of olestra in men prevents declines in serum vitamins A and E but not carotenoids. J Nutr. 2005;135(6):1456-61. DOI: 10.1093/jn/135.6.1456. PMID: 15930452.
- Lovejoy JC, Bray GA, Lefevre M, Smith SR, Most MM, Denkins YM, Volaufova J, Rood JC, Eldridge AL, Peters JC, Ole S. Consumption of a controlled low-fat diet containing olestra for 9 months improves health risk factors in conjunction with weight loss in obese men: the Ole' Study. Int J Obes Relat Metab Disord. 2003;27(10):1242-9. DOI: 10.1038/sj.ijo.0802373. PMID: 14513073.
- Burns AA, Livingstone MB, Welch RW, Dunne A, Reid CA, Rowland IR. The effects of yoghurt containing a novel fat emulsion on energy and macronutrient intakes in non-overweight, overweight and obese subjects. Int J Obes Relat Metab Disord. 2001;25(10):1487-96. DOI: 10.1038/sj.ijo.0801720. PMID: 11673771.
- Diepvens K, Steijns J, Zuurendonk P, Westerterp-Plantenga MS. Short-term effects of a novel fat emulsion on appetite and food intake. Physiol Behav. 2008;95(1-2):114-7. DOI: 10.1016/j.physbeh.2008.05.006. PMID: 18571210.
- Rebello CJ, Martin CK, Johnson WD, O'Neil CE, Greenway FL. Efficacy of Olibra: a 12-week randomized controlled trial and a review of earlier studies. J Diabetes Sci Technol. 2012;6(3):695-708. DOI: 10.1177/193229681200600326. PMID: 22768902.
- Falchi M, El-Sayed Moustafa JS, Takousis P, Pesce F, Bonnefond A, Andersson-Assarsson JC, Sudmant PH, Dorajoo R, Al-Shafai MN, Bottolo L, Ozdemir E, So HC, Davies RW, Patrice A, Dent R, Mangino M, Hysi PG, Dechaume A, Huyvaert M, Skinner J, Pigeyre M, Caiazzo R, Raverdy V, Vaillant E, Field S, Balkau B, Marre M, Visvikis-Siest S, Weill J, Poulain-Godefroy O, Jacobson P, Sjostrom L, Hammond CJ, Deloukas P, Sham PC, McPherson R, Lee J, Tai ES, Sladek R, Carlsson LM, Walley A, Eichler EE, Pattou F, Spector TD, Froguel P. Low copy number of the salivary amylase gene predisposes to obesity. Nat Genet. 2014;46(5):492-7. DOI: 10.1038/ng.2939. PMID: 24686848.
- Nakajima K, Muneyuki T, Munakata H, Kakei M. Revisiting the cardiometabolic relevance of serum amylase. BMC Res Notes. 2011;4:419. DOI: 10.1186/1756-0500-4-419. PMID: 22004561.
- Heianza Y, Sun D, Wang T, Huang T, Bray GA, Sacks FM, Qi L. Starch Digestion-Related Amylase Genetic Variant Affects 2-Year Changes in Adiposity in Response to Weight-Loss Diets: The POUNDS Lost Trial. Diabetes. 2017;66(9):2416-2423. DOI: 10.2337/db16-1482. PMID: 28659346.
- Coniff RF, Shapiro JA, Seaton TB, Bray GA. Multicenter, placebo-controlled trial comparing acarbose (BAY g 5421) with placebo, tolbutamide, and tolbutamide-plus-acarbose in non-insulin-dependent diabetes mellitus. Am J Med. 1995;98(5):443-51. DOI: 10.1016/S0002-9343(99)80343-X. PMID: 7733122.
- William-Olsson T. alpha-Glucosidase inhibition in obesity. Acta Med Scand Suppl. 1985;706:1-39. DOI: 10.1111/j.0954-6820.1986.tb19118.x. PMID: 3914827.
- Wolever TM, Chiasson JL, Josse RG, Hunt JA, Palmason C, Rodger NW, Ross SA, Ryan EA, Tan MH. Small weight loss on long-term acarbose therapy with no change in dietary pattern or nutrient intake of individuals with non-insulin-dependent diabetes. Int J Obes Relat Metab Disord. 1997;21(9):756-63. DOI: 10.1038/sj.ijo.0800468. PMID: 9376887.
- Erlanson-Albertsson C, York D. Enterostatin--a peptide regulating fat intake. Obes Res. 1997;5(4):360-72. DOI: 10.1002/j.1550-8528.1997.tb00565.x. PMID: 9285845.
- Erlanson-Albertsson C, Mei J, Okada S, York D, Bray GA. Pancreatic procolipase propeptide, enterostatin, specifically inhibits fat intake. Physiol Behav. 1991;49(6):1191-4. DOI: 10.1016/0031-9384(91)90350-w. PMID: 1896501.
- Okada S, York DA, Bray GA, Erlanson-Albertsson C. Enterostatin (Val-Pro-Asp-Pro-Arg), the activation peptide of procolipase, selectively reduces fat intake. Physiol Behav. 1991;49(6):1185-9. DOI: 10.1016/0031-9384(91)90349-s. PMID: 1896500.
- Lin L, York DA. Chronic ingestion of dietary fat is a prerequisite for inhibition of feeding by enterostatin. Am J Physiol. 1998;275(2):R619-23. DOI: 10.1152/ajpregu.1998.275.2.R619. PMID: 9688701.
- Rossner S, Barkeling B, Erlanson-Albertsson C, Larsson P, Wahlin-Boll E. Intravenous enterostatin does not affect single meal food intake in man. Appetite. 1995;24(1):37-42. DOI: 10.1016/s0195-6663(95)80004-2. PMID: 7741534.