ABSTRACT
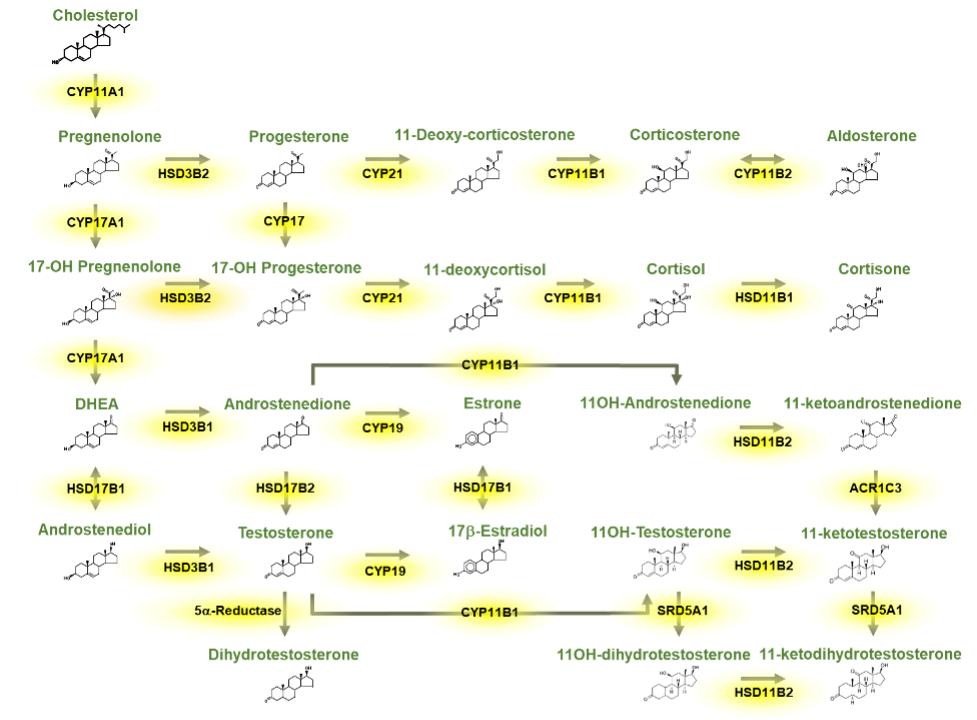
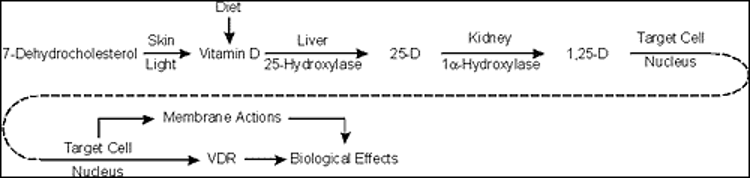
Vitamin D is produced in the skin under the influence of UVB portion of the light spectrum in sunlight. This form of vitamin D is cholecalciferol or vitamin D3. Vitamin D may also be part of the diet either in certain foods such as fatty fish or as supplements either ingested separately or in supplemented food such as milk. Such supplements can be either vitamin D3 or vitamin D2, the form produced in plants from UVB radiation of ergosterol. For the purposes of this chapter, we will make no distinction between these forms of vitamin D, although the differences in the side chain (double bond at C22-23 and methyl group at C24 of vitamin D2) do alter both the binding of vitamin D2 to vitamin D binding protein (DBP) and the metabolism of vitamin D2. Vitamin D then must be metabolized to its most active form, 1,25 dihydroxyvitamin D (1,25(OH)2D), by a series of steps involving first the conversion to 25 hydroxyvitamin D (25OHD), the principal circulating form of vitamin D, principally by the enzyme CYP2R1 in the liver, then to 1,25(OH)2D principally in the kidney by the enzyme CYP27B1. That said there are a number of enzymes with 25-hydroxylase activity found in a number of tissues, and although CYP27B1 is essentially the only 1-hydroxylase, this enzyme is found in numerous tissues where it is thought to play primarily a paracrine/autocrine role. Putting a check to the production of 25OHD and 1,25(OH)2D is the 24-hydroxylase CYP24A1, that metabolizes both 25OHD and 1,25(OH)2D to inactive products. Like CYP27B1, CYP24A1 is widely distributed. Vitamin D and its metabolites are carried in blood tightly bound to DBP, such that only a small fraction (<1%) is free to enter most cells unless those cells express the megalin/cubilin complex which facilitates the transport of the DBP bound metabolites into the cell. DBP is produced in the liver, and its levels can vary in patients with limited hepatic synthetic capacity. Moreover, DBP is an acute phase reactant so a variety of conditions can alter DBP levels and thus the measurement of vitamin D and its metabolites. 1,25(OH)2D is the major ligand for the vitamin D receptor (VDR), a nuclear transcription factor found in most if not all cells of the body and known to regulate thousands of genes in a cell specific manner. Although numerous physiologic functions have been attributed to vitamin D and its metabolites, clinically its major action is to control the availability of calcium and phosphate from the diet for the proper mineralization of the skeleton. Thus, disorders in vitamin D availability, metabolism, and action manifest first and foremost in the skeleton that when severe result in rickets in growing children and osteomalacia in adults in whom the growth plates have closed. After a brief review of vitamin D metabolism and molecular mechanisms of action this chapter will describe the pathologic changes in bone when the supply of mineral to bone is inadequate due to disorders in vitamin D action whether from dietary deficiency, metabolism, or mechanism of action. We will then look in depth at the causes of vitamin D deficiency including mutations resulting in altered metabolism, then discuss the range of effects different mutations in the VDR have on its function including the example of alopecia, which is best known function of VDR that is independent of its ligand 1,25(OH)2D.
INTRODUCTION
Vitamin D derived from endogenous production in the skin or absorbed from the gut is transformed into its active form by two successive steps: hydroxylation in the liver to 25-hydroxyvitamin D [25(OH)D] primarily by the enzyme CYP2R1 followed by 1a-hydroxylation in the renal proximal tubule to 1,25-dihydroxyvitamin D (1,25(OH)2D-calcitriol) by the enzyme CYP27B. Other cells exhibit 1α-hydroxylase activity including placental decidual cells, epithelial cells including keratinocytes, macrophages, dendritic cells, parathyroid cells, and some tumor cells. The role of the extrarenal production of 1,25(OH)2D is thought to play a paracrine and/or autocrine function and under normal conditions does not significantly contribute to the circulating levels of the hormone. Hydroxylation at carbon 24 to produce 24,25-dihydroxyvitamin D [24,25(OH)2D] or 1,24,25-trihydroxyvitamin D by the enzyme CYP24A1 is performed in a wide range of normal tissues and is believed to be important in the removal of vitamin D metabolites. CYP27B1 and CYP24A1 are mitochondrial mixed function oxidase containing cytochrome P450 with ferredoxin and heme-binding domains whereas CYP2R1 is likewise a P450 mixed function oxidase in the microsomes. The sequences of these genes are known contributing to an understanding of the mutations that affect their function (1-9).
The 25-hydroxylation of vitamin D has been thought to be primarily substrate dependent but recently studies found that it is under metabolic control (10). Obesity induced by a high fat diet reduces the levels of hepatic CYP2R1 leading to reduced levels of 25OHD (11). CYP27B1 is stringently regulated by parathyroid hormone (PTH) through a cAMP mediated pathway; calcitonin in a different region of the proximal tubule, apparently not via a rise in cAMP; 1,25(OH)2D through its receptor; calcium in part via its regulation of PTH and by phosphorus mainly via Fibroblast Growth Factor 23 (FGF-23) production in osteocytes and osteoblasts. Of these the stimulation by PTH and inhibition by FGF23 and 1,25(OH)2D are best studied and likely the dominant regulators. CYP24A1is likewise regulated by PTH and FGF23 but opposite to that of CYP27B1 and is markedly induced by 1,25(OH)2D. All vitamin D metabolites are fat soluble and circulate principally bound to vitamin D binding protein (DBP), an a-globulin produced primarily in the liver. Most of the body pool of vitamin D is in the body fat while only a small fraction of the pools of 25(OH)D or 1,25(OH)2D are in fat. Normal daily turnover of vitamin D and 25(OH)D are approximately 30 and 15 mg per day respectively with 1,25(OH)2D daily production about 1 mg. The fractional conversion of vitamin D to 1,25(OH)2D is much higher in vitamin D deficient states. It is difficult to measure vitamin D in blood. Circulating vitamin D metabolites measured in clinical practice are 25(OH)D and 1,25(OH)2D. As 25(OH)D synthesis is primarily substrate dependent, serum levels of this metabolite are taken as a measure of vitamin D status.
1,25(OH)2D biologic actions are mediated via a high-affinity intracellular vitamin D receptor (VDR). VDR acts as a ligand-modulated transcription factor that belongs to the steroid, thyroid, and retinoic acid receptors gene family (12-15). VDR is found in most if not all tissues, leading to the increased recognition of multiple target organs and actions of the hormone. However, the degree to which vitamin D impacts physiologic processes outside the musculoskeletal system remains a subject of substantial investigation and controversy.
1,25(OH)2D is the most powerful physiological agent that stimulates active transport of calcium, and to a lesser degree phosphorus and magnesium, across the small intestine (16,17). Thus, disorders in vitamin D action will lead to a decrease in the net flux of mineral to the extracellular compartment causing hypocalcemia and secondary hyperparathyroidism. Hypophosphatemia will also ensue, as a result of both increased renal phosphate clearance due to secondary hyperparathyroidism and reduced absorption of phosphorus due to deficient 1,25(OH)2D action on the gut. Low concentrations of calcium and phosphorus in the extracellular fluid will lead to defective mineralization of organic bone matrix. Defective bone matrix mineralization of the newly formed bone and growth plate cartilage will produce the characteristic morphological and clinical signs of rickets, while at sites of bone remodeling, it will cause osteomalacia. Disorders in vitamin D action may impair the differentiation of osteoblasts and thus their functional capacity to mineralize bone matrix; this may be an additional mechanism of minor importance that contributes to rickets and osteomalacia (18,19). Continuous bone remodeling takes place in the growing skeleton, thus children with rickets will have osteomalacia as well. The clinical term used in this chapter to describe defective bone matrix mineralization in general will be rickets and/or osteomalacia.
Mineralization of the newly formed organic matrix of bone is a complex and highly ordered process. The essential components for normal mineralization include appropriate extracellular concentrations of calcium, phosphorous and normal functions of the bone forming cells. Disturbances in any of these components will lead to a stereotypic response of disturbed mineralization. The histological, radiological and most of the clinical features characteristic of rickets and/or osteomalacia will be the same regardless of the primary disorder. Thus, defective bone matrix mineralization can be caused by: a) calcium deficiency (20) (e.g. a rare nutritional deficiency of the element seen primarily in children or the more common form of calcium deficiency secondary to disorders of vitamin D metabolism); b) phosphorous deficiency (e.g. increased renal phosphate clearance as in X-linked hypophosphatemia (for detailed discussion see Endotext chapter entitled Primary Disorders of Phosphate Metabolism); c) primary defects in local bone processes (e.g. hypophosphatasia).
Serum concentrations of calcium, phosphorous, PTH, 25(OH)D, biochemical markers of bone turnover and 24 hour urinary calcium excretion will differentiate among the different primary disturbances leading to defective bone matrix mineralization (Table 1).
|
Table 1. Biochemical Parameters of Mineral and Bone Metabolism in Patients with Rickets and/or Osteomalacia, by Etiology |
|||||
|
Serum levels |
|
||||
|
Etiology |
Calcium |
Phosphorous |
iPTH |
Bone specific alk.phos* |
|
|
Hypocalcemic e.g., vitamin D deficiency |
Low to low normal |
Low |
Elevated |
Elevated |
Low |
|
Hypophosphatemic e.g., X-linked hypophosphatemia |
Normal |
Low |
Normal to |
Elevated |
Low to elevated |
|
Primary defects in bone e.g., hypophosphatasia |
Normal |
Normal |
Normal |
Low |
Normal |
* Alk. phos. alkaline phosphatase activity
This chapter will deal with hypocalcemic rickets and/or osteomalacia. A primary calcium deficiency state due to extremely low calcium intake was reported in growing children (20) but is very uncommon. Thus, the discussion will concentrate on calcium deficiency secondary to disorders in vitamin D action. Such disorders can be due to disturbances along the cascade of 1,25(OH)2D synthesis leading to a deficiency of the hormonal form of the vitamin (vitamin D deficiency states) or defects in the interaction of 1,25(OH)2D and its target tissues (resistance to 1,25(OH)2D).
As all disorders in vitamin D action will lead to the same clinical, radiological, histological, and most of the biochemical aberrations, those common features will be discussed first.
CLINICAL AND RADIOLOGICAL FEATURES OF RICKETS AND OSTEOMALACIA
The clinical features of rickets due to disorders in vitamin D action are weakness, bone pain, bone deformity, and fracture. The most rapidly growing bones show the most striking abnormalities. Thus, the localization and severity of the clinical features will depend on the age of onset. In children with acquired disorders in vitamin D action, signs and symptoms of rickets will vary with the age in which the deficiency is being manifested. Children with hereditary disorders of vitamin D action will appear normal at birth as calcium and phosphorous levels in fetal plasma are sustained by placental transport from maternal plasma that is not regulated by the fetal vitamin D system. These children usually develop the characteristic features of rickets within the first 2 years of life. Defects in bone mineralization are particularly evident in regions of rapid bone growth, including, during the first year of life, the cranium, wrist, and ribs. Rickets at this time will lead to widened cranial sutures, frontal bossing, posterior flattening of the skull (craniotabes), widening of the wrists, bulging of costochondral junction (rachitic rosary), and indentation of the ribs at the diaphragmatic insertion (Harrison’s groove). The rib cage may be so deformed that it contributes to respiratory failure. Dental eruption is delayed, and teeth show enamel hypoplasia. Muscle weakness and hypotonia are severe and result in a protuberant abdomen, that may contribute to respiratory failure, and may result in the inability to walk without support. After the first year of life with the acquisition of erect posture and rapid linear growth, the deformities are most severe in the legs. Bow legs (genu varum) or knock-knee (genu valgum) deformities of various severity develop as well as widening of the end of long bones. If not treated, rickets may cause severe lasting deformities, compromise adult height, and increase susceptibility to pathological fractures (Figure 1).
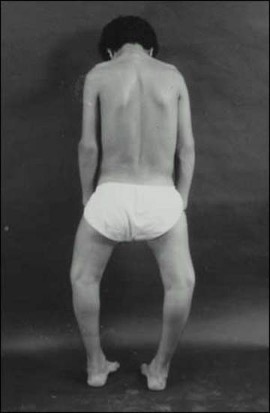
Figure 1. Teenage male with rickets. Note deformities of legs (bow legs) and compromised height.
The specific radiographic features of rickets reflect the failure of cartilage calcification and endochondral ossification and therefore are best seen in the metaphysis of rapidly growing bones (Figures 2 and 3). The metaphyses are widened, uneven, concave, or cupped and because of the delay in or absence of calcification, the metaphyses could become partially or totally invisible on x-ray. In more severe forms or in patients untreated for prolonged periods, rarefaction and thinning of the cortex of the entire shaft, sparse bone trabecularization, and bone deformities will become evident. Greenstick fractures may appear as well.
