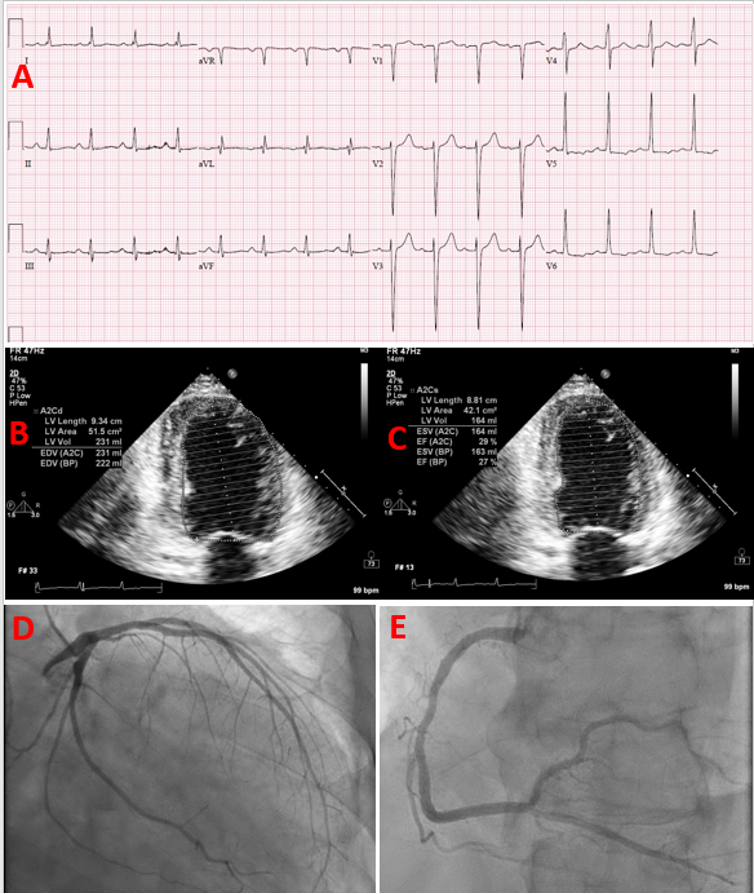
ABSTRACT
Atherosclerotic cardiovascular disease (ASCVD) is a major cause of morbidity and mortality in both men and women with T1DM and T2DM. In patients with T1DM, intensive glycemic control results in a reduction in ASCVD. However, intensive glycemic control does not have a major impact in reducing ASCVD in patients with T2DM. Metformin, pioglitazone, SGLT2 inhibitors, and certain GLP-1 receptor agonists have been shown to decrease major cardiovascular events in patients with T2DM to a greater extent than other treatment modalities. In patients with T2DM other risk factors including, hypertension and dyslipidemia, play a major role in inducing ASCVD and control of these risk factors is paramount. In patients with T1DM in good glycemic control, the lipid profile is very similar to the general population. In contrast, in patients with T2DM, even with good glycemic control, there are frequently lipid abnormalities (elevated TG and non-HDL-C, decreased HDL-C, and an increase in small dense LDL). In both T1DM and T2DM, poor glycemic control increases TG levels and decreases HDL-C levels with modest effects on LDL-C levels. Extensive studies have demonstrated that statins decrease ASCVD in patients with diabetes. Treatment with high doses of potent statins reduces ASCVD events to a greater extent than low dose statin therapy. Adding fibrates or niacin to statin therapy has not been shown to further decrease ASCVD events. In contrast, studies have shown that the combination of a statin and ezetimibe or a statin and a PCSK9 inhibitor result in a greater decrease in ASCVD events than statins alone. Studies have suggested that EPA, an omega-3-fatty acid, when added to statins also reduces ASCVD events but this result is controversial. In statin intolerant patients with T2DM bempedoic acid decreases ASCVD events. Current recommendations state that most patients with diabetes should be on statin therapy. In certain patients with diabetes ezetimibe, PCSK9 inhibitors, and bempedoic acid can play a role in reducing ASCVD.
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) is a major cause of morbidity and mortality in both men and women with diabetes (1-5). In addition to coronary disease, ASCVD includes stroke and peripheral arterial disease (PAD). PAD is common in diabetes, may be the first presentation of ASCVD and should be recognized as needing aggressive treatment of risk factors. The risk of ASCVD is increased approximately 2-fold in men and 3-4-fold in women (2-4,6,7). In the Framingham study, the annual rate of ASCVD was similar in men and women with diabetes, emphasizing that woman with diabetes need as aggressive preventive treatment as men with diabetes (2,6). In addition, several but not all studies, have shown that patients with diabetes with no history of ASCVD have a similar risk of having a myocardial infarction as non-diabetic patients who have a history of ASCVD, i.e., diabetes is an equivalent risk factor as a history of a previous cardiovascular event (8,9). The duration of diabetes and the presence of other risk factors or complications of diabetes likely determine whether a patient with diabetes has a risk equivalent to patients with a history of previous ASCVD events (10,11). In one study patients with T2DM who had the following risk factors within the target range, HbA1c, LDL-C, albuminuria, smoking, and blood pressure, the risk of an acute myocardial infarction or stroke was similar to individuals without diabetics (12). Moreover, numerous studies have shown that patients with diabetes who have ASCVD are at a very high risk of having another event, indicating that this population of patient’s needs especially aggressive preventive measures (1,8). This increased risk for the development of ASCVD in patients with diabetes is seen both in populations where the prevalence of ASCVD is high (Western societies) and low (for example, Japan) (2). However, in societies where the prevalence of ASCVD is low, the contribution of ASCVD as a cause of morbidity and mortality in patients with diabetes is relatively low compared to Western societies (2).
While the database is not as robust, the evidence indicates that patients with T1DM are also at high risk for the development of ASCVD (1,13-15). Interestingly, women with T1DM have twice the excess risk of fatal and nonfatal vascular events compared to men with T1DM (16,17). Additionally, developing T1DM at a young age increases the risk of ASCVD to a greater degree than late onset T1DM (17). Approximately 50% of patients with T1DM are obese or overweight and between 8% and 40% meet the criteria for the metabolic syndrome, which increases their risk of developing ASCVD (18).
While the development of diabetes at a young age increases the risk of ASCVD in patients with both T1DM and T2DM the deleterious impact is greater in patients with T2DM (19). Lastly, in patients with both T1DM and T2DM the presence of renal disease increases the risk of ASCVD (4,14). Of note is that the risk of developing ASCVD events in patients with diabetes has decreased recently, most likely due to better lipid and blood pressure control, which again reinforces the need to aggressively treat these risk factors in patients with diabetes (5,7,20).
ROLE OF GLYCEMIC CONTROL
Epidemiological studies have shown an association between the level of glycemic control and the development of ASCVD in both T1DM and T2DM (1,4,5,21,22). However, the association of glycemic control with ASCVD is considerably weaker than the association of glycemic control with the microvascular complications of diabetes, such as retinopathy and nephropathy (4). It must be recognized that epidemiological studies can only demonstrate associations and that confounding variables could account for the association between poor glycemic control and ASCVD. For example, patients with poor glycemic control may not undertake other preventive measures that could reduce ASCVD such as exercise, healthy diet, etc. Furthermore, the patients with poor glycemic control may have less compliance with therapies that reduce lipids and blood pressure. Therefore, randomized studies are essential in determining the role of glycemic control on ASCVD.
Early randomized studies, such as the University Group Diabetes Project (UGDP) and VA cooperative study, did not demonstrate a reduction in cardiovascular events in patients who were aggressively treated for glucose control (23-25). In fact, the data from these early studies suggested that improvements in glycemic control (VA cooperative study) or the use of certain drugs to treat diabetes (oral sulfonylureas in UGDP) may actually increase the risk of ASCVD.
Diabetes Control and Complications Trial (DCCT) and Kumamoto Studies
Latter studies, the DCCT in patients with T1DM and the Kumamoto study in patients with T2DM, while finding a decrease in ASCVD events (DCCT 41% decrease) in the subjects randomized to improved glycemic control did not have enough ASCVD events to demonstrate a statistically significant reduction (DCCT studied a population at low risk for ASCVD and the Kumamoto study had a very small number of subjects) (26-28). In contrast, both the DCCT and the Kumamoto study clearly demonstrated that improvements in glycemic control resulted in a reduction in microvascular disease (26-28). However, the long- term follow-up of the DCCT has demonstrated that those in the intensive glycemic control group had a decrease in ASCVD in subsequent years (29,30). The initial DCCT compared intensive vs. conventional therapy for a mean of 6.5 years. At the end of the study, a very large proportion of subjects agreed to participate in a follow-up observational study (Epidemiology of Diabetes Interventions and Complications- EDIC). During this follow-up period, glycemic control was relatively similar between the intensive therapy and conventional therapy group (glycosylated hemoglobin 7.9% vs. 7.8%) but during the actual trial there was a large difference in glycosylated hemoglobin levels (7.4% vs. 9.1%). After a mean 17 years of observation, the risk of any cardiovascular event was reduced by 42% and the risk of nonfatal myocardial infarction (MI), stroke, or death from ASCVD was reduced by 57% in the intensive control group. This study demonstrates that being in the intensive glycemic control group (for 6.5 of the 17 years of observation) is sufficient to have long-term beneficial effects on the risk of developing ASCVD in patients with T1DM. This beneficial effect was not entirely due to the prevention of microvascular complications as the differences between the intensive and conventional treatment groups for ASCVD persisted after adjusting for microalbuminuria and albuminuria. When an outcome of improved glycemic control is seen, or persists for years after the trial is over the phenomenon is called a “metabolic memory” effect.
UK Prospective Diabetes Study (UKPDS)
A similar finding has been reported with regard to T2DM. The UKPDS studied a large number of newly diagnosed patients with T2DM at risk for ASCVD. In this study improved glycemic control, with either insulin or sulfonylureas, reduced ASCVD by 16%, which just missed being statistically significant (p=0.052) (31). In the UKPDS, the improvement in glycemic control was modest (HbA1c reduced by approximately 0.9%) and the 16% reduction in ASCVD was in the range predicted based on epidemiological studies. The results of a 10-year follow-up of the UKPDS study have been reported (total duration of observation 25 years) (32). After termination of the study, glycosylated hemoglobin levels became very similar between the control and treatment groups. Nevertheless, risk reductions for MI became statistically significant for the insulin and the sulfonylurea group compared to controls (15% decrease, p=0.01).
DiGami Studies
Similarly, the DiGami study, which used insulin infusion during the peri-MI period to improve glycemic control followed by long-term glycemic control, demonstrated that survival post MI was significantly improved by good glycemic control (33). While this study focused on a highly-selected population and time period (patients undergoing a MI), the results are consistent with the hypothesis that improvements in glycemic control will reduce ASCVD. However, the DiGami 2 study did not confirm the benefits of tight glucose control beginning in the peri-MI period on outcomes (34). It must be noted though that the differences in glucose control achieved in DiGami 2 were much smaller than planned and the number of patients recruited was less than anticipated. Together these deficiencies could account for the failure to demonstrate significant differences in ASCVD events in this study.
ACCORD Study
Because of the need for more definitive data on the effect of glycemic control on ASCVD in T2DM, three large randomized trials, the ACCORD, ADVANCE, and VA Diabetes Trial, have been carried out. Much to everyone’s surprise and disappointment, improvement in glycemic control did not clearly result in a significant reduction in ASCVD in these trials.
The ACCORD study randomized 10,251 subjects with T2DM in the US and Canada with either a history of ASCVD or at increased risk for the development of ASCVD (35). Multiple different treatment protocols were used with the goal of achieving an A1c level < 6% in the intensive group and between 7-7.9% in the standard glycemic control group. During the trial the A1c levels were 6.4% in the intensive group and 7.5% in the standard group. As expected, the use of insulin therapy was much greater in the intensive group, as was the occurrence of hypoglycemia and weight gain. After a mean duration of 3.5 years this study was stopped early by the data safety monitoring board due to an increased all-cause mortality in the intensive treatment group (1.41% vs. 1.14% per year; hazard ratio 1.22 CI 1.01- 1.46). The primary outcome (MI, stroke, ASCVD death) was reduced by 10% in the intensive control group but this was not statistically significant (p=0.16). Of note, intensive glycemic control reduced the incidence of any MI (i.e. fatal or non-fatal) by 16%, nonfatal MI by 19%, coronary revascularization by 16%, and unstable angina by 19% (36).The explanation for the increased death rate in the intensive treatment arm remains unknown, but it has been speculated that the increased deaths might have been due to hypoglycemia, weight gain, too rapidly lowering A1c levels, or unrecognized drug toxicity. Long term follow-up of the ACCORD study did not reveal any beneficial effects on the primary outcome (nonfatal MI, nonfatal stroke, or cardiovascular death), death from any cause, and an expanded composite outcome that included all-cause death in the intensive glycemic control group (37).
ADVANCE Study
The ADVANCE study randomized 11,140 subjects with T2DM in Europe, Asia, Australia/New Zealand, and Canada who either had ASCVD or at least one other risk factor for ASCVD (38). In the intensive group the goal A1c was <6.5%. The achieved A1c levels during the trial were 6.3% in the intensive group and 7.3% in the standard treatment group. Of note is that compared to the ACCORD study, less insulin use was required to achieve these A1c levels. With regard to macrovascular disease (MI, stroke, and cardiovascular death), no significant differences were observed between the intensive and standard treatment groups (HR 0.94, CI 0.84-1.06, p=0.32). In contrast to the ACCORD trial, no increase in overall or cardiovascular mortality in the intensive treatment group was observed in the ADVANCE study. Long term follow-up did not demonstrate a decrease in the risk of death from any cause or major macrovascular events between the intensive-glucose-control group and the standard-glucose-control group (39).
VA Diabetes Trial
The VA Diabetes Trial randomized 1,791 subjects with poor glycemic control on maximal oral agent therapy or insulin (entry A1c 9.4%) (40). In the intensive group, the goal A1c was <6.0%. The achieved A1c levels during the trial were 6.9% in the intensive group and 8.5% in the standard treatment group. Similar to the other trials, a significant reduction in ASCVD was not observed in the intensive glycemic control group (HR 0.88, CI 0.74-1.05, p=0.12). Notably there were more ASCVD deaths and sudden deaths in the intensive treatment group, but this increase was not statistically significant. With long-term follow-up (approximately 10 years), the intensive-therapy group had a significantly lower risk of MI, stroke, congestive heart failure, amputation for ischemic gangrene, or cardiovascular-related death than did the standard-therapy group (hazard ratio, 0.83; P=0.04 (41). However, there was no reduction in cardiovascular or total mortality. Furthermore, with a longer period of follow-up (15 years) the risks of major cardiovascular events or death were not lower in the intensive-therapy group than in the standard-therapy group (42). In a careful analysis it was noted that that the risk of cardiovascular events was 17% lower in the intensive treatment group than in the standard control group during the approximate 10-year period when there was a separation of the glycated hemoglobin curves between the two groups, suggesting that glycemic control was reducing the risk of cardiovascular events (42).
Meta-analyses
In a meta-analysis of 6 randomized studies (UKPDS, Kumamoto, VA Feasibility study, ACCORD, ADVANCE, and VA Diabetes Trial) of intensive vs. conventional glycemic control in patients with T2DM (27,654 patients) there was no significant effect of tight blood glucose control on all-cause mortality (RR 1.03; 95% CI 0.90-1.17), cardiovascular mortality (RR 1.04; 95% CI 0.83-1.29), or nonfatal stroke (RR 1.02; 95% CI 0.88-1.17) but tight glucose control reduced the risk for nonfatal MI (RR 0.85; 95% CI 0.76-0.95) (43). In a meta-analysis of 4 studies (UKPDS, ACCORD, ADVANCE, and VA Diabetes Trial) the primary outcome was the composite of death from cardiovascular causes (including sudden death), non-fatal MI and non-fatal stroke, which was decreased by 9% (HR 0.91, 95% CI 0.84–0.99) in the intensive control group (44). Of note the risk of non-fatal/fatal MI was reduced by 15% (HR 0.85, 95% CI 0.76–0.94) in the intensive group without significant reductions in the risk of non-fatal/fatal stroke, fatal heart failure, all-cause mortality, or cardiovascular death.
Limitations of Cardiovascular Outcome Studies
Thus, while the epidemiological data strongly suggests that glycemic control would favorably impact ASCVD the recent randomized trials that were designed specifically to prove this hypothesis have failed to definitively demonstrate a clear link. There are several explanations for why these trials may not have worked as planned.
First, in the ACCORD, ADVANCE, and VA Diabetes Trial, other ASCVD risk factors were aggressively treated (lipid and BP lowering, ASA therapy). As a result of these treatments, the actual number of ASCVD events was considerably less than expected in these trials. The lower event rate may have reduced the ability to see a beneficial effect of glucose control. Additionally, the beneficial effects of glucose control maybe more robust if other risk factors are not aggressively controlled. In this regard, it is worth noting that in the earlier UKPDS, which showed that improved glycemic control reduced ASCVD events, both BP and lipids were not aggressively treated by current standards (systolic BP 135-140mm Hg, LDL-C 135-142mg/dL), which could be why this older trial demonstrated a benefit of improving glycemic control on ASCVD.
Second, these three recent trials were comparing relatively low A1c levels in both the intensive and usual control groups (A1c in intensive from ~6.4-6.9% and usual control group from ~7.0-8.4%). It is likely that both levels are on the “flatter” portion of the glycemic control-cardiovascular risk curve and that if one compared patients with intensive glycemic control with a control group with higher A1c values one would see more impressive results. If the difference in A1c levels were greater in the intensive and control groups the likelihood of seeing a reduction in cardiovascular events in the intensive group would be enhanced.
Third, all three trials were carried out by initiating tight control in patients with long standing diabetes who either had pre-existing ASCVD or were at high risk for ASCVD. It is possible that patients with a different clinical profile would be more likely to benefit from intensive glucose control. Subgroup analysis from these trials have suggested that patients with a shorter duration of diabetes, less severe diabetes, or the absence of pre-existing ASCVD actually benefited from intensive control. It may be that glycemic control is most important prior to the development of significant atherosclerosis. Clearly additional studies on different types of patients (i.e., newly diagnosed without evidence of ASCVD) will be necessary to definitively determine the role of glycemic control in different diabetic populations.
Fourth, the duration of these studies was relatively short and it is possible that a much longer duration of glycemic control is required to show benefits on ASCVD. In the UKPDS study the beneficial effects of intensive glucose control was not statistically significant at the end of the study but with an extended duration of follow-up (15-25 years) became statistically significant.
Fifth, it may be that glycemic control will be more important in patients with T1DM where abnormalities in glucose metabolism are a major reason for the increased risk of ASCVD. In contrast, patients with T2DM have multiple risk factors for ASCVD (dyslipidemia, hypertension, inflammation, insulin resistance, coagulation disorders, etc.) and glucose may play only a minor role in the increased risk. The differences in other cardiovascular risk factors could account for why intensive glycemic control produced a marked reduction in ASCVD in the DCCT (T1DM trial) and had only minimal effects in the trials carried out in patients with T2DM.
Finally, it is possible that certain treatments have side effects that mask the beneficial effects of glucose control. For example, hypoglycemia and weight gain could counterbalance the beneficial effects of improvements in glycemic control. It is possible that different treatment strategies could lead to more profound benefits (see below).
Summary
Thus, the currently available data do not definitively indicate that glycemic control will have major effects on reducing ASCVD in patients with T2DM. Furthermore, there are concerns that too tight control in patients with advanced disease could be harmful. In contrast, in patients with T1DM intensive glucose control appears to have a major beneficial effect on ASCVD based on the results of the DCCT.
THE EFFECT OF GLUCOSE LOWERING DRUGS ON ASCVD
Metformin
In the UKPDS, metformin, while producing a similar improvement in glycemic control as insulin or sulfonylureas, markedly reduced ASCVD by approximately 40% (45). In the ten-year follow-up the patients randomized to metformin in the UKPDS continued to show a reduction in MI and all-cause mortality (32). Two other randomized controlled trials have also demonstrated cardiovascular benefits with metformin therapy.
A study by Kooy et al compared the effect of adding metformin or placebo in overweight or obese patients already on insulin therapy (46). After a mean follow-up of 4.3 years this study observed a reduction in macrovascular events (HR 0.61 CI- 0.40-0.94, p=0.02), which was partially accounted for by metformin’s beneficial effects on weight. In this study the difference in A1c between the metformin and placebo group was only 0.3%.
Hong et al randomized non-obese patients with coronary artery disease to glipizide vs. metformin therapy for three years (47). A1c levels were similar, but there was a marked reduction in cardiovascular events in the metformin treated group (HR 0.54 CI 0.30- 0.90, p=0.026).
In contrast, long term follow-up (21 years) of individuals in the Diabetes Prevention Program with “pre-diabetes” did not demonstrate a reduction in cardiovascular event in individuals treated with metformin (48). A reduction in the use of metformin when the formal study ended after 3 years coupled with out-of-study metformin use over time may have diluted the potential effects of metformin therapy.
Support for the beneficial effects of metformin on atherosclerosis comes from long term follow-up of the Diabetes Prevention Program, which compared the effect of lifestyle changes or metformin in patients at high risk of developing diabetes (49). Coronary artery calcium scores were measured on average 13-14 years after randomization (49). There were no differences in coronary artery calcium scores between the lifestyle and placebo groups. However, in males, coronary artery calcium scores were significantly lower in the metformin group vs. the placebo group. In females treated with metformin coronary artery calcium scores were similar to the placebo group. The absence of a beneficial effect of metformin in women could be due to the lower baseline coronary artery calcium scores making it more difficult to demonstrate a beneficial effect. In HIV-infected patients with the metabolic syndrome metformin similarly reduced the progression of coronary artery calcium scores (50).
Thus, while there are no large cardiovascular outcome trials with metformin, together, the above results suggest that metformin may reduce ASCVD and that this effect is not due to improving glucose control. Metformin decreases weight or prevents weight gain and lowers lipid levels and these or other non-glucose effects may account for the beneficial effects on ASCVD.
Sulfonylureas
Based on the University Group Diabetes Project (UGDP) sulfonylureas carry a warning regarding an increased risk of ASCVD (24,25). However, the UKPDS studied a large number of newly diagnosed patients with T2DM at risk for ASCVD and in this study improved glycemic control with sulfonylureas reduced ASCVD by approximately 16%, which just missed being statistically significant (p=0.052) (31). In the UKPDS, A1c was reduced by approximately 0.9% and the 16% reduction in ASCVD was in the range predicted based on epidemiological studies. Thus, the reduction in cardiovascular events was likely due to improvements in glycemic control and not a direct benefit of sulfonylurea treatment. In support of this conjecture is that in the UKPDS, insulin treatment resulted in a similar decrease in A1c levels and reduction in cardiovascular events (31). Additionally, a large randomized cardiovascular outcome study (Carolina Study) reported that linagliptin, a DPP-4 inhibitor, and glimepiride, a sulfonylurea, had similar effects on cardiovascular events (hazard ratio 0.98) (51). Similarly, in the ADVANCE trial patients in the intensive therapy group were randomized to gliclazide and as noted above the occurrence of cardiovascular events was similar to the control group (38).Taken together these results suggest that sulfonylureas have a neutral effect on ASCVD.
Meglitinides
The Navigator study was a double-blind, randomized clinical trial in 9,306 individuals with impaired glucose tolerance and either ASCVD or cardiovascular risk factors who received nateglinide (up to 60 mg three times daily) or placebo (52). After 5 years, nateglinide administration did not alter the incidence of cardiovascular outcomes suggesting that meglitinides do not have an adverse or beneficial effect on cardiovascular events.
Thiazolidinediones
Studies with pioglitazone have suggested a beneficial effect on ASCVD. The PROactive study was a randomized controlled trial that examined the effect of pioglitazone vs. placebo over a 3-year period in T2DM with pre-existing macrovascular disease (53). With regard to the primary endpoint (a composite of all-cause mortality, non-fatal MI including silent MI, stroke, acute coronary syndrome, endovascular or surgical intervention in the coronary or leg arteries, and amputation above the ankle), there was a 10% reduction in events in the pioglitazone group but this difference was not statistically significant (p=0.095). It should be noted that both leg revascularization and leg amputations are not typical primary end points in ASCVD trials and these could be affected by pioglitazone induced edema. When one focuses on standard ASCVD endpoints, the pioglitazone treated group did demonstrate a 16% reduction in the main secondary endpoint (composite of all-cause mortality, non-fatal MI, and stroke) that was statistically significant (p=0.027). In the pioglitazone treated group, blood pressure, A1c, triglyceride, and HDL-C levels were all improved compared to the placebo group making it very likely that the mechanism by which pioglitazone decreased vascular events was multifactorial.
A multicenter, double-blind trial (IRIS Trial), randomly assigned 3,876 patients with insulin resistance (defined as score of more than 3.0 on the homeostasis model assessment of insulin resistance [HOMA-IR] index) but without diabetes and a recent ischemic stroke or TIA to treatment with either pioglitazone (target dose, 45 mg daily) or placebo (54). After 4.8 years, the primary outcome of fatal or nonfatal stroke or MI occurred in 9.0% of the pioglitazone group and 11.8% of the placebo group (hazard ratio 0.76; P=0.007). All components of the primary outcome were reduced in the pioglitazone treated group. Fasting glucose, fasting TG, and systolic and diastolic blood pressure were lower while HDL-C and LDL-C levels were higher in the pioglitazone group than in the placebo group. Although this study excluded patients with diabetes the results are consistent with and support the results of a protective effect of pioglitazone observed in the PROactive study.
In contrast, the TOSCA.IT study compared the effect of pioglitazone vs. sulfonylurea on ASCVD and did not observe a reduction in events with pioglitazone treatment (55). Patients with T2DM (n= 3028), inadequately controlled with metformin monotherapy (2-3 g per day), were randomized to pioglitazone or sulfonylurea and followed for a median of 57 months. Only 11% of the participants had a previous cardiovascular event. The primary outcome, was a composite of first occurrence of all-cause death, non-fatal MI, non-fatal stroke, or urgent coronary revascularization and occurred in 6.8% of the patients treated with pioglitazone and 7.2% of the patients treated with a sulfonylurea (HR 0.96; NS) (55). Limitations of this study are the small number of events due to the low-risk population studied and the relatively small number of participants. Additionally, 28% of the subjects randomized to pioglitazone prematurely discontinued the medication. Furthermore, it should be noted that when patients in this study were analyzed based on the risk of developing ASCVD those at high risk had a marked reduction in events when treated with pioglitazone compared to the sulfonylurea (56).Thus, the results of this study should be interpreted with caution.
Further support for the beneficial effects of pioglitazone on atherosclerosis is provided by studies that have examined the effect of pioglitazone on carotid intima-medial thickness. Both the Chicago and Pioneer studies demonstrated favorable effects on carotid intima-medial thickness in patients treated with pioglitazone compared to patients treated with sulfonylureas (57,58). Similarly, Periscope, a study that measured atheroma volume in the coronary arteries by intravascular ultrasonography, also demonstrated less atherosclerosis in the pioglitazone treated group compared to patients treated with sulfonylureas (59).
