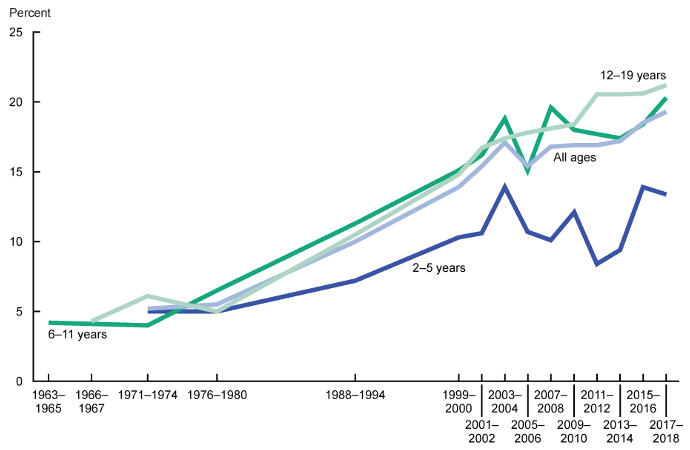
ABSTRACT
Resistance to thyroid hormone (RTH), a syndrome of reduced responsiveness of target tissues to thyroid hormone (TH) was identified in 1967 (1). An early report proposed various mechanisms including defects in TH transport, metabolism and action (2). However, with the identification of TH receptor beta (THRB) gene mutations in 1989 (3, 4), the term RTH became synonymous with defects of this specific gene (5). Subsequent discoveries of genetic defects that reduce the effectiveness of TH through altered cell membrane transport (6, 7) and metabolism (8) have broadened the definition of TH hyposensitivity to encompass all defects that can interfere with the biological activity of a chemically intact hormone secreted in normal or even excessive amounts. In this chapter, we have retained the acronym RTH to denote the syndrome produced by reduced intracellular action of the active TH, triiodothyronine (T3). However, with the identification of mutations in the TH receptor alpha (THRA) gene (9), RTH syndromes are designated as RTHα and RTHß. The term of impaired sensitivity to TH (ISTH) has been therefore proposed (10-12) to denote altered effectiveness of TH in a broader sense.
Normal TH action requires 1) adequate synthesis and secretion of TH, 2) its transport across cell membranes, 3) hormone activation through intracellular metabolism, 4) cytosolic processing and nuclear translocation, 5) binding to receptors, and 6) interaction of the receptors with co-regulators or other post receptor effects mediating the TH effect. In addition to nuclear actions of TH, non-genomic actions are also of physiological relevance (13, 14).
Maintenance of TH supply is insured by a feedback control mechanism involving the hypothalamus, pituitary, and thyroid gland (Figure 1A). A decrease in the circulating TH concentration induces a hypothalamus-mediated stimulation of thyroid stimulating hormone (TSH) secretion mediated by TSH-releasing hormone (TRH) from the pituitary thyrotrophs, which stimulates the thyroid follicular cells to synthesize and secrete more hormone. In contrast, TH excess attenuates or suppresses the system through the same pathway, in order to maintain homeostasis. This centrally regulated system, does not respond to changing requirements for TH in a particular organ or cell.
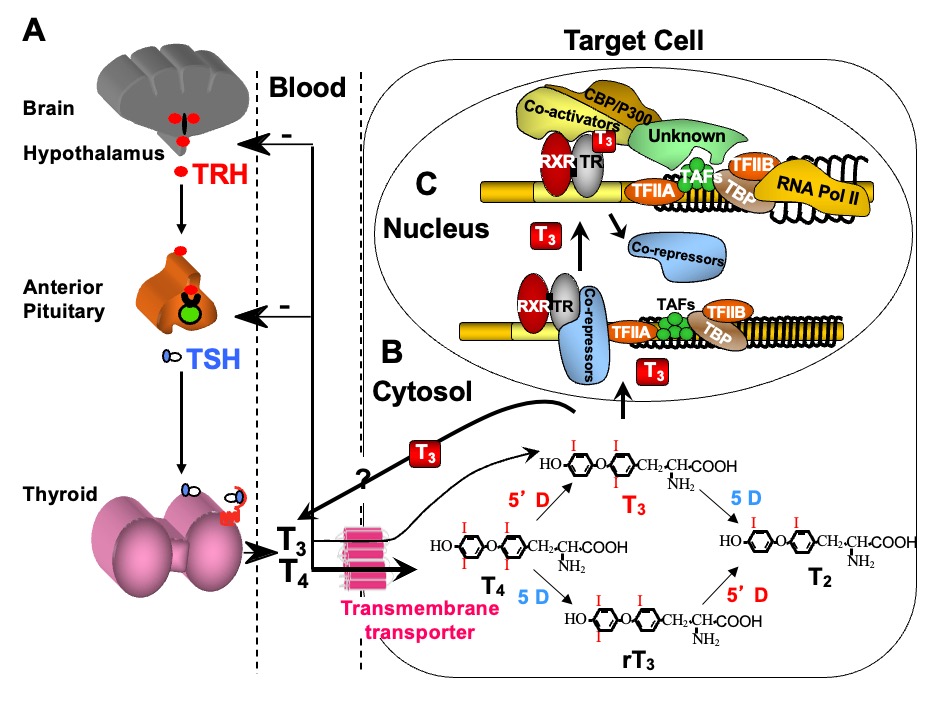
Figure 1. Regulation of TH supply, metabolism and genomic action. (A) Feedback control that regulates the amount of TH in blood. (B) Intracellular metabolism of TH, regulating TH bioactivity. (C) Genomic action of TH. For details see text. CBP/P300, cAMP-binding protein/general transcription adaptor; TFIIA and TFIIB, transcription intermediary factor II, A and B; TBP, TATA-binding protein; TAF, TBP-associated factor.
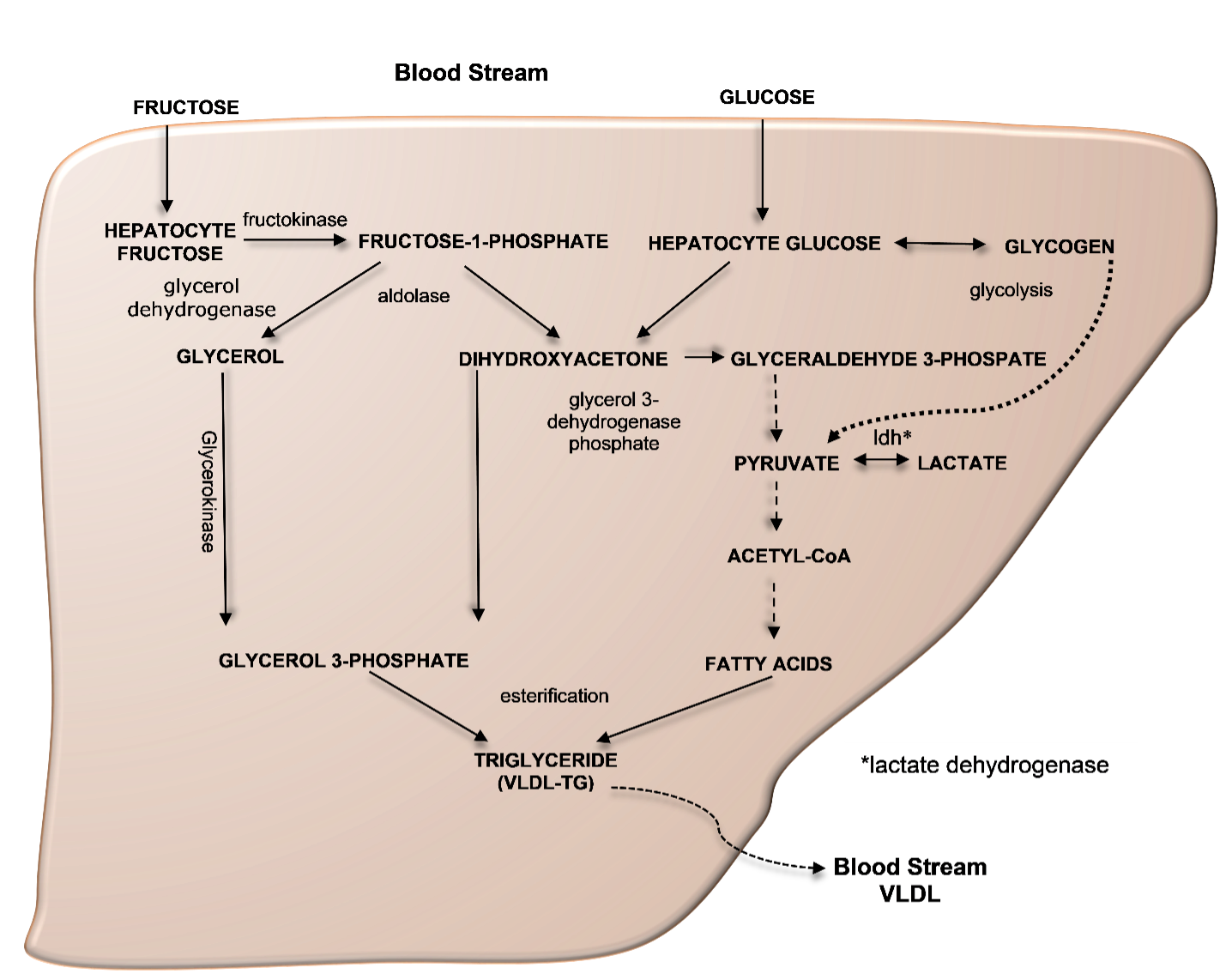
Additional systems operate to accommodate for local TH requirements. One such system is the control of TH entry into the cell through active transmembrane transporters (15). Another is the activation of the hormone precursor thyroxine (T4) by removal of the outer ring iodine (5’-deiodination) to form T3 or, inactivation of T4 and T3 by removal of the inner ring iodine (5-deiodination) to form reverse T3 (rT3) and T2, respectively (Chapter 6) (Figure 1B). Cell specific adjustment in deiodinase activity allows for additional local regulation of hormone supply (16).
Finally, the types and abundance of TH receptors (TRs), through which TH action is mediated, determine the nature and degree of the response. TH action takes place in the cytosol as well as in the nucleus (13). The latter, known as genomic or type 1 effect, has been more extensively studied (14, 17, 18) (Figure 1C). TRs are ligand-regulated transcription factors that bind to DNA of genes whose expression they regulate either positively or negatively.
THE PARADOX OF COEXISTING MANIFESTTIONS OF THYROID HORMONE DEFICIENCY AND EXCESS
TH deficiency and excess are associated with typical symptoms and signs reflecting the global effects of lack and excess of the hormone, respectively, on all organs. A departure from this became apparent with the identification of the RTHß syndrome. Subjects with RTHß have high serum TH levels without TSH suppression. This paradox encompasses biochemical and clinical observations suggesting TH deficiency, sufficiency, and excess, depending on the degree and nature of the TR protein abnormality in affected individuals (5). The syndrome of TH cell membrane transport defect (THCMTD) presents a similar paradox, as subjects have high serum T3 concentration but the cellular uptake of TH is not uniform in all tissues and cell types (19).
THYROID HORMONE ACTION DEFECTS KNOWN AS RESISTANCE TO THYROID HORMONE (RTH)
The first syndrome recognized to impair the sensitivity to TH was that of reduced TH action at the cellular level (1), and it was described as Resistance to Thyroid Hormone (RTH) (2). After the clinical recognition of the syndrome, it took 22 years until the molecular defect could be unraveled by demonstrating mutations in the THRB gene in 1989 (3). Twenty-three years after this discovery, mutations in the THRA gene led to the recognition of a distinct syndrome, RTHα, in 2012 (9, 20). In addition to these two syndromes, RTHß and RTHα, other causes that impair the sensitivity to TH have been identified during the last two decades.
RESISTANCE TO THYROID HORMONE-BETA (RTHß)
Patients with RTHß are identified by their persistent elevation of circulating free TH associated with non-suppressed serum TSH levels, in the absence of intercurrent illnesses, drugs, or alterations of TH transport serum proteins. In addition, laboratory testing reveals that unusually high doses of exogenous TH are required to produce the expected suppressive effect on the secretion of pituitary TSH and the expected metabolic responses in peripheral tissues.
Although the apparent resistance to TH may vary in severity, it is always partial. The variability in clinical manifestations may be due to the severity of the hormonal resistance, the effectiveness of compensatory mechanisms, the presence of modulating genetic factors, and the effects of prior therapy. The magnitude of the hormonal resistance is, in turn, dependent on the nature of the underlying genetic defect, most commonly, a mutation in the THRB gene (5, 21)
Despite a variable clinical presentation, the common features characteristic of the RTHß syndrome are: 1) elevated serum levels of free T4 and to a lesser degree T3, particularly in older individuals, 2) normal or slightly increased serum TSH levels that respond to thyrotropin releasing hormone (TRH), 3) an absence of the usual symptoms and metabolic consequences of TH excess, and 4) goiter.
Clinical Classification
The diagnosis is based on the clinical findings and standard laboratory tests and confirmed by genetic studies. Before THRB gene defects were recognized, the proposed sub-classification of RTHß was based on symptoms, signs and laboratory parameters of tissue responses to TH (22). Notwithstanding the assessment of TSH feedback regulation by TH, the measurements of most other responses to TH have low sensitivity and are relatively nonspecific. For this reason, all tissues other than the pituitary have been grouped together under the term peripheral tissues, on which the impact of TH was roughly assessed by a combination of clinical observation and laboratory tests.
The majority of patients who appear to be eumetabolic and maintain a near normal serum TSH concentration have been classified as having Generalized Resistance to TH (GRTH). In such individuals, the defect seems to be compensated by the high levels of TH. In contrast, patients with equally high serum levels of TH and non-suppressed TSH levels, who appear to be hypermetabolic, because they have signs such as sinus tachycardia, are classified as having selective pituitary resistance to TH (PRTH). TSH-producing pituitary adenomas caused by somatic mutations or isoform specific TRßs mutants also fall into this category of RTH (23, 24). Finally, the occurrence of isolated peripheral tissue resistance to TH (PTRTH) was reported in a single patient (25). No mutation in the THRB gene of this patient could be identified (26), and no similar cases have been reported. Thus, it is uncertain whether PTRTH exists as a true entity. The earliest suggestion that PRTH may not constitute an entity distinct from GRTH was reported by Beck-Peccoz et al. (27). A comprehensive study involving 312 patients with GRTH and 72 patients with PRTH, has conclusively shown a significant overlap in all parameters examined including tachycardia, emotional disturbance and hyperactivity in the two categories (28). More importantly, identical mutations were found in individuals classified as having GRTH and PRTH, many of whom belonged to the same family (29). This led to the conclusion that these two seemingly different forms of RTH are in fact related to a spectrum of subjective symptoms, as well as the individual’s target organ susceptibility to changes of TH, a phenomenon also observed in subjects with thyroid dysfunction in the absence of RTH (See section on the Molecular Basis of the Defect).
Incidence and Inheritance
The precise incidence of RTH is unknown. Because most routine neonatal screening programs are based on the determination of TSH, RTHß is rarely identified by this means (30). A limited neonatal survey by measuring blood T4 concentrations suggested the occurrence of one case per 40,000 live births (31, 32). Known cases with THRBgene mutations surpass 4,000 affected individuals.
Although most thyroid diseases occur more commonly in women, RTHß has been found with equal frequency in females and males. The condition appears to have wide geographic distribution and has been reported in Caucasians, Africans, Asians and Amerindians. The prevalence may vary among different ethnic groups.
Familial occurrence of RTHß has been documented in approximately 75% of cases. Taking into account only those families in whom both parents of the affected subjects have been studied, the true incidence of sporadic cases or de novo mutations is 19% (Table 1). Not uncommonly, hypothyroid adults given supraphysiological amounts of TH on subjective basis are labeled as having acquired RTH. Such individuals have no mutations in the THRB gene but a compensatory upregulation of the TH degrading enzyme deiodinase 3. This can be demonstrated by the very high concentration of reverse T3 but normal T3 (33).
|
Table 1. Types of TRß Gene Mutations
|
|
Type
|
Number of occurrences
|
Number of families
|
Effect on TRß
|
|
qt different sites
|
(total)
|
(authors)’
|
|
Substitution
|
Single nucleotide
|
190
|
610
|
286
|
Single a.a. substitution;
|
|
|
5
|
15
|
9
|
Premature stop: C434*, K443*, E445* C446*, E449*
|
|
|
Dinucleotide
|
3
|
3
|
1
|
Single a.a. substitution: P453N, P453Y;
Premature stop: F451*
|
|
Deletion
|
Single nucleotide
|
2
|
2
|
2
|
FrSh at codon 438 and 440; stop at codon 442*
|
|
|
Trinucleotide
|
6
|
10
|
4
|
Single a.a. deletion: E248Δ, I276Δ, T337Δ, M430Δ , G432Δ, P452Δ
|
|
|
Eight nucleotides
|
1
|
1
|
0
|
FrSh at codon 443 normal stop at codon 462
|
|
|
Eleven nucleotides
|
1
|
2
|
1
|
FrSh at codon 449 stop at codon 459
|
|
|
All coding sequence
|
1
|
1
|
1
|
Complete deletion
|
|
Insertion
|
Single nucleotide
|
8
|
20
|
12
|
FrSh at codons: 436, 443, 448, 451, 454, 456 stop at 464)
|
|
|
Trinucleotide
|
1
|
0
|
1
|
Single a.a. insertion (328S)
|
|
Duplication
|
Seven nucleotides
|
1
|
1
|
0
|
At codon 452 FrSh and a.a. 464 (extended with 2 a.a.)
|
|
TOTAL
|
|
219
|
665
|
317
|
|
|
Mutations at CpG dinucleotides
|
20
|
212a
|
111a
|
35% of families with single nucleotide substitution
and 39% of similar families studied in the authors’ laboratory
|
|
De novo mutations
|
|
b
|
60c
|
19% of families studied in the authors’ laboratory
|
a.a., amino acid. FrSh, frame shift
a Not included are 10 families in which the mutation did not follow the rule of G to A or C to T transition.
b Not counted as publications do not always include parental genotype
c Families with THRB gene mutations excluding those with a single affected individual when both parents were not tested.
The inheritance of RTHβ is typically autosomal dominant. Transmission was only recessive in a single family (1, 34). The biallelic expression of the mutant TRß due to consanguinity in three families with dominant inheritance of RTHß, as well as the possible deletion of the paternal allele in another family, has led to very severe clinical manifestations in the affected children (35, 36).
Etiology and Genetics
Using the technique of restriction fragment length polymorphism, Usala et al. (37) were first to demonstrate linkage between the THRB locus on chromosome 3 and the RTHß phenotype. Subsequent studies at the University of Chicago and at the National Institutes of Health have identified distinct point mutations in the THRB gene of two unrelated families with RTHß (3, 4). In both families, only one of the two THRB alleles was mutated, which was consistent with the dominant mode of inheritance.
Mutations in the THRB gene have now been identified in subjects with RTHß of 665 families (Table 1). They comprise 219 different mutations including the initially reported index family, which was found to have complete deletion of the THRB gene (34), a finding that contrasts with the usually observed point mutations. Forty-eight of the known mutations have not been published (33). The majority of the families, 625, have single nucleotide substitutions resulting in single amino acid replacements: in 15 families, mutations leading to premature stop codons result in truncated TRß proteins. In the remaining 40 families, the sequence alterations consisted of dinucleotide substitutions, insertions, deletions of nucleotides ranging from a single base pair to 11 nucleotides, and a duplication of 7 bases (for details see Table 1).
Given that there are 446 more families than the 219 different mutations, 64 of the mutations are shared by more than one family. Haplotyping of intragenic polymorphic markers showed that, in most instances, identical mutations have developed independently in different families (38). These occur more often, though not exclusively, in CpG dinucleotide hot spots. In fact, de novo mutations are twice as frequent in CpG dinucleotides with the largest number of families, namely 41 harboring the same mutation, R338W. In addition, different mutations resulting in more than one amino acid substitution at the same codon have been found at 44 different sites. Mutations in codons 345 and 451 result both in 5 different amino acid replacements (G345R,S,A,V,D; F451I,L,S,C,*), and those in codon 453 include 8 different substitutions (P453T,S,A,N,Y,H,L,R), as well as an insertion and a deletion, and a total of 74 families harbor mutations of this particular codon.
The detected mutations are located in the last four exons of the gene and include 8, 27, 89 and 90 mutations in exons 7, 8, 9 and 10, respectively. These involve 56, 40, 292 and 278 families (Figure 2). The following mutations have been identified in more than 20 families: R243Q, A317T, R320C, R338W, R438H and P453T. Of note the first five are in CpG dinucleotides and the last in a stretch of six cytidines.
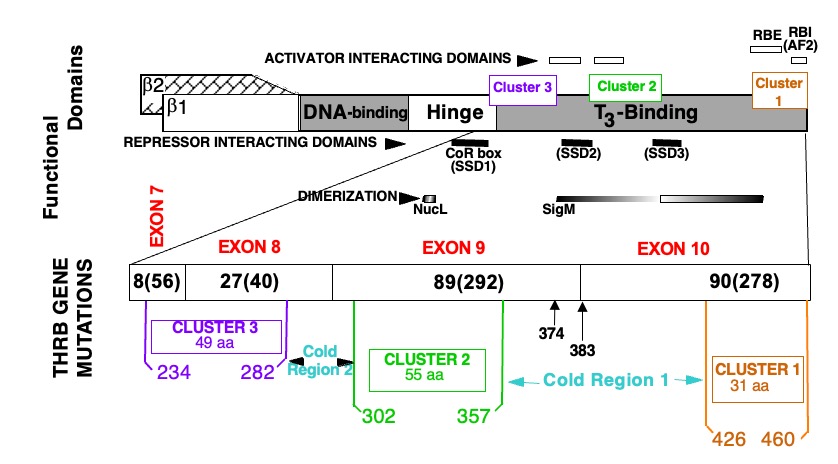
Figure 2. Location of mutations in the TRß protein in subjects with RTHß.
TOP PORTION: Schematic representation of TRß and its functional domains for interaction with TREs (DNA-binding), with hormone (T3-binding), with activating (298), repressing (299-301) cofactors and with nuclear receptor partners (dimerization) (74, 302, 303). Note their relationship to the three clusters of natural mutations.
BOTTOM PORTION: The T3-binding domain and distal end of the hinge region, which contain the three mutation clusters, are expanded. The four terminal exons containing all so far identified mutation are shown with the number different mutation and number of families in parenthesis (published and our unpublished data). Amino acids are numbered consecutively starting at the amino terminus of the TRß1 molecule according to the consensus statement of the First International Workshop on RTH (304). TRß2 has 15 additional residues at the N-terminus. Mutations occur in three clusters as indicated. A silent region between cluster 1 and 2, located in the dimerization domain contains two mutations (Q374K and R383H), indicated with arrows.
AF2, Hormone-dependent activation function (12th amphipathic helix) (305, 306); RBE, corepressor-binding enhancer; RBI, corepressor-binding inhibitor (306); SSD, silencing subdomain (301); NucL, nuclear localization (307); SigM, signature motif (308). aa, amino acid.
All THRB gene mutations are located in the functionally relevant T3-binding domain and its adjacent hinge region. Three mutational clusters have been identified with intervening cold regions (Figure 2). No mutations have been identified in the DNA binding domain or in the amino termini characterizing TRß1 and TRß2. A report of a putative mutation, C36Y, in the amino terminus (39) represents a polymorphism that does not alter the biological properties of the TRß1 molecule (40). With the exception of the family with THRB gene deletion, the inheritance of all others is autosomal dominant.
Somatic mutations in the THRB gene have been identified in some TSH-secreting pituitary tumors (23, 41). These mutations can be identical to those occurring in the germline. However, which affects the negative regulation of TSH by TH, is responsible for the development of the pituitary tumor.
In 14% of families, RTHß occurs in the absence of mutations in the THRB genes (nonTRß-RTH) (42). Such individuals may have a defect in one of the cofactors involved in the mediation of TH action (see Animal Models of RTH, below).
Molecular Mechanisms of TR Action
The two TH receptor genes located on chromosome 17 and 3 encode TRα and TRß, which have substantial structural and sequence similarities. Both genes produce two isoforms, α1 and α2 by alternative splicing, and ß1 and ß2 by different transcription start points. TRα2 binds to TH response elements (TREs) but due to a sequence difference in the ligand-binding domain (LBD), it does not bind TH (43) and appears to have a weak antagonistic effect (44). Additional TR isoforms, including a TRß with a shorter amino terminus (TRß3), a truncated TRß3, TRα1 and TRα2 lacking the DNA-binding domain (DBD) have been identified in rodents (45, 46), and TRß4 that lacks the LBD has been reported in selected human tissues (47). The significance of these variants in humans remains unknown (48). Finally, a p43 protein, translated from a downstream AUG of TRα1, is believed to mediate the TH effect in mitochondria (49).
The relative expression of the two TR genes and the distribution of the encoded proteins vary among tissues and during different stages of development (50-52). The abundance of several splice variants involving the 5'-untranslated region of the human TRß1 (53, 54) is regulated developmentally and varies among tissues. Although TRß and TRα are interchangeable (55, 56) to a certain degree, the absence of one or the other receptor does not produce equivalent phenotypes. Some TH effects are entirely TR isoform specific (see Animal Models of RTH, below).
TREs, located in TH regulated genes, consist of half-sites that contain the consensus sequence AGGTCA, and vary in number, spacing and orientation (57, 58). Each half-site usually binds a single TR molecule (monomer), two half-sites bind two TRs (dimer) or one TR and a heterologous partner (heterodimer), the most prominent being the retinoid X receptor g (RXR). Dimer formation is facilitated by the presence of an intact "leucine zipper" motif located in the middle of the LBD of TRs. Occupation of TREs by unliganded TRs, also known as aporeceptors, inhibits the constitutive expression of genes that are positively regulated by TH (59) through association with corepressors such as the nuclear corepressor (NCoR) or the silencing mediator of retinoic acid and TH receptors (SMRT) (60). Transcriptional repression is mediated through the recruitment of the mammalian homologue of the Saccaromyces transcriptional corepressor (mSin3A) and histone deacetylases (HDAC) (61). This latter activity compacts nucleosomes into a tight and inaccessible structure, effectively shutting down gene expression (Figure 1C). This effect is relieved by the addition of TH, which releases the corepressor, reduces the binding of TR dimers to TRE, enhances the occupation of TREs by TR/RXR heterodimers (62) and recruits coactivators (CoA) such as p/CAF (CREB binding protein-associated factor) and the nuclear coactivator (NCoA) (63) with histone acetylation (HAT) activity (60, 64). This results in the loosening of the nucleosome structure making the DNA more accessible to transcription factors (Figure 1C). The ligand-dependent association with TR associated proteins, in conjunction with the general coactivators PC2 and PC4, act to mediate transcription by RNA polymerase II and general initiation factors (65). Furthermore, it is believed that T3 exerts its effect by inducing conformational changes of the TR molecule and that TR associated proteins (TRAP) stabilize the association of TRs with TREs.
In addition to these genomic effects, TH can also act at the cell membrane and in the cytosol through non-genomic actions (13, 66). These non-genomic type 2 effects (14) include oxidative phosphorylation and mitochondrial gene transcription and involve the generation of intracellular secondary messengers with induction of [Ca(2+)] (I), cyclic adenosine monophosphate (cAMP) AMP or protein kinase signaling cascades.
Properties of Mutant TRß Receptors and Associated Dominant Negative Effect
THRB gene mutations produce two forms of RTH. The less common, described in only one family (1), is caused by deletion of all coding sequences of the THRB gene and is inherited as an autosomal recessive trait (34). The complete lack of TRß in these individuals results in severe deafness and is associated with mutism (1), as well as monochromatic vision (67) because TRß is required for the cochlear maturation and the development of cone photoreceptors that mediate color vision (68) (see Animal Models of RTH, below). Heterozygous individuals that express a single THRB gene have no clinical or laboratory abnormalities. This is not due to compensatory overexpression of the single normal allele of the THRB gene nor that of the THRA gene (69). However, because subjects with complete TRß gene deletion preserve some TH responsiveness, it is logical to conclude that TRα1 is capable of partially substituting for the function of TRß (see Animal Models of RTH, below).
The more common form of RTHß is inherited in an autosomal dominant fashion and is characterized by defects in one allele of the THRB gene, principally missense mutations. This contrasts with the lack of a phenotype in individuals that express a single THRB allele. The RTHß phenotype does not result from a lack of a functional allele (haploinsufficiency) caused by the mutant TRßs (mutTRs) but by interfering with the function of the wild-type (WT) TR (dominant negative effect, DNE). This has been clearly demonstrated in experiments in which mutTRs are coexpressed with WT TRs (70, 71).
Studies have established two basic requirements for mutTRs to exert a DNE: 1) preservation of binding to TREs on DNA and 2) the ability to dimerize with a homologous (72, 73) or heterologous partner (74, 75). These criteria apply to mutTRs with predominantly impaired T3-binding activity (Figure 3). In addition, a DNE can be exerted through impaired association with a cofactor even in the absence of important impairment of T3-binding. Increased affinity of a mutTR for a corepressor (CoR) (76, 77), or reduced association with a coactivator (CoA) (78-80), have been found to play a role in the dominant expression of RTH. The introduction in a mutTR of an additional artificial mutation that abolishes either DNA binding, dimerization or the association with a CoR results in the abrogation of its DNE (75, 81, 82).