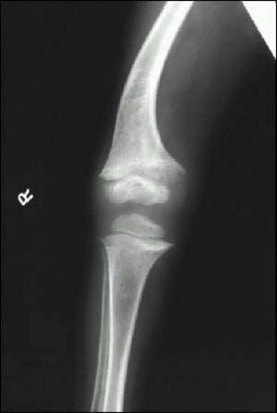
Figure 2. Radiograph of distal femur and proximal tibia and fibula of a patient with rickets. Note widening of epiphysis, resorption of provisional zone of calcification, flaring of metaphysis and bone deformity.
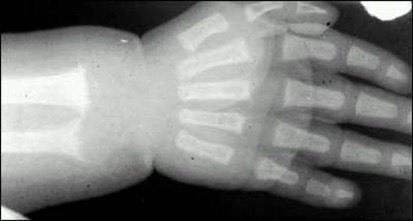
Figure 3. Radiograph of the wrist and hand of a patient with rickets. Note scalloping, widening and irregularity of metaphyseal end of the ulna; apparent widening of the joint space as a result of absence of epiphyseal provisional calcification.
The clinical features of osteomalacia in adults are subtle and could be manifested as bone pain or low back pain of varying severity in some cases. Severe muscle weakness and hypotonia may be a prominent feature in adults with vitamin D deficiency. Improvement of myopathy occurs after low doses of vitamin D.
The first clinical presentation could be an acute fracture of the long bones, pubic ramii, ribs, or spine. The radiographic manifestations could be mild, e.g., generalized, nonspecific osteopenia or more specific, such as pseudofractures, commonly seen at the medial edges of the shafts of long bones (Figure 4).
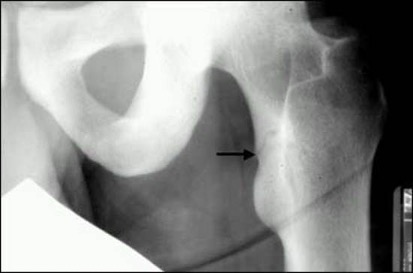
Figure 4. Young male with osteomalacia. Note a pseudofracture in the medial edge of the upper femoral shaft (arrow).
In hypocalcemic rickets and/or osteomalacia, as is the case in disorders in vitamin D action, there may exist radiographic features of secondary hyperparathyroidism such as subperiosteal resorption and cysts of the long bones.
HISTOLOGICAL FEATURES OF RICKETS AND OSTEOMALACIA
The characteristic histological feature of rickets and osteomalacia is deficiency or lack of mineralization of the organic matrix of bone. Osteomalacia is defined as excess osteoid (hyperosteoidosis) and a quantitative dynamic proof of defective bone matrix mineralization obtained by analysis of time-spaced tetracycline labeling (21,22).
Bone Biopsy and Bone Histomorphometry
GENERAL
Transiliac bone biopsy is taken at a standard location: 2cm behind the antero-superior iliac spine and just below the crest, with a terpine 7.5mm inner diameter or 5mm for children. The biopsy should contain the inner and outer cortex and intact trabecular bone in between. Double tetracycline labeling is performed by giving the drug: e.g., dimethyl-chlortetracycline 1g/day in two divided doses, for 2 days, followed by a second administration typically of a different tetracycline with different fluorescent color after 10 to 14 days of drug free interval. The bone biopsy is performed 5 to 7 days following the last dose of tetracycline. The biopsy is sectioned undecalcified and is evaluated unstained or with different stains and techniques to obtain qualitative and quantitative histomorphometric parameters (Figure 5). The following are the commonly used quantitative histomorphometric parameters (22): trabecular bone volume (BV/TV), osteoid volume (OV/BV), osteoid surface (OS/BS), osteoid thickness (O. Th), osteoblast surface (Ob.s/BS), osteoclast surface (Oc.S/BS), osteoclast number (N.Oc/TA), double labeled surface (dLS/BS), single labeled surface (sLS/BS), mineral appositional rate (MAR), bone formation rate (BFR/BS), and mineralization lag time (MLT).
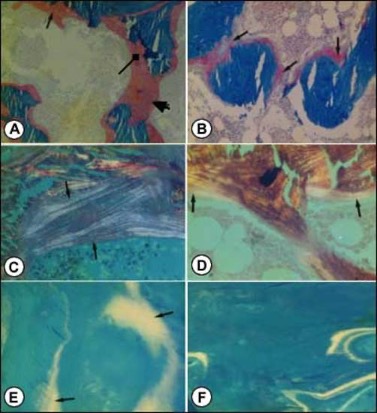
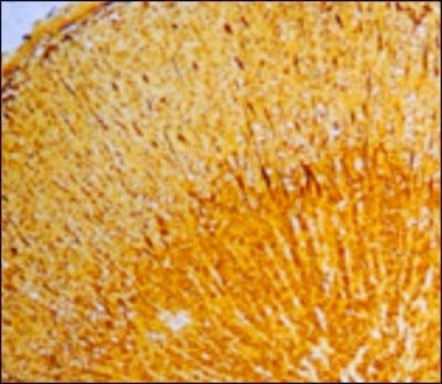
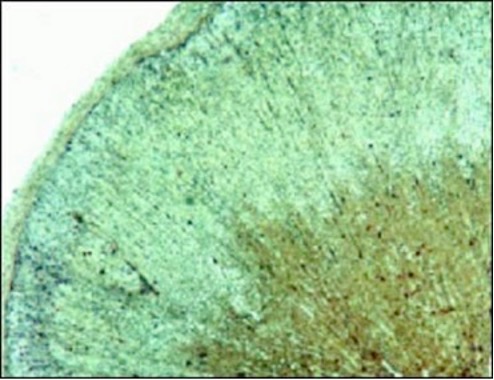
Figure 5. Photomicrographs of transilial bone biopsies of patients with rickets and osteomalacia (A, C and E) and siblings without bone disease (B, D and F) (Courtesy of Drs. D. Gazit and I.A. Bab). A and B: Modified Mason stain; magnification x130. Note in A: broad osteoid seams (arrow), osteoid trabeculae (heavy arrow) and irregular mineralization front (rectangular arrow).C and D: Polarized light; Von Kossa toluidine blue stain; magnification x360. Note in C: increased number of osteoid lamellae (arrows). E and F: Fluorescent photomicrograph, unstained; magnification x200. Note in E wide fluorescent bands (arrows), no double or single tetracycline labels and ground glass appearance.
OSTEOMALACIA
Histologically patients with osteomalacia with or without rickets will represent an abundance of unmineralized matrix, sometimes to the extent that whole trabeculae appear to be composed of only osteoid (Figure 5A). This will be depicted by quantitative histomorphometry as increases in osteoid volume, surface and thickness (OV/BV, OS/BS and O.TH). However, hyperosteoidosis could be observed in a number of bone diseases with a high turnover. The osteomalacic nature of the hyperosteoidosis is being demonstrated by defective mineralization; irregularity of mineralization fronts (Figure 5A); high number of osteoid lamellae (Figure 5C); broad single tetracycline fluorescent labels (Figure 5E) or no label at all, in contrast to the normal double tetracycline fluorescent labels (Figure5F). These qualitative observations have to be supported by the unequivocal changes in quantitative histomorphometry, i.e., decreases in a double and single tetracycline labeled surface (dLS/BS and sLS/BS) and in mineral apposition rate (MAR) as well as prolongation of mineralization lag time (MLT).
BIOCHEMICAL FEATURES OF RICKETS AND OSTEOMALACIA
The biochemical parameters characterizing disorders in vitamin D action can be divided into those associated with vitamin D status, the primary disturbance in mineral homeostasis and the respective compensatory mechanisms, and changes in bone metabolism. Changes in mineral and bone metabolism will be similar in all states of disorders in vitamin D action, while serum levels of vitamin D metabolites will characterize each of the classes delineated in the introduction (Table 2).
|
Table 2. Serum Levels of Vitamin D Metabolites in Patients with Disorders in Vitamin D Action, by Etiology |
||
|
Serum levels |
||
|
*Etiology |
25(OH)D |
1,25(OH)2D |
|
Vitamin D deficiency |
Low |
Low to normal |
|
1,25(OH)2D deficiency |
Normal to elevated |
Very low |
|
Resistance to 1,25(OH)2D |
Normal to elevated |
Markedly elevated |
* See section on Definitions and Terminology
As previously discussed, disorders in vitamin D action will lead to hypocalcemia and secondary hyperparathyroidism. Thus, the characteristic biochemical features are low to low-normal concentrations of serum calcium (depending on compensatory parathyroid activity), low urinary calcium excretion, hypophosphatemia, increased serum immunoreactive parathyroid hormone (iPTH) levels, increased urinary cyclic AMP excretion, and decreased tubular reabsorption of phosphate (the last two measures reflecting the biological activity of elevated iPTH). Biochemical markers associated with increased osteoid production as bone specific alkaline phosphatase and osteocalcin will be elevated in states of rickets and osteomalacia (23).
DEFINITIONS AND TERMINOLOGY
Terminology applied to disorders in vitamin D action has led to much confusion. This is principally because the terms were coined prior to our detailed understanding of vitamin D metabolism and mechanism of action. The following will define and clarify the terms deficiency and resistant and point to the limitations and ambiguity of these terms as being currently used as well as additional widely used terms such as pseudodeficiency and dependency.
Deficiency States
It is important to specify the localization of a defect as precisely as possible. The description of a deficiency state should indicate the most proximal metabolite in the vitamin D biosynthetic pathway that is deficient, emphasizing, if possible, that the concentrations of the immediate precursor of the metabolite specified are normal. For example, low serum levels of 1,25(OH)2D could be the end result of vitamin D deficiency or defects in the activity of the renal tubular enzyme 25 (OH)D1α-hydroxylase. Only in the latter case will it be appropriate to define it as calcitriol deficiency. Based on our current understanding of vitamin D biosynthesis, three states of deficiency could be envisioned involving the major metabolites: vitamin D, 25 (OH) D, and 1,25(OH)2D. As mentioned, before it is difficult to measure vitamin D in blood. Thus, in clinical practice the diagnosis of vitamin D deficiency states is usually established by measurements of serum 25(OH)D levels. 25(OH)D measurements as a marker of vitamin D status is due to relatively long half-life of 25(OH)D and levels in the blood relative to vitamin D plus a conversion of vitamin D to 25(OH)D that has been regarded as not tightly regulated. That said, recent findings indicate that CYP2R1, the major hepatic 25-hydroxylase, is influenced by disorders in metabolism (10) such as the negative impact of obesity induced by a diet high in fat on CYP2R1 expression. (24). Moreover, mutations in CYP2R1 have also been described leading to osteomalacia accompanied by low 25(OH)D but generally normal 1,25(OH)2D (25).
Based on the commonly available measurement of only two vitamin D metabolites, two deficiency states could be defined in clinical practice: low serum 25(OH) levels implying vitamin D and/or 25(OH)D deficiency, but referred mainly to define a vitamin D deficiency with the limitations mentioned above; and calcitriol deficiency, based on low 1,25(OH)2D and normal or high 25(OH)D serum levels (Table 2).
In theory, the pathogenic mechanism leading to deficiency of any of these metabolites could be categorized to either defective synthesis or increased clearance, with the exception of the parent vitamin D in which the additional possibility of deficient intake has to be taken into account. Each category: deficient synthesis or increased clearance could have a hereditary or acquired etiology and each subcategory could be further divided into simple (implying that the defect is localized only to vitamin D metabolism) or complex (implying that the defect is a part of a more generalized disturbance affecting other metabolic pathways).
To demonstrate the practical use and usefulness of the terminology, the following are some examples. Calcitriol deficiency could be caused by hereditary or acquired defects of the renal tubular enzyme 25(OH)D 1α-hydroxylase. If a hereditary defect involves only the vitamin D metabolic pathway it will be termed simple hereditary calcitriol deficiency. If the defect is a part of a more generalized hereditary disturbance, involving other metabolic pathways as well, e.g., Fanconi syndrome, renal tubular acidosis, X-linked hypophosphatemic rickets, or all states of increased FGF-23 secretion, the disorder will be defined as a complex hereditary 1,25(OH)2D deficiency. Decreased calcitriol synthesis could be caused by acquired diseases that usually affect additional metabolic pathways, e.g., chronic renal failure, hypoparathyroidism, tumor induced osteomalacia, thus the appropriate term will be complex acquired 1,25(OH)2D deficiency due to defective synthesis. Some drugs as barbiturates and the antiepileptic-hydantoin may affect the liver cytochrome P450 enzyme system and increase vitamin D catabolism. It may accelerate vitamin D deficiency usually in patients that have vitamin D insufficiency. Such disturbances will be termed complex acquired vitamin D deficiency due to increased clearance. Intestinal malabsorption either hereditary or acquired interferes with the absorption and enterohepatic recirculation of vitamin D and its metabolites and thus may lead to complex hereditary or acquired vitamin D deficiency due to decreased input. In the skin the substrate for vitamin D, 7-dehydrocholesterol, (7-DHC), is on the Kandutsch-Russell pathway of cholesterol synthesis with the last step of conversion of 7-DHC to cholesterol being performed by 7-DHC reductase (DHCR7). Mutations of this enzyme including polymorphisms as well a number metabolites including vitamin D and cholesterol regulate this enzyme thus also providing for complex hereditary of acquired alterations in vitamin D availability due to changes in its precursor 7-DHC (25).
Resistant States
Resistance to a factor may be defined as a state where normal levels of the factor are associated with subnormal bioeffects. This definition has two important determinants, the factor and the bioeffect. In the case of the vitamin D system, the factor can be the parent vitamin D, the 25-hydroxylated metabolite, or calcitriol. Based on vitamin D metabolism and mechanism of action, the term resistance should only be used with the most distal metabolite or the active hormonal form of vitamin D, i.e., 1,25(OH)2D. For example, patients with a renal defect in calcitriol synthesis are resistant to vitamin D or 25(OH)D caused by defective calcitriol production, but have a completely normal response to physiological replacement doses of 1a-hydroxylated vitamin D metabolites.
Several bioeffects in vivo have potential relevance in evaluating states of resistance to 1,25(OH)2D. These include the effects on bone matrix mineralization and the effect on calcium transport through the small intestine. However, as discussed before, rickets and/or osteomalacia are stereotypic responses of the mineralizing bone matrix to a variety of perturbations not all of which are correlated to disturbances in 1,25(OH)2D action. Two clinical examples are presented to clarify this point: a. X-linked hypophosphatemic rickets (XLH) was and still is termed vitamin D resistant rickets. However, the unresponsiveness to vitamin D of the bone disease in XLH is due to the fact that the defective mineralization is caused mainly by phosphorus deficiency though there is some suppression of calcitriol synthesis caused by increased FGF-23 secretion; b. Hereditary hypophosphatemic rickets with hypercalciuria is a disease characterized by defective bone matrix mineralization, low serum phosphorus and elevated serum 1,25(OH)2D levels, taken together, it may look as a resistance to the hormone. However, these patients have increased intestinal absorption of calcium and hypercalciuria. The primary defect in this disease is an isolated defect in a renal phosphate transporter leading to increased renal phosphate clearance, hypophosphatemia, increased 1,25(OH)2D production and the appropriate physiological response i.e., increased intestinal calcium absorption. The rickets and/or osteomalacia are the result of phosphorus deficiency rather than unresponsiveness to calcitriol (for details see Endotext chapter Primary Disorders of Phosphate Metabolism).
