ABSTRACT
This chapter summarizes the intimate relationship between the hypothalamus and the anterior pituitary with respect to the secretion of ACTH and GH from the physiological viewpoint. Other chapters in Endotext cover the hormones prolactin, LH, FSH, TSH and the posterior pituitary. Adrenocorticotropic hormone (ACTH) and growth hormone (GH) are both peptide hormones secreted from the anterior pituitary. ACTH is derived from cleavage of the precursor hormone pro-opiomelanocortin (POMC) by prohormone convertase enzymes. Classically, it activates the production and release of cortisol from the zona fasciculata of the adrenal cortex via the melanocortin receptor MC2R. The major hypophysiotropic factor controlling ACTH expression and secretion is corticotropin-releasing hormone (CRH), in conjunction with arginine vasopressin (AVP). Key physiological features of the hypothalamo-pituitary-adrenal (HPA) axis are discussed, including the ultradian pulsatility of CRH, AVP and ACTH secretion, the circadian pattern of secretion, the negative feedback of cortisol on the HPA axis, the stress response, and the effects of aging and gender. GH is secreted mainly by somatotrophs in the anterior pituitary, but it is also expressed in other parts of the brain. Similarly, to ACTH, the release of GH is pulsatile with diurnal variation, under a negative feedback auto-regulatory loop, and can be affected by various factors. Activities that affect secretion of GH include sleep and exercise, and physical stresses such as fasting and hypoglycemia, hyperglycemia, hypovolemic shock, and surgery. GH secretion demonstrates differences between the sexes, with male ‘pulsatile’ secretion versus female ‘continuous’ secretion. In addition, the level of secretion also declines with age, a phenomenon termed the ‘somatopause’. All these are discussed in detail in this chapter.
THE HYPOTHALAMO-PITUITARY INTERFACE
The hypothalamus and pituitary serve as the body’s primary interface between the nervous system and the endocrine system. This interface takes the form of:
- Amplification from femto (10-15) and pico (10-12)-molar concentrations of hypophysiotropic hormones to nano (10-9) molar concentrations of pituitary hormones.
- Temporal smoothing from ultradian pulsed secretion of hypophysiotropic hormones to circadian rhythms of pituitary hormone secretion (1).
The function of this interface is modified by feedback, usually negative, via the nervous system and via the endocrine system.
REGULATION OF ACTH
Cells of Origin
ACTH is released from corticotrophs in the human pituitary, constituting 15-20% of the cells of the anterior pituitary (see Endotext chapter- Development and Microscopic Anatomy of the Pituitary Gland). They are distributed in the median wedge, anteriorly and laterally, and posteriorly adjacent to the pars nervosa. These cells are characteristically identified from their basophil staining and PAS-positivity due to the high glycoprotein content of the N-terminal glycopeptide of pro-opiomelanocortin (vide infra), as well as ACTH immunopositivity. Scattered ACTH-positive cells are also present in the human homologue of the intermediate lobe. Some of these appear to extend into the posterior pituitary, the so-called “basophilic invasion” (2).
ACTH/POMC
POMC GENE STRUCTURE
ACTH is derived from a 266 amino acid precursor, pro-opiomelanocortin (POMC: Figure 1). POMC is encoded by a single-copy gene on chromosome 2p23.3 over 8 kb (3). It contains a 5′ promoter and three exons. Apart from the hydrophobic signal peptide and 18 amino acids of the N-terminal glycopeptide, the rest of POMC is encoded by the 833 bp exon 3.
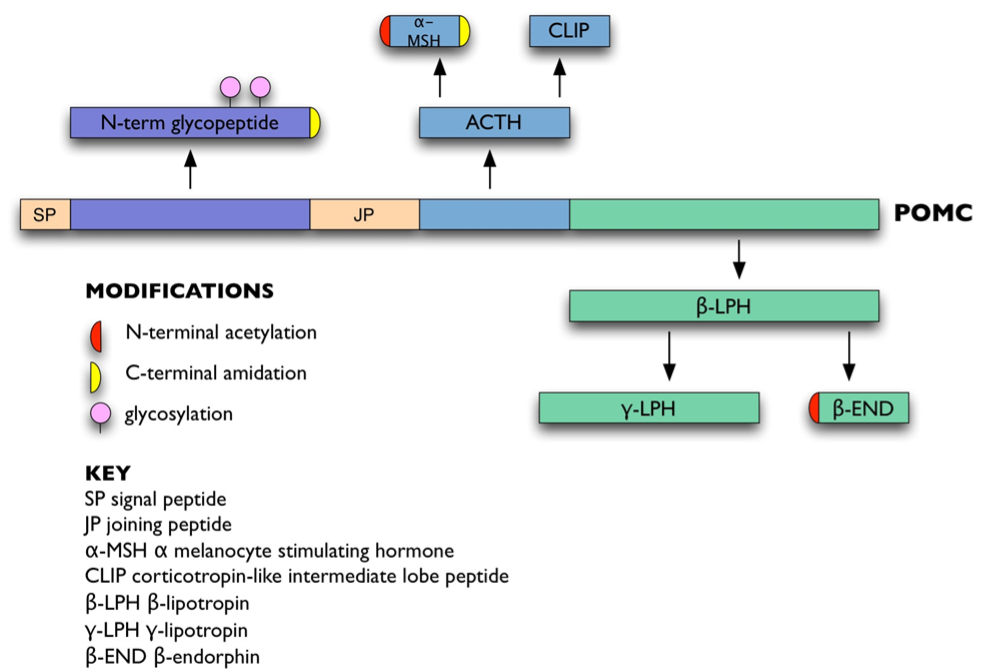
Figure 1. POMC and its derivatives.
The promoter of POMC has most extensively been studied in rodents (4). Common transcription elements such as a TATA box, a CCAAT box, and an AP-1 site are found within the promoter (5,6). Corticotroph and melanotroph-specific transcription of POMC appears to be dependent on a CANNTG element motif synergistically binding corticotroph upstream transcription element-binding (CUTE) proteins (7). These include neurogenic differentiation 1 factor (NeuroD1) (8), pituitary homeobox 1 (Pitx1 or Ptx1) (9), and Tpit (10,11). NeuroD1 is a member of the NeuroD family and forms heterodimers with other basic-helix-loop-helix (bHLH) proteins, activating transcription of genes that contain an E-box, in this case POMC. This highly restricted pattern of expression in the nervous and endocrine systems is important during development. NeuroD1 is expressed in corticotrophs but not melanotrophs, thus indicating that there are some differences between the operations of the transcriptional mechanisms of these two POMC-expressing cell types (8). Tpit is a transcription factor of the T-box family and it plays an important role in late-stage cell determination of corticotrophs and melanotrophs (10). Pitx1 is a homeoprotein belonging to a class of transcription factors that are involved in organogenesis and cell differentiation. Both Tpit and Pitx1 bind to their respective responsive elements and are involved in controlling the late differentiation of POMC gene expression, maintaining a basal level of POMC transcription and participating in hormone-induced POMC expression (12). To summarize the respective roles of the CUTE proteins, Pitx1 confers pituitary specificity in the broadest sense, Tpit confers the POMC lineage identity common to corticotrophs and melanotrophs, whereas NeuroD1 expression confers corticotroph identity (4). However, CUTE proteins are not the only method by which POMC expression is differentiated between corticotrophs and melanotrophs. The Pax7 transcription factor has been shown to be a key determinant of melanotroph identity, and it works by remodeling chromatin prior to Tpit expression, opening key areas of chromatin to allow Tpit and other transcription factors access to enhancers, resulting in melanotroph specification (13).
Ikaros transcription factors, which had previously been characterized as being essential for B and T cell development, have been demonstrated to bind and regulate the POMC gene in mice. Moreover, Ikaros knockout mice demonstrate impaired corticotroph development in their pituitaries, as well as reduced circulating ACTH, MSH, and corticosterone levels (14), suggesting a role in corticotroph development.
POMC transcription is positively regulated by corticotrophin releasing hormone (CRH). CRH acts via its G-protein coupled receptor to activate adenylate cyclase, increase intracellular cAMP and stimulate protein kinase-A (15). Transcription stimulation is mediated by an upstream element (PCRH-RE) binding a novel transcription factor (PCRH-REB) containing protein kinase-A phosphorylation sites (16). CRH also stimulates the transcription of c-Fos, FosB and JunB, as well as binding to the POMC AP-1 site (17). Another secondary messenger pathway that controls POMC expression involves intracellular Ca2+ ions (18). Both cAMP and intracellular Ca2+ pathways cross-talk with each other(19). These findings further support the importance of cAMP and Ca2+ in the intracellular signaling of corticotrophs and melanotrophs. Interestingly, there is a remarkable absence of cAMP-responsive elements (CRE) and Ca2+ responsive elements (CaRE) in the promoter region of POMC despite the demonstrated importance of cAMP and Ca2+ in the intracellular signaling of corticotrophs and melanotrophs. Other, more indirect strategies have evolved to translate cAMP signals into changes in POMC gene expression involving a CREB/c-Fos/AP-1 signaling cascade activating POMC transcription via an activator protein-1 (AP-1) site in exon 1. Similarly, intracellular Ca2+ may signal via the Ca2+ binding repressor DREAM (downstream response element-antagonist modulator) and modulation of c-Fos expression (20).
CRH also activates POMC expression through a Nur response element which binds the related orphan nuclear receptors Nur77, Nurr1, and NOR1 (21). The pituitary adenylate cyclase-activating peptide (PACAP) also stimulates cAMP synthesis and POMC transcription, presumably through a common pathway with CRH (22).