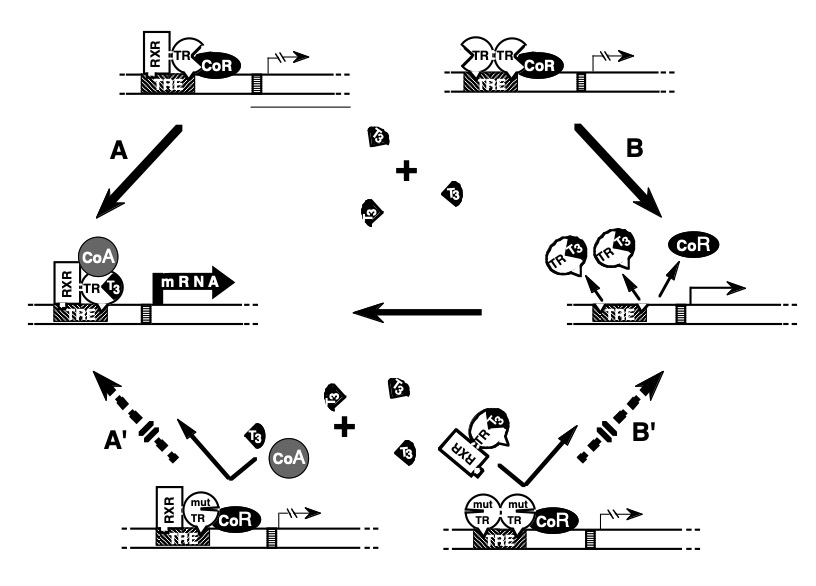
Figure 3. Mechanism of the dominant expression of RTH: In the absence of T3, occupancy of TRE by TR heterodimers (TR-TRAP) or dimers (TR-TR) suppresses transactivation through association with a corepressor (CoR). (A) T3-activated transcription mediated by TR-TRAP heterodimers involves the release of the CoR and association with coactivators (CoA) as well as (B) the removal of TR dimers from TRE releases their silencing effect and liberates TREs for the binding of active TR-TRAP heterodimers. The dominant negative effect of a mutant TR (mutTR), that does not bind T3, can be explained by the inhibitory effect of mutTR-containing-dimers and heterodimers that occupy TRE. Thus, T3 is unable to activate the mutTR-TRAP heterodimer (A') or release TREs from the inactive mutTR homodimers (B'). [Modified from Refetoff et al. (5)].
The distribution of THRB gene mutations associated with RTHß reveals a conspicuous absence of mutations in regions of the molecule that are important for dimerization, for the binding to DNA, and for the interaction with CoRs (Figure 2). These "cold regions" contain CpG hot spots, suggesting that they may not be devoid of naturally occurring mutations. Rather, mutations would escape detection owing to their failure to produce clinically significant RTHß in heterozygotes, as tested in vitro (83). This was recently confirmed in a study of a family in which one member has been fortuitously identified to have a mutation in the cold region (84). Nevertheless, mutation in other regions of the TRß could also be phenotypically silent, particularly if not occurring near the T3 binding pocket (85). Structural studies of the DBD and LBD have provided further understanding about the clustered distribution of mutTRßs associated RTH and defects in the association with cofactors (86-89).
Based on the early finding that RTHß is associated with mutations confined to the LBD of the TRß, it was anticipated that the clinical severity of RTHß would correlate with the degree of T3-binding impairment. While this was true in 12 different natural mutTRßs, in 5 others, the severity of the resistance was less pronounced despite virtually complete absent T3-binding. This is explained by the reduced dominant negative potency due to diminished ability to form homodimers (for example R316H and R338W) (90). Weakened association of TRß with DNA or CoR can produce the same effect.
Less evident was the observation of relatively severe interference with the function of the WT TRß, despite very mild impairment or no T3-binding defect at all. This was the case when hormone-binding was tested in two mutTRßs, located in the hinge region of the receptor (R243Q and R243W) (91). However, reduced T3-binding could be demonstrated after binding of the mutTRß to TRE, indicating a change in the mutTRß configuration when bound to genomic DNA (91, 92). Other mechanisms and examples of DNE in the presence of normal or slightly attenuated T3-binding include a decreased interaction of L454V with CoA (78), and a delay of R383H to release CoR (93).
In general, the relative degree of impaired function among various mutTRßs is similar whether tested using TRE-containing reporter genes that are negatively or positively regulated by T3. Exceptions to this rule are the mutTRßs R383H and R429Q that show greater impairment of transactivation on negatively rather than positively regulated promoters (90, 93, 94). In this respect, these two mutTRßs are candidates for a predominant PRTH phenotype, even though they have been clinically described as producing GRTH (95), as well as PRTH (96, 97). Substitution of these charged amino acids (in this case arginine) disrupts the unique property of TRß2 to bind certain coactivators through multiple contact surfaces (98). The result is a decrease in the normal T3-mediated feedback suppression mediated by TRß2 through the conversion of TRß2 to a TRß1-like molecule with altered CoA binding. As a consequence, the mutation affects predominantly TRß2 mediated action. In vivo support for a TRß2 predominant impairment of the mutTRß R429Q was also obtained in mice (99). Another putative mechanism for isolated PRTH was illustrated by the occurrence of a double-hit combining in cis the THRB mutation R338W and a single nucleotide polymorphism (SNP) located in an intronic enhancer shown to play a critical role in the pituitary expression of the TRβ2 isoform (100). The presence of a thymidine in this SNP, leads to over-expression of the mutant allele in GH3 pituitary-derived cells, thus having the potential to generate a tissue-specific dominant-negative condition. However, the T/C nucleotides of this SNP have not been correlated with the clinical presentation in individuals with this most common TRß R338W mutation. No mutations specific to the TRß2 involved in the hypothalamic-pituitary feedback regulation have been identified.
Molecular Basis of the Variable RTHβ Phenotype
The extremes of the RTHß phenotype have a readily apparent molecular basis. Subjects heterozygous for a THRBgene deletion are normal because the expression of a single THRB allele is sufficient for normal function. RTHß manifests in homozygotes completely lacking the THRB gene and in heterozygotes that express a mutTRß with DNE. The most severe form of RTHß, with extremely high TH levels and signs of both hypothyroidism and thyrotoxicosis, occur in homozygous individuals expressing only mutTRßs (35, 36). The severe hypothyroidism manifesting in bone and brain of such subjects can be explained by the silencing effect of a double dose of mutTR and its interference with the function of TRα (72), a situation which does not occur in homozygous subjects who lack TRß. In contrast, the manifestation of thyrotoxicosis in other tissues, such as the heart, may be explained by the effect of high TH levels on tissues that normally express predominantly TRα1 (101, 102) (see Animal Models of RTH, below). For this same reason, tachycardia is a relatively common finding in RTHß (103).
Various mechanisms can be postulated to explain the tissue differences in TH resistance within the same subject and among individuals. The distribution of receptor isoforms varies from tissue to tissue (50, 104, 105). This likely accounts for greater hormonal resistance of the liver as compared to the heart. Differences in the degree of resistance among individuals harboring the same mutTRß could be explained by the relative level of mutant and WT TR expression. Such differences have been found in one study using cultured fibroblasts (106) but not in another (69). Various reasons for a predominant TRß2 dysfunction have been presented in the preceding section.
Although in a subset of mutTRßs a correlation was found between their functional impairment and the degree of thyrotroph hyposensitivity to TH, this correlation is not maintained in terms of hormonal resistance in peripheral tissues (90). Subjects with the same mutations, even belonging to the same family, show different degrees of hormone resistance. A most striking example is that of family G.H. in which the mutTRß R316H did not co-segregate with the RTHß phenotype in all family members (107). This variability of clinical and laboratory findings was not observed in affected members of two other families with the same mutation (29, 108). A study in a large family with the mutTRß R320H, suggests that genetic variability of factors other than TR may modulate the phenotype of RTHß (109).
RTHß Without THRB Gene Mutations (nonTRß-RTH)
The molecular basis of nonTRß-RTH remains unknown. Since the first demonstration of nonTRß-RTH (21), more than 75 families with the phenotype of RTHß have been identified, in which affected individuals did not harbor germline mutations in the THRB, 39 of which in the authors’ laboratory (42, 110-113). The phenotype is indistinguishable from that in subjects with THRB gene mutations. Distinct features are an increased female to male ratio and a high prevalence of sporadic cases. While it has been postulated that nonTRß-RTH is likely caused by a defect in one of the cofactors involved in the mediation of TH action, proof supporting this contention is lacking (114). Recently, in-depth targeted new generation sequencing revealed mosaicism of previously reported THRB gene mutations in 19% of families (33) as previously identified in one family (115). Two families with more than one affected individual were found to harbor a THRB gene mutation that had been missed when early sequencing required cloning of amplified fragments into plasmids (33).
Animal Models of RTHß
Understanding the action of TH in vivo, and the mechanisms underlying the abnormalities observed in patients with RTHß, has been bolstered by observations made in genetically manipulated mice. Three types of genetic manipulations have been applied: (a) transgenic mice that overexpress a receptor; (b) deletion of the receptor (knockout or KO); and (c) introduction of mutations in the receptor (knockin or KI). The latter two types of gene manipulation, species differences notwithstanding, have yielded true models of the recessively and dominantly inherited forms of RTHß (116).
The features of RTHß found in patients homozygous for TRß deletion also manifest in the TRß deficient mouse (117-119). Special features, such as sensorineural deafness and monochromatic vision are characteristic and shared by mouse (120) (121) and man (1, 122). The mouse model allowed for investigations in greater depth into the mechanisms responsible for the development of these abnormalities. Thus, TRß deficiency retards the expression of fast-activating potassium conductance in inner hair cells of the cochlea that transforms the immature cells from spiking pacemakers to high-frequency signal transmitters (123). TRß2 interacts with transcription factors providing timed and spatial order for cone differentiation. Its absence results in the selective loss of M-opsin (121). The down regulation of hypothalamic TRH is also TRß2 specific (124). Mice deficient in TRß have increased heart rate that can be decreased to the level of the WT mouse by reduction on the TH level (119). This finding, together with the lower heart rate in mice selectively deficient in TRα1 (101), indicates that TH dependent changes in heart rate are mediated through TRα, and explains the tachycardia observed in some patients with RTHß.
The combined deletion of TRα1 and α2, produces no important alterations in TH or TSH concentrations in serum (55). The complete lack of TRs, both α and ß, is compatible with life (55, 56). This contrasts with the complete lack of TH which, in the athyreotic Pax8 deficient mouse, results in death prior to weaning, unless rescued by TH treatment (125). The survival of mice deficient in both TRα and ß is not due to expression of a yet unidentified TR but to the absence of the noxious silencing effects of aporeceptors. Indeed, removal of the Thrα gene rescues the Pax8 KO mice from death (126). The combined Thrß and Thrα deficient mice have serum TSH levels that are 500-fold higher than those of the WT mice, and T4 concentrations 12-fold above the average normal mean (55). Thus, the presence of an aporeceptor does not seem to be required for the upregulation of TSH but no amount of TH causes its downregulation.
The first animal model of the dominantly inherited organ-limited RTHß utilized somatic transfer of a mutTRß1 G345Rmutation by means of a recombinant adenovirus (127). The liver of these mice was resistant to TH, and overexpression of the WT TRß increased the severity of hypothyroidism, confirming that the unliganded TR has a constitutive effect both in vivo as in vitro. True mouse models of dominantly inherited RTHß have been generated by targeted mutations in the Thrß gene (128, 129). The mutations were modeled on those identified in humans with RTH [frame-shift resulting in 16 carboxylterminal nonsense amino acids (PV mouse) and T337D]. As in humans, the phenotype seen in the heterozygous KI animals was more severe in mice lacking both Thrß alleles.
NcoA (SRC-1) deficient mice have RTHß with typical increase in T4, T3 and TSH concentrations (130). A milder form of RTHß was identified in mice deficient in RXRg (131). These animals show reduced sensitivity to L-T3 in terms of TSH downregulation but not in metabolic rate. These data indicate that abnormalities in cofactors can produce RTHß.
Pathogenesis
The reduced sensitivity to TH in subjects with RTHß is shared to a variable extent by most tissues. The hyposensitivity of the pituitary thyrotrophs results in a non-suppressed serum TSH, which in turn, increases the synthesis and secretion of TH. The persistence of TSH secretion in the face of high levels of serum free TH contrasts with the low TSH levels in the more common forms of TH hypersecretion that are TSH-independent. This apparent paradoxical dissociation between TH and TSH is responsible for the wide use of the term "inappropriate secretion of TSH" to designate the syndrome. However, TSH hypersecretion is not at all inappropriate, given the fact that the response to TH is reduced. It is compensatory and appropriate for the level of TH action mediated through a defective TRß. As a consequence, most patients are eumetabolic, though the compensation is variable among affected individuals and among tissues in the same individual. However, the level of tissue responses does not correlate with the level of TH, probably owing to discordance between the hormonal effect on the pituitary and other body tissues. Thyroid gland enlargement occurs with chronic, though minimal TSH hypersecretion due to increased biological potency of this glycoprotein through increased sialylation (132). Administration of supraphysiological doses of TH is required to suppress TSH secretion without induction of global thyrotoxic changes in peripheral tissues.
Thyroid-stimulating antibodies, which are responsible for the hyperactivity of the thyroid gland in Graves' disease, have been conspicuously absent in patients with RTHß. Another potential thyroid stimulator, human chorionic gonadotropin, has not been found in serum of subjects with RTHß (133, 134).
The selectivity of the resistance to TH has been convincingly demonstrated. When tested at the pituitary level, both thyrotrophs and lactotrophs were less sensitive only to TH. Thyrotrophs responded normally to the suppressive effects of the dopaminergic drugs L-dopa and bromocriptine (135, 136), as well as to glucocorticoids (136-138). Studies carried out in cultured fibroblasts confirm the in vivo findings of selective resistance to TH. The responsiveness to dexamethasone, measured in terms of glycosaminoglycan (139) and fibronectin synthesis (140), was preserved in the presence of T3 insensitivity.
Several of the clinical features encountered in some patients with RTHß may be the manifestation of selective tissue deprivation of TH during early stages of development. These clinical features include retarded bone age, stunted growth, mental retardation or learning disability, emotional disturbances, attention deficit/hyperactivity disorder (ADHD), hearing defects, and nystagmus (5). A variety of associated somatic abnormalities appear to be unrelated pathogenically and may be the result of involvement of other genes such as in major deletions of DNA sequences (34). However, no gross chromosomal abnormalities have been detected on karyotyping (1, 141).
Pathology
Little can be said about the pathologic findings in tissues other than the thyroid. Electron microscopic examination of striated muscle obtained by biopsy from one patient revealed mitochondrial swelling, also known to be encountered in thyrotoxicosis (1). This is compatible with the predominant expression of TRα in muscle, responding to the excessive amount of circulating TH (142). Light microscopy of skin fibroblasts stained with toluidine blue showed moderate to intense metachromasia (2) as described in myxedema. However, in contrast to patients with TH deficiency, treatment with the hormone failed to induce the disappearance of the metachromasia in fibroblasts from patients with RTHß.
Thyroid tissue, obtained by biopsy or at surgery, revealed various degrees of hyperplasia of the follicular epithelium (136, 143-145). Specimens have been described as "adenomatous goiters", "colloid goiters”, and normal thyroid tissue. When present, lymphocytic infiltration is due to the coexistence of thyroiditis (146).
Clinical Features
Characteristic of the RTHß syndrome is the paucity of specific clinical manifestations. When present, manifestations are variable from one patient to another. Investigations leading to the diagnosis of RTHß have been undertaken because of the presence of goiter, hyperactive behavior or learning disabilities, developmental delay and sinus tachycardia (Figure 4). Fortuitous detection of RTHß on laboratory testing can become more common with the increased frequency of routine thyroid testing. The finding of elevated serum TH levels in association with a non-suppressed TSH is usually responsible for the pursuit of further studies leading to the diagnosis.
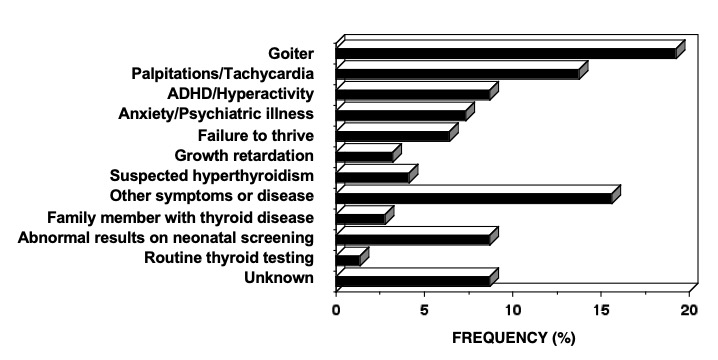
Figure 4. The reasons prompting further investigation of the index member of each family with RTHß.
The degree of compensation for tissue hyposensitivity by the increased levels of TH is variable among individuals, as well as in different tissues. As a consequence, clinical and laboratory evidence of TH deficiency and excess often coexist. For example, RTHß can present with a mild to moderate growth retardation, delayed bone maturation and learning disabilities suggestive of hypothyroidism, alongside with hyperactivity and tachycardia compatible with thyrotoxicosis. The common clinical features and their frequency are given in Table 2. Frank symptoms of hypothyroidism are more common in those individuals who, because of erroneous diagnosis, have received treatment to normalize their circulating TH levels.
|
Table 2. Clinical Features of RTHß
|
|
FINDINGS
|
FREQUENCY (%)
|
|
Thyroid gland
|
|
Goiter
|
66-95
|
|
Nervous System
|
|
Hyperkinetic behavior
|
33-68
|
|
Attention deficit hyperactivity disorder
|
40-60
|
|
Learning disability
|
30
|
|
Mental retardation (IQ <70)
|
4-16
|
|
Hearing loss (sensorineural)
|
10-22
|
|
Growth and Development
|
|
Short stature (<5th percentile)
|
18-25
|
|
Delayed bone age >2 SD
|
29-47
|
|
Low body mass index (in children)
|
33
|
|
Recurrent Ear and Throat Infections
|
55
|
Goiter is the most common abnormality. It has been reported in 66-95% of cases and is almost always detected by ultrasonography. The enlargement of the gland is usually diffuse; nodular changes and gross asymmetry are found in recurrent goiters after surgery. Goiter is more often present in children with RTHß born to normal than to affected mothers (96).
Sinus tachycardia is also very common, with some studies reporting a frequency as high as 80% (28). Palpitations often bring the patient to medical attention, and the finding of tachycardia is the most common reason for the erroneous diagnosis of autoimmune thyrotoxicosis or the suspicion of PRTH.
About one-half of subjects with RTHß have some degree of learning disability with or without ADHD (5, 147). One-quarter have intellectual quotients (IQ) less than 85, but frank mental retardation (IQ <60) has been found only in 3% of cases. Impaired mental function was found to be associated with impaired or delayed growth (<5th percentile) in 20% of subjects, although isolated growth retardation is rare (4%) (5). Despite the high prevalence of ADHD in patients with RTH, the occurrence of RTHß in children with ADHD must be very rare, none having been detected in 330 such children studied (148, 149). The higher prevalence of low IQ scores appear to confer a higher likelihood for subjects with RTHß to exhibit ADHD symptoms (108). A retrospective survey has shown an increased miscarriage rate and low birth weight of normal infants born to mothers with RTHß (150). These same individuals, exposed to high TH levels during embryonic life, develop reduced sensitivity to TH as adults despite the absence of THRB gene mutations. This epigenetic effect is transmitted along the male line for at least three generations (151).
A variety of physical defects that cannot be explained on the basis of TH deprivation or excess have been recorded. These include major or minor somatic defects, such as winged scapulae, vertebral anomalies, pigeon breast, prominent pectoralis, birdlike facies, scaphocephaly, short 4th metacarpals, as well as Besnier's prurigo, congenital ichthyosis, and bull's eye type macular atrophy (5). Some may be related to the severity of the hormonal resistance as they manifest in homozygotes (36). An infant compound heterozygous for a THRB gene mutation (R338W and R429W) presented with a cone photoreceptor disorder associated with severe thyroid hormone resistance (152).
Course of Disease
The course of the disease is as variable as is its presentation. Most subjects have normal growth and development, and lead a normal life at the expense of high TH levels and a small goiter. Others present variable degrees of mental and growth retardation. Symptoms of hyperactivity tend to improve with age as it does in subjects with ADHD only.
Goiter has recurred in every patient who underwent incomplete thyroid surgery. As a consequence, some subjects have been submitted to several consecutive thyroidectomies or treatments with radioiodine (145, 153-155). Thyroid cancer has been rarely reported in individuals with RTHß and, when occurring, the outcome has not been unfavorable despite incomplete TSH suppression (156).
Laboratory Findings
In the untreated patient, elevation in the concentration of serum free T4 is a sine qua non requirement for the diagnosis of RTHß. It is often accompanied by high serum levels of T3, but less so with advancing age. Serum thyroid binding globulin (TBG) and transthyretin (TTR) concentrations are normal. The resin T3 uptake is usually high as is the case in patients with thyrotoxicosis.
Serum T4 and T3 values range from just above to several fold the upper limit of normal. Although the levels may vary in the course of time in the same patient (28), the T3:T4 ratio remains normal (5). This contrasts with the disproportionate increase in serum T3 concentration characteristic of autoimmune thyrotoxicosis (157).
Reverse T3 concentration is also high in patients with RTHß as is that of another product of T4 degradation, 3,3'-T2 (144). Serum thyroglobulin level tends also to be high and the degree of its elevation reflects the level of TSH induced thyroid gland hyperactivity.
In vivo turnover kinetics of T4 showed a normal or slightly increased volume of distribution and fractional disappearance rate of the hormone. However, because of the large extrathyroidal pool, the absolute daily production of T4 and T3 are increased by about two- to four-fold (2, 153, 158, 159), but the extrathyroidal conversion of T4 to T3 remains normal (159).
THYROTROPIN AND OTHER THYROID STIMULATORS
A characteristic feature of the syndrome is the preservation of the TSH response to TRH despite the elevated TH levels (160). In most cases, the basal serum TSH concentration is normal and the circadian rhythm is unaltered (161, 162). TSH values above 6 mU/L indicate a decrease in thyroidal reserve due to treatment directed to the thyroid or associated thyroid disease. The severity of the central RTHß can be quantitated, even in the presence of reduced thyroidal reserve, using the thyrotroph T4 resistance index (TT4RI); the product of serum FT4, expressed as percent of the upper limit of normal, and the TSH level (91).
TSH has increased biological activity (132, 163) and the free alpha subunit (α-SU) is not disproportionately high. Antibodies against thyroglobulin and thyroid peroxidase indicating the presence of autoimmune thyroid disease, have a higher prevalence in RTHß (164).
THYROID GLAND ACTIVITY AND INTEGRITY OF HORMONE SYNTHESIS
The fractional uptake of radioiodine by the thyroid gland is high as is the absolute amount of accumulated iodide. The latter is normally organified as demonstrated by the retention of radioiodine following the administration of perchlorate (1, 153, 165).
IN VIVO EFFECTS OF TH
The impact of TH on peripheral tissues, assessed in vivo by a variety of tests, suggests a reduced biologic response to the hormone in some tissues but not in others. Early studies measuring the metabolic rate (BMR) evaluated by measurement of oxygen consumption showed normal results (2). However resting energy expenditure, measured subsequently by indirect calorimetry was increased, but not the rate of ATP synthesis, measured by magnetic resonance spectroscopy (166). This indicates that in subjects with RTHß, the basal mitochondrial substrate oxidation is increased and energy production in the form of ATP synthesis is decreased. Yet, the metabolic response to the administration of TH is reduced compared to normal individuals (5). With the exception of increased resting pulse rate in about one half of the patients with RTHß, the cardiac function is only minimally altered. Two-dimensional and Doppler echocardiography showed findings consistent with a mild excess of TH on cardiac systolic and diastolic function whereas other parameters, such as ejection and shortening fractions of the left ventricle, systolic diameter, and left ventricle wall thickness, were normal (103). Findings suggestive of hypothyroidism have also been reported (167). The Achilles tendon reflex relaxation time has been normal or slightly prolonged.
In contrast to overt thyrotoxicosis, serum parameters of TH action on peripheral tissues are usually in the reference range. These include, serum cholesterol, carotene, triglycerides, creatine kinase, alkaline phosphatase, angiotensin-converting enzyme, sex hormone-binding globulin (SHBG), ferritin and osteocalcin. Urinary excretion of magnesium, hydroxyproline, creatine, creatinine, carnitine, and cyclic adenosine monophosphate (cAMP), all found to be elevated in thyrotoxicosis, have been normal or low, suggesting normal or slightly reduced TH effect. The prolactin hyper-responsiveness in some patients with RTHß may be due to the functional TH deprivation at the level of the lactotrophs (160).
Radiological evidence of delayed bone maturation has been observed in one-half of patients with RTHß diagnosed during infancy or childhood (5). However, the majority achieve normal adult stature.
Evaluation of endocrine function by a variety of tests has failed to reveal significant defects other than those related to the thyroid (5).
In Vitro Tests of Thyroid Hormone Action
Cultured skin fibroblasts from patients with RTHß showed reduced responses to L-T3 added to the medium in terms of degradation rate of lipoproteins (155), synthesis of glycosaminoglycans (139) and fibronectin (140). This was also true for L-T3-induced changes on specific messenger ribonucleic acid (mRNA) (168). The normal responses of dexamethasone were preserved indicating that the activity of the glucocorticoid receptor was preserved.
Responses to the Administration of Thyroid Hormone
Because reduced responsiveness to TH is central in the pathogenesis of the syndrome, patients have been given TH in order to observe their responses and thereby establish the presence of hyposensitivity to the hormone. Unfortunately, the data have been discrepant, not only because of differences in the relative degree of resistance to TH among patients, but also because of differences in the manner in which tests have been carried out.
The dose of TH that suppresses the TSH secretion, and eventually abolishes the TSH response to TRH, is greater than that required for unaffected individuals. The decreased TSH secretion during the administration of supraphysiological doses of TH is accompanied by a reduction in the thyroidal radioiodine uptake and, when exogenous L-T3 is given, a reduction in the pretreatment level of serum T4 (133, 134, 145, 153, 155).
Various responses of peripheral tissues to the administration of TH have been quantitated. Most notable are measurements of the BMR, pulse rate, reflex relaxation time, serum cholesterol, lipids, enzymes, osteocalcin and SHBG, and urinary excretion of hydroxyproline, creatine, and carnitine. Either no significant changes were observed, or they were much reduced relative to the amount of TH given (5).
Of great importance are observations on the catabolic effect of exogenous TH. In some subjects with RTHß, L-T4 given in doses of up to 1000 µg/day, and L-T3 up to 400 µg/day, failed to produce weight loss without a change in caloric intake, nor did they induce a negative nitrogen balance (2, 133, 136). In contrast, administration of these large doses of TH over a prolonged period of time was apparently anabolic as evidenced by a dramatic increase in growth rate and accelerated bone maturation (30, 136).
Effects of Other Drugs
As expected, administration of the TH analogue, 3,5,3'-triiodo-L-thyroacetic acid (TRIAC) to patients with RTHß produced attenuated responses (2, 162, 169).
Administration of glucocorticoids promptly reduced the TSH response to TRH and the serum T4 concentration (133, 136, 137, 143, 158).
Administration of L-dopa and bromocriptine produced a prompt suppression of TSH secretion, as well as a diminution of the thyroidal radioiodine uptake and serum T3 level (135, 136, 143). Domperidone, a dopamine antagonist, caused a rise in the serum TSH level when given to patients with RTHß (162). These observations indicate that, in this syndrome, the normal inhibitory effect of dopamine on TSH is intact.
The response to antithyroid drugs has shown some variability. Methimazole and propylthiouracil, in doses usually effective in reducing the high serum TH level of autoimmune hyperthyroidism, had no effect in two patients (2). However, in other cases of RTHß, antithyroid drugs induced some decrease in the circulating level of TH, producing a reciprocal change in the TSH concentration (3, 141, 165, 170). Administration of 100 mg of iodine daily had a similar effect in one patient (134), but 4 mg potassium iodide per day produced no changes in another (2).
The ß adrenergic blockers, propranolol and atenolol, produce a significant reduction in heart rate.