There is no doubt about the pivotal role of calcitriol on net calcium transport across the intestinal epithelium. Thus, intestinal calcium absorption measured directly or assessed by secondary changes in serum and urine calcium and iPTH levels can be utilized as a bioeffect in evaluating states of resistance to 1,25(OH)2D. When the defect in bone matrix mineralization is caused by calcium deficiency, rickets and/or osteomalacia could be used as an additional bioeffect to define calcitriol resistance.
Resistance to 1,25(OH)2D is defined here as a state in which normal or high levels of the hormone are associated with hypo-or normocalcemia, secondary hyperparathyroidism, rickets and/or osteomalacia caused by calcium deficiency.
Pseudovitamin D Deficiency and Vitamin D Dependency
The term pseudovitamin D deficiency refers to a state with biochemical and tissue features of vitamin D deficiency (calcium deficiency, secondary hyperparathyroidism, impaired bone matrix mineralization) with no history of vitamin D or calcium deficiency or low serum levels of 25(OH)D. This is an ambiguous term as it includes two different diseases:1,25(OH)2D deficiency and resistance to 1,25(OH)2D, the so-called pseudovitamin D deficiency type I and II respectively, and does not include the known etiology and pathogenesis of these disturbances.
The term vitamin D dependency has been used interchangeably with pseudovitamin D deficiency. It meant to describe patients capable or responding to, and thus dependent on, supraphysiological doses of vitamin D. This is the situation in patients with simple hereditary 1,25(OH)2D deficiency due to defects in the renal enzyme 25(OH)vitamin D 1α-hydroxylase. Patients with this disease have a complete clinical remission on physiological replacement doses of calcitriol. The term vitamin D dependency type II was applied to describe patients with simple hereditary resistance to 1,25(OH)2D due to mutations in the vitamin D receptor (VDR), the majority of whom are unresponsive to any dose of vitamin D or its active metabolites, and therefore are not dependent on vitamin D.
DISORDERS IN VITAMIN D ACTION
Three disorders in vitamin D action will be discussed in this chapter: vitamin D deficiency, mainly simple acquired vitamin D deficiency (i.e., involving only the vitamin D metabolic pathway); simple hereditary 1,25(OH)2D deficiency resulting from defects in calcitriol biosynthesis; simple hereditary resistance to 1,25(OH)2D caused by defects in calcitriol receptor-effector system. While acquired vitamin D deficiency is the most common disorder, the other two rare inborn errors in vitamin D metabolism will be discussed because of its immense contribution to the understanding of vitamin D metabolism and mode of action in human beings.
Vitamin D Deficiency
PATHOGENESIS
Vitamin D deficiency can be caused by decreased input or increased clearance. Decreased input of vitamin D may be the end result of: a) defective intake due to dietary (nutritional) deficiency or intestinal malabsorption; b) deficient photosynthesis of vitamin D in the skin. Increased vitamin D clearance could result from increased catabolism, mainly in the liver, or increased loss via the kidneys or intestine. As discussed, each of these disturbances could be, at least in theory, hereditary or acquired, and further subcategorized as simple (involving only the vitamin D metabolic pathway) or complex (implying a more generalized disturbance). Thus, whenever the diagnosis of vitamin D deficiency is established, there is a need to define the etiology and pathophysiology of the disorder and categorize it accordingly.
Vitamin D deficiency due to increased clearance is relatively uncommon in clinical practice and usually is part of more general diseases (i.e., complex disturbances) e.g., protein losing nephropathy, intestinal malabsorption, and increased liver catabolism sometimes caused by certain drugs (e.g., barbiturates, antiepileptics). The rest of this section will deal with vitamin D deficiency caused by decreased input.
Vitamin D content of various unfortified food substances is very low with the exception of fatty fish such as herring, mackerel, or cod liver oil. It is estimated that under normal unfortified food consumption, less than 20% of the total circulating 25(OH)D is contributed by nutritional vitamin D. In some countries the dietary content of vitamin D is higher due to vitamin D supplementation of some food products. In the United States, milk is being fortified by 400IU per quart. Increased vitamin D intake could result from habitual use of multivitamins that usually contain 400IU of vitamin D per tablet, or some calcium salt preparations that contain vitamin D as well. These supplements will of course increase the relative contribution of dietary vitamin D intake to the total body vitamin D pool in general, and when cutaneous production of vitamin D is limited, in particular.
Gastrointestinal malabsorption will interfere with vitamin D input from the gut and it may affect the enterohepatic recirculation of vitamin D metabolites as well. Thus, intestinal malabsorption may contribute to vitamin D deficiency by decreasing the input and increasing the clearance of the vitamin and its metabolites. Vitamin D deficiency and its clinical and biochemical consequences may be the first sign of occult-malabsorption due, for example, to non-tropical sprue.
Vitamin D synthesis in the skin is produced from 7-DHC under the influence of UV light with a maximal effective wave length between 290-310 nm. Thus, levels of 7-DHC regulated by DHCR7 influences the amount of vitamin D that can be produced. Cutaneous vitamin D production is also affected by the intensity of the UV light that reaches the body, the surface area of the skin exposed and intrinsic properties of the epidermis. In northern latitudes, during the winter, almost no UV reaches the ground. In the northern parts of the United States, Canada and Northwestern Europe, between October to March, very little or practically no vitamin D is produced in exposed skin. Clothing affected by religious or cultural habits, glass, plastic and sunscreens that are widely used due to concerns of skin cancer effectively block UV radiation and prevent cutaneous vitamin D synthesis. Vitamin D production is much less effective in dark skin, melanin absorbs UV radiation, and in elderly relative to young people. However, even in the elderly, dermal vitamin D production remains very effective. It has been estimated that in summer, a 10-minute exposure, three times a week of the unprotected skin of the head and arms is adequate to prevent vitamin D deficiency in the elderly (for detailed discussion see Endotext chapter Vitamin D: Production, Metabolism, and Mechanisms of Actionand references (26-28).
DIAGNOSIS
The diagnosis of vitamin D deficiency is established by low serum concentrations of 25(OH)D. As discussed, circulating levels of 25(OH)D are a good and reliable measure of vitamin D status in most clinically relevant situations although as mentioned obesity and diabetes mellitus can alter the conversion from vitamin D to 25(OH)D. All additional biochemical parameters as well as clinical signs and symptoms reflect the primary and secondary perturbations in mineral and bone metabolism caused by vitamin D deficiency and are common to all disorders in vitamin D action and calcium deficiency (Tables 1,2). Those parameters include low to low normal serum calcium levels, hypocalciuria, secondary hyperparathyroidism, hypophosphatemia, increased levels of biochemical markers of bone turnover, rickets and/or osteomalacia. Therefore, all these measures can be used to support but not to establish the diagnosis, and mainly to assess the relative severity of the vitamin D deficiency and the response to treatment. It is important to note that in vitamin D deficiency, circulating levels of 1,25(OH)2D could vary from low to elevated (Table 2) and thus are useless for the diagnosis.
25(OH)D serum levels are currently being determined in clinical practice by methods that have become more accurate and reproducible, though still with some variability. Reference values of serum 25(OH)D that were population based introduce uncertainty. These reference values differed according to geography, season, dress habits, bed or housebound situations, and age, all of which may affect sunshine exposure and thus vitamin D synthesis, as well as eating habits, local regulation on food fortification, and customs of vitamin supplementation, all of which will affect vitamin D intake. An alternative approach to obtain clinically meaningful reference values was to define health-based parameters, i.e., values of serum 25(OH)D levels below which adverse health outcome may occur. In actuality it is an intervention threshold reference value, below which therapy may prevent adverse effects on the musculoskeletal system (29). This approach is based on the current notion that though severe vitamin D deficiency will lead to rickets and/or osteomalacia, milder vitamin D deficiency states will not affect bone matrix mineralization, but via its effect on mineral homeostasis will cause secondary hyperparathyroidism, increased bone turnover and bone loss. These aberrations in mineral and bone metabolism will contribute to the development and acceleration of osteoporosis and together with the non-skeletal effects of vitamin D on physical function, will increase the risk of low impact skeletal fractures (28,30). The relationship between serum concentrations of 25(OH) D and iPTH in various vitamin D states was analyzed in multiple studies (28,31-33). The aim was to define serum 25(OH)D concentrations below which serum iPTH levels started to increase, or base line 25(OH)D levels above which vitamin D supplementation caused a significant decrease of iPTH serum concentrations. Both approaches yielded similar functional thresholds of serum 25(OH)D levels that affect circulating iPTH concentrations, although the relationship between 25(OH)D and PTH levels is substantially affected by calcium intake.
Based on these studies, a diagnostic staging of vitamin D deficiency states, based on serum 25(OH)D levels and secondary perturbation in mineral and bone metabolism has been proposed (28). Mild vitamin D deficiency (or vitamin D insufficiency) is diagnosed when serum 25(OH)D levels are below 50 nmol/liter (20 ng/ml). This is associated with mild elevations of serum iPTH and biochemical markers of bone turnover. Moderate vitamin D deficiency is defined when serum 25(OH)D levels are below 25 nmol/liter (10 ng/ml). Serum iPTH concentration is moderately increased with high bone turnover. Severe vitamin D deficiency occurs when serum 25(OH)D levels are lower than 12.5 nmol/liter (5 ng/ml), iPTH circulating concentration may be markedly increased and rickets and/or osteomalacia may occur. It is obvious that according to this scheme, vitamin D sufficiency is defined as serum 25(OH)D levels above 50 nmol/liter (20 ng/ml). That said these cut off values remain controversial.
A somewhat similar definition was suggested recently by an expert panel for the Institute of Medicine (IOM) (current name National Academy of Medicine (NAM)) recommending that a level of 50 nmol/liter (20 ng/ml) of 25(OH)D was sufficient for 97.5% of the adult USA population, although up to 125 nmol/liter (50 ng/ml) was considered safe (34). At the same time, another group of experts, from the Endocrine Society, suggested that the most advantageous value of 25(OH)D for musculoskeletal health is 75 nmol/liter (30 ng/ml) (35).
PREVALENCE
Serum 25(OH)D levels vary widely among different populations. On average serum 25(OH)D concentration are lower in European countries than in the USA and even lower in some countries in the Middle East and the Asia-Pacific region. A multinational study (with the exception of North America), in post-menopausal women treated for osteoporosis, revealed that 30-90% (depending on location and season), had serum levels of 25(OH)D below 75 nmol/liter (30 ng/ml), (the highest frequency of women below this level was observed in the Middle East and Asia (two countries in each region)). If the threshold is taken as 50 nmol/liter (20 ng/ml), 31% of the women fell below that threshold. Based on more than 40 studies from Europe, North America, the Middle East and the Asian-Pacific regions (28), it seems that a gradual decline in mean serum 25(OH)D levels is apparent starting from healthy adults to independent elderly, institutionalized elderly, and the lowest value in patients with hip fracture. Several studies of patients hospitalized for non-traumatic fractures, revealed that few (about 5%) of these subjects had serum 25(OH)D levels above 50 nmol/liter (20 ng/ml); the majority of them had vitamin D deficiency of various severity.
As discussed, vitamin D status will be determined by cutaneous photosynthesis and intake. The populations at risk to develop vitamin D deficiency are those who have limited sunshine exposure and no vitamin D supplementation. Thus, vitamin D insufficiency and deficiency will be more prevalent in populations that are not exposed to sunshine due to geography-latitude, cultural, religious, and life-style habits, or are less efficient in cutaneous vitamin D synthesis due to age or pigmentation. The most vulnerable populations are those who are unable to move freely, bedridden or their mobility is severely diminished. This is more common at the beginning or the end of the life cycle, i.e., infants and the elderly, but of course it can happen at any age when free movement is limited due to physical or mental handicap.
The prevalence of osteomalacia is much lower and will depend on the criteria used for diagnosis, i.e., clinical, biochemical, bone histology or quantitative bone histomorphometry. In a review by Lips (28) of 19 publications describing bone histomorphometry of the femoral head or iliac crest biopsy in a total of about 1400 patients with hip fracture, the frequency of osteomalacia ranged from none to more than 30%. This difference may reflect different populations but also the histological criteria used to define osteomalacia. The very high incidence of vitamin D deficiency recorded in patients with hip fractures supports the notion that increased fracture risk is not just the outcome of frank osteomalacia but may result from secondary hyperparathyroidism, increased bone turnover, bone loss, and increased risk of low impact fractures caused by vitamin D deficiency. This is further supported by the positive relationship observed between serum 25(OH)D levels (below a certain threshold) and hip BMD (28,36,37), and the negative relationship observed between hip BMD and serum iPTH (28,36-38). Moreover, a daily dose of 700-800 IU and above of vitamin D and calcium supplementation caused a significant reduction in the incidence of hip and other nonvertebral fractures in women with post-menopausal osteoporosis, including nursing home residents (29,30,39).
TREATMENT
Adequate intake of vitamin D was defined recently in the USA by two working groups (NAM and the Endocrine Society) as discussed above. Both reports agree on the recommended dose for infants (400 IU/day) and children (600 IU/day). There is a difference in the recommendations for adults (600 IU/day) until the age of 70 (including pregnant and lactating women) and 800 IU/day above this age (NAM report), while the Endocrine Society recommendations are 1,500-2,000 IU/day for all of the adult population (34,35). In Europe, vitamin D intake recommendations are similar or somewhat lower than the NAM. The upper tolerable daily doses of vitamin D were suggested to be 4,000 IU, 10,000 IU, and 2,000 IU by the NAM, the Endocrine Society guidelines and the European Commission, respectively. There is an unresolved debate on the equipotency of vitamin D2 versus D3, but as vitamin D3 is widely available, all recommendations are actually for vitamin D3. These recommended intakes of vitamin D are usually unachievable if widely consumed food substances are not fortified with vitamin D. Thus, the population at large and the elderly population in particular are dependent on adequate cutaneous vitamin D synthesis, i.e., sunshine exposure, or vitamin D supplementation. In the United States and a few additional countries, milk is fortified with vitamin D, 400 IU per quart or liter, and the consumption of multivitamins containing vitamin D as well is relatively common, but not uniform or mandatory. Thus, in most countries and even in the USA, the elderly and especially the immobile, housebound elderly are prone to develop vitamin D deficiency.
Treatment has to be targeted towards the population with the highest risk to develop vitamin D deficiency. For a long period, vitamin D supplementation for infants up to 1 year of age, is mandatory in many countries and the daily dose recommendations was increased from 200 IU/day to 400 IU/day (34,35). Unfortunately, this has not yet become common practice for the elderly population. As discussed, nursing home residents, institutionalized and hospitalized elderly, and patients with hip and other non-traumatic fractures and neurological disorders are among the ones with the highest risk of having and developing vitamin D deficiency. Treatment with the recommended doses of vitamin D and calcium (see below) can be initiated even before biochemical screening of 25(OH)D serum levels and indices of mineral and bone metabolism is obtained. This is based on the very high incidence of vitamin D deficiency in this population of patients and on the fact that with the recommended vitamin D doses and the tight physiological control of 1.25(OH)2D production, no toxicity is likely to occur at doses of up to 4,000 IU/day for a limited period.
It is important to remember that in concordance with vitamin D treatment, the recommended daily calcium allowance must be achieved as well, usually by calcium salt supplementation (see Endotext chapter Osteoporosis: Prevention and Treatment).
The usual recommended oral doses of vitamin D as discussed above are 600 to 1,500 IU daily. In severe vitamin D deficiency, doses of 4,000 IU- to 6,000 IU per day (or the equivalent weekly or twice weekly dose) could be given for the first 4-6 weeks, followed by dose adjustment in accordance with the biochemical response, with the final aim to achieve the recommended maintenance dose. Because vitamin D is stored in fat and released slowly and the half-life of 25(OH)D is 2-3 weeks, the vitamin can be given orally once a week. Studies with administration of vitamin D in megadoses at intervals greater than monthly have shown increased risk of falls and should be avoided (28,40-43).