While the data from a variety of different types of studies strongly suggests that pioglitazone is anti-atherogenic, the results with rosiglitazone are different. Several meta-analyses of small and short-duration rosiglitazone trials suggested that rosiglitazone was associated with an increased risk of adverse cardiovascular outcomes (60,61). However, the final results of the RECORD study, a randomized trial that was specifically designed to compare the effect of rosiglitazone vs. either metformin or sulfonylurea therapy as a second oral drug in those receiving either metformin or a sulfonylurea on ASCVD events, have been published and did not reveal a difference in ASCVD death, MI, or stroke (62-64). Similarly, an analysis of patients on rosiglitazone in the BARI 2D trial also did not suggest an increase or decrease in ASCVD events in the patients treated with rosiglitazone (65). Thus, while the available data suggests that pioglitazone is anti-atherogenic, the data for rosiglitazone suggests a neutral effect. Whether these differences between pioglitazone and rosiglitazone are accounted for by their differential effects on lipid levels are unknown (see below for information on the effects of these drugs on lipid levels).
Numerous studies have shown that both pioglitazone and rosiglitazone increase the risk of heart failure (66).
DPP4 Inhibitors
Because of the importance of ASCVD in patients with diabetes the FDA is requiring manufacturers of new drugs to treat diabetes to carry out studies addressing ASCVD endpoints. The effect of the DPP4 inhibitors saxagliptin, alogliptin, sitagliptin, and linagliptin on ASCVD endpoints has been reported. In the saxagliptin study (SAVOR‐TIMI 53 trial), 16,492 patients with T2DM who had a history of cardiovascular events or who were at high risk were randomized to saxagliptin or placebo for 2.1 years (67). Saxagliptin did not increase or decrease cardiovascular death, MI, or ischemic stroke. Interestingly more patients treated with saxagliptin were admitted to the hospital for heart failure. The risk of heart failure with saxagliptin was greatest in patients at a high overall risk of heart failure (i.e., history of heart failure, impaired renal function, or elevated baseline levels of NT-proBNP) (68). Additionally, in the patients treated with saxagliptin the increase in heart failure was an early event with a 6-month rate of 1.1% vs. 0.6% in the placebo group (HR 1.80, p=0·001) and a 12-month rate of 1·9% vs. 1·3% (1.46; p=0.002) (68). In contrast, after 12 months no difference in the rate of heart failure was observed in the saxagliptin and placebo groups indicating that the development of heart failure is an early event (68).
In the alogliptin trial (EXAMINE), 5,380 patients with either an acute MI or unstable angina within the previous 15-90 days were randomized to alogliptin or placebo and followed for a median of 18 months (69). As seen in the saxagliptin study the rates of ASCVD events were similar in the alogliptin and placebo groups. The risk of hospitalization for heart failure was not statistically increased in the entire subset of patients treated with alogliptin (70). However, the hazard ratio for the subgroup of patients without heart failure at baseline was 1.76, p=0.026) (70).
In the sitagliptin trial (TECOS), 14,671 patients with established ASCVD were randomized to sitagliptin or placebo for 3 years (71). Sitagliptin did not decrease the risk of major adverse cardiovascular events or increase hospitalization for heart failure. Finally, in the linagliptin trial (CARMELINA), 6,979 patients at high risk for ASCVD were randomized to linagliptin or placebo for a median follow-up of 2.2 years (72). As in the other DPP4 inhibitor studies, linagliptin did not have a beneficial effect on ASCVD events. Additionally, linagliptin did not increase the risk of hospitalization for heart failure. Thus, these results indicate that DPP4 inhibitors do not increase or decrease ASCVD. The extent to which specific DPP4 inhibitors affect heart failure needs further investigation.
SGLT2 Inhibitors
EMPA-REG OUTCOME TRIAL
The effects of empagliflozin on cardiovascular morbidity and mortality in patients with T2DM has been reported (73). In this study, 7,020 patients at high risk for ASCVD were randomly assigned to receive 10 mg or 25 mg of empagliflozin or placebo once daily and were followed for 3.1 years. In the combined empagliflozin treated groups there was a statistically significant 14% reduction in the primary outcome (death from cardiovascular causes, nonfatal MI, or nonfatal stroke). As compared with placebo, empagliflozin treatment did not result in a significant difference in the occurrence of non-fatal MI or strokes. However, empagliflozin resulted in a significantly lower risk of death from cardiovascular causes (hazard ratio, 0.62), death from any cause (hazard ratio, 0.68), and hospitalization for heart failure (hazard ratio, 0.65). The beneficial effects of empagliflozin were noted to occur very rapidly and the beneficial effects on heart failure appeared to be the dominant effect compared to effects on ASCVD events. Decreases in cardiovascular outcomes and mortality with empagliflozin occurred across the range of cardiovascular risk (74). Additionally, the reduction in hospitalizations for heart failure and cardiovascular death were observed both in patients with and without heart failure at baseline (75).
CANVAS TRIAL
The effects of placebo vs. canagliflozin were determined in two combined trials involving a total of 10,142 participants with T2DM and high cardiovascular risk (76). The primary outcome was a composite of death from cardiovascular causes, nonfatal MI, or nonfatal stroke and the mean follow-up was 188 weeks. The primary outcome was reduced in the canagliflozin group (hazard ratio, 0.86; P=0.02). Death from any cause (hazard ratio 0.87; 95% CI 0.74-1.01) and death from ASCVD (hazard ratio 0.87; 95% CI 0.72-1.06) were reduced but were not statistically significant. Similarly, canagliflozin treatment did not result in a significant difference in non-fatal strokes or non-fatal MI (hazard ratio 0.90 for stroke and 0.85 for myocardial infarction). As seen with empagliflozin, hospitalization for heart failure was markedly reduced (hazard ratio 0.67; 95% CI 0.52-0.87) and this beneficial effect occurred rapidly. Notably, there was an increased risk of amputation (hazard ratio, 1.97; 95% CI, 1.41 to 2.75), which were primarily at the level of the toe or metatarsal. The basis for the increase in amputations is unknown. In other SGLT2 inhibitor studies an increase in amputations was not noted.
CREDENCE TRIAL
In a second canagliflozin trial that focused on kidney disease, a decrease in cardiovascular events was also observed (77). In this double-blind trial 4,401 patients with chronic kidney disease and T2DM were randomized to canagliflozin 100mg per day or placebo and followed for a median of 2.62 years. All the patients had an eGFR of 30 to <90 ml per minute per 1.73 m2 and albuminuria (ratio of albumin [mg] to creatinine [g], >300 to 5000). In this trial hospitalization for heart failure was reduced by 39%. The relative benefits of canagliflozin for cardiovascular outcomes was similar in individuals across the spectrum of eGFR levels (78). In contrast to the CANVAS trial, an increased risk of amputations was not observed.
DECLARE–TIMI 58 TRIAL
The effect of dapagliflozin on cardiovascular events has also been reported (79). 17,160 patients, including 10,186 without ASCVD were randomized to dapagliflozin or placebo and followed for a median of 4.2 years. The primary outcome was a composite of major adverse cardiovascular events (MACE), defined as cardiovascular death, MI, or ischemic stroke. The primary efficacy outcomes were MACE and a composite of cardiovascular death or hospitalization for heart failure. Dapagliflozin did not result in a lower rate of major adverse cardiovascular events (8.8% in the dapagliflozin group and 9.4% in the placebo group; hazard ratio, 0.93; P=0.17) but did result in a lower rate of cardiovascular death or hospitalization for heart failure (4.9% vs. 5.8%; hazard ratio, 0.83; P=0.005), which reflected a lower rate of hospitalization for heart failure (hazard ratio, 0.73; 95% CI, 0.61 to 0.88). Interestingly, in the patients with a history of a previous MI, dapagliflozin reduced the risk of a MACE (HR 0.84; P=0.039), whereas there was no effect in patients without a previous MI (80). Additionally, there was no increase in lower extremities amputations in the dapagliflozin treated group.
VERTIS CV
Patients with ASCVD and T2DM were randomized to ertugliflozin 5mg (n=2752), 15mg (2747), or placebo (n=2747) and the primary composite outcome of cardiovascular death and non-fatal MI or stroke was determined after a mean duration of follow-up of 3.5 years (81). This trial did not demonstrate a significant difference in the primary endpoint (MACE) nor any components of the primary endpoint. However, heart failure hospitalizations were significantly reduced by 30% in the patients treated with ertugliflozin (HR 0.70; CI 0.54–0.90). The benefits on heart failure were observed in both patients with a history of heart failure (decreased 37%) and patients without a history of heart failure (decreased 21%) (82).
SUMMARY
Thus, all four SGLT2 inhibitor studies demonstrated a decrease in heart failure with SGLT2 inhibitor therapy without consistent effects on ASCVD events. For additional information on the beneficial effects of SGLT2 inhibitors and SGLT1/SGLT2 inhibitors on ASCVD and heart failure see the Endotext chapter entitled “Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes” (83).
GLP-1 Receptor Agonists
The effect of six GLP-1 receptor agonists on ASCVD has been reported.
ELIXA
In the ELIXA trial 6,068 patients with T2DM who recently had a MI or been hospitalized for unstable angina were randomized to placebo or lixisenatide and followed for a median of 25 months (84). The primary end point of cardiovascular death, MI, stroke, or hospitalization for unstable angina was similar in the placebo or lixisenatide groups.
LEADER TRIAL
In contrast, the LEADER trial has shown that liraglutide decreased cardiovascular events (85). In this trial 9,340 patients at high cardiovascular risk were randomly assigned to receive liraglutide or placebo. After a median time of 3.5 years, the primary outcome of death from cardiovascular causes, nonfatal MI, or nonfatal stroke occurred in significantly fewer patients in the liraglutide group (13.0%) than in the placebo group (14.9%) (hazard ratio, 0.87, P=0.01). Additionally, deaths from cardiovascular causes (hazard ratio 0.78, P=0.007) or any cause was lower in the liraglutide group than in the placebo group (hazard ratio, 0.85; P=0.02). Interestingly patients with established ASCVD or decreased renal function (eGFR < 60) appeared to derive the greatest benefit of liraglutide treatment (86,87). As expected, weight and blood pressure were decreased in the liraglutide treated group and A1c levels were also decreased by 0.4%.
SUSTAIN 6 TRIAL
In support of the beneficial effects of GLP1 receptor agonists to reduce cardiovascular events, semaglutide, a long acting GLP-1 receptor agonist, has been shown to also reduce cardiovascular events (88). In this trial, 3,297 patients with T2DM with established ASCVD, chronic heart failure, chronic kidney disease, or age >60 with at least one cardiovascular risk factor were randomized to receive once-weekly semaglutide (0.5 mg or 1.0 mg) or placebo for 104 weeks. The primary outcome of cardiovascular death, nonfatal MI, or nonfatal stroke occurred in 6.6% of the semaglutide group and 8.9% of the placebo group (hazard ratio, 0.74; P = 0.02). In this study, both body weight and A1c levels were decreased in the patients treated with semaglutide.
PIONEER 6
In the PIONEER 6 study 3,183 patients with T2DM at high cardiovascular risk (age ≥50 years with established cardiovascular or chronic kidney disease, or age ≥60 years with cardiovascular risk factors) were randomly assigned to receive oral semaglutide or placebo (89). After a median time of 15.9 months, major adverse cardiovascular events, the primary outcome, occurred in 3.8% of the subjects treated with oral semaglutide and 4.8% of the placebo group (HR 0.79; 95% CI 0.57 to 1.11). Deaths from cardiovascular causes were 0.9% in the oral semaglutide group and 1.9% in the placebo group (HR 0.49; 95% CI, 0.27 to 0.92) while death from any cause occurred in 1.4% in the oral semaglutide group and 2.8% in the placebo group (HR 0.51; 95% CI, 0.31 to 0.84). It should be noted that the primary outcome was not statistically decreased in this study, which may be due to the relatively small number of subjects studied and the short duration of the study that together resulted in a small number of events. Additionally, more patients in the placebo group received treatment with an SGLT2 inhibitor than in the oral semaglutide group and SGLT2 inhibitors are well recognized to reduce cardiovascular events, which could also have diminished the ability to observe a decrease in events in the oral semaglutide group. Because the direction of change in cardiovascular events in PIONEER 6 and glucose lowering, weight loss, and many other effects of oral semaglutide are very similar to injected semaglutide many experts consider the effects on cardiovascular to also be similar.
EXSCEL TRIAL
The effect of once weekly exenatide vs. placebo on cardiovascular outcomes was tested in 14,752 patients, 73% who had ASCVD (90). The primary outcome was the occurrence of death from cardiovascular causes, nonfatal MI, or nonfatal stroke. After a median follow-up of 3.2 years (duration of drug exposure 2.4 years) the primary outcome was reduced in the exenatide treated group but this difference just missed achieving statistical significance (hazard ratio 0.91; 95% CI 0.83-1.00; p=0.06). While not statistically significant these results are consistent with the results observed with liraglutide and semaglutide treatment. It should be recognized that a high percentage of patients discontinued exenatide therapy in this trial (>40%) and this could have adversely affected the ability of exenatide treatment to favorably effect ASCD outcomes.
HARMONY OUTCOMES TRIAL
The effect of once weekly albiglutide vs. placebo was tested in 9,463 patients with ASCVD (91). The primary outcome was first occurrence of cardiovascular death, MI, or stroke. After a median follow-up of 1.6 years a 22% decrease in the primary endpoint was observed in the albiglutide group (hazard ratio 0·78, p<0·0001). It should be noted that albiglutide is no longer available as it was removed from the market due to commercial considerations by Glaxo.
REWIND TRIAL
REWIND was a randomized study of weekly subcutaneous injection of dulaglutide (1.5 mg) or placebo in 9,901 patients with T2DM who had either a previous cardiovascular event or cardiovascular risk factors (approximately 70% of patients did not have prior ASCVD) (92). During a median follow-up of 5.4 years the primary outcome of non-fatal MI, non-fatal stroke, or death from cardiovascular causes was decreased by 12% in the dulaglutide treated group (HR 0.88, p=0.026). The decrease in events was similar in participants with and without previous ASCVD. In an analysis that focused on stroke it was noted that dulaglutide reduced ischemic stroke by 25% compared to placebo but had no effect on hemorrhagic stroke (93).
SUMMARY
Thus, four studies have clearly demonstrated that treatment with GLP-1 receptor agonists reduces cardiovascular events, two studies has provided data consistent with these results, and one study failed to demonstrate benefit (Table 1). In a meta-analysis of these seven trials it was observed that cardiovascular death, stroke, or MI was decreased by 12% (HR 0.88, p<0.0001), death from cardiovascular causes by 12% (HR 0.88, p=0.003), fatal or non-fatal stroke by 16% (HR 0.84, p<0.0001) and fatal and non-fatal MI by 9% (HR 0.91, p=0.043) (Table 1) (94). Why there are differences in results between these studies is unknown but could be due to differential effects of the GLP-1 receptor agonists, differences in the patient populations studied, or other unrecognized variables. For additional information on the beneficial effects of GLP-1 receptor agonists on ASCVD see the Endotext chapter entitled “Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes” (83).
|
Table 1. Summary of GLP-1 Receptor Agonist Cardiovascular Outcome Trials |
||||||
|---|---|---|---|---|---|---|
|
Number |
|
HbA1c |
Mean Follow-up (years) |
Hazard Ratio* (95% CI) |
P value |
|
|
ELIXA |
6068 |
100% |
7.7% |
2.1 |
1.02 |
0.78 |
|
LEADER |
9340 |
81% |
8.7% |
3.8 |
0.87 |
0.015 |
|
SUSTAIN 6 |
3297 |
83% |
8.7% |
2.1 |
0.74 |
0.016 |
|
EXSCEL |
14,752 |
73% |
8.0% |
3.2 |
0.91 |
0.061 |
|
HARMONY |
9463 |
100% |
8.7% |
1.6 |
0.78 |
<0.001 |
|
REWIND |
9901 |
31% |
7.3% |
5.4 |
0>88 |
0.026 |
|
PIONEER 6** |
3183 |
85% |
8.2% |
1.3 |
0.79 |
0.17 |
|
Overall (94) |
0.88 |
<0.001 |
||||
*CVD death, MI, Stroke.
The mechanism accounting for this decrease in ASCVD is uncertain but could be related to reductions in HbA1c, body weight, systolic blood pressure, postprandial triglyceride levels, or the direct effect of activation of GLP-1 receptors on the atherosclerotic process such as improving endothelial function (95).
Tirzepatide
Tirzepatide activates both GLP-1 and GIP receptors. Long term cardiovascular trials with tirzepatide are underway. In a meta-analysis of seven randomized controlled trials with a duration of at least 26 weeks with 4,887 participants treated with tirzepatide and 2,328 control participants a 20% decrease in cardiovascular events was observed in the tirzepatide group (HR 0.80; 95% CI 0.57-1.11) suggesting that the effect of tirzepatide will be similar to the GLP-1 receptor agonists (96).
Acarbose
In the STOP-NIDDM trial 1,429 subjects with impaired glucose tolerance were randomized to placebo vs. acarbose and followed for 3.3 years (97). In the acarbose group a 49% relative risk reduction in the development of ASCVD events (hazard ratio 0.51; P =0.03) was observed. Among cardiovascular events, the major reduction was in the risk of MI (HR, 0.09; P =.02). In a smaller trial, 135 patients hospitalized for the acute coronary syndrome who were newly diagnosed with IGT were randomly assigned to acarbose or placebo (98). During a mean follow-up of 2.3 years the risk of recurrent major adverse cardiovascular event was decreased significantly in the acarbose group compared with that in control group (26.7% versus 46.9%, P < 0.05).
Despite these favorable observations a large trial failed to demonstrate a beneficial effect of acarbose in Chinese patients with impaired glucose tolerance (99). In a randomized trial acarbose vs. placebo was compared in 6,522 patients with coronary heart disease and impaired glucose tolerance. The primary outcome was cardiovascular death, non-fatal MI, non-fatal stroke, hospital admission for unstable angina, and hospital admission for heart failure and patients were followed for a median of 5 years. The primary outcome was similar in the acarbose and placebo groups (hazard ratio 0.98; 95% CI 0.86-1.11, p=0·73). No significant differences were seen for death from any cause, cardiovascular death, fatal or non-fatal MI, fatal or non-fatal stroke, hospital admission for unstable angina, hospital admission for heart failure, or impaired renal function.
Thus, whether acarbose favorably affects ASCVD in patients at high risk for developing diabetes is uncertain. Moreover, the effect of acarbose on ASCVD in patients with diabetes is unknown.
Cycloset
Cycloset is a quick-release bromocriptine formulation (bromocriptine-QR) that activates the D2 dopamine receptor and is approved for the treatment of diabetes. A 52 week, randomized, double-blind, multicenter trial evaluated cardiovascular safety in 3,095 patients with T2DM treated with bromocriptine-QR or placebo (100). The composite end point of first MI, stroke, coronary revascularization, or hospitalization for angina or congestive heart failure occurred in 1.8% of the bromocriptine-QR treated vs. 3.2% of the placebo-treated patients resulting in a 40% decrease in cardiovascular events (HR 0.60; CI 0.37– 0.96). Clearly further studies to confirm this finding and to elucidate the mechanism of this beneficial effect are required.
Bile Acid Sequestrants
Colesevelam is a non-absorbed, polymeric, LDL-C lowering and glucose lowering agent that is a high-capacity bile acid-binding molecule. This drug was developed primarily to lower LDL-C levels and was later noted to have favorable effects on blood glucose levels and was approved for improving glycemic control in patients with T2DM (101).
There have been no randomized studies that have examined the effect of bile acid sequestrants on cardiovascular end points in subjects with diabetes. In non-diabetic-subjects bile acid sequestrants have reduced cardiovascular events(102,103). Since bile acid sequestrants have a similar beneficial impact on LDL-C levels in diabetic and non-diabetic subjects one would anticipate that these drugs would also result in a reduction in events in the diabetic population.
Insulin
As described above in patients with T1DM the DCCT trial and in T2DM in the UKPDS trial demonstrated that insulin therapy reduced cardiovascular events by improving glycemic control (29-32). In the Origin Trial 12,537 people with cardiovascular risk factors plus impaired fasting glucose, impaired glucose tolerance, or T2DM were randomized to receive insulin glargine or standard care (104). The cardiovascular outcomes, which included nonfatal MI, nonfatal stroke, death from cardiovascular causes, revascularization, or hospitalization for heart failure, were similar in the glargine and placebo groups. Extended follow-up also did not demonstrate favorable effects on cardiovascular events in the glargine treated patients (105). Additionally, in patients with T2DM at high risk for cardiovascular events the occurrence of major cardiovascular events was similar in patients treated with degludec insulin or glargine insulin (106). These studies demonstrate that insulin does not accelerate atherosclerosis and by lowering glucose levels may decrease atherosclerosis, although the protective effects are mainly observed in patients with T1DM over a protracted period of time.
Other Studies
Finally, the Bari 2D study compared the effect of insulin sensitizers (metformin/TZD- mostly rosiglitazone) vs. insulin provision therapy (sulfonylureas/insulin) on cardiovascular outcomes in patients with T2DM and coronary artery disease (> 50% stenosis and positive stress test or > 70% stenosis and classic angina) (107,108). In this study, no differences in survival or cardiovascular endpoints were observed between metformin/TZD therapy vs. sulfonylurea/insulin therapy for the entire study. However, in the group with more severe coronary artery disease who were selected for coronary artery bypass surgery, the combination of coronary artery bypass and treatment with insulin sensitizers was associated with a lower rate of cardiovascular events. Why the metformin/TZD group only derived an enhanced benefit in the coronary artery bypass patients in this study is unknown. It should be noted that the vast majority of patients on TZD therapy were treated with rosiglitazone and, as discussed above, the effects of rosiglitazone on ASCVD do not appear to be as beneficial as pioglitazone.
Summary
These studies clearly demonstrate that the method by which one improves glycemic control may be very important with different drugs having effects in addition to glucose lowering that reduce cardiovascular events (table 2). While previous treatment algorithms have primarily focused on the effect of drugs on glycemic control, current treatment recommendations for patients with diabetes are using the results of these ASCVD trials to decide which drugs should be employed. For example, the ADA is recommending that in patients with high risk or established ASCVD an SGLT inhibitor or GLP1 receptor agonist with proven cardiovascular benefit should be part of the initial treatment regimen independent of A1c levels (109).
|
Table 2. Effect of Glucose Lowering Drugs on Atherosclerotic ASCVD |
|
|---|---|
|
Metformin |
Studies suggest benefit |
|
Sulfonylureas |
No effect |
|
Meglitinides |
No effect |
|
Thiazolidinediones |
Rosiglitazone no effect; Pioglitazone- studies suggest benefit |
|
DPP4 Inhibitors |
No effect on atherosclerosis. |
|
SGLT2 Inhibitors |
Marginal effect on ASCVD, Large effect on heart failure |
|
GLP-1 Receptor Agonists |
Decrease events |
|
Tirzepatide |
Study ongoing |
|
Acarbose |
No effect |
|
Cycloset |
Further studies required |
|
Bile Acid Sequestrants |
Decrease events, further studies required |
|
Insulin |
No effect |
Thiazolidinediones clearly increase the risk of heart failure while saxagliptin and alogliptin may increase risk of heart failure. SGLT2 inhibitors decrease the risk of heart failure.
ROLE OF OTHER RISK FACTORS IN ASCVD
Numerous studies have demonstrated that the traditional risk factors for ASCVD play an important role in patients with diabetes (2,4,5,110). Patients with diabetes without other risk factors have a relatively low risk of ASCVD (in most studies higher than similar non-diabetic patients), whereas the increasing prevalence of other risk factors markedly increases the risk of developing ASCVD (2). The major reversible traditional risk factors are hypertension, cigarette smoking, and lipid abnormalities (2,4,5,14,111). Other risk factors include obesity (particularly visceral obesity), insulin resistance, small dense LDL, elevated TG, low HDL-C, procoagulant state (increased PAI-1, fibrinogen), family history of early ASCVD, homocysteine, Lp (a), renal disease, albuminuria, and inflammation (C-reactive protein, SAA, cytokines) (2,4,5,110,111). In the last decade, it has become clear that to reduce the risk of ASCVD in patients with diabetes, one will not only need to improve glycemic control but also address these other cardiovascular risk factors. In the remainder of this chapter, I will focus on the dyslipidemia that occurs in patients with diabetes.
ROLE OF LIPIDS IN ASCVD
As in non-diabetic populations, epidemiological studies have shown that increased LDL-C and non-HDL-C levels and decreased HDL-C levels are associated with an increased risk of ASCVD in patients with diabetes (2,4,110,111). In the UKPDS cohort LDL-C levels were the strongest predictor of coronary artery disease (112). While it is universally accepted that elevated levels of LDL-C and non-HDL-C cause atherosclerosis and ASCVD the role of HDL-C is uncertain. Genetic studies and studies of drugs that raise HDL-C have not supported low HDL-C levels as a causative factor for atherosclerosis (113). Rather it is currently thought that HDL function is associated with atherosclerosis risk and that this does not precisely correlate with HDL-C levels (113). In patients with diabetes, elevations in serum triglyceride (TG) levels also are associated with an increased risk of ASCVD (4,111,114). With regard to TG, it is not clear whether they are a causative factor for ASCVD or whether the elevation in TG is a marker for other abnormalities (4,111,114,115). Recent Mendelian randomization studies have provided support for the hypothesis that elevated TG levels play a causal role in atherosclerosis (115,116). Unfortunately, as will be discussed later in this chapter lowering TG levels in patients on statin therapy has not decreased cardiovascular events.
LIPID ABNORMALITIES IN PATIENTS WITH DIABETES
In patients with T1DM in good glycemic control, the lipid profile is very similar to lipid profiles in the general population (110). In some studies HDL-C levels are modestly increased in patients with T1DM (117). In contrast, in patients with T2DM, even when in good glycemic control, there are abnormalities in lipid levels (118-121). It is estimated that 30-60% of patients with T2DM have dyslipidemia (5,122). Specifically, patients with T2DM often have an increase in serum TG levels, increased VLDL and IDL, and decreased HDL-C levels. Non-HDL-C levels are increased due to the increase in VLDL and IDL. LDL-C levels are typically not markedly different than in normal subjects but there is an increase in small dense LDL, a lipoprotein particle that may be particularly pro-atherogenic (123). As a consequence, there are more LDL particles, which coupled with the increases in VLDL and IDL, leads to an increase in apolipoprotein B levels (118-121). Additionally, the postprandial increase in serum TG is accentuated and elevations in postprandial lipids may increase the risk of ASCVD (118-121).
It should be recognized that the lipid changes in patients with T2D are characteristic of the alterations in lipid profile seen in obesity and the metabolic syndrome (insulin resistance syndrome) (124). Since a high percentage of patients with T2DM are obese, insulin resistant, and have the metabolic syndrome, it is not surprising that the prevalence of increased TG and small dense LDL and decreased HDL-C is common in patients with T2DM even when these patients are in good glycemic control. Obesity is also accompanied by increased systemic inflammation. The increasing prevalence of obesity/overweight in patients with T1D will likely result in an increased prevalence of dyslipidemia in this population.
Studies have shown that the anti-oxidant and anti-inflammatory functions of HDL isolated from patients with T1DM and T2DM are reduced (117,125). Additionally, the ability of HDL to facilitate cholesterol efflux is reduced in patients with T1DM and T2DM (126,127). Together these findings indicate that HDL-C levels per se may not fully reflect risk of ASCVD in patients with diabetes and that HDL function is perturbed in patients with diabetes.