Differential Diagnosis
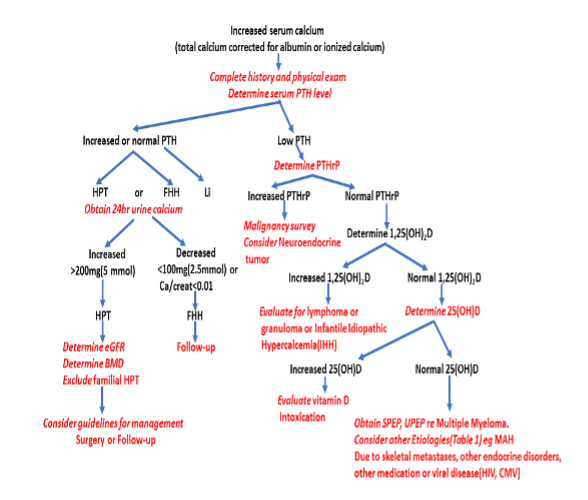
Because the clinical presentation of RTHß is variable, detection requires a high degree of suspicion. The differential diagnosis includes all possible causes of hyperthyroxinemia (Table 3). The sequence of diagnostic procedures listed in Table 4 is suggested.
|
Table 3. Serum Thyroid Function Tests in the Differential Diagnosi 0f Impaired Sensitivity to Thyroid Hormone
|
|
Defect
|
T4
|
T3
|
rT3
|
T3/rT3ratio
|
TSH
|
FT4 Dialysis
|
Other common manifestations
|
|
RTHß
|
↑
|
↑ or N
|
↑
|
N
|
N or ↑
|
↑
|
tachycardia, goiter, ADHD
|
|
RTHα
|
N or sl↓
|
N or sl↑
|
N sl↓
|
↑
|
N or sl↑
|
N or sl↓
|
growth and mental delay, constipation
|
|
TSHoma
|
↑
|
↑
|
↑
|
N
|
sl↑ or N
|
↑
|
thyrotoxicosis
|
|
MCT8 mut
|
N or ↓
|
↑↑
|
↓↓
|
↑↑
|
N or sl↑
|
↓
|
neuropsychomotor delay
|
|
SBP2 muta
|
↑
|
↓
|
↑
|
↓↓
|
N or sl↑
|
↑
|
growth delay
|
|
FDH (ALBmut)
|
↑
|
N or sl↑b
|
↑
|
↓
|
N
|
N or ↑
|
none
|
|
TBG excess
|
↑
|
↑
|
↑
|
N
|
N
|
N
|
none
|
|
Acute NTI
|
↑
|
↓↓
|
↑
|
↓
|
N
|
N or ↑
|
variable depending on illness
|
sl: slightly; N: normal; ↑: increased; ↓: decreased; mut: mutation
ADHD: attention deficit hyperactivity disorder; NTI: non-thyroidal illness
FDH: familial dysalbuminemic hyperthyroxinemia
a Low serum selenium
b High in ALB L66P
|
Table 4. Suggested Sequence of Diagnostic Procedures in Suspected RTHß
|
|
1. 1. Usual presentation: high serum levels of free T4 with non-suppressed TSH.
|
|
2. Confirm the elevated serum level of free T4 and exclude interfering substance, such as antibodies to T4, and other serum TH transport defects, especially if T3 is normal and obtain free T4 measurement by equilibrium dialysis
|
|
3. Obtain tests of thyroid function in first-degree relatives; parents, sibs and children
|
|
4. Sequence the THRB gene and if a mutation is detected shown to have an impaired function, the diagnosis of RTHß is secured
|
|
5. In the absence of THRB gene mutation and lack of abnormal thyroid function tests in other family members, the presence of a TSHoma should be excluded by measurement of the α-SU in serum and other appropriate tests (T3-suppression, TRH stimulation and MRI).
|
|
6. 6. Demonstrate a blunted TSH-suppression and metabolic response to the administration of
supraphysiological doses of TH (see response to L-T3 protocol, Figure 5).
|
|
7. 7. Blunted TSH response to L-T3 with absence of THRB gene mutation in indicates nonTRß-RTH, which includes possible THRB mosaicism.
|
The presence of an elevated serum T4 concentration with a non-suppressed TSH needs to be confirmed by repeated testing. The possibility of an inherited or acquired increase in T4-binding to serum TH transport proteins must be excluded by direct measurement and by estimation of the circulating free T4 level. The presence of a high serum T3 is helpful, though normal levels do not exclude RTHß. Examples of instances when serum T3 is not high are: transiently during the administration of some drugs, or with concomitant nonthyroidal illnesses (see other Endotext chapters), and permanently with advanced age, familial dysalbuminemic hyperthyroxinemia (FDH) and inherited defects of iodothyronine metabolism (see the THMD Section in this Chapter). In FDH, free T4 measured by automated direct methods but not by equilibrium dialysis may be falsely elevated. A rare cause of elevated serum T4 and T3 level is the endogenous production of antibodies directed against these iodothyronines, which can be excluded by direct testing.
Measurement of the serum TSH is an absolute requirement. Under most circumstances, patients with high concentrations of circulating free TH have virtually undetectable serum TSH levels, which fail to respond to TRH. This is true even when the magnitude of TH excess is minimal and therefore subclinical, either on physical examination or by other laboratory tests. The combination of elevated serum levels of free TH and non-suppressed TSH, narrows the differential diagnosis to one of the syndromes of reduced sensitivity to TH and autonomous hypersecretion of TSH associated with pituitary tumors (TSHomas). The clinical and laboratory findings of the latter mimic those of RTHß with a few exceptions. TSHomas have: 1) a disproportionate abundance in serum free α-SU relative to whole TSH (171); 2) lack similar thyroid test abnormalities in parents and first degree relatives; 3) with rare exceptions (172), their serum TSH fails to respond to TRH or suppress with supra-physiologic doses of TH; 4) often have concomitant hypersecretion of growth hormone and or prolactin; and 5) in the majority of cases, tumors can be demonstrated by computerized tomography (CT) or by magnetic resonance imaging (MRI) of the pituitary.
Rarely, subjects with autoimmune thyrotoxicosis may have endogenous antibodies to TSH or some of the test components, that can give rise to false increase in serum TSH values (173). Ectopic production of TSH and endogenous TRH hypersecretion could theoretically result in TSH-induced hyperthyroidism. The presence of high serum free T3 or free T4 only, in the presence of a non-suppressed TSH, is characteristic of the syndromic abnormalities of TH cell transport and metabolism, respectively (see the THCMTD and THMD Sections in this Chapter).
Proving the existence of isolated peripheral tissue resistance to TH is not simple. Lack of clinical symptoms and signs of hypermetabolism are insufficient to establish the diagnosis of RTHß and symptoms suggestive of thyrotoxicosis are not uncommon in RTHß. Because resistance to the hormone is variable in different tissues, no single test measuring a particular response to TH is diagnostic. Furthermore, results of most tests that measure the effect of TH on peripheral tissues show considerable overlap among thyrotoxic, euthyroid and hypothyroid subjects. The value of these tests is enhanced if measurements are obtained before and following the administration of supraphysiological doses of TH.
While the demonstration of a THRB gene mutation is sufficient to establish the diagnosis of RTHß, lack of cosegregation of the THRB haplotype with the phenotype has been used to exclude the involvement of TRß in the individuals suspected of having RTHß (174). This does not exclude mosaicism (115) when a single member of the family is affected (see nonTRß-RTH Section in this Chapter). In such cases, in vivo demonstration of tissue resistance to TH is required. A standardized diagnostic protocol, using short-term administration of incremental doses of L-T3, and outlined in Figure 5, is recommended. It is designed to assess several parameters of central and peripheral tissue effects of TH in the basal state and in comparison to those determined following the administration of L-T3. The three doses, given to adults in sequence, are a replacement dose of 50 µg/day and two supraphysiological doses of 100 and 200 µg/day. The hormone is administered in a split dose every 12 hours and each incremental dose is given for the period of 3 days. Doses are adjusted in children and in adults of unusual size to achieve the same level of serum T3 (for details see reference (5)). L-T3, rather than L-T4, is used because of its direct effect on tissues, bypassing potential defects of T4 transport and metabolism, which may also produce attenuated responses. In addition, the more rapid onset and shorter duration of T3 action reduces the period required to complete the evaluation and shortens the duration of symptoms that may arise in individuals with normal responses to the hormone. Responses to each incremental dose of L-T3 are expressed as increments and decrements or as a percent of the value measured at baseline. The results of such a study are shown in Figure 6.
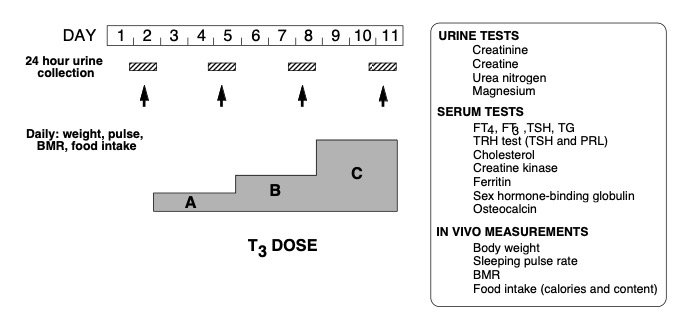
Figure 5. Schematic representation of a protocol for the assessment of the sensitivity to TH using incremental doses of L-T3. For details see text.
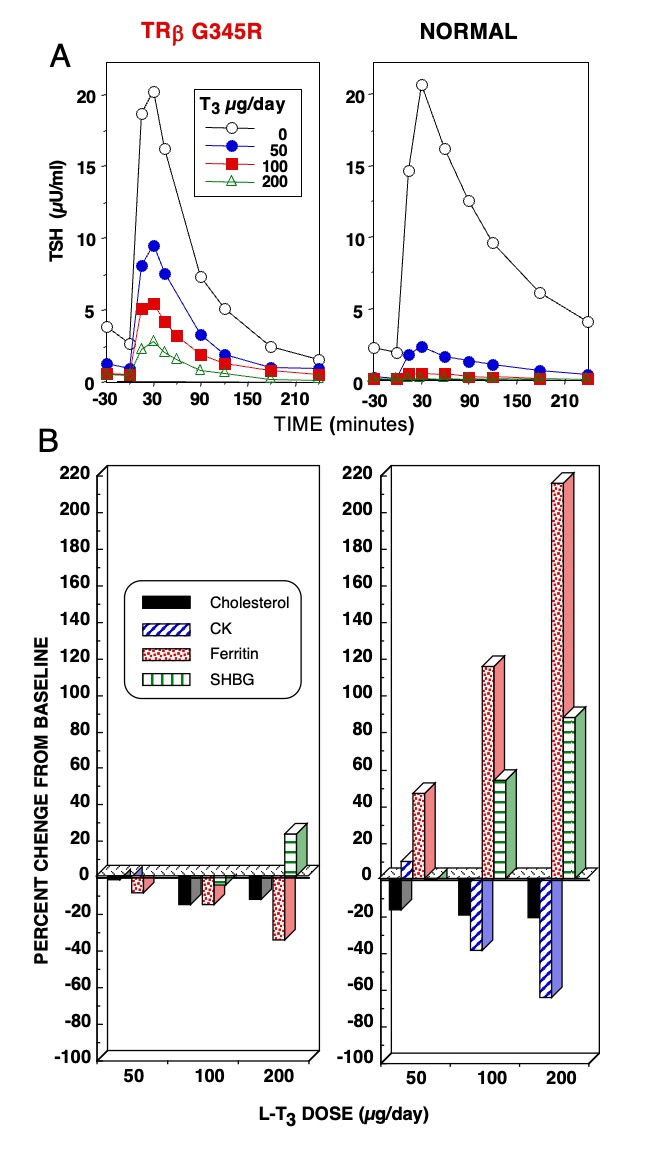
Figure 6. Responses to the administration of L-T3 in a subject with RTHß harboring TRß G345R mutant and an unaffected individual. The hormone was given in three incremental doses, each for 3 days as illustrated in Figure 5. Results are shown at baseline and after each dose of L-T3. (A) TSH responses to TRH stimulation. Note the reduced suppression of the TSH response by L-T3 in the individual with RTHß. (B) Responses of peripheral tissues. Note the stimulation of ferritin and sex hormone binding globulin (SHBG) and the suppression of cholesterol and creatine kinase (CK) in the normal subject. Responses in the affected subject were blunted or paradoxical.
The diagnosis of RTHß is particularly challenging when the latter is associated with other thyroid diseases, such as autoimmune thyrotoxicosis that suppresses the TSH level (175) or with congenital (176, 177) or acquired (178)hypothyroidism. Failure to differentiate RTHß from ordinary thyrotoxicosis continues to result in inappropriate treatments. The diagnosis requires awareness of the possible presence of RTHß, usually suspected when high levels of circulating TH are not accompanied by a suppressed TSH.
Treatment
No specific treatment is available to fully and specifically correct the defect. Theoretically, an ideal treatment for RTHß caused by mutant TRßs with altered TH binding would be to design mutation-specific TH analogues that overcome the binding defect (179). The ability to identify specific mutations in the THRB gene provides a means for prenatal diagnosis and appropriate family counseling. This is particularly important for families whose affected members show evidence of growth or mental retardation. Fortunately, in most cases of RTHß, the partial tissue resistance to TH appears to be adequately compensated for by an increase in the endogenous supply of TH. Thus, treatment need not be given to such patients. This is not the case in patients who have undergone ablative therapy or have a concomitant condition limiting their thyroidal reserve. In these patients, the serum TSH level can be used as a guideline for hormone dosage.
Not infrequently, some peripheral tissues in patients with RTHß appear to be relatively more resistant than the pituitary. Thus, compensation for the defect at the level of peripheral tissues is incomplete. In such instances, judicious administration of supraphysiological doses of the hormone is indicated. Since the dose varies greatly among cases, it should be individually determined by assessing tissue responses. In childhood, particular attention must be paid to growth, bone maturation and mental development. It is suggested that TH be given in incremental doses and that the BMR, nitrogen balance, serum SHBG and osteocalcin be monitored at each dose, and bone age and growth on a longer term. Development of a catabolic state is an indication of overtreatment.
The exact criteria for treatment of RTHß in infancy have not been established. This is often an issue when the diagnosis is made at birth or in early infancy. In infants with elevated serum TSH levels, subclinical hypothyroidism may be more harmful than treatment with TH. Indications for treatment may include a TSH level above the upper limit of the reference range, retarded bone development, and failure to thrive. This may not apply to children homozygous for a mutant TRß. The outcome of affected older members of the family who did not receive treatment may serve as a guideline. Longer follow-up and psychological testing of infants who have been given treatment will determine the efficacy of early intervention.
It is unclear at this time whether intervention during early gestation is appropriate. However, limited experience suggests that the T4 of mothers with RTHß carrying a normal embryo should not be allowed to be higher than 50% the upper limit of normal in order to prevent low birth weight (180). The concept of in utero treatment is questionable (181, 182).
Patients with more severe thyrotroph resistance and symptoms of thyrotoxicosis may require therapy. Usually, symptomatic treatment with an adrenergic ß blocking agent, preferably atenolol, would suffice. Treatments with antithyroid drugs or thyroid gland ablation increase TSH secretion and may result in thyrotroph hyperplasia. Development of true pituitary tumors, even after long periods of thyrotroph overactivity, is extremely rare (183).
Treatment with supraphysiological doses of L-T3, given as a single dose every other day, is successful in reducing goiter size without causing side effects (184). Such treatment is preferable considering that postoperative recurrence of goiter is the rule. The L-T3 dose must be adjusted until TSH and TG are suppressed and reduction of goiter size is observed. L-T3 has been also used with some success in the treatment of ADHD in an individual with RTHß resistant to conventional treatments with stimulants (185).
Among the TH analogues used to alleviate symptoms of apparent TH excess (186), TRIAC has had the widest use(187, 188). It has a relatively greater affinity than T3 for some mutant TRßs (189). In general, TRIAC’s short half-life produces greater effect centrally than on peripheral tissues. This, in turns, reduces TSH and TH secretion with apparent amelioration of hypermetabolism. The value of treatment with D-T4 is questionable. Theoretically, the ideal treatment is development of mutant-specific TH analogues that would rescue the dominant negative effect of the mutant TRß (179).
Patients with presumed isolated peripheral tissue resistance to TH present a most difficult therapeutic dilemma. The problem is, in reality, diagnostic rather than therapeutic. Many, if not most patients falling into this category, are habitual users of TH preparations. Gradual reduction of the TH dose and psychotherapy are recommended.
RESISTANCE TO THYROID HORMONE-ALPHA (RTHα)
Background
Following the identification of a mutation in the THRB gene in 1989, finding one in the THRA gene was implicit. However, this did not come to pass for the next 23 years. With the development of mice deficient in Trα (knockout) (55) and further, mice harboring Thrα gene mutations (knockin), modeled after human mutations known to occur in the THRB gene (190, 191), a phenotype was defined to aid in the identification of similar THRA gene defects in humans. Yet, laboratories searching for mutations in the THRA gene in individuals with low IQ and short without growth hormone deficiency did not succeed. It is through whole exome sequencing that in 2012 the first few families with THRA gene mutations were identified (9, 20, 192, 193). In retrospect, the failure to identify THRA gene mutation using the candidate gene approach was the lack of signature serum thyroid test abnormalities characteristic to individuals with THRB gene mutations.
Incidence and Inheritance
The precise incidence of RTHα is unknown. Because most routine neonatal screening programs are based on the determination of TSH, as is the case of RTHß, subjects with RTHα cannot be detected owing to their normal blood TSH. The RTHα in key cases of half of the reported families has been caused by de novo mutations (194). While the ethnic origin in most reported cases is not specified, white European is presumed based on pictures.
Etiology and Genetics
Mutations in the THRA gene have now been identified in 32 subjects with RTHα belonging to 19 families. They comprise 18 different mutations, 15 of which have been published (9, 195-203) and reviewed (194, 204). E403*, located in a CpG hot-spot was found in two unrelated families (9, 200). All are located in the ligand-binding domain of TRα and 6 of the 18 mutations involve both TRα1 and TRα2, but none affect the REV-RRBα gene transcription from the opposite strand of the THRA locus. Given the 85% amino acid homology between the hinge region and ligand binding domains of TRα1 and TRß with the exception of THRA N359Y, all have been found to have corresponding mutations in the homologous regions of the THRB and five are located in CpG hot spots. As expected, in three of them (A263V/S, R384C/H and E403K/*) mutations have produced more than one codon alteration (Figure 7).
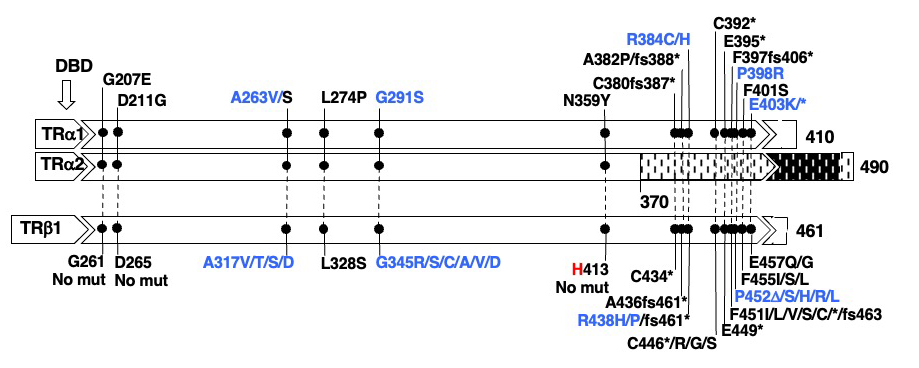
Figure 7. Mutations in TRα1 and TRα2 and in the corresponding amino acid mutations in TRß1 are aligned according to amino acid sequence homology. The single difference is indicated in red. In blue are mutations occurring in hot spots (CpG or CG-rich regions. The ligand binding domain (LBD) containing the mutations is expanded and the locations of mutation is in scale. The DNA binding domain DLBD) is upstream of the mutations. Sequences from amino acid 370 to 490 of the TRα2 diverge from those of TRα1 due to alternative splicing. Data on THRA gene mutations courtesy of Carla Moran, University of Cambridge, United Kingdom.
Molecular Basis of the Defect and Properties of the Mutant TRα
The THRA gene is located on chromosome 17. It generates two protein isoforms, TRα1 and TRα2 by alternative splicing. TRα2 is devoid of a ligand binding and its physiological function through binding on DNA remains unclear. TRα1 functions in the same manner as TRß but there are some differences in the interaction with cofactors and in tissue distribution. It is the latter that is responsible for the differences in the phenotype in individuals harboring mutations in these two transcription factors. By virtue of TRα1 expression predominantly in the central nervous system, bone, intestine and heart, manifestations in these organs dominate. Differences in the severity among mutations can be explained by the degree of loss of function and by the amount of L-T3 required to demonstrate reversibility in vitro (197).
The mechanism causing the defect were convincingly demonstrated in the first reported patient with RTHα harboring a nonsense mutation, produces a truncated TRα1 (E403*) that lacks the C-terminal alpha-helix of the molecule (9). As a consequence, in addition to negligible T3-binding, the mutation promoted corepressor binding while abolishing binding of the coactivator, both contributing to a strong DNE as demonstrated ex vivo. The 6-year-old girl, harboring this mutation, presented with chronic constipation noted upon weaning at 7 months of age, and growth and developmental delay. Hypothyroidism manifested in organs expressing predominantly TRα, including bone, gastrointestinal tract, heart, striated muscle and central nervous system. More specifically X-rays showed patent cranial sutures with Wormian bones, delayed dentition, femoral epiphyseal dysgenesis and retarded bone age. In addition, diminished colonic motility with megacolon, slow heart rate, reduced muscle strength were suggestive of hypothyroidism, as was her placid affect, slow monotonous speech and cognitive impairment. Thyroid function tests, were of subtle nature somewhat reminiscent of MCT8 defects, presumably due to alterations in iodothyronine metabolism (Table 3) (see the THCMTD Section in this Chapter).
Animal Models of RTHα (See also animal models under RTHß, above)
The question of why mutations in the THRA gene have not been identified earlier in man was partially answered by the study of mice with targeted gene manipulations. As stated in an earlier section, THRA gene deletions, total or only α1, failed to produce a RTHß-type serum phenotype. Several human mutations known to occur in the THRB gene were targeted in homologous regions of the Thrα gene of the mouse. These are, the PV frame-shift mutation, Trα1 R384C (equivalent to Trß R438C), Trα P398H (equivalent to Trß P452H) and Trα L400R (corresponding to Trß454) (205). While the resulting phenotypes were somewhat variable, none exhibited thyroid test abnormalities characteristic of RTHß. Common features in heterozygous mice were retarded post-natal development and growth, decreased heart rate, and difficulty in reproducing. Also, all were lethal in the homozygous state, in accordance with the noxious effect of unliganded Trα1.
Clinical Features
The earliest clinical observations are poor feeding, coarse cry and macroglossia. Growth retardation with shorter lower limbs was also noted in infancy (9, 20). Other somatic abnormalities and clinical findings are listed in Table 5. Unusual somatic defects including clavicle agenesis, marked abnormalities of fingers, toes and elbow joints were observed in a single patient with a THRA N359Y mutation (195). It is unlikely that these findings are related to the THRA gene defect. Constipation is a common finding that results in fecal impaction. Bowel dilatation is seen on X-rays. Decreased peristalsis and delayed intestinal transit have been documented. In general, symptoms and signs are compatible with hypothyroidism. These include delayed fontanel closure, slow mentation and motor activity, reduced global IQ, and bradycardia.
|
Table 5. List of Clinical Features of RTHα
|
|
System
|
Infant and Child
|
Adult
|
|
Early features
|
poor feeding; coarse cry; umbilical hernia
|
|
|
Developmental
|
delayed milestones; growth retardation
|
short statute (short limbs)
|
|
Somatic defects
(Dysmorphism)
|
macroglossia, broad and coarse face, broad face, thick lips, flat nasal bridge
|
|
|
skin tags
|
|
Skeletal
|
delayed fontanel closure
epiphyseal dysgenesis
|
serpiginous cranial sutures
cranial and cortical hyperostosis
|
|
Gastrointestinal
|
constipation; bowel dilatation
|
constipation
|
|
Cardiovascular
|
bradycardia
|
bradycardia, low blood pressure
|
|
Neurological
|
delayed speech; dyspraxia
|
dysarthria, slow motor initiation
ataxia, dysdiadochokinesis, low IQ
|
|
Metabolic
|
low metabolic Low metabolic rate, reduced resting energy expenditure, peripheral markers of TH action compatible with hormone deprivation
|
|
Hematological
|
mild anemia
|
mild anemia
|
Data derived from references (193, 298)
Laboratory Findings
Thyroid test abnormalities are not as prominent as in other syndromes of impaired sensitivity to TH and explain the failure to readily recognize the defect. There is a trend for serum T4 and rT3 to be low and T3 to be high. However, in most instances the concentrations of these iodothyronines are not outside of the reference range. Yet, the T3/rT3 ratio is consistently high and a good laboratory biomarker (Table 3). TSH is usually within the upper part of the reference range. A number of test results are compatible with reduced TH action in peripheral thyroid tissues but not centrally. There is a trend for insulin like growth factor I (IGF-1) to be low and for low density lipoprotein (LDL) cholesterol and creatine kinase to be elevated. Bone mineral density is increased. Anemia is a common finding and has been observed as early as 7 months of age (33). Anemia is normocytic with normal reticulocyte count, hemolytic indices, vitamin B12 and folate.
Differential Diagnosis
Suspicion for the presence of RTHα is based on clinical rather than laboratory findings. The serum thyroid test abnormalities are subtle and their relative pattern somewhat reminiscent of those found in MCT8 deficiency. However, individuals with THRA gene mutations do not manifest the severe neuropsychomotor defect characteristic of MCT8 deficiency. Symptoms of hypothyroidism are disproportionate to the thyroid function test abnormalities and TSH is not elevated as in primary hypothyroidism. The fact that in most cases diagnosis was made by whole exome sequencing attests to the difficulty in identifying patients based on clinical and standard laboratory evaluation. Clinical findings suggestive of congenital hypothyroidism without elevated TSH and a high T3/rT3 ratio should rise a high degree of suspicion.
Treatment
The majority of individuals with THRA gene mutations have received L-T4 treatment. The treatment has shown beneficial effect on symptoms and signs caused by functional thyroid hormone deprivation in peripheral tissues. These include, constipation and bradycardia but not anemia. In children, L-T4 treatment resulted in catch-up growth, motor development and in one case reduced hypotonia (201). The effectiveness of L-T4 treatment in children appears to depend on the severity of the defect [frame shift mutations (203)] and time of treatment initiation. There is no experience with treatment beginning in early infancy.
L-T4 treatment resulted in normalization of low serum T4 and rT3 levels but T3 concentration remained elevated. Serum TSH, on the other hand was suppressed, even with physiological L-T4 doses and FT4 levels within the reference range. This confirms an intact hypothalamic-pituitary axis, which may even be hypersensitive to TH. Peripheral tissue markers of TH action, such as serum SHBG, LDL cholesterol and creatine kinase also responded appropriately to L-T4 treatment.
No treatments with TH analogues have been reported. The use of TRα-specific agonist may be helpful but, being directed to the WT-receptor it will not abrogate the DNE of the mutant TRα. The development of mutant specific analogues, as suggested for RTHß, may be an option.
THYROID HORMONE CELL MEMBRANE TRANSPORTER DEFECT (THCMTD)
Patients with THCMTD caused by X-linked MCT8 deficiency are usually boys identified in infancy or in early childhood with feeding difficulties, severe cognitive deficiency, infantile hypotonia and poor head control. They develop progressive spastic quadriplegia, diminished muscle mass with weakness, joint contractures, and dystonia. Early and characteristic thyroid abnormalities are high serum T3, low T4, and a slightly elevated TSH.
The neurological phenotype is severe and incapacitating in affected individuals, with minimal variability across families. Most importantly, this phenotype is not consistent with classical generalized hyperthyroidism or hypothyroidism. Depending on the type of TH transporters expressed, different tissues manifest the consequences of TH excess or deprivation. Tissues expressing other transporters than MCT8 respond to the high circulating T3 level, resulting in a hyperthyroid state, while tissues dependent on MCT8 for TH transport, are hypothyroid. This complicates treatment as standard TH replacement fails to reach some tissues, while it worsens the hyperthyroidism in others.
The complex and severe neurodevelopmental phenotype together with the pathognomonic thyroid tests including high serum T3, low rT3, low or low normal serum T4 concentrations and normal of slightly elevated serum TSH levels represent the characteristic presentation of the MCT8 deficiency syndrome.
Cell Membrane Transporters of TH
The identification and characterization of several classes of molecules that transport TH across membranes (206), has changed the previously held paradigm of passive TH diffusion into cells (207). These proteins belong to different families of solute carriers: 1) Na+/taurocholate cotransporting polypeptide (NTCP) (208); 2) fatty acid translocase(209); 3) multidrug resistance-associated proteins (210); 4) L-amino acid transporters (LAT) (211), among which LAT1 and LAT2 have been shown to transport TH; 5) members of the organic anion-transporting polypeptide (OATP) family (212), of which OATP1B1 and OATP1B3 are exclusively expressed in liver and transport the sulfated iodothyronines, T4S, T3S, and rT3S and less efficiently the corresponding non-sulfated analogues, whereas OATP1C1 is localized preferentially in brain capillaries and shows a high specificity and affinity towards T4. The latter suggests that OATP1C1 may be important for transport of T4 across the blood-brain barrier (213); and 6) within the monocarboxylate transporter (MCT) family (214), MCT8 and MCT10 are specific TH transporters (215, 216). Differences in tissue distribution and transport kinetics of TH and of other ligands, impart their distinctive roles in the cell-specific delivery of TH.