The effect of Nuclear transcription factor kappa B (NF-κB) on POMC expression is unclear. Although NF-κB is mostly associated with an activation of gene expression, it has been shown to inhibit POMC gene expression by binding to the promoter region (23). In keeping with this finding, CRH treatment blocks this binding, leading to an increase in POMC expression. On the contrary, it has also been shown that more pertinent high glucose (metabolic stress condition) elevates POMC transcription in AtT-20 cells through, or at least in part, the NF-κB responsive element and AP-1 sites (24).
POMC mRNA transcription in corticotrophs is negatively regulated by glucocorticoids (25), although glucocorticoids increase expression of POMC in the hypothalamus (26). The inhibitory effect of glucocorticoids on corticotroph POMC expression appears, in the rat POMC promoter, to be dependent on a glucocorticoid response element partially overlapping the CCAAT box (27). The element binds the glucocorticoid receptor as a homodimer plus a monomer on the other side of the DNA helix (28). Glucocorticoid regulation of corticotroph POMC transcription is also indirectly mediated via other mechanisms such as down-regulation of c-jun expression and direct protein-protein mediated inhibition of CRH-induced AP-1 binding (29), inhibition of CRH receptor transcription (30), inhibition of CRH/cAMP induced activation of Tpit/Pitx1, inhibition of CRH action via the Nur response element (12), and suppression of NeuroD1 expression which in turn inhibits the positive NeuroD1/E-box interaction in the POMC promoter (31).
There are also some other nuclear receptors and respective ligands that show potential roles in POMC regulation. All-trans retinoic acid (ATRA), a stereoisomeric form of retinoic acid, has been shown to inhibit POMC transactivation and ACTH secretion in murine corticotroph tumor AtT20 cells via inhibition of AP-1 and Nur transcriptional activities (32). Mutations in the retinoic acid receptor-related orphan receptors (ROR) also result in enhanced corticosterone secretion and ACTH response as well as a lack of diurnal variation compared to wild-type mice (33). As for the thyroid hormone and its receptor, there appears to be no reported direct interaction with the POMC promoter, although POMC-/- animals are known to display primary hyperthyroidism (34). More studies are needed to elucidate the potential roles of different nuclear receptors and ligands in POMC regulation. It is also important to note that most of these studies were conducted using tumor cells or in vitro models, as some of the global knockout models can be lethal or difficult.
Leukemia Inhibitory Factor (LIF), a pro-inflammatory cytokine expressed in corticotrophs, has also been shown to stimulate POMC transcription via activation of the Jak-STAT pathway (35,36). This stimulation is synergistic with CRH. Deletional analysis of the POMC promoter has identified a LIF-responsive region from –407 to –301. A STAT binding site that stimulates POMC transcription and which partly overlaps with the Nur response element has been identified within the POMC promoter (37). This pathway might form an interface between the immune system and regulation of the pituitary-adrenal axis, particularly during chronic inflammation, where pro-inflammatory cytokines such as LIF might stimulate STAT3 expression and therefore POMC transcription (38). Another interface between the immune system and POMC expression involves Toll-like receptor (most likely TLR4) recognition of lipopolysaccharide, which is a component of the bacterial cell wall. This appears to act via activation of c-Fos and AP-1 expression (39).
The POMC promoter sits within a CpG island, defined as the regions in the genome which the G and C content exceed 50%. These genomic regions are important controllers of gene expression as hypermethylation of the cytosine leads to silencing of gene expression via remodeling of the chromatin structure to favor heterochromatinization (40). Hypermethylation of the POMC promoter leads to repression of POMC expression in non-expressing tissues. In contrast, hypomethylation leads to de-repression of the POMC promoter in POMC expressing tissues (e.g. corticotrophs). Notably, a small cell lung carcinoma cell line, which expresses POMC and ACTH, possesses a hypomethylated POMC promoter, suggesting that ectopic ACTH secretion by tumors may be due to hypomethylation at a relatively early stage in carcinogenesis (41).
BIOGENESIS OF ACTH
Prohormone convertase enzymes PC1 and PC2 process POMC at pairs of basic residues (Lys-Lys or Lys-Arg). This generates ACTH, the N-terminal glycopeptide, joining peptide, and beta-lipotropin (beta-LPH) (Figure 1). ACTH can be further processed to generate alpha-melanocyte stimulating hormone (alpha-MSH) and corticotropin-like intermediate lobe peptide (CLIP), whereas beta-LPH can be processed to generate gamma-LPH and beta-endorphin (42). In corticotrophs, POMC is mainly processed to the N-terminal glycopeptide, joining peptide, ACTH, and beta-LPH; smaller amounts of the other peptides are present (43). Other post-translational modifications include glycosylation of the N-terminal glycopeptide (44), C-terminal amidation of N-terminal glycopeptide, joining peptide and alpha-MSH (45,46), and N-terminal acetylation of ACTH, alpha-MSH and beta-endorphin (47,48).
HYPOPHYSIOTROPIC HORMONES AFFECTING ACTH RELEASE
Corticotropin Releasing Hormone (CRH)
This 41 amino acid neuropeptide (49) is derived from a 196-amino acid prohormone (50). CRH is likely to be involved in all the three types of stress responses: behavioral, autonomic and hormonal. CRH immunoreactivity is mainly found in the paraventricular nuclei (PVN) of the hypothalamus, often co-localized with AVP (51). CRH is part of a family of neuropeptides together with the urocortins 1, 2 and 3 (52).
CRH binds to G-protein coupled seven-transmembrane domain receptors (53,54), which are classically coupled to adenylate cyclase via Gs, stimulating cAMP synthesis and PK-A activity. However, it is increasingly clear that CRH receptors also couple to Gi (inhibiting adenylate cyclase) and Gq (stimulating phospholipase C, the processing of phosphatidylinositol 4,5-bisphosphate into inositol trisphosphate and diacylglycerol and intracellular Ca2+ release), as well as the recruitment of beta-arrestins which counter-regulate CRH-R function via G-protein decoupling and receptor internalization/desensitization (52).
To date, two CRH receptor genes have been identified in humans. CRH-R1 mediates the action of CRH at corticotrophs by binding to CRH; it also binds urocortin 1. CRH-R1 is most extensively expressed in the CNS. CRH-R2 binds to all three urocortins, while binding CRH at a far lower affinity (52). CRH-R2 is predominantly expressed in the heart and has profound effects on the regulation of the cardiovascular system and blood pressure (55,56).
Besides stimulating POMC transcription and ACTH biogenesis, CRH stimulates the release of ACTH from corticortophs via CRH-R1 leading to a biphasic response with the fast release of a pre-synthesized pool of ACTH, and the slower and sustained release of newly-synthesized ACTH (57). Figure 2 describes the stimulation of ACTH release by CRH (58). It is clear that CRH and CRH-R1 is the ‘main line’ of the HPA axis with major defects in this axis with CRH (59) and CRH-R1 knockout (60). Although urocortin 1 can also activate CRH-R1, urocortin 1 knockout mice appear to have normal HPA axis function, suggesting that urocortin 1 does not have a significant regulatory role on the axis (61). Indeed, knocking out all three urocortins does not have any major effect on basal corticosterone levels (62) although female urocortin 2 knockout mice exhibit a more subtle dysregulation with elevated basal ACTH and corticosterone secretion which is modulated by their estrogen status (63).
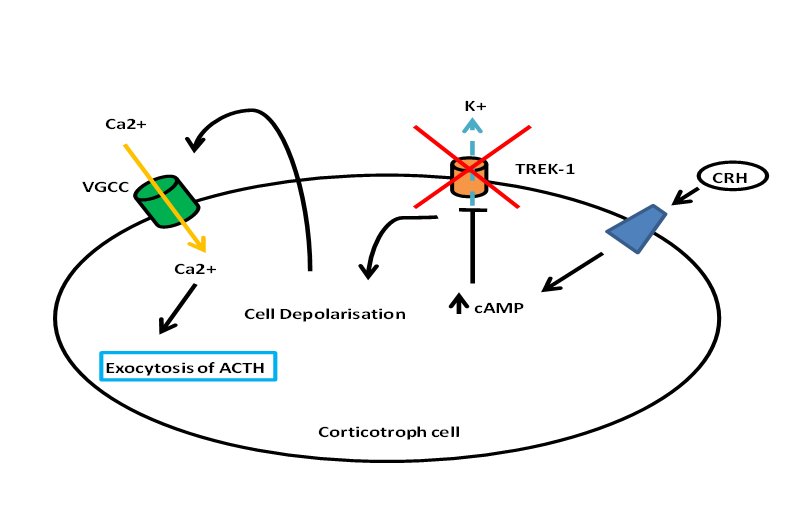
Figure 2. Diagram showing the release of ACTH from corticotroph cells. CRH binds to a particular receptor that leads to activation of cAMP. The rise in cAMP inhibits TREK-1, thus leading to the depolarization of the cell and subsequently influx of calcium via VGCC. The rise in intracellular calcium leads to the exocytosis and release of ACTH.
CRH secretion is also regulated by other neurotransmitters and cytokines. These include acetylcholine, norepinephrine/noradrenaline, histamine, serotonin, gamma-aminobutyric acid (GABA), interleukin-1beta, and tumor necrosis factor. All of these factors increase hypothalamic CRH expression, except for GABA which is inhibitory.
Arginine Vasopressin (AVP)
In the anterior pituitary, AVP principally binds to the seven-transmembrane domain V1b receptor, also known as the V3 receptor (64). The receptor is coupled to phospholipase C, phosphatidyl inositol generation, and activation of protein kinase-C (65,66) and not via adenylate cyclase and cAMP (15). AVP stimulates ACTH release weakly by itself, but synergizes with the effects of CRH on ACTH release (67). Downregulation of protein kinase C by phorbol ester treatment abolishes the synergistic effect of AVP on ACTH release by CRH (68). AVP does not stimulate POMC transcription either by itself or in synergism with CRH (69). Between the two neuropeptide effects on ACTH release, CRH is the more dominant effect although there is some residual HPA axis activation in female CRH knockout mice (59).