In both T1DM and T2DM, poor glycemic control increases serum TG levels, VLDL, and IDL, and decreases HDL-C levels (119). Poor glycemic control can also result in a modest increase in LDL-C, which because of the elevation in TG is often in the small dense LDL subfraction. It is therefore important to optimize glycemic control in patients with diabetes because this will have secondary beneficial effects on lipid levels.
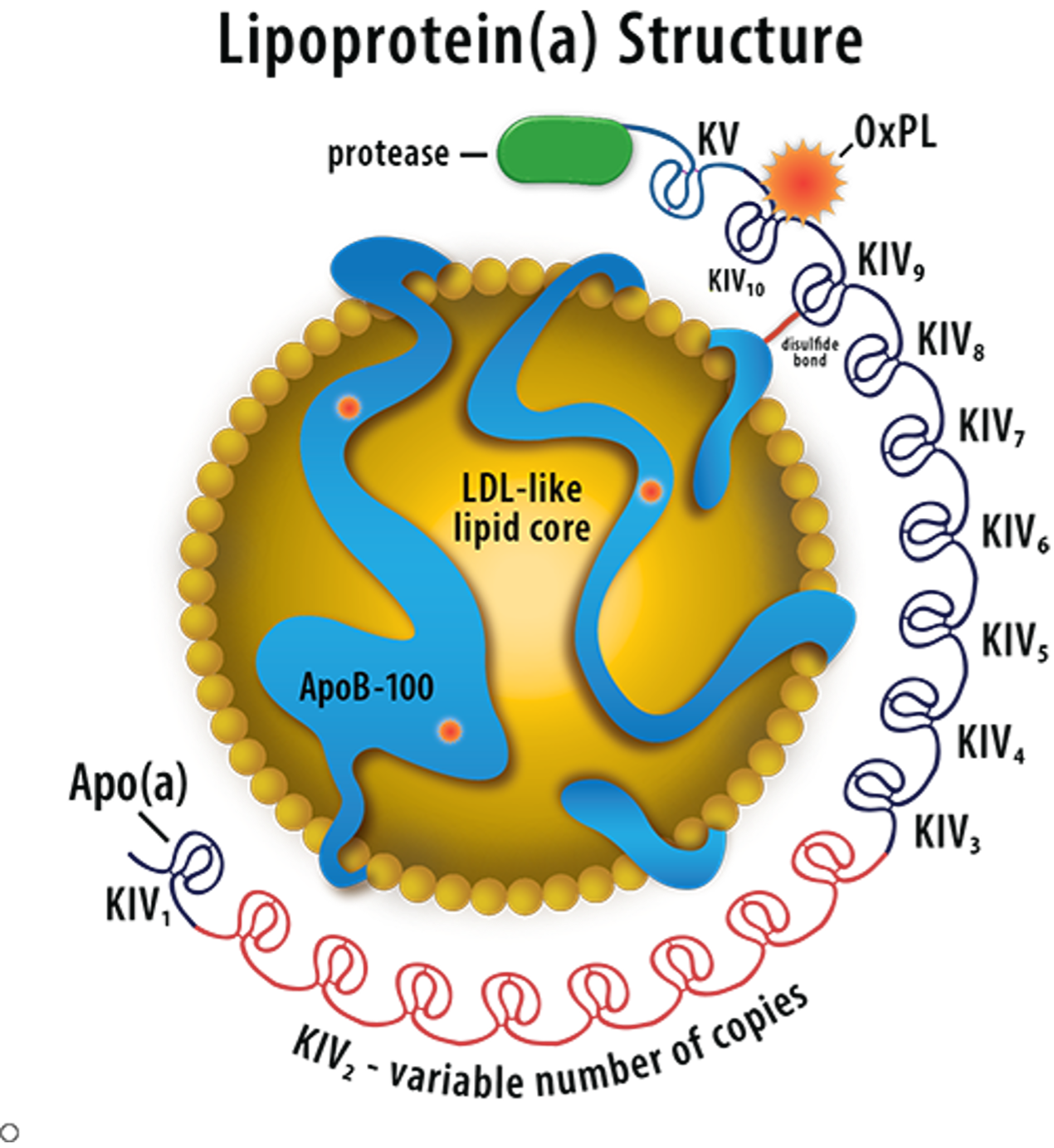
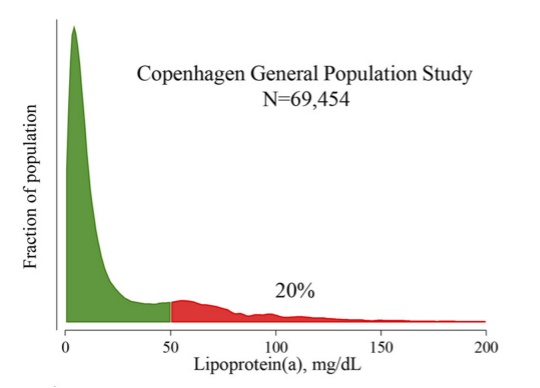
Lp(a) levels are usually within the normal range in patients with T1DM and T2DM (128). Some studies have observed no impact of diabetes mellitus on Lp(a) concentrations while other studies reported an elevation or a decrease in Lp(a) concentrations (128). The development of microalbuminuria and the onset of renal disease are associated with an increase in Lp (a) levels (129). Of note low Lp(a) levels are associated with an increased risk of developing T2DM (128). A recent very large case control study found that an Lp(a) concentration in the bottom 10% increases T2DM risk (130).
|
Table 3. Lipid Abnormalities in Patients with Diabetes |
|
|---|---|
|
T1DM |
Lipid profile is similar to controls if glycemic control is good |
|
T2DM |
Increased TG, VLDL, IDL, and non-HDL-C. Decreased HDL-C. Normal LDL-C but increase in small dense LDL, LDL particle number, and apolipoprotein B. |
|
Poor glycemic control |
Increased TG, VLDL, IDL, and non-HDL-C. Decreased HDL-C. Modest increase in LDL-C with increase in small dense LDL, LDL particle number, and apolipoprotein B. |
EFFECT OF GLUCOSE LOWERING DRUGS ON LIPIDS
Some therapies used to improve glycemic control may have an impact on lipid levels above and beyond their effects on glucose metabolism. In reviewing the literature, it is often very difficult to separate improvements in glycemic control vs. direct effects of drugs. Additionally, many of the changes induced by drug therapy result in only small changes in LDL-C, HDL-C, and TG levels, are variable from study to study, and are of questionable clinical significance. Insulin, sulfonylureas, meglinitides, DPP4 inhibitors, and alpha-glucosidase inhibitors do not appear to markedly alter fasting lipid profiles other than by improving glucose control (there are data indicating that DPP4 inhibitors and acarbose decrease postprandial triglyceride excursions, but they do not markedly alter fasting lipid levels) (131). In contrast, metformin, thiazolidinediones, GLP1 receptor agonists, bromocriptine-QR, and SGLT2 inhibitors have effects independent of glycemic control on serum lipid levels.
Metformin may decrease serum TG levels and LDL-C levels without altering HDL-C levels (131). In a meta-analysis of 37 trials with 2,891 patients, metformin decreased TG by 11.4mg/dL when compared with control treatment (p=0.003) (132). In an analysis of 24 trials with 1,867 patients, metformin decreased LDL-C by 8.4mg/dL compared to control treatment (p<0.001) (132). In contrast, metformin did not significantly alter HDL-C levels (132). It should be noted that in the Diabetes Prevention Program 3,234 individuals with impaired glucose metabolism were randomized to placebo, intensive lifestyle, or metformin therapy. In the metformin therapy group no significant changes were noted in TG, LDL-C, or HDL-C levels compared to the placebo group (133). Thus, metformin may have small effects on lipid levels.
The effect of thiazolidinediones depends on which agent is used. Rosiglitazone increases serum LDL-C levels, increases HDL-C levels, and only decreases serum TG if the baseline TG levels are high (131). In contrast, pioglitazone has less impact on LDL-C levels, but increases HDL-C levels, and decreases TG (131). In the PROactive study, a large randomized cardiovascular outcome study, pioglitazone decreased TG levels by approximately 10%, increased HDL-C levels by approximately 10%, and increased LDL-C by 1-4% (134). It should be noted that reductions in the small dense LDL subfraction and an increase in the large buoyant LDL subfraction are seen with both thiazolidinediones (131). In a randomized head-to-head trial, it was shown that pioglitazone decreased TG levels and increased serum HDL-C levels to a greater degree than rosiglitazone treatment (135,136). Additionally, pioglitazone increased LDL-C levels less than rosiglitazone. In contrast to the differences in lipid parameters, both rosiglitazone and pioglitazone decreased A1c and C-reactive protein to a similar extent. The mechanism by which pioglitazone induces more favorable changes in lipid levels than rosiglitazone despite similar changes in glucose levels is unclear, but differential actions of ligands for nuclear hormone receptors are well described.
Treatment with SGLT2 inhibitors results in a small increase in LDL-C and HDL-C levels (131). In a meta-analysis of 48 randomized controlled trials SGLT2 inhibitors significantly increased LDL-C (3.8mg/dL, p < 0.00001), HDL-C (2.3mg/dL, p < 0.00001), and decreased TG levels (8.8mg/dL, p < 0.00001) (137). The mechanism for these increases in LDL and HDL cholesterol is unknown but could be due to a decrease in plasma volume. The decrease in TG levels could be secondary to weight loss.
Bromocriptine-QR (Cycloset) treatment decreases TG levels but has no significant effect on LDL-C or HDL-C levels (138,139). The decrease in TG levels is thought to be due to a decrease in hepatic TG synthesis, likely due to a decrease in adipose tissue lipolysis resulting in decreased blood free fatty acid levels and reduced delivery of fatty acids to the liver for TG synthesis (140).
Colesevelam, a bile acid sequestrant that is approved for glucose lowering, lowers LDL-C levels by 15-20% and has only a modest effect on HDL-C levels (101,141). The effect of bile acid sequestrants on TG levels varies (141). In patients with normal TG levels, bile acid sequestrants increase TG levels by a small amount. However, as baseline TG levels increase, the effect of bile acid sequestrants on TG levels becomes greater, and can result in substantial increases in TG levels (141). In patients with TG > 500mg/dL the use of bile acid sequestrants is contraindicated (141).
Finally, GLP-1 receptor agonists can favorably affect the lipid profile by inducing weight loss (decreasing TG and very modestly decreasing LDL-C levels) (131). In a review by Nauck and colleagues it was noted that GLP-1 receptor agonists lowered TG levels by 18 to 62mg/dL depending upon the specific GLP-1 receptor agonist while decreasing LDL-C by 3-8mg/dL and increasing HDL-C by less than 1mg/dL (142). Additionally, GLP-1 receptor agonists reduce postprandial TG by reducing circulating chylomicrons by decreasing intestinal lipoprotein production (131,142). DPP4 inhibitors have a similar effect on postprandial TG levels as GLP-1 receptor agonists while having minimal effects on fasting lipid levels (142).
In the SURPASS trials, tirzepatide studies TG levels were consistently decreased by 13-25% (83,143). In most studies with the exception of SURPASS 5, HDL cholesterol levels increased by 3-11% (83,143). Total cholesterol and LDL cholesterol levels were modestly decreased in most studies (83,143). Not unexpectedly given the decrease in TG levels small LDL particles were decreased. For details see the Endotext chapter Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes (83).
|
Table 4. Effect of Glucose Lowering Drugs on Lipid Levels |
|
|---|---|
|
Metformin |
Modestly decrease TG and LDL-C |
|
Sulfonylureas |
No effect |
|
DPP4 inhibitors |
Decrease postprandial TG |
|
GLP1 analogues |
Decrease fasting and postprandial TG, modestly decrease LDL-C |
|
Tirzepatide |
Decrease TG, modestly decrease LDL-C, increase HDL-C |
|
Acarbose |
Decrease postprandial TG |
|
Pioglitazone Rosiglitazone |
Decrease TG and increase HDL-C. Small increase LDL-C but a decrease in small dense LDL |
|
SGLT2 inhibitors |
Small increase in LDL-C and HDL-C |
|
Colesevelam |
Decrease LDL-C. May increase TG |
|
Bromocriptine-QR |
Decrease TG |
|
Insulin |
No effect |
PATHOPHYSIOLOGY OF THE DYSLIPIDEMIA OF DIABETES
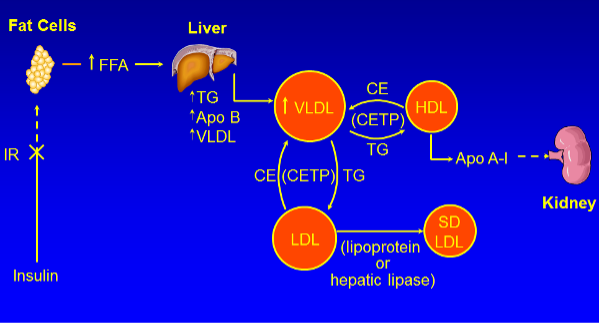
Figure 1. Pathophysiology of the Dyslipidemia of Diabetes
Multiple mechanisms account for the dyslipidemia seen in patients with T2DM, which are affected both by the level of glucose control and by factors such as obesity and inflammation that also contribute to dyslipidemia.
Increase in TG
There are a number of different abnormalities that contribute to the dyslipidemia seen in patients with T2DM and obesity (figure 1) (119-122,144-146).
OVERPRODUCTION OF VLDL BY THE LIVER
A key abnormality is the overproduction of VLDL by the liver, which is a major contributor to the elevations in serum TG levels. The rate of secretion of VLDL is highly dependent on TG availability, which is determined by the levels of fatty acids available for the synthesis of TG in the liver. An abundance of TG prevents the intra-hepatic degradation of Apo B-100 allowing for increased VLDL formation and secretion. There are three major sources of fatty acids in the liver all of which may be altered in patients with T2DM. First, the flux of fatty acids from adipose tissue to the liver is increased. An increased mass of adipose tissue, particularly visceral stores, results in increased fatty acid delivery to the liver. Additionally, insulin suppresses the lipolysis of TG to free fatty acids in adipose tissue; thus, in patients with either poorly controlled diabetes due to a decrease in insulin or a decrease in insulin activity due to insulin resistance, the inhibition of TG lipolysis is blunted and there is increased TG breakdown leading to increased fatty acid deliver to the liver. A second source of fatty acids in the liver is de novo fatty acid synthesis. Numerous studies have shown that fatty acid synthesis is increased in the liver in patients with T2DM. This increase may be mediated by the hyperinsulinemia seen in patients with insulin resistance. While the liver is resistant to the effects of insulin on carbohydrate metabolism, the liver remains sensitive to the effects of insulin stimulating lipid synthesis. Specifically, insulin stimulates the activity of SREBP-1c, a transcription factor that increases the expression of the enzymes required for the synthesis of fatty acids. Thus, while the liver is resistant to the effects of insulin on carbohydrate metabolism the liver remains sensitive to the effects of insulin stimulating lipid synthesis. Additionally, in the presence of hyperglycemia, glucose can induce another transcription factor, carbohydrate responsive element binding protein (ChREBP), which also stimulates the transcription of the enzymes required for fatty acid synthesis. The third source of fatty acids is the uptake of TG rich lipoproteins by the liver. Studies have shown an increase in intestinal fatty acid synthesis and the enhanced secretion of chylomicrons in animal models of T2DM. This increase in chylomicrons leads to the increased delivery of fatty acids to the liver. The increase in hepatic fatty acids produced by these three pathways results in an increase in the synthesis of TG in the liver and the protection of Apo B-100 from degradation resulting in the increased formation and secretion of VLDL. Finally, insulin stimulates the post translational degradation of Apo B-100 in the liver and a decrease in insulin activity in patients with T2DM also allows for the enhanced survival of Apo B-100 promoting increased VLDL formation.
DECREASED DEGRADATION OF TRIGLYCERIDE RICH LIPOPROTEINS
While the overproduction of triglyceride rich lipoproteins by the liver and intestine are important contributors to the elevations in serum TG levels in patients with T2DM, there are also abnormalities in the metabolism of these TG rich lipoproteins. First, there is a modest decrease in lipoprotein lipase activity, the key enzyme that metabolizes TG rich lipoproteins. The expression of lipoprotein lipase is stimulated by insulin and decreased insulin activity in patients with T2DM results in a decrease in lipoprotein lipase, which plays a key role in the hydrolysis of the TG carried in chylomicrons and VLDL. Additionally, patients with T2DM have an increase in Apo C-III levels, a key regulator of TG rich lipoprotein clearance. Glucose stimulates and insulin suppresses Apo C-III expression; thus, diabetes with hyperglycemia and either insulin deficiency or insulin resistance contribute to an increase in Apo C-III. Apo C-III is an inhibitor of lipoprotein lipase activity and thereby reduces the clearance of TG rich lipoproteins. In addition, Apo C-III also inhibits the cellular uptake of lipoproteins. Studies have shown that loss of function mutations in Apo C-III lead to lower serum TG levels and a reduced risk of ASCVD (147,148). Interestingly, inhibition of Apo C-III expression results in a decrease in serum TG levels even in patients deficient in lipoprotein lipase, indicating that the ability of Apo C-III to modulate serum TG levels is not dependent solely on regulating lipoprotein lipase activity (149). Lastly, insulin resistance is associated with an increase in Angptl3, an inhibitor of LPL (150). Thus, in patients with diabetes, a decrease in clearance of TG rich lipoproteins also contributes to the elevation in serum triglyceride levels.
Mechanism for the Increase in Small Dense LDL and Decrease in HDL
The elevation in TG rich lipoproteins in turn has effects on other lipoproteins. Specifically, cholesterol ester transfer protein (CETP) mediates the exchange of TG from TG rich VLDL and chylomicrons to LDL and HDL. The increase in TG rich lipoproteins per se leads to an increase in CETP mediated exchange, increasing the TG content of both LDL and HDL. The TG on LDL and HDL is then hydrolyzed by hepatic lipase and lipoprotein lipase leading to the production of small dense LDL and small HDL. Notably hepatic lipase activity is increased in patients with T2DM, which will also facilitate the removal of TG from LDL and HDL resulting in small lipoprotein particles. The affinity of Apo A-I for small HDL particles is reduced, leading to the disassociation of Apo A-I, which in turn leads to the accelerated clearance and breakdown of Apo A-I by the kidneys. Additionally, the production of Apo A-I may be reduced in patients with diabetes. High glucose levels can activate ChREBP and this transcription factor inhibits Apo A-I expression. Furthermore, insulin stimulates Apo A-I expression and a reduction in insulin activity due to insulin resistance or decreased insulin levels may also lead to a decrease in Apo A-I expression. The net result is lower levels of Apo A-I and HDL-C levels in patients with T2DM.
Role of Poor Glycemic Control
The above-described changes lead to the typical dyslipidemia observed in patients with T2DM (increased TG, decreased HDL-C, and an abundance of small dense LDL and small HDL). In patients with both Type 1 and T2DM, poor glycemic control can further adversely affect lipid and lipoprotein metabolism. As noted above the expression of lipoprotein lipase is stimulated by insulin. If insulin activity is very low the expression of lipoprotein lipase is severely suppressed and the metabolism of TG rich lipoproteins is markedly impaired. This leads to the delayed clearance of both chylomicrons and VLDL and elevations of TG rich lipoproteins. Additionally, insulinopenia results in a marked increase in lipolysis in adipose tissue, leading to the release of free fatty acids into the circulation. This increase in serum fatty acids results in the increased delivery of fatty acids to the liver, enhanced TG synthesis in the liver, and the increased production and secretion of VLDL. Whereas patients with T1DM who are well controlled and not obese or overweight typically have normal serum lipid profiles, if their control deteriorates, they will develop hypertriglyceridemia. In patients with T2DM deterioration of glycemic control will further exacerbate their underlying dyslipidemia resulting in greater increases in TG levels. If the synthesis of new VLDL is increased sufficiently this can result in an increase in LDL-C levels. HDL-C levels may decrease due to the formation of small HDL that are more susceptible to accelerated clearance. Improvements in glycemic control can markedly lower TG levels and may increase serum HDL-C levels. In patients with poorly controlled diabetes improvements in glycemic control may also lower LDL-C levels.
Role of Obesity and Inflammation
Most patients with T2DM and many patients with T1D are obese or overweight. Obesity is a pro-inflammatory state due to the macrophages that infiltrate adipose tissue. The cytokines produced by these macrophages and the adipokines that are produced by fat cells also alter lipid metabolism (151,152). The pro-inflammatory cytokines, TNF and IL-1, decrease the expression of lipoprotein lipase and increase the expression of angiopoietin like protein 4, an inhibitor of lipoprotein lipase. Together these changes decrease lipoprotein lipase activity, thereby delaying the clearance of TG rich lipoproteins. In addition, pro-inflammatory cytokines stimulate lipolysis in adipocytes increasing circulating free fatty acid levels, which will provide substrate for hepatic TG synthesis. In the liver, pro-inflammatory cytokines stimulate de novo fatty acid and TG synthesis. These alterations will lead to the increased production and secretion of VLDL. Thus, increases in the levels of pro-inflammatory cytokines will stimulate the production of TG rich lipoproteins and delay the clearance of TG rich lipoproteins, which together will contribute to the increase in serum TG that occurs in obese patients.
Obesity and the increase in pro-inflammatory cytokines may also affect HDL-C levels (153-155). First, pro-inflammatory cytokines inhibit the production of Apo A-I, the main protein constituent of HDL. Second, in many tissues pro-inflammatory cytokines decrease the expression of ABCA1 and ABCG1, which will lead to a decrease in the efflux of phospholipids and cholesterol from the cell to HDL decreasing the formation of mature HDL. Third, pro-inflammatory cytokines inhibit the production and activity of LCAT, which will limit the conversion of cholesterol to cholesterol esters in HDL. This conversion step is required for the formation of a normal spherical HDL particle and is crucial for the ability of HDL to increase the efflux of cholesterol from cells (including macrophages). Together these effects may lead to a decrease in HDL-C levels and a decrease in reverse cholesterol transport. Reverse cholesterol transport plays an important role in preventing cholesterol accumulation in macrophages and thereby reduces atherosclerosis.
Inflammation also decreases other important functions of HDL, such as its ability to prevent LDL oxidation (156). This reduction in the ability of HDL to protect from oxidation may be mediated in part by inflammation inducing lower levels of the enzyme paraoxonase, which is commonly seen in patients with diabetes (151,157). In parallel inflammation increases the oxidation of LDL and the amount of small dense LDL that is more susceptible to oxidation.
Role of Adipokines
Adipokines, such as leptin, adiponectin, and resistin, regulate lipid metabolism and the levels are altered in obese patients. Obesity increases serum leptin levels and leptin stimulates lipolysis in adipocytes which will increase serum free fatty acid levels (158). The circulating levels of adiponectin are decreased in subjects who are obese (159). Decreased adiponectin levels are associated with elevations in serum TG levels and decreases in HDL-C levels (159). This association is thought to be causal as studies in mice have shown that overexpressing adiponectin (transgenic mice) decreases TG and increases HDL-C levels while conversely, adiponectin knock-out mice have increased TG and decreased HDL-C levels (159). The adiponectin induced decrease in TG levels is mediated by an increased catabolism of TG rich lipoproteins due to an increase in lipoprotein lipase activity and a decrease Apo C-III, an inhibitor of lipoprotein lipase (159). The increase in HDL-C levels induced by adiponectin is mediated by an increase in hepatic Apo A-I and ABCA1, which results in the increased production of HDL particles (159).
Resistin is increased in subjects who are obese and the levels of resistin directly correlate with plasma TG levels (160). Moreover, resistin has been shown to stimulate hepatic VLDL production and secretion due to an increase in the synthesis of Apo B, TG, and cholesterol (160,161). Finally, resistin is associated with a decrease in HDL-C and Apo A-I levels (160).
EFFECT OF LIPID LOWERING ON ASCVD EVENTS IN PATIENTS WITH DIABETES
Monotherapy Studies
STATINS
The Cholesterol Treatment Trialists analyzed data from 18,686 subjects with diabetes (mostly T2DM) from 14 randomized trials (162). In the statin treated group there was a 9% decrease in all-cause mortality, a 13% decrease in vascular mortality, and a 21% decrease in major vascular events per 39mg/dL (1mmol/L) reduction in LDL-C. The beneficial effect of statin therapy was seen in both primary and secondary prevention patients. The effect of statin treatment on cardiovascular events in patients with diabetes was similar to that seen in non-diabetic subjects. Thus, these studies indicate that statins are beneficial in reducing ASCVD in patients with diabetes. Because of the large number of patients with diabetes included in the Heart Protection Study (HPS) and CARDS these two studies will be discussed in greater depth.
The HPS was a double-blind randomized trial that focused on patients at high risk for the development of cardiovascular events, including patients with a history of MIs, other atherosclerotic lesions, diabetes, and/or hypertension (163,164). Patients were between 40 and 80 years of age and had to have total serum cholesterol levels greater than 135mg/dL (thus very few patients were excluded because they did not have a high enough cholesterol level). The major strength of this trial was the large number of patients studied (>20,000). The diabetes subgroup included 5,963 subjects and thus was as large as many other prevention trials. The study was a 2x2 study design comparing simvastatin 40mg a day vs. placebo and anti-oxidant vitamins (vitamin E 600mg, vitamin C 250mg, and beta-carotene 20mg) vs. placebo and lasted approximately 5 years. Analysis of the group randomized to the anti-oxidant vitamins revealed no beneficial or harmful effects. In contrast, simvastatin therapy (40mg per day) reduced cardiovascular events, including MIs and strokes, by approximately 25% in all participants and to a similar degree in the diabetic subjects (total ASCVD reduced 27%, coronary mortality 20%, MI 37%, stroke 24%). Further analysis of the subjects with diabetes revealed that the reduction in cardiovascular events with statin therapy was similar in individuals with diabetes diagnosed for a short duration (<6 years) and for a long duration (>13 years). Similarly, subjects with diabetes in good control (HbA1c <7%) and those not in ideal control (HbA1c >7%) also benefited to a similar degree with statin therapy. Moreover, both T1DM and T2DM patients had a comparable reduction in ASCVD with simvastatin therapy. The decrease in cardiovascular events in patients with T1DM was not statistically significant because of the small number of subjects. Nevertheless, this is the only trial that included patients with T1DM and suggests that patients with T1DM will benefit from statin therapy similar to T2DM. In general, statin therapy reduced ASCVD in all subgroups of subjects with diabetes (females, males, older age, renal disease, hypertension, high TG, low HDL, ASA therapy, etc.) i.e., statin therapy benefits all patients with diabetes (note this study did not include patients with end stage renal disease but other studies have failed to show benefits of statin therapy in patients with diabetes and end stage renal disease (165)).