Early studies using the expression of rat Mct8 in an heterologous system, showed that it potentiated the uptake of T4, T3, rT4, and 3,3′-T2 by 10-fold, but it had no effect on the uptake of sulfated T4, the aromatic amino acids phenylalanine, tyrosine, and tryptophan, or lactate (216). Furthermore, transfection of human MCT8 in mammalian cells enhanced the metabolism of iodothyronines by endogenous deiodinases (217). These studies demonstrated the potent and iodothyronine-specific cell membrane transport function of MCT8.
The importance of MCT8 was most convincingly demonstrated by the identification of the first inherited THCMTD caused by mutations in the MCT8 gene (6, 7). Although presence of the defect is suspected on the basis of clinical findings and standard laboratory tests, genetic confirmation is mandatory. Recently a novel neurodegenerative disease associated with a homozygous missense mutation in the T4 transporter OATP1C1 was reported (218). The patient was a 15.5-year-old girl with normal development during the first year of life, who gradually developed dementia with spasticity and intolerance to cold. Brain imaging demonstrated gray and white matter degeneration and severe glucose hypometabolism. When studied in vitro, decreased uptake of T4 was shown, however, serum thyroid function tests were normal (218). At this point it cannot be excluded that the deficit in another substrate is responsible for or contributes to the phenotype.
Inheritance and Incidence
MCT8 deficiency is a recessive X-linked defect that affects males, while females harboring a mutation are carriers. There is 100% penetrance in males that inherit a deleterious mutation. They manifest neuro-psychomotor and characteristic thyroid test abnormalities, whereas carrier females may show only mild thyroid test abnormalities (6, 219, 220). A female with typical features of MCT8-specific THCMTD had a de novo translocation disrupting the MCT8gene and unfavorable nonrandom X-inactivation (221). No affected male has reproduced. The defect has been reported in individuals of all races and diverse ethnic origins.
The exact incidence of this defect is not known. As most routine neonatal screening programs are based on the determination of TSH, MCT8 deficiency is rarely identified at birth by this mean. In neonatal screening programs based on T4 measurements, a low concentration could potentially identify new cases. The yield is expected to be low given the high frequency of low T4 levels in newborns.
The identification of more than 200 families with MCT8 gene defects during the last 16 years indicates that this syndrome is more common than initially suspected. MCT8 gene mutations can be maintained in the population because carrier females are asymptomatic and fertile, which precludes negative selection. Familial occurrence of MCT8 defects has been documented in many instances, however, genetic information on mothers of affected males is not always available.
Etiology
The clinical condition was first recognized in 1944, in a large family with X chromosomal mental retardation presenting with motor abnormalities (222), a form of syndromic X-linked mental retardation, subsequently named the Allan-Herndon-Dudley syndrome (AHDS). In 1990, the syndrome was mapped to a locus on chromosome Xq21 (223). Following the identification of MCT8, gene mutations in subjects with thyroid abnormalities and neuropsychomotor manifestations were identified (6, 7), and subsequently MCT8 mutations were found in other affected males, including the original family described in 1944 (224). The affected subjects have the characteristic thyroid test abnormalities, which went unnoticed in the past.
A large-scale screening of 401 males with X-linked mental retardation has identified MCT8 gene mutations in only 3 subjects, and two of them had the characteristic thyroid phenotype. The other one had a normal serum T3, but the mutation was also found in an unaffected relative (221). This underscores the importance of performing thyroid tests prior to undertaking gene sequencing, in individuals suspected of having a MCT8 defect on the basis of the neurological phenotype.
Given the existence of other types of TH transporters and their different tissue distributions, it is anticipated that defects in such transport molecules would result in distinct phenotypes, the nature of which is difficult to predict. However, as mice deficient in specific TH transporters become available, some predictions about the nature of such diseases may be deduced despite species constraints. In this regard, mice with targeted inactivation of the Lat2(Slc7a8), Mct10 (Slc16a10) and Oapt1c1 (Slco1c1 or Oatp14) TH transporters showed normal development, growth and normal thyroid function tests (225-227). The distribution of Oatp1c1 differs in the brain of mice compared to humans, with mouse Oatp1c1 being predominantly localized in capillary endothelial and only weak OATP1C1 staining being detected by immunohistochemistry in capillary endothelial cells in human brain sections (228). No mutations have been reported in humans in the LAT2 and MCT10 transporters, however a human case of OATP1C1 defect has been recently reported (218).
The MCT8 Gene and Mutations
The MCT8 gene was first cloned during the physical characterization of the Xq13.2 region known to contain the X-inactivation center (229). It has 6 exons and a large first intron that encompasses >100 kb. It belongs to a family of genes, named SLC16, the products of which catalyze proton-linked transport of monocarboxylates, such as lactate, pyruvate and ketone bodies. The deduced products of the MCT8 (SLC16A2) gene are proteins of 613 and 539 amino acids (translated from two in-frame start sites) containing 12 transmembrane domains (TMD) with both amino- and carboxyl- ends located within the cell (230). The furthest upstream translation start site is absent in most species, including mouse and rat. Thus, the importance of the additional N-terminal sequence of the longer human MCT8 protein is unknown. The demonstration that the rat homologue is a specific transporter of TH into cells in 2003 opened the field to clinical and genetic investigation (215).
Over 150 different MCT8 gene mutations are known (33, 231 ) without counting the larger deletions that are not uncommon in MCT8 due to the large intron 1. Mutations are distributed throughout the coding region of the gene. At the protein level, amino acids in the extracellular and intracellular loops are relatively underrepresented. One could speculate that missense mutations in these domains could putatively result in a milder phenotype, escaping detection, as sequences in these regions are less conserved across species compared to the TMD regions (232).
The types of MCT8 gene mutations other than the large deletions involving one or more exons are shown in Figure 8. Coding single nucleotide substitutions are most common at 66%, while insertions/deletions (in/dels) causing frameshift or in frame insertions or deletions represent 30% and only 4% of defects are located at splice sites. At the protein level, missense mutations are most common at 46%, followed by in/del frameshifts with premature termination at 26%, and nonsense mutations at 20%. A similar small proportion of 4% are in frame in/dels and defects with abnormal splicing. Some mutations have occurred in more than one family, for example, three different single amino acid in frame deletions (F229D, F501D and F554D) have occurred in more than 5 families. Different mutations in the same codon have been found to produce 2 or 3 mutant amino acids for example codon 224 (GCC) was mutated as A224T, V and E. In the families studied in our laboratory, 10% of the mutations occurred de novo, while 52% were present in at least one female carrier and one third occurred in mutation hotspots such as CpG dinucleotides C repeats or A repeats (33).
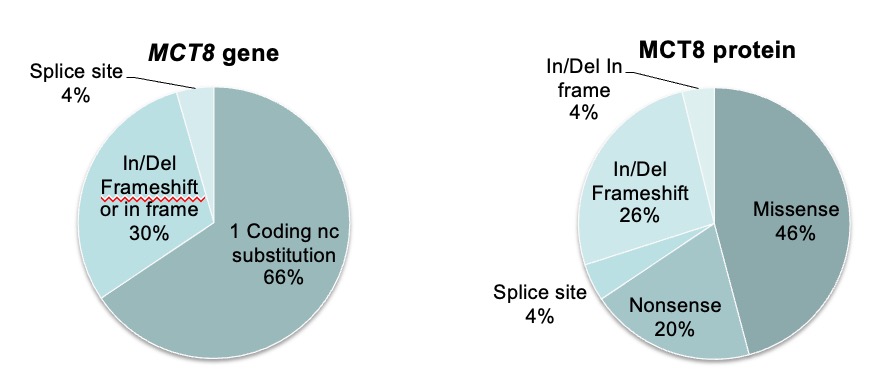
Figure 8. Representation of A. MCT8 gene mutations and B. resulting mutant MCT8 proteins associated with THCMTD, using both data reported by other groups and our published and unpublished cases, for a total of 157 different mutations.
More recently, variants of unknown significance (VUS) in the MCT8 gene have been identified by whole exome sequencing performed in individuals with less typical neurodevelopmental abnormalities. The clinical relevance of these variants has been questioned, especially if the thyroid function tests are not characteristic of MCT8 deficiency. This remains an area of investigation that could further expand the knowledge on genotype-phenotype correlations of VUS in the MCT8 gene.
Clinical Features and Course of the Disease
Male subjects that are later found to have MCT8 gene mutations, are referred for medical investigation during infancy or early childhood because of neurodevelopmental abnormalities. The clinical presentation of affected males with MCT8 gene mutations is very similar, with characteristic thyroid test abnormalities and severe psychomotor retardation.
Newborns have normal Apgar scores and in most cases there is a history of normal gestation. However, polyhydramnios and reduced fetal movements have been reported (33, 224, 233). It is unclear whether this is an intrauterine manifestation of the syndrome. At birth there were no typical signs of hypothyroidism.
Truncal hypotonia and feeding problems are the most common early signs of the defect, appearing in the first 6 months of life. Only in a few cases they manifested within the first few days of life. Characteristically the neurological manifestations progress from flaccidity to limb rigidity and impairment of psychomotor development, leading to spastic quadriplegia with advancing age. With the exception of a few, subjects are unable to walk, stand or sit independently and they do not develop speech. To date, the ability to walk or talk has been reported only in the members of three families (224, 234). These are patients harboring L568P, L434W and F501del mutations who walked with ataxic gait or support, and had a limited and dysarthric speech. A possible explanation for the milder neurological phenotype in these patients is a residual 15-37% TH-binding activity of their mutant MCT8 molecules (235).
Dystonia and purposeless movements are common and characteristic paroxysms of kinesigenic dyskinesias have been reported in several patients, particularly severe in one boy, who presented up to 150 dyskinetic episodes per day (236). These are usually triggered by somatosensory stimuli, such as changing clothes or lifting the child. The attacks consist of extension of the body, opening of the mouth, and stretching or flexing of the limbs lasting for 2 or less than a minute (237). In addition to these non-epileptic events, true seizures can also occur. An altered sleep pattern with difficulty falling asleep and frequent awakening, can represent an important clinical issue for caregivers (236). Reflexes are usually brisk, clonus is often present, but nystagmus and extension plantar responses are less common.
With advancing age, weight gain lags and microcephaly becomes apparent, while linear growth proceeds normally (238). Muscle mass is diminished and there is generalized muscle weakness with typical poor head control, originally described as “limber neck” (222). A common and pronounced feature in MCT8 deficient patients is the failure to thrive, which can be severe, requiring the placement of gastric feeding tubes in some cases. Possible causes for low weight and muscle wasting are the neurologic dysphagia, and increased metabolism due to the thyrotoxic state of peripheral tissues as indicated by reduced cholesterol, and increased transaminases, SHBG, and lactate levels found in some patients with MCT8 mutations (236, 239-241).
Common facial findings that may be attributed to the prenatal and infantile hypotonia include ptosis, open mouth, and a tented upper lip. Ear length is above the 97th centile in about half of adults. Cup-shaped ears, thickening of the nose and ears, upturned earlobes, and a decrease in facial creases have been also reported. Pectus excavatum and scoliosis are common, most likely the result of hypotonia and reduced muscle mass.
While the cognitive impairment is severe, MCT8 deficient patients tend to present a non-aggressive behavior. Generally, affected individuals are attentive, friendly, and docile. Death during childhood or teens is not uncommon, usually caused by recurrent infections and/or aspiration pneumonia. However in a few instances of more mild neurologic involvement, survival beyond age 70 years has been observed (224). Data accumulated by a parent support group between 2009 and 2018 on the causes of death in 24 children and adolescents with MCT8 deficiency with age ranging between 1 day and 23 years, shows respiratory causes with aspiration, pneumonia, sometimes leading to septic shock or death during sleep as being common causes of demise (Figure 9).
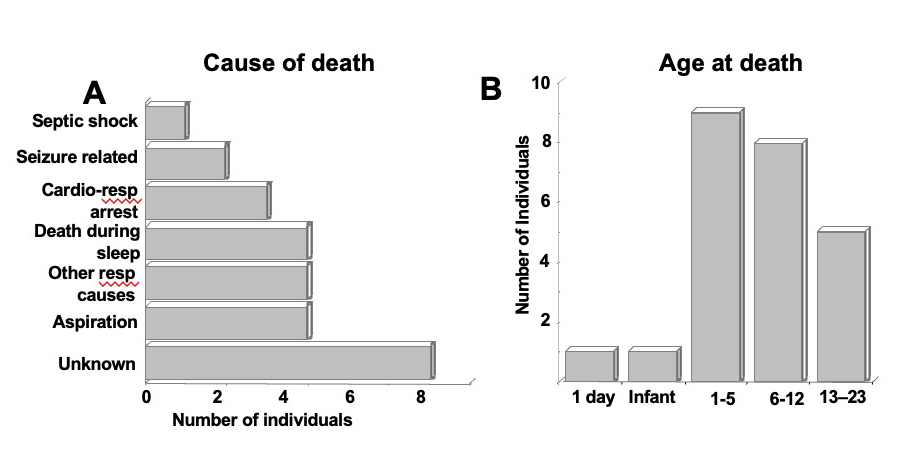
Figure 9. Death in 24 children and adolescents with MCT8 deficiency, between 2009-2018, age range 1 day to 23 years causes (A) and age (B).
Female carriers do not manifest any of the psychomotor abnormalities described above. However, intellectual delay and frank mental retardation have been reported in six carrier females (6, 221, 224, 240). Although an unfavorable nonrandom X-inactivation could alter the phenotype in these females (224), cognitive impairment can be due to a variety of causes. Thus, the causative link of MCT8 mutations in heterozygous females with cognitive impairment remains to be proven (220).
Laboratory Findings
SERUM TESTS OF THYROID FUNCTION
Most characteristic, if not pathognomonic, are the high serum total and free T3 and low rT3 concentrations. T4 is reduced in most cases and TSH levels can be slightly elevated but rarely above 6 mU/L (Figure 10).
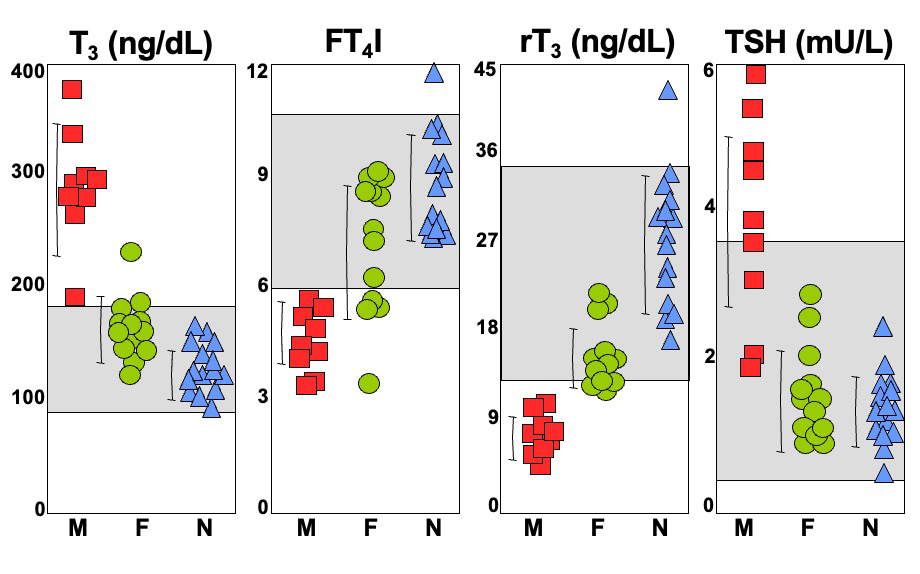
Figure 10. Thyroid function tests in several families with MCT8 deficiency studied in the authors’ laboratory. Grey regions indicate the normal range for the respective test. Hemizygous males (M) are represented as red squares, heterozygous carrier females (F), as green circles and unaffected members of the families, as blue triangles (N). With the exception of TSH, mean values of iodothyronines in carrier females are significantly different than those in affected males and normal relatives.
TSH was normal at neonatal screening in most cases. Information about neonatal T4 levels available in 8 cases revealed low values in 6 and normal levels in 2 subjects (33, 224, 236). However, low T4 concentrations at birth are not uncommon, and are more often associated with low levels of T4-binding protein and prematurity. Information regarding the T3 and rT3 concentration in the first days of life is not available. However, within one month the typical thyroid test abnormalities of MCT8 deficiency become apparent. In infants and children, tests results should be interpreted using age-specific reference ranges (see Chapter 62). This is particularly important for T3 and rT3, which are higher than those in adults. The ratio of T3 to rT3 is characteristically high in MCT8 deficiency while, with the exception of RTHα, it is low in other more common causes of abnormal T3 and rT3 levels, such as binding defects, iodine deficiency and non-thyroidal illness (Table 3).
Heterozygous female carriers can have iodothyronine concentrations that are on average intermediate between affected males and unaffected family members (6, 224, 240). While they are significantly different compared to both affected and unaffected individuals, overlapping values are observed in both groups. Serum TSH concentrations are, however, normal (Figure 10).
OTHER SERUM TESTS
Some patients have undergone extensive testing prior to the diagnosis of MCT8 deficiency. Results are summarized here and in the subsequent sections. Urinary organic acids, serum amino acids and fatty acids, CSF neurotransmitters, glucose and lactate were normal. Other test results were abnormal only in some patients. These included, elevated serum SHBG, transaminases, ammonia, lactate and pyruvate, mildly elevated medium chain products in plasma acylcarnitine profile, elevated hydroxybutyric acid in urine (33, 233, 240) and reduced serum cholesterol. While the relation of some test abnormalities with MCT8 deficiency is unclear, others can be ascribed to the effect of the high serum T3 levels on peripheral tissues. These are reduced cholesterol, and increased SHBG, and lactate.
Other endocrine tests, including pituitary function were normal when tested in a few individuals. However, administration of incremental doses of L-T3, using the protocol devised for the study of patients with RTHß, showed reduced pituitary sensitivity to the hormone (33). This is probably due the reduced feedback effect of T3 on the hypothalamo-pituitary axis, as well as the reduced incremental effect of the hormone on peripheral tissues already exposed to high levels of T3.
X-RAYS AND IMAGING
Bone age has been inconsistently reported, and was found to be delayed in four cases and was slightly advanced in one (33, 240, 242, 243). The consequences of the MCT8 defects on bone are not clear at this time.
Mild to severe delayed myelination or dysmyelination (33, 244, 245) is a common finding when brain MRI is performed in early life. However, this can be missed as the delay in myelination is usually less apparent by approximately 4 years of age, and an adequate MRI technique is required for optimal interpretation. This distinguishes MCT8 deficiency from other leukodystrophies in which the myelination defect is persistent. Other reported MRI abnormalities in single cases might be non-specific and include subtle cortical and subcortical atrophy (239), mild cerebellar atrophy (240), putaminal lesions (246), and a small corpus callosum (33). Increased choline and myoinositol levels and decreased N-acetyl aspartate were detected by MR-spectroscopy, and these abnormalities in brain metabolism were associated with the degree of dysmyelination according to MRI findings (247).
A recent study aimed to better define the spectrum of motor disability in MCT8 deficiency and to elucidate the neuroanatomic basis of the motor impairments using clinical observation and brain MRI in a cohort of 6 affected pediatric patients (248). T1- and T2-weighted brain MRI sequences revealed hypomyelination involving the subcortical U-fibers and periventricular white matter tracts that became more conspicuous over time in all 6 patients. In contrast, the callosal and cortical spinal tracts showed near normal myelination. Diffusion tensor imaging, performed in 3 of the patients, showed poor definition of the white matter association tracts relative to normal controls, suggesting the presence of subtle microstructural abnormalities. The same 3 subjects had a normal-appearing corpus callosum. These findings are consistent with the presence of dystonia in the MCT8 deficient patients studied. These imaging biomarkers suggest that rehabilitation efforts targeting dystonia may be more beneficial than those targeting spasticity in the prepubertal pediatric MCT8 deficiency population as the combination of hypotonia and dystonia presents a neurorehabilitation challenge for these patients and therapies directed only toward spasticity have commonly produced suboptimal responses (248).
TESTS IN TISSUES
Altered activity of mitochondrial complexes II and IV was identified in muscle biopsies from two cases (33, 249). It is unclear if this is due to the abnormal TH status of the muscle or to a yet unidentified effect of MCT8 on the mitochondria.
Cultured skin fibroblasts from males with MCT8 deficiency showed a significant reduction of T4 and T3 uptake while D2 enzymatic activity was higher, compared to fibroblasts from normal individuals (33, 234). Fibroblasts from carrier females gave results intermediate to those of affected males and normal individuals. Cellular T3-uptake of cell lines transfected with different mutant MCT8 molecules (235), demonstrated or predicted complete inactivation in about 2/3 of mutations, while in the remaining 1/3, T3-uptake ranged from 8.6 to 33% that of the WT MCT8. In particular, three missense mutations, S194F, L434W, and L598P showed significant residual transport capacity of more than 15% of normal MCT8, which may underlie the relatively milder phenotype observed in patients with these mutations (see section on Clinical Features and Course of the Disease, above).
Histological analysis of brain sections from a 30th gestational week male fetus and an 11-year-old boy with MCT8 deficiency was performed using as controls, brain tissue from a 30th and 28th gestational week male and female fetuses, respectively, and a 10-year-old girl and a 12-year-old boy (250). The MCT8-deficient fetus showed a delay in cortical and cerebellar development and myelination (Figure 11), loss of parvalbumin expression, abnormal calbindin-D28k content, impaired axonal maturation, and diminished biochemical differentiation of Purkinje cells. The 11-year-old boy showed altered cerebellar structure with deficient myelination, deficient synaptophysin and parvalbuminexpression, and abnormal calbindin-D28k expression. This study showed that brain damage in MCT8 deficiency is diffuse, without evidence of focal lesions, that was present from fetal stages despite apparent normality at birth, and the deficient hypomyelination was found to persist at 11 years of age (250).
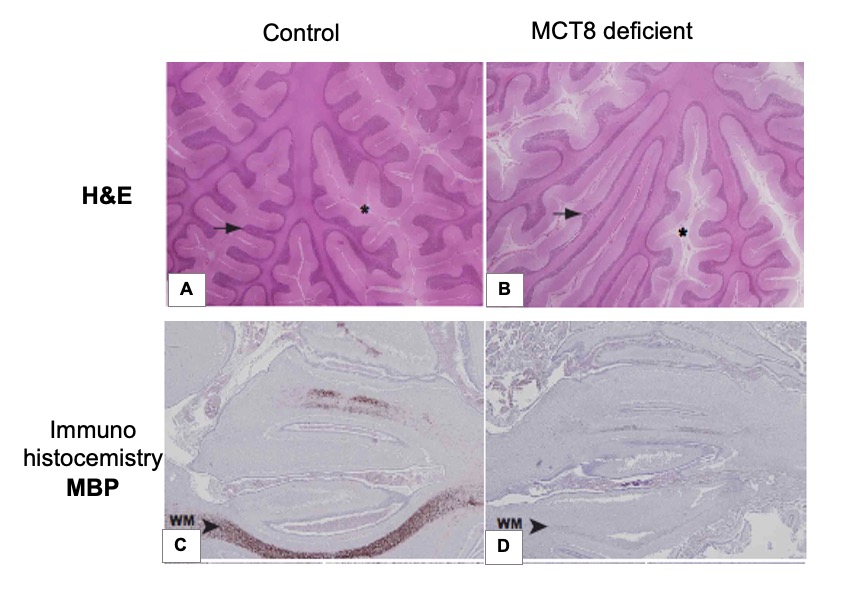
Figure 11. Structure and myelination of the fetal and juvenile cerebellum. Representative images showing hematoxylin-eosin (H&E) staining (A, B) and myelin basic protein (MBP) immunostaining (C, D) of tissue sections from the cerebellar vermis from 10-year-old control child (A), and 11-year-old MCT8 deficient child (B), control fetus and (C) MCT8-deficient fetus (D), both 30 weeks gestational age. In panels A and B, asterisks indicate the subarachnoid space in the cerebellum (wider in the MCT8-deficient subject) and arrows point to cerebellar folia (thinner size in the cerebellum of the MCT8-deficient boy). In panels C and D, arrowheads indicate immunopositive axons (lower proportion of immunopositive axons in the vermis from the MCT8-deficient fetus) WM, white matter. [Reproduced with permission from reference (250)].
GENETIC TESTING
By definition, a defect in the MCT8 gene is present in all patients. Genetic testing by sequencing is available in commercial laboratories and can detect nucleotide substitutions and small deletions and insertions. However, larger deletions and splice defects may require application of more in-depth genetic investigations, such as Southern blotting and haplotyping, available in research laboratories. Carrier testing for relatives at-risk and prenatal testing of pregnant carriers should be offered to families (251).
Animal Models of MCT8 Deficiency
Mct8-deficient recombinant (Mct8KO) mice (19, 252) replicate the characteristic thyroid test abnormalities found in humans and, thus, helped in understanding the mechanisms responsible for the thyroid phenotype (253). Measurements of tissue T3 content showed the variable availability of the circulating hormone to tissues, depending on the redundant presence of TH cell membrane transporters. In Mct8KO mice, tissues such as the liver, that express other transporters than Mct8 (15), have high T3 concentrations reflecting the high levels in serum and are, therefore, “thyrotoxic” as demonstrated by an increase in the D1 enzymatic activity (Figure 12A). In accordance with a thyrotoxic state, serum cholesterol concentration is decreased and serum alkaline phosphatase is increased. In contrast, tissues with limited redundancy in cell membrane TH transporters, such as the brain (15), have decreased T3 content in Mct8KO mice, which together with the increase in D2, indicate “hypothyroidism” in this tissue (Figure 12B). The role of D2 is to maintain local levels of T3 in the context of TH deficiency and its activity is inversely regulated by TH availability (16). These findings of coexistent T3 excess and deficiency in the Mct8KO mouse tissues explain, in part, the mechanisms responsible for the tissue specific manifestation of TH deficiency and excess in humans with MCT8 deficiency.
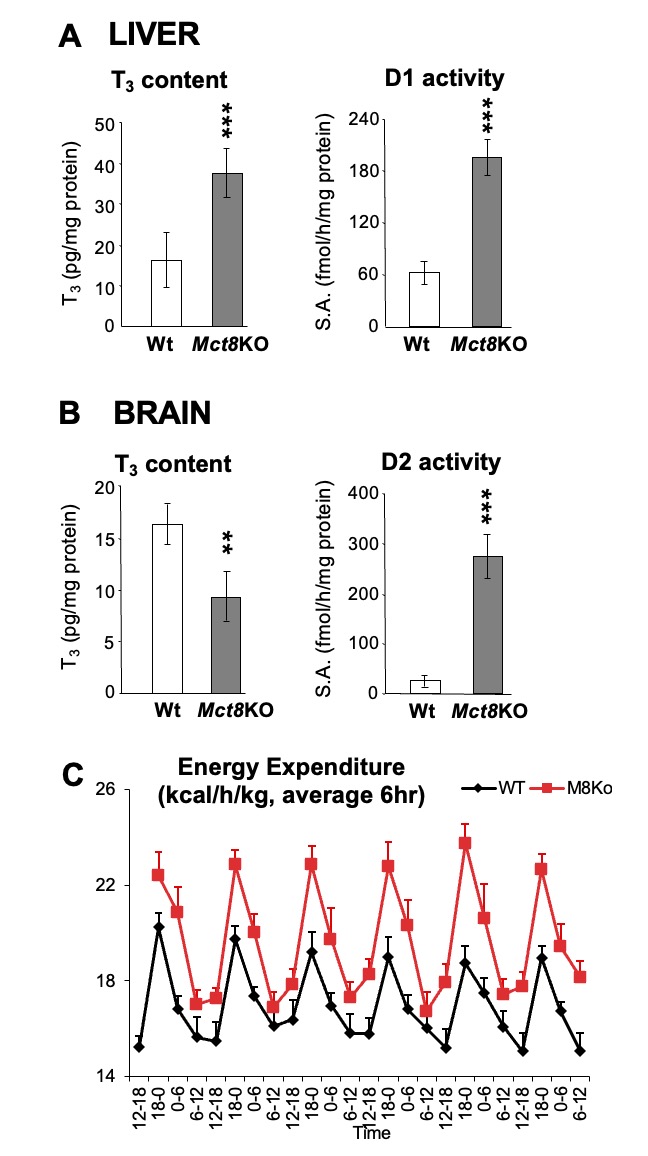
Figure 12. Data from Mct8KO vs Wt mice. A. T3 content and D1 enzymatic activity in liver. B. T3 content and D2 enzymatic activity in brain. Data from Mct8KO mice are represented as grey bars and those from Wt littermates are in open bars. ** p-value <0.01, *** p-value <0.001. C. Total energy expenditure (TEE) flow chart represented as 6h average over 6 days, Mct8KO mice are shown in red.