The association between AVP and ACTH release suggests that measurement of AVP levels might be useful for assessing anterior pituitary function. However, direct measurement of plasma AVP is technically difficult due to its small molecular size and binding to platelets. Copeptin is a 39-amino acid glycosylated peptide which is derived from the C-terminal part of the AVP precursor at an equimolar amount to AVP. It remains stable for several days at room temperature in serum or plasma, and its measurement is reliable and reproducible, making it a biomarker of AVP release (70). The copeptin increment during glucagon stimulation testing correlates well with the ACTH increment in healthy controls, but not in patients with pituitary disease (71). Interestingly, there appears to be a sexual dimorphism in terms of the correlation between copeptin and ACTH/cortisol release under the conditions of insulin tolerance testing, with a positive correlation observed in women but no significant correlation in men, i.e. copeptin cannot be used as a universal marker of HPA axis stimulation (72).
Other Influences on ACTH Release
Glucocorticoids rapidly travel through circulation to inhibit the HPA axis at the level of the hypothalamus (release of CRH) (73-76) and anterior pituitary (release of ACTH) (77,78) when synthesized. There is an inherent short delay in this dynamic relationship between the hypothalamus-pituitary-adrenal system but nevertheless it is one of the main influences on ACTH release.
The mineralocorticoid system has always been closely linked to the glucocorticoid system. The endogenous glucocorticoids bind to the mineralocorticoid receptors with a 10-fold greater affinity than to the glucocorticoid receptors (79-81). The mineralocorticoid receptors have a more restricted expression profile throughout the body, with notably high levels of expression in the kidney and adipose tissue, although it is also expressed in certain parts of the brain (82). Administration of mineralocorticoid antagonists intracerebroventricularly or intrahippocampal infusion have been shown to increase the basal HPA axis activity as well as potentiate the initial rise of ACTH in response to stress (83,84).
Oxytocin and AVP have been co-localized to the PVN and supraoptic nuclei of the hypothalamus (85). Oxytocin controversially inhibits ACTH release in man (86-88) by competing for AVP receptor binding (89), but its more dominant effect seems to be a potentiation of the effects of CRH on ACTH release (90,91).
Vasoactive intestinal peptide (VIP) and its relative, peptide histidine isoleucine (PHI), have been shown to activate ACTH secretion (92). This is most probably mediated indirectly via CRH (93).
Atrial natriuretic peptide (ANP) 1-28 has been localized to the PVN and supraoptic nuclei (94). In healthy males, infusion of ANP 1-28 was reported to attenuate the ACTH release induced by CRH (95,96), but this only occurs under highly specific conditions and is not readily reproducible. In physiological doses, ANP 1-28 does not appear to affect CRH-stimulated ACTH release (97).
Opiates and opioid peptides inhibit ACTH release (98). There does not seem to be a direct action at the pituitary level. It is likely that these act by modifying release of CRH at the hypothalamic level (99). Opiate receptor antagonists such as naloxone or naltrexone cause ACTH release by blocking tonic inhibition by endogenous opioid peptides (100).
The endocannabinoid system has recently appeared as a key player in regulating the baseline tone and stimulated peaks of ACTH release. The seven-transmembrane cannabinoid receptor type 1 (CB1) is found on corticotrophs, and the endocannabinoids anandamide and 2-arachidonoylglycerol can be detected in normal pituitaries (101). Antagonism of CB1 causes a dose-dependent rise in corticosterone levels in mice (102). CB1-/- knockout mice demonstrate higher corticosterone levels compared to wild-type CB1+/+ littermates, although the circadian rhythm is preserved. Treatment of the CB1-/- mice with low-dose dexamethasone did not significantly suppress their corticosterone levels and surprisingly caused a paradoxical rise in ACTH levels when compared to the wild-type, although high-dose dexamethasone suppressed corticosterone and ACTH to the same degree in both CB1-/- and CB1+/+ mice. These CB1-/- mice have: (1) higher CRH mRNA expression in the PVN; (2) lower glucocorticoid receptor mRNA expression in the CA1 hippocampal region, but not in the dentate gyrus or the PVN; (3) significantly higher baseline ACTH secretion from primary pituitary cell cultures as well as augmented ACTH responses to stimulation with CRH or forskolin (103). It has also been known for some time that the administration of the cannabinoid agonist delta-9-tetrahydrocannabinol (THC) for 14 days suppresses the cortisol response to hypoglycemia in normal humans (104). Thus, the endocannabinoids appear to negatively regulate basal and stimulated ACTH release at multiple levels of the hypothalamo-pituitary-adrenal axis.
Catecholamines act centrally via alpha1-adrenergic receptors to stimulate CRH release. Peripheral catecholamines do not affect ACTH release at the level of the pituitary in humans (105).
Nitric oxide (NO) and carbon monoxide negatively modulate the HPA axis by reducing CRH release, at least in vitro(106,107). Endotoxin administered into isolated rat hypothalamus led to generation of NO and CO, which subsequently led to significant decrease in CRH and vasopressin secretion (107).
GH secretagogues such as ghrelin and the synthetic GH secretagogue hexarelin stimulate ACTH release, probably via stimulating AVP release with a much lesser effect on CRH (108-111). GH-releasing peptide-2 (GHRP-2) has also been shown to cause ACTH release in humans (112,113). GH releasing hormone (GHRH) has been shown to potentiate the ACTH and cortisol response to insulin-induced hypoglycemia, but not to potentiate the ACTH and cortisol response after administration of CRH/AVP (114).
Obestatin, a 23 amino acid amidated peptide, is derived from preproghrelin, which is the same precursor as ghrelin (Figure 3). Obestatin is found to suppress food intake and have opposing metabolic effects to ghrelin when administered intraperitoneally in mice (115). An early study showed that intravenous or intracerebroventricular obestatin had no effects on pituitary hormone release (GH, prolactin, ACTH and TSH) in male rats (116), consistent with the fact that the obestatin receptor GPR39 is not expressed in the pituitary (115,117,118). A study in mice and non-human primates (baboon) again showed no effects of obestatin on prolactin, LH, FSH and TSH expression and release. However, obestatin was shown to stimulate POMC expression and ACTH release in vitro and in vivo, and in this study the authors found GPR39 expression in pituitary tissue and primary pituitary cell cultures, contrary to the above-mentioned studies. This effect was mediated by the adenylyl cyclase and MAPK pathways. The increase in ACTH release was also associated with an increase in pituitary CRH receptor expression. Interestingly, obestatin did not inhibit the stimulatory effect of ghrelin on ACTH release (119). Therefore, the effects of obestatin on pituitary hormone secretions remain controversial.

Figure 3. Schematic diagram showing the synthesis of ghrelin and obestatin from the same precursor, preproghrelin. Preproghrelin is a 117 amino acid precursor encoded at chromosome 3. Cleavage of this protein leads to the production of ghrelin, a 28 amino acid peptide, and obestatin, a 23 amino acid protein. Ghrelin can be present as both des-acyl- and acyl-ghrelin (figure modified from (291)).
Angiotensin II (Ang II) is able to stimulate ACTH release in vitro from pituitary cells (120). Central Ang II is likely to stimulate CRH release via its receptors in the median eminence, as passive immunization with anti-CRH can abolish the effect of Ang II (121). Intracerebroventricular Ang II can stimulate ACTH release in rats (122) and is able to stimulate the synthesis of CRH and POMC mRNA (123). Conversely, blockade of Ang II subtype 1 (AT1) receptors with candesartan is able to decrease the CRH, ACTH, and cortisol response to isolation stress in rats (124,125). There is some controversy as to whether peripheral Ang II can modulate ACTH secretion. It is likely that the ACTH rise seen after Ang II infusion into rats is mediated via circumventricular organ stimulation, as blockade of Ang II effects on the circumventricular organs with simultaneous infusion of saralasin blocks this rise (122).
In vitro studies have shown an inhibitory effect of somatostatin on ACTH release in AtT-20 pituitary cell lines from rats, which is mediated via somatostatin receptor (SSTR) subtypes 2 and 5 (126). This inhibitory effect is dependent on the absence of glucocorticoids in the culture medium, but is more prominent when somatostatin analogues targeting SSTR 5 are used (127,128). In rodents, pasireotide, a somatostatin analogue capable of activating SSTRs 1, 2, 3, and 5, is capable of inhibiting CRH-stimulated ACTH release in contrast to octreotide (selective for SSTRs 2 and 5), which was less efficacious (129). Early in vivo studies in humans showed no effect of somatostatin on basal or CRH-stimulated ACTH release (130), although somatostatin does decrease basal secretion in the context of Addison’s disease (131). It is unlikely, therefore, that somatostatin itself is an inhibitor of ACTH release in normal human physiology. Corticotroph adenomas express the somatostatin receptor (SSTR) subtype 5 (132) and ACTH secretion from cultured corticotroph adenomas is inhibited by pasireotide (133). This is the basis for the use of pasireotide to treat Cushing’s disease (134). Octreotide is clinically ineffective in this context (135), but may be effective if glucocorticoids are lowered.
The role of TRH in ACTH release is in dispute. Although there is evidence that prepro-TRH 178-199 can inhibit both basal and CRH-stimulated ACTH release in AtT-20 cell lines and rat anterior pituitary cells (136,137), other investigators have not been able to confirm this (138). There has also been another study showing that TRH is able to induce ACTH release from AtT-20/NYU-1 cells (139), but no in vivo studies exist to substantiate a physiological role.
Tumor necrosis factor-alpha (TNFalpha) is a macrophage-derived pleiotropic cytokine that has been shown to stimulate plasma ACTH and corticosterone secretion in a dose-dependent manner (140). The primary site of action of TNFalpha effect on the HPA axis is likely to be on hypothalamic CRH-secreting neurons. The effects are abolished with CRH antiserum treatment, thus suggesting that CRH is a major mediator of the HPA axis response to TNFalpha.