The response to treatment with vitamin D will depend on the degree and severity of vitamin D deficiency and the secondary changes in mineral and bone metabolism. In severe vitamin D deficiency with osteomalacia, a dramatic response in the signs, symptoms, and laboratory parameters will be observed. Bone pain and muscle weakness will improve quickly, pseudofractures will show signs of healing on x-ray, and serum calcium, iPTH and biochemical markers of bone turnover will return towards the normal range. In moderate or mild vitamin D deficiency or insufficiency, the response to treatment is more subtle. Muscle weakness and bone pain may improve, serum 25(OH)D levels will increase towards the normal, serum iPTH and biochemical markers of bone turnover will return towards normal (this will be a function of the severity of the initial vitamin D deficiency). In the long run, bone mineral density may increase somewhat and the incidence of fractures may decrease. These results are based on responses of groups of patients in clinical trials and not individuals (28).
1,25 Dihydroxyvitamin D Deficiency
As discussed, 1,25(OH)2D deficiency is defined as low circulating levels of this metabolite with normal or elevated (depending on preceding vitamin D therapy) serum concentrations of 25(OH)D. In theory, it could be the end result of decreased production or increased clearance. Increased clearance of 1,25(OH)2D is uncommon and usually a part of increased clearance of additional vitamin D metabolites such as 25(OH)D, thus it would fit the definition of vitamin D deficiency.
Decreased production of 1,25(OH)2D could be hereditary or acquired and each one can be subcategorized as simple or complex. Acquired 1,25(OH)2D deficiency due to defective synthesis is usually part of a more general disease (i.e., complex) as chronic renal failure, acquired Fanconi syndrome, hypoparathyroidism, or tumor induced osteomalacia, that will affect mineral and bone metabolism in a multiple and complex ways that is beyond the scope of this chapter. Thus, the entity to be discussed here will be simple hereditary 1,25(OH)2D deficiency caused by defective synthesis of calcitriol.
Prader et al. (44) were the first to describe two young children who showed all the usual clinical features of vitamin D deficiency despite adequate intake of the vitamin, thus coining the name “pseudovitamin D deficiency.” Complete remission was dependent on continuous therapy with high doses of vitamin D, thus the term “vitamin D-dependent rickets.” However, remission of the disease could be achieved by continuous therapy with physiological (microgram) doses of 1a-hydroxylated vitamin D metabolites (45,46).
Family studies have revealed it to be an autosomal recessive disease. Linkage analysis in a subset of French-Canadian families assigned the gene responsible for the disease to chromosome 12q13 (47,48). The gene encoding the 25(OH)D-1α-hydroxylase (CYP27B1) of mouse kidney, human keratinocytes, and peripheral mononuclear cells was localized on chromosome 12q 13.1-13.3 which maps to the disease locus of simple hereditary 1,25(OH)2D deficiency (49-55). Cloning and sequencing of the enzyme has enabled the demonstration of its decreased expression in patients with this disease (56) as well as enabling studies showing that the same enzyme is widely distributed in tissues and not limited to the kidney (56,57). The sequence of the human CYP27B1 gene from keratinocytes and peripheral blood mononuclear cells has been shown to be identical with the renal gene (49,51-55), thus supporting the use of these accessible cells as a proxy to study the renal tubular enzymatic defect. That said regulation of extrarenal CYP27B1 differs from that of the kidney, with its regulation in keratinocytes and macrophages responding to cytokines rather than PTH and FGF23 (25). Subjects with mutations in CYP27B1 have circulating levels of 25(OH)D that are normal or elevated, depending on previous vitamin D treatment; serum concentrations of 1,25(OH)2D are very low (Table 2); while massive doses (100-300 times the daily recommended dose) of vitamin D or 25(OH)D are required to maintain remission of rickets, physiological replacement doses of calcitriol are sufficient to achieve the same effect. The 1α-hydroxylase gene from more than 25 families with simple hereditary 1,25(OH)2D deficiency and some of their first-degree healthy relatives were analyzed by direct sequencing, site directed mutagenesis, and cDNA expression in transfected cells (49,51-55). All patients had homozygous mutations while parents or other healthy siblings were heterozygous for the mutation. Most patients of French-Canadian origin had the same mutation causing a frameshifting and a premature stop codon in the putative heme-binding domain (51). The same mutation was observed in additional families of diverse origin (55). All other patients had either a base-pair deletion causing premature termination codon upstream from the putative ferredoxin and heme-binding domains, or missense mutations (56,58-61). No 1α-hydroxylase activity was detected when the mutant enzyme was expressed in various cells. Taken together, these observations support the notion that the etiology of this hereditary disease is a defect in CYP27B1.
The beneficial therapeutic effect of high serum concentrations of 25(OH)D in patients treated with massive doses of vitamin D, while 1,25(OH)2D levels remain low, may have several possible explanations. First, high levels of 25(OH)D may activate the VDR whose affinity for this metabolite is approximately two orders of magnitude lower than for 1,25(OH)2D. Second, a metabolite of 25(OH)D may act directly on target tissue, and finally, high levels of 25(OH)D may drive the production of 1,25(OH)2D if the mutated enzyme has some residual function.
The differential diagnosis of simple hereditary 1,25(OH)2D deficiency from other hereditary or acquired forms of hypocalcemic rickets and/or osteomalacia especially the one associated with defects in the vitamin D receptor-effector system is based on serum concentrations of calcitriol and the response to treatment with 1-alpha hydroxylated vitamin D metabolites (Table 2).
A similar syndrome has been described and studied in a mutant strain of pigs where the mode of inheritance as well as the clinical and biochemical features are similar to the human disease (62,63). Piglets affected by the disease have rickets, elevated 25(OH)D with low or undetectable 1,25(OH)2D serum concentrations, normal specific tissue binding sites for tritiated 1,25(OH)2D, and no detectable activity of 25(OH)D-1α-hydroxylase in renal cortical homogenates. Thus, there is strong evidence that the disease state in the pig is caused solely by an inherited defect in the porcine CYP27B1. An animal model in which the gene encoding 25(OH)D-1α-hydroxylase was knocked out by homologous recombination reproduced all the clinical and biochemical features of this disease including undetectable serum 1,25(OH)2D levels (64).
Simple Hereditary Resistance to 1,25 Dihydroxy-Vitamin D
This is a rare disorder and only about 60 patients have been reported (65) Brooks et al. (66) described an adult patient with hypocalcemic osteomalacia and elevated serum concentration of 1,25(OH)2D. Treatment with vitamin D, causing a further increase in serum calcitriol levels, cured the patient. The term vitamin D-dependent rickets type II was suggested to describe this disorder. However, reports on additional patients, about half of whom did not respond to vitamin D therapy, as well as in vivo and in vitro studies to be discussed below, seem to prove that vitamin D dependency is a misnomer. The term hereditary vitamin D resistant rickets (HVDRR) is the current most widely used name of this disorder.
CLINICAL AND BIOCHEMICAL FEATURES
General Features
The clinical, radiological, histological, and biochemical features (except serum levels of vitamin D metabolites) are typical of hypocalcemic rickets and/or osteomalacia as previously discussed with one exception. Many of these patients develop alopecia in early childhood, not seen in even severe cases of vitamin D deficiency. In these patients there is no history of vitamin D deficiency and no clinical or biochemical response to physiological doses of vitamin D or its 1a-hydroxylated active metabolites. Serum levels of 25(OH)D are normal or elevated (depending on preceding vitamin D therapy); 1,25(OH)2D concentrations are markedly elevated before or during therapy with vitamin D preparations (Table 2).
The disease manifests itself as an active metabolic bone disease in early childhood. However, late onset at adolescence and adulthood was documented in several sporadic cases including the first report by Brooks et al. (66,67). These patients represented the mildest form of the disease and had a complete remission when treated with vitamin D or its active metabolites. It is unclear if the adult-onset patients belong to the same hereditary entity, as no further studies on their VDR status have been published.
Ectodermal Anomalies
A peculiar clinical feature of the patients, appearing in more than half of the subjects, is total alopecia or sparse hair (Figure 6). Alopecia usually appears during the first year of life and in one patient, at least, has been associated with additional ectodermal anomalies (68) (Figure 6).
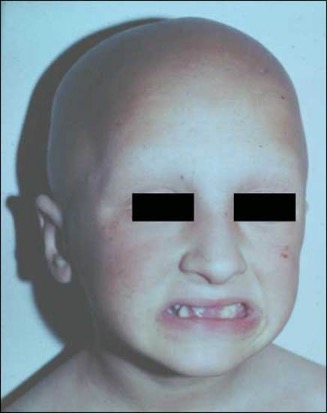
Figure 6. Patient with mutation in VDR demonstrating both alopecia and changes in dentition.
It seems that alopecia is a marker of a more severe form of the disease as judged by the earlier onset, the severity of the clinical features, the proportion of patients who do not respond to treatment with high doses of vitamin D or its active metabolites, and the extremely elevated levels of serum 1,25(OH)2D recorded during therapy (69,70). Though some patients with alopecia could achieve clinical and biochemical remission of their bone disease, none have shown hair growth. The notion that total alopecia is probably a consequence of a defective vitamin D receptor-effector system is supported by the following: alopecia has only been associated with hereditary defects in the VDR system, i.e., with end-organ resistance to the action of the hormone, and has not been recorded with hereditary deficiency in 1,25(OH)2D synthesis, i.e., low circulating levels of the hormone (71); alopecia is present in kindreds with different defects in the VDRs; high-affinity uptake of tritiated 1,25(OH)2D3 in the nucleus of the outer root sheath of the hair follicle of rodents has been demonstrated by autoradiography (72). Of interest is that mutations in Hairless likewise cause alopecia with histologic features similar to those seen with VDR mutations. Hairless and VDR interact in the keratinocyte suggesting their codependence for regulation of hair follicle cycling (73). High dietary or infusions of calcium can reverse the skeletal changes but do not reverse the alopecia (74). Finally, alopecia developed in homozygote VDR knockout mice (75-77). Taken together, it could be hypothesized that an intact VDR-effector system is important for the differentiation of the hair follicle in the fetus, which is unrelated to mineral homeostasis.
VITAMIN D METABOLISM
Serum concentrations of 1,25(OH)2D range from upper normal values to markedly elevated before therapy, but on vitamin D treatment may reach the highest levels found in any living system (100 times and more than the upper normal range) (70). These values may represent the end results of four different mechanisms acting synergistically to stimulate strongly the renal 25(OH)D-1α-hydroxylase. Three of the mechanisms are hypocalcemia, secondary hyperparathyroidism, and hypophosphatemia. The fourth mechanism may be a failure of the negative feedback loop by which the hormone inhibits the renal enzyme activity caused by the basic defect in the VDR-effector system. This was demonstrated in a patient in remission (normal serum levels of calcium, phosphorus and PTH) in whom a load of 25(OH)D3 had caused a marked increase in serum 1,25(OH)2D3 concentration (69,78). It was reported that the 1α-hydroxylase gene expression was not suppressed by 1,25(OH)2D3 in renal tubular cells from VDR knock out mice while it was suppressed in cells with normal VDR or heterozygote for the null mutation (6,79).
MODE OF INHERITANCE
In approximately half of the reported kindreds, parental consanguinity and multiple siblings with the same defect suggest autosomal recessive mode of inheritance (69). Parents or siblings of patients who are expected to be obligate heterozygotes have been reported to be normal, i.e., no bone disease or alopecia and normal blood biochemistry. However, studies on cells (cultured dermal fibroblasts, Epstein-Barr transformed lymphoblasts, and mitogen-stimulated lymphocytes) obtained from parents or siblings of affected children revealed decreased bioresponses, decreased normal VDR protein and its mRNA, and a heterozygote genotype exhibiting both normal and mutant DNA alleles (80-83). There is a striking clustering of patients around the Mediterranean, including patients reported from Europe and America who originated from the same area. A notable exception is a cluster of some kindreds from Japan (67,84-89).
CELLULAR AND MOLECULAR DEFECTS
Methods
The near ubiquity of a similar if not identical VDR-effector system among various cell types including cells originating from tissues easily accessible for sampling made feasible studies on the nature of the intracellular and molecular defects in patients with simple hereditary resistance to 1,25(OH)2D. The cells used were mainly fibroblasts derived from skin biopsies (69,78,80-82,87,90-104) and peripheral blood mononuclear (PBM) cells. PBM cells contain high-affinity receptors for 1,25(OH)2D3 that are expressed constitutively in monocytes and are induced in mitogen-stimulated T-lymphocytes and Epstein-Barr (EB) transformed lymphoblasts. All cells have been used to assess most of the steps in 1,25(OH)2D3 action from cellular and subcellular uptake of the hormone to bioresponse as well as to elucidate the molecular aberrations in the VDR protein, RNA, and DNA.
The hormone-receptor interaction has been analyzed by several methods including binding characteristics of 3H 1,25(OH)2D3 to intact cells, nuclei or high salt cellular soluble extracts (69,78,80-83,90-92,95-99,105-110); measurements of VDR protein content by monoclonal antibodies with radiological immunoassay or Western blot analysis (81,82); by immunocyto-chemical methods in whole cells (111); characterization of the hormone-receptor complex on continuous sucrose gradient and nonspecific DNA-cellulose columns (69,78,80,90-92,94-97,106,110,112).
The cloning and nucleotide sequencing of the human VDR gene made feasible studies of the molecular defects in patients with this disease. The methods used included, among others, isolation, amplification, and sequencing of genomic VDR DNA, as well as cloning and sequencing of VDR complementary DNA (cDNA). The mutant DNA was recreated in vitro and was transfected into cells that do not express endogenous VDR. Post-transcriptional action of 1,25(OH)2D was tested in cells originating from patients or in cells co-transfected with VDR (either mutant or wild type) fused to a promoter containing vitamin D response element (VDRE).
Studies with the above-mentioned methods in cells originating from a variety of patients revealed heterogeneity of the cellular and molecular defects in the VDR-effector system. Based on the known functional properties of the VDR, different classes of defects could be identified.