Metabolic and body composition studies showed that Mct8KO mice were leaner and had increased total energy expenditure (TEE) with increased food and water intake, while the activity level was normal (254). T3-treated Wt mice also showed increased food intake and TEE, and increased T3 content, TH action and increased glucose metabolism in skeletal muscle similar to Mct8KO mice. Thus, the hypermetabolic state in MCT8 deficiency is due to the high serum T3 and is responsible for the failure to maintain weight despite adequate caloric intake. Normalizing serum T3level by deleting deiodinase 1 in the combined Mct8Dio1KO mice was able to improve body composition and the metabolic alterations caused by the Mct8 deficiency (254). Treatment of adult mice with the TH analog diiodothyropropionic acid (DITPA), which enters cells independently of Mct8 transport, revealed amelioration of the thyrotoxic state in liver and improving the hypermetabolism of the Mct8KO mice (255), making this thyromimetic chemical suitable for the treatment of the hypermetabolism in patients with MCT8 deficiency.
Mct8 also has a role in TH efflux in the kidney and secretion from the thyroid gland (256, 257). The content of T4 and T3 in kidney is increased and their local actions increase D1 activity which enhances the local generation of T3. In the thyroid, Mct8 is localized at the basolateral membrane of thyrocytes. Thyroidal T4 and T3 content is increased in Mct8KO mice as is the rate of their secretion and appearance in serum is reduced (257).
These observations from the Mct8 deficient mice have helped understanding the mechanisms involved in producing the thyroid abnormalities in mice and humans. The increased D1 and D2 activities, stimulated by opposite states of intracellular TH availability, have an additive consumptive effect on T4 levels and result in increased T3 generation. The important contribution of D1 in maintaining a high serum T3 level is supported by the observation in mice deficient in both Mct8 and D1. These mice have a normal serum T3 and rT3 (258). The low serum T4 in Mct8 deficiency is not only the result of attrition through deiodination but also due to reduced secretion from the thyroid gland and possibly increased renal loss.
In MCT8 deficient subjects serum TSH is usually modestly increased, a finding that may be compatible with the decreased serum T4 concentration but not with the elevated serum T3 level. However, MCT8 is expressed in the hypothalamus and pituitary, and its inactivation likely interferes with the negative feedback of TH at both sites (259). In Mct8KO mice, hypothalamic TRH expression is markedly increased and high T3 doses are needed to suppress it, indicating T3 resistance particularly at the hypothalamic level.
Mct8KO mice have been valuable in testing thyromimetic compounds for their potential to bypass the Mct8 defect in tissues. One such TH analogue, DITPA has been tested. It was found to be effective in equal doses in the Mct8KO and Wt animal to replace centrally (pituitary and brain) and peripherally (liver) the TH requirements in animals rendered hypothyroid (260). In contrast, 2.5 and 8-fold higher doses of L-T4 and L-T3, respectively, were required to produce a central effect in the Mct8KO compared to that in Wt animal. These high doses of TH produced “hyperthyroidism” in peripheral tissues of the Mct8KO mice. Importantly, DITPA given to pregnant mice carrying Mct8-deficient embryos was able to cross the placenta and to have similar thyromimetic action on the expression of TH-dependent genes in brain of Mct8KO and Wt pups at similar DITPA serum levels (261). The similar serum TSH in Mct8KO vs Wt pups prenatally treated with DITPA, demonstrated better accessibility of DITPA at the pituitary compared to L-T4 thus making DITPA a candidate for the prenatal treatment of MCT8 deficiency (261).
Mct8KO mice were also used to test the possibility of gene therapy in MCT8 deficiency. Normal MCT8 was delivered using an AAV9 vector, injected either intravenously (IV) and/or intracerebroventricularly (ICV) into postnatal day 1 Mct8KO and Wt mice (262). Analysis of brains at 28 postnatal days, after L-T3 injection for four days showed that MCT8 delivery to the blood brain barriers by IV but not ICV injection is necessary for its proper function and resulted in increased T3 in the brain tissue triggering expression of known TH-regulated genes (262). These studies have introduced the consideration for gene therapy in the patients with MCT8 deficiency.
The lack of a neurological phenotype in Mct8KO mice limits their use as a model for understanding the mechanisms of the neurological manifestations in patients with MCT8 deficiency. If combined with deficiencies of other TH transporters in brain, Mct8 has the potential of producing an obvious neurological phenotype. Thus, mice deficient in both Mct8 and another TH transporter such as Lat2, Mct10 and Oatp1c1 were generated (225, 226, 228). From them, the Mct8/Oatp1c1 double KO (DKO) mice showed alterations in peripheral TH homeostasis that were similar to those in Mct8KO mice; while, uptake of both T3 and T4 into the brain of Mct8/Oatp1c1 DKO mice was strongly reduced (228). TH deprivation in the CNS of Mct8/Oatp1c1 DKO mice with marked decrease in brain TH content and in TH target gene expression manifested with delayed cerebellar development, reduced myelination and compromised differentiation of GABAergic interneurons in the cerebral cortex (228). Mct8/Oatp1c1 DKO mice displayed pronounced locomotor abnormalities and currently are being used to assess the pathogenic mechanisms underlying the neurologic phenotype in Mct8 deficiency and as models to test the effect treatment modalities on neurodevelopmental defects observed in humans.
Molecular Basis of the Disorder
In vitro studies using mutant MCT8 alleles as well as observations from animals deficient in Mct8, serve to explain the mechanism leading to the defect. All mutant MCT8 alleles tested by transfection or in fibroblast derived from affected individuals show absent or greatly reduced ability to transport iodothyronines, primarily T3 (235). Although MCT8 mRNA is widely expressed in human and rat tissues, including brain, heart, liver, kidney, adrenal gland, and thyroid (263, 264), repercussions due to its absence manifest primarily in tissues and cells in which MCT8 is the principal, if not unique TH transporter.
Analysis of the MCT8 mRNA expression pattern in the mouse brain by in situ hybridization revealed a distinct localization of this transporter in specific neuronal populations known to be highly dependent on proper TH supply, indicating that a defective MCT8 will perturb T3-dependent neuronal function. Moreover, high transcript levels for MCT8 were observed in choroid plexus structures and in capillary endothelial cells, suggesting that MCT8 also contributes to the passage of THs via the blood-brain barrier and/or via the blood-cerebrospinal fluid barrier (265, 266). In the thyroid, it has been demonstrated that MCT8 is involved in the secretion of TH into the bloodstream (257, 267).
The magnitude of serum T3 elevation does not correlate with the degree of T3 transport defect produced by a particular MCT8 mutation. This is probably due to the important contribution of the concomitant perturbation in iodothyronine metabolism on the production of T3, as demonstrated in the Mct8KO mice (see the section above). Similarly, there is no correlation between the magnitude of serum T3 elevation or rT3 reduction in affected males compared to their carrier mothers (33). Some imperfect correlation does appear to exist between the degree of the MCT8 defect and clinical consequences. Patients that are least severely affected and capable of some locomotion have mutations with partial preservation of T3 transport function (see Clinical Features and Course of the Disease, above). In contrast, early death is more common in patients with mutations that completely disrupt the MCT8 molecule. However, it should be kept in mind that genetic factors, variability in tissue expression of MCT8, and other iodothyronine cell membrane transporters could be responsible for the lack of a stronger phenotype/genotype correlation. The possibility that MCT8 is involved in the transport of other ligands, or has functions other than TH transport, cannot be excluded.
There has been a great deal of effort in trying to understand how MCT8 transports TH into cells, and how MCT8 distinguishes TH substrates from structurally closely related compounds is not known. One starting point used was the fact that T3 is bound between a His-Arg clamp in the crystal structure of the T3 receptor/T3 complex. To investigate whether such a motif might potentially be relevant for T3 recognition in MCT8, several mutations occurring in patients, or generated in vitro have been tested in combination with amino acid specific chemical modification. Molecular modeling has demonstrated a perfect fit of T3 poised into the substrate channel between His415 and Arg301 and observing the same geometry as in the T3 receptor (268). Similarly, cysteine residues Cys481 and Cys497 were found to probably be located at or near the substrate-recognition site in MCT8 (269). The question whether MCT8 functions as a monomer or as an oligomer has also been investigated. Although several mutations have been shown to affect oligomerization in vitro, currently, it is not known whether there is an obligatory functional need for dimerization of MCT8, or whether there is substrate-induced or constitutive oligomerization versus monomerization (270), However, different from the tight interaction of TH with receptors as ligand and with the deiodinases as substrate, the transporters have to achieve specific interactions with TH, while avoiding tight binding, as this would stall the transport process. Characterizing the mechanism is of fundamental interest and will be key for the design of specific MCT8 modulators.
Differential Diagnosis
MCT8-dependent THCMTD is syndromic, presenting a thyroid and a neuropsychomotor component. However, the majority of patients come to medical attention because of retarded development, and neurological deficits. Although the thyroid abnormalities are most characteristic, they escape detection by neonatal screening. TSH concentration is not elevated above the diagnostic cut off level and although T4 is commonly low, it more often accompanies premature births and low levels of TH-binding serum proteins. Studies in Mct8KO mice suggest that rT3 could turn out to be a good marker for the early detection of MCT8 defects in humans.
Hypotonia is an early manifestation, but is not specific. Reduced myelin, documented by brain MRI, places MCT8 in the category of other diseases showing reduced myelination, among them Pelizaeus–Merzbacher disease (PMD; OMIM# 312080). The latter is also X-linked, and is a leukodystrophy caused by an inborn error of myelin formation due to defects in the PLP1 gene located on Xq22. In fact, a survey of 53 families affected by hypomyelinating leukodystrophies of unknown etiology, classified as PMD, resulted in the identification of MCT8 gene mutations in 11% (244), and the affected subjects were subsequently found to have the typical thyroid test abnormalities. Patients with PMD do not exhibit the thyroid phenotype of MCT8 deficiency and their myelination defect is persistent, rather than transient.
All children above the age of 1 month found to have MCT8 gene mutations show the characteristic thyroid test abnormalities. This underscores the importance of performing thyroid tests in patients diagnosed with mental retardation or syndromic X-linked phenotypes suggestive of a MCT8 defect, prior to sequencing the MCT8 locus. Most useful is the finding of a high serum T3 and low rT3. A reduced (at the low limit or below normal) serum total or free T4, and a normal or slightly elevated TSH are also present. In cases with increased T3 due to other causes, calculating the ratio of T3/rT3 is helpful in differentiating them from cases of MCT8 defects, in whom the ratio will be above 10.
Treatment
Treatment options for patients with MCT8 gene mutations are currently limited (251). Supportive measures include the use of braces to prevent mal-positioned contractures that may ultimately require orthopedic surgery. Intensive physical, occupational, and speech therapies have subjective but limited objective beneficial effects. Diet should be adjusted to prevent aspiration and a permanent gastric feeding tube may be placed to avert malnutrition. Dystonia could be ameliorated with medications such as anticholinergics, L-DOPA carbamazepine and lioresol. Drooling might be improved with glycopyrolate or scopolamine. Seizures should be treated with standard anticonvulsants. When refractory to the latter, a ketogenic diet, as well as administration of supraphysiological doses of L-T4, has been successful. Experience with such treatments is, however, limited to only a few cases.
Detection of low T4 levels by neonatal screening has led to treatment with L-T4 in several infants. As expected, no improvement has been noted when used in physiological doses, because of the impaired uptake of the hormone by MCT8-dependent tissues. Under these circumstances it would be logical to treat with supraphysiological doses of L-T4, thereby increasing the availability of TH to the brain. However, the presence of an already increased D1, as observed in Mct8 deficient mice (see Animal Models in a preceding section of this Chapter), is likely to aggravate the hypermetabolic state of the patient by generating more T3 from the exogenous L-T4. Therefore, high L-T4 dose treatment has been used in combination with propylthiouracil (PTU), which is a specific inhibitor of D1. This results in reduction of the conversion of T4 to T3 in peripheral tissues by D1, while it allows the local generation of T3 by D2 in tissues. Although this treatment allowed an increase in serum L-T4 level without increasing the hypermetabolism and weight loss, it did not improve the neuropsychomotor deficit (33, 241).
Other possible treatments that have been tested include the administration of the thyromimetic drug DITPA, that seems to be effectively transported into mouse brain in the absence of Mct8 (260) (see Animal Models in a preceding section of this Chapter). DITPA given to children with MCT8 deficiency in doses of 1–2 mg/kg/d fully normalizes the serum thyroid tests, and reduces the hypermetabolism with improvement in the nutritional status with no objective change in the neuropsychiatric deficit (271).
In vitro treatment with DITPA of oligodendrocytes derived from a human embryonic stem cell reporter line expressing MCT8 (272) was found to up-regulate oligodendrocytes differentiation transcription factors and myelin gene expression and to promote myelination of retinal ganglion axons in co-culture. Pharmacological and genetic blockade of MCT8 induced significant oligodendrocyte apoptosis, impairing myelination, and DITPA treatment was able to limit this effect (272). As MCT8 seems to be essential for TH transport in human oligodendrocytes development, DITPA has the potential to be a promising treatment for developmentally regulated myelination in AHDS (272).
Other TH metabolites, such as TRIAC and its precursor tetraiodothyroacetic acid (TETRAC) have been tested.Recently, a multicenter, open-label, single-arm, phase 2, clinical trial investigated the effectiveness and safety of oral TRIAC in male pediatric and adult patients with MCT8 deficiency assessed at baseline and after 12 months of TRIAC administered in a predefined escalating dose schedule at a dose ranging from 23–48 μg/kg per day (273). All serum thyroid tests decreased, including T3; TSH, total free T4, and reverse T3 further decreased from their already low baseline levels in patients with MCT8 deficiency. While the reduction in serum T3 concentrations was associated with improvements in body weight, cardio-vascular status, and markers of TH action in different tissues, causality cannot be proven directly because the open-label study design did not include a control group. Moreover, there are concerns that the further reduction in circulating T3 concentrations under TRIAC treatment aggravates the hypothyroid state in the brain in individuals with MCT8 deficiency (273).
Another treatment option tested in vitro consists of the use of chemical and pharmacological chaperones on the expression and transport activity of several MCT8 mutant proteins. The chemical chaperone sodium phenylbutyrate (NaPB), has been used to treat patients with cystic fibrosis and urea cycle defects for extended periods of time. In vitro testing showed that NaPB could rescue the expression and activities of MCT8 mutations that retain some residual activity (274). Testing of another chemical chaperone, 4-phenylbutyric acid (PBA), demonstrated that it was effective in potentiating the T3 uptake in transiently transfected COS-1 cells with WT MCT8 and the F501Δ mutant, but only minor effects were observed in F501Δ fibroblasts (275). Thus, the applicability of chemical and pharmacological chaperones may be limited to only a small number of patients with certain mutations. In addition, because the magnitude of the effect of chaperone therapy strongly depends on the disease model, more extensive preclinical studies are warranted before clinically available chaperones should be considered as a treatment option in patients with MCT8 deficiency.
Treatments that reduce the high serum T3 level, including combining PTU with L-T4, DITPA or TRIAC, have all beneficial effect on the hypermetabolism and reduce weight loss. This often obviates the need for feeding through a G-tube. However, no demonstrable effect on the neuropsychomotor abnormalities has been observed even when treatment was initiated during infancy. It is noteworthy that none of the treatments discussed above have been initiated before the age of 6 months. It is possible that for any TH mediated treatment to be effective on brain development, it will have to be initiated before birth. However, intra-amniotic treatment with L-T4 of an embryo beginning at 17 weeks of gestation failed to prevent the neurological complications, even though myelination proceeded normally (33). This suggests that earlier brain damage, unrelated to myelin formation, is involved and that treatment should begin at 10 weeks of gestation, when brain TH receptors becomes fully active (276).
Use of thyromimetic drugs is supported by the defect in the transport of authentic THs. However, it is possible that a deficiency in a different substrate or that the loss of a putative constitutive effect harbored by the intact MCT8, play a role in the observed brain morbidity.
Recessive mutation in the SBP2 (SECISBP2) gene encoding selenocysteine insertion sequence-binding protein 2 is an inherited condition causing a TH metabolism defect (THMD). The mutations affect selenoprotein synthesis including the deiodinases, which are selenoenzymes. Only twelve families with this defect have been identified so far. Affected individuals present with short stature and characteristic thyroid test abnormalities with high serum T4, low T3, high rT3 and normal or slightly elevated serum TSH. In addition, they also have decreased serum selenium (Se) and decreased selenoprotein levels and activities in serum and tissues. The overall clinical phenotype is complex. Affected individuals may have delayed growth and puberty, and in severe cases failure to thrive, developmental delay, infertility, myopathy, hearing impairment, photosensitivity, and immune deficits.
Another inherited condition also reported to cause THMD is the mutation in the TRU-TCA1-1 gene encoding the selenocysteine transfer RNA (tRNASec), which has a role in selenoprotein synthesis. The affected individual had high serum T4, high rT3, normal T3, normal TSH with low Se level (277)
Mutations in the in selenocysteine synthase (SEPSECS) have also been identified, however affected individuals do not manifest abnormal thyroid function tests (278). Affected children manifest a complex neurodevelopmental and neurodegenerative disorder involving, among other features, microcephaly, delayed motor and intellectual development, spasticity, and seizures.
The requirement for TH varies among tissues, cell types, and the timing in development. In order to provide the proper intracellular hormone supply, TH entry into cells is controlled by membrane transporters, and further fine-tuned by its intracellular metabolism, regulated by three selenoprotein iodothyronine deiodinases (D1, D2, D3). D1 and D2 are 5’-iodothyronine deiodinases that catalyze TH activation by converting T4 to T3. D3, a 5-deiodinase is the main TH inactivator through conversion of T4 to rT3 and T3 to T2 (Figure 1B) (see other Endotext chapters for details)
Deiodinases are selenoproteins containing the rare amino acid selenocysteine (Sec), present in the active center of the molecule and required for their enzymatic activity. They are differentially expressed in tissues and in response to alterations in the intracellular environment, further regulated at the level of transcription, translation and metabolism (16). D2 activity can change very rapidly as its half-life is more than 15-fold shorter than that of D1 and D3. T4accelerates D2 inactivation through ubiquitination, a reversible process that can regenerate active D2 enzyme through de-ubiquitination.
Deiodinases share with other selenoproteins the synthesis through a unique mode of translation. The codon used for insertion of Sec is UGA, which under most circumstances serves as a signal to stop synthesis. This recoding of UGA is directed by the presence of a selenocysteine insertion sequence (SECIS) in the 3’-untranslated region of the selenoprotein messenger RNA. It is the SECIS-binding protein 2 (in short SBP2) that recognizes the SECIS and recruits an elongation factor (EFSec) and the specific selenocysteine transfer RNA (tRNASec) for addition of Sec at this particular UGA codon (Figure 13) (279).
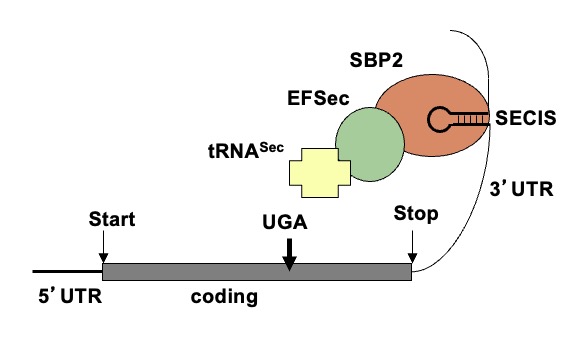
Figure 13. Components involved in Sec incorporation central in the synthesis of selenoproteins. Elements present in the mRNA of selenoproteins: an in frame UGA codon and Sec incorporation sequence (SECIS) element, a stem loop structure located in the 3’UTR (untranslated region). SBP2 binds SECIS and recruits the Sec-specific elongation factor (EFSec) and Sec-specific tRNA (tRNASec) thus resulting in the recoding of the UGA codon and Sec incorporation.
Etiology and Genetics
Abnormalities in TH metabolism have first been observed in humans as acquired condition, the “low T3 syndrome”, which occurs during non-thyroidal illness or starvation (see Chapter 14) (280). The first inherited disorder of TH metabolism in humans was reported in 2005 (8), and was found to be caused by a mutation in the SBP2 gene. Later, mutations in TRU-TCA1-1 gene were also reported to cause THMD in a single individual (277). Both genes are involved in selenoprotein synthesis, thus the mutations result in the defect of selenoproteins including the deiodinases. It is anticipated that mutations in other genes causing defective TH metabolism may have different phenotypes. So far, no humans have been reported with mutations in the deiodinase genes or in other proteins involved in selenoprotein synthesis.
Incidence and Inheritance
The incidence of THMD caused by SBP2 deficiency is unknown. Ten additional families have been identified since the description of the initial two families (33, 281-285). The inheritance is autosomal recessive and males and females are equally affected. For this reason, the likelihood of being affected is less than that for autosomal dominant or X-linked conditions. The ethnic origins of the reported patients include Bedouin from Saudi Arabia, African, Irish, Brazilian, English, Japanese, Turkish and Argentinian individuals.
THE SBP2 GENE AND MUTATIONS
The human SBP2 gene, cloned in 2002, is located on chromosome 9 and encodes a protein of 854 amino acids widely expressed in most tissues (286). The C-terminal domain encompassing codons 399-774 of the protein is the minimal functional protein domain required for SECIS binding, ribosome binding and Sec incorporation (287), which is mandatory for SBP2 function. The role of the N-terminal region is not fully understood. In vitro studies have characterized a nuclear localization signal located in the N-terminal part and nuclear export signal in the C-terminal part. These domains enable SBP2 to shuttle between the nucleus and the cytoplasm (288), and they play a role in the function of SBP2 in the nucleus in vivo.
The finding of SBP2 defects was made possible by extensive genetic studies of a large family with three affected and four unaffected children (8). Although affected individuals had clinical evidence of abnormal TH metabolism, the sequences of the deiodinase genes, as well as those of genes encoding proteins involved in the ubiquitination and de-ubiquitination of the deiodinase 2 were normal. Subsequently, mutational analysis of the SBP2 gene revealed that the affected individuals were homozygous for a R540Q mutation, while both parents were heterozygous carriers. It is likely that the parents, which were not directly related but both members of the same Bedouin tribe, had a common ancestor. The affected child of the 2nd family, of mixed African/European background, was compound heterozygous for a paternal nonsense mutation (K438*), and a maternal mutation located in intron 8 (+29bp G->A), causing alternative splicing, but allowing 24% expression of a normal transcript. Since this initial report, at least ten more families with mutations in SBP2 have been identified (33, 281-285). Affected individuals harbor homozygous or compound heterozygous mutations. The mutations are summarized in Table 6 and are schematically represented in Figure 14. Among the 20 mutations identified, four are missense mutations located within the functional domain (codon 399-774) causing deleterious effects on protein function, while the other six result in a prematurely truncated protein disrupting the functional domain. Nine mutations cause either transcripts with abnormal splicing or truncations, affecting the N-terminal part of the protein. The generation of shorter isoforms translated from downstream ATGs at codons 139, 233, and 300, which all contain the intact C-terminal functional domain results in preservation of partial SBP2 function (281). One remaining mutation, Q782*, results in a truncated protein downstream of the functional domain 399-774 and is likely subject to nonsense-mediated decay.
|
Table 6. Mutations in SBP2 Gene
|
|
Family (# affected)
|
Mutations
|
Protein change
|
Comments on putative defect
|
Status
|
Ref.
|
|
1 (3)
|
c.1619G>A
|
R540Q
|
Predicted damaging (PolyPhen-2 score 1)
|
Homozygous
|
(8)
|
|
2 (1)
|
c.1312A>T
|
K438*
|
Truncated functional domain
|
Compound heterozygous
|
(8)
|
|
c.1283+29G>A Abnormal splicing
|
Frameshift
|
Truncated functional domain
|
|
3 (1)
|
c.382C>T
|
R128*
|
Shorter isoformsa
|
Homozygous
|
(280)
|
|
4 (1)
|
c.358C>T
|
R120*
|
Shorter isoformsa
|
Compound heterozygous
|
(281)
|
|
c.2308C>T
|
R770*
|
Truncated functional domain
|
|
5 (1)
|
c.668delT
|
F223Ffs*32
|
Shorter isoforma
|
Compound heterozygous
|
(282)
|
|
c.881-155T>A, abnormal splicing
|
Frameshift
|
Shorter isoforma
|
|
6 (1)
|
c.2071T>C
|
C691R
|
Predicted damaging (PolyPhen-2 score 1)
|
Compound heterozygous
|
(282)
|
|
Intronic SNP, abnormal splicing
|
Frameshift
|
Shorter isoformsa
|
|
7 (1)
|
c.1529_1541dup CCAGCGCCCCACT
|
M515Qfs*48
|
Truncated functional domain
|
Compound heterozygous
|
(283)
|
|
c.235C>T
|
Q79*
|
Shorter isoformsa
|
|
8 (1)
|
c.800_801insA
|
K267Kfs*2
|
Shorter isoforma
|
Homozygous
|
(284)
|
|
9 (1)
|
c.589C>T
|
R197*
|
Shorter isoformsa
|
Compound heterozygous
|
(37)
|
|
c.2037G>T
|
E679D
|
Predicted damaging (PolyPhen-2 score 1)
|
|
10 (1)
|
c.2344C>T
|
Q782*
|
Truncated after functional domain, NMD
|
Compound heterozygous
|
(37)
|
|
c.2045_2048delAACA
|
K682Tfs*2
|
Truncated functional domain
|
|
11 (2)
|
c.1588A>G
|
T530A
|
Predicted damaging (PolyPhen-2 score 1)
|
Compound heterozygous
|
(37)
|
|
c.1711C>T
|
Q571*
|
Truncated functional domain
|
|
12 (1)
|
c.283delT
|
Y95Ifs*31
|
Shorter isoformsa
|
Compound heterozygous
|
(37)
|
|
c.589C>T
|
R197*
|
Shorter isoformsa
|
The nomenclature as transcript ID ENST00000375807.7 (854-amino acid long)
a Shorter isoform(s) generated from downstream ATG(s) (M139, M233, M300) containing C-terminal functional domain
NMD, nonsense mediated decay
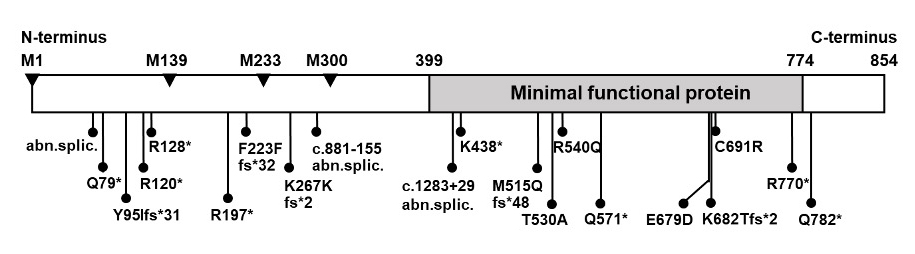
Figure 14. Schematic representation of human SBP2 showing the location of the mutations. Region of minimal functional protein encompassing codon 399-774 is showed in gray. The positions of methionine used as alternative translational initiation sites (M1, M139, M233, M300) are indicated as arrowheads.
Clinical Features and Course of the Disease
Age at presentation ranged from 4 months to 14 years, with the exception of one adult patient who had been identified at the age of 35 years (283). Delayed growth and bone maturation are the main manifestations that brought the affected individuals to medical attention. The severity of growth delay varies among patients. Some patients developed failure to thrive during infancy, while some presented with short stature during mid-childhood. Delayed motor and intellectual milestones were also recognized in seven cases (33, 282-284). Affected individuals started walking and talking around the age of 1.5 to 3 years. More severe developmental delay was found in one patient who started to walk, but still was unable to talk, at the age of 4.5 years (33).
Congenital myopathy with characteristic MRI findings was reported in five cases (282-285). A patient developed hypotonia and muscle weakness early in life and still had hip girdle weakness, impaired motor coordination, waddling gait, and positive Gower’s sign when she was 11 years old (282). The adult patient was reported to have delayed motor milestones during childhood and still had difficulty walking and running during adolescence, with genua valga and external rotation of the hip requiring orthotic footwear (283). The other two patients were evaluated for muscle weakness during mid-childhood (284, 285) while another came to medical attention at the age of 2 years (283).
Sensorineural hearing loss was reported in three cases (282, 283) and conductive hearing loss following recurrent exudative otitis media was reported in one another case (284). Rotatory vertigo was also found (283, 284). Obesity was documented in two cases (282, 285). In addition, another two cases had an increased fat mass index (283), paradoxically associated with low fasting insulin with enhanced insulin sensitivity, elevated adiponectin levels, and a favorable lipid profile. One of them, a 2-year-old boy, had recurrent fasting nonketotic hypoglycemia with low insulin levels requiring supplemental enteral nutrition.