The CARDS trial specifically focused on subjects with diabetes (166). The subjects in this trial were males and females with T2DM between the ages of 40 to 75 years of age who were at high risk of developing ASCVD based on the presence of hypertension, retinopathy, renal disease, or current smoking. Of particular note, the subjects did not have any evidence of clinical atherosclerosis (myocardial disease, stroke, peripheral vascular disease) at entry and hence this study is a primary prevention trial. Inclusion criteria included LDL-C levels less than 160mg/dL and TG levels less than 600mg/dL. It is important to recognize that the average LDL-C in this trial was approximately 118mg/dL, indicating relatively low LDL-C levels. A total of 2,838 T2DM subjects were randomized to either placebo or atorvastatin 10mg a day. Atorvastatin therapy resulted in a 40% decrease in LDL-C levels with over 80% of patients achieving LDL-C levels less than 100mg/dL. Most importantly, atorvastatin therapy resulted in a 37% reduction in cardiovascular events. In addition, strokes were reduced by 48% and coronary revascularization by 31%. As seen in the HPS, subjects with relatively low LDL-C levels (LDL <120mg/dL) benefited to a similar extent as subjects with higher LDL-C levels (>120mg/dL).
HPS and CARDS, in combination with the other statin trials, provide conclusive evidence that statin therapy will reduce cardiovascular events in patients with diabetes. Importantly, the benefits of statin therapy are seen in patients with diabetes in both primary and secondary prevention trials.
Effect of Aggressive LDL-C Lowering with Statins
Studies have compared reductions of LDL-C to approximately 100mg/dL to more aggressive reductions in LDL-C on atheroma volume. The Reversal Trial studied 502 symptomatic coronary artery disease patients with an average LDL-C of 150mg/dL (167). Approximately 19% of the patients in this trial had diabetes. Patients were randomized to moderate LDL lowering therapy with pravastatin 40mg per day or to aggressive lipid lowering with atorvastatin 80mg per day. As expected, LDL-C levels were considerably lower in the atorvastatin treated group (pravastatin LDL= 110mg/dL vs. atorvastatin LDL= 79mg/dL). Most importantly, when one analyzed the change in atheroma volume determined after 18 months of therapy using intravascular ultrasound, the group treated aggressively with atorvastatin had a much lower progression rate than the group treated with pravastatin. Compared with baseline values, patients treated with atorvastatin had no change in atheroma burden (there was a very slight regression of lesions), whereas patients treated with pravastatin showed progression of lesions. When one compares the extent of the reduction in LDL-C to the change in atheroma volume, a 50% reduction in LDL (LDL-C levels of approximately 75mg/dL) resulted in the absence of lesion progression. This study suggests that lowering the LDL-C to levels well below 100mg/dL is required to prevent disease progression as measured by intravascular ultrasound. Other studies, such as Asteroid, have shown that marked reductions in LDL-C (in Asteroid the mean LDL-C levels were 61mg/dL) can also result in the regression of coronary artery atherosclerosis determined by intravascular ultrasound measurements (168). Additionally, the Saturn trial demonstrated that aggressive lipid lowering with either atorvastatin 80mg or rosuvastatin 40mg would induce regression of coronary artery atherosclerosis to a similar degree in patients with and without diabetes if the LDL-C levels were reduced to less than 70mg/dL (169). Together these trials indicate that aggressive lowering of LDL-C levels to below 70mg/dL can induce regression of atherosclerotic lesions.
The Prove-It trial determined in patients recently hospitalized for an acute coronary syndrome whether aggressively lowering of LDL-C with atorvastatin 80mg per day vs. moderate LDL-C lowering with pravastatin 40mg per day would have a similar effect on cardiovascular end points such as death, MI, documented unstable angina requiring hospitalization, revascularization, or stroke (170,171). In this trial, approximately 18% of the patients were diabetic. As expected, the on-treatment LDL-C levels were significantly lower in patients aggressively treated with atorvastatin compared to the moderate treated pravastatin group (atorvastatin LDL-C = approximately 62 vs. pravastatin LDL-C = approximately 95mg/dL). Of great significance, death or major cardiovascular events was reduced by 16% over the two years of the study in the group aggressively treated with atorvastatin. Moreover, the risk reduction in the patients with diabetes in the aggressive treatment group was similar to that observed in non-diabetics.
In the treating to new targets trial (TNT) patients with stable coronary heart disease and LDL-C levels less than 130mg/dL were randomized to either 10mg or 80mg atorvastatin and followed for an average of 4.9 years (172,173). Approximately 15% of the patients had diabetes. As expected, LDL-C levels were lowered to a greater extent in the patients treated with 80mg atorvastatin than with 10mg atorvastatin (77mg/dL vs. 101mg/dL). Impressively, the occurrence of major cardiovascular events was reduced by 22% in the group treated with atorvastatin 80mg (p<0.001). In the patients with diabetes events were reduced by 25% in the high dose statin group.
Finally, the IDEAL trial was a randomized study that compared atorvastatin 80mg vs. simvastatin 20-40mg in 8,888 patients with a history of ASCVD (174). Approximately 12% of the patients had diabetes. As expected, LDL-C levels were reduced to a greater extent in the atorvastatin treated group than the simvastatin treated group (approximately 81mg/dL vs. 104mg/dL). Once again, the greater reduction in LDL-C levels was associated with a greater reduction in cardiovascular events. Specifically, major coronary events defined as coronary death, nonfatal MI, or cardiac arrest was reduced by 11% (p=0.07), while nonfatal acute MI were reduced by 17% (p=0.02).
Combining the results of the Heart Protection Study, CARDS, Reversal, Saturn, Asteroid, Prove-It, TNT, and IDEAL leads one to the conclusion that aggressive lowering of LDL-C with statin therapy will be beneficial and suggests that in high-risk patients lowering the LDL to levels well below 100mg/dL is desirable. Moreover, the Cholesterol Treatment Trialists reviewed five trials with 39,612 subjects that were designed to determine the effect of usual vs. aggressive reductions in LDL-C (175). They reported that intensive control (approximately a 19mg/dL difference in LDL-C) resulted in a 15% decrease in major vascular events, a 13% reduction in coronary death or non-fatal MI, a 19% decrease in coronary revascularization, and a 16% decrease in strokes. As will be discussed below treatment guidelines reflect the results of these studies. Additionally, as described in detail below, studies of the addition of either ezetimibe or PCSK9 inhibitors to statins further demonstrates that aggressive lowering of LDL-C levels further reduces cardiovascular events
FIBRATES
The beneficial effect of monotherapy with fibrates (e.g., gemfibrozil, fenofibrate) on ASCVD in patients with diabetes is shown in Table 5. The results of these randomized trials suggest that monotherapy with this class of drug might reduce cardiovascular events in patients with diabetes, but the data is not very robust. The largest trial was the Field Trial (176). In this trial, 9,795 patients with T2DM between the ages of 50 and 75 not taking statin therapy were randomized to fenofibrate or placebo and followed for approximately 5 years. Fenofibrate therapy resulted in a 12% decrease in LDL-C, a 29% decrease in TG, and a 5% increase in HDL-C levels. The primary outcome was coronary events (coronary heart disease death and non-fatal MI), which were reduced by 11% in the fenofibrate group but did not reach statistical significance (p= 0.16). However, there was a 24% decrease in non-fatal MI in the fenofibrate treated group (p=0.01) and a non-significant increase in coronary heart disease mortality. Total ASCVD events (coronary events plus stroke and coronary or carotid revascularization) were reduced 11% (p=0.035). These beneficial effects of fenofibrate therapy on ASCVD were observed in patients without a previous history of ASCVD. In patients with a previous history of ASCVD no benefits were observed. Additionally, the beneficial effect of fenofibrate therapy was seen only in those subjects less than 65 years of age. The beneficial effects of fenofibrate in this study may have been muted by the increased use of statins in the placebo group, which reduced the differences in lipid levels between the placebo and fenofibrate groups. If one adjusted for the addition of lipid-lowering therapy, fenofibrate reduced the risk of coronary heart disease events by 19% (p=0.01) and of total ASCVD events by 15% (p=0.004). Thus, while the results of this large trial are intriguing they do not clearly show a benefit of fibrate therapy reducing ASCVD events. The number of patients with diabetes in the other fibrate trials are relatively small (table 4).
While the results of the monotherapy fibrate trials have been very heterogeneous it should be noted that fibrate trials in patients with elevated TG levels have reported a greater reduction of cardiovascular events (177). Additionally, subgroup analysis of several fibrate trials has also suggested that the benefit of fibrates was greatest in patients with elevated TG levels (177,178).
The mechanism by which fibrates may reduce cardiovascular events is unclear. These drugs lower serum TG levels and increase HDL-C, but it should be recognized that the beneficial effects of fibrates could be due to other actions of these drugs. Specifically, these drugs activate the nuclear hormone receptor PPAR alpha, which is present in the cells that comprise the atherosclerotic lesions, and it is possible that these compounds directly affect lesion formation and development. In addition, fibrates are anti-inflammatory. In fact, analysis of the VA-HIT study suggested that much of the benefit of fibrate therapy was not due to changes in serum lipoprotein levels (179,180).
To summarize, while in general the studies suggest that monotherapy with fibrates may reduce ASCVD in patients with diabetes, the results are not very robust or consistent as seen in the statin trials. Of note fibrate therapy appeared to be most effective in patients with increased TG levels and decreased HDL levels, a lipid profile typically seen in patients with T2DM. However, as will be presented in detail below (combination therapy section) the addition of fibrates to statins does not reduce ASCVD.
|
Table 5. Effect of Fibrate Monotherapy on Cardiovascular Outcomes |
||||
|---|---|---|---|---|
|
Study |
Drug |
#Diabetic subjects |
%Decrease controls |
% Decrease diabetics |
|
Helsinki Heart Study (181) |
Gemfibrozil |
135 |
34 |
60* |
|
VA-HIT (180) |
Gemfibrozil |
620 |
24 |
24 |
|
DIAS (182) |
Fenofibrate |
418 |
- |
23* |
|
Sendcap (183) |
Bezafibrate |
164 |
- |
70 |
|
Field (176) |
Fenofibrate |
9795 |
- |
11* |
* Not statistically significant
NIACIN
A single randomized trial, the Coronary Drug Project, has examined the effect of niacin monotherapy on cardiovascular outcomes (184). This trial was carried out from 1966 to 1974 (before the introduction of statin therapy) in men with a history of a prior MI and demonstrated that niacin therapy reduced cardiovascular events. The results of this study were re-analyzed to determine the effect of niacin therapy in subjects with varying baseline fasting and 1-hour post meal glucose levels (185). It was noted that 6 years of niacin therapy reduced the risk of coronary heart disease death or nonfatal MI by approximately 15-25% regardless of baseline fasting or 1-hour post glucose challenge glucose levels. Particularly notable is that reductions in events were seen in the subjects who had a fasting glucose level >126mg/dL or 1-hour glucose levels >220mg/dL (i.e., patients with diabetes). Thus, based on this single study, niacin monotherapy reduces cardiovascular events both in normal subjects and patients with diabetes. However, as will be presented in detail below (combination therapy section) the addition of niacin to statins does not reduce ASCVD.
EZETIMIBE
A multicenter, randomized trial in Japan examined the efficacy of ezetimibe in patients aged ≥75 years with elevated LDL-C (≥140 mg/dL) without a history of coronary artery disease who were not taking lipid lowering drugs (186). Patients were randomized to ezetimibe (n=1716) or usual care (n=1695) and followed for 4.1 years. The primary outcome was a composite of sudden cardiac death, MI, coronary revascularization, or stroke. In the ezetimibe group LDL-C was decreased by 25.9% and non-HDL-C by 23.1% while in the usual care group LDL-C was decreased by 18.5% and non-HDL-C by 16.5% (p<0.001 for both lipid parameters). By the end of the trial 9.6% of the patients in the usual care group and 2.1% of the ezetimibe group were taking statins. Ezetimibe reduced the incidence of the primary outcome by 34% (HR 0.66; P=0.002). Additionally, composite cardiac events were reduced by 60% (HR 0.60; P=0.039) and coronary revascularization by 62% (HR 0.38; P=0.007) in the ezetimibe group vs. the control group. There was no difference in the incidence of stroke or all-cause mortality between the groups. Approximately 25% of the patients in this trial had diabetes and the beneficial effects were similar in the diabetic and non-diabetic subjects. It should be noted that the reduction in cardiovascular events was much greater than one would expect based on the absolute difference in LDL-C levels (121mg/dL in ezetimibe group vs. 132mg/dL). As stated by the authors “Given the open-label nature of the trial, its premature termination, and issues with follow-up, the magnitude of benefit observed should be interpreted with caution.” Nevertheless, this study provides suggestive evidence that ezetimibe monotherapy may reduce cardiovascular events in patients with diabetes.
BEMPEDOIC ACID
A multicenter study of bempedoic acid in statin intolerant patients with ASCVD or at high risk for ASCVD was recently reported (187). Patients were randomized to bempedoic acid, 180 mg daily (n=6992), or placebo (n=6978) and the primary end point was death from cardiovascular causes, nonfatal MI, nonfatal stroke, or coronary revascularization. Bempedoic acid therapy reduced LDL-C and hsCRP levels by approximately 22% compared to the placebo group. The primary composite endpoint was reduced by 13% in the bempedoic acid group (HR 0.87; 95% CI, 0.79 to 0.96; P = 0.004). The four individual components of the primary endpoint were also significantly reduced in the bempedoic acid treatment group. In this trial approximately 45% of the patients had diabetes. In an analysis of the patients without clinical ASCVD, (i.e., primary prevention) (bempedoic acid n = 2100 or placebo n = 2106), there was a 30% decrease in cardiovascular events (HR 0.70: 95% CI, 0.55-0.89; P = .002) (188). In this subgroup analysis 66% of the patients had diabetes. This study clearly indicates that monotherapy with bempedoic acid will reduce cardiovascular events.
OTHER DRUGS
With regard to PCSK9 inhibitors and bile acid sequestrants there have been no randomized monotherapy studies that have examined the effect of these drugs on cardiovascular end points in subjects with diabetes. In non-diabetic subjects, monotherapy with bile acid sequestrants have reduced cardiovascular events (102,103). Since bile acid sequestrants have a similar beneficial impact on serum lipid levels in diabetic and non-diabetic subjects one would anticipate that these drugs would also result in a reduction in events in the diabetic population. Additionally, bile acid sequestrants improve glycemic control (101). However, bile acid sequestrants can raise TG levels and therefore must be used with caution in hypertriglyceridemic patients. There are no outcome studies with PCSK9 inhibitor monotherapy in patients with diabetes but given that these drugs reduce LDL-C levels and in combination with statins reduce cardiovascular events one would anticipate that PCSK9 inhibitor monotherapy will also reduce cardiovascular events.
Combination Therapy
The studies with statins have been so impressive that most patients with diabetes over the age of 40 are routinely treated with statin therapy and younger patients with diabetes at high risk for ASCVD are also typically on statin therapy (see Current Guidelines Section). Therefore, a key issue is whether the addition of other lipid lowering drugs to statins will result in a further reduction in cardiovascular events. A difficulty with such studies is that the reduction in cardiovascular events induced by statin therapy is so robust that very large trials may be required to see additional benefit.
STATINS + FIBRATES
The ACCORD-LIPID trial was designed to determine if the addition of fenofibrate to aggressive statin therapy would result in a further reduction in ASCVD in patients with T2DM (189). In this trial, 5,518 patients on statin therapy were randomized to placebo or fenofibrate therapy. The patients had diabetes for approximately 10 years and either had pre-existing ASCVD or were at high risk for developing ASCVD. During the trial, LDL-C levels were approximately 80mg/dL in both groups. There was only a small difference in HDL-C with the fenofibrate groups having a mean HDL-C of 41.2mg/dL while the control group had an HDL-C of 40.5mg/dL. Differences in TG levels were somewhat more impressive with the fenofibrate group having a mean TG level of 122mg/dL while the control group had a TG level of 144mg/dL. First occurrence of nonfatal MI, nonfatal stroke, or death from cardiovascular causes was the primary outcome and there was no statistical difference between the fenofibrate treated group and the placebo group. Additionally, there were also no statistically significant differences between the groups with regards to any of the secondary outcome measures of ASCVD. Of note, the addition of fenofibrate to statin therapy did not result in an increase in either muscle or liver side effects. On further analysis, there was a possible benefit of fenofibrate therapy in the patients in whom the baseline TG levels were elevated (>204mg/dL) and HDL-C levels decreased (<34mg/dL). Finally, similar to what has been reported in other trials, fenofibrate had beneficial effects on the progression of microvascular disease (190,191).
The PROMINENT trial studied the effect of pemafibrate, a new selective PPAR-alpha activator, in reducing cardiovascular outcomes in 10,497 patients (66.9% with previous ASCVD) with diabetes (192). This was a double-blind, randomized, controlled trial, in patients with T2DM, with mild-to-moderate hypertriglyceridemia (TG level, 200 to 499 mg/dL), LDL-C < 100mg/dL, and HDL-C levels < 40 mg/dL) who received either pemafibrate (0.2-mg tablets twice daily) or placebo in addition to statin therapy (96% on statins). The primary end point was a composite of nonfatal MI, ischemic stroke, coronary revascularization, or death from cardiovascular causes. Baseline fasting TG was 271 mg/dL, HDL-C 33 mg/dL, and LDL-C 78 mg/dL. Compared with placebo, pemafibrate decreased TG by 26.2%, while HDL-C increased 5.1% and LDL-C increased 12.3%. Notably non-HDL-C levels were unchanged and Apo B levels increased 4.8%. The primary endpoint was similar in the pemafibrate and placebo group (HR 1.03; 95% CI 0.91 to 1.15). The increase in LDL-C and Apo B levels likely account for the failure to reduce cardiovascular events.
Taken together the ACCORD study and the PROINENT trial indicate that the addition of fibrate therapy to statin therapy will not result in a reduction in cardiovascular events in patients with diabetes.
STATIN + NIACIN
The AIM-HIGH trial was designed to determine if the addition of Niaspan to aggressive statin therapy would result in a further reduction in cardiovascular events in patients with pre-existing ASCVD (193). In this trial 3,314 patients were randomized to Niaspan vs. placebo. Approximately 33% of the patients had diabetes. On trial, LDL-C levels were in the 60-70mg/dL range in both groups. As expected, HDL-C levels were increased in the Niaspan treated group (approximately 44mg/dL vs. 38mg/dL), while TG were decreased (approximately 121mg/dL vs. 155mg/dL). However, there were no differences in the primary endpoint between the control and Niaspan treated groups (Primary endpoint consisted of the first event of death from coronary heart disease, nonfatal MI, ischemic stroke, hospitalization for an acute coronary syndrome, or symptom-driven coronary or cerebral revascularization). There were also no differences in secondary endpoints except for a possible increase in strokes in the Niaspan treated group. The addition of Niaspan to statin therapy did not result in a significant increase in either muscle or liver toxicity. Thus, this study does not provide support for the addition of niacin to statins. However, it should be recognized that this was a relatively small study and a considerable number of patients stopped taking the Niaspan during the course of the study (25.4% of patients discontinued Niaspan therapy). In addition, most of the patients included in this study did not have a lipid profile that one would typically consider treating with niacin therapy. In the subset of patients with TG > 198mg/dL and HDL-C < 33mg/dL niacin showed a trend towards benefit (hazard ratio 0.74; p=0.073), suggesting that if the appropriate patient population was studied the results may have been positive (194).
HPS 2 Thrive also studied the effect of niacin added to statin therapy (195). This trial utilized extended-release niacin combined with laropiprant, a prostaglandin D2 receptor antagonist that reduces the flushing side effect of niacin treatment. HPS 2 Thrive was a very large trial with over 25,000 patients randomized to either niacin therapy or placebo. Approximately 32% of the patients in this trial had diabetes. The LDL-C level was 63mg/dL, the HDL-C 44mg/dL, and the TG 125mg/dL at baseline. As expected, niacin therapy resulted in a modest reduction in LDL-C (10mg/dL), a modest increase in HDL-C (6mg/dL), and a larger reduction in TG (33mg/dL). However, despite these lipid changes there were no significant differences in major cardiovascular events between the niacin and control group (risk ratio 0.96 CI 0.90- 1.03). It is unknown whether laropiprant, the prostaglandin D2 receptor antagonist, might have effects that worsen atherosclerosis and increase event rates. Similar to the AIM-HIGH study, the group of patients included in the HPS 2 Thrive trial were not the ideal patient population to test for the beneficial effects of niacin treatment added to statin therapy. Ideally, patients with high TG and high non-HDL-C levels coupled with low HDL-C levels should be studied. Nevertheless, the results of the AIM-HIGH and HPS 2 Thrive trials do not provide support for the addition of niacin to statin therapy in patients with diabetes.
STATIN + EZETIMIBE
The IMPROVE-IT trial tested whether the addition of ezetimibe to statin therapy would provide an additional beneficial effect in patients with the acute coronary syndrome (196). This was a large trial with over 18,000 patients randomized to statin therapy vs. statin therapy + ezetimibe. Approximately 27% of the patients in this trial had diabetes. On treatment LDL-C levels were 70mg/dL in the statin alone group vs. 53mg/dL in the statin + ezetimibe group. There was a small but significant 6.4% decrease in major cardiovascular events (Cardiovascular death, MI, documented unstable angina requiring re-hospitalization, coronary revascularization, or stroke) in the statin + ezetimibe group (HR 0.936 CI (0.887, 0.988) p=0.016). Cardiovascular death, non-fatal MI, or non-fatal stroke were reduced by 10% (HR 0.90 CI (0.84, 0.97) p=0.003). These beneficial effects were particularly pronounced in the patients with diabetes (Primary endpoint hazard ratio, 0.85; 95% confidence interval, 0.78-0.94) (197,198). This trial provides evidence that the combination of a statin + ezetimibe that results in a greater reduction in LDL-C levels will lead to a larger decrease in cardiovascular events than statin alone. It should be noted that the observed reduction in events was in the range expected based on the decrease in LDL-C levels.
The RACING trial compared rosuvastatin 10 mg plus ezetimibe 10 mg (combination therapy) vs. rosuvastatin 20mg in 3,780 patients (1,398 patients (37.0%) with diabetes) at 26 centers in South Korea (199). In the patients with diabetes the baseline LDL-C levels was 74mg/dL and during the study the median LDL-C was 53mg/dL in the combination therapy group and 61mg/dL in the high-intensity statin group (P < 0.001). After a median follow-up of 3 years the rate of cardiovascular events in patients with diabetes was 10.0% in the combination therapy group and 11.3% in the high-intensity statin group (HR: 0.89; 95% CI: 0.64–1.22; P = 0.460). Interestingly the rate of discontinuation or dose reduction of the study drug due to intolerance was lower in the combination therapy group than in the high-intensity statin group (5.2 vs. 8.7%; P = 0.014). This study demonstrates that cardiovascular outcomes were comparable between those receiving combination therapy vs. high-intensity statin monotherapy and that combination therapy significantly reduced the rate of drug discontinuation or dose reduction due to intolerance.
STATIN + PCSK9 INHIBITORS
The FOURIER trial was a randomized, double-blind, placebo-controlled trial of evolocumab vs. placebo in 27,564 patients with atherosclerotic ASCVD and an LDL-C level of 70 mg/dL or higher who were on statin therapy (200). Approximately 40% of the patients had diabetes (201). The primary end point was cardiovascular death, MI, stroke, hospitalization for unstable angina, or coronary revascularization and the key secondary end point was cardiovascular death, MI, or stroke. The median duration of follow-up was 2.2 years. Baseline LDL-C levels were 92mg/dL and evolocumab resulted in a 59% decrease in LDL-C levels (LDL-C level on treatment approximately 30mg/dL). Evolocumab treatment significantly reduced the risk of the primary end point (HR 0.85; 95% CI 0.79 to 0.92; P<0.001) and the key secondary end point (HR 0.80; 95% CI 0.73 to 0.88; P<0.001). The results were consistent across key subgroups, including the subgroup of patients in the lowest quartile for baseline LDL-C levels (median, 74 mg/dL). Of note, a similar decrease in cardiovascular events occurred in patients with diabetes treated with evolocumab and glycemic control was not altered (202). Further analysis has shown that in the small number of patients with a baseline LDL-C level less than 70mg/dL, evolocumab reduced cardiovascular events to a similar degree as in the patients with an LDL-C greater than 70mg/dL (203). Finally, the lower the on-treatment LDL-C levels (down to levels below 20mg/dL), the lower the cardiovascular event rate, suggesting that greater reductions in LDL-C levels will result in greater reductions in ASCVD (204).
The ODYSSEY trial was a multicenter, randomized, double-blind, placebo-controlled trial involving 18,924 patients who had an acute coronary syndrome 1 to 12 months earlier, an LDL-C level of at least 70 mg/dL, a non-HDL-C level of at least 100 mg/dL, or an Apo B level of at least 80 mg/dL while on high intensity statin therapy or the maximum tolerated statin dose (205). Approximately 29% of the patients had diabetes. Patients were randomly assigned to receive alirocumab 75 mg every 2 weeks or matching placebo. The dose of alirocumab was adjusted to target an LDL-C level of 25 to 50 mg/dL. The primary end point was a composite of death from coronary heart disease, nonfatal MI, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization. During the trial LDL-C levels in the placebo group was 93-103mg/dL while in the alirocumab group LDL-C levels were 40mg/dL at 4 months, 48mg/dL at 12 months, and 66mg/dL at 48 months (the increase with time was due to discontinuation of alirocumab or a decrease in dose). The primary endpoint was reduced by 15% in the alirocumab group (HR 0.85; 95% CI 0.78 to 0.93; P<0.001). In addition, total mortality was reduced by 15% in the alirocumab group (HR 0.85; 95% CI 0.73 to 0.98). The absolute benefit of alirocumab was greatest in patients with a baseline LDL-C level > than 100mg/dL. In patients with an LDL-C level > than 100mg/dL the number needed to treat with alirocumab to prevent an event was only 16. It should be noted that similar to the FOURIER trial the duration of this trial was very short (median follow-up 2.8 years) which may have minimized the beneficial effects. Additionally, because alirocumab 75mg every 2 weeks was stopped if the LDL-C level was < 15mg/dL on two consecutive measurements the beneficial effects may have been blunted (7.7% of patients randomized to alirocumab were switched to placebo).
It should be noted that that the duration of the PCSK9 outcome trials were relatively short and it is well recognized from previous statin trials that the beneficial effects of lowering LDL-C levels takes time with only modest effects observed during the first year of treatment. In the FOURIER trial the reduction of cardiovascular death, MI, or stroke was 16% during the first year but was 25% beyond 12 months. In the ODYSSEY trial the occurrence of cardiovascular events was similar in the alirocumab and placebo group during the first year of the study with benefits of alirocumab appearing after year one. Thus, the long-term benefits of treatment with a PCSK9 inhibitor may be greater than that observed during these relatively short-term studies.
Additional support for the benefits of further lowering of LDL-C levels with a PCSK9 inhibitor added to statin therapy is seen in the GLAGOV trial (206). This trial was a double-blind, placebo-controlled, randomized trial of evolocumab vs. placebo in 968 patients presenting for coronary angiography. Approximately 20-21% of the patients had diabetes. The primary efficacy measure was the change in percent atheroma volume (PAV) from baseline to week 78, measured by serial intravascular ultrasonography (IVUS) imaging. Secondary efficacy measures included change in normalized total atheroma volume (TAV) and percentage of patients demonstrating plaque regression. As expected, there was a marked decrease in LDL-C levels in the evolocumab group (Placebo 93mg/dL vs. evolocumab 37mg/dL; p<0.001). PAV increased 0.05% with placebo and decreased 0.95% with evolocumab (P < .001) while TAV decreased 0.9 mm3 with placebo and 5.8 mm3 with evolocumab (P < .001). There was a linear relationship between achieved LDL-C and change in PAV (i.e., the lower the LDL-C the greater the regression in atheroma volume down to an LDL-C of 20mg/dL). Additionally, evolocumab induced plaque regression in a greater percentage of patients than placebo (64.3% vs 47.3%; P < .001 for PAV and 61.5% vs 48.9%; P < .001 for TAV). The results in the patients with diabetes were similar to the non-diabetic patients.