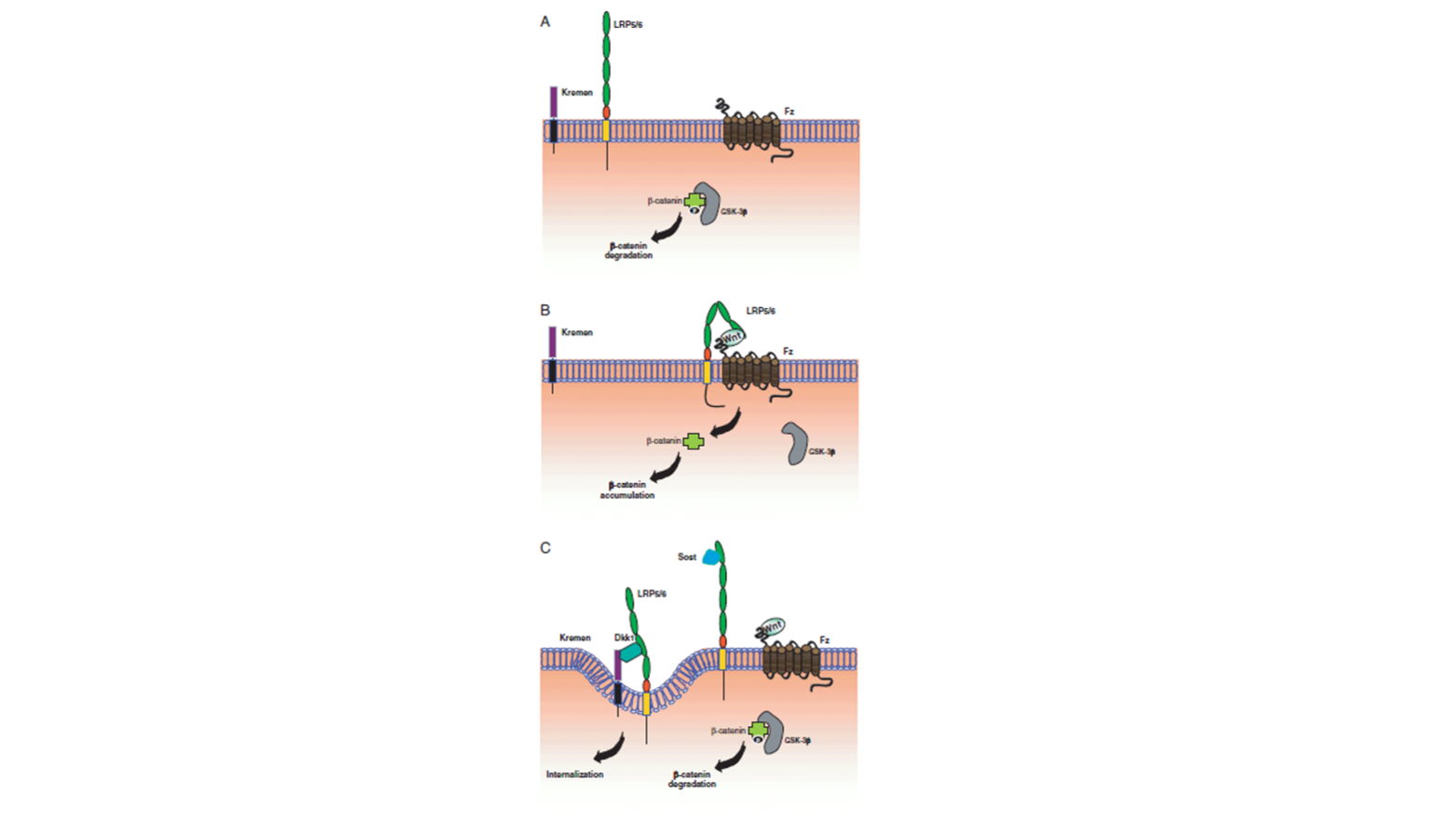
Defect in the Hormone Binding Region (Including Heterodimerization)
Deficient Hormone Binding. There are three subgroups in this class:
(i) No Hormone Binding. This is the most common abnormality observed and is characterized by unmeasurable specific binding of 3H 1,25(OH)2D3 to either intact cells, nuclei, or cell extracts (69,78,81,82,91,96,100-102,105,107,112-116). Studies in several kindreds with this defect (including an extended kindred with 8 patients studied) revealed undetectable levels of VDR by immunoblots on an immunoradiometric assay in most kindreds (81,82,102,116,117). DNA from these affected subjects exhibited a single base mutation that was different in each kindred resulting in a premature stop codon. The truncated VDRs produced lacked hormone binding or both hormone and DNA binding domains (Figure 7) (113-115,117). The recreated mutant VDR cDNA was expressed in mammalian cells, and the resulting mutant VDR was demonstrated to be the truncated protein that exhibited no specific hormone binding. In cells cultured from parents of some patients, expected to be obligate heterozygotes, binding of 3H 1,25(OH)2D3, VDR protein, and mRNA content of cells ranged from the lower limit of normal to about half the normal level.
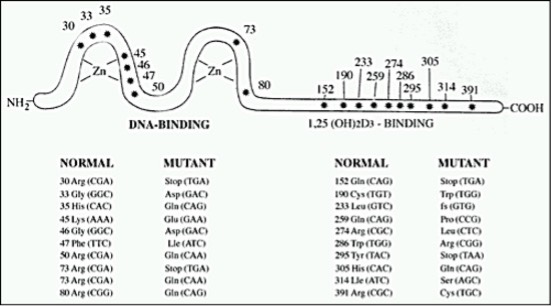
Figure 7. Schematic presentation of homozygous mutation in the VDR protein in simple hereditary resistance to 1,25(OH)2D. The asterisks depict sites of amino acid substitutions due to point mutation and codon changes, using the numbering system of Baker et (13); fs-frame shift.
In one patient representing a kindred with no hormone binding, a missense mutation resulted in the substitution of the hydrophobic basic arginine 274 by the hydrophilic nonpolar leucine in the hormone binding region (116) (Figure 7). In this patient, normal transcription could be elicited by 1000-fold higher concentrations of calcitriol than needed for the wild-type receptor. However, no in vivo or in vitro stimulation of 25(OH)D-24-hydroxylase could be obtained by high concentrations of 1,25(OH)2D3 (see below).
Two siblings without alopecia and no response to any dose of 1,25(OH)2D in vivo and in vitro (118) had a missense mutation that caused a substitution of tryptophan by arginine at amino acid 286 of the VDR (102). This substitution in a normal size VDR abolished completely the binding of 1,25(OH)2D to its receptor. The tryptophan in this position is critical for the positioning of calcitriol in the VDR as was unveiled by the three-dimensional arrangement of the VDR and its ligand based on its crystal structure.
(ii) Defective Hormone Binding Capacity. In a patient representing one kindred, the number of binding sites in nuclei and soluble cell extracts was 10% of control, with an apparent normal affinity (78,105,112). Recently, a boy with total alopecia, severe rickets and growth retardation was found to have two heterozygote different molecular defects in the ligand binding domain (9). The patient’s VDR had a low hormone binding capacity, 10% to 30% of controls, with normal affinity and a markedly deficient stimulation of 25(OH)D3-24-hydroxylase. The recreated mutations, each one tested separately in vitro, showed also deficient heterodimerization as well as different transactivation of two gene promoters. This patient, similar to another one described more than 20 years ago, could be completely cured by very high doses of 25(OH) vitamin D, 250 μg a day initially, followed by 100 μg/day and then 75 μg/day as a maintenance dose for years, plus modest calcium supplementation. In both patients, it could be shown that during remission (normocalcemia, normophosphatemia, normal iPTH), 1,25(OH)2D production is driven by the substrate, i.e., 25(OH) vitamin D concentrations.
An additional patient with hereditary resistance to 1,25(OH)2D and alopecia was found to have compound heterozygous mutations in the VDR (119). This girl’s cultured fibroblasts were found to have about 30% binding sites of 1,25(OH)2D in comparison to her parents or normal controls. However, RXR heterodimerization, co-activator interaction and gene transactivation were completely abolished, and no response in vitro to high doses of 1,25(OH)2D3 was observed. Treatment with 3,000mg of calcium carbonate orally per day and 3μg/kg of calcitriol caused some improvement in the clinical features and biochemical parameters.
(iii) Defective Hormone Binding Affinity. Binding affinity of tritiated calcitriol was reduced 20- to 30-fold, with normal capacity in soluble dermal fibroblast extracts from one kindred (98). An additional patient, representing a different kindred had a modest decrease of VDR affinity when measured at 0°C (120).
Deficient Nuclear Uptake. The following features characterize the hormone-receptor-nuclear interaction in this defect: normal or near normal binding capacity and affinity of 3H 1,25(OH)2D3 to soluble cell extracts with low to unmeasurable hormone uptake into nuclei of intact cells (87,90,105,121,122). These features were demonstrated in skin-derived fibroblasts in all kindreds, in cells cultured from a bone biopsy of one patient (94), and in EB-transformed lymphoblasts of one patient (121). Occupied VDR obtained from fibroblast extracts of two kindreds demonstrated normal binding to nonspecific DNA cellulose (80). Immuno-cytological studies in fibroblasts of a patient with this defect showed that immediately after 1,25(OH)2D3 treatment, VDR accumulated along the nuclear membrane with no nuclear translocation (123). Patients with this defect included a kindred with normal hair and several kindreds with total alopecia. Finally, almost all patients responded with a complete clinical remission to high doses of vitamin D and its active 1a-hydroxylated metabolites.
Attempts to characterize the molecular defect were carried in six kindreds. In three of them, no mutation in the coding region of the VDR gene was observed (121). Studies in three kindreds, revealed a normal molecular mass and quantitative expression of the VDR (122). Complete sequencing of the VDR coding region revealed a different single nucleotide mutation in each kindred in a region that is considered to play a role in heterodimerization of VDR with RXR (Figure 7) (115,122). Thus, it has been suggested that these patients’ receptors have defects that compromise RXR heterodimerization, which is essential for nuclear localization and probably for recognition of the vitamin D responsive element as well. The fact that no mutation in the VDR coding region was observed in three additional kindreds with the same phenotypical defect may suggest that the genetic defect affects another component of the receptor effector system that is essential for the VDR function as a nuclear transcription factor. It has been shown that coactivation complexes are essential for the ligand induced transactivation of VDR (124).
Deficient coactivators of the calcitriol-VDR complex. A patient with simple hereditary resistance to 1,25(OH)2D without alopecia was described (104). Sequencing of the VDR-DNA revealed a missense mutation in the ligand binding domain that caused a substitution of glutamic acid to lysine at amino acid 120. This receptor exhibits many normal properties including calcitriol binding, dimerization and binding to vitamin D response elements in the DNA, but a marked impairment in binding coactivators that are essential for the transactivation of the hormone receptor complex and the initiation of the physiological response (104).
Defects in the DNA Binding Region
Deficient Binding to DNA. Cell preparations derived from patients with this defect demonstrate normal or near normal binding capacity and affinity for 3H 1,25(OH)2D3 to nuclei of intact cells and soluble cell extracts, as well as normal molecular size. Hormone receptor complexes, however, have decreased affinity to nonspecific DNA (88,89,97,108,109). A single nucleotide missense mutation within exon 2 or 3, encoding the DNA binding domain of the VDR, was demonstrated in genomic DNA isolated from dermal fibroblasts and/or EB-transformed lymphoblasts from ten unrelated kindreds (80,88,89,95,110,114,125,126). Eight different single nucleotide mutations were found in the ten kindreds (Figure 7). Two apparently unrelated kindreds share the same mutation (88,110). All point mutations caused a single substitution of an amino acid that resides in the region of the two zinc fingers of the VDR protein that are essential for the functional interaction of the hormone-receptor complex with DNA. Interestingly, all these altered amino acids are highly conserved in the steroid receptor superfamily that includes the receptors for steroid hormones, thyroid hormones and retinoic acid.
Studies on cells obtained from parents and some of the siblings of these patients revealed, as expected, a heterozygous state, i.e., expression of both normal and defective forms of VDR as well as normal and mutant gene sequences (80,102,109), but without any clinical and biochemical abnormalities.
In Vitro Post Transcriptional Effect of 1,25(OH)2D3
In vitro bioeffects of the hormone on various cells in patients with simple hereditary resistance to 1,25(OH)2D have been assayed mainly by two procedures, induction of 25(OH)D-24-hydroxylase and inhibition of mitogen stimulated PBM cells.
1,25(OH)2D3 induces 25(OH)D-24 hydroxylase activity in skin-derived fibroblasts (78,81,82,89,91-93,96-104,109,110,121,126,127), mitogen-stimulated lymphocytes (83), and cells originating from bone (94,128) in a dose-dependent manner. In cells from normal subjects, maximal and half-maximal induction of the enzyme was achieved by 10-8and 10-9M concentrations of 1,25(OH)2D3, respectively. Dermal fibroblast or PBM cells from patients with no calcemic response to maximal doses of vitamin D or its metabolites in vivo did not show any 25(OH)D-24-hydroxylase response to very high concentration of 1,25(OH)2D3 in vitro, while dermal fibroblasts from patients with a calcemic response to high doses of vitamin D or its metabolites in vivo showed inducible 24-hydroxylase with supraphysiological concentrations of 1,25(OH)2D3 in vitro. Physiological concentrations of 1,25(OH)2D3 partially inhibit mitogen-induced DNA synthesis in peripheral lymphocytes with a half maximal inhibition achieved at 10-10M of the hormone (85,107). Mitogen stimulated lymphocytes from several kindreds with defects characterized as no hormone binding or deficient binding to DNA, with no calcemic response to high doses of vitamin D and its metabolites in vivo, showed no inhibition of lymphocyte proliferation in vitro with concentrations of up to 10-6M 1,25(OH)2D3. Additional methods to measure bioeffects of 1,25(OH)2D3 on various cells in vitro were carried out only in a few patients and included inhibition of dermal fibroblast proliferation (93), induction of osteocalcin synthesis in cells derived from bone (128), a mitogenic effect on dermal fibroblasts (123), and stimulation of cGMP production in cultured skin fibroblasts (129). It is noteworthy that in all assays mentioned and without exception, each patient’s cell showed severely deficient responses.
With the elucidation of the molecular defects in simple hereditary resistance to 1,25(OH)2D, the transactivation abilities of naturally occurring mutant or recreated mutant VDRs were evaluated in a transcriptional activation assay. In this assay a gene promoter responsive to VDR is fused to a gene reporter that its message is easily measured, and the plasmid is transfected into patients or normal cells (81,82,88,89,109,116,121). Treating normal transfected cells with 1,25(OH)2D3 caused a concentration-dependent induction of transcription. No induction of transcription was observed in cells originating from patients with defects characterized as no hormone binding (130,131)(116) or deficient binding to DNA (88,89,109,110). Moreover, in a cotransfection assay, the addition of a normal human VDR cDNA expression vector to the transfected plasmid that directed synthesis of a normal VDR, restored hormone responsiveness of resistant cells. Finally, in a patient characterized as deficient nuclear uptake defect, no mutation was identified within the coding region of the VDR gene; no induction of 25(OH)D-24-hydroxylase activity by up to 10-6 1,25(OH)2D3 was observed in cultured skin fibroblasts, but there was normal transactivation by 1,25(OH)2D3 in the transcriptional activation assay (121).
CELLULAR DEFECTS AND CLINICAL FEATURES
Normal hair was described with most phenotypes of the cellular defects, the exception being patients with deficient hormone binding capacity and affinity, but this could be due to the fact that only one or two kindreds were described per subgroup. Normal hair is usually associated with a milder form of the disease, as judged by the age of onset, severity of the clinical features, and usually the complete clinical and biochemical remission on high doses of vitamin D or its metabolites. Notable exceptions are 3 kindreds (4 patients), two of them without alopecia that displayed resistance both in vivo (no clinical remission on circulating calcitriol level up to 100 times the mean normal adult values) and in vitro (no induction of 25(OH)D-24-hydroxylase activity in dermal fibroblasts by up to 10-8 M 1,25(OH)2D3) (99,102,104). Only approximately half of the patients with alopecia have shown satisfactory clinical and biochemical remission to high doses of vitamin D or its active 1a-hydroxylated metabolites, but the dose requirement is ~10-fold higher than in patients with normal hair (70).
It seems that patients’ defects characterized as deficient hormone binding affinity and deficient nuclear uptake achieve complete clinical and biochemical remission on high doses of vitamin D or its active 1a-hydroxylated metabolites. Most of the patients with other types of defects could not be cured with high doses of vitamin D or its metabolites. However, it should be emphasized that not all of the patients received treatment for a long enough period of time and with sufficiently high doses (see Treatment, below).