Pubertal development was documented only in 4 cases, while the information is not available in the remaining cases. In females, an affected girl had Tanner stage II breasts when she was 11 years old (33) and she had her menarche at the age of 13 years. In males, one affected boy was prepubertal at the age of 14 years but had a pubertal growth spurt within the following 2 years (8), another one was in Tanner stage III with a testicular volume of 10 mL at the age of 11.5 years (285). The only adult affected individual was reported to have normal pubertal development, despite unilateral orchidectomy at age 15 years following an episode of testicular torsion. He had infertility in adulthood and the investigations revealed azoospermia despite the presence of a sonographically normal remaining testis (283). Other manifestations found in the adult patient were severe Raynaud disease, skin photosensitivity with evidence of enhanced UV-mediated oxidative DNA damage and mild reduction in red blood cell and total lymphocyte counts with impaired T cell proliferation and abnormal cytokine production (283). This, together with the variety of the aforementioned manifestations, suggests that the defects in the SBP2 gene result in a multi-organ involvement. The consequences of SBP2 deficiency could still be underestimated since the features associated with oxidative damage such as neoplasia, neurodegeneration and premature aging may develop later in life. A detailed longitudinal evaluation of affected individuals is needed to further understand and characterize the spectrum of the clinical manifestations in SBP2 deficiency.
Laboratory Findings
The characteristic thyroid test abnormalities in subjects with SBP2 gene mutations are high total and free T4, low T3, high rT3, and a normal or slightly elevated serum TSH (8) (Figure 15A). In vivo studies assessing the hypothalamo-pituitary-thyroid axis show that compared to normal siblings, affected children require higher doses of L-T4 and higher serum concentrations of T3, but not T3, to reduce their TSH levels compared to unaffected siblings, suggesting impaired conversion of T4 to T3 (Figure 15B). None of the affected individuals was reported to have an enlarged thyroid gland on physical examination. Thyroid ultrasonography showed a normal thyroid gland in one patient (282)and a hypoplastic thyroid gland in another patient (284). Delay in bone age was reported in the majority of the patients investigated, except one patient who presented with obesity and a bone age of 11 years at the chronological age of 10 years (285).
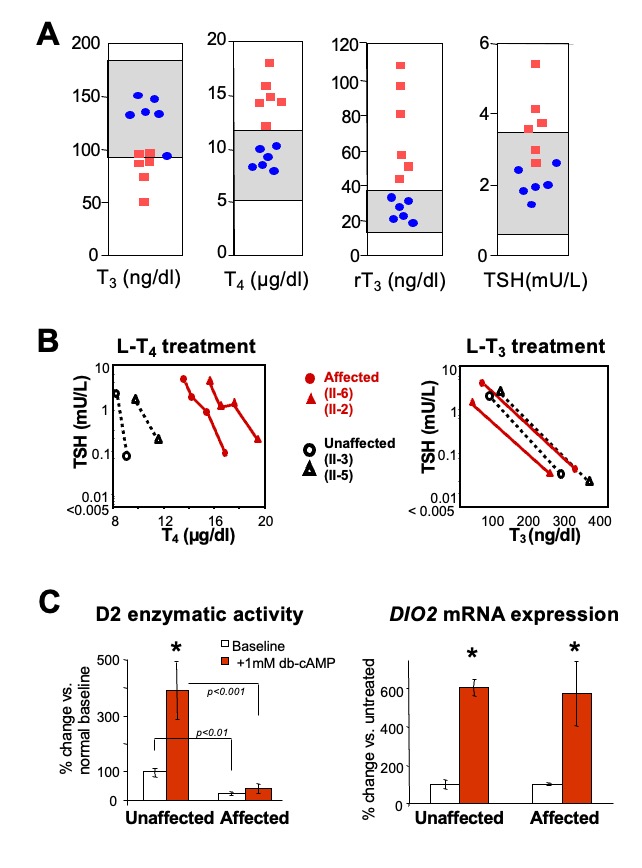
Figure 15. Thyroid function tests in several families with SBP2 deficiency studied in the authors’ laboratory. Grey regions indicate the normal range for the respective test. Affected individuals are represented as red squares and unaffected members of the families, as blue circles. B. In vivo studies: Serum TSH and corresponding serum T4 and T3 levels, before and during the oral administration of incremental doses of L-T4 and L-T3. Note the higher concentrations of T4 required to reduce serum TSH in the affected subjects; C. In vitro studies: Deiodinase 2 enzymatic activity and mRNA expression in cultured fibroblasts. Baseline and stimulated D2 activity is significantly lower in affected. There is a significant increase of DIO2 mRNA with dibutyryl cyclic adenosine monophosphate [(db)-cAMP)], in both unaffected and affected (*p <0.001) while there are no significant differences in baseline (db)-cAMP stimulated DIO2 mRNA in affected versus the unaffected.
Skin fibroblasts obtained from affected individuals and propagated in cell culture, showed reduced baseline and cAMP-stimulated D2 enzymatic activity, compared to fibroblasts from unaffected individuals. However, baseline and cAMP-stimulated D2 mRNA levels were not different compared to those in fibroblast from normal individuals (Figure 15C).
As SBP2 is epistatic to selenoprotein synthesis, SBP2 deficiency is expected to affect multiple selenoproteins. Indeed, serum concentrations of selenium, selenoprotein P and other selenoproteins are reduced, and skin fibroblasts have decreased D2 and glutathione peroxidase (Gpx) activities in affected individuals (8).
Thigh MRI of affected patients with muscle weakness showed muscle hypotrophy and increased signal in T1-weighted images suggesting of connective tissue / fatty infiltration in the thigh muscles, predominantly in the adductor magnus, biceps femoris and sartorius, with relative sparing of other muscle groups (282, 283, 285). This pattern is similar to that of individuals with myopathies caused by selenoprotein N1 (SEPN1) deficiency, suggesting that SEPN1 is also affected in the individuals with SBP2 deficiency. Detailed evaluation in the adult patient with multi-systemic involvement also demonstrated deficiencies in multiple selenoproteins: lack of testis-enriched selenoproteins resulting in failure of the latter stages of spermatogenesis and azoospermia, cutaneous deficiencies of antioxidant selenoenzymes causing increased cellular reactive oxygen species (ROS), and reduced selenoproteins in peripheral blood cells resulting in immune deficits (283).
Deficiencies of other selenoproteins of unknown function, such as SELH, SELT, SELW, SELI, were found and their consequences are as yet unknown (283). In some of these patients, multiple tissues and organs show damage mediated by reactive oxygen species, and it is conceivable that other pathologies linked to oxidative damage such as neoplasia, neurodegeneration, premature ageing, may manifest with time.
Molecular Basis of the Disorder
Clinical and laboratory investigations have established that the mutations in the SBP2 gene fully explain the observed abnormalities, as SBP2 is a major determinant in the incorporation of Sec during selenoprotein synthesis. Complete lack of SBP2 function is predicted to be lethal, as its immunodepletion eliminates Sec incorporation. The absence of lethality in the reported patients with SBP2 deficiency is attributed to the preservation of partial SBP2 activity and the hierarchy in the synthesis of selenoproteins.
The thyroid test abnormalities in subjects with SBP2 deficiency are consistent with a defect in TH metabolism due to the deficiency in deiodinases and have been found in all cases, even those with a relative mild phenotype. The mutant R540Q SBP2 behaves as a hypomorphic allele in in vitro studies using the corresponding R531Q mutation of the rat Sbp2 (289). The mutant molecule showed no binding to some, but not all SECIS elements, resulting in selective loss in the expression of a subset of selenoproteins. The affected child of another family was compound heterozygous and expressed ~24% of a normal transcript. In the case of the homozygous R128* mutation, smaller SBP2 isoforms translated from downstream ATGs were preserved which contained the intact C-terminus functional domains.
As the human selenoproteome comprises at least 25 selenoproteins (290, 291) it is not surprising that the phenotype of SBP2 deficiency is complex and goes beyond the thyroid test abnormalities that dominate the mild cases. The more severe phenotype reported in three families is due to a more extensive impairment in SBP2 function (292). In the patient with two nonsense mutations (282), the R770* mutation truncates the C-terminal functional domain in all the isoforms and likely abolishes SBP2 function. However, the R120* allele likely generates smaller functionally active SBP2 isoforms, but the overall amount would be less than that of the homozygous R128* patient (281), thus explaining the more severe phenotype. Low expression of functional SBP2 also explains the phenotype of the two patients from the United Kingdom. Increased proteasomal degradation was demonstrated for the C691R mutation, and Western blotting of skin fibroblasts from both probands showed lack of full length SBP2 protein expression (283)
THMD Caused by Mutation in the TRU-TCA1-1 Gene
Another inherited condition also reported to cause THMD is the mutation in the TRU-TCA1-1 gene encoding selenocysteine transfer RNA (tRNASec) (277), an essential molecule in the Sec-incorporation pathway. The first and only patient reported to date was found to have elevated serum T4 and rT3 levels, together with a normal serum T3, suggestive of impaired deiodinase activity, during the investigations for his abdominal pain, fatigue and muscle weakness. A low plasma selenium level was also found, together with undetectable red cell and plasma GPX, low plasma selenoprotein P (SEPP1), mild signaling intensity change in muscle imaging, and negligible SEPN1 expression in fibroblasts. However, SBP2 protein expression was normal and no mutation in the SBP2 gene was identified, leading to the suspicion of a defect in another gene involved in the selenoprotein synthesis pathway. A homozygous missense mutation in the TRU-TCA1-1 gene, resulting in the amino acid substitution C65G, was then identified in the proband and segregated with the phenotype in the family. Primary cells from the proband showed marked reduction of the mcm5Um isoform of tRNASec, needed mainly for the synthesis of stress-related selenoproteins, whereas the mcm5U isoform of tRNASec, mainly responsible for the synthesis of housekeeping selenoproteins, was relatively preserved.
Animal Models of THMD
To bypass the embryonic lethality of lacking Sbp2 (293), a global partial Sbp2 deficiency mouse model, Sbp2conditional knockout (Sbp2 iCKO) was generated using tamoxifen-inducible Cre-ER induced at P35 (294). The Sbp2iCKO mice replicate most of the characteristic thyroid function tests in patients, with high serum T4, rT3 and TSH, whereas serum T3 is normal (Figure 16A). The enzymatic activity of D1 in the liver, and the D2 enzymatic activity and Dio3 mRNA expression in the cerebrum were decreased in Sbp2 iCKO compared to Wt littermates (Figure 16B) (294). In addition to the affected selenoenzymes deiodinases, Sbp2 iCKO mice also had decreased expression and/or activity of other selenoproteins in the liver, cerebrum and serum (294). Decreased body weight was observed in Sbp2iCKO mice by 2 weeks after tamoxifen injection, similar to the failure-to-thrive phenotype in patients. Other phenotypes are being further investigated in Sbp2 iCKO mice in order to understand the mechanisms of SBP2 deficiency as a multi-organ syndrome.
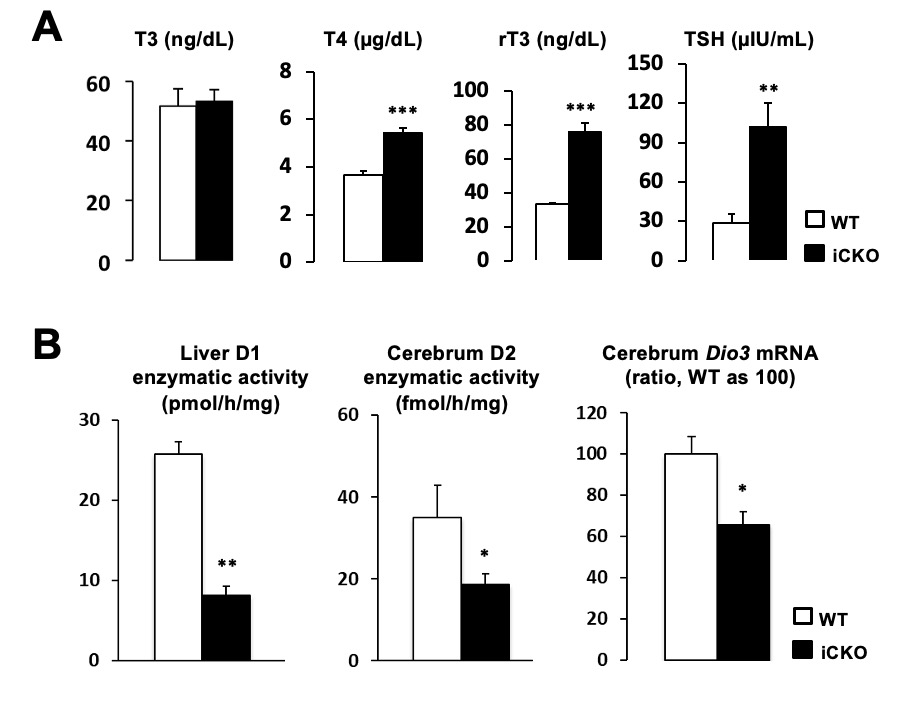
Figure 16. Data from Sbp2 iCKO vs Wt male mice. A. Serum TFTs in Sbp2 iCKO vs Wt male mice. B. Enzymatic activity of the D1 in liver, D2 in cerebrum and mRNA expression of Dio3 in cerebrum. Sbp2 iCKO mice represented as black bars and Wt littermates in open bars *, P <0.05; **, P <0.01; ***, P <0.001.
Differential Diagnosis
From the point of view of the thyroid phenotype, the combination of non-suppressed (normal or slightly elevated) serum TSH with increased concentrations of T4 and decreased T3 levels, is characteristic for the TH metabolism defects due to SBP2 deficiency. An elevated TSH and a general medical evaluation would help distinguishing the thyroid test abnormalities from those encountered in acute non-thyroidal illness, which in terms of iodothyronines could be similar. It is important to confirm the abnormalities by repeat testing several weeks or months apart because the consequence of a variety of non-thyroidal illnesses and some drugs are often transient. For a comprehensive thyroid evaluation, it is recommended to perform the entire panel of thyroid tests, including the free TH levels by dialysis, to exclude abnormalities in serum TH-binding proteins.
Because the clinical presentations of a THMD can be variable, broad and non-specific, including short stature and growth delay, the differential diagnosis can be extensive. Obtaining thyroid tests in first-degree relatives is important in determining the inheritance pattern of the phenotype, and identification of other affected individuals can help in categorizing the symptoms and signs. Given the recessive mode of inheritance, investigation of relatives is helpful in large families and when the patient has multiple siblings. For SBP2 deficiency in particular, measuring serum Se and SePP levels as well as GPX activity can avoid more invasive tests such are skin or muscle biopsies.
Finding a mutation in the SBP2 gene can be sufficient to provide a diagnosis if the mutation is predicted and/or demonstrated to result in a functionally defective protein or results in failure to synthesize the protein. Linkage analysis in smaller families is particularly helpful in excluding the involvement of SBP2. Failure to detect a SBP2 mutation by sequencing only coding regions of the gene is not sufficient, as putative mutations can exist in introns and regulatory elements. In this case, measuring the TSH responses to incremental doses of L-T4 and/or L-T3 could be used to confirm the biochemical diagnosis of TH metabolism defect, as described in the section on Laboratory Tests.
Treatment
Identification of the metabolic pathway responsible for the phenotype in these patients and the demonstration of defects in the SBP2 gene provided further insight into targeted treatment possibilities. Three such options, namely, administration of Se, TH and vitamin E were tested (270, 281, 295).
Administration of up to 400 mcg of selenium (295), in the form of selenomethionine but not selenite, normalized the serum selenium concentration but not selenoprotein P levels and did not restore the TH metabolism dysfunction. Se supplementation in form of selenomethionine contained in Se-rich yeast seems to be more effective as it can be incorporated nonspecifically into all circulating serum proteins (296), whereas selenite is metabolized and inserted as selenocysteine into the growing peptide chain of selenoproteins (297), therefore resulting in different Se bioavailability.
The effect of L-T3 administration was tested in three patients as it was demonstrated to equally suppress serum TSH concentration in affected and unaffected subjects, bypassing the defect (8). Delayed linear growth can be improved with L-T3 supplementation (281), but experience with TH administration in these patients is limited.
As increased oxidative stress state was documented in SBP2 deficiency, treatment with vitamin E was evaluated in a patient. The level of 7β-hydroxycholesterol, a free radical-mediated lipid peroxidation product, was found to be elevated in the patient at baseline, and was reduced to control levels after 2 weeks of α-tocopherol acetate treatment. The effect persisted during 2 years of treatment and at least 7 months after withdrawal (270). Other clinical features of SBP2 defects are treated symptomatically. Physical, occupational and speech therapy was required in some of these patients with developmental delay.
ACKNOWLEDGMENTS
Supported in part by Grants DK15070 and DK110322, from the National Institutes of Health.
Reproduced, in part, with permission from Dumitrescu AM, Korwutthikulrangsri M, and Refetoff S: Reduced sensitivity to Thyroid Hormone: Defects of Transport, Metabolism and Action (Chapter 64). In Werner & Ingbar's The Thyroid: A Fundamental and Clinical Text. Braverman, L.E., and Cooper D.S. (eds.), Wolters Kluver / Lippincott, Williams & Wilkins Publications, Philadelphia, PA., pp. 845-873, 2021, with permission.
REFERENCES
- Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stippled epiphyses, goiter, and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27:279-94.
- Refetoff S, DeGroot LJ, Benard B, DeWind LT. Studies of a sibship with apparent hereditary resistance to the intracellular action of thyroid hormone. Metabolism. 1972;21:723-56.
- Sakurai A, Takeda K, Ain K, Ceccarelli P, Nakai A, Seino S, et al. Generalized resistance to thyroid hormone associated with a mutation in the ligand-binding domain of the human thyroid hormone receptor b. Proc Natl Acad Sci (USA). 1989;86:8977-81.
- Usala SJ, Tennyson GE, Bale AE, Lash RW, Gesundheit N, Wondisford FE, et al. A base mutation of the c-erbAb thyroid hormone receptor in a kindred with generalized thyroid hormone resistance. Molecular heterogeneity in two other kindreds. J Clin Invest. 1990;85:93-100.
- Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14:348-99.
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated with Mutations in a Monocarboxylate Transporter Gene. Am J Hum Genet. 2004 Jan;74(1):168-75. PubMed PMID: 14661163.
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004 Oct 16;364(9443):1435-7. PubMed PMID: 15488219.
- Dumitrescu AM, Liao X-H, Abdullah SYM, Lado-Abeal J, Abdul-Majed F, Moeller LC, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005 Nov;37(11):1247-52. PubMed PMID: 16228000.
- Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, et al. A Mutation in the Thyroid Hormone Receptor Alpha Gene. N Engl J Med. 2012 Jan 19;366(3):243-9. PubMed PMID: 22168587.
- Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Thyroid. 2014 Mar;24(3):407-9. PubMed PMID: 24588711. Epub 2014/03/05. eng.
- Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. J Clin Endocrinol Metab. 2014 Mar;99(3):768-70. PubMed PMID: 24823702. Epub 2014/05/16. eng.
- Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Eur Thyroid J. 2014 Mar;3(1):7-9. PubMed PMID: 24847459. Epub 2014/05/23. eng.
- Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol. 2003 Dec 31;213(1):1-11. PubMed PMID: 15062569.
- Flamant F, Cheng SY, Hollenberg AN, Moeller LC, Samarut J, Wondisford FE, et al. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology. 2017 Jul 01;158(7):2052-7. PubMed PMID: 28472304.
- Friesema EC, Jansen J, Milici C, Visser TJ. Thyroid hormone transporters. Vitam Horm. 2005;70:137-67. PubMed PMID: 15727804.
- Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23(1):38-89. PubMed PMID: 11844744.
- Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439-66. PubMed PMID: 10845098.
- Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol. 2006 Feb 26;246(1-2):121-7. PubMed PMID: 16442701.
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue specific thyroid hormone deprivation and excess in Mct8 deficient mice. Endocrinology. 2006 Sep(147):4036-43. PubMed PMID: 16709608.
- van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, et al. Clinical phenotype and mutant TRalpha1. N Engl J Med. 2012 Apr 12;366(15):1451-3. PubMed PMID: 22494134. Epub 2012/04/13. eng.
- Weiss RE, Hayashi Y, Nagaya T, Petty KJ, Murata Y, Tunka H, et al. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptors a or ß genes may be due to a defective co-factor. J Clin Endocrinol Metab. 1996 Dec;81(12):4196-203. PubMed PMID: 8954015.
- Weintraub BD, Gershengorn MC, Kourides IA, Fein H. Inappropriate secretion of thyroid stimulating hormone. Ann Intern Med. 1981;95:339-51.
- Ando S, Sarlis NJ, Krishan J, Feng X, Refetoff S, Zhang MQ, et al. Aberrant alternative splicing of thyroid hormone receptor in a TSH-secreting pituitary tumor is a mechanism for hormone resistance. Mol Endocrinol. 2001;15(9):1529-38.
- Tagami T, Usui T, Shimatsu A, Beniko M, Yamamoto H, Moriyama K, et al. Aberrant expression of thyroid hormone receptor beta isoform may cause inappropriate secretion of TSH in a TSH-secreting pituitary adenoma. J Clin Endocrinol Metab. 2011 Jun;96(6):E948-52. PubMed PMID: 21430027.
- Kaplan MM, Swartz SL, Larsen PR. Partial peripheral resistance to thyroid hormone. Am J Med. 1981;70(5):1115-21. PubMed PMID: 7234877.
- Usala SJ. Molecular diagnosis and characterization of thyroid hormone resistance syndromes. Thyroid. 1991;1:361-7.
- Beck-Peccoz P, Roncoroni R, Mariotti S, Medri G, Marcocci C, Brabant G, et al. Sex hormone-binding globulin measurement in patients with inappropriate secretion of thyrotropin (IST):Evidence against selective pituitary thyroid hormone resistance in nonneoplastic IST. J Clin Endocrinol Metab. 1990;71:19-25.
- Beck-Peccoz P, Chatterjee VKK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid. 1994;4:225-32.
- Collingwood TN, Adams M, Tone Y, Chatterjee VKK. Spectrum of transcriptional, dimerization, and dominant negative properties of twenty different mutant thyroid hormone ß-receptors in thyroid hormone resistance syndrome. Mol Endocrinol. 1994;8:1262-77.
- Weiss RE, Balzano S, Scherberg NH, Refetoff S. Neonatal detection of generalized resistance to thyroid hormone. JAMA. 1990;264:2245-50. PubMed PMID: 2120481.
- LaFranchi SH, Snyder DB, Sesser DE, Skeels MR, Singh N, Brent GA, et al. Follow-up of newborns with elevated screening T4 concentrations. J Pediatr. 2003 Sep;143(3):296-301. PubMed PMID: 14517508.
- Tajima T, Jo W, Fujikura K, Fukushi M, Fujieda K. Elevated free thyroxine levels detected by a neonatal screening system. Pediatr Res. 2009 Sep;66(3):312-6. PubMed PMID: 19542904.
- Personal-Observation.
- Takeda K, Sakurai A, DeGroot LJ, Refetoff S. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-ß gene. J Clin Endocrinol Metab. 1992 Jan;74(1):49-55. PubMed PMID: 1727829.
- Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. Homozygosity for a "dominant negative" thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1991;73:990-4.
- Ferrara AM, Onigata K, Ercan O, Woodhead H, WeissRE, Refetoff S. Homozygous thyroid hormone receptor beta gene mutations in resistance to thyroid hormone: Three new cases and review of the literature. J Clin Endocrinol Matab. 2012 Apr;97(4):1328-36. PubMed PMID: 22319036. eng.
- Usala SJ, Bale AE, Gesundheit N, Weinberger C, Lash RW, Wondisford FE, et al. Tight linkage between the syndrome of generalized thyroid hormone resistance and the human c-erbAb gene. Mol Endocrinol. 1988;2:1217-20.
- Weiss RE, Weinberg M, Refetoff S. Identical mutations in unrelated families with generalized resistance to thyroid hormone occur in cytosine-guanine-rich areas of the thyroid hormone receptor beta gene: Analysis of 15 families. J Clin Invest. 1993;91:2408-15.
- Zhou Z, Yang C, Lv F, Liu W, Yan S, Zang H, et al. Novel THRB mutation analysis in congenital hypothyroidism with thyroid dysgenesis. J Cell Biochem. 2018 Nov;119(11):9474-82. PubMed PMID: 30074255.
- Takeda K, Nemoto KI, Hayashi Y, Yamamoto M, Sakuta R, Kimura T, et al. Two Mutations in Thyroid Hormone Receptor Beta Gene (P453A and C36Y) in a Family with Resistance to Thyroid Hormone with Comorbid Myotonic Dystrophy. Thyroid. 2019 Apr;29(4):607-8. PubMed PMID: 30672388.
- Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001;86(11):5572-6. PubMed PMID: 11701737.
- Sadow P, Reutrakul S, Weiss RE, Refetoff S. Resistnace to thyroid hormone in the absence of mutations in the thyroid hormone receptor genes. Curr Opin Endocrinol Diabetes. 2000;7:253-9.
- Mitsuhashi T, Tennyson GE, Nikodem VM. Alternative splicing generates messages encoding rat c-erbA proteins that do not bind thyroid hormones. Proc Natl Acad Sci USA. 1988;85:5804-5.
- Macchia PE, Takeuchi Y, Kawai T, Cua K, Gauthier K, Chassande O, et al. Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor alpha. Proc Natl Acad Sci (USA). 2001 Jan 2;98(1):349-54. PubMed PMID: 11120878.
- Chassande O, Fraichard A, Gauthier K, Flamant F, Legrand C, Savatier P, et al. Identification of transcripts initiated from an internal promoter in the c-erb-Aa locus that encode inhibitors of retinoic acid receptor-a and triiodothyronine receptor activities. Mol Endocrinol. 1997;11:1278-90.
- Williams GR. Cloning and Characterization of Two Novel Thyroid Hormone Receptor beta Isoforms. Mol Cell Biol. 2000;20(22):8329-42.
- Tagami T, Yamamoto H, Moriyama K, Sawai K, Usui T, Shimatsu A, et al. Identification of a novel human thyroid hormone receptor beta isoform as a transcriptional modulator. Biochem Biophys Res Commun. 2010 Jun 11;396(4):983-8. PubMed PMID: 20470753. Epub 2010/05/18. eng.
- Gauthier K, Plateroti M, Harvey CB, Williams GR, Weiss RE, Refetoff S, et al. Genetic analysis reveals different functions for the products of the thyroid hormone receptor alpha locus. Mol Cell Biol. 2001;21(14):4748-60.
- Casas F, Rochard P, Rodier A, Cassar-Malek I, Marchal-Victorion S, Wiesner RJ, et al. A variant form of the nuclear triiodothyronine receptor c-ErbAalpha1 plays a direct role in regulation of mitochondrial RNA synthesis. Mol Cell Biol. 1999 Dec;19(12):7913-24. PubMed PMID: 10567517.
- Hodin RA, Lazar MA, Chin WW. Differential and tissue-specific regulation of the multiple rat c-erbA messenger RNA species by thyroid hormone. J Clin Invest. 1990;85:101-5.
- Macchia E, Nakai A, Janiga A, Sakurai A, Fisfalen ME, Gardner P, et al. Characterization of site-specific polyclonal antibodies to c-erbA peptides recognizing human thyroid hormone receptors a1, a2, and ß and native 3,5,3'-triiodothyronine receptor, and study of tissue distribution of the antigen. Endocrinology. 1990;126:3232-9.
- Strait KA, Schwartz HL, Perez-Castillo A, Oppenheimer JH. Relationship of c-erbA mRNA content to tissue triiodothyronine nuclear binding capacity and function in developing and adult rats. J Biol Chem. 1990;265:10514-21.
- Mannavola D, Moeller LC, Beck-Peccoz P, Persani L, Weiss RE, Refetoff S. A novel splice variant involving the 5' untranslated region of the thyroid hormone receptor ß1 (TRß1). J Endocrinol Invest. 2004;27:318-22.
- Frankton S, Harvey CB, Gleason LM, Fadel A, Williams GR. Multiple mRNA variants regulate cell-specific expression of human thyroid hormone receptor ß1. Mol Endocrinol. 2004;18(7):1931-642.
- Gauthier K, Chassande O, Platerotti M, Roux J-P, Legrand C, Rousset B, et al. Different functions for the thyroid hormone receptors TRa and TRß in the control of thyroid hormone production and post-natal development. EMBO J. 1999;18:623-31.
- Göthe S, Wang Z, Ng L, Kindblom JM, Campos barros A, Ohlsson C, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes & Dev. 1999;13:1329-41.
- Forman BM, Casanova J, Raaka BM, Ghysdael J, Samuels HH. Half-site spacing and orientation determines whether thyroid hormone and retinoic acid receptors and related factors bind to DNA response elements as monomers, homodimers, or heterodimers. Mol Endocrinol. 1992;6:429-42.
- Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers and heterodimers. Endocr Rev. 1994;15:391-407.
- Brent GA, Dunn MK, Harney JW, Gulick T, Larsen PR, Moore DD. Thyroid hormone aporeceptor represses T3-inducible promoters and blocks activity of the retinoic acid receptor. New Biologist. 1989;1:329-36.
- Koenig RJ. Thyroid hormone receptor coactivators and corepressors. Thyroid. 1998;8:703-13.
- Pazin MJ, Kadonaga JT. What's up and down with histone deacetylation and transcription? Cell. 1997;89:325-8.
- Yen PM, Darling DS, Carter RL, Forgione M, Umeda PK, Chin WW. Triiodothyronine (T3) decreases binding to DNA by T3-receptor homodimers but not receptor-auxiliary protein heterodimers. J Biol Chem. 1992;267:3565-8.