Taken together these trials demonstrate that further lowering LDL-C levels with PCSK9 monoclonal antibodies in patients taking statins will have beneficial effects on ASCVD outcomes. A study of the effect of inclisiran on ASCVD endpoints is currently in progress.
The results of the ezetimibe and PCSK9 trials have several important implications. First, it demonstrates that combination therapy may have benefits above and beyond statin therapy alone. Second, it provides further support for the hypothesis that lowering LDL per se will reduce cardiovascular events. Third, it suggests that lowering LDL levels to much lower levels than usual will have significant benefits. These results have implications for determining goals of therapy.
STATINS + LOW DOSE OMEGA-3-FATTY ACIDS
ORIGIN was a double-blind study in 12,536 patients at high risk for ASCVD who had impaired fasting glucose, impaired glucose tolerance, or diabetes (207). Patients were randomized to receive a 1-gram capsule containing at least 900mg of ethyl esters of omega-3 fatty acids (EPA 465mg and DHA 375mg) or placebo for approximately 6 years. Greater than 50% of the patients were on statin therapy. The primary outcome was death from cardiovascular causes. TG levels were reduced by 14.5mg/dL in the group receiving omega-3-fatty acids compared to the placebo group (P<0.001), without a significant effect on other lipids. The incidence of the primary outcome was not significantly decreased among patients receiving omega-3-fatty acids as compared with those receiving placebo. The use of omega-3-fatty acids also had no significant effect on the rates of major vascular events, death from any cause, or death from arrhythmia.
A Study of Cardiovascular Events in Diabetes (ASCEND) was a randomized, placebo controlled, double blind trial of 1-gram omega-3-fattys acids (400mg EPA and 300mg DHA ethyl esters) vs. olive oil placebo in 15,480 patients with diabetes without a history of ASCVD (primary prevention trial) (208). Approximately 75% of patients were on statin therapy. The primary end point was serious vascular events (non-fatal MI, non-fatal stroke, transient ischemic attack, or vascular death). Total cholesterol, HDL-C, and non-HDL-C levels were not significantly altered by omega-3-fatty acid treatment (changes in TG levels were not reported). After a mean follow-up of 7.4 years the composite outcome of a serious vascular event or revascularization occurred in 882 patients (11.4%) on omega-3-fatty acids and 887 patients (11.5%) on placebo (rate ratio, 1.00; 95% CI, 0.91 to 1.09). Serious adverse events were similar in placebo and omega-3-fatty acid treated groups.
Taken together these studies indicate that low dose omega-3-fatty acids do not reduce cardiovascular events in patients with diabetes. Studies in non-diabetics have also found little effect of low dose omega-3-fatty acids on ASCVD (209).
STATINS + HIGH DOSE OMEGA-3-FATTY ACIDS
The Japan EPA Lipid Intervention Study (JELIS) was an open label non-placebo controlled study in patients on statin therapy with total cholesterol levels > 254mg/dL with (n= 3664) or without ASCVD (n=14,981) who were randomly assigned to be treated with 1800 mg of EPA (Vascepa) + statin (n=9326) or statin alone (n= 9319) with a 5 year follow-up (210). Approximately 16% of the patients had diabetes. The mean baseline TG level was 153mg/dL. The primary endpoint was any major coronary event, including sudden cardiac death, fatal and non-fatal MI, and other non-fatal events including unstable angina pectoris, angioplasty, stenting, or coronary artery bypass grafting. On treatment total cholesterol, LDL-C, and HDL-C levels were similar in the two groups but plasma TG were modestly decreased in the EPA treated group (5% decrease in EPA group compared to controls; p = 0.0001). In the EPA + statin group the primary endpoint occurred in 2.8% of the patients vs. 3.5% of the patients in the statin alone group (19% decrease; p = 0.011). Unstable angina and non-fatal coronary events were also significantly reduced in the EPA group but in this study sudden cardiac death and coronary death did not differ between groups. Unstable angina was the main component contributing to the primary endpoint and this is a more subjective endpoint than other endpoints such as a MI, stroke, or cardiovascular death. A subjective endpoint has the potential to be an unreliable endpoint in an open label study and is a major limitation of the JELIS Study. The reduction in events was similar in the subgroup of patients with diabetes. In patients with TG levels >150mg/dL and HDL-C levels < 40mg/dL there was a 53% decrease in events (211). In the EPA group, small increases in the occurrence of bleeding (1.1% vs. 0.6%, p=0.0006), gastrointestinal disturbance (3.8%% vs. 1.7%, p<0.0001) and skin abnormalities (1.7 vs. 0.7%, p<0.0001) were seen.
The Reduction of Cardiovascular Events with EPA – Intervention Trial (REDUCE-IT) was a randomized, double blind trial of 2 grams twice per day of EPA ethyl ester (icosapent ethyl) (Vascepa) vs. placebo (mineral oil) in 8,179 patients with hypertriglyceridemia (135mg/dL to 499mg/dL) and established ASCVD or high ASCVD risk (diabetes plus one risk factor) who were on stable statin therapy (212). Approximately 60% of the patients in this trial had diabetes. The primary end point was a composite of cardiovascular death, nonfatal MI, nonfatal stroke, coronary revascularization, or unstable angina. At baseline, the median LDL-C level was 75.0 mg/dL, HDL-C level was 40.0 mg/dL, and TG level was 216.0 mg/dL. The median change in TG level from baseline to 1 year was a decrease of 18.3% (−39.0 mg/dL) in the EPA group and an increase of 2.2% (4.5 mg/dL) in the placebo group. After a median of 4.9 years the primary end-point occurred in 17.2% of the patients in the EPA group vs. 22.0% of the patients in the placebo group (HR 0.75; 95% CI 0.68 to 0.83; P<0.001), indicating a 25% decrease in events. The beneficial effects were similar in patients with and without diabetes. The number needed to treat to avoid one primary end-point event was 21. The reduction in cardiovascular events was noted after approximately 2 years of EPA treatment. Additionally, the risk of cardiovascular death was decreased by 20% in the EPA group (HR 0.80; 95% CI, 0.66 to 0.98; P=0.03). The cardiovascular benefits of EPA were similar across baseline levels of TG (<150, ≥150 to <200, and ≥200 mg/dL). Moreover, the cardiovascular benefits of EPA appeared to occur irrespective of the attained TG level at 1 year (≥150 or <150 mg/dL), suggesting that the cardiovascular risk reduction was not associated with attainment of a normal TG levels. An increase in hospitalization for atrial fibrillation or flutter (3.1% vs. 2.1%, P=0.004) occurred in the EPA group. In addition, serious bleeding events occurred in 2.7% of the patients in the EPA group and in 2.1% in the placebo group (P=0.06). There were no fatal bleeding events in either group and the rates of hemorrhagic stroke, serious central nervous system bleeding, and serious gastrointestinal bleeding were not significantly higher in the EPA group.
These results demonstrate that EPA treatment reduces ASCVD events. Of note the reduction in TG levels is relatively modest and would not be expected to result in the magnitude of the decrease in ASCVD observed in the JELIS and REDUCE-IT trials. Other actions of EPA, such as decreasing platelet function, anti-inflammation, decreasing lipid oxidation, stabilizing membranes, etc. could account for or contribute to the reduction in cardiovascular events (213). It is likely that the beneficial effects of EPA seen in the JELIS and REDUCE-IT trials are multifactorial.
The Statin Residual Risk Reduction with Epanova in High Risk Patients with Hypertriglyceridemia (STRENGTH) trial was a randomized, placebo controlled, double blind trial of 4 grams per day of omega-3-fatty acids (Epanova) (carboxylic acid formulation of EPA and DHA) vs. placebo (corn oil) in 13,000 patients on statins with hypertriglyceridemia (180-500mg/dL), optimal LDL-C levels (< 100mg/dL or on maximal statin therapy), low HDL-C (<42mg/dL in men and < 47mg/dL in women), and either ASCVD or high risk for ASCVD (214). The primary outcome was major atherosclerotic cardiovascular events (cardiovascular death, MI, stroke, coronary revascularization or hospitalization for unstable angina). The primary end point occurred in 785 patients (12.0%) treated with omega-3 CA vs 795 (12.2%) treated with corn oil (HR, 0.99: [95% CI, 0.90-1.09]; P = .84) (215). Thus, in contrast to EPA alone this omega-3-fatty acid formulation failed to show benefits despite reducing TG levels (18% decrease) to a similar degree as in the REDUCE-IT trial.
Whether EPA has special properties that resulted in the reduction in cardiovascular events in the REDUCE-IT trial or there were flaws in the trial design (the use of mineral oil as the placebo) is uncertain and debated. It should be noted that in the REDUCE-IT trial LDL-C and non-HDL-C levels were increased by approximately 10% (LDL-C by approximately 9mg/dL and non-HDL-C by approximately 10mg/dL) in the mineral oil placebo group (212). Additionally, Apo B levels were increased by 7% (6mg/dL) by mineral oil (212). Finally, an increase in hsCRP (20-30%) and other biomarkers of atherosclerosis (oxidized LDL-C, IL-6, IL-1 beta, and lipoprotein-associated phospholipase A2) were noted in the mineral oil group (212,216). In the STRENGTH trial there were no differences in LDL-C, Non-HDL-C, HDL-C, Apo B, or hsCRP levels between the treated vs. placebo groups (215). Whether EPA has special properties compared to DHA leading to a reduction in cardiovascular events or the mineral oil placebo resulted in adverse changes increasing ASCVD in the placebo resulting in an artifactual decrease in the EPA group is debated (217,218). Ideally, another large randomized cardiovascular trial with EPA ethyl ester (icosapent ethyl) (Vascepa) using a placebo other than mineral oil would resolve this controversy.
CURRENT GUIDELINES FOR SERUM LIPIDS
There are several different guidelines for treating lipids in patients with diabetes. While they all focus on lowering LDL-C there are differences between the various guidelines.
American Diabetes Association Guidelines
The 2023 American Diabetes Association (ADA) recommends that adult patients with diabetes have their lipid profile determined at the time of diabetes diagnosis and at least every 5 years thereafter or more frequently if indicated (219). This profile includes total cholesterol, HDL-C, TG, and calculated LDL-C. A lipid panel should be obtained immediately prior to initiating statin therapy. Once a patient is on statin therapy testing should be carried out 4-12 weeks after initiating therapy and annually thereafter to monitor adherence and efficacy. Lifestyle modifications including a reduction in saturated fat, trans fat, and cholesterol intake, weight loss if indicated, an increase in omega-3-fatty acids, viscous fiber, and plant stanols /sterol intake, and increased physical activity is indicated in all patients with diabetes. A focus on a Mediterranean style diet or Dietary Approaches to Stop Hypertension (DASH) diet should be encouraged. In patients with elevated TG levels glycemic control is beneficial and dietary changes and lifestyle changes including weight loss and abstinence from alcohol should be undertaken. Secondary disorders and medications that raise TG levels should be evaluated. Optimize glycemic control to improve TG and HDL-C levels. The recommendations for lipid lowering therapy are shown in table 6. If one follows these recommendations almost all patients with diabetes over the age of 40 will be on statin therapy and many under the age of 40 will also be treated with statins. The addition of ezetimibe should be considered to further lower LDL-C levels in high-risk primary prevention patients. In very high-risk patients with ASCVD if the LDL-C level on statin therapy is greater than 70mg/dL the use of ezetimibe or a PCSK9 inhibitor should be considered. The use of fibrates or niacin with statins were generally not recommended as there is no evidence of benefit. However, in patients with ASCVD or other cardiovascular risk factors on a statin with controlled LDL-C but elevated TG levels (135-499mg/dL) the addition of icosapent ethyl can be considered. Finally, in patients with fasting TG levels greater than 500mg/dL an evaluation for secondary causes of hypertriglyceridemia should be initiated and consideration of drug therapy to reduce the risk of pancreatitis.
|
Table 6. ADA Recommendations for Lipid Lowering Therapy |
|---|
|
Primary Prevention |
|
Age 20-39: With additional risk factors may be reasonable to initiate statin therapy |
|
Age 40-75: Use moderate-intensity statin therapy* in addition to lifestyle therapy |
|
Age 40-75: If at higher cardiovascular risk, including those with one or more ASCVD risk factors, it is recommended to use high intensity statin therapy to reduce LDL cholesterol by >50% and to target an LDL-C <70 mg/dL |
|
Age 40-75: If at higher cardiovascular risk, especially those with multiple ASCVD risk factors and an LDL-C >70 mg/dL, it may be reasonable to add ezetimibe or a PCSK9 inhibitor to maximum tolerated statin therapy |
|
Age > 75: Initiating moderate intensity statin therapy is reasonable after discussion and in patient already on statin therapy it is reasonable to continue statin therapy |
|
Secondary Prevention |
|
All ages: High intensity statin therapy**/maximally tolerated stain |
|
For people with diabetes and ASCVD, treatment with high intensity statin therapy is recommended to target an LDL-C reduction of >50% and an LDL-C l goal of <55 mg/dL. Addition of ezetimibe or a PCSK9 inhibitor is recommended if this goal is not achieved |
*Moderate intensity statin- atorvastatin 10-20mg, rosuvastatin 5-10mg, simvastatin 20-40mg, pravastatin 40-80mg, lovastatin 40mg, Fluvastatin XL 80mg, pitavastatin 3-4mg.
**High Intensity statin- atorvastatin 40-80mg, rosuvastatin 20-40mg.
American College of Cardiology and American Heart Association Guidelines
The 2018 American College of Cardiology and American Heart Association (ACC/AHA) guidelines recommend the following (220). “In patients 40 to 75 years of age with diabetes mellitus and LDL-C ≥70 mg/dL (≥1.8 mmol/L), start moderate-intensity statin therapy without calculating 10-year ASCVD risk. In patients with diabetes mellitus at higher risk, especially those with multiple risk factors or those 50 to 75 years of age, it is reasonable to use a high-intensity statin to reduce the LDL-C level by ≥50%.” In patients with diabetes and ASCVD they recommend “In patients with clinical ASCVD, reduce LDL-C with high-intensity statin therapy or maximally tolerated statin therapy. The more LDL-C is reduced on statin therapy, the greater will be subsequent risk reduction. Use a maximally tolerated statin to lower LDLC levels by ≥50%. In very high-risk ASCVD, use an LDL-C threshold of 70 mg/dL (1.8 mmol/L) to consider addition of non-statins to statin therapy. Very high-risk includes a history of multiple major ASCVD events or 1 major ASCVD event and multiple high-risk conditions. In very high-risk ASCVD patients, it is reasonable to add ezetimibe to maximally tolerated statin therapy when the LDL-C level remains ≥70 mg/dL (≥1.8 mmol/L). In patients at very high risk whose LDL-C level remains ≥70 mg/dL (≥1.8 mmol/L) on maximally tolerated statin and ezetimibe therapy, adding a PCSK9 inhibitor is reasonable, although the long-term safety (>3 years) is uncertain and cost effectiveness is low at mid-2018 list prices.” With regards to testing they recommend “Assess adherence and percentage response to LDL-C–lowering medications and lifestyle changes with repeat lipid measurement 4 to 12 weeks after statin initiation or dose adjustment, repeated every 3 to 12 months as needed”. Finally, there are several diabetes specific risk enhancers that are independent of other risk factors that should be considered in deciding the risk of cardiovascular events in a patient with diabetes (Table 7).
|
Table 7. Diabetes Specific Risk Enhancers That are Independent of Other Risk Factors in Diabetes |
|---|
|
Long duration (≥10 years for type 2 diabetes mellitus or ≥20 years for type 1 diabetes mellitus Albuminuria ≥30 mcg of albumin/mg creatinine eGFR <60 mL/min/1.73 m2 Retinopathy Neuropathy ABI <0.9 |
ABI indicates ankle-brachial index
American Association of Clinical Endocrinologists/American College of Endocrinology Guidelines
The American Association of Clinical Endocrinologists and American College of Endocrinology guidelines consider individuals with T2DM to be at high, very high, or extreme risk for ASCVD (221,222). Patients with T1DM and a duration of diabetes of more than 15 years or two or more risk factors, poorly controlled A1c, or insulin resistance with metabolic syndrome should be considered to have an equivalent risk to patients with T2DM (221). The recommended treatment goals are shown in Table 8.
|
Table 8. ASCVD Risk Categories and Treatment Goals |
|||||
|---|---|---|---|---|---|
|
Risk Category |
Risk Factors/10-year risk |
LDL-C mg/dL |
Non-HDL-C mg/dL |
Apo B mg/dL |
TG mg/dL |
|
Extreme Risk |
Diabetes and clinical ASCVD |
<55 |
<80 |
<70 |
<150 |
|
Very High Risk |
Diabetes with one or more risk factors |
<70 |
<100 |
<80 |
<150 |
|
High Risk |
Diabetes and no other risk factors |
<100 |
<130 |
<90 |
<150 |
European Society of Cardiology and European Atherosclerosis Society Guidelines
The European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) has 2019 guidelines for the treatment of lipids in patients with diabetes (223). These guidelines classify patients with diabetes as very high risk, high risk, or moderate risk (table 9). The recommended goals of therapy based on risk classification are shown in table 10. As with other guidelines intensification of statin therapy should be considered before the introduction of combination therapy. If the goal is not reached, statin combination with ezetimibe should be considered next.
|
Table 9. ESC/EAS Classification of Risk in Patients with Diabetes |
|---|
|
Very High Risk- target organ damage, or at least three major risk factors, or early onset of T1DM of long duration (>20 years) |
|
High Risk- without target organ damage, with DM duration >10 years or another additional risk factor |
|
Moderate Risk- Young patients (T1DM <35 years; T2DM <50 years) with DM duration <10 years, without other risk factors. Calculated SCORE >1 % and <5% for 10-year risk of fatal CVD |
|
Table 10. ESC/EAS Goals of Therapy in Patients with Diabetes |
|||
|---|---|---|---|
|
|
LDL-C |
Non-HDL-C |
Apo B |
|
Very High Risk |
>50% reduction and <55mg/dL (<1.4mmol/L) |
<85mg/d; |
<65mg/dL |
|
High Risk |
>50% reduction and <70mg/dL (<1.8mmol/L) |
<100mg/dL |
<80mg/dL |
|
Moderate Risk |
<100mg/dL |
<130mg/dL |
<100mg/dL |
European Society of Cardiology
The ESC has updated their guidelines in 2023 (224). An important new recommendation is that in patients with T2DM without symptomatic ASCVD or severe target organ damage, it is recommended to estimate 10-year CVD risk using SCORE2-Diabetes (225). This has resulted in a new classification of risk in patients with T2DM (table 9). The LDL-C goals for each category are shown in table 11.
|
Table 11. Cardiovascular Risk Categories and Goals in Patients with Type 2 Diabetes |
||
|---|---|---|
|
Very high risk |
Clinically established ASCVD or Severe target organ damage or 10-year risk of CVD> 20% using SCORE2-Diabetes |
LDL-C < 55mg/dl Non-HDL-C <85mg/dL |
|
High risk* |
10-year risk of CVD 10% to < 20% using SCORE2-Diabetes |
LDL-C < 70mg/dL Non-HDL-C <100mg/dL |
|
Moderate risk* |
10-year risk of CVD 5% to <10% using SCORE2-Diabetes |
LDL-C < 100mg/dL |
|
Low Risk* |
10-year risk of CVD <5% using SCORE2-Diabetes |
no recommendations |
*Patients not meeting the very high-risk category.
Severe target organ damage is defined as eGFR <45 mL/min/1.73 m2 irrespective of albuminuria; or eGFR 45–59 mL/min/1.73 m2 and microalbuminuria (UACR 30–300 mg/g; stage A2); or proteinuria (UACR>300 mg/g; stage A3), or presence of microvascular disease in at least three different sites [e.g., microalbuminuria (stage A2) plus retinopathy plus neuropathy].
My Guideline Recommendations
Thus, different organizations have proposed somewhat different recommendations for the treatment of lipids in patients with diabetes. Despite these differences it is clear that the vast majority of patients with diabetes will need to be treated with statins regardless of which guidelines one elects to follow.
The approach I use is to combine these recommendations (Tables 12 and 13). In patients with diabetes who have pre-existing ASCVD I initiate intensive statin therapy. I prefer LDL or non-HDL-C goals over percent reduction goals. Given the extensive data showing that the lower the LDL-C the greater the reduction in cardiovascular events most secondary prevention patients would benefit from the addition of ezetimibe to maximize LDL-C lowering without markedly increasing costs (226). In patients with diabetes 40-75 years of age without pre-existing ASCVD I calculate the 10-year risk of developing ASCVD (http://www.cvriskcalculator.com/) and identify risk enhancing factors (Table 7) and other factors that increase risk that are not included in the calculator (for example family history, inflammatory disorders, etc.). I initiate intensive statin therapy if the 10-year risk is > 7.5% or if there are multiple risk factors/risk enhancers. I initiate moderate statin therapy if the risk is < 7.5% without multiple risk factors/enhancers. Four to twelve weeks after initiating statin therapy I obtain a lipid panel to determine if the LDL and non-HDL-C levels are at goal. In patients with pre-existing ASCVD or multiple risk factors/risk enhancers (i.e., very high-risk patients) my goal is an LDL-C < 55mg/dL and a non-HDL-C < 80mg/dL. In patients that are at high-risk the goal my goal is an LDL-C < 70mg/dL and a non-HDL-C < 100mg/dL. In patients with moderate risk an LDL-C goal of < 100mg/dL and a non-HDL c < 130mg/dL is appropriate. If the levels are not at goal, I first adjust the statin dose until the patient is taking the maximally tolerated statin dose and then consider adding additional medications. In patients with diabetes who are less than 40 years of age I initiate statin therapy if the patient has overt ASCVD, long standing diabetes, or risk factors/risk enhancers for ASCVD and the LDL and non-HDL-C levels are not at goal. In these younger patients I also calculate the life time risk of ASCVD events to start a discussion of beginning early therapy given the abundance of data indicating that initiating LDL-C lowering therapy early has great potential in markedly lowering ASCVD risk (226,227). In patients over 75 years of age with a reasonable life expectancy I begin moderate statin therapy and adjust based on response. When there is difficulty classifying a patient’s risk, I will obtain a coronary calcium score and use the score to help stratify the patient’s risk. In all cases the benefits and risks of lipid lowering therapy needs to be discussed with patients and the patient’s personnel preferences taken into account.
|
Table 12. ASCVD Risk Categories and Treatment Goals |
|||
|---|---|---|---|
|
Risk Category |
Risk Factors/10-year risk |
LDL-C mg/dL |
Non-HDL-C mg/dL |
|
Very High Risk |
Diabetes and clinical ASCVD, multiple risk factors, 10-year risk > 20% |
<55 |
<85 |
|
High Risk |
Diabetes with one or more risk factors, 10-year risk >7.5% to <20% |
<70 |
<100 |
|
Moderate Risk |
Diabetes and no other risk factors. 10-year risk <7.5% |
<100 |
<130 |
|
Table 13. Drug Therapy According to Risk Category that is Typically Required |
|
|---|---|
|
Very High Risk |
Intensive statin therapy + ezetimibe. Add PCSK9 if not close to goal |
|
High Risk |
Intensive statin therapy. Add ezetimibe if not at goal |
|
Moderate Risk |
Moderate statin therapy. Increase to intensive statin therapy if not at goal |
TREATMENT OF LIPID ABNORMALITIES IN PATIENT WITH DIABETES
Life Style Changes and Weight Loss
Initial treatment of lipid disorders should focus on lifestyle changes. There is little debate that exercise is beneficial and that all patients with diabetes should, if possible, exercise for at least 150 minutes per week (for example 30 minutes 5 times per week). Exercise will decrease serum TG levels and increase HDL-C levels (an increase in HDL-C requires vigorous exercise) (124). Exercise increases fitness and improves insulin resistance even with limited weight loss; reductions in obesity are even more beneficial. It should be noted that many patients with diabetes may have substantial barriers to participating in exercise programs, such as comorbidities that limit exercise tolerance, risk of hypoglycemia, and presence of microvascular complications (visual impairment, neuropathy) that make exercise difficult.
Diet is debated to a greater extent and for detailed information on nutrition therapy for adults with diabetes see the consensus report by the American Diabetes Association (228). Everyone agrees that weight loss in obese patients is essential (124). But how this can be achieved is hotly debated with many different "experts" advocating different dietary approaches. The wide diversity of approach is likely due to the failure of any approach to be effective in the long term for the majority of obese patients with diabetes. If successful, weight loss will decrease serum TG levels, increase HDL-C levels, and modestly reduce LDL-C (124,229). To reduce LDL-C levels, it is important that the diet decrease saturated fat, trans fatty acids, and cholesterol intake. Increasing soluble fiber is also helpful.
It is debated whether a low fat, high complex carbohydrate diets vs. a high monounsaturated fat diet is ideal for obese patients with diabetes (124). One can find "experts" in favor of either of these approaches and there are pros and cons to each approach. It is essential to recognize that both approaches reduce simple sugars, saturated fat, trans fatty acids, and cholesterol intake. The high complex carbohydrate diet will increase serum TG levels in some patients and if the amount of fat in the diet is markedly reduced serum HDL-C levels may decrease. In obese patients, it has been postulated that a diet high in monounsaturated fats, because of the increase in caloric density, will lead to an increase in weight gain. Both diets reduce saturated fat and cholesterol intake that will result in reductions in LDL-C levels. Additionally, both diets also reduce trans-fatty acid intake, which will have a beneficial effect on LDL and HDL-C levels and simple sugars, which will have a beneficial effect on TG levels. Very high levels of TG (>1000mg/dL), require diets that are very low in fat.
The available data do not indicate that any particular diet is best for inducing weight loss and it is essential to adapt the diet to fit the food preferences of the patient. Ultimately no weight loss diet will be successful if the patient cannot follow the diet for the long term and therefore the diet needs to be tailored to the specific preferences of the patient. For more detailed information on the effect of diet on lipid and lipoprotein levels and cardiovascular disease see the Endotext chapter “The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels” (229).
While it is widely accepted that lifestyle changes will decrease ASCVD events it should be recognized that the Look Ahead trial failed to demonstrate a reduction in ASCVD events (230). In this trial, over 5,000 overweight or obese patients with T2DM were randomized to either an intensive lifestyle intervention group that promoted weight loss through decreased caloric intake and increased physical activity or to a group that received diabetes support and education (control group). After a median follow-up of 9.6 years there was no difference in cardiovascular events (hazard ratio in the intervention group, 0.95; 95% CI 0.83 to 1.09; P=0.51). A major limitation of this study was that while the weight difference between groups was impressive during the first year of the trial, over time the differences greatly narrowed such that at the end of the trial the intensive group had a 6.0% weight loss while the control group had a 3.5% weight loss. This very modest weight difference demonstrates the difficulty in sustaining long term lifestyle changes. Thus, while weight loss and diet therapy are likely to be beneficial in reducing cardiovascular events, in clinical practice they are seldom sufficient because long-term life style changes are very difficult for most patients to maintain.