Interleukins IL-1, IL-6 and possibly IL-2 appear to stimulate ACTH release (141-143). There seem to be multiple mechanisms for interleukins to stimulate ACTH release, but most of the acute effects of these agents are almost certainly via the hypothalamus (144).
Leukemia Inhibitory Factor is able to stimulate POMC synthesis, as noted above.
Endothelial Growth Factor (EGF) is a pituitary cell growth factor that is previously known to induce production of prolactin (145). Both EGF and its receptor (EGFR) are expressed in normal pituitary tissue (146). More recently, EGF has been found to regulate the transcription of POMC and production of ACTH (147-149). The mechanism behind this is still unclear, although mutations in ubiquitin-specific protease 8 (USP8), a deubiquitinase enzyme with various targets including EGFR, leading to hyperactivation of this enzyme and subsequent increased EGFR deubiquitination and recirculation to the cell surface, enhance the release of ACTH (147,150). A significant percentage of corticotroph adenomas harbor somatic mutations in USP8, and a germline mutation case have also been described and can develop Cushing’s disease (147,150,151). These findings further provide evidence that EGF and EGFR can regulate production of ACTH.
PHYSIOLOGY OF ACTH RELEASE
Pulsatility of ACTH Release
Frequent sampling of ACTH with deconvolution analysis reveals that it is secreted in pulses from the corticotroph with 40 pulses ± 1.5 measured per 24 hours, on analysis of 10-minute sampling data. These pulses temporally correlate with the pulsed secretion of cortisol, allowing for a 15 minute delay in secretion, and correlate in amplitude (152). Pulse concordance has been measured at 47% (ACTH to cortisol) and 60% (cortisol to ACTH) in one study (153), and 90% (ACTH to cortisol) in another (154). Although the pulsatility of ACTH secretion may result from pulsatile CRH release, there is evidence that isolated human pituitaries intrinsically release ACTH in a pulsatile fashion (155). In addition, studies in rats have shown that constant CRH infusion still resulted in oscillations of ACTH and glucocorticoid release (156). However, the pulsatile activities of ACTH and glucocorticoid are entirely dependent on the level, rather than the pattern, of CRH secretion (156).
The pulsatile release of ACTH induces pulses of glucocorticoid secretion. In rats with HPA axis suppression, constant infusion of ACTH did not induce pulsatile glucocorticoid secretion (157). It was shown in vitro using ZF cell lines that constant ACTH treatment led to larger increase in pCREB and steroidogenic gene transcription at the start of treatment but the cells became unresponsive to the stimuli over time (158). The responsiveness of cells to the ACTH treatment could only be maintained with pulsatile ACTH treatment, further supporting the importance of pulsatile release of ACTH physiologically.
Recent developments in automated sampling of blood (159) and tissue interstitial fluid via microdialysis (160) have also uncovered the ultradian rhythms in plasma ACTH which correlate well with plasma and tissue steroid (cortisol and cortisone) concentrations, indicating that the ultradian rhythms in blood ACTH and cortisol/cortisone translate well to tissue exposure to these steroids.
Circadian Rhythm
In parallel with cortisol, ACTH levels vary in an endogenous circadian rhythm, reaching a peak between 06.00-09.00h, declining through the day to a nadir between 23.00h-02.00h, and beginning to rise again at about 02.00-03.00h. An increase in ACTH pulse amplitude rather than frequency is responsible for this rhythm (152). The circadian rhythm in glucocorticoid secretion is a key mechanism for re-entraining behavior in the face of external perturbations such as an abrupt phase shift of light conditions, i.e. a model of ‘jet lag’ (161).
The circadian rhythm is mediated via a master oscillator in the supra-chiasmatic nucleus (SCN). A lesion in the SCN eliminates the glucocorticoid circadian rhythm (162). An autoregulatory negative transcription-translation loop feedback system involving cyclical synthesis of the period proteins Per1-3, Clock/BMAL1, and Cry1/2 acts as the basic molecular oscillator, where the Clock/BMAL1 heterodimer acts to activate the transcription of Per and Cry proteins (the so-called ‘positive limb’). In turn, the Per and Cry proteins complex together, translocate back into the nucleus and inhibit Clock/BMAL1-mediated transcription (the so-called ‘negative limb’). The system is reset by phosphorylation, ubiquitination and proteasomal degradation of the Per/Cry repressor complexes (163,164). Entrainment of the oscillator is achieved by light input from the retina, mediated via the retino-hypothalamic tract. Light-activated transcription of immediate-early genes such as c-fos and JunB (165,166) causes activation of PER1 gene transcription as well as modification of the acetylation pattern of histone tails. The latter are implicated in the control of chromatin structure and accessibility of genes to transcription (167). The impact of a period protein gene deletion on circulating glucocorticoids depends on which side of the clock feedback loop is affected (164). Knockout mice with mutations in the components of positive limb of the oscillator (Clock or BMAL1) suffer from hypocortisolism and lose circadian cyclicity (168,169). The deletion of Per2, which affects the negative limb of the oscillator, also results in hypocortisolism (170). However, Cry1 knockout (also affecting the negative limb) leads to hypercortisolism (171,172).
Is a circadian rhythm in CRH secretion responsible for the ACTH rhythm? Although there is a report of a circadian rhythm in CRH secretion (173), and in situ hybridization studies show that there is a circadian rhythm in CRH expression in the suprachiasmatic nucleus (174), other reports do not confirm this (175). Moreover, the circadian rhythm persists despite a continuous infusion of CRH, suggesting that other factors are responsible for the modulation of ACTH pulses (176). The most likely alternative candidate is AVP: immunocytochemical studies show a circadian rhythm in AVP expression (177) and Clock knockout mice show a loss of the circadian rhythm in AVP RNA expression in the SCN (178). In addition, metyrapone and CRH infusion in normal individuals showed a persistence of the HPA circadian rhythm, thus further supporting the role of AVP in regulating ACTH rhythm (176).
However, rhythmic HPA axis activity is not the be-all and end-all of the circadian rhythm of glucocorticoid release. For example, the adrenal rhythm of cortisol secretion persists after hypophysectomy (179). Indeed, light pulses can induce glucocorticoid secretion independent of ACTH secretion. This HPA axis-independent pathway is mediated by the sympathetic nervous system innervation of the adrenals (180). The adrenal glands also possess an independent circadian oscillator: oscillatory Clock/BMAL1, Per1-3 and Cry1 expression is seen in the outer adrenal cortex (zona glomerulosa and zona fasciculata). This adrenal circadian clock appears to ‘gate’ the response to ACTH, i.e. it defines a time window during which ACTH is most able to stimulate glucocorticoid secretion (181). Exogenous ACTH is capable of phase-dependently resetting glucocorticoid rhythms (182), suggesting that the adrenal circadian clock can be entrained by the ACTH rhythm. This illustrates a general principle of circadian system organization, namely that there is a hierarchical system with the SCN master clock entraining and coordinating peripheral and non-SCN tissue clocks via endocrine and neuronal signals.
Stress
Stress, both physical and psychological, induces the release of ACTH and cortisol, particularly via CRH and AVP (183,184), and increases the turnover of these neurohypophysiotropic factors by increasing the transcription of CRH and AVP (185).
During acute stress, an immediate activation of the autonomic nervous system takes place, followed by a delayed response via the HPA axis-mediated release of glucocorticoids (164). During the initial stage, there is an immediate increase of catecholamines via activation of the sympathetic preganglionic neurons in the spinal cord, which in turn stimulates adrenal medulla production of catecholamines via splanchnic nerve innervation. The catecholamines released will also collectively affect peripheral effector organs where they are translated into the classical fight-or-flight response. The delayed response of stress involves activation of the HPA axis, leading to an increase in glucocorticoid level, which in turn can terminate the effects of the sympathetic response together with the reflex parasympathetic activation. It is important to note that this neurohormonal stress response has an additional endocrine leg in the form of glucagon: together, one of the important effects of this trio is to enhance the release of glucose, amino acids and fatty acids, a coordinated catabolic response to stress (186).
Stress paradigms studied in humans include hypoglycemia during the insulin tolerance test (Figure 4), and venipuncture (187). Elective surgery has also long been used as a paradigm of the stress response in humans (188-190): the magnitude of cortisol rise correlates positively with the severity of surgery (191). Experimentally, other stress paradigms such as hemorrhage, oxidative stress, intraperitoneal hypertonic saline, restraint/immobilization, foot shock, forced swimming, or shaking are used to study the stress responses in animals. Importantly, different stress paradigms can have differential effects on CRH and AVP. In situ hybridization with intronic and exonic probes can be used to study the transcription of heterogenous nuclear RNA (hnRNA), followed by its processing (including splicing, capping and polyadenylation) to messenger RNA (mRNA) within 1-2 hours. CRH and AVP hnRNA levels in rats subjected to restraint show significant increases at 1 and 2 hours after the induction of stress, followed by significant increases in mRNA levels at 4 hours (192). In contrast, intraperitoneal hypertonic saline causes a rapid 8.6-fold increase in CRH hnRNA and mRNA within 15 minutes, returning to basal levels by 1 hour. AVP hnRNA responses are slower, peaking at 11.5-fold increase by 2 hours, followed by a prolonged elevation of AVP mRNA levels from 4 hours onwards (193). As previously noted, serum copeptin can be used as a more stable biomarker of AVP secretion and copeptin increments correlate well with cortisol secretion in a glucagon stimulation test paradigm (71), but exhibit a sexual dimorphism in the context of the insulin tolerance test (72).