DIAGNOSIS
Clinical features of early onset rickets with no history of vitamin D deficiency, total alopecia, parental consanguinity, additional siblings with the same disease, serum biochemistry of hypocalcemic rickets, elevated circulating levels of 1,25(OH)2D, and normal to high levels of 25(OH)D (Table 2) support the diagnosis of simple hereditary resistance to 1,25(OH)2D. The issue becomes more complicated when the clinical features are atypical, i.e., late onset of the disease, sporadic cases, and normal hair. Failure of a therapeutic trial with calcium and/or physiological replacement doses of vitamin D or its metabolites may support the diagnosis but the final direct proof requires the demonstration of a cellular, molecular, and functional defect in the VDR-effector system.
Based on the clinical and biochemical features, the following additional disease states should be considered: (1) extreme calcium deficiency: a seemingly rare situation described in a group of children from a rural community in South Africa, who consumed an exceptionally low calcium diet of 125 mg/day (20). All had severe bone disease with histologically proven osteomalacia, biochemical features of hypocalcemic rickets with elevated serum levels of 1,25(OH)2D, and sufficient vitamin D. Calcium repletion caused complete clinical and biochemical remission. Nutritional history and the response to calcium supplementation support this diagnosis. (2) Severe vitamin D deficiency: during initial stages of vitamin D therapy in children with severe vitamin D-deficient rickets, the biochemical picture may resemble 1,25(OH)2D resistance, i.e., hypocalcemic rickets with elevated serum calcitriol levels. This may represent a “hungry bone syndrome,” i.e., high calcium demands of the abundant osteoid tissue becoming mineralized. This is a transient condition that may be differentiated from simple hereditary resistance to 1,25(OH)2D by a history of vitamin D deficiency and the final therapeutic response to replacement doses of vitamin D.
TREATMENT
In about half of the kindreds with simple hereditary resistance to 1,25(OH)2D, the bioeffects of 1,25(OH)2D3 were measured in vitro (see above). An invariable correlation (with one exception) was documented between the in vitro effect and the therapeutic response in vivo; i.e., patients with no calcemic response to high levels of serum calcitriol showed no effects of 1,25(OH)2D3 on their cells in vitro (either induction of 25(OH))D-24-hydroxylase or inhibition of lymphocyte proliferation) and vice versa. If the predictive therapeutic value of the in vitro cellular response to 1,25(OH)2D3 could be substantiated convincingly, it may eliminate the need for time consuming and expensive therapeutic trials with massive doses of vitamin D or its active metabolites. In the meantime, it is mandatory to treat every patient with this disease irrespective of the type of receptor defect.
An adequate therapeutic trial must include vitamin D at a dose that is sufficient to maintain high serum concentrations of 1,25(OH)2D3 as the patients can produce high hormone levels if supplied with enough substrate. If high serum calcitriol levels are not achieved, it is advisable to treat with 1a-hydroxylated vitamin D metabolites in daily doses of up to 6 mg/kg weight or a total of 30-60 mg and calcium supplementation of up to 3 g of elemental calcium daily; therapy must be maintained for a period sufficient to mineralize the abundant osteoid (usually 3-5 months). Therapy may be considered a failure if no change in the clinical, radiological, or biochemical parameters occurs during continuous and frequent follow up while serum 1,25(OH)2D concentrations are maintained at ~100 times the mean normal range.
In some patients with no response to adequate therapeutic trials with vitamin D or its metabolites, a remarkable clinical and biochemical remission of their bone disease, including catch-up growth, was obtained by treatment with large amounts of calcium as noted previously. This was achieved by long-term (months) intracaval infusions of up to 1000 mg of calcium daily (102,128,132-134). Another way to increase calcium input into the extra cellular compartment is to increase net gut absorption, independent of vitamin D, by increasing calcium intake (135). This approach is limited by dose and patient tolerability and was actually used successfully in only a few patients.
Several patients have shown unexplained fluctuations in response to therapy or in presentation of the disease. One patient after a prolonged remission became completely unresponsive to much higher doses of active 1a-hydroxylated vitamin D metabolites (78), and another patient seemed to show amelioration of resistance to 1,25(OH)2D3 after a brief therapeutic trial with 24,25(OH)2D (68). In several patients, spontaneous healing occurred in their teens (106) or rickets did not recur for 14 years after cessation of therapy (87).
ANIMAL MODELS
Some New World primates (marmoset and tamarins) that develop osteomalacia in captivity are known to have high nutritional requirements for vitamin D and maintain high serum levels of 1,25(OH)2D, thus exhibiting a form of end-organ resistance to 1,25(OH)2D (136-139). Cultured dermal fibroblasts and EB-virus transformed lymphoblast have shown deficient hormone binding capacity and affinity (136,137). It has been observed that marmoset lymphoblasts contain a soluble protein of 50-60 kDa that binds 1,25(OH)2D3 with a low affinity but high capacity and thus may serve as a sink that interferes with the hormone binding and its cognate receptor (140). The same group described another protein present in the nuclear extract of these cells capable of inhibiting normal VDR-RXR binding to the vitamin D response element (141). It is of interest that these New World primates also exhibit a compensated hereditary end-organ resistance to the true steroid hormones including glucocorticoids, estrogens, and progestins (142). This of course raises the interesting possibility that the defect in the hormone-receptor-effector system involves an element shared by all the members of this superfamily of ligand modulated transcription factors. Subsequent studies have identified the proteins involved as members of the heterogeneous nuclear ribonucleoprotein family (143-145).
VDR knock out mice have been created by targeted ablation of the first or second zinc finger (75,76). Only the homozygote mice were affected. Though phenotypically normal at birth, after weaning however, they become hypocalcemic, develop secondary hyperparathyroidism, rickets, osteomalacia and progressive alopecia. The female mice with ablation of the first zinc finger are infertile and show uterine hypoplasia and impaired folliculogenesis. Otherwise, both VDR cell mutant mice show clinical, radiological, histological and biochemical features that are identical to the human disease. Supplementation with a calcium enriched diet can prevent or treat most of the disturbances in mineral and bone metabolism in these animal models except alopecia (77).
It is of interest that targeting expression of the human VDR to keratinocytes of VDR null mice prevented alopecia without correcting the mineral disorder (146). Further evidence that the role of VDR in hair follicle cycling is ligand independent comes from the observation that mice in which Cyp27b1, the enzyme solely responsible for producing 1,25(OH)2D, has been deleted do not lose hair (147)
CONCLUDING REMARKS
Acquired vitamin D deficiency, especially moderate or mild (sometimes referred to as vitamin D insufficiency), is much more common than previously appreciated. It may affect more than 50% of populations with limited sunshine exposure and no vitamin D supplementation. In the less severe forms of vitamin D deficiency, there is no defective bone matrix mineralization, but an increased bone loss secondary to the perturbations in calcium homeostasis and secondary hyperparathyroidism, which accelerates the development of osteoporosis in post-menopausal women, the elderly population at large in general, and some subgroups in particular such as those on drugs that contribute to bone loss or deficient in calcium intake. Osteomalacia, which marks severe vitamin D deficiency, may affect some of these patients as well. Based on these observations, it is highly recommended for anyone taking care of the elderly, especially the house or bed bound, institutionalized or patients with physical or mental deficiencies that limit their free movement, to consider and evaluate their vitamin D status. It seems to be good clinical practice and cost effective to recommend vitamin D supplementation for populations at risk, as a measure to prevent the deleterious effects of vitamin D deficiency on the musculoskeletal system.
Hereditary deficiencies in vitamin D action are rare disorders. The importance of studying these diseases stems from the fact that they represent a naturally occurring experimental model that helps to elucidate the function and importance of vitamin D and the VDR-effector system in human beings in vivo.
VDRs are abundant and widely distributed among most tissues studied and multiple effects of calcitriol are observed on various cell functions in vitro. Yet, the clinical and biochemical features in patients with simple hereditary 1,25(OH)2D deficiency and resistance seems to demonstrate that the most important disturbances of clinical relevance are perturbations in mineral and bone metabolism. This emphasizes the pivotal role of 1,25(OH)2D in transepithelial net calcium fluxes. Moreover, the fact that in patients with extreme end-organ resistance to calcitriol, calcium infusions correct the disturbances in mineral homeostasis and cure the bone disease may support the notion that defective bone matrix mineralization is secondary to disturbances in mineral homeostasis. That said, the widespread distribution of the VDR and the enzymes metabolizing vitamin D to its active metabolites have led to numerous studies into its non-skeletal actions with the hope that vitamin D may play a role in the treatment of diseases such as cancer, heart disease, respiratory illness, inflammatory diseases, and diabetes. Characterization of the molecular, cellular, and functional defects of the different natural mutants of the human VDR in simple hereditary 1,25(OH)2D deficiency and resistance, demonstrates the essentiality of the VDR as the mediator of calcitriol action and the importance and function of its different domains, i.e., binding of the hormone, an RXR isoform, and binding to a specifically defined DNA region, as well as coactivators and corepressor complexes. Thus, studies in patients with hereditary deficiencies in vitamin D action are the essential link between molecular defects and physiological relevant effects in human beings.
REFERENCES
- Ohyama Y, Noshiro M, Eggertsen G, Gotoh O, Kato Y, Bjoerkhem I, Okuda K. Structural characterization of the gene encoding rat 25-hydroxyvitamin D3 24-hydroxylase. Biochemistry. 1993;32(1):76-82.
- Akeno N, Saikatsu S, Kawane T, Horiuchi N. Mouse Vitamin D-24-Hydroxylase: Molecular Cloning, Tissue Distribution, and Transcriptional Regulation by 1α,25-Dihydroxyvitamin D3*. Endocrinology. 1997;138(6):2233-2240.
- Fu GK, Portale AA, Miller WL. Complete Structure of the Human Gene for the Vitamin D 1α-Hydroxylase, P450c1α. DNA and Cell Biology. 1997;16(12):1499-1507.
- Monkawa T, Yoshida T, Wakino S, Shinki T, Anazawa H, Deluca HF, Suda T, Hayashi M, Saruta T. Molecular Cloning of cDNA and Genomic DNA for Human 25-hydroxyvitamin D31α-hydroxylase. Biochemical and Biophysical Research Communications. 1997;239(2):527-533.
- Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca HF, Suda T. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(24):12920-12925.
- Takeyama K-i, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D <sub>3</sub> 1α-Hydroxylase and Vitamin D Synthesis. Science. 1997;277(5333):1827-1830.
- Jehan F, Ismail R, Hanson K, DeLuca HF. Cloning and expression of the chicken 25-hydroxyvitamin D3 24-hydroxylase cDNA. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 1998;1395(3):259-265.
- Jones G, Ramshaw H, Zhang A, Cook R, Byford V, White J, Petkovich M. Expression and Activity of Vitamin D-Metabolizing Cytochrome P450s (CYP1α and CYP24) in Human Nonsmall Cell Lung Carcinomas*. Endocrinology. 1999;140(7):3303-3310.
- Nguyen M, d'Alesio A, Pascussi JM, Kumar R, Griffin MD, Dong X, Guillozo H, Rizk-Rabin M, Sinding C, Bougnères P, Jehan F, Garabédian M. Vitamin D-Resistant Rickets and Type 1 Diabetes in a Child With Compound Heterozygous Mutations of the Vitamin D Receptor (L263R and R391S): Dissociated Responses of the CYP-24 and rel-B Promoters to 1,25-Dihydroxyvitamin D3. Journal of Bone and Mineral Research.2006;21(6):886-894.
- Bouillon R, Bikle D. Vitamin D Metabolism Revised: Fall of Dogmas. J Bone Miner Res. 2019;34(11):1985-1992.
- Roizen JD, Long C, Casella A, O'Lear L, Caplan I, Lai M, Sasson I, Singh R, Makowski AJ, Simmons R, Levine MA. Obesity Decreases Hepatic 25-Hydroxylase Activity Causing Low Serum 25-Hydroxyvitamin D. J Bone Miner Res. 2019;34(6):1068-1073.
- Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889-895.
- Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, Shine J, O'Malley BW. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(10):3294-3298.
- Burmester JK, Maeda N, DeLuca HF. Isolation and expression of rat 1,25-dihydroxyvitamin D3 receptor cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(4):1005-1009.
- Ozono K, Sone T, Pike JW. Perspectives: The genomic mechanism of action of 1,25-dihydroxyvitamin D3. Journal of Bone and Mineral Research. 2009;6(10):1021-1027.
- Deluca HF. The Metabolism, Physiology, and Function of Vitamin D. Vitamin D: Springer US; 1984:1-68.
- Mayer E, Kadowaki S, Williams G, Norman AW. Mode of Action of 1α,25-Dihydroxyvitamin D. Vitamin D: Springer US; 1984:259-302.
- Reichel H, Koeffler HP, Norman AW. The Role of the Vitamin D Endocrine System in Health and Disease. New England Journal of Medicine. 1989;320(15):980-991.
- Owen TA, Aronow MS, Barone LM, Bettencourt B, Stein GS, Lian JB. Pleiotropic Effects of Vitamin D on Osteoblast Gene Expression Are Related to the Proliferative and Differentiated State of the Bone Cell Phenotype: Dependency upon Basal Levels of Gene Expression, Duration of Exposure, and Bone Matrix Competency in Normal Rat Osteoblast Cultures*. Endocrinology. 1991;128(3):1496-1504.
- Pettifor JM, Ross FP, Travers R, Glorieux FH, Deluca HF. Dietary calcium deficiency : A syndrome associated with bone deformities and elevated serum 1,25-Dihyroxyvitamin D concentrations. Metabolic Bone Disease and Related Research. 1981;2(5):301-305.
- Teitelbaum SL, Bullough PG. The pathophysiology of bone and joint disease. Am J Pathol. 1979;96(1):282-354.
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: Standardization of nomenclature, symbols, and units: Report of the asbmr histomorphometry nomenclature committee. Journal of Bone and Mineral Research. 2009;2(6):595-610.
- Cole DEC, Carpenter TO, Gundberg CM. Serum osteocalcin concentrations in children with metabolic bone disease. The Journal of Pediatrics. 1985;106(5):770-776.
- Roizen JD, Levine MA. Response to: Obesity and Vitamin D Metabolism Modifications. J Bone Miner Res.2019;34(7):1384.
- Bikle DD. Vitamin D: Newer Concepts of Its Metabolism and Function at the Basic and Clinical Level. J Endocr Soc. 2020;4(2):bvz038.
- Holick MF. McCollum Award Lecture, 1994: Vitamin D—new horizons for the 21st century. The American Journal of Clinical Nutrition. 1994;60(4):619-630.
- Holick MF. Environmental factors that influence the cutaneous production of vitamin D. The American Journal of Clinical Nutrition. 1995;61(3):638S-645S.
- Lips P. Vitamin D Deficiency and Secondary Hyperparathyroidism in the Elderly: Consequences for Bone Loss and Fractures and Therapeutic Implications. Endocrine Reviews. 2001;22(4):477-501.
- Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. The American Journal of Clinical Nutrition.2006;84(1):18-28.
- Bischoff-Ferrari HA, Willett WC, Wong JB, Stuck AE, Staehelin HB, Orav EJ, Thoma A, Kiel DP, Henschkowski J. Prevention of Nonvertebral Fractures With Oral Vitamin D and Dose Dependency. Archives of Internal Medicine. 2009;169(6):551.