In contrast to the failure of lifestyle therapy in the Look Ahead trial to reduce cardiovascular events, the PREDIMED trial employing a Mediterranean diet (increased monounsaturated fats) did reduce the incidence of major ASCVD events (231,232). In this multicenter trial center trial, carried out in Spain, over 7,000 patients at high risk for developing ASCVD were randomized to three diets (primary prevention trial). A Mediterranean diet supplemented with extra-virgin olive oil, a Mediterranean diet supplemented with mixed nuts, or a control diet. Approximately 50% of the patients in this trial had T2DM. In the patients assigned to the Mediterranean diets there was 29% decrease in the primary end point (MI, stroke, and death from ASCVD). Subgroup analysis demonstrated that the Mediterranean diet was equally beneficial in patients with and without diabetes. The Mediterranean diet resulted in only a small but significant increase in HDL-C levels and a small decrease in both LDL-C and TG levels, suggesting that the beneficial effects were not mediated by changes in lipids (233).
The CORDIOPREV study was a single center randomized trial that compared a Mediterranean diet to a low-fat diet in 1,002 patients with ASCVD (234). Approximately 50% of the patients had diabetes. The Mediterranean diet contained a minimum of 35% of the calories as fat (22% monounsaturated fatty acids, 6% polyunsaturated fatty acids, and <10% saturated fat), 15% proteins, and a maximum of 50% carbohydrates while the low-fat diet contained less than 30% of total fat (<10% saturated fat, 12–14% monounsaturated fatty acids, and 6–8% polyunsaturated fatty acids), 15% protein, and a minimum of 55% carbohydrates. The risk of an ASCVD event was reduced by approximately 25-30% in the Mediterranean diet group. Whether these diets differed in their effects on fasting lipid levels is unknown.
Finally, another secondary prevention trial of a Mediterranean diet has also demonstrated a reduction in cardiovascular events. The Lyon Diet Heart Study randomized 584 patients who had a MI within 6 months to a Mediterranean type diet vs usual diet (235,236). There was a marked reduction in events in the group of patients randomized to the Mediterranean diet (cardiac death and nonfatal MI rate was 4.07 per 100 patient years in the control diet vs. 1.24 in the Mediterranean diet; p<0.0001). Unfortunately, there is no indication of the number of patients with diabetes in the Lyon Diet Heart Study or whether patients with diabetes responded similar to the entire group. Lipid levels were similar in both groups in this trial (235).
The results of these three randomized trials indicate that we should be encouraging our patients to follow a Mediterranean type diet. It is likely that the beneficial effects of the Mediterranean diet on ASCVD is mediated by multiple mechanisms with alterations in lipid levels making only a minor contribution.
For additional information on the effect of diets on lipid levels and ASCVD see the chapter entitled “The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels” in the Lipids and Lipoproteins section of Endotext (229).
Bariatric surgery can have profound effects on weight and can result in marked improvements in lipid profiles with a decrease in TG and LDL-C and an increase in HDL-C (124,229) Additionally, observational studies have shown a decrease in cardiovascular events following bariatric surgery in patients with and without diabetes (237-241). For additional information see the chapter entitled “Obesity and Dyslipidemia” (124).
Ethanol and simple sugars, in particular fructose, increase serum TG levels in susceptible patients. In patients with hypertriglyceridemia efforts should be made to reduce the intake of ethanol, simple sugars, and fructose (229).
Lastly, in the past some "experts" advocated the addition of fish oil supplements to reduce cardiovascular events. However, both the Origin Trial and the ASCEND Trial did not demonstrate that fish oil supplements were beneficial in patients with T2DM or patients at high risk for the development of T2DM (207,208). It should be recognized that higher doses of fish oil are required to lower serum triglyceride levels (~ 3-4 grams of DHA/EPA per day) and are useful in treating patients with high TG levels (242). Most studies of fish oil supplements in patients with diabetes have demonstrated that this is a safe approach and that worsening of glycemic control does not occur in patients with diabetes treated with fish oil supplements (242). Additionally, in some patient's high dose fish oil increases LDL-C levels, particularly when serum TG levels are very high (242). For additional information on fish oil see the chapter on Triglyceride Lowering Drugs (209).
Drug Therapy
The effect of statins, fibrates, niacin, ezetimibe, omega-3-fatty acids, bile acid sequestrants, bempedoic acid, and PCSK9 inhibitors on lipid levels in patients with diabetes is virtually identical to that seen in non-diabetic patients (Table 14). Below we will highlight issues particularly relevant to the use of these drugs in patients with diabetes. For detailed information on lipid lowering drugs see the chapters on Triglyceride Lowering Drugs and Cholesterol Lowering Drugs (141,209).
STATINS
Statins are easy to use and generally well tolerated by patients with diabetes. However, statins can adversely affect glucose homeostasis. In patients without diabetes the risk of developing diabetes is increased by approximately 10% with higher doses of statin causing a greater risk than more moderate doses (243,244). The mechanism for this adverse effect is unknown but older, obese patients with higher baseline glucose levels are at greatest risk. In patients with diabetes, an analysis of 9 studies with over 9,000 patients with diabetes reported that the patients randomized to statin therapy had a 0.12% higher HbA1c than the placebo group indicating that statin therapy is associated with only a very small increase in HbA1c levels in patients with diabetes, which is unlikely to be clinically significant (245). Individual studies such as CARDS and the Heart Protection Study have also shown only a very modest effect of statins on HbA1c levels in patients with diabetes (163,166,246). Muscle symptoms occur in patients with diabetes similar to what is observed in patients without diabetes.
EZETIMIBE
Ezetimibe is easy to use and generally well tolerated by patients with diabetes. Ezetimibe does not appear to increase the risk of new onset diabetes (199,247,248).
FIBRATES
Fibrates are easy to use and generally well tolerated by patients with diabetes. When combining fibrates with statin therapy it is best to use fenofibrate as the risk of inducing myositis is much less than when statins are used in combination with gemfibrozil, which can inhibit statin metabolism (249). In the ACCORD-LIPID Trial the incidence of muscle disorders was not increased in the statin + fenofibrate group compared to statin alone (189). The dose of fenofibrate needs to be adjusted in patients with renal disease and fenofibrate itself can induce a reversible increase in serum creatinine levels. It should be noted that marked reductions in HDL-C levels can occur in some patients treated with both fenofibrate and a TZD (250).
Diabetic Retinopathy
Fenofibrate has been shown to have beneficial effects on diabetic eye disease. The FIELD study, described earlier, was a randomized trial of fenofibrate vs. placebo in patients with T2DM. Laser treatment for retinopathy was significantly lower in the fenofibrate group than in the placebo group (3.4% patients on fenofibrate vs 4.9% on placebo; p=0.0002) (191). Fenofibrate therapy reduced the need for laser therapy to a similar extent for maculopathy (31% decrease) and for proliferative retinopathy (30% decrease). In the ophthalmology sub-study (n=1012), the primary endpoint of 2-step progression of retinopathy grade did not differ significantly between the fenofibrate and control groups (9.6% patients on fenofibrate vs 12.3% on placebo; p=0.19). In patients without pre-existing retinopathy there was no difference in progression (11.4% vs 11.7%; p=0.87). However, in patients with pre-existing retinopathy, significantly fewer patients on fenofibrate had a 2-step progression than did those on placebo (3.1% patients vs 14.6%; p=0.004). A composite endpoint of 2-step progression of retinopathy grade, macular edema, or laser treatments was significantly reduced in the fenofibrate group (HR 0.66, 95% CI 0.47-0.94; p=0.022).
In the ACCORD Study a subgroup of participants was evaluated for the progression of diabetic retinopathy by 3 or more steps on the Early Treatment Diabetic Retinopathy Study Severity Scale or the development of diabetic retinopathy necessitating laser photocoagulation or vitrectomy over a four-year period (190). At 4 years, the rates of progression of diabetic retinopathy were 6.5% with fenofibrate therapy (n=806) vs. 10.2% with placebo (n=787) (adjusted odds ratio, 0.60; 95% CI, 0.42 to 0.87; P = 0.006). Of note, this reduction in the progression of diabetic retinopathy was of a similar magnitude as intensive glycemic treatment vs. standard therapy.
Taken together these results indicate that fibrates have beneficial effects on the progression of diabetic retinopathy. The mechanisms by which fibrates decrease diabetic retinopathy are unknown.
Diabetic Nephropathy
The Diabetes Atherosclerosis Intervention Study (DAIS) evaluated the effect of fenofibrate therapy (n= 155) vs. placebo (n=159) on changes in urinary albumin excretion in patients with T2DM (251). Fenofibrate significantly reduced the worsening of albumin excretion (fenofibrate 8% vs. placebo 18%; P < 0.05). This effect was primarily due to reduced progression from normal albumin excretion to microalbuminuria (fenofibrate 3% vs. 18% placebo; P < 0.001).
In the FIELD trial, fenofibrate reduced urine albumin/creatinine ratio by 24% vs 11% in placebo group (p < 0.001), with 14% less progression and 18% more albuminuria regression (p < 0.001) in the fenofibrate group than in participants on placebo (252). As expected, fenofibrate therapy acutely increased plasma creatinine levels and decreased eGFR but over the long term, the increase in plasma creatinine was decreased in the fenofibrate group compared to the placebo group (14% decrease; p=0.01). Similarly, there was a slower annual decrease in eGFR in the fenofibrate group (1.19 vs 2.03 mL/min/1.73m2 annually, p < 0.001). The effect of fenofibrate on kidney function was greater in those with higher TG and lower HDL levels. End-stage renal disease, dialysis, renal transplant, and renal death were similar in the fenofibrate and placebo groups, but the incidence was low.
In the ACCORD-LIPID trial the post-randomization incidence of microalbuminuria was 38.2% in the fenofibrate group and 41.6% in the placebo group (p=0.01) and post-randomization incidence of macroalbumuria was 10.5% in the fibrate group and 12.3% in the placebo group (p=0.04) indicating a modest reduction in the development of proteinuria in patients treated with fenofibrate (189). There was no significant difference in the incidence of end-stage renal disease or need for dialysis between the fenofibrate group and the placebo group.
These studies suggest that fibrates may have a beneficial effect on diabetic kidney disease. One should recognize that reducing proteinuria is a surrogate marker and may not indicate a reduction in the development of end stage renal disease. The mechanisms accounting for decreased in proteinuria are unknown.
Amputations
In the FIELD study the risks of first amputation were decreased by 36% (p=0.02) and minor amputation events without known large-vessel disease by 47% (p=0.027) in the fenofibrate treated group (253). The reduction in amputations was independent of glucose control or dyslipidemia. No difference between the risks of major amputations was seen in the placebo and fenofibrate groups. The basis for this reduction in amputations is unknown.
Do Fibrates have an Independent Effect on Microvascular Disease?
Multiple studies cited above have now shown that fenofibrate decreases retinopathy, nephropathy, and amputation in the absence of large vessel disease. The effects are independent of blood glucose control. Given that there also was no effect of fenofibrate on cardiovascular (macrovascular) disease, these results may suggest that fenofibrate has an independent effect on microvascular disease. Further studies are warranted, but these results should be taken into account when considering treatment of marked hypertriglyceridemia in patients with diabetes.
BILE ACID SEQUESTRANTS
Bile acid sequestrants are relatively difficult to take due to GI toxicity (mainly constipation) (141). Diabetic subjects have an increased prevalence of constipation, which may be exacerbated by the use of bile acid sequestrants. On the other hand, in diabetic patients with diarrhea, the use of bile acid sequestrants may be advantageous. Bile acid sequestrants may also increase serum TG levels, which can be a problem in patients with diabetes who are already hypertriglyceridemic (141). An additional difficulty in using bile acid sequestrants is their potential for binding other drugs (141). Many drugs should be taken either two hours before or four hours after taking bile acid sequestrants to avoid the potential of decreased drug absorption. Patients with diabetes are frequently on multiple drugs for glycemic control, hypertension, etc., and it can sometimes be difficult to time the ingestion of bile resin sequestrants to avoid these other drugs. Colesevelam (Welchol) is a bile acid sequestrant that comes in pill, powder, or chewable bars and causes fewer side effects and has fewer interactions with other drugs than other preparations (254). The usual dose is 3.75 grams per day and can be given as tablets (take 6 tablets once daily or 3 tablets twice daily), oral suspension (take one packet once daily), or chewable bars (take one bar once daily). Of particular note is that a number of studies have shown that colesevelam improves glycemic control in patients with diabetes resulting in an approximately 0.5% decrease in A1c levels (255).
NIACIN
Niacin is well known to cause skin flushing and itching and GI upset (256). Additionally, niacin reduces insulin sensitivity (i.e., causes insulin resistance), which can worsen glycemic control (256). Studies have shown that niacin is usually well tolerated in diabetic subjects who are in good glycemic control (257,258). In patients with poor glycemic control, niacin is more likely to adversely impact glucose levels. In the HPS2-Thrive trial, niacin therapy significantly worsened glycemic control in patients with diabetes and induced new onset diabetes in 1.3% of subjects that were non-diabetic (195). High doses of niacin are more likely to adversely affect glycemic control. Niacin can also increase serum uric acid levels and induce gout, both of which are already common in obese patients with T2DM (256). Additionally recent trials have reported an increased incidence of infection and bleeding with niacin therapy (256). However, niacin is the most effective drug in increasing HDL-C levels, which are frequently low in patients with diabetes.
OMEGA-3-FATTY ACIDS
A Cochrane review of fish oil in patients with diabetes have demonstrated that this is a safe approach and does not result in worsening of glycemic control in patients with diabetes (242). Fish oil effectively lowers TG levels but, in some patients, particularly those with significant hypertriglyceridemia, high dose fish oil increases LDL-C levels (242). It should be noted that fish oil products that contain just EPA (Vascepa) do not adversely affect LDL-C levels (259). When using fish oil to lower serum TG levels it is important to recognize that one is aiming to provide 3-4 grams of DHA/EPA per day. The quantity of these active omega-3-fatty acids can vary greatly from product to product. Prescription fish oil products contain large amounts of these active ingredients whereas the amount of DHA/EPA in food supplements can vary greatly and in some products levels are very low. Additionally, while prescription omega-3-fatty acid preparations have high levels of quality control, omega-3-fish oil food supplements may have contaminants and the dosage may not be precisely controlled.
PCSK9 INHIBITORS
The beneficial effects of PCSK9 inhibitors in patients with diabetes is similar to what is observed in non-diabetic patients. Additionally, except for local reactions at the injection sites PCSK9 inhibitors do not seem to cause major side effects. PCSK9 inhibitors do not appear to increase the risk of developing new-onset diabetes (260,261).
BEMPEDOIC ACID
The effect of bempedoic acid on LDL-C levels in patients with diabetes are similar to the decreases seen on non-diabetics. Patients with T2DM often have elevated uric acid levels and an increased risk of gouty attacks and a major side effect of bempedoic acid is elevating uric acid levels (141). In clinical trials, 26% of bempedoic acid-treated patients with normal baseline uric acid values experienced hyperuricemia one or more times versus 9.5% in the placebo group. Elevations in blood uric acid levels may lead to the development of gout and gout was reported in 1.5% of patients treated with bempedoic acid vs. 0.4% of patients treated with placebo. The risk for gout attacks were higher in patients with a prior history of gout (11.2% for bempedoic acid treatment vs. 1.7% in the placebo group). In patients with no prior history of gout only 1% of patients treated with bempedoic acid and 0.3% of the placebo group had a gouty attack.
In a meta-analysis, bempedoic acid therapy was associated with a decrease in the onset of diabetes and worsening of diabetes (RR 0.65, p = 0.03) (7/100 vs 6.4/100 patient years) (262). In a study focusing solely on the development of new onset diabetes it was reported that new-onset diabetes/hyperglycemia occurred less frequently with bempedoic acid vs placebo (263). In the bempedoic acid cardiovascular outcome trial (CLEAR Outcomes) the development of diabetes and worsening of pre-existing diabetes was similar in the bempedoic acid and placebo groups (187).
|
Table 14. Effect of Lipid Lowering Drugs |
|||
|---|---|---|---|
|
|
LDL-C |
HDL-C |
TG |
|
Statins |
↓ 20-60% |
↑ 5-15% |
↓ 0-35%* |
|
Bile acid sequestrants |
↓ 10-30% |
↑ 0-10% |
↑ 0-10%** |
|
Fibrates |
↓ 0-15%*** |
↑ 5-15% |
↓ 20-50% |
|
Niacin |
↓ 10-25% |
↑ 10-30% |
↓ 20-50% |
|
Ezetimibe |
↓ 15-25% |
↑ 1-3% |
↓ 10-20% |
|
PCSK9 Inhibitors |
↓ 50-60% |
↑ 5-15% |
↓ 5-20% |
|
Bempedoic Acid |
↓ 15-25% |
↓ 5-6% |
No change |
|
High Dose Fish Oil |
↑ 0- 50%*** |
↑ 4- 9% |
↓ 20- 50%* |
*Patients with elevated TG have largest decrease
** In patients with high TG may cause marked increase
*** In patients with high TG may increase LDL
Therapeutic Approach
FIRST PRIORITY- LDL-C
The first priority in treating lipid disorders in patients with diabetes is to lower the LDL-C levels to goal, unless TG are markedly elevated (> 500- 1000mg/dL), which increases the risk of pancreatitis. LDL-C is the first priority because the database linking lowering LDL-C with reducing ASCVD is extremely strong and we now have the ability to markedly decrease LDL-C levels. Dietary therapy is the initial step but, in almost all patients, will not be sufficient to achieve the LDL-C goals. If patients are willing and able to make major changes in their diet it is possible to achieve significant reductions in LDL-C levels but this seldom occurs in clinical practice (264).
Statins are the first-choice drugs to lower LDL-C levels and the vast majority of diabetic patients will require statin therapy. There are several statins currently available in the US and they are available as generic drugs and therefore relatively inexpensive. The particular statin used may be driven by price, ability to lower LDL-C levels, and potential drug interactions. Patients with ASCVD (secondary prevention patients) should be started on intensive statin therapy (atorvastatin 40-80mg per day or rosuvastatin 20-40mg per day). Given the extensive data showing that the lower the LDL-C the greater the reduction in ASCVD events most secondary prevention patients would benefit from the addition of ezetimibe to maximize LDL-C lowering. Ezetimibe is now a generic drug and therefore this strategy will not markedly increase costs. Similarly, primary prevention patients who are at high risk for cardiovascular events will also benefit from the use of high intensity statin therapy in combination with ezetimibe. Primary prevention patients at moderate risk can be started on moderate intensity statin therapy.
If a patient is unable to tolerate statins or statins as monotherapy are not sufficient to lower LDL-C to goal the second-choice drug is either ezetimibe or a PCSK9 inhibitor. Ezetimibe can be added to any statin. PCSK9 inhibitors can also be added to any statin and are the drug of choice if a large decrease in LDL-C is required to reach goal (PCSK9 inhibitors will lower LDL-C levels by 50-60% when added to a statin, whereas ezetimibe will only lower LDL-C by approximately 20%). Bile acid sequestrants and bempedoic acid are alternatives with the use of a bile acid sequestrant particularly useful if a reduction in A1c level is also needed. It should be noted that in statin intolerant patients with ASCVD or at high risk (approximately 45% with diabetes), bempedoic acid has been shown to reduce cardiovascular events by 13% (HR 0.87; 95% CI 0.79 to 0.96; P = 0.004) (187). Ezetimibe, PCSK9 inhibitors, bempedoic acid, and bile acid sequestrants additively lower LDL-C levels when used in combination with a statin, because these drugs increase hepatic LDL receptor levels by different mechanisms, thereby resulting in a reduction in serum LDL-C levels (141). Niacin and the fibrates also lower LDL-C levels but are not usually employed to lower LDL-C levels.
SECOND PRIORITY- NON-HDL-C
The second priority should be non-HDL-C (non-HDL-C = total cholesterol – HDL-C), which is particularly important in patients with elevated TG levels (>150mg/dL). Non-HDL-C is a measure of all the pro-atherogenic apolipoprotein B containing particles. Numerous studies have shown that non-HDL-C is a strong risk factor for the development of ASCVD (265). The non-HDL-C goals are approximately 30mg/dL greater than the LDL-C goals. For example, if the LDL goal is <100mg/dL then the non-HDL-C goal would be <130mg/dL. Drugs that reduce either LDL-C or TG levels will reduce non-HDL-C levels. To lower TG levels initial therapy should focus on glycemic control and lifestyle changes including weight loss if indicated and a decrease in simple sugars and ethanol intake. Additionally, if possible, discontinue medications that increase triglyceride levels. As discussed above, studies with the omega-3-fatty acid icosapent ethyl (EPA; Vascepa) added to statin therapy have reduced the risk of cardiovascular events. The National Lipid Association has recommended “that for patients aged ≥45 years with clinical ASCVD, or aged ≥50 years with diabetes mellitus requiring medication plus ≥1 additional risk factor, with fasting TGs 135 to 499 mg/dL on high-intensity or maximally tolerated statin therapy (±ezetimibe), treatment with icosapent ethyl is recommended for ASCVD risk reduction” (266). As noted earlier in this chapter there is controversy regarding the benefits of icosapent ethyl on cardiovascular outcomes with some experts interpreting the beneficial results of the REDUCE-IT trial as being due to the adverse effects of the mineral oil placebo (217).
VERY HIGH TG
Patients with very high TG levels (> 500-1000 mg/dL) are at risk of pancreatitis and therefore lifestyle interventions including diet, exercise, and weight loss if indicated should be initiated early. Treatment is a low-fat diet and glycemic control. Additionally, decreasing simple sugars and avoiding alcohol is beneficial. When the TG levels are very elevated (> 1000mg/dL) a very low-fat diet (5-20% of calories as fat) should be the primary treatment until the TG levels decrease to < 1000mg/dL. Treating secondary disorders that raise TG levels and when possible, stopping drugs that increase TG levels is essential. If the TG levels remain above 500mg/dL the addition of fenofibrate or omega-3-fatty acids is indicated.
LOW HDL-C
While there is strong epidemiologic data linking low HDL-C levels with ASCVD there is no clinical trials demonstrating that increasing HDL-C levels reduce ASCVD. Thus, the use of drugs such as niacin to raise HDL-C levels is not recommended.
CONCLUSION
Patients with diabetes, particularly T2DM, often have dyslipidemia. Modern therapy of patients with diabetes demands that we aggressively treat lipids to reduce the high risk of ASCVD in this susceptible population and in those with very high TG to reduce the risk of pancreatitis.