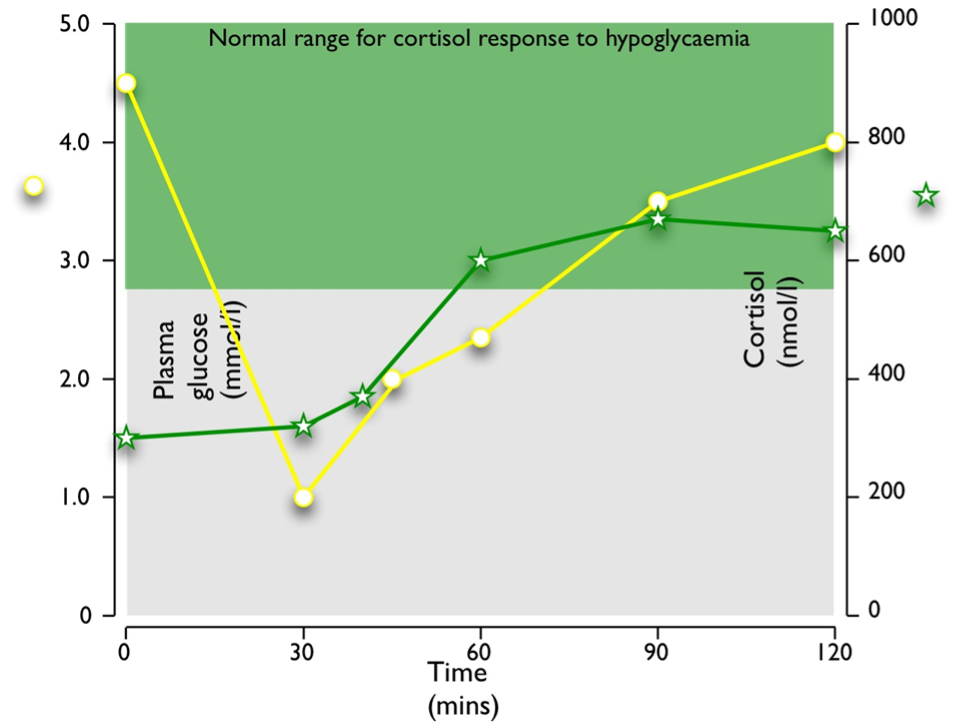
Figure 4. Typical response to hypoglycemia (≤2.2 mmol/l) induced by 0.15 U/kg Actrapid i.v. in a normal subject. Peak cortisol is ≥550 nmol/l.
Various stressors are known to stimulate oxytocin release which in turn, at least acutely, appears to potentiate CRH-induced ACTH secretion and therefore cortisol release (90). There are also roles for endogenous nitric oxide (NO) and carbon monoxide (CO) in modulating the ACTH response to stress (194). Neuronal NO synthase co-localizes with AVP and to some extent CRH in paraventricular neurons (195,196). Knockout mice lacking wild-type and neuronal NO synthase have much reduced quantities of POMC immunoreactivity in their arcuate nuclei and pituitaries compared to wild-type mice (195,197). In general, inflammatory stressors appear to activate an endogenous inhibitory pathway, whereby NO and CO attenuate the stimulated secretion of CRH and AVP. These effects can also be seen in terms of circulating AVP. However, the regulation of the pituitary-adrenal axis by other stressors may involve an activating role for these gaseous neurotransmitters. CRH-R2, as noted above, binds the urocortins 1, 2 and 3, and appears to mediate a down-regulatory role in the HPA response to stress: knockout mice exhibit a ‘hypersensitive’ acute ACTH and corticosterone response (198) and a defective recovery from stress with a slower drop in corticosterone (199).
Repetitive stress causes variable effects, enhancement or desensitization, on ACTH responses, depending on the stress paradigm involved. This appears to be positively correlated with changes in AVP binding to V1b receptors, reflecting changes in the number of binding sites and not their affinities. It is at present unclear whether this is due to changes in transcription of the V1b gene, alterations in mRNA stability, translational control or recruitment of receptors from intercellular pools (200). With chronic stress, oxytocin is thought to have a longer term stress-antagonistic function, partially via cortisol-mediated negative feedback on CRH, partially via GABAergic inhibition of CRH neuron function and partially via a direct inhibitory effect of oxytocin on CRH expression (90).
As noted above, circadian rhythms in adrenal ACTH responsiveness, controlled by local oscillator circuits, gate’ the glucocorticoid output in response to a certain level of ACTH. In the case of stress, this leads to markedly different glucocorticoid responses depending on when (during the active or inactive phase) the experimental stress is applied to experimental animals. Moreover, the timing of repetitive stress application can lead to differences in the behavioral and metabolic responses to repetitive/chronic stress. Lastly, it is also known that stress can influence clock function at the level of the SCN and also at the level of the adrenal circadian oscillator leading to phase shifts (164). In humans, stressors such as illness leads to abolition of the diurnal variation of cortisol, which appears to be ACTH independent (201,202). This change in the diurnal regulation of cortisol secretion is linked to regulation of immune responses which is likely to be adaptive in the acute context, but which may be maladaptive with chronic stress (203).
FEEDBACK REGULATION OF THE HPA AXIS
Glucocorticoid feedback occurs at multiple levels: at the pituitary, at the hypothalamus, and most importantly, centrally at the level of the hippocampus, which contains the highest concentration of glucocorticoid receptors in the central nervous system. Multiple effects mediate this feedback (Figure 5), including:
- inhibition of CRH and AVP synthesis and release in the PVN (204,205).
- inhibition of POMC transcription (as outlined above).
- inhibition of ACTH release induced by CRH and AVP (206).
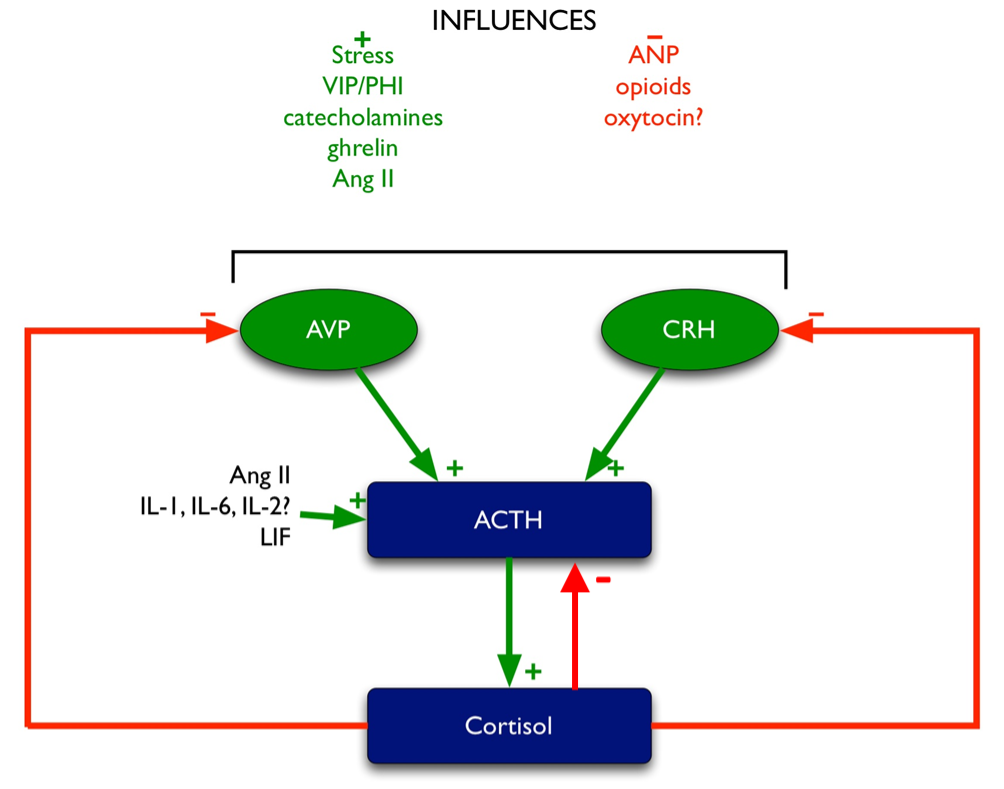
Figure 5. Regulation of ACTH. Green arrows denote stimulatory influences, red arrows denote inhibitory influences.
Fast feedback occurs within seconds to minutes and involves inhibition of ACTH release by the corticosteroids, mediated through the glucocorticoid receptor (GR). For example, an injection of prednisolone inhibits ovine CRH-stimulated ACTH release within 20 minutes (207). In vitro this appears to involve inhibition of CRH-stimulated ACTH release, and CRH release, but basal secretion is not affected. Protein synthesis is not required, implying that the glucocorticoid effect is non-genomic (208,209). Cell membrane-associated GR has recently been shown to directly mediate fast feedback inhibition by inhibition of Src phosphorylation in corticotrophs (210), but other work implicates the GC-induced secretion of annexin 1/lipocortin1 from folliculostellate cells as a paracrine mechanism for inhibition of ACTH release (211). In addition, receptors for ACTH (MC2R) are present in normal corticotrophs, allowing ‘ultra-fast’ feedback regulation of the HPA axis (212). The receptor expression is lost in the corticotroph adenomas of patients with Cushing’s disease, which could be the potential mechanism of resistance to feedback of the HPA axis seen in these patients (212).
Intermediate feedback occurs within 4 hours’ time frame and involves inhibition of CRH synthesis and release from CRH neurons, not affecting ACTH synthesis (209). However, it is thought that this is a relatively minor contributor to negative feedback (73). Slow feedback occurs over longer timeframes and involves inhibition of POMC transcription (209), via GR antagonism of Nur response element activation of POMC transcription by CRH. The molecular mechanism involves a GR-dependent recruitment of the histone deacetylase HDAC2 to a trans-repressor complex with Brg1, histone H4 deacetylation, and chromatin remodeling (213,214).