- Yu J, Koenig RJ. Regulation of Hepatocyte Thyroxine 5'-Deiodinase by T3 and Nuclear Receptor Coactivators as a Model of the Sick Euthyroid Syndrome. J Biol Chem. 2000;275(49):38296-301.
- Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Current Opinion Cell Biol. 1997;9:222-32.
- Fondell JD, Guermah M, Malik S, Roeder RG. Thyroid hormone receptor-associated proteins and general positive cofactors mediate thyroid hormone receptor function in the absence of the TATA box-binding protein-associated factors of TFIID. Proc Natl Acad Sci USA. 1999;96:1959-64.
- Moeller LC, Cao X, Dumitrescu AM, Seo H, Refetoff S. Thyroid hormone mediated changes in gene expression can be initiated by cytosolic action of the thyroid hormone receptor beta through the phosphatidylinositol 3-kinase pathway. Nucl Recept Signal. 2006;4:e020. PubMed PMID: 16862226.
- Newell FW, Diddie KR. [Typical monochromacy, congenital deafness, and resistance to intracellular action of thyroid hormone (author's transl)]. Klinische Monatsblatter fur Augenheilkunde. 1977 Nov;171(5):731-4. PubMed PMID: 304503. Typische Monochromasie, angeborene Taubheit und Resistenz gegenuber der intrazellularen Wirkung des Thyreoideahormons.
- Jones I, Srinivas M, Ng L, Forrest D. The thyroid hormone receptor beta gene: structure and functions in the brain and sensory systems. Thyroid. 2003 Nov;13(11):1057-68. PubMed PMID: 14651789.
- Hayashi Y, Janssen OE, Weiss RE, Murata Y, Seo H, Refetoff S. The relative expression of mutant and normal thyroid hormone receptor genes in patients with generalized resistance to thyroid hormone determined by estimation of their specific messenger ribonucleic acid products. J Clin Endocrinol Metab. 1993;76(1):64-9. PubMed PMID: 8421105.
- Sakurai A, Miyamoto T, Refetoff S, DeGroot LJ. Dominant negative transcriptional regulation by a mutant thyroid hormone receptor ß in a family with generalized resistance to thyroid hormone. Mol Endocrinol. 1990;4:1988-94.
- Chatterjee VKK, Nagaya T, Madison LD, Datta S, Rantoumis A, Jameson JL. Thyroid hormone resistance syndrome.Inhibition of normal receptor function by mutant thyroid hormone receptors. J Clin Invest. 1991;87:1977-84.
- Yen PM, Sugawara A, Refetoff S, Chin WW. New insights on the mechanism(s) of the dominant negative effect of mutant thyroid hormone receptor in generalized resistance to thyroid hormone. J Clin Invest. 1992;90:1825-31.
- Piedrafita FJ, Ortiz MA, Pfahl M. Thyroid hormone receptor-ß mutants, associated with generalized resistance to thyroid hormone show defects in their ligand-sensitive repression function. Mol Endocrinol. 1995;9:1533-48.
- Au-Fliegner M, Helmer E, Casanova J, Raaka BM, Samuels HT. The conserved ninth C-terminal heptad in thyroid hormone and retinoic acid receptors mediates diverse responses by affecting heterodimer but not homodimer formation. Molecular and Cellular Biology. 1993;13(9):5725-37.
- Nagaya T, Jameson JL. Thyroid hormone receptor dimerization is required for the dominant negative inhibition by mutations that cause thyroid hormone resistance. J Biol Chem. 1993;268(21):15766-71.
- Yoh SM, Chatterjee VKK, Privalsky ML. Thyroid hormone resistance syndrome manifests as an aberrant interaction between mutant T3 receptor and transcriptional corepressor. Mol Endocrinol. 1997;11:470-80.
- Tagami T, Gu W-X, Peairs PT, West BL, Jameson JL. A novel natural mutation in the thyroid hormone receptor defines a dual functional domain that exchanges nuclear receptor corepressors and coactivators. Mol Endocrinol. 1998;12:1888-902.
- Collingwood TN, Rajanayagam O, Adams M, Wagner R, Cavaillès V, Kalkhoven E, et al. A natural transactivation mutation in the thyroid hormone ß receptor: Impaired interaction with putative transcriptional mediators. Proc Natl Acad Sci USA. 1997;94:248-53.
- Liu Y, Takeshita A, Misiti S, Chin WW, Yen PM. Lack of coactivator interaction can be a mechanism for dominant negative activity by mutant thyroid hormone receptors. Endocrinology. 1998;139:4197-204.
- Collingwood TN, Wagner R, Matthews CH, Clifton-Bligh RJ, Mark G, Rajanayagam O, et al. A role for helix 3 of the TRß ligand-binding domain in coactivator recruitment identified by characterization of a third cluster of mutations in resistance to thyroid hormone. Embo J. 1998;17(Aug 17):4760-70.
- Nagaya T, Madison LD, Jameson JL. Thyroid hormone receptor mutants that cause resistance to thyroid hormone. Evidence for receptor competition for DNA sequences in target genes. J Biol Chem. 1992;267:13014-9.
- Nagaya T, Fujieda N, Seo H. Requirement of corepressor binding of thyroid hormone receptor mutants for dominant negative inhibition. BiochemBiophys Res Commun. 1998;247:620-3.
- Hayashi Y, Sunthornthepvarakul T, Refetoff S. Mutations of CpG dinucleotides located in the triiodothyronine (T3)-binding domain of the thyroid hormone receptor (TR) ß gene that appears to be devoid of natural mutations may not be detected because they are unlikely to produce the clinical phenotype of resistance to thyroid hormone. J Clin Invest. 1994;94:607-15.
- Korwutthikulrangsri M, Dosiou C, Dumitrescu AM, Refetoff S. A novel G385E variant in the cold region of T3-binding domain of THRB gene and investigations to assess its clinical significance. Europ Thyroid J. 2019:in press.
- Larsen CC, Dumitrescu A, Guerra-Arguero LM, Gallego-Suarez C, Vazquez-Mellado A, Vinogradova M, et al. Incidental Identification of a Thyroid Hormone Receptor Beta (THRB) Gene Variant in a Family with Autoimmune Thyroid Disease. Thyroid. 2013 Dec;23(12):1638-43. PubMed PMID: 23806029. Pubmed Central PMCID: 3868256. Epub 2013/06/29. eng.
- Rastinejad F, Perlmann T, Evans F, Sigler P. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203-11.
- Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;138:690-7.
- Marimuthu A, Feng W, Tagami T, Nguyen H, Jameson JL, Fletterick RJ, et al. TR surfaces and conformations required to bind nuclear receptor corepressor. Mol Endocrinol. 2002;16(2):271-86. PubMed PMID: 11818500.
- Huber BR, Desclozeaux M, West BL, Cunha-Lima ST, Nguyen HT, Baxter JD, et al. Thyroid hormone receptor-beta mutations conferring hormone resistance and reduced corepressor release exhibit decreased stability in the N-terminal ligand-binding domain. Mol Endocrinol. 2003 Jan;17(1):107-16. PubMed PMID: 12511610.
- Hayashi Y, Weiss RE, Sarne DH, Yen PM, Sunthornthepvarakul T, Marcocci C, et al. Do clinical manifestations of resistance to thyroid hormone correlate with the functional alteration of the corresponding mutant thyroid hormone-ß receptors? J Clin Endocrinol Metab. 1995;80:3246-56.
- Yagi H, Pohlenz J, Hayashi Y, Sakurai A, Refetoff S. Resistance to thyroid hormone caused by two mutant thyroid hormone receptor ß, R243Q and R243W, with marked impairment of function that cannot be explained by altered in-vitro 3,5,3'-triiodothyronine binding affinity. J Clin Endocrinol Metab. 1997;82:1608-14.
- Safer JD, Cohen RN, Hollenberg AN, Wondisford FE. Defective release of corepressor by hinge mutants of the thyroid hormone receptor found in patients with resistance to thyroid hormone. J Biol Chem. 1998;273:30175-82.
- Clifton-Bligh RJ, de Zegher F, Wagner RL, Collingwood TN, François I, Van Helvoirt M, et al. A novel mutation (R383H) in resistance to thyroid hormone syndrome predominantly impairs corepressor release and negative transcriptional regulation. Mol Endocrinol. 1998 May;12(5):609-21. PubMed PMID: 9605924.
- Flynn TR, Hollenberg AN, Cohen O, Menke JB, Usala SJ, Tollin S, et al. A novel C-terminal domain in the thyroid hormone receptor selectively mediates thyroid hormone inhibition. J Biol Chem. 1994;629(52):32713-6.
- Taniyama M, Ishikawa N, Momotani N, Ito K, Ban Y. Toxic multinodular goitre in a patient with generalized resistance to thyroid hormone who harbours the R429Q mutation in the thyroid hormone receptor beta gene. Clin Endocrinol (Oxf). 2001;54(1):121-4.
- Brucker-Davis F, Skarulis MC, Grace MB, Benichou J, Hauser P, Wiggs E, et al. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. The National Institutes of Health prospective study. Ann Intern Med. 1995;123:573-83.
- Safer JD, O'Connor MG, Colan SD, Srinivasan S, Tollin SP, Wondisford FE. The thyroid hormone receptor-ß gene mutation R383H is associated with isolated central resistance to thyroid hormone. J Clin Endocrinol Metab. 1999;84:3099-109.
- Lee S, Young BM, Wan W, Chan IH, Privalsky ML. A Mechanism for Pituitary-Resistance to Thyroid Hormone (PRTH) Syndrome: a Loss in Cooperative Coactivator Contacts by Thyroid Hormone Receptor (TR)[beta]2. Mol Endocrinol. 2011 Jul;25(7):1111-25. PubMed PMID: 21622532.
- Machado DS, Sabet A, Santiago LA, Sidhaye AR, Chiamolera MI, Ortiga-Carvalho TM, et al. A thyroid hormone receptor mutation that dissociates thyroid hormone regulation of gene expression in vivo. Proc Natl Acad Sci U S A. 2009 Jun 9;106(23):9441-6. PubMed PMID: 19439650.
- Alberobello AT, Congedo V, Liu H, Cochran C, Skarulis MC, Forrest D, et al. An intronic SNP in the thyroid hormone receptor beta gene is associated with pituitary cell-specific over-expression of a mutant thyroid hormone receptor beta2 (R338W) in the index case of pituitary-selective resistance to thyroid hormone. J Transl Med. 2011;9:144. PubMed PMID: 21871106. Pubmed Central PMCID: 3170239. Epub 2011/08/30. eng.
- Johansson C, Göthe S, Forrest D, Vennsrtöm B, Thorén P. Cardiovascular phenotype and temperature controlin mice lacking tyroid hormone receptor ß or both a1 and ß. Am J Physiol. 1999;276:H2006-H12.
- Gloss B, Trost SU, Bluhm WF, Swanson EA, Clark R, Winkfein R, et al. Cardiac Ion Channel Expression and Contractile Function in Mice with Deletion of Thyroid Hormone Receptor alpha or beta. Endocrinology. 2001;142(2):544-50.
- Kahaly GJ, Matthews CH, Mohr-Kahaly S, Richards CA, Chatterjee VK. Cardiac involvement in thyroid hormone resistance. J Clin Endocrinol Metab. 2002;87(1):204-122. PubMed PMID: 11788648.
- Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14:184-93.
- Falcone M, Miyamoto T, Fierro-Renoy F, Nacchia E, DeGroot LJ. Antipeptide polyclonal antibodies specifically recognize each human thyroid hormone receptor isoform. Endocrinology. 1992;131:2419-29.
- Mixson AJ, Hauser P, Tennyson G, Renault JC, Bodenner DL, Weintraub BD. Differential expression of mutant and normal beta T3 receptor alleles in kindreds with generalized resistance to thyroid hormone. J Clin Invest. 1993;91:2296-300.
- Geffner ME, Su F, Ross NS, Hershman JM, Van Dop C, Menke JB, et al. An arginine to histidine mutation in codon 311 of the C-erbAß gene results in a mutant thyroid hormone receptor that does not mediate a dominant negative phenotype. J Clin Invest. 1993;91:538-46.
- Weiss RE, Stein MA, Duck SC, Chyna B, Phillips W, O'Brien T, et al. Low intelligence but not attention deficit hyperactivity disorder is associated with resistance to thyroid hormone caused by mutation R316H in the thyroid hormone receptor ß gene. J Clin Endocrinol Metab. 1994 Jun;78(6):1525-8. PubMed PMID: 8200958.
- Weiss RE, Marcocci C, Bruno-Bossio G, Refetoff S. Multiple genetic factors in the heterogeneity of thyroid hormone resistance. J Clin Endocrinol Metab. 1993;76:257-9.
- Vlaeminck-Guillem V, Margotat A, Torresani J, D'Herbomez M, Decoulx M, Wemeau JL. Resistance to thyroid hormone in a family with no TRß gene anomaly: pathogenic hypotheses. Ann Endocrinol (Paris). 2000;61(3):194-9.
- Parikh S, Ando S, Schneider A, Skarulis MC, Sarlis NJ, Yen PM. Resistance to thyroid hormone in a patient without thyroid hormone receptor mutations. Thyroid. 2002;12(1):81-6.
- Lado Abeal J, Albero Gamboa R, Araujo Vilar D, Barca Mallo O, Bernabeu Moron I, Calvo MT, et al. [Clinical and molecular study of five families with resistance to thyroid hormones.]. Med Clin (Barc). 2011 Nov 12;137(12):551-4. PubMed PMID: 21703645. Epub 2011/06/28. Estudio clinico y molecular de cinco familias con resistencia a la accion de las hormonas tiroideas. Spa.
- Macchia E, Lombardi M, Raffaelli V, Piaggi P, Macchia L, Scattina I, et al. Clinical and genetic characteristics of a large monocentric series of patients affected by thyroid hormone (Th) resistance and suggestions for differential diagnosis in patients without mutation of Th receptor beta. Clin Endocrinol (Oxf). 2014 Dec;81(6):921-8. PubMed PMID: 25040256.
- Reutrakul S, Sadow PM, Pannain S, Pohlenz J, Carvalho G, Macchia PE, et al. Search for abnormalities of nuclear corepressors, coactivators and a coregulator in families with resistance to thyroid hormone without thyroid hormone receptor ß or a genes mutations. J Clin Endocrinol Metab. 2000;85:3609-17.
- Mamanasiri S, Yesil S, Dumitrescu AM, Liao XH, Demir T, Weiss RE, et al. Mosaicism of a Thyroid Hormone Receptor (TR) Beta Gene Mutation in Resistance to Thyroid Hormone (RTH). J Clin Endocrinol Metab. 2006 Jun 27;91(9):3471-7. PubMed PMID: 16804041.
- Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab. 2003 Mar;14(2):85-90. PubMed PMID: 12591179.
- Forrest D, Hanebuth E, Smeyne RJ, Evereds N, Stewart CL, Wehner JM, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor ß: evidence for tissue-specific modulation of receptor function. EMBO J. 1996;15:3006-15.
- Weiss RE, Forrest D, Pohlenz J, Cua K, Curran T, Refetoff S. Thyrotropin regulation by thyroid hormone in thyroid hormone receptor ß-deficient mice. Endocrinology. 1997;138:3624-9.
- Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. Thyroid hormone action on liver, heart and energy expenditure in thyroid hormone receptor ß deficient mice. (Erratum, 141:4767, 2000). Endocrinology. 1998 Dec;139:4945-52. PubMed PMID: 9832432.
- Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor ß is essential for development of auditory function. Nature Genet. 1996;13:354-7.
- Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27(1):94-8.
- Newell FW, Diddie KR. Typische Monochromasie, angeborene Taubheit und Resistenz gegenüber der intrazellulären Wikung des Thyroideahormons. Klin Mbl Augenheilk. 1977;171:731-4.
- Rüsch A, Erway LC, Oliver D, Vennström B, Forrest D. Thyroid hormone receptor ß-dependent expression of a potassium conductance in inner hair cells at the onset of hearing. Proc Natl Acad Sci (USA). 1998;95(26):15758-62.
- Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, et al. Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291-300.
- Mansouri A, Chawdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nature Genet. 1998;19:87-90. PubMed PMID: 9590297.
- Flamant F, Poguet AL, Plateroti M, Chassande O, Gauthier K, Streichenberger N, et al. Congenital Hypothyroid Pax8(-/-) Mutant Mice Can Be Rescued by Inactivating the TRalpha Gene. Mol Endocrinol. 2002;16(1):24-32.
- Hayashi Y, Mangoura D, Refetoff S. A mouse model of resistance to thyroid hormone produced by somatic gene transfer of a mutant thyroid hormone receptor. Mol Endocrinol. 1996;10:100-6.
- Kaneshige M, Kaneshige K, Zhu X-g, Dace A, Garrett L, Carter TA, et al. Mice with a targeted mutation in the thyroid hormone ß receptor gene exhibit impaired growth and resistanceto thyroid hormone. Proc Natl Acad Sci (USA). 2000;97:13209-14.
- Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci (USA). 2001;98:3998-4003.
- Weiss RE, Xu J, Ning G, Pohlenz J, O'Malley BW, Refetoff S. Mice deficient in the steroid receptor coactivator-1 (SRC-1) are resistant to thyroid hormone. EMBO J. 1999;18(7):1900-4. PubMed PMID: 10202153.
- Brown NS, Smart A, Sharma V, Brinkmeier ML, Greenlee L, Camper SA, et al. Thyroid hormone resistance and increased metabolic rate in the RXR-g-deficient mouse. J Clin Invest. 2000;106:73-9.
- Persani L, Borgato S, Romoli R, Asteria C, Pizzocardo A, Beck-Peccoz P. Changes in the degree of sialylation of carbohydrate chain modify the biological properties of circulating thyrotropin isoforms in various physiological and pathological states. J Clin Endocrinol Metab. 1998;83:2486-92.
- Bode HH, Danon M, Weintraub BD, Maloof F, Crawford JD. Partial target organ resistance to thyroid hormone. J Clin Invest. 1973;52:776-82.
- Tamagna EI, Carlson HE, Hershman JM, Reed AW. Pituitary and peripheral resistance to thyroid hormone. Clin Endocrinol. 1979;10:431-41.
- Bajorunas DR, Rosner W, Kourides IA. Use of bromocriptine in a patient with generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1984;58:731-5.
- Refetoff S, Salazar A, Smith TJ, Scherberg NH. The consequences of inappropriate treatment because of failure to recognize the syndrome of pituitary and peripheral tissue resistance to thyroid hormone. Metabolism. 1983;32:822-34. PubMed PMID: 6865780.
- Refetoff S, DeGroot LJ, Barsano CP. Defective thyroid hormone feedback regulation in the syndrome of peripheral resistance to thyroid hormone. J Clin Endocrinol Metab. 1980;51(1):41-5. PubMed PMID: 6769941.
- Kaplowitz PB, D'Ercole AJ, Utiger RD. Peripheral resistance to thyroid hormone in an infant. J Clin Endocrinol Metab. 1981;53:958-63.
- Murata Y, Refetoff S, Horwitz AL, Smith TJ. Hormonal regulation of glycosaminoglycan accumulation in fibroblasts from patients with resistance to thyroid hormone. J Clin Endocrinol Metab. 1983;57(6):1233-9. PubMed PMID: 6630416.
- Ceccarelli P, Refetoff S, Murata Y. Resistance to thyroid hormone diagnosed by the reduced response of fibroblasts to the triiodothyronine induced suppression of fibronectin synthesis. J Clin Endocrinol Metab. 1987;65(2):242-6. PubMed PMID: 3597704.
- Mäenpää J, Liewendahl K. Peripheral insensitivity to thyroid hormones in a euthyroid girl with goitre. Arch Dis Child. 1980;55:207-12.
- White P, Burton KA, Fowden AL, Dauncey MJ. Developmental expression analysis of thyroid hormone receptor isoforms reveals new insights into their essential functions in cardiac and skeletal muscles. Faseb J. 2001 Jun;15(8):1367-76. PubMed PMID: 11387234.
- Cooper DS, Ladenson PW, Nisula BC, Dunn JF, Chapman EM, Ridgway EC. Familial thyroid hormone resistance. Metabolism. 1982;31:504-9.
- Lamberg B-A, Liewendahl K. Thyroid hormone resistance. Ann Clin Res. 1980;12:243-53.
- Vandalem JL, Pirens G, Hennen G. Familial inappropriate TSH secretion: evidence suggesting a dissociated pituitary resistance to T3 and T4. J Endocrinol Invest. 1981;4:413-22.
- Lamberg BA, Sandström R, Rosengård S, Säarinen P, Evered DC. Sporadic and familial partial peripheral resistance to thyroid hormone. In: Harland WA, Orr JS, editors. Thyroid Hormone Metabolism. London: Academic Press; 1975. p. 139-61.
- Hauser P, Zametkin AJ, Martinez P, Vitiello B, Matochik JA, Mixson AJ, et al. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N Engl J Med. 1993;328:997-1001.
- Weiss RE, Stein MA, Trommer B, Refetoff S. Attention-deficit hyperactivity disorder and thyroid function. J Pediatr. 1993;123:539-45.
- Elia J, Gulotta C, Rose SR, Martin G, Rapoport J. Thyroid function and attention-deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1994;33:169-72.
- Anselmo J, Cao D, Karrison T, Weiss RE, Refetoff S. Fetal loss associated with excess thyroid hormone exposure. Jama. 2004 Aug 11;292(6):691-5.
- Anselmo J, Scherberg NH, Dumitrescu AM, Refetoff S. Reduced Sensitivity to Thyroid Hormone as a Transgenerational Epigenetic Marker Transmitted Along the Human Male Line. Thyroid. 2019 Jun;29(6):778-82. PubMed PMID: 30938226.
- Weiss AH, Kelly JP, Bisset D, Deeb SS. Reduced L- and M- and increased S-cone functions in an infant with thyroid hormone resistance due to mutations in the THRbeta2 gene. Ophthalmic Genet. 2012 May 2. PubMed PMID: 22551329. Epub 2012/05/04. Eng.
- Lamberg BA. Congenital euthyroid goitre and partial peripheral resistance to thyroid hormones. Lancet. 1973;1:854-7.
- Bantle JP, Seeling S, Mariash CN, Ulstrom RA, Oppenheimer JH. Resistance to thyroid hormones: A disorder frequently confused with Graves' disease. Arch Intern Med. 1982;142:1867-71.
- Chait A, Kanter R, Green W, Kenny M. Defective thyroid hormone action in fibroblasts cultured from subjects with the syndrome of resistance to thyroid hormones. J Clin Endocrinol Metab. 1982;54:767-72.
- Ünlütürk U, Sriphrapradang C, Erdoğan MF, Emral R, Güldiken S, Refetoff S, et al. Management of differentiated thyroid cancer in the presence of resistance to thyroid hormone and TSH producing adenomas: A report of four cases and review of the literatures. J Clin Endocrinol Metab. 2013 Apr 3;98(6):2210-17. PubMed PMID: 23553855. Epub 2013/04/05. Eng.
- Schimmel M, Utiger R. Thyroidal and peripheral production of thyroid hormones:Review of recent findings and their clinical implications. Ann Intern Med. 1977;87:760-8.
- Gómez-Sáez JM, Fernández Castañer M, Navarro MA, Bonnin MR, Soler Ramón J, Roca A. Resistencia parcial a las hormonas tiroideas con bocio y eutiroidismo. Med Clin. 1981;76:412-6.
- Gheri RG, Bianchi R, Mariani G, Toccafondi R, Cappelli G, Brat A, et al. A new case of familial partial generalized resistance to thyroid hormone: Study of 3,5,3'-triiodothyronine (T3) binding to lymphocyte and skin fibroblast nuclei and in vivo conversion of thyroxine to T3. J Clin Endocrinol Metab. 1984;58:563-9.
- Sarne DH, Sobieszczyk S, Ain KB, Refetoff S. Serum thyrotropin and prolactin in the syndrome of generalized resistance to thyroid hormone:Responses to thyrotropin-releasing hormone stimulation and triiodothyronine suppression. J Clin Endocrinol Metab. 1990;70:1305-11.
- Kasai Y, Aritaki S, Utsunomiya M, Matsuno T. Twin sisters with Refetoff's Syndrome. J Jap Pediat Assoc. 1983;87:1203-12.
- Hughes IA, Ichikawa K, DeGroot LJ, John R, Jones MK, Hall R, et al. Non-adenomatous inappropriate TSH hypersecretion and euthyroidism requires no treatment. Clin Endocrinol. 1987;27:475-83. PubMed PMID: 3124992.
- Persani L, Asteria C, Tonacchera M, Vitti P, Chatterjee VKK, Beck-Peccoz P. Evidence for secretion of thyrotropin with enhanced bioactivity in syndromes of thyroid hormone resistance. J Clin Endocrinol Metab. 1994;78:1034-9.
- Barkoff MS, Kocherginsky M, Anselmo J, Weiss RE, Refetoff S. Autoimmunity in Patients with Resistance to Thyroid Hormone. J Clin Endocrinol Metab. 2010 Jul;95(7):3189-93. PubMed PMID: 20444926.
- David L, Blanc JF, Chatelain P, Rouchon A, François R. Goitre congéntal avec résistance péripherique partielle aux hormones thyroïdiennes.Ou syndrome de pseudo-hyperthyroïdie. Pédiatrie. 1979;34:443-9.
- Mitchell CS, Savage DB, Dufour S, Schoenmakers N, Murgatroyd P, Befroy D, et al. Resistance to thyroid hormone is associated with raised energy expenditure, muscle mitochondrial uncoupling, and hyperphagia. J Clin Invest. 2010 Apr;120(4):1345-54. PubMed PMID: 20237409.
- Pulcrano M, Palmieri EA, Ciulla DM, Campi I, Covelli D, Lombardi G, et al. Impact of resistance to thyroid hormone on the cardiovascular system in adults. J Clin Endocrinol Metab. 2009 Aug;94(8):2812-6. PubMed PMID: 19435825.
- Moeller LC, Dumitrescu AM, Walker RL, Meltzer PS, Refetoff S. Thyroid hormone responsive genes in cultured human fibroblasts. J Clin Endocrinol Metab. 2005 Feb;90(2):936-43. PubMed PMID: 15507505.
- Kunitake JM, Hartman N, Henson LC, Lieberman J, Williams DE, Wong M, et al. 3,5,3'-triiodothyroacetic acid therapy for thyroid hormone resistance. J Clin Endocrinol Metab. 1989;69:461-6.
- Magner JA, Petrick P, Menezes-Ferreira MM, Stelling M, Weintraub BD. Familial generalized resistance to thyroid hormones: report of three kindreds and correlation of patterns of affected tissues with the binding of [125I] triiodothyronine to fibroblast nuclei. J Endocrinol Invest. 1986;9:459-70.
- Beck-Peccoz P, Persani L, Faglia G. Glycoprotein hormone a-subunit in pituitary adenomas. Trends Endocrinol Metab. 1992;3:41-5.
- Mornex R, Tommasi M, Cure M, Farcot J, Orgiazzi J, Rousset B. Hyperthyroidie associee a un hypopituitarisme au cours de l'evolution d'une tumeur hypophysaire secretant T.S.H. Ann d'Endocrinol (Paris). 1972;33(4):390-6. PubMed PMID: 4197701.
- Hattori N, Ishihara T, Yamagami K, Shimatsu A. Macro TSH in patients with subclinical hypothyroidism. Clin Endocrinol (Oxf). 2015 Dec;83(6):923-30. PubMed PMID: 25388002. Epub 2014/11/13. Eng.
- Pohlenz J, Weiss RE, Macchia P, E., Pannain S, Lau IT, Ho H, et al. Five new families with resistance to thyroid hormone not caused by mutations in the thyroid hormone receptor ß gene. J Clin Endocrinol Metab. 1999;84:3919-28. PubMed PMID: 10566629.
- Shiwa T, Oki K, Awaya T, Nakanishi S, Yamane K. Resistance to thyroid hormone accompanied by graves' disease. Intern Med. 2011;50(18):1977-80. PubMed PMID: 21921380. Epub 2011/09/17. eng.
- Grasberger H, Ringkananont U, Croxson M, Refetoff S. Resistance to thyroid hormone in a patient with thyroid dysgenesis. Thyroid. 2005 Jul;15(7):730-3. PubMed PMID: 16053391.
- Borck G, Seewi O, Jung A, Schonau E, Kubisch C. Genetic causes of goiter and deafness: Pendred syndrome in a girl and cooccurrence of Pendred syndrome and resistance to thyroid hormone in her sister. J Clin Endocrinol Metab. 2009 Jun;94(6):2106-9. PubMed PMID: 19318451.
- Sabet A, Pallotta JA. Dichotomous responses to thyroid hormone treatment in a patient with primary hypothyroidism and thyroid hormone resistance. Thyroid. 2011 May;21(5):559-61. PubMed PMID: 21595517.
- Hassan AQ, Koh JT. Selective chemical rescue of a thyroid-hormone-receptor mutant, TRbeta (H435Y), identified in pituitary carcinoma and resistance to thyroid hormone. Angew Chem Int Ed Engl. 2008;47(38):7280-3. PubMed PMID: 18683837.