REFERENCES
- Milicevic Z, Raz I, Beattie SD, Campaigne BN, Sarwat S, Gromniak E, Kowalska I, Galic E, Tan M, Hanefeld M. Natural history of cardiovascular disease in patients with diabetes: role of hyperglycemia. Diabetes Care 2008; 31 Suppl 2:S155-160
- Feingold KR, Siperstein MD. Diabetic vascular disease. Adv Intern Med 1986; 31:309-340
- Regensteiner JG, Golden S, Huebschmann AG, Barrett-Connor E, Chang AY, Chyun D, Fox CS, Kim C, Mehta N, Reckelhoff JF, Reusch JE, Rexrode KM, Sumner AE, Welty FK, Wenger NK, Anton B. Sex Differences in the Cardiovascular Consequences of Diabetes Mellitus: A Scientific Statement From the American Heart Association. Circulation 2015; 132:2424-2447
- Fox CS, Golden SH, Anderson C, Bray GA, Burke LE, de Boer IH, Deedwania P, Eckel RH, Ershow AG, Fradkin J, Inzucchi SE, Kosiborod M, Nelson RG, Patel MJ, Pignone M, Quinn L, Schauer PR, Selvin E, Vafiadis DK. Update on Prevention of Cardiovascular Disease in Adults With Type 2 Diabetes Mellitus in Light of Recent Evidence: A Scientific Statement From the American Heart Association and the American Diabetes Association. Diabetes Care 2015; 38:1777-1803
- Low Wang CC, Hess CN, Hiatt WR, Goldfine AB. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus - Mechanisms, Management, and Clinical Considerations. Circulation 2016; 133:2459-2502
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979; 241:2035-2038
- Members Writing Group, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive Summary: Heart Disease and Stroke Statistics--2016 Update: A Report From the American Heart Association. Circulation 2016; 133:447-454
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229-234
- Evans JM, Wang J, Morris AD. Comparison of cardiovascular risk between patients with type 2 diabetes and those who had had a myocardial infarction: cross sectional and cohort studies. BMJ 2002; 324:939-942
- Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Sattar N. Impact of diabetes on cardiovascular disease risk and all-cause mortality in older men: influence of age at onset, diabetes duration, and established and novel risk factors. Arch Intern Med 2011; 171:404-410
- Howard BV, Best LG, Galloway JM, Howard WJ, Jones K, Lee ET, Ratner RE, Resnick HE, Devereux RB. Coronary heart disease risk equivalence in diabetes depends on concomitant risk factors. Diabetes Care 2006; 29:391-397
- Rawshani A, Rawshani A, Franzen S, Sattar N, Eliasson B, Svensson AM, Zethelius B, Miftaraj M, McGuire DK, Rosengren A, Gudbjornsdottir S. Risk Factors, Mortality, and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2018; 379:633-644
- Lind M, Svensson AM, Kosiborod M, Gudbjornsdottir S, Pivodic A, Wedel H, Dahlqvist S, Clements M, Rosengren A. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med 2014; 371:1972-1982
- de Ferranti SD, de Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, Magge SN, Marx N, McGuire DK, Orchard TJ, Zinman B, Eckel RH. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Diabetes Care 2014; 37:2843-2863
- Maahs DM, Daniels SR, de Ferranti SD, Dichek HL, Flynn J, Goldstein BI, Kelly AS, Nadeau KJ, Martyn-Nemeth P, Osganian SK, Quinn L, Shah AS, Urbina E. Cardiovascular disease risk factors in youth with diabetes mellitus: a scientific statement from the American Heart Association. Circulation 2014; 130:1532-1558
- Huxley RR, Peters SA, Mishra GD, Woodward M. Risk of all-cause mortality and vascular events in women versus men with type 1 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 2015; 3:198-206
- Rawshani A, Sattar N, Franzen S, Rawshani A, Hattersley AT, Svensson AM, Eliasson B, Gudbjornsdottir S. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study. Lancet 2018; 392:477-486
- Chillaron JJ, Flores Le-Roux JA, Benaiges D, Pedro-Botet J. Type 1 diabetes, metabolic syndrome and cardiovascular risk. Metabolism 2014; 63:181-187
- Constantino MI, Molyneaux L, Limacher-Gisler F, Al-Saeed A, Luo C, Wu T, Twigg SM, Yue DK, Wong J. Long-term complications and mortality in young-onset diabetes: type 2 diabetes is more hazardous and lethal than type 1 diabetes. Diabetes Care 2013; 36:3863-3869
- Preis SR, Hwang SJ, Coady S, Pencina MJ, D'Agostino RB, Sr., Savage PJ, Levy D, Fox CS. Trends in all-cause and cardiovascular disease mortality among women and men with and without diabetes mellitus in the Framingham Heart Study, 1950 to 2005. Circulation 2009; 119:1728-1735
- Matuleviciene-Anangen V, Rosengren A, Svensson AM, Pivodic A, Gudbjornsdottir S, Wedel H, Kosiborod M, Haraldsson B, Lind M. Glycaemic control and excess risk of major coronary events in persons with type 1 diabetes. Heart 2017; 103:1687-1695
- Miller RG, Orchard TJ, Costacou T. 30-Year Cardiovascular Disease in Type 1 Diabetes: Risk and Risk Factors Differ by Long-term Patterns of Glycemic Control. Diabetes Care 2022; 45:142-150
- Abraira C, Colwell JA, Nuttall FQ, Sawin CT, Nagel NJ, Comstock JP, Emanuele NV, Levin SR, Henderson W, Lee HS. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care1995; 18:1113-1123
- Goldner MG, Knatterud GL, Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. 3. Clinical implications of UGDP results. JAMA 1971; 218:1400-1410
- Meinert CL, Knatterud GL, Prout TE, Klimt CR. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. II. Mortality results. Diabetes 1970; 19:Suppl:789-830
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977-986
- Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, Kojima Y, Furuyoshi N, Shichiri M. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995; 28:103-117
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23 Suppl 2:B21-29
- Lachin JM, Orchard TJ, Nathan DM, Group DER. Update on cardiovascular outcomes at 30 years of the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care2014; 37:39-43
- Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B, Diabetes C, Complications Trial/Epidemiology of Diabetes I, Complications Study Research G. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643-2653
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837-853
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577-1589
- Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ 1997; 314:1512-1515
- Mellbin LG, Malmberg K, Norhammar A, Wedel H, Ryden L, Investigators D. The impact of glucose lowering treatment on long-term prognosis in patients with type 2 diabetes and myocardial infarction: a report from the DIGAMI 2 trial. Eur Heart J 2008; 29:166-176
- Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, Goff DC, Jr., Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr., Probstfield JL, Simons-Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545-2559
- Gerstein HC, Miller ME, Ismail-Beigi F, Largay J, McDonald C, Lochnan HA, Booth GL, ACCORD STUDY Group. Effects of intensive glycaemic control on ischaemic heart disease: analysis of data from the randomised, controlled ACCORD trial. Lancet 2014; 384:1936-1941
- ACCORD STUDY Group. Nine-Year Effects of 3.7 Years of Intensive Glycemic Control on Cardiovascular Outcomes. Diabetes Care 2016; 39:701-708
- Advance Collaborative Group, Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560-2572
- Zoungas S, Chalmers J, Neal B, Billot L, Li Q, Hirakawa Y, Arima H, Monaghan H, Joshi R, Colagiuri S, Cooper ME, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Lisheng L, Mancia G, Marre M, Matthews DR, Mogensen CE, Perkovic V, Poulter N, Rodgers A, Williams B, MacMahon S, Patel A, Woodward M, Group A-OC. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N Engl J Med 2014; 371:1392-1406
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, Warren SR, Goldman S, McCarren M, Vitek ME, Henderson WG, Huang GD, Investigators V. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129-139
- Hayward RA, Reaven PD, Wiitala WL, Bahn GD, Reda DJ, Ge L, McCarren M, Duckworth WC, Emanuele NV, Investigators V. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med2015; 372:2197-2206
- Reaven PD, Emanuele NV, Wiitala WL, Bahn GD, Reda DJ, McCarren M, Duckworth WC, Hayward RA, Investigators V. Intensive Glucose Control in Patients with Type 2 Diabetes - 15-Year Follow-up. N Engl J Med2019; 380:2215-2224
- Buehler AM, Cavalcanti AB, Berwanger O, Figueiro M, Laranjeira LN, Zazula AD, Kioshi B, Bugano DG, Santucci E, Sbruzzi G, Guimaraes HP, Carvalho VO, Bordin SA. Effect of tight blood glucose control versus conventional control in patients with type 2 diabetes mellitus: a systematic review with meta-analysis of randomized controlled trials. Cardiovasc Ther 2013; 31:147-160
- Control Group, Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, Evans GW, Gerstein HC, Holman RR, Moritz TE, Neal BC, Ninomiya T, Patel AA, Paul SK, Travert F, Woodward M. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia 2009; 52:2288-2298
- Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:854-865
- Kooy A, de Jager J, Lehert P, Bets D, Wulffele MG, Donker AJ, Stehouwer CD. Long-term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch Intern Med 2009; 169:616-625
- Hong J, Zhang Y, Lai S, Lv A, Su Q, Dong Y, Zhou Z, Tang W, Zhao J, Cui L, Zou D, Wang D, Li H, Liu C, Wu G, Shen J, Zhu D, Wang W, Shen W, Ning G. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care 2013; 36:1304-1311
- Goldberg RB, Orchard TJ, Crandall JP, Boyko EJ, Budoff M, Dabelea D, Gadde KM, Knowler WC, Lee CG, Nathan DM, Watson K, Temprosa M. Effects of Long-term Metformin and Lifestyle Interventions on Cardiovascular Events in the Diabetes Prevention Program and Its Outcome Study. Circulation 2022; 145:1632-1641
- Goldberg RB, Aroda VR, Bluemke DA, Barrett-Connor E, Budoff M, Crandall JP, Dabelea D, Horton ES, Mather KJ, Orchard TJ, Schade D, Watson K, Temprosa M. Effect of Long-Term Metformin and Lifestyle in the Diabetes Prevention Program and Its Outcome Study on Coronary Artery Calcium. Circulation 2017; 136:52-64
- Fitch K, Abbara S, Lee H, Stavrou E, Sacks R, Michel T, Hemphill L, Torriani M, Grinspoon S. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. AIDS 2012; 26:587-597
- Rosenstock J, Kahn SE, Johansen OE, Zinman B, Espeland MA, Woerle HJ, Pfarr E, Keller A, Mattheus M, Baanstra D, Meinicke T, George JT, von Eynatten M, McGuire DK, Marx N. Effect of Linagliptin vs Glimepiride on Major Adverse Cardiovascular Outcomes in Patients With Type 2 Diabetes: The CAROLINA Randomized Clinical Trial. JAMA 2019;
- Navigator Study Group, Holman RR, Haffner SM, McMurray JJ, Bethel MA, Holzhauer B, Hua TA, Belenkov Y, Boolell M, Buse JB, Buckley BM, Chacra AR, Chiang FT, Charbonnel B, Chow CC, Davies MJ, Deedwania P, Diem P, Einhorn D, Fonseca V, Fulcher GR, Gaciong Z, Gaztambide S, Giles T, Horton E, Ilkova H, Jenssen T, Kahn SE, Krum H, Laakso M, Leiter LA, Levitt NS, Mareev V, Martinez F, Masson C, Mazzone T, Meaney E, Nesto R, Pan C, Prager R, Raptis SA, Rutten GE, Sandstroem H, Schaper F, Scheen A, Schmitz O, Sinay I, Soska V, Stender S, Tamas G, Tognoni G, Tuomilehto J, Villamil AS, Vozar J, Califf RM. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N Engl J Med 2010; 362:1463-1476
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005; 366:1279-1289
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, Guarino PD, Lovejoy AM, Peduzzi PN, Conwit R, Brass LM, Schwartz GG, Adams HP, Jr., Berger L, Carolei A, Clark W, Coull B, Ford GA, Kleindorfer D, O'Leary JR, Parsons MW, Ringleb P, Sen S, Spence JD, Tanne D, Wang D, Winder TR. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N Engl J Med 2016; 374:1321-1331
- Vaccaro O, Masulli M, Nicolucci A, Bonora E, Del Prato S, Maggioni AP, Rivellese AA, Squatrito S, Giorda CB, Sesti G, Mocarelli P, Lucisano G, Sacco M, Signorini S, Cappellini F, Perriello G, Babini AC, Lapolla A, Gregori G, Giordano C, Corsi L, Buzzetti R, Clemente G, Di Cianni G, Iannarelli R, Cordera R, La Macchia O, Zamboni C, Scaranna C, Boemi M, Iovine C, Lauro D, Leotta S, Dall'Aglio E, Cannarsa E, Tonutti L, Pugliese G, Bossi AC, Anichini R, Dotta F, Di Benedetto A, Citro G, Antenucci D, Ricci L, Giorgino F, Santini C, Gnasso A, De Cosmo S, Zavaroni D, Vedovato M, Consoli A, Calabrese M, di Bartolo P, Fornengo P, Riccardi G. Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): a randomised, multicentre trial. Lancet Diabetes Endocrinol2017; 5:887-897
- Vaccaro O, Lucisano G, Masulli M, Bonora E, Del Prato S, Rivellese AA, Giorda CB, Mocarelli P, Squatrito S, Maggioni AP, Riccardi G, Nicolucci A. Cardiovascular Effects of Pioglitazone or Sulfonylureas According to Pretreatment Risk: Moving Toward Personalized Care. J Clin Endocrinol Metab 2019; 104:3296-3302
- Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D'Agostino RB, Sr., Perez A, Provost JC, Haffner SM. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. JAMA 2006; 296:2572-2581
- Pfutzner A, Marx N, Lubben G, Langenfeld M, Walcher D, Konrad T, Forst T. Improvement of cardiovascular risk markers by pioglitazone is independent from glycemic control: results from the pioneer study. J Am Coll Cardiol2005; 45:1925-1931
- Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, Jure H, De Larochelliere R, Staniloae CS, Mavromatis K, Saw J, Hu B, Lincoff AM, Tuzcu EM. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA2008; 299:1561-1573
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007; 356:2457-2471
- Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA 2007; 298:1189-1195
- Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet 2009; 373:2125-2135
- Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes--an interim analysis. N Engl J Med 2007; 357:28-38
- Mahaffey KW, Hafley G, Dickerson S, Burns S, Tourt-Uhlig S, White J, Newby LK, Komajda M, McMurray J, Bigelow R, Home PD, Lopes RD. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J 2013; 166:240-249 e241
- Bach RG, Brooks MM, Lombardero M, Genuth S, Donner TW, Garber A, Kennedy L, Monrad ES, Pop-Busui R, Kelsey SF, Frye RL. Rosiglitazone and outcomes for patients with diabetes mellitus and coronary artery disease in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation 2013; 128:785-794
- Feingold KR. Oral and Injectable (Non-Insulin) Pharmacological Agents for Type 2 Diabetes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317-1326
- Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, Udell JA, Mosenzon O, Im K, Umez-Eronini AA, Pollack PS, Hirshberg B, Frederich R, Lewis BS, McGuire DK, Davidson J, Steg PG, Bhatt DL. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation2014; 130:1579-1588
- White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369:1327-1335
- Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Lam H, White WB. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015; 385:2067-2076
- Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2015; 373:232-242
- Rosenstock J, Perkovic V, Johansen OE, Cooper ME, Kahn SE, Marx N, Alexander JH, Pencina M, Toto RD, Wanner C, Zinman B, Woerle HJ, Baanstra D, Pfarr E, Schnaidt S, Meinicke T, George JT, von Eynatten M, McGuire DK. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2018;
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 2015; 373:2117-2128
- Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, Sambevski S, Kaspers S, Pfarr E, George JT, Zinman B. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019; 139:1384-1395
- Fitchett D, Butler J, van de Borne P, Zinman B, Lachin JM, Wanner C, Woerle HJ, Hantel S, George JT, Johansen OE, Inzucchi SE. Effects of empagliflozin on risk for cardiovascular death and heart failure hospitalization across the spectrum of heart failure risk in the EMPA-REG OUTCOME(R) trial. Eur Heart J 2018; 39:363-370
- Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med 2017; 377:644-657
- Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 2019; 380:2295-2306
- Jardine MJ, Zhou Z, Mahaffey KW, Oshima M, Agarwal R, Bakris G, Bajaj HS, Bull S, Cannon CP, Charytan DM, de Zeeuw D, Di Tanna GL, Greene T, Heerspink HJL, Levin A, Neal B, Pollock C, Qiu R, Sun T, Wheeler DC, Zhang H, Zinman B, Rosenthal N, Perkovic V. Renal, Cardiovascular, and Safety Outcomes of Canagliflozin by Baseline Kidney Function: A Secondary Analysis of the CREDENCE Randomized Trial. J Am Soc Nephrol 2020; 31:1128-1139
- Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med2018;
- Furtado RHM, Bonaca MP, Raz I, Zelniker TA, Mosenzon O, Cahn A, Kuder J, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Nicolau JC, Gause-Nilsson IAM, Fredriksson M, Langkilde AM, Sabatine MS, Wiviott SD. Dapagliflozin and Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus and Previous Myocardial Infarction. Circulation 2019; 139:2516-2527
- Cannon CP, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, Masiukiewicz U, Charbonnel B, Frederich R, Gallo S, Cosentino F, Shih WJ, Gantz I, Terra SG, Cherney DZI, McGuire DK. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N Engl J Med 2020; 383:1425-1435
- Cosentino F, Cannon CP, Cherney DZI, Masiukiewicz U, Pratley R, Dagogo-Jack S, Frederich R, Charbonnel B, Mancuso J, Shih WJ, Terra SG, Cater NB, Gantz I, McGuire DK. Efficacy of Ertugliflozin on Heart Failure-Related Events in Patients With Type 2 Diabetes Mellitus and Established Atherosclerotic Cardiovascular Disease: Results of the VERTIS CV Trial. Circulation 2020; 142:2205-2215
- Feingold KR. Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2022.
- Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Kober LV, Lawson FC, Ping L, Wei X, Lewis EF, Maggioni AP, McMurray JJ, Probstfield JL, Riddle MC, Solomon SD, Tardif JC. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N Engl J Med 2015; 373:2247-2257
- Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016; 375:311-322
- Mann JFE, Fonseca V, Mosenzon O, Raz I, Goldman B, Idorn T, von Scholten BJ, Poulter NR. Effects of Liraglutide Versus Placebo on Cardiovascular Events in Patients With Type 2 Diabetes Mellitus and Chronic Kidney Disease. Circulation 2018; 138:2908-2918
- Verma S, Poulter NR, Bhatt DL, Bain SC, Buse JB, Leiter LA, Nauck MA, Pratley RE, Zinman B, Orsted DD, Monk Fries T, Rasmussen S, Marso SP. Effects of Liraglutide on Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus With or Without History of Myocardial Infarction or Stroke. Circulation 2018; 138:2884-2894
- Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsboll T. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2016;
- Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, Jeppesen OK, Lingvay I, Mosenzon O, Pedersen SD, Tack CJ, Thomsen M, Vilsboll T, Warren ML, Bain SC. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2019; 381:841-851
- Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, Chan JC, Choi J, Gustavson SM, Iqbal N, Maggioni AP, Marso SP, Ohman P, Pagidipati NJ, Poulter N, Ramachandran A, Zinman B, Hernandez AF. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2017; 377:1228-1239
- Hernandez AF, Green JB, Janmohamed S, D'Agostino RB, Sr., Granger CB, Jones NP, Leiter LA, Rosenberg AE, Sigmon KN, Somerville MC, Thorpe KM, McMurray JJV, Del Prato S. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 2018; 392:1519-1529
- Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, Probstfield J, Riesmeyer JS, Riddle MC, Ryden L, Xavier D, Atisso CM, Dyal L, Hall S, Rao-Melacini P, Wong G, Avezum A, Basile J, Chung N, Conget I, Cushman WC, Franek E, Hancu N, Hanefeld M, Holt S, Jansky P, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Cardona Munoz EG, Pirags V, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WH, Temelkova-Kurktschiev T. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019; 394:121-130
- Gerstein HC, Hart R, Colhoun HM, Diaz R, Lakshmanan M, Botros FT, Probstfield J, Riddle MC, Ryden L, Atisso CM, Dyal L, Hall S, Avezum A, Basile J, Conget I, Cushman WC, Hancu N, Hanefeld M, Jansky P, Keltai M, Lanas F, Leiter LA, Lopez-Jaramillo P, Munoz EGC, Pogosova N, Raubenheimer PJ, Shaw JE, Sheu WH, Temelkova-Kurktschiev T. The effect of dulaglutide on stroke: an exploratory analysis of the REWIND trial. Lancet Diabetes Endocrinol 2020; 8:106-114
- Kristensen SL, Rorth R, Jhund PS, Docherty KF, Sattar N, Preiss D, Kober L, Petrie MC, McMurray JJV. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol 2019; 7:776-785
- Sposito AC, Berwanger O, de Carvalho LSF, Saraiva JFK. GLP-1RAs in type 2 diabetes: mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc Diabetol 2018; 17:157
- Sattar N, McGuire DK, Pavo I, Weerakkody GJ, Nishiyama H, Wiese RJ, Zoungas S. Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med 2022; 28:591-598
- Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. JAMA 2003; 290:486-494
- Yun P, Du AM, Chen XJ, Liu JC, Xiao H. Effect of Acarbose on Long-Term Prognosis in Acute Coronary Syndromes Patients with Newly Diagnosed Impaired Glucose Tolerance. J Diabetes Res 2016; 2016:1602083
- Holman RR, Coleman RL, Chan JCN, Chiasson JL, Feng H, Ge J, Gerstein HC, Gray R, Huo Y, Lang Z, McMurray JJ, Ryden L, Schroder S, Sun Y, Theodorakis MJ, Tendera M, Tucker L, Tuomilehto J, Wei Y, Yang W, Wang D, Hu D, Pan C. Effects of acarbose on cardiovascular and diabetes outcomes in patients with coronary heart disease and impaired glucose tolerance (ACE): a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2017; 5:877-886
- Gaziano JM, Cincotta AH, O'Connor CM, Ezrokhi M, Rutty D, Ma ZJ, Scranton RE. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010; 33:1503-1508
- Younk LM, Davis SN. Evaluation of colesevelam hydrochloride for the treatment of type 2 diabetes. Expert Opin Drug Metab Toxicol 2012; 8:515-525
- The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA 1984; 251:351-364
- The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA 1984; 251:365-374
- Investigators OT, Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, Pogue J, Probstfield J, Ramachandran A, Riddle MC, Ryden LE, Yusuf S. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med 2012; 367:319-328
- Origin Trial Investigators. Cardiovascular and Other Outcomes Postintervention With Insulin Glargine and Omega-3 Fatty Acids (ORIGINALE). Diabetes Care 2016; 39:709-716
- Marso SP, McGuire DK, Zinman B, Poulter NR, Emerson SS, Pieber TR, Pratley RE, Haahr PM, Lange M, Brown-Frandsen K, Moses A, Skibsted S, Kvist K, Buse JB. Efficacy and Safety of Degludec versus Glargine in Type 2 Diabetes. N Engl J Med 2017; 377:723-732
- Chaitman BR, Hardison RM, Adler D, Gebhart S, Grogan M, Ocampo S, Sopko G, Ramires JA, Schneider D, Frye RL. The Bypass Angioplasty Revascularization Investigation 2 Diabetes randomized trial of different treatment strategies in type 2 diabetes mellitus with stable ischemic heart disease: impact of treatment strategy on cardiac mortality and myocardial infarction. Circulation 2009; 120:2529-2540
- Bari D. Study Group, Frye RL, August P, Brooks MM, Hardison RM, Kelsey SF, MacGregor JM, Orchard TJ, Chaitman BR, Genuth SM, Goldberg SH, Hlatky MA, Jones TL, Molitch ME, Nesto RW, Sako EY, Sobel BE. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med 2009; 360:2503-2515
- ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Seley JJ, Stanton RC, Gabbay RA, on behalf of the American Diabetes A. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes-2023. Diabetes Care 2023; 46:S140-S157
- de Ferranti SD, de Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, Magge SN, Marx N, McGuire DK, Orchard TJ, Zinman B, Eckel RH. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Circulation 2014; 130:1110-1130
- Martin-Timon I, Sevillano-Collantes C, Segura-Galindo A, Del Canizo-Gomez FJ. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J Diabetes 2014; 5:444-470
- Turner RC, Millns H, Neil HA, Stratton IM, Manley SE, Matthews DR, Holman RR. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823-828
- Hovingh GK, Rader DJ, Hegele RA. HDL re-examined. Curr Opin Lipidol 2015; 26:127-132
- Brunzell JD, Davidson M, Furberg CD, Goldberg RB, Howard BV, Stein JH, Witztum JL. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811-822
- Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, Laufs U, Oliver-Williams C, Wood AM, Butterworth AS, Di Angelantonio E, Danesh J, Nicholls SJ, Bhatt DL, Sabatine MS, Catapano AL. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA 2019; 321:364-373
- Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res 2016; 118:547-563
- Ganjali S, Dallinga-Thie GM, Simental-Mendia LE, Banach M, Pirro M, Sahebkar A. HDL functionality in type 1 diabetes. Atherosclerosis 2017; 267:99-109
- Ginsberg HN, MacCallum PR. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: Part I. Increased cardiovascular disease risk and the importance of atherogenic dyslipidemia in persons with the metabolic syndrome and type 2 diabetes mellitus. J Cardiometab Syndr 2009; 4:113-119
- Goldberg IJ. Clinical review 124: Diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab2001; 86:965-971
- Krauss RM. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004; 27:1496-1504
- Wu L, Parhofer KG. Diabetic dyslipidemia. Metabolism 2014; 63:1469-1479
- Taskinen MR, Boren J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis2015; 239:483-495
- Feingold KR, Grunfeld C, Pang M, Doerrler W, Krauss RM. LDL subclass phenotypes and triglyceride metabolism in non-insulin-dependent diabetes. Arterioscler Thromb 1992; 12:1496-1502
- Feingold KR. Obesity and Dyslipidemia. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, eds. Endotext. South Dartmouth (MA)2023.
- Morgantini C, Natali A, Boldrini B, Imaizumi S, Navab M, Fogelman AM, Ferrannini E, Reddy ST. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011; 60:2617-2623
- Apro J, Tietge UJ, Dikkers A, Parini P, Angelin B, Rudling M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler Thromb Vasc Biol 2016; 36:787-791
- Manjunatha S, Distelmaier K, Dasari S, Carter RE, Kudva YC, Nair KS. Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metabolism 2016; 65:1421-1431
- Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res 2016; 57:1111-1125
- Durrington PN, Schofield JD, Siahmansur T, Soran H. Lipoprotein (a): gene genie. Curr Opin Lipidol 2014; 25:289-296
- Gudbjartsson DF, Thorgeirsson G, Sulem P, Helgadottir A, Gylfason A, Saemundsdottir J, Bjornsson E, Norddahl GL, Jonasdottir A, Jonasdottir A, Eggertsson HP, Gretarsdottir S, Thorleifsson G, Indridason OS, Palsson R, Jonasson F, Jonsdottir I, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Danielsen R, Matthiasson SE, Kristmundsdottir S, Halldorsson BV, Hreidarsson AB, Valdimarsson EM, Gudnason T, Benediktsson R, Steinthorsdottir V, Thorsteinsdottir U, Holm H, Stefansson K. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J Am Coll Cardiol 2019; 74:2982-2994
- Ferrannini E, DeFronzo RA. Impact of glucose-lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J 2015; 36:2288-2296
- Wulffele MG, Kooy A, de Zeeuw D, Stehouwer CD, Gansevoort RT. The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: a systematic review. J Intern Med 2004; 256:1-14
- Ratner R, Goldberg R, Haffner S, Marcovina S, Orchard T, Fowler S, Temprosa M, Diabetes Prevention Program Research Group. Impact of intensive lifestyle and metformin therapy on cardiovascular disease risk factors in the diabetes prevention program. Diabetes Care 2005; 28:888-894
- Spanheimer R, Betteridge DJ, Tan MH, Ferrannini E, Charbonnel B. Long-term lipid effects of pioglitazone by baseline anti-hyperglycemia medication therapy and statin use from the PROactive experience (PROactive 14). Am J Cardiol 2009; 104:234-239
- Deeg MA, Buse JB, Goldberg RB, Kendall DM, Zagar AJ, Jacober SJ, Khan MA, Perez AT, Tan MH. Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2007; 30:2458-2464
- Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, Tan MH, Khan MA, Perez AT, Jacober SJ. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2005; 28:1547-1554
- Sanchez-Garcia A, Simental-Mendia M, Millan-Alanis JM, Simental-Mendia LE. Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol Res 2020; 160:105068
- Lamos EM, Levitt DL, Munir KM. A review of dopamine agonist therapy in type 2 diabetes and effects on cardio-metabolic parameters. Prim Care Diabetes 2016; 10:60-65
- Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab 2010; 12:1048-1057
- Raskin P, Cincotta AH. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: developmental basis and therapeutic profile summary. Expert Rev Endocrinol Metab 2016; 11:113-148
- Feingold KR. Cholesterol Lowering Drugs. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular Actions and Clinical Outcomes With Glucagon-Like Peptide-1 Receptor Agonists and Dipeptidyl Peptidase-4 Inhibitors. Circulation 2017; 136:849-870
- Kanbay M, Copur S, Siriopol D, Yildiz AB, Gaipov A, van Raalte DH, Tuttle KR. Effect of tirzepatide on blood pressure and lipids: A meta-analysis of randomized controlled trials. Diabetes Obes Metab 2023;
- Ginsberg HN. Diabetic dyslipidemia: basic mechanisms underlying the common hypertriglyceridemia and low HDL cholesterol levels. Diabetes 1996; 45 Suppl 3:S27-30
- Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring)2006; 14 Suppl 1:41S-49S
- Klop B, Elte JW, Cabezas MC. Dyslipidemia in obesity: mechanisms and potential targets. Nutrients 2013; 5:1218-1240
- Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32-41
- Tg,Hdl Working Group of the Exome Sequencing Project, National Heart LungBlood, Institute, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, Masca N, Stirrups K, Kanoni S, Do R, Jun G, Hu Y, Kang HM, Xue C, Goel A, Farrall M, Duga S, Merlini PA, Asselta R, Girelli D, Olivieri O, Martinelli N, Yin W, Reilly D, Speliotes E, Fox CS, Hveem K, Holmen OL, Nikpay M, Farlow DN, Assimes TL, Franceschini N, Robinson J, North KE, Martin LW, DePristo M, Gupta N, Escher SA, Jansson JH, Van Zuydam N, Palmer CN, Wareham N, Koch W, Meitinger T, Peters A, Lieb W, Erbel R, Konig IR, Kruppa J, Degenhardt F, Gottesman O, Bottinger EP, O'Donnell CJ, Psaty BM, Ballantyne CM, Abecasis G, Ordovas JM, Melander O, Watkins H, Orho-Melander M, Ardissino D, Loos RJ, McPherson R, Willer CJ, Erdmann J, Hall AS, Samani NJ, Deloukas P, Schunkert H, Wilson JG, Kooperberg C, Rich SS, Tracy RP, Lin DY, Altshuler D, Gabriel S, Nickerson DA, Jarvik GP, Cupples LA, Reiner AP, Boerwinkle E, Kathiresan S. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371:22-31
- Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, Witztum JL. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014; 371:2200-2206
- Arca M, D'Erasmo L, Minicocci I. Familial combined hypolipidemia: angiopoietin-like protein-3 deficiency. Curr Opin Lipidol 2020; 31:41-48
- Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res2004; 45:1169-1196
- Lara-Castro C, Fu Y, Chung BH, Garvey WT. Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr Opin Lipidol 2007; 18:263-270
- Feingold KR, Grunfeld C. The acute phase response inhibits reverse cholesterol transport. J Lipid Res 2010; 51:682-684
- Feingold KR, Grunfeld C. Effect of inflammation on HDL structure and function. Curr Opin Lipidol 2022; 27:521-530
- Lu B, Moser A, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response stimulates the expression of angiopoietin like protein 4. Biochem Biophys Res Commun 2010; 391:1737-1741
- Feingold KR, Grunfeld C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, eds. Endotext. South Dartmouth (MA)2019.