There is evidence that ACTH can inhibit CRH synthesis in the context of elevated CRH levels due to Addison’s disease or hypopituitarism, although not in the context of normal human subjects (215). Immunohistochemical studies of the paraventricular nuclei in adrenalectomized or hypophysectomized rats show a reduction of CRH and AVP positive cells when these rats are given ACTH infusions (216).
Glucocorticoids have also been shown to control the cell cycle in corticotrophs. This occurs via feedback repression of the positive cell-cycle regulators L-Myc, N-Myc, and E2F2, plus activation of the negative cell-cycle regulators Gadd45b, GADD45g, and Cables1. In this way, glucocorticoids negatively regulate corticotroph proliferation, a key influence which appears to be lost in corticotroph adenomas (217).
Eating
Cortisol is well known to rise after eating (218,219). This rise is provoked by two mechanisms: (i) by direct stimulation of the HPA axis; and (ii) via regeneration of cortisone to cortisol by stimulation of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) (220). The postprandial rise in cortisol has been shown to be mediated via increased pituitary ACTH secretion, which is in turn is modulated by central stimulant alpha-1 adrenoreceptors (221). The cortisol response to food is also enhanced in obese subjects compared to normal BMI individuals (222).
There also appear to be key differences between the effects of individual macronutrients, where carbohydrates lead to equal stimulation of the HPA axis and 11βHSD1, and where fat and protein led to greater stimulation of the HPA axis compared to 11βHSD1. Direct intravenous infusion of macronutrients such as Intralipid and amino acids does not stimulate cortisol secretion (223,224). The most likely candidates for the factors that mediate stimulation of the HPA axis after eating are the gut hormones which are released in response to enteral nutrients. For example, glucagon-like peptide-17-36 (GLP-17-36) has been shown to stimulate cortisol and ACTH secretion, suggesting a direct effect on the hypothalamus/pituitary (225-227). Gastric inhibitory peptide (GIP), however, has not been shown to stimulate cortisol secretion except in the special case of ectopic GIP receptors in bilateral adrenal hyperplasia, causing food-stimulated Cushing’s syndrome (228). 11HSD1 activity appears to be inhibited by GIP (229), therefore suggesting the GIP is not a key player in mediating the post-prandial rise in cortisol. Although ghrelin has been shown to increase cortisol secretion when given in infusion (108-110), ghrelin is suppressed after eating, making it an unlikely mediator of the post-prandial cortisol response.
AGING OF THE HPA AXIS
Studies in humans and experimental animals have shown evidence that hyperactivity of the HPA axis contributes to neuronal and peripheral deterioration associated with aging (230,231). Hyperactivity of the HPA axis can be caused by stress and is necessary as part of physiological adaptation. However, there must be mechanisms to limit the response to stress, especially during chronic stress, in order to avoid the damaging effects of prolonged exposure to stress hormones such as CRH and corticosterone.
High basal levels of glucocorticoids and loss of circadian rhythm have been associated with greater cognitive decline at a given age (232). Aging is associated with high basal levels of circulating corticosteroids, although there is not always a correlation between plasma ACTH and corticosteroids (233-235). In addition, there is also an alteration to the circadian rhythm of the HPA axis, as demonstrated by studies using a feeding-associated circadian rhythm paradigm. It was found that it took 1 week for young rats and 3 weeks for older rats to entrain the secretion of corticosterone in response to a restricted feeding schedule where they were fed for 2 hours per day. After the rats were shifted to a different pattern of feeding, the entrained circadian rhythm of corticosterone secretion persisted much longer in young rats than in older rats. This suggests that the aged HPA axis appears to take longer to adjust to changes in circadian rhythm, but such adjustments do not ‘stick’ as well as compared to the younger HPA axis (236,237).
When the expression of CRH in the SCN was examined using in situ hybridization, younger 3-4-month-old Sprague-Dawley rats exposed to light from 04.00h to 18.00h have a clear diurnal rhythm with higher expression seen in samples taken at 03.00h versus 23.00h. This rhythm was lost in older 17-20 month old rats with equal expression seen in samples from 03.00h and 23.00h (174). Fetal grafts containing the SCN have been shown to restore the circadian rhythm in old Sprague-Dawley rats, thereby suggesting that the altered diurnal variation of HPA axis probably involves alterations in the function of the suprachiasmatic nuclei (238).
Aging is also associated with an increase in expression of 11HSD1 both in brain and peripheral tissues (239,240). Such changes could conceivably expose tissues to elevated levels of glucocorticoids and contribute to the aging process.
The effects of aging on CRH regulation and whether CRH influences the course of aging are still unclear. Studies have reported increased, unchanged, or reduced hypothalamic CRH release and expression during aging (232).
GENDER DIFFERENCES IN HPA AXIS REGULATION
Endogenous glucocorticoid responses to stress are significantly elevated (in an estrogen-dependent fashion) in females as compared with males (241-244). This estrogen dependence is likely mediated through estrogen-response elements within the promoter regions of CRH (245). As previously noted, there is also a sexual differential in the relationship between AVP release and the ACTH/cortisol response during insulin tolerance testing where the serum levels of copeptin (as a marker of AVP release) positively correlate with ACTH/cortisol release in women but not men (72). However, the sexual dimorphism of the stress response is not seen with exercise-induced stress (246) nor acute psychological stress (247).
PSYCHONEUROENDOCRINOLOGY OF HPA
The link between the HPA axis and psychophysiopathology has long been speculated (248). Neuropsychological disturbances are well observed in humans and study models with abnormal or aberrant HPA axis.
Depression is associated with increased inflammation and given that HPA axis is strictly implicated in inflammation, it is hypothesized that alteration in HPA axis is associated with increase in pro-inflammatory cytokines causing depression, at least with a subgroup of individuals with depression (249). In depression, it is hypothesized that the regulation of ACTH and cortisol secretory activity are altered, along with impaired corticosteroid receptor signaling (250,251). Dysregulation of the HPA axis contributes to suppression of transcription of the brain-derived neurotrophic factor (BDNF) gene, thereby reducing the synthesis and secretion of BDNF protein, a nerve growth factor family (252). This leads to neurodegenerative changes, most prominently in hippocampus, observed in depression. Chronic excess of cortisol in the brain may also lead to serotonin deficiency due to decreased availability of tryptophan, the substrate for serotonin production, and reduction of density and reactivity of serotonin receptors (252). The use of antidepressants targeting this aspect of neurotransmission has shown to normalize the activity of HPA axis, decreasing the levels of CRH and consequently also ACTH and cortisol (252).
Some depressed patients were also reported to have enlarged adrenal glands (253,254) and impaired negative feedback with the hypercortisolemia, thereby suggesting that the level of impairment is at the glucocorticoid receptor-dependent negative feedback, either centrally or at the level of pituitary (255,256). However, when a study looking at 24-hour automated blood cortisol sampling study in depressed premenopausal women was conducted, it found only 6 patients (24%) of a cohort of 25 to have hypercortisolemia (257). This suggests that not all depressed patients will have hypercortisolemia as the main feature of dysregulation of HPA axis. The impairment of the negative feedback was hypothesized to be due to the diminished sensitivity of the glucocorticoid receptors (the ‘glucocorticoid resistance’ theory) secondary to reduced receptor function and expression, shown in large number of experimental, biological and molecular studies (258).
In bipolar disorder, an increase in cortisol secretion may be seen in the manic phase (259). Interestingly, a weaker cortisol awakening response is observed in patients with depression, mania and partial remission against those of healthy control subjects (260), thereby indicating dysregulation of HPA axis in bipolar disorder subjects.
In schizophrenia, individuals who developed or at risk of developing psychosis have been observed to have elevated levels of cortisol measured upon waking up (261,262). The disturbance is more pronounced in individuals not treated with antipsychotic medications. Elevated cortisol levels appear to be correlated with the risk of a first psychotic episode (263), but symptom severity is only correlated with cortisol levels during the initial phase of psychosis (264-266).
REGULATION OF GH RELEASE
Somatotroph Development and Differentiation
Somatotrophs make up approximately 50% of the cell population of the anterior pituitary and generally are concentrated in the lateral wings of the pituitary gland. These cells are characteristically acidophilic, polyhedral and immunopositive for GH and Pit-1. A smaller number of such cells are mammo-somatotrophs, i.e. immunopositive for GH and prolactin (267).
During the process of cell differentiation in the Rathke’s pouch primordium, a cascade of transcription factors is activated to specify anterior pituitary cell types. The two factors particularly involved in differentiation of the lactotroph, somatotroph, and thyrotroph lineages are Prop-1 (Prophet of Pit-1) and Pit-1, also known as GHF-1 and Pou1f1. Prop-1 is a paired-like homeodomain transcription factor; mutations in this gene cause combined GH, prolactin, and TSH deficiency. Mutations of Prop-1 will also give abnormalities of gonadotroph function and, occasionally, corticotroph reserve. Interestingly, these deficiencies are often progressive over time. Pit-1 is part of the POU homeodomain family of transcription factors that includes unc-86, Oct-1, and Oct-2 (268). Pit-1 is a key transcription factor that activates GH gene transcription in the somatotroph (vide infra).
The transcription factor Foxo1 (forkhead box transcription factor) is expressed in 40% of somatotrophs. Foxo1 is involved in the development of various other tissues slow-twitch muscle fibers, bone and pancreas, and a global knockout is lethal. A pituitary-specific knockout of Foxo1 causes a delay in the terminal differentiation of somatotrophs but does not affect commitment of pituitary progenitor cells to the somatotroph lineage (269). Foxo1 exerts its effect via stimulation of NeuroD4 expression which is also important to the terminal differentiation of somatotrophs (270).
Growth Hormone (GH)
GH GENOMIC LOCUS
Human GH was first isolated in 1956 (271) and the structure of the peptide was elucidated fifteen years later (272). Human GH is a 191 amino acids single chain peptide with two disulphide bonds and molecular weight of 22,000 daltons. The GH locus, a 66 kb region of DNA, is located on chromosome 17q22-q24 and consists of 5 homologous genes, which appear to have been duplicated from an ancestral GH-like gene (Table 1) (273,274).
|
Table 1. The Five Genes in the GH Locus
|
|
Gene
|
Product
|
Variant(s)
|
Expressed in
|
References
|
|
hGH-N or GH1
|
Normal GH
|
2 alternatively spliced variants (97):
22 kDa (full-length 191 aa).
20 kDa (lacking residues 32-46)
|
Anterior pituitary
|
(275)
|
|
hGH-V or GH2
|
Variant GH detectable in pregnancy from mid-term to delivery (276,277)
|
20 kDa
|
Placental syncytiotrophoblast cells
|
(278)
|
|
CSH-1, CSH-2
|
Chorionic somatotropin/human placental lactogen
|
22 kDa
|
Placental syncytiotrophoblast cells
|
(279,280)
|
|
CSH-like gene CSHL-1
|
Non-functional proteins
|
Many alternatively spliced variants
|
|
(281)
|
Because of their origin from an ancestral GH-like gene, all five genes in the GH genomic locus share 95% sequence identity including their promoters (282): proximal elements in the promoter bind Pit-1/GHF-1 (283-286). Pit-1 plays a central role in controlling the expression of hGH-NN gene. Inactivation or lack of functional Pit-1 expression in both mice and human inhibits the differentiation and proliferation of the pituitary cells (287). Although Pit-1 is necessary for transcription of transfected GH1 genes in rat pituitary cells, it is not sufficient (288). Other transcription factors such as Sp1, CREB, and the thyroid hormone receptor are involved (285,289,290).
A placenta-specific enhancer found downstream of the CSH genes (291) as well as pituitary-specific repressor sequences found upstream of GH2, CSH-1 and -2, and CSHL-1 may serve to limit transcription of these particular genes to the placenta (292).
A locus control region consisting of two DNase-I hypersensitive regions (HS), specifically HG-I site, 14.5 and 30 kb upstream of GH1 appears to be required for pituitary-specific GH1 expression (293). This region, which also binds Pit-1 (294), activates histone acetyltransferase, which controls chromatin structure and the accessibility of the GH locus to transcription factors (295,296). The acetylated histone domain potentiates GH transcription and, more recently, HS-I was also shown to be crucial for establishing a domain of non-coding polymerase II transcription necessary for gene activation (297).
Pit-1 is mainly expressed in the pituitary somatotrophs, but it has also notably been demonstrated to be expressed in extrapituitary tissues. Pit-1 regulates local GH expression in the mammary gland and may be involved in mammary development and possibly the pathogenesis of breast carcinoma (298).
GROWTH HORMONE STRUCTURE
This is a 191 amino acid single chain polypeptide hormone that occurs in various modified forms in the circulation. During spontaneous pulses of secretion, the majority full-length isoform of 22 kDa makes up 73%, the alternatively spliced 20 kDa isoform contributes 16%, while the ‘acidic’ desamido and N-alpha acylated isoforms make up 10%. During basal secretion between pulses other forms (30 kDa, 16 kDa and 12 kDa) can also be identified which consist of immunoreactive fragments of GH (299-301).
Higher molecular weight forms of GH exist in the circulation, representing GH bound to growth hormone binding proteins (GHBP) (302). The high-affinity GHBP consists of the extracellular domain of the hepatic GH receptor, and this binds the 22 kDa GH isoform preferentially (303). This high-affinity GHBP is released into circulation by proteolytic processing of the GH receptor by the metalloprotease TACE/ADAM-17 (304). The low-affinity GHBP binds the 20 kDa isoform preferentially (305). Binding of GH to GHBP prolongs the circulation time of GH as the complex is not filtered by the glomeruli (300). GH/GHBP interactions may also compete for GH binding to its surface receptors (306).
GH is also expressed in other areas of the brain, such as the cortex, hippocampus, cortex, caudate nucleus, and retinal areas (307), as is the GH receptor, IGF-1, and the IGF-1 receptor, where it is thought that these mediate neuroprotective and regenerative functions (308).
HYPOPHYSIOTROPIC HORMONES AFFECTING GH RELEASE
GHRH
GHRH was originally isolated from a pancreatic tumor taken from a patient that presented with acromegaly and somatotroph hyperplasia (309). GHRH is derived from a 108 amino acid prepro-hormone to give GHRH (1-40) and (1-44) (Figure 6), which are both found in the human hypothalamus (310,311). The C-terminal 30-44 residues appear to be dispensable, as residues 1-29 show full bioactivity. GHRH binds to a seven-transmembrane domain G-protein coupled receptor that activates adenylate cyclase (312), which stimulates transcription of the GH gene as well as release of GH from intracellular pools (313,314). No other hormone is released by GHRH, although GHRH has homology to other neuropeptides such as PHI, glucagon, secretin and GIP (315).
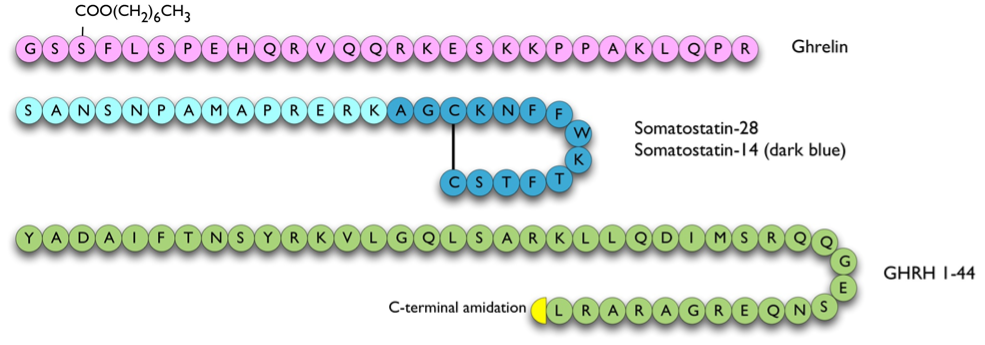
Figure 6. Hypophysiotrophic hormones influencing GH release. The pathway of GPR101 leading to GH release is currently unclear therefore not shown on this figure.
Somatostatin
Somatostatin (a.k.a. somatotropin release inhibitory factor or SRIF) is derived from a 116 amino acid prohormone to give rise to two principal forms, somatostatin-28 and -14 (316). Both of these are cyclic peptides due to an intramolecular disulphide bond (Figure 6). Somatostatin has multiple effects on anterior pituitary as well as pancreatic, liver and gastrointestinal function:
- It inhibits GH secretion directly from somatotrophs (317,318) and antagonizes the GH secretagogue activity of ghrelin (319).
- It inhibits GH secretion indirectly via antagonizing GHRH secretion.
- It inhibits GH secretion indirectly via inhibiting the secretion of ghrelin from the stomach (320-322).
- It inhibits secretion of TSH and TRH stimulation of TSH secretion from the pituitary (323,324).
- It inhibits the secretion of CCK, glucagon, gastrin, secretin, GIP, insulin and VIP from the pancreas (325).
Somatostatin binds to specific seven-transmembrane domain G-protein coupled receptors (SSTRs), of which there are at least 5 subtypes. SSTRs 2 and 5 are the most abundant in the pituitary (326). An immunohistochemical study on fetal pituitaries has shown that SSTR 2 is present from 13 weeks gestation, mainly on thyrotrophs and gonadotrophs. SSTR 5 is mainly found on somatotrophs and develops relatively late in gestation at 35-38 weeks of gestation, suggesting that SSTR 2 regulates TSH, LH and FSH whereas SSTR 5 regulates GH (327). The somatostatin receptors couple to various 2nd messenger systems such as adenylate cyclase, protein phosphatases, phospholipase C, cGMP dependent protein kinases, potassium, and calcium ion channels (328).
Ghrelin
Ghrelin is an orexigenic (appetite-stimulatory) peptide that was isolated from stomach and can stimulate the release of GH. It is derived from preproghrelin, a 117 amino acid peptide, by cleavage and n-octanoylation at the third residue to give a 28 amino acid active peptide (Figure 3 and Figure 6). Ghrelin is the endogenous ligand of the GH secretagogue receptor (GHS-R) 1a, another member of the seven-transmembrane receptor family G-protein coupled to the phospholipase C-phosphoinositide pathway (329,330). This variant of GHS-R has been shown to transduce the GH-releasing effect of synthetic growth hormone secretagogues (GHSs) as well as ghrelin and also plays a role in neuroendocrine and appetite-stimulating activities centrally. Both ghrelin and GHS-R1a have corresponding widespread tissue expression (331). The other GHS-R variant, GHS-R1b, is a 289 amino acid G-protein coupled receptor with five transmembrane domains. The biological function of GHS-R1b is unclear. It has widespread expression throughout the body (331) but does not bind to ghrelin or other GHSs. However, it was shown to have counter-regulatory attenuating role on GHS-R1a signaling, possibly via the formation of heterodimers with GHS-R1a (332).
The majority of circulating ghrelin exists as the des-octanoylated (des-acyl) form: octanoylated ghrelin constitutes approximately 1.8% of the total amount of circulating ghrelin (333). Octanoylation appears to be essential for GH secretagogue activity, as des-acyl ghrelin is inactive for GH release (329). The enzyme that octanoylates ghrelin has recently been identified as ghrelin O-acyltransferase (GOAT) (334). GOAT is a porcupine-like enzyme belonging to the super-family of membrane-bound O-acyltransferase 4 (MBOAT4) and has widespread tissue expression corresponding to ghrelin (335). Historically, the earliest GH secretagogues discovered such as GHRP-1, GHRP-2, GHRP-6, and hexarelin were synthetic and derived from the enkephalins (336).