- Krall EA, Sahyoun N, Tannenbaum S, Dallal GE, Dawson-Hughes B. Effect of Vitamin D Intake on Seasonal Variations in Parathyroid Hormone Secretion in Postmenopausal Women. New England Journal of Medicine.1989;321(26):1777-1783.
- Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, Vamvakas EC, Dick IM, Prince RL, Finkelstein JS. Hypovitaminosis D in Medical Inpatients. New England Journal of Medicine.1998;338(12):777-783.
- Lips P, Duong T, Oleksik A, Black D, Cummings S, Cox D, Nickelsen T. A Global Study of Vitamin D Status and Parathyroid Function in Postmenopausal Women with Osteoporosis: Baseline Data from the Multiple Outcomes of Raloxifene Evaluation Clinical Trial. The Journal of Clinical Endocrinology & Metabolism.2001;86(3):1212-1221.
- Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Mayne ST, Rosen CJ, Shapses SA. The 2011 Dietary Reference Intakes for Calcium and Vitamin D: What Dietetics Practitioners Need to Know⁎⁎This article is a summary of the Institute of Medicine report entitled Dietary Reference Intakes for Calcium and Vitamin D (available at http://www.iom.edu/Reports/2010/Dietary-Reference-Intakes-for-Calcium-and-Vitamin-D.aspx) for dietetics practitioners; a similar summary for clinicians has also been published (Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Mayne ST, Rosen CJ, Shapses SA. The 2011 report on Dietary Reference Intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J Clin Endocrinol Metab. 2011;96:53-58). Journal of the American Dietetic Association. 2011;111(4):524-527.
- Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: an Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2011;96(7):1911-1930.
- Khaw KT, Sneyd MJ, Compston J. Bone density parathyroid hormone and 25-hydroxyvitamin D concentrations in middle aged women. BMJ. 1992;305(6848):273-277.
- Mezquita-Raya P, Muñoz-Torres M, De Dios Luna J, Luna V, Lopez-Rodriguez F, Torres-Vela E, Escobar-Jiménez F. Relation Between Vitamin D Insufficiency, Bone Density, and Bone Metabolism in Healthy Postmenopausal Women. Journal of Bone and Mineral Research. 2001;16(8):1408-1415.
- Collins D, Jasani C, Fogelman I, Swaminathan R. Vitamin D and bone mineral density. Osteoporosis International. 1998;8(2):110-114.
- Chapuy MC, Arlot ME, Duboeuf F, Brun J, Crouzet B, Arnaud S, Delmas PD, Meunier PJ. Vitamin D<sub>3</sub>and Calcium to Prevent Hip Fractures in Elderly Women. New England Journal of Medicine.1992;327(23):1637-1642.
- Davies M, Mawer EB, Hann JT, Stephens WP, Taylor JL. VITAMIN D PROPHYLAXIS IN THE ELDERLY: A SIMPLE EFFECTIVE METHOD SUITABLE FOR LARGE POPULATIONS. Age and Ageing. 1985;14(6):349-354.
- Weisman Y, Schen RJ, Eisenberg Z, Amarilio N, Graff E, Edelstein-Singer M, Goldray D, Harell A. Single Oral High-Dose Vitamin D<sub>3</sub>Prophylaxis in the Elderly. Journal of the American Geriatrics Society.1986;34(7):515-518.
- Khaw KT, Scragg R, Murphy S. Single-dose cholecalciferol suppresses the winter increase in parathyroid hormone concentrations in healthy older men and women: a randomized trial. The American Journal of Clinical Nutrition. 1994;59(5):1040-1044.
- Heikinheimo RJ, Inkovaara JA, Harju EJ, Haavisto MV, Kaarela RH, Kataja JM, Kokko AM-L, Kolho LA, Rajala SA. Annual injection of vitamin D and fractures of aged bones. Calcified Tissue International. 1992;51(2):105-110.
- Prader A, Illig R, Heierli E. [An unusual form of primary vitamin D-resistant rickets with hypocalcemia and autosomal-dominant hereditary transmission: hereditary pseudo-deficiency rickets]. Helv Paediatr Acta.1961;16:452-468.
- Fraser D, Kooh SW, Kind HP, Holick MF, Tanaka Y, DeLuca HF. Pathogenesis of Hereditary Vitamin-D-Dependent Rickets. New England Journal of Medicine. 1973;289(16):817-822.
- Delvin EE, Glorieux FH, Marie PJ, Pettifor JM. Vitamin D dependency: Replacement therapy with calcitriol. The Journal of Pediatrics. 1981;99(1):26-34.
- Labuda M, Morgan K, Glorieux FH. Mapping autosomal recessive vitamin D dependency type I to chromosome 12q14 by linkage analysis. Am J Hum Genet. 1990;47(1):28-36.
- Braekeleer MD, Larochelle J. Population genetics of vitamin D-dependent rickets in northeastern Quebec. Annals of Human Genetics. 1991;55(4):283-290.
- Fu GK, Lin D, Zhang MYH, Bikle DD, Shackleton CHL, Miller WL, Portale AA. Cloning of Human 25-Hydroxyvitamin D-1α-Hydroxylase and Mutations Causing Vitamin D-Dependent Rickets Type 1. Molecular Endocrinology. 1997;11(13):1961-1970.
- St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH. The 25-Hydroxyvitamin D 1-Alpha-Hydroxylase Gene Maps to the Pseudovitamin D-Deficiency Rickets (PDDR) Disease Locus. Journal of Bone and Mineral Research. 1997;12(10):1552-1559.
- Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, Portale AA, Miller WL. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63(6):1694-1702.
- Yoshida T, Monkawa T, Tenenhouse HS, Goodyer P, Shinki T, Suda T, Wakino S, Hayashi M, Saruta T. Two novel 1α-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type I. Kidney International. 1998;54(5):1437-1443.
- Kitanaka S, Takeyama K-i, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. Inactivating Mutations in the 25-Hydroxyvitamin D<sub>3</sub>1α-Hydroxylase Gene in Patients with Pseudovitamin D–Deficiency Rickets. New England Journal of Medicine. 1998;338(10):653-662.
- Kitanaka S, Murayama A, Sakaki T, Inouye K, Seino Y, Fukumoto S, Shima M, Yukizane S, Takayanagi M, Niimi H, Takeyama K-i, Kato S. No Enzyme Activity of 25-Hydroxyvitamin D3 1α-Hydroxylase Gene Product in Pseudovitamin D Deficiency Rickets, Including That with Mild Clinical Manifestation. The Journal of Clinical Endocrinology & Metabolism. 1999;84(11):4111-4117.
- Smith SJ, Rucka AK, Berry JL, Davies M, Mylchreest S, Paterson CR, Heath DA, Tassabehji M, Read AP, Mee AP, Mawer EB. Novel Mutations in the 1α-Hydroxylase (P450c1) Gene in Three Families with Pseudovitamin D-Deficiency Rickets Resulting in Loss of Functional Enzyme Activity in Blood-Derived Macrophages. Journal of Bone and Mineral Research. 1999;14(5):730-739.
- Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, Portale AA. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961-1970.
- Glorieux FH, Arabian A, Delvin EE. Pseudo-vitamin D deficiency: absence of 25-hydroxyvitamin D 1 alpha-hydroxylase activity in human placenta decidual cells. The Journal of Clinical Endocrinology & Metabolism. 1995;80(7):2255-2258.
- Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338(10):653-661.
- Yoshida T, Monkawa T, Tenenhouse HS, Goodyer P, Shinki T, Suda T, Wakino S, Hayashi M, Saruta T. Two novel 1alpha-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type I1. Kidney Int. 1998;54(5):1437-1443.
- Kitanaka S, Murayama A, Sakaki T, Inouye K, Seino Y, Fukumoto S, Shima M, Yukizane S, Takayanagi M, Niimi H, Takeyama K, Kato S. No enzyme activity of 25-hydroxyvitamin D3 1alpha-hydroxylase gene product in pseudovitamin D deficiency rickets, including that with mild clinical manifestation. J Clin Endocrinol Metab.1999;84(11):4111-4117.
- Smith SJ, Rucka AK, Berry JL, Davies M, Mylchreest S, Paterson CR, Heath DA, Tassabehji M, Read AP, Mee AP, Mawer EB. Novel mutations in the 1alpha-hydroxylase (P450c1) gene in three families with pseudovitamin D-deficiency rickets resulting in loss of functional enzyme activity in blood-derived macrophages. J Bone Miner Res. 1999;14(5):730-739.
- Winkler I, von Grabe C, Harmeyer J. Pseudo vitamin D deficiency rickets in pigs: in vitro measurements of renal 25-hydroxycholecalciferol-1-hydroxylase activity. Zentralbl Veterinarmed A. 1982;29(2):81-88.
- Fox J, Maunder EMW, Randall VA, Care AD. Vitamin D-dependent rickets type I in pigs. Clinical Science.1985;69(5):541-548.
- Dardenne O, Prud’homme Je, Arabian A, Glorieux FH, St-Arnaud R. Targeted Inactivation of the 25-Hydroxyvitamin D3-1α-Hydroxylase Gene (CYP27B1) Creates an Animal Model of Pseudovitamin D-Deficiency Rickets*. Endocrinology. 2001;142(7):3135-3141.
- Malloy PJ, Tasic V, Taha D, Tutunculer F, Ying GS, Yin LK, Wang J, Feldman D. Vitamin D receptor mutations in patients with hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab. 2014;111(1):33-40.
- Brooks MH, Bell NH, Love L, Stern PH, Orfei E, Queener SF, Hamstra AJ, DeLuca HF. Vitamin-D-Dependent Rickets Type II. New England Journal of Medicine. 1978;298(18):996-999.
- Fujita T, Nomura M, Okajima S, Furuya H. Adult-Onset Vitamin D-Resistant Osteomalacia with the Unresponsiveness to Parathyroid Hormone. The Journal of Clinical Endocrinology & Metabolism.1980;50(5):927-931.
- Liberman UA, Halabe A, Samuel R, Kauli R, Edelstein S, Weisman Y, Papapoulos SE, Fraher LJ, Clemens TL, O'Riordan JLH. END-ORGAN RESISTANCE TO 1,25-DIHYDROXYCHOLECALCIFEROL. The Lancet.1980;315(8167):504-507.
- Marx SJ, Liberman UA, Eil C, Gamblin GT, Degrange DA, Balsan S. Hereditary Resistance to 1,25-Dihydroxyvitamin D. Proceedings of the 1983 Laurentian Hormone Conference: Elsevier; 1984:589-620.
- Marx SJ, Bliziotes MM, Nanes M. ANALYSIS OF THE RELATION BETWEEN ALOPECIA AND RESISTANCE TO 1,25-DIHYDROXYVITAMIN D. Clinical Endocrinology. 1986;25(4):373-381.
- Bikle DD, Chang S, Crumrine D, Elalieh H, Man MQ, Choi EH, Dardenne O, Xie Z, Arnaud RS, Feingold K, Elias PM. 25 Hydroxyvitamin D 1 α-Hydroxylase Is Required for Optimal Epidermal Differentiation and Permeability Barrier Homeostasis. Journal of Investigative Dermatology. 2004;122(4):984-992.
- Stumpf WE, Sar M, Reid FA, Tanaka Y, DeLuca HF. Target Cells for 1,25-Dihydroxyvitamin D <sub>3</sub> in Intestinal Tract, Stomach, Kidney, Skin, Pituitary, and Parathyroid. Science. 1979;206(4423):1188-1190.
- Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem. 2003;278(40):38665-38674.
- Balsan S, Garabedian M, Larchet M, Gorski AM, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77(5):1661-1667.
- Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(18):9831-9835.
- Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, Masushige S, Fukamizu A, Matsumoto T, Kato S. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nature Genetics.1997;16(4):391-396.
- Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139(10):4391-4396.
- Balsan S, Garabedian M, Liberman UA, Bourdeau A, Eil C, Guillozo H, Grimberg R, Deunff MJL, Lieberherr M, Guimbaud P, Broyer M, Marx SJ. Rickets and Alopecia with Resistance to 1,25-Dihydroxyvitamin D: Two Different Clinical Courses with Two Different Cellular Defects*. The Journal of Clinical Endocrinology & Metabolism. 1983;57(4):803-811.
- Murayama A, Takeyama K-i, Kitanaka S, Kodera Y, Kawaguchi Y, Hosoya T, Kato S. Positive and Negative Regulations of the Renal 25-Hydroxyvitamin D3 1α-Hydroxylase Gene by Parathyroid Hormone, Calcitonin, and 1α,25(OH)2D3 in Intact Animals*. Endocrinology. 1999;140(5):2224-2231.
- Malloy PJ, Hochberg Z, Pike JW, Feldman D. Abnormal Binding of Vitamin D Receptors to Deoxyribonucleic Acid in a Kindred With Vitamin D-Dependent Rickets, Type II*. The Journal of Clinical Endocrinology & Metabolism. 1989;68(2):263-269.
- Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. The Journal of clinical investigation.1990;86(6):2071-2079.
- Ritchie HH, Hughes MR, Thompson ET, Malloy PJ, Hochberg Z, Feldman D, Pike JW, O'Malley BW. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(24):9783-9787.
- Takeda E, Yokota I, Ito M, Kobashi H, Saijo T, Kuroda Y. 25-Hydroxyvitamin D-24-Hydroxylase in Phytohemagglutinin-Stimulated Lymphocytes: Intermediate Bioresponse to 1,25-Dihydroxyvitamin D<sub>3</sub>of Cells from Parents of Patients with Vitamin DDependent Rickets type II*. The Journal of Clinical Endocrinology & Metabolism. 1990;70(4):1068-1074.
- Tsuchiya Y, Matsuo N, Cho H, Kumagai M, Yasaka A, Suda T, Orimo H, Shiraki M. An Unusual Form of Vitamin D-Dependent Rickets in a Child: Alopecia and Marked End-Organ Hyposensitivity to Biologically Active Vitamin D. The Journal of Clinical Endocrinology & Metabolism. 1980;51(4):685-690.
- Takeda E, Kuroda Y, Saijo T, Toshima K, Naito E, Kobashi H, Iwakuni Y, Miyao M. Rapid diagnosis of vitamin D-dependent rickets type II by use of phytohemagglutinin-stimulated lymphocytes. Clinica Chimica Acta.1986;155(3):245-250.
- Takeda E, Kuroda Y, Saijo T, Naito E, Kobashi H, Yokota I, Miyao M. 1α-Hydroxyvitamin D3 Treatment of Three Patients With 1,25-Dihydroxyvitamin D-Receptor-Defect Rickets and Alopecia. Pediatrics.1987;80(1):97-101.
- Takeda E, Yokota I, Kawakami I, Hashimoto T, Kuroda Y, Arase S. Two siblings with vitamin-D-dependent rickets type II: No recurrence of rickets for 14 years after cessation of therapy. European Journal of Pediatrics.1989;149(1):54-57.
- Saijo T, Ito M, Takeda E, Huq AH, Naito E, Yokota I, Sone T, Pike JW, Kuroda Y. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Am J Hum Genet.1991;49(3):668-673.
- Yagi H, Ozono K, Miyake H, Nagashima K, Kuroume T, Pike JW. A new point mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor in a kindred with hereditary 1,25-dihydroxyvitamin D-resistant rickets. The Journal of Clinical Endocrinology & Metabolism. 1993;76(2):509-512.
- Eil C, Liberman UA, Rosen JF, Marx SJ. A Cellular Defect in Hereditary Vitamin-D-Dependent Rickets Type II: Defective Nuclear Uptake of 1,25-Dihydroxyvitamin D in Cultured Skin Fibroblasts. New England Journal of Medicine. 1981;304(26):1588-1591.