- Rosenblat M, Karry R, Aviram M. Paraoxonase 1 (PON1) is a more potent antioxidant and stimulant of macrophage cholesterol efflux, when present in HDL than in lipoprotein-deficient serum: relevance to diabetes. Atherosclerosis 2006; 187:74-81
- Ahima RS. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006; 14 Suppl 5:242S-249S
- Christou GA, Kiortsis DN. Adiponectin and lipoprotein metabolism. Obes Rev 2013; 14:939-949
- Rashid S, Kastelein JJ. PCSK9 and resistin at the crossroads of the atherogenic dyslipidemia. Expert Rev Cardiovasc Ther 2013; 11:1567-1577
- Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: sequelae of insulin-resistant adipose tissue. Circ Res 2005; 96:1042-1052
- Cholesterol Treatment Trialists, Collaborato, Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Peto R, Armitage J, Baigent C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117-125
- Collins R, Armitage J, Parish S, Sleigh P, Peto R, Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005-2016
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7-22
- Wanner C, Krane V, Marz W, Olschewski M, Mann JF, Ruf G, Ritz E,German, Diabetes
Dialysis Study, Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238-248
- Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet2004; 364:685-696
- Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, Crowe T, Howard G, Cooper CJ, Brodie B, Grines CL, DeMaria AN. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071-1080
- Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, Davignon J, Erbel R, Fruchart JC, Tardif JC, Schoenhagen P, Crowe T, Cain V, Wolski K, Goormastic M, Tuzcu EM. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556-1565
- Stegman B, Puri R, Cho L, Shao M, Ballantyne CM, Barter PJ, Chapman MJ, Erbel R, Libby P, Raichlen JS, Uno K, Kataoka Y, Nissen SE, Nicholls SJ. High-intensity statin therapy alters the natural history of diabetic coronary atherosclerosis: insights from SATURN. Diabetes Care 2014; 37:3114-3120
- Ahmed S, Cannon CP, Murphy SA, Braunwald E. Acute coronary syndromes and diabetes: Is intensive lipid lowering beneficial? Results of the PROVE IT-TIMI 22 trial. Eur Heart J 2006; 27:2323-2329
- Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM, Pravastatin or Atorvastatin E, Infection Therapy-Thrombolysis in Myocardial Infarction I. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495-1504
- LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, Gotto AM, Greten H, Kastelein JJ, Shepherd J, Wenger NK, Treating to New Targets I. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425-1435
- Shepherd J, Barter P, Carmena R, Deedwania P, Fruchart JC, Haffner S, Hsia J, Breazna A, LaRosa J, Grundy S, Waters D. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220-1226
- Pedersen TR, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, Larsen ML, Bendiksen FS, Lindahl C, Szarek M, Tsai J, Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437-2445
- Cholesterol Treatment Trialists Colloboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670-1681
- Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, Forder P, Pillai A, Davis T, Glasziou P, Drury P, Kesaniemi YA, Sullivan D, Hunt D, Colman P, d'Emden M, Whiting M, Ehnholm C, Laakso M. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005; 366:1849-1861
- Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ, Grobbee DE, Cass A, Chalmers J, Perkovic V. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet 2010; 375:1875-1884
- Sacks FM, Carey VJ, Fruchart JC. Combination lipid therapy in type 2 diabetes. N Engl J Med 2010; 363:692-694; author reply 694-695
- Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, McNamara JR, Kashyap ML, Hershman JM, Wexler LF, Rubins HB. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA 2001; 285:1585-1591
- Rubins HB, Robins SJ, Collins D, Nelson DB, Elam MB, Schaefer EJ, Faas FH, Anderson JW. Diabetes, plasma insulin, and cardiovascular disease: subgroup analysis from the Department of Veterans Affairs high-density lipoprotein intervention trial (VA-HIT). Arch Intern Med 2002; 162:2597-2604
- Koskinen P, Manttari M, Manninen V, Huttunen JK, Heinonen OP, Frick MH. Coronary heart disease incidence in NIDDM patients in the Helsinki Heart Study. Diabetes Care 1992; 15:820-825
- Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet 2001; 357:905-910
- Elkeles RS, Diamond JR, Poulter C, Dhanjil S, Nicolaides AN, Mahmood S, Richmond W, Mather H, Sharp P, Feher MD. Cardiovascular outcomes in type 2 diabetes. A double-blind placebo-controlled study of bezafibrate: the St. Mary's, Ealing, Northwick Park Diabetes Cardiovascular Disease Prevention (SENDCAP) Study. Diabetes Care 1998; 21:641-648
- Clofibrate and niacin in coronary heart disease. JAMA 1975; 231:360-381
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254-257
- Ouchi Y, Sasaki J, Arai H, Yokote K, Harada K, Katayama Y, Urabe T, Uchida Y, Hayashi M, Yokota N, Nishida H, Otonari T, Arai T, Sakuma I, Sakabe K, Yamamoto M, Kobayashi T, Oikawa S, Yamashita S, Rakugi H, Imai T, Tanaka S, Ohashi Y, Kuwabara M, Ito H. Ezetimibe Lipid-Lowering Trial on Prevention of Atherosclerotic Cardiovascular Disease in 75 or Older (EWTOPIA 75): A Randomized, Controlled Trial. Circulation 2019; 140:992-1003
- Nissen SE, Lincoff AM, Brennan D, Ray KK, Mason D, Kastelein JJP, Thompson PD, Libby P, Cho L, Plutzky J, Bays HE, Moriarty PM, Menon V, Grobbee DE, Louie MJ, Chen CF, Li N, Bloedon L, Robinson P, Horner M, Sasiela WJ, McCluskey J, Davey D, Fajardo-Campos P, Petrovic P, Fedacko J, Zmuda W, Lukyanov Y, Nicholls SJ. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med 2023; 388:1353-1364
- Nissen SE, Menon V, Nicholls SJ, Brennan D, Laffin L, Ridker P, Ray KK, Mason D, Kastelein JJP, Cho L, Libby P, Li N, Foody J, Louie MJ, Lincoff AM. Bempedoic Acid for Primary Prevention of Cardiovascular Events in Statin-Intolerant Patients. JAMA 2023; 330:131-140
- Group AS, Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr., Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563-1574
- ACCORD Study Group, Group AES, Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, Hubbard L, Esser BA, Lovato JF, Perdue LH, Goff DC, Jr., Cushman WC, Ginsberg HN, Elam MB, Genuth S, Gerstein HC, Schubart U, Fine LJ. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 2010; 363:233-244
- Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d'Emden MC, Crimet DC, O'Connell RL, Colman PG. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007; 370:1687-1697
- Das Pradhan A, Glynn RJ, Fruchart JC, MacFadyen JG, Zaharris ES, Everett BM, Campbell SE, Oshima R, Amarenco P, Blom DJ, Brinton EA, Eckel RH, Elam MB, Felicio JS, Ginsberg HN, Goudev A, Ishibashi S, Joseph J, Kodama T, Koenig W, Leiter LA, Lorenzatti AJ, Mankovsky B, Marx N, Nordestgaard BG, Pall D, Ray KK, Santos RD, Soran H, Susekov A, Tendera M, Yokote K, Paynter NP, Buring JE, Libby P, Ridker PM. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med 2022; 387:1923-1934
- Investigators AIM-HIGH, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255-2267
- Guyton JR, Slee AE, Anderson T, Fleg JL, Goldberg RB, Kashyap ML, Marcovina SM, Nash SD, O'Brien KD, Weintraub WS, Xu P, Zhao XQ, Boden WE. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1580-1584
- Hps Thrive Collaborative Group, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. The New England journal of medicine 2014; 371:203-212
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RMI. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med 2015; 372:2387-2397
- Bohula EA, Morrow DA, Giugliano RP, Blazing MA, He P, Park JG, Murphy SA, White JA, Kesaniemi YA, Pedersen TR, Brady AJ, Mitchel Y, Cannon CP, Braunwald E. Atherothrombotic Risk Stratification and Ezetimibe for Secondary Prevention. J Am Coll Cardiol 2017; 69:911-921
- Giugliano RP, Cannon CP, Blazing MA, Nicolau JC, Corbalan R, Spinar J, Park JG, White JA, Bohula EA, Braunwald E. Benefit of Adding Ezetimibe to Statin Therapy on Cardiovascular Outcomes and Safety in Patients With Versus Without Diabetes Mellitus: Results From IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2018; 137:1571-1582
- Lee YJ, Cho JY, You SC, Lee YH, Yun KH, Cho YH, Shin WY, Im SW, Kang WC, Park Y, Lee SY, Lee SJ, Hong SJ, Ahn CM, Kim BK, Ko YG, Choi D, Hong MK, Jang Y, Kim JS. Moderate-intensity statin with ezetimibe vs. high-intensity statin in patients with diabetes and atherosclerotic cardiovascular disease in the RACING trial. Eur Heart J 2023; 44:972-983
- Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017; 376:1713-1722
- Sabatine MS, Leiter LA, Wiviott SD, Giugliano RP, Deedwania P, De Ferrari GM, Murphy SA, Kuder JF, Gouni-Berthold I, Lewis BS, Handelsman Y, Pineda AL, Honarpour N, Keech AC, Sever PS, Pedersen TR. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol 2017;
- Sabatine MS, Leiter LA, Wiviott SD, Giugliano RP, Deedwania P, De Ferrari GM, Murphy SA, Kuder JF, Gouni-Berthold I, Lewis BS, Handelsman Y, Pineda AL, Honarpour N, Keech AC, Sever PS, Pedersen TR. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol 2017; 5:941-950
- Giugliano RP, Keech A, Murphy SA, Huber K, Tokgozoglu SL, Lewis BS, Ferreira J, Pineda AL, Somaratne R, Sever PS, Pedersen TR, Sabatine MS. Clinical Efficacy and Safety of Evolocumab in High-Risk Patients Receiving a Statin: Secondary Analysis of Patients With Low LDL Cholesterol Levels and in Those Already Receiving a Maximal-Potency Statin in a Randomized Clinical Trial. JAMA Cardiol 2017; 2:1385-1391
- Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni-Berthold I, Lopez-Miranda J, Schiele F, Mach F, Ott BR, Kanevsky E, Pineda AL, Somaratne R, Wasserman SM, Keech AC, Sever PS, Sabatine MS. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet 2017; 390:1962-1971
- Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, Jukema JW, Lecorps G, Mahaffey KW, Moryusef A, Pordy R, Quintero K, Roe MT, Sasiela WJ, Tamby JF, Tricoci P, White HD, Zeiher AM. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med 2018; 379:2097-2107
- Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016; 316:2373-2384
- ORIGIN Trial Investigators OT, Bosch J, Gerstein HC, Dagenais GR, Diaz R, Dyal L, Jung H, Maggiono AP, Probstfield J, Ramachandran A, Riddle MC, Ryden LE, Yusuf S. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med 2012; 367:309-318
- Ascend Study Collaborative Group, Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, Barton J, Murphy K, Aung T, Haynes R, Cox J, Murawska A, Young A, Lay M, Chen F, Sammons E, Waters E, Adler A, Bodansky J, Farmer A, McPherson R, Neil A, Simpson D, Peto R, Baigent C, Collins R, Parish S, Armitage J. Effects of n-3 Fatty Acid Supplements in Diabetes Mellitus. N Engl J Med 2018; 379:1540-1550
- Feingold KR. Triglyceride Lowering Drugs. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K, Shirato K. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007; 369:1090-1098
- Saito Y, Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K, Shirato K. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis 2008; 200:135-140
- Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT, Jr., Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med 2018;
- Mason RP, Libby P, Bhatt DL. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arterioscler Thromb Vasc Biol 2020; 40:1135-1147
- Nicholls SJ, Lincoff AM, Bash D, Ballantyne CM, Barter PJ, Davidson MH, Kastelein JJP, Koenig W, McGuire DK, Mozaffarian D, Pedersen TR, Ridker PM, Ray K, Karlson BW, Lundstrom T, Wolski K, Nissen SE. Assessment of omega-3 carboxylic acids in statin-treated patients with high levels of triglycerides and low levels of high-density lipoprotein cholesterol: Rationale and design of the STRENGTH trial. Clin Cardiol 2018; 41:1281-1288
- Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, Davidson MH, Kastelein JJP, Koenig W, McGuire DK, Mozaffarian D, Ridker PM, Ray KK, Katona BG, Himmelmann A, Loss LE, Rensfeldt M, Lundstrom T, Agrawal R, Menon V, Wolski K, Nissen SE. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020;
- Ridker PM, Rifai N, MacFadyen J, Glynn RJ, Jiao L, Steg PG, Miller M, Brinton EA, Jacobson TA, Tardif JC, Ballantyne CM, Mason RP, Bhatt DL. Effects of Randomized Treatment With Icosapent Ethyl and a Mineral Oil Comparator on Interleukin-1beta, Interleukin-6, C-Reactive Protein, Oxidized Low-Density Lipoprotein Cholesterol, Homocysteine, Lipoprotein(a), and Lipoprotein-Associated Phospholipase A2: A REDUCE-IT Biomarker Substudy. Circulation 2022; 146:372-379
- Goff ZD, Nissen SE. N-3 polyunsaturated fatty acids for cardiovascular risk. Curr Opin Cardiol 2022; 37:356-363
- Mason RP, Sherratt SCR, Eckel RH. Omega-3-fatty acids: Do they prevent cardiovascular disease? Best Pract Res Clin Endocrinol Metab 2023; 37:101681
- ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Das SR, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Kosiborod M, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Seley JJ, Stanton RC, Gabbay RA, on behalf of the American Diabetes A. 10. Cardiovascular Disease and Risk Management: Standards of Care in Diabetes-2023. Diabetes Care 2023; 46:S158-S190
- Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, Goldberg R, Heidenreich PA, Hlatky MA, Jones DW, Lloyd-Jones D, Lopez-Pajares N, Ndumele CE, Orringer CE, Peralta CA, Saseen JJ, Smith SC, Jr., Sperling L, Virani SS, Yeboah J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019; 139:e1082-e1143
- Jellinger PS, Handelsman Y, Rosenblit PD, Bloomgarden ZT, Fonseca VA, Garber AJ, Grunberger G, Guerin CK, Bell DSH, Mechanick JI, Pessah-Pollack R, Wyne K, Smith D, Brinton EA, Fazio S, Davidson M. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Dyslipidemia and Prevention of Cardiovascular Disease. Endocr Pract 2017; 23:1-87
- Handelsman Y, Jellinger PS, Guerin CK, Bloomgarden ZT, Brinton EA, Budoff MJ, Davidson MH, Einhorn D, Fazio S, Fonseca VA, Garber AJ, Grunberger G, Krauss RM, Mechanick JI, Rosenblit PD, Smith DA, Wyne KL. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Management of Dyslipidemia and Prevention of Cardiovascular Disease Algorithm - 2020 Executive Summary. Endocr Pract 2020; 26:1196-1224
- Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, Graham IM, Halliday A, Landmesser U, Mihaylova B, Pedersen TR, Riccardi G, Richter DJ, Sabatine MS, Taskinen MR, Tokgozoglu L, Wiklund O. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020; 41:111-188
- Marx N, Federici M, Schutt K, Muller-Wieland D, Ajjan RA, Antunes MJ, Christodorescu RM, Crawford C, Di Angelantonio E, Eliasson B, Espinola-Klein C, Fauchier L, Halle M, Herrington WG, Kautzky-Willer A, Lambrinou E, Lesiak M, Lettino M, McGuire DK, Mullens W, Rocca B, Sattar N. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur Heart J 2023;
- SCORE2-Diabetes Working Group. SCORE2-Diabetes: 10-year cardiovascular risk estimation in type 2 diabetes in Europe. Eur Heart J 2023; 44:2544-2556
- Feingold KR. Maximizing the benefits of cholesterol-lowering drugs. Curr Opin Lipidol 2019; 30:388-394
- Feingold KR, Chait A. Approach to patients with elevated low-density lipoprotein cholesterol levels. Best Pract Res Clin Endocrinol Metab 2023; 37:101658
- Evert AB, Dennison M, Gardner CD, Garvey WT, Lau KHK, MacLeod J, Mitri J, Pereira RF, Rawlings K, Robinson S, Saslow L, Uelmen S, Urbanski PB, Yancy WS, Jr. Nutrition Therapy for Adults With Diabetes or Prediabetes: A Consensus Report. Diabetes Care 2019; 42:731-754
- Feingold KR. The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2021.
- Look ARG, Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, Coday M, Crow RS, Curtis JM, Egan CM, Espeland MA, Evans M, Foreyt JP, Ghazarian S, Gregg EW, Harrison B, Hazuda HP, Hill JO, Horton ES, Hubbard VS, Jakicic JM, Jeffery RW, Johnson KC, Kahn SE, Kitabchi AE, Knowler WC, Lewis CE, Maschak-Carey BJ, Montez MG, Murillo A, Nathan DM, Patricio J, Peters A, Pi-Sunyer X, Pownall H, Reboussin D, Regensteiner JG, Rickman AD, Ryan DH, Safford M, Wadden TA, Wagenknecht LE, West DS, Williamson DF, Yanovski SZ. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369:145-154
- Estruch R, Ros E, Salas-Salvado J, Covas MI, Corella D, Aros F, Gomez-Gracia E, Ruiz-Gutierrez V, Fiol M, Lapetra J, Lamuela-Raventos RM, Serra-Majem L, Pinto X, Basora J, Munoz MA, Sorli JV, Martinez JA, Martinez-Gonzalez MA. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med2013; 368:1279-1290
- Estruch R, Ros E, Salas-Salvado J, Covas MI, Corella D, Aros F, Gomez-Gracia E, Ruiz-Gutierrez V, Fiol M, Lapetra J, Lamuela-Raventos RM, Serra-Majem L, Pinto X, Basora J, Munoz MA, Sorli JV, Martinez JA, Fito M, Gea A, Hernan MA, Martinez-Gonzalez MA. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med 2018; 378:e34
- Estruch R, Martinez-Gonzalez MA, Corella D, Salas-Salvado J, Ruiz-Gutierrez V, Covas MI, Fiol M, Gomez-Gracia E, Lopez-Sabater MC, Vinyoles E, Aros F, Conde M, Lahoz C, Lapetra J, Saez G, Ros E. Effects of a Mediterranean-style diet on cardiovascular risk factors: a randomized trial. Ann Intern Med 2006; 145:1-11
- Delgado-Lista J, Alcala-Diaz JF, Torres-Pena JD, Quintana-Navarro GM, Fuentes F, Garcia-Rios A, Ortiz-Morales AM, Gonzalez-Requero AI, Perez-Caballero AI, Yubero-Serrano EM, Rangel-Zuniga OA, Camargo A, Rodriguez-Cantalejo F, Lopez-Segura F, Badimon L, Ordovas JM, Perez-Jimenez F, Perez-Martinez P, Lopez-Miranda J. Long-term secondary prevention of cardiovascular disease with a Mediterranean diet and a low-fat diet (CORDIOPREV): a randomised controlled trial. Lancet 2022; 399:1876-1885
- de Lorgeril M, Renaud S, Mamelle N, Salen P, Martin JL, Monjaud I, Guidollet J, Touboul P, Delaye J. Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet 1994; 343:1454-1459
- de Lorgeril M, Salen P, Martin JL, Monjaud I, Delaye J, Mamelle N. Mediterranean diet, traditional risk factors, and the rate of cardiovascular complications after myocardial infarction: final report of the Lyon Diet Heart Study. Circulation 1999; 99:779-785
- Adams TD, Gress RE, Smith SC, Halverson RC, Simper SC, Rosamond WD, Lamonte MJ, Stroup AM, Hunt SC. Long-term mortality after gastric bypass surgery. N Engl J Med 2007; 357:753-761
- Romeo S, Maglio C, Burza MA, Pirazzi C, Sjoholm K, Jacobson P, Svensson PA, Peltonen M, Sjostrom L, Carlsson LM. Cardiovascular events after bariatric surgery in obese subjects with type 2 diabetes. Diabetes Care2012; 35:2613-2617
- Sjostrom L, Peltonen M, Jacobson P, Sjostrom CD, Karason K, Wedel H, Ahlin S, Anveden A, Bengtsson C, Bergmark G, Bouchard C, Carlsson B, Dahlgren S, Karlsson J, Lindroos AK, Lonroth H, Narbro K, Naslund I, Olbers T, Svensson PA, Carlsson LM. Bariatric surgery and long-term cardiovascular events. JAMA 2012; 307:56-65
- Sheng B, Truong K, Spitler H, Zhang L, Tong X, Chen L. The Long-Term Effects of Bariatric Surgery on Type 2 Diabetes Remission, Microvascular and Macrovascular Complications, and Mortality: a Systematic Review and Meta-Analysis. Obes Surg 2017; 27:2724-2732
- Fisher DP, Johnson E, Haneuse S, Arterburn D, Coleman KJ, O'Connor PJ, O'Brien R, Bogart A, Theis MK, Anau J, Schroeder EB, Sidney S. Association Between Bariatric Surgery and Macrovascular Disease Outcomes in Patients With Type 2 Diabetes and Severe Obesity. JAMA 2018; 320:1570-1582
- Farmer A, Montori V, Dinneen S, Clar C. Fish oil in people with type 2 diabetes mellitus. Cochrane Database Syst Rev 2001:CD003205
- Preiss D, Sattar N. Statins and the risk of new-onset diabetes: a review of recent evidence. Curr Opin Lipidol2011; 22:460-466
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735-742
- Erqou S, Lee CC, Adler AI. Statins and glycaemic control in individuals with diabetes: a systematic review and meta-analysis. Diabetologia 2014; 57:2444-2452
- Livingstone SJ, Looker HC, Akbar T, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Fuller JH, Colhoun HM. Effect of atorvastatin on glycaemia progression in patients with diabetes: an analysis from the Collaborative Atorvastatin in Diabetes Trial (CARDS). Diabetologia 2016; 59:299-306
- Chiu SW, Pratt CM, Feinn R, Chatterjee S. Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors and Ezetimibe on Risk of New-Onset Diabetes: A Systematic Review and Meta-Analysis of Large, Double-Blinded Randomized Controlled Trials. J Cardiovasc Pharmacol Ther 2020; 25:409-417
- Shah NP, McGuire DK, Cannon CP, Giugliano RP, Lokhnygina Y, Page CB, Tershakovec AM, Braunwald E, Blazing MA. Impact of Ezetimibe on New-Onset Diabetes: A Substudy of IMPROVE-IT. J Am Heart Assoc 2023; 12:e029593
- Kellick KA, Bottorff M, Toth PP, The National Lipid Association's Safety Task F. A clinician's guide to statin drug-drug interactions. J Clin Lipidol 2014; 8:S30-46
- Linz PE, Lovato LC, Byington RP, O'Connor PJ, Leiter LA, Weiss D, Force RW, Crouse JR, Ismail-Beigi F, Simmons DL, Papademetriou V, Ginsberg HN, Elam MB. Paradoxical reduction in HDL-C with fenofibrate and thiazolidinedione therapy in type 2 diabetes: the ACCORD Lipid Trial. Diabetes Care 2014; 37:686-693
- Ansquer JC, Foucher C, Rattier S, Taskinen MR, Steiner G. Fenofibrate reduces progression to microalbuminuria over 3 years in a placebo-controlled study in type 2 diabetes: results from the Diabetes Atherosclerosis Intervention Study (DAIS). Am J Kidney Dis 2005; 45:485-493
- Davis TM, Ting R, Best JD, Donoghoe MW, Drury PL, Sullivan DR, Jenkins AJ, O'Connell RL, Whiting MJ, Glasziou PP, Simes RJ, Kesaniemi YA, Gebski VJ, Scott RS, Keech AC, Event Lowering in Diabetes Study i. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia 2011; 54:280-290
- Rajamani K, Colman PG, Li LP, Best JD, Voysey M, D'Emden MC, Laakso M, Baker JR, Keech AC. Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet 2009; 373:1780-1788
- Aldridge MA, Ito MK. Colesevelam hydrochloride: a novel bile acid-binding resin. Ann Pharmacother 2001; 35:898-907
- Bays HE. Colesevelam hydrochloride added to background metformin therapy in patients with type 2 diabetes mellitus: a pooled analysis from 3 clinical studies. Endocr Pract 2011; 17:933-938
- Song WL, FitzGerald GA. Niacin, an old drug with a new twist. J Lipid Res 2013; 54:2586-2594
- Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA 2000; 284:1263-1270
- Grundy SM, Vega GL, McGovern ME, Tulloch BR, Kendall DM, Fitz-Patrick D, Ganda OP, Rosenson RS, Buse JB, Robertson DD, Sheehan JP, Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of niaspan trial. Archives of internal medicine 2002; 162:1568-1576
- Weintraub H. Update on marine omega-3 fatty acids: management of dyslipidemia and current omega-3 treatment options. Atherosclerosis 2013; 230:381-389
- Khan SU, Rahman H, Okunrintemi V, Riaz H, Khan MS, Sattur S, Kaluski E, Lincoff AM, Martin SS, Blaha MJ. Association of Lowering Low-Density Lipoprotein Cholesterol With Contemporary Lipid-Lowering Therapies and Risk of Diabetes Mellitus: A Systematic Review and Meta-Analysis. J Am Heart Assoc 2019; 8:e011581
- Carugo S, Sirtori CR, Corsini A, Tokgozoglu L, Ruscica M. PCSK9 Inhibition and Risk of Diabetes: Should We Worry? Curr Atheroscler Rep 2022; 24:995-1004
- Wang X, Zhang Y, Tan H, Wang P, Zha X, Chong W, Zhou L, Fang F. Efficacy and safety of bempedoic acid for prevention of cardiovascular events and diabetes: a systematic review and meta-analysis. Cardiovasc Diabetol2020; 19:128
- Bays HE, Banach M, Catapano AL, Duell PB, Gotto AM, Jr., Laufs U, Leiter LA, Mancini GBJ, Ray KK, Bloedon LT, Sasiela WJ, Ye Z, Ballantyne CM. Bempedoic acid safety analysis: Pooled data from four phase 3 clinical trials. J Clin Lipidol 2020; 14:649-659 e646
- Jenkins DJ, Jones PJ, Lamarche B, Kendall CW, Faulkner D, Cermakova L, Gigleux I, Ramprasath V, de Souza R, Ireland C, Patel D, Srichaikul K, Abdulnour S, Bashyam B, Collier C, Hoshizaki S, Josse RG, Leiter LA, Connelly PW, Frohlich J. Effect of a dietary portfolio of cholesterol-lowering foods given at 2 levels of intensity of dietary advice on serum lipids in hyperlipidemia: a randomized controlled trial. JAMA 2011; 306:831-839
- Feingold KR. Utility of Advanced Lipoprotein Testing in Clinical Practice. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, eds. Endotext. South Dartmouth (MA)2023.
- Orringer CE, Jacobson TA, Maki KC. National Lipid Association Scientific Statement on the use of icosapent ethyl in statin-treated patients with elevated triglycerides and high or very-high ASCVD risk. J Clin Lipidol 2019; 13:860-872