In the circulation, ghrelin appears to be bound to a subfraction of HDL particles containing clusterin and the A-esterase paraoxonase. It has been suggested that paraoxonase may be responsible for catalyzing the conversion of ghrelin to des-acyl ghrelin (337). However, inhibition of paraoxonase in human serum does not inhibit the de-acylation of ghrelin, and there is a negative correlation in these sera between the paraoxonase activity and ghrelin degradation. Instead, it is more likely that butyrylcholinesterase and other B-esterases are responsible for this activity (338).
Ghrelin is present in the arcuate nucleus of the hypothalamus and in the anterior pituitary (339). Immunofluorescence studies show that ghrelin is localized in somatotrophs, thyrotrophs, and lactotrophs, but not in corticotrophs or gonadotrophs, suggesting that ghrelin may be acting in a paracrine fashion in the anterior pituitary (340). It stimulates GH release in vitro directly from somatotrophs (329) and also when infused in vivo, although the latter action appears to require the participation of an intact GHRH system (319). Ghrelin stimulates GH secretion in a synergistic fashion when co-infused with GHRH (110). Both GHS and ghrelin have been shown to stimulate the release of GH in a dose-related pattern which is more marked in humans than in animals (341,342).
Besides its GH releasing activity, ghrelin has orexigenic activity (343,344), and stimulates insulin secretion (345), ACTH and prolactin release (346). Knocking out the preproghrelin gene in mice does not seem to affect their size, growth rate, food intake, body composition, and reproduction, indicating that proghrelin products (acyl- or desacyl-ghrelin, obestatin) are not dominantly and critically involved in mouse viability, appetite regulation, and fertility (347), although subtle reductions in the amplitude of secretory GH peaks can be detected in these knockout mice during their youth: these differences recede with aging (348). Ghrelin null mice show an increased utilization of fat as an energy substrate when placed on a high-fat diet, which may indicate that ghrelin is involved in modulating the use of metabolic substrates (349). GHS-R knockout mice have the same food intake and body composition as their wild-type littermates, although their body weight is decreased in comparison. However, treatment of GHS-R null mice with ghrelin does not stimulate GH release or food intake, confirming that these properties of ghrelin are mediated through the GHS-R (350).
Although it is clear that acyl-ghrelin activates GH secretion when injected into mice and men, the specific contribution of acyl-ghrelin to physiological pulsatile GH release is less clear. This question has been studied by knocking out GOAT: these mice showed an overall decline in the amount of GH release compared to age matched wild-type mice. The alteration of the GH release observed did not coincide with alterations in the pituitary GH content and GHRH, somatostatin, neuropeptide Y, or GHS-R mRNA expression. However, an increase in pulse number and greater irregularity of GH pulses was observed in these mice. Although other mutations that cause derangement of GH secretion have been previously associated with the ‘feminization’ of the expression of GH-dependent sexually divergent liver genes in male animals, there was no evidence of this in the Goat-/- mice. An increase in IGF-1 in the circulation, in the liver and also in the muscle was observed in the Goat-/- mice, either as a result of the disordered GH pulse pattern, or because there was a failure of the elevated IGF-1 levels to feedback on GH release. Overall, the data suggest that acyl-ghrelin has a regulatory role in the patterning of GH secretion, but the absence of acyl-ghrelin does not fatally knock out GH production (351).
To complicate things further, des-acyl ghrelin may have biological effects of its own. It has been shown to inhibit apoptosis and cell death in primary cardiomyocyte and endothelial cell cultures (352), to have varying effects on the proliferation of various prostate carcinoma cell lines (353), to inhibit isoproterenol-induced lipolysis in rat adipocyte cultures (354), and to induce hypotension and bradycardia when injected into the nucleus tractus solitarii of rats (355). More controversially, intracerebroventricular or peripherally administered des-acyl ghrelin causes a decrease in food consumption in fasted mice and inhibits gastric emptying. Des-acyl ghrelin overexpression in transgenic mice causes a decrease in body weight, food intake, fat pad mass weight, and decreased linear growth compared to normal littermates (356). These observations were not replicated by other researchers, who found no effect of des-acyl ghrelin on feeding (357). The effects of des-acyl ghrelin appear not to be mediated via the type 1a or 1b GHS-R (352-354). The effects of peripherally administered des-acyl ghrelin on stomach motility can be inhibited by intracerebrovascular CRH receptor type 2 antagonists, suggesting that CRH receptor type 2 is involved, but there is no direct evidence that des-acyl ghrelin binds this receptor (358)
As noted above, the GH-stimulatory actions of ghrelin in vivo seem to require an intact GHRH system, as immunoneutralization of GHRH blocks ghrelin-induced GH secretion (319). The actions of GH secretagogues are blocked by hypothalamo-pituitary disconnection, which suggests that in vivo ghrelin’s stimulatory actions are indirect and mediated by GHRH (359). However, GHRH cannot be the sole mediator of ghrelin’s actions as the GH response to ghrelin is greater than that to GHRH (360), and, as noted above, ghrelin synergistically potentiates GH release by a maximal dose of GHRH (110). There is no evidence to suggest that ghrelin decreases somatostatinergic tone as immunoneutralization of somatostatin does not block ghrelin’s ability to release GH (319). There may therefore be another mediator, the so-called ‘U’ factor, released by ghrelin, which causes GH secretion (361).
Macimorelin (also known as Ghryelin) is an orally available ghrelin receptor (GHSR) agonist which is now validated for stimulation testing for GH reserve (362).
LEAP2
Liver-expressed antimicrobial peptide 2 (LEAP2) has recently been discovered as an endogenous antagonist to GHSR(363). It is produced in the small intestines, mainly in the jejunum (363). Level of LEAP2 declines with fasting, as opposed to the level of ghrelin which goes up (363,364). In addition, the expression of LEAP2 is significantly upregulated following bariatric surgery, which is currently the most effective treatment for obesity (363). In vivo studies have shown that LEAP2 is capable of inhibiting the effects of ghrelin on GH secretion and food intake (363). LEAP2 is also shown to bind to GHSR in a non-competitive manner to ghrelin, thereby suggesting the presence of an allosteric site on the receptor (363).
Obestatin
As mentioned earlier, the effects of obestatin on pituitary hormones release remain controversial. Initial study has shown that intravenous or intracerebrovascular treatment of obestatin did not affect the release of growth hormone in male rats (116). However, a more recent study has shown that obestatin treatment inhibits both basal and ghrelin-induced GH release and expression, both in vitro and in vivo in non-human primates and in mice (119). This inhibitory effect is mediated by the adenylyl-cyclase and MAPK pathways. Obestatin treatment causes a reduction in Pit-1 and GHRH-R mRNA levels in the pituitary as well as a decrease in hypothalamic GHRH and ghrelin expression. Obestatin also reduces the expression of pituitary somatostatin receptors, namely SSTR subtypes 1 and 2 (119).
OTHER INFLUENCES ON GROWTH HORMONE RELEASE
Glucocorticoids and Sex Hormones
Glucocorticoid treatment has a biphasic effect on GH secretion: an initial acute stimulation in 3 hours, followed by suppression within 12 hours (365,366). The latter is the clinically important effect, as excess endogenous and exogenous glucocorticoids are well known to suppress growth in children (367). The inhibitory effect of glucocorticoids on GH release is possibly mediated by increase in expression of somatostatin (368).
Sex hormones are also involved in regulating GH release particularly during puberty and also later in life. They affect GH release by acting at hypothalamic, pituitary, and peripheral levels. Both estrogen and testosterone increase GH secretion in humans by amplifying secretory burst mass and reduce the orderliness of GH secretion (369). Estrogen affects GH secretion mainly by interacting with the estrogen receptor-alpha expressed in the GHRH neurons and in the GH-secreting pituitary cells. The stimulatory effects of estrogen on GH secretion are possibly mediated by the release of GHRH and/or by enhancing the sensitivity to ghrelin released from the hypothalamus (370). Estrogen increases the irregularity in pulsatility and lowers total and free IGF-1. Although estrogen increases the secretion of GH, it is also known to counter-regulate itself by reducing GH sensitivity in the liver and other peripheral organs, hence decreasing the secretion of IGF-1. The mechanism of this effect is via upregulating the SOCS-2 protein which in turn inhibits the JAK1-STAT5 signal transduction pathway of the GHR (371). GH deficient patients started on estrogen therapy therefore require a higher dose of GH replacement therapy to achieve a particular target IGF-1 level (372). The route of estrogen replacement is an important influence on GH requirement and those on oral estrogen are clearly more GH resistant than women using transdermal preparations (373,374). Testosterone, on the other hand, increases basal GH secretion and IGF-1 concentrations, thus relieving the negative feedback on GH secretion (369).
Leptin
Leptin is a 167 amino acid anorexigenic peptide primarily produced by white adipose tissue (375), regulates body fat mass (376) by feedback inhibition of the appetite centers of the hypothalamus (377). Leptin and its receptor have been detected both by RT-PCR and immunohistochemistry in surgical pituitary adenoma specimens and in normal pituitary tissue (378,379). However, pituitary adenoma cells in culture do not secrete GH in response to leptin treatment (379,380).
Leptin increases GH secretion in the short term, mainly via an increase in GHRH secretion and decrease in somatostatin expression. In the long term, it leads to a decrease in GH secretion, probably reducing GHRH sensitivity (381). In obese subjects, in whom which plasma leptin levels are persistently elevated, GH secretion and responsiveness are reduced in both animals and humans (382). However, if leptin-deficient obese subjects are studied in parallel with sex and BMI-matched leptin-replete obese subjects, it is found that their GH responses to GHRH and GHRP-6 are equally blunted suggesting that the leptin is not influential in mediating the hyposomatotropinism of obesity (