- Feldman D, Chen T, Cone C, Hirst M, Shani S, Benderli A, Hochberg, Zeev. VITAMIN D RESISTANT RICKETS WITH ALOPECIA: CULTURED SKIN FIBROBLASTS EXHIBIT DEFECTIVE CYTOPLASMIC RECEPTORS AND UNRESPONSIVENESS TO 1,25(OH)2D3. The Journal of Clinical Endocrinology & Metabolism. 1982;55(5):1020-1022.
- Clemens TL, Adams JS, Horiuchi N, Gilchrest BA, Cho H, Tsuchiya Y, Matsuo N, Suda T, Holick MF. Interaction of 1,25-Dihydroxyvitamin-D<sub>3</sub>with Keratinocytes and Fibroblasts from Skin of Normal Subjects and a Subject with Vitamin-D-Dependent Rickets, Type II: A Model for Study of the Mode of Action of 1,25-Dihydroxyvitamin D<sub>3</sub>*. The Journal of Clinical Endocrinology & Metabolism.1983;56(4):824-830.
- Griffin JE, Zerwekh JE. Impaired stimulation of 25-hydroxyvitamin D-24-hydroxylase in fibroblasts from a patient with vitamin D-dependent rickets, type II. A form of receptor-positive resistance to 1,25-dihydroxyvitamin D3. The Journal of clinical investigation. 1983;72(4):1190-1199.
- Liberman UA, Eil C, Holst P, Rosen JF, Marx SJ. Hereditary Resistance to 1,25-Dihydroxyvitamin D: Defective Function of Receptors for 1,25-Dihydroxy vitamin D in Cells Cultured from Bone*. The Journal of Clinical Endocrinology & Metabolism. 1983;57(5):958-962.
- Malloy PJ, Weisman Y, Feldman D. Hereditary 1 alpha,25-dihydroxyvitamin D-resistant rickets resulting from a mutation in the vitamin D receptor deoxyribonucleic acid-binding domain. The Journal of Clinical Endocrinology & Metabolism. 1994;78(2):313-316.
- Chen TL, Hirst MA, Cone CM, Hochberg Z, Tietze H-U, Feldman D. 1,25-Dihydroxyvitamin D Resistance, Rickets, and Alopecia: Analysis of Receptors and Bioresponse in Cultured Fibroblasts from Patients and Parents*. The Journal of Clinical Endocrinology & Metabolism. 1984;59(3):383-388.
- Hirst MA, Hochman H, Feldman D. Vitamin D Resistance and Alopecia: A Kindred with Normal 1,25-Dihydroxyvitamin D Binding, but Decreased Receptor Affinity for Deoxyribonucleic Acid*. The Journal of Clinical Endocrinology & Metabolism. 1985;60(3):490-495.
- Castells S, Greig F, Fusi MA, Finberg L, Yasumura S, Liberman UA, Eil C, Marx SJ. Severely Deficient Binding of 1,25-Dihydroxyvitamin D to Its Receptors in a Patient Responsive to High Doses of This Hormone*. The Journal of Clinical Endocrinology & Metabolism. 1986;63(1):252-256.
- Fraher LJ, Karmali R, Hinde FRJ, Hendy GN, Jani H, Nicholson L, Grant D, O'Riordan JLH. Vitamin D-dependent rickets type II: extreme end organ resistance to 1,25-dihydroxy vitamin D3 in a patient without alopecia. European Journal of Pediatrics. 1986;145(5):389-395.
- Zhu W, Malloy PJ, Delvin E, Chabot G, Feldman D. Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets Due to an Opal Mutation Causing Premature Termination of the Vitamin D Receptor. Journal of Bone and Mineral Research. 1998;13(2):259-264.
- Malloy PJ, Zhu W, Zhao X-Y, Pehling GB, Feldman D. A Novel Inborn Error in the Ligand-Binding Domain of the Vitamin D Receptor Causes Hereditary Vitamin D-Resistant Rickets. Molecular Genetics and Metabolism.2001;73(2):138-148.
- Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, Lagier P, Palix C, Garnier JM, Garabedian M. Tryptophan Missense Mutation in the Ligand-Binding Domain of the Vitamin D Receptor Causes Severe Resistance to 1,25-Dihydroxyvitamin D. Journal of Bone and Mineral Research.2002;17(9):1728-1737.
- Malloy PJ, Pike JW, Feldman D. The Vitamin D Receptor and the Syndrome of Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets*. Endocrine Reviews. 1999;20(2):156-188.
- Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A Novel Mutation in Helix 12 of the Vitamin D Receptor Impairs Coactivator Interaction and Causes Hereditary 1,25-Dihydroxyvitamin D-Resistant Rickets without Alopecia. Molecular Endocrinology. 2002;16(11):2538-2546.
- Liberman UA, Eil C, Marx SJ. Resistance to 1,25-dihydroxyvitamin D. Association with heterogeneous defects in cultured skin fibroblasts. The Journal of clinical investigation. 1983;71(2):192-200.
- Hochberg Z, Benderli A, Levy J, Vardi P, Weisman Y, Chen T, Feldman D. 1,25-dihydroxyvitamin D resistance, rickets, and alopecia. The American Journal of Medicine. 1984;77(5):805-811.
- Koren R, Ravid A, Liberman UA, Hochberg Z, Weisman Y, Novogrodsky A. Defective binding and function of 1,25-dihydroxyvitamin D3 receptors in peripheral mononuclear cells of patients with end-organ resistance to 1,25-dihydroxyvitamin D. The Journal of clinical investigation. 1985;76(5):2012-2015.
- Liberman UA, Eil C, Marx SJ. Receptor-Positive Hereditary Resistance to 1,25-Dihydroxyvitamin D: Chromatography of Hormone-Receptor Complexes on Deoxyribonucleic Acid-Cellulose Shows Two Classes of Mutation*. The Journal of Clinical Endocrinology & Metabolism. 1986;62(1):122-126.
- Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the Human Vitamin D Receptor Gene Associated with Hypocalcemic Rickets. Science. 1988;242(4886):1702-1705.
- Sone T, Marx SJ, Liberman UA, Pike JW. A Unique Point Mutation in the Human Vitamin D Receptor Chromosomal Gene Confers Hereditary Resistance to 1,25-Dihydroxyvitamin D<sub>3</sub>. Molecular Endocrinology. 1990;4(4):623-631.
- Barsony J, Pike JW, DeLuca HF, Marx SJ. Immunocytology with microwave-fixed fibroblasts shows 1 alpha,25-dihydroxyvitamin D3-dependent rapid and estrogen-dependent slow reorganization of vitamin D receptors. J Cell Biol. 1990;111(6 Pt 1):2385-2395.
- Sone T, Scott RA, Hughes MR, Malloy PJ, Feldman D, O'Malley BW, Pike JW. Mutant vitamin D receptors which confer hereditary resistance to 1,25-dihydroxyvitamin D3 in humans are transcriptionally inactive in vitro. Journal of Biological Chemistry. 1989;264(34):20230-20234.
- Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, O'Riordan JLH, Farrow SM. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clinical Endocrinology.1996;45(1):85-92.
- Mechica JB, Leite MOR, Mendonca BB, Frazzatto EST, Borelli A, Latronico AC. A Novel Nonsense Mutation in the First Zinc Finger of the Vitamin D Receptor Causing Hereditary 1,25-Dihydroxyvitamin D<sub>3</sub>-Resistant Rickets. The Journal of Clinical Endocrinology & Metabolism. 1997;82(11):3892-3894.
- Cockerill FJ, Hawa NS, Yousaf N, Hewison M, O’Riordan JLH, Farrow SM. Mutations in the Vitamin D Receptor Gene in Three Kindreds Associated with Hereditary Vitamin D Resistant Rickets. The Journal of Clinical Endocrinology & Metabolism. 1997;82(9):3156-3160.
- Kristjansson K, Rut AR, Hewison M, O'Riordan JL, Hughes MR. Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1,25 dihydroxyvitamin D3. The Journal of clinical investigation. 1993;92(1):12-16.
- Wiese RJ, Goto H, Prahl JM, Marx SJ, Thomas M, Al-Aqeel A, DeLuca HF. Vitamin D-dependency rickets type II: truncated vitamin D receptor in three kindreds. Molecular and Cellular Endocrinology. 1993;90(2):197-201.
- Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The Crystal Structure of the Nuclear Receptor for Vitamin D Bound to Its Natural Ligand. Molecular Cell. 2000;5(1):173-179.
- Zhou Y, Wang J, Malloy PJ, Dolezel Z, Feldman D. Compound heterozygous mutations in the vitamin D receptor in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research.2009;24(4):643-651.
- Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. The Journal of clinical investigation. 1997;99(2):297-304.
- Hewison M, Rut AR, Kristjansson K, Walker RE, Dillon MJ, Hughes MR, O'Riordan JLH. Tissue resistance to 1,25-dihydroxyvitamin D without a mutation of the vitamin D receptor gene. Clinical Endocrinology.1993;39(6):663-670.
- Whitfield GK, Selznick SH, Haussler CA, Hsieh JC, Galligan MA, Jurutka PW, Thompson PD, Lee SM, Zerwekh JE, Haussler MR. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Molecular Endocrinology. 1996;10(12):1617-1631.
- Barsony J, McKoy W, DeGrange DA, Liberman UA, Marx SJ. Selective expression of a normal action of the 1,25-dihydroxyvitamin D3 receptor in human skin fibroblasts with hereditary severe defects in multiple actions of that receptor. The Journal of clinical investigation. 1989;83(6):2093-2101.
- Freedman LP. Increasing the Complexity of Coactivation in Nuclear Receptor Signaling. Cell. 1999;97(1):5-8.
- Lin NU, Malloy PJ, Sakati N, al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. The Journal of Clinical Endocrinology & Metabolism. 1996;81(7):2564-2569.
- Rut AR, Hewlson Y, Kristjansson K, Luisl B, Hughes MR, O'Riordan JLH. Two mutations causing vitamin D resistant rickets: modelling on the basis of steroid hormone receptor DNA-binding domain crystal structures. Clinical Endocrinology. 1994;41(5):581-590.
- Gamblin GT, Liberman UA, Eil C, Downs RW, Jr., DeGrange DA, Marx SJ. Vitamin D-dependent rickets type II. Defective induction of 25-hydroxyvitamin D3-24-hydroxylase by 1,25-dihydroxyvitamin D3 in cultured skin fibroblasts. The Journal of clinical investigation. 1985;75(3):954-960.
- Balsan S, Garabédian M, Larchet M, Gorski AM, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. The Journal of clinical investigation. 1986;77(5):1661-1667.
- Barsony J, Marx SJ. Receptor-mediated rapid action of 1 alpha,25-dihydroxycholecalciferol: increase of intracellular cGMP in human skin fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(4):1223-1226.
- Ritchie HH, Hughes MR, Thompson ET, Malloy PJ, Hochberg Z, Feldman D, Pike JW, O'Malley BW. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proc Natl Acad Sci U S A. 1989;86(24):9783-9787.
- Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest. 1990;86(6):2071-2079.
- Lin JP, Uttley WS. Intra-atrial calcium infusions, growth, and development in end organ resistance to vitamin D. Arch Dis Child. 1993;69(6):689-692.
- Bliziotes M, Yergey AL, Nanes MS, Muenzer J, Begley MG, Vieira NE, Kher KK, Brandi ML, Marx SJ. Absent Intestinal Response to Calciferols in Hereditary Resistance to 1,25-Dihydroxyvitamin D: Documentation and Effective Therapy With High Dose Intravenous Calcium Infusions*. The Journal of Clinical Endocrinology & Metabolism. 1988;66(2):294-300.
- Weisman Y, Bab I, Gazit D, Spirer Z, Jaffe M, Hochberg Z. Long-term intracaval calcium infusion therapy in end-organ resistance to 1,25-dihydroxyvitamin D. The American Journal of Medicine. 1987;83(5):984-990.
- Sakati N, Woodhouse NJY, Niles N, Harfi H, de Grange DA, Marx S. Hereditary Resistance to 1,25-Dihydroxyvitamin D: Clinical and Radiological Improvement during High-Dose Oral Calcium Therapy. Hormone Research. 1986;24(4):280-287.
- Adams JS, Gacad MA. Phenotypic Diversity of the Cellular 1,25-Dihydroxyvitamin D<sub>3</sub>-Receptor Interaction Among Different Genera of New World Primates. The Journal of Clinical Endocrinology & Metabolism. 1988;66(1):224-229.
- Liberman UA, de Grange D, Marx SJ. Low affinity of the receptor for 1α, 25-dihydroxyvitamin D<sub>3</sub> in the marmoset, a new world monkey. FEBS Letters. 1985;182(2):385-388.
- Shinki T, Shiina Y, Takahashi N, Tanioka Y, Koizumi H, Suda T. Extremely high circulating levels of 1α,25-dihydroxyvitamin D3 in the marmoset, a new world monkey. Biochemical and Biophysical Research Communications. 1983;114(2):452-457.
- Takahashi N, Suda S, Shinki T, Horiuchi N, Shiina Y, Tanioka Y, Koizumi H, Suda T. The mechanism of end-organ resistance to 1 alpha,25-dihydroxycholecalciferol in the common marmoset. Biochem J.1985;227(2):555-563.
- Gacad MA, Adams JS. Identification of a competitive binding component in vitamin D-resistant new world primate cells with a low affinity but high capacity for 1,25-dihydroxyvitamin D3. Journal of Bone and Mineral Research. 2009;8(1):27-35.
- Arbelle JE, Chen H, Gaead MA, Allegretto EA, Pike JW, Adams JS. Inhibition of vitamin D receptor-retinoid X receptor-vitamin D response element complex formation by nuclear extracts of vitamin D-resistant New World primate cells. Endocrinology. 1996;137(2):786-789.
- Lipsett MB, Chrousos GP, Tomita M, Brandon DD, Loriaux DL. The Defective Glucocorticoid Receptor in Man and Nonhuman Primates. Proceedings of the 1984 Laurentian Hormone Conference: Elsevier; 1985:199-247.
- Gacad MA, Chen H, Arbelle JE, LeBon T, Adams JS. Functional characterization and purification of an intracellular vitamin D-binding protein in vitamin D-resistant new world primate cells. Amino acid sequence homology with proteins in the hsp-70 family. J Biol Chem. 1997;272(13):8433-8440.
- Chen H, Hu B, Gacad MA, Adams JS. Cloning and expression of a novel dominant-negative-acting estrogen response element-binding protein in the heterogeneous nuclear ribonucleoprotein family. J Biol Chem.1998;273(47):31352-31357.
- Chen H, Hewison M, Hu B, Adams JS. Heterogeneous nuclear ribonucleoprotein (hnRNP) binding to hormone response elements: a cause of vitamin D resistance. Proc Natl Acad Sci U S A. 2003;100(10):6109-6114.
- Chen CH, Sakai Y, Demay MB. Targeting Expression of the Human Vitamin D Receptor to the Keratinocytes of Vitamin D Receptor Null Mice Prevents Alopecia. Endocrinology. 2001;142(12):5386-5389.
- Bikle DD, Chang S, Crumrine D, Elalieh H, Man MQ, Choi EH, Dardenne O, Xie Z, Arnaud RS, Feingold K, Elias PM. 25 Hydroxyvitamin D 1 alpha-hydroxylase is required for optimal epidermal differentiation and permeability barrier homeostasis. J Invest Dermatol. 2004;122(4):984-992.