- Pappa T, Anselmo J, Mamanasiri S, Dumitrescu AM, Weiss RE, Refetoff S. Prenatal Diagnosis of Resistance to Thyroid Hormone and Its Clinical Implications. J Clin Endocrinol Metab. 2017 Oct 01;102(10):3775-82. PubMed PMID: 28938413.
- Asteria C, Rajanayagam O, Collingwood TN, Persani L, Romoli R, Mannavola D, et al. Prenatal diagnosis of thyroid hormone resistance. J Clin Endocrinol Metab. 1999;84:405-10.
- Weiss RE, Refetoff S. Treatment of resistance to thyroid hormone--primum non nocere. J Clin Endocrinol Metab. 1999;84(2):401-4.
- Safer JD, Colan SD, Fraser LM, Wondisford FE. A pituitary tumor in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11(3):281-91. PubMed PMID: 11327621.
- Anselmo J, Refetoff S. Regression of a large goiter in a patient with resistance to thyroid hormone by every other day treatment with triiodothyronine. Thyroid. 2004 Jan;14(1):71-4. PubMed PMID: 15009917.
- Weiss RE, Stein MA, Refetoff S. Behavioral effects of liothyronine (L-T3) in children with attention deficit hyperactivity disorder in the presence an absence of resistance to thyroid hormone. Thyroid. 1997 jun;7(3):389-93. PubMed PMID: 9226208.
- Iglesias P, Diez JJ. [Therapeutic possibilities in patients with selective pituitary resistance to thyroid hormones.]. Med Clin (Barc). 2008 Mar 15;130(9):345-50. PubMed PMID: 18373914.
- Beck-Peccoz P, Piscitelli G, Cattaneo MG, Faglia G. Successful treatment of hyperthyroidism due to nonneoplastic pituitary TSH hypersecretion with 3,5,3'-triiodothyroacetic acid (TRIAC). J Endocrinol Invest. 1983;6:217-23.
- Radetti G, Persani L, Molinaro G, Mannavola D, Cortelazzi D, Chatterjee VKK, et al. Clinical and hormonal outcome after two years of triiodothyroacetic acid treatment in a child with thyroid hormone resistance. Thyroid. 1997;7:775-8.
- Takeda T, Suzuki S, Liu R-T, DeGroot LJ. Triiodothyroacetic acid has unique potential for therapy of resistance to thyroid hormone. J Clin Endocrinol Metab. 1995;80:2033-40.
- Kaneshige M, Suzuki H, Kaneshige K, Cheng J, Wimbrow H, Barlow C, et al. A targeted dominant negative mutation of the thyroid hormone alpha 1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci (USA). 2001;98:15095-100. PubMed PMID: 11734632.
- Tinnikov A, Nordstrom K, Thoren P, Kindblom JM, Malin S, Rozell B, et al. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor alpha1. Embo J. 2002 Oct 1;21(19):5079-87. PubMed PMID: 12356724.
- Liu YY, Schultz JJ, Brent GA. A thyroid hormone receptor alpha gene mutation (P398H) Is associated with visceral adiposity and impaired catecholamine-stimulated lipolysis in mice. J Biol Chem. 2003 Jul 16;278:38913-20. PubMed PMID: 12869545.
- Quignodon L, Vincent S, Winter H, Samarut J, Flamant F. A Point Mutation in the Activation Function 2 Domain of Thyroid Hormone Receptor {alpha}1 Expressed after CRE-Mediated Recombination Partially Recapitulates Hypothyroidism. Mol Endocrinol. 2007 Oct;21(10):2350-60. PubMed PMID: 17622582.
- van Gucht ALM, Moran C, Meima ME, Visser WE, Chatterjee K, Visser TJ, et al. Resistance to Thyroid Hormone due to Heterozygous Mutations in Thyroid Hormone Receptor Alpha. Curr Top Dev Biol. 2017;125:337-55. PubMed PMID: 28527577.
- Espiard S, Savagner F, Flamant F, Vlaeminck-Guillem V, Guyot R, Munier M, et al. A novel mutation in THRA gene associated with an atypical phenotype of resistance to thyroid hormone. J Clin Endocrinol Metab. 2015 Jun 2:jc20151120. PubMed PMID: 26037512.
- Korkmaz O, Ozen S, Ozdemir TR, Goksen D, Darcan S. A novel thyroid hormone receptor alpha gene mutation, clinic characteristics, and follow-up findings in a patient with thyroid hormone resistance. Hormones (Athens). 2019 Jon;18(2):223-7. PubMed PMID: 30747412.
- Moran C, Agostini M, McGowan A, Schoenmakers E, Fairall L, Lyons G, et al. Contrasting Phenotypes in Resistance to Thyroid Hormone Alpha Correlate with Divergent Properties of Thyroid Hormone Receptor alpha1 Mutant Proteins. Thyroid. 2017 Jul;27(7):973-82. PubMed PMID: 28471274. Pubmed Central PMCID: PMC5561448.
- Moran C, Agostini M, Visser WE, Schoenmakers E, Schoenmakers N, Offiah AC, et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)alpha1 and TRalpha2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. 2014 Jun 23. PubMed PMID: 24969835. Epub 2014/06/28. Eng.
- Moran C, Schoenmakers N, Agostini M, Schoenmakers E, Offiah A, Kydd A, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. J Clin Endocrinol Metab. 2013 Nov;98(11):4254-61. PubMed PMID: 23940126. Epub 2013/08/14. eng.
- Tylki-Szymanska A, Acuna-Hidalgo R, Krajewska-Walasek M, Lecka-Ambroziak A, Steehouwer M, Gilissen C, et al. Thyroid hormone resistance syndrome due to mutations in the thyroid hormone receptor alpha gene (THRA). Journal of medical genetics. 2015 May;52(5):312-6. PubMed PMID: 25670821.
- van Gucht AL, Meima ME, Zwaveling-Soonawala N, Visser WE, Fliers E, Wennink JM, et al. Resistance to Thyroid Hormone Alpha in an 18-Month-Old Girl: Clinical, Therapeutic, and Molecular Characteristics. Thyroid. 2016 Mar;26(3):338-46. PubMed PMID: 26782358.
- van Gucht ALM, Meima ME, Moran C, Agostini M, Tylki-Szymanska A, Krajewska MW, et al. Anemia in Patients With Resistance to Thyroid Hormone alpha: A Role for Thyroid Hormone Receptor alpha in Human Erythropoiesis. J Clin Endocrinol Metab. 2017 Sep 01;102(9):3517-25. PubMed PMID: 28911146.
- Demir K, van Gucht AL, Buyukinan M, Catli G, Ayhan Y, Nijat Bas V, et al. Diverse Genotypes and Phenotypes of Three Novel Thyroid Hormone Receptor Alpha Mutations. J Clin Endocrinol Metab. 2016 May 4:jc20161404. PubMed PMID: 27144938.
- Moran C, Chatterjee K. Resistance to Thyroid Hormone alpha-Emerging Definition of a Disorder of Thyroid Hormone Action. J Clin Endocrinol Metab. 2016 Jul;101(7):2636-9. PubMed PMID: 27381958.
- Mittag J, Wallis K, Vennstrom B. Physiological consequences of the TRalpha1 aporeceptor state. Heart Fail Rev. 2010 Mar;15(2):111-5. PubMed PMID: 19009345. Epub 2008/11/15. eng.
- Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001 Aug;22(4):451-76. PubMed PMID: 11493579.
- Ekins R. The free hormone hypothesis and measurement of free hormones. Clin Chem. 1992;38:1289-93.
- Friesema EC, Docter R, Moerings EP, Stieger B, Hagenbuch B, Meier PJ, et al. Identification of thyroid hormone transporters. Biochem Biophys Res Commun. 1999 Jan 19;254(2):479-501. PubMed PMID: 9918867.
- van der Putten HH, Friesema EC, Abumrad NA, Everts ME, Visser TJ. Thyroid hormone transport by the rat fatty acid translocase. Endocrinology. 2003 Apr;144(4):1315-23. PubMed PMID: 12639914.
- Mitchell AM, Tom M, Mortimer RH. Thyroid hormone export from cells: contribution of P-glycoprotein. J Endocrinol. 2005 Apr;185(1):93-8. PubMed PMID: 15817830. Epub 2005/04/09. eng.
- Taylor PM, Ritchie JW. Tissue uptake of thyroid hormone by amino acid transporters. Best Pract Res Clin Endocrinol Metab. 2007 Jun;21(2):237-51. PubMed PMID: 17574006. Epub 2007/06/19. eng.
- Hagenbuch B. Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab. 2007 Jun;21(2):209-21. PubMed PMID: 17574004. Epub 2007/06/19. eng.
- Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol. 2002 Oct;16(10):2283-96. PubMed PMID: 12351693. Epub 2002/09/28. eng.
- Visser WE, Friesema EC, Jansen J, Visser TJ. Thyroid hormone transport by monocarboxylate transporters. Best Pract Res Clin Endocrinol Metab. 2007 Jun;21(2):223-36. PubMed PMID: 17574005.
- Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003 Oct 10;278:40128-35. PubMed PMID: 12871948.
- Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008 Jun;22(6):1357-69. PubMed PMID: 18337592.
- Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006 Nov;20(11):2761-72. PubMed PMID: 16887882.
- Stromme P, Groeneweg S, Lima de Souza EC, Zevenbergen C, Torgersbraten A, Holmgren A, et al. Mutated Thyroid Hormone Transporter OATP1C1 Associates with Severe Brain Hypometabolism and Juvenile Neurodegeneration. Thyroid. 2018 Nov;28(11):1406-15. PubMed PMID: 30296914.
- Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, Refetoff S. A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics. 2008 Jan;121(1):e199-202. PubMed PMID: 18166539.
- Schwartz CE, Stevenson RE. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab. 2007 Jun;21(2):307-21. PubMed PMID: 17574010.
- Frints SG, Lenzner S, Bauters M, Jensen LR, Van Esch H, des Portes V, et al. MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet. 2008 Sept;16(9):1029-37. PubMed PMID: 18398436.
- Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic. 1944;48:325-34.
- Schwartz CE, Ulmer J, Brown A, Pancoast I, Goodman HO, Stevenson RE. Allan-Herndon syndrome. II. Linkage to DNA markers in Xq21. Am J Hum Genet. 1990 Sep;47(3):454-8. PubMed PMID: 2393020.
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, et al. Allan-Herndon-Dudley Syndrome and the Monocarboxylate Transporter 8 (MCT8) Gene. Am J Hum Genet. 2005 Jul;77(1):41-53. PubMed PMID: 15889350.
- Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, et al. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J. 2011 Oct 15;439(2):249-55. PubMed PMID: 21726201. Epub 2011/07/06. eng.
- Muller J, Mayerl S, Visser TJ, Darras VM, Boelen A, Frappart L, et al. Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology. 2014 Jan;155(1):315-25. PubMed PMID: 24248460. Epub 2013/11/20. eng.
- Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology. 2012 Mar;153(3):1528-37. PubMed PMID: 22294745. Epub 2012/02/02. eng.
- Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest. 2014 May;124(5):1987-99. PubMed PMID: 24691440. Pubmed Central PMCID: 4001533. Epub 2014/04/03. eng.
- Lafreniere RG, Carrel L, Willard HF. A novel transmembrane transporter encoded by the XPCT gene in Xq13.2. Hum Mol Genet. 1994 Jul;3(7):1133-9. PubMed PMID: 7981683.
- Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004 Feb;447(5):619-28. PubMed PMID: 12739169.
- Korwutthikulrangsri M, Raimondi C, Dumitrescu AM, editors. A novel compound heterozygous SBP2 gene mutations in a boy with developmental delay and failure to thrive The 13th International Workshop on Resistance to Thyroid Hormone; Sept 2018; Doorn, Netherlands.
- Kleinau G, Schweizer U, Kinne A, Kohrle J, Gruters A, Krude H, et al. Insights into molecular properties of the human monocarboxylate transporter 8 by combining functional with structural information. Thyroid Res. 2011;4 Suppl 1:S4. PubMed PMID: 21835051. Pubmed Central PMCID: 3155110. Epub 2011/08/13. eng.
- Holden KR, Zuniga OF, May MM, Su H, Molinero MR, Rogers RC, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005 Oct;20(10):852-7. PubMed PMID: 16417886.
- Visser WE, Jansen J, Friesema EC, Kester MH, Mancilla E, Lundgren J, et al. Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat. 2008 Jul 17;30:29-38. PubMed PMID: 18636565.
- Friesema EC, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. 2010 Jan 18. PubMed PMID: 20083155.
- Boccone L, Mariotti S, Dessi V, Pruna D, Meloni A, Loudianos G. Allan-Herndon-Dudley syndrome (AHDS) caused by a novel SLC16A2 gene mutation showing severe neurologic features and unexpectedly low TRH-stimulated serum TSH. Eur J Med Genet. 2010 Nov-Dec;53(6):392-5. PubMed PMID: 20713192.
- Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005 Jun;252(6):663-6. PubMed PMID: 15834651.
- Visser WE, Friesema EC, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. 2011 Jan;25(1):1-14. PubMed PMID: 20660303.
- Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005 Sep;153(3):359-66. PubMed PMID: 16131597.
- Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, et al. Unexpected Peripheral Markers of Thyroid Function in a Patient with a Novel Mutation of the MCT8 Thyroid Hormone Transporter Gene. Horm Res. 2007 Sep 15;67(1):1-6. PubMed PMID: 16974106.
- Wemeau JL, Pigeyre M, Proust-Lemoine E, d'Herbomez M, Gottrand F, Jansen J, et al. Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008 Jun;93(6):2084-8. PubMed PMID: 18334584.
- Fuchs O, Pfarr N, Pohlenz J, Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009 Mar;51(3):240-4. PubMed PMID: 19018842. Epub 2008/11/21. eng.
- Namba N, Etani Y, Kitaoka T, Nakamoto Y, Nakacho M, Bessho K, et al. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. 2008 Sep 25;167(7):785-91. PubMed PMID: 17899191.
- Vaurs-Barriere C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Vinas JM, et al. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009 Jan;65(1):114-8. PubMed PMID: 19194886.
- Gika AD, Siddiqui A, Hulse AJ, Edwards S, Fallon P, McEntagart ME, et al. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med & Child Neurol. 2010 May;52(5):475-82. PubMed PMID: 19811520.
- Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. 2005 Oct;147(4):552-4. PubMed PMID: 16227048.
- Sijens PE, Rodiger LA, Meiners LC, Lunsing RJ. 1H magnetic resonance spectroscopy in monocarboxylate transporter 8 gene deficiency. J Clin Endocrinol Metab. 2008 May;93(5):1854-9. PubMed PMID: 18319316. Epub 2008/03/06. eng.
- Korwutthikulrangsri M, Fujisawa H, Fu J, Liao X-H, Dumitrescu AM, , editors. Contributions of the hypothalamus and pituitary in expressing the thyroid phenotype of Sbp2 deficiency: other regulatory tiers in addition to the thyroid gland. The 101st Annual Meeting of the Endocrine Society, ; 2019; New Orleans, LA.
- Crushell E, Reardon W. Elevated TSH levels in a mentally retarded boy. Eur J Pediatr. 2009 May;169(5):573-5. PubMed PMID: 19936787.
- Lopez-Espindola D, Morales-Bastos C, Grijota-Martinez C, Liao XH, Lev D, Sugo E, et al. Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab. 2014 Dec;99(12):E2799-804. PubMed PMID: 25222753. Epub 2014/09/16. eng.
- Dempsey MA, Dumitrescu AM, Refetoff S. MCT8 (SLS16A2)-Specific thyroid hormone cell transporter deficiency. GeneReviews. 2010. PubMed PMID: 20301789. Epub 2010/03/20. eng.
- Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007 Mar 1;117(3):627-35. PubMed PMID: 17318265.
- Bernal J. Role of Monocarboxylate Anion Transporter 8 (MCT8) in Thyroid Hormone Transport: Answers from Mice. Endocrinology. 2006 Sep;147(9):4034-5. PubMed PMID: 16912236.
- Di Cosmo C, Liao XH, Ye H, Ferrara AM, Weiss RE, Refetoff S, et al. Mct8-deficient mice have increased energy xpenditure and reduced fat mass that is abrogated by normalization of serum T3 levels. Endocrinology. 2013 Dec;154(12):4885-95. PubMed PMID: 24029243. Epub 2013/09/14. Eng.
- Ferrara AM, Liao XH, Ye H, Weiss RE, Dumitrescu AM, Refetoff S. The Thyroid Hormone Analog DITPA Ameliorates Metabolic Parameters of Male Mice With Mct8 Deficiency. Endocrinology. 2015 Nov;156(11):3889-94. PubMed PMID: 26322373. Pubmed Central PMCID: 4606752.
- Trajkovic-Arsic M, Visser TJ, Darras VM, Friesema EC, Schlott B, Mittag J, et al. Consequences of Monocarboxylate Transporter 8 Deficiency for Renal Transport and Metabolism of Thyroid Hormones in Mice. Endocrinology. 2010 Feb;151(2):802-9. PubMed PMID: 19996182.
- Di Cosmo C, Liao XH, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S. Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest. 2010 Sept 1;120(9):3377-88. PubMed PMID: 20679730.
- Liao XH, Di Cosmo C, Dumitrescu AM, Hernandez A, Van Sande J, St Germain DL, et al. Distinct Roles of Deiodinases on the Phenotype of Mct8 Defect: A Comparison of Eight Different Mouse Genotypes. Endocrinology. 2011 Mar;152(3):1180-91. PubMed PMID: 21285310.
- Fliers E, Unmehopa UA, Alkemade A. Functional neuroanatomy of thyroid hormone feedback in the human hypothalamus and pituitary gland. Mol Cell Endocrinol. 2006 Jun 7;251(1-2):1-8. PubMed PMID: 16707210.
- Di Cosmo C, Liao XH, Dumitrescu AM, Weiss RE, Refetoff S. A thyroid hormone analogue with reduced dependence on the monocarboxylate transporter 8 (MCT8) for tissue transport. Endocrinology. 2009 Jun 4;150(9):4450-8. PubMed PMID: 19497976.
- Ferrara AM, Liao XH, Gil-Ibanez P, Bernal J, Weiss RE, Dumitrescu AM, et al. Placenta Passage of the Thyroid Hormone Analog DITPA to Male Wild-Type and Mct8-Deficient Mice. Endocrinology. 2014 Oct;155(10):4088-93. PubMed PMID: 25051435. Epub 2014/07/23. eng.
- Iwayama H, Liao XH, Braun L, Barez-Lopez S, Kaspar B, Weiss RE, et al. Adeno Associated Virus 9-Based Gene Therapy Delivers a Functional Monocarboxylate Transporter 8, Improving Thyroid Hormone Availability to the Brain of Mct8-Deficient Mice. Thyroid. 2016 Sep;26(9):1311-9. PubMed PMID: 27432638.
- Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2008;23(1):22-44. PubMed PMID: 18305372. Epub 2008/02/29. eng.
- Price NT, Jackson VN, Halestrap AP. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochem J. 1998 Jan 15;329 ( Pt 2):321-8. PubMed PMID: 9425115. Pubmed Central PMCID: 1219047. Epub 1998/02/28. eng.
- Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone sensitive neuron populations. Endocrinology. 2005 Jan 20. PubMed PMID: 15661862.
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters MCT8 (SLC16A2) and OATP14 (SLCO1C1) at the blood-brain barrier. Endocrinology. 2008 Dec;149(12):6251-61. PubMed PMID: 18687783.
- Trajkovic-Arsic M, Muller J, Darras VM, Groba C, Lee S, Weih D, et al. Impact of Monocarboxylate Transporter-8 Deficiency on the Hypothalamus-Pituitary-Thyroid Axis in Mice. Endocrinology. 2010 Oct;151(10):5053-62. PubMed PMID: 20702572.
- Braun D, Lelios I, Krause G, Schweizer U. Histidines in Potential Substrate Recognition Sites Affect Thyroid Hormone Transport by Monocarboxylate Transporter 8 (MCT8). Endocrinology. 2013 Apr 16. PubMed PMID: 23592749. Epub 2013/04/18. Eng.
- Lima de Souza EC, Groeneweg S, Visser WE, Peeters RP, Visser TJ. Importance of Cysteine Residues in the Thyroid Hormone Transporter MCT8. Endocrinology. 2013 May;154(5):1948-55. PubMed PMID: 23546606. Epub 2013/04/03. eng.
- Saito Y, Shichiri M, Hamajima T, Ishida N, Mita Y, Nakao S, et al. Enhancement of lipid peroxidation and its amelioration by vitamin E in a subject with mutations in the SBP2 gene. Journal of lipid research. 2015 Nov;56(11):2172-82. PubMed PMID: 26411970. Pubmed Central PMCID: 4617404.
- Verge CF, Konrad D, Cohen M, Di Cosmo C, Dumitrescu AM, Marcinkowski T, et al. Diiodothyropropionic Acid (DITPA) in the Treatment of MCT8 Deficiency. J Clin Endocrinol Metab. 2012 Dec;97(12):4515-23. PubMed PMID: 22993035. Epub 2012/09/21. eng.
- Lee JY, Kim MJ, Deliyanti D, Azari MF, Rossello F, Costin A, et al. Overcoming Monocarboxylate Transporter 8 (MCT8)-Deficiency to Promote Human Oligodendrocyte Differentiation and Myelination. EBioMedicine. 2017 Nov;25:122-35. PubMed PMID: 29111262. Pubmed Central PMCID: 5704066.
- Groeneweg S, Peeters RP, Moran C, Stoupa A, Auriol F, Tonduti D, et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019 Sep;7(9):695-706. PubMed PMID: 31377265.
- Braun D, Schweizer U. The Chemical Chaperone Phenylbutyrate Rescues MCT8 Mutations Associated With Milder Phenotypes in Patients With Allan-Herndon-Dudley Syndrome. Endocrinology. 2017 Mar 1;158(3):678-91. PubMed PMID: 27977298.
- Groeneweg S, van den Berge A, Meima ME, Peeters RP, Visser TJ, Visser WE. Effects of Chemical Chaperones on Thyroid Hormone Transport by MCT8 Mutants in Patient-Derived Fibroblasts. Endocrinology. 2018 Mar 1;159(3):1290-302. PubMed PMID: 29309566.
- Bernal J, Pekonen F. Ontogenesis of the nuclear 3,5,3'-triiodothyronine receptor in the human fetal brain. Endocrinology. 1984 Feb;114(2):677-9. PubMed PMID: 6317365.
- Schoenmakers E, Carlson B, Agostini M, Moran C, Rajanayagam O, Bochukova E, et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J Clin Invest. 2016 Mar 1;126(3):992-6. PubMed PMID: 26854926. Pubmed Central PMCID: 4767355.
- Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. 2010 Oct 8;87(4):538-44. PubMed PMID: 20920667. Pubmed Central PMCID: 2948803.
- Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr. 2003;23:17-40. PubMed PMID: 12524431.
- Koenig RJ. Regulation of type 1 iodothyronine deiodinase in health and disease. Thyroid. 2005 Aug;15(8):835-40. PubMed PMID: 16131326.
- Di Cosmo C, McLellan N, Liao XH, Khanna KK, Weiss RE, Papp L, et al. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J Clin Endocrinol Metab. 2009 0CT;94(10):4003-9. PubMed PMID: 19602558.
- Azevedo MF, Barra GB, Naves LA, Ribeiro Velasco LF, Godoy Garcia Castro P, de Castro LC, et al. Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J Clin Endocrinol Metab. 2010 Aug;95(8):4066-71. PubMed PMID: 20501692.
- Schoenmakers E, Agostini M, Mitchell C, Schoenmakers N, Papp L, Rajanayagam O, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. 2010 Dec 1;120(12):4220-35. PubMed PMID: 21084748.
- Hamajima T, Mushimoto Y, Kobayashi H, Saito Y, Onigata K. Novel compound heterozygous mutations in the SBP2 gene: characteristic clinical manifestations and the implications of GH and triiodothyronine in longitudinal bone growth and maturation. Eur J Endocrinol. 2012 Apr;166(4):757-64. PubMed PMID: 22247018. Epub 2012/01/17. eng.
- Catli G, Fujisawa H, Kirbiyik O, Mimoto MS, Gencpinar P, Ozdemir TR, et al. A Novel Homozygous Selenocysteine Insertion Sequence Binding Protein 2 (SECISBP2, SBP2) Gene Mutation in a Turkish Boy. Thyroid. 2018 Sep;28(9):1221-3. PubMed PMID: 29882503.
- Lescure A, Allmang C, Yamada K, Carbon P, Krol A. cDNA cloning, expression pattern and RNA binding analysis of human selenocysteine insertion sequence (SECIS) binding protein 2. Gene. 2002 May 29;291(1-2):279-85. PubMed PMID: 12095701.
- Copeland PR. Regulation of gene expression by stop codon recoding: selenocysteine. Gene. 2003 Jul 17;312:17-25. PubMed PMID: 12909337.
- Papp LV, Lu J, Striebel F, Kennedy D, Holmgren A, Khanna KK. The redox state of SECIS binding protein 2 controls its localization and selenocysteine incorporation function. Mol Cell Biol. 2006 Jul;26(13):4895-910. PubMed PMID: 16782878.
- Bubenik JL, Driscoll DM. Altered RNA-binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in Sec insertion sequence binding protein 2. J Biol Chem. 2007 Sep 27;282:34653-62. PubMed PMID: 17901054.
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, et al. Characterization of mammalian selenoproteomes. Science. 2003 May 30;300(5624):1439-43. PubMed PMID: 12775843.
- Papp LV, Lu J, Holmgren A, Khanna KK. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid Redox Signal. 2007 Jul;9(7):775-806. PubMed PMID: 17508906.
- Dumitrescu A, Di Cosmo C, Liao XH, Weiss R, Refetoff S. The syndrome of inherited partial SBP2 deficiency in humans. Antioxid Redox Signal. 2010 April 1;12(7):905-20. PubMed PMID: 19769464.
- Seeher S, Atassi T, Mahdi Y, Carlson BA, Braun D, Wirth EK, et al. Secisbp2 is essential for embryonic development and enhances selenoprotein expression. Antioxid Redox Signal. 2014 Aug 20;21(6):835-49. PubMed PMID: 24274065. Pubmed Central PMCID: 4116110. Epub 2013/11/28. eng.
- Fu J, Fujisawa H, Follman B, Liao XH, Dumitrescu AM. Thyroid Hormone Metabolism Defects in a Mouse Model of SBP2 Deficiency. Endocrinology. 2017 Dec 1;158(12):4317-30. PubMed PMID: 29029094. Pubmed Central PMCID: PMC5711384.
- Schomburg L, Dumitrescu AM, Liao XH, Bin-Abbas B, Hoeflich J, Kohrle J, et al. Selenium supplementation fails to correct the selenoprotein synthesis defect in subjects with SBP2 gene mutations. Thyroid. 2009 Mar;19(3):277-81. PubMed PMID: 19265499.
- Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol Biomarkers Prev. 2006 Apr;15(4):804-10. PubMed PMID: 16614127. Epub 2006/04/15. eng.
- Xu J, Yang F, An X, Hu Q. Anticarcinogenic activity of selenium-enriched green tea extracts in vivo. J Agric Food Chem. 2007 Jun 27;55(13):5349-53. PubMed PMID: 17542612. Epub 2007/06/05. eng.
- Feng W, Ribeiro RCJ, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, et al. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747-9.
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454-7.
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397-404.
- Busch K, Martin B, Baniahmad A, Renkawitz R, Muller M. At least three subdomains of v-erbA are involved in its silencing function. Mol Endocrinol. 1997;11:379-89.
- Forman BM, Samuels HH. Interactions among a subfamily of nuclear hormone receptors: the regulatory zipper model. Mol Endocrinol. 1990;4:1293-301.
- Kurokawa R, Yu VC, Naar A, Kyakumoto S, Han Z, Silverman S, et al. Differential orientations of the DNA-binding domain and carboxy-terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes & Development. 1993;7:1423-35.
- Beck-Peccoz P, Chatterjee VKK, Chin WW, DeGroot LJ, Jameson JL, Nakamura H, et al. Nomenclature of thyroid hormone receptor ß-gene mutations in resistance to thyroid hormone: Consensus statement from the first workshop on thyroid hormone resistance, July 10-11, 1993, Cambridge, United Kingdom. JClin Endocrinol Metab. 1994 Apr; 78(4):990-3. PubMed PMID: 8157732.
- Tone Y, Collingwood TN, Adams M, Chatterjee VK. Functional analysis of a transactivation domain in the thyroid hormone ß receptor. J Biol Chem. 1994;269:31157-61.
- Baniahmad A, Leng X, Burris TP, Tsai SY, Tsai M-J, O'Malley BW. The tau 4 activation domain of the thyroid hormone receptor is required for release of a putative corepressor(s) necessary for transcriptional silencing. Mol Cell Biol. 1995;15:76-86.
- Hamy F, Helbeque NJ, Henichart P. Comparison between synthetic nuclear localisation signal peptides from the steroid thyroid hormone receptor superfamily. Biochem Byophys Res Commun. 1992;183:289-93.
- Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D. A canonical strusture for the ligand-binding domain of the nuclear receptors. Nature Struct Biol. 1996;3:87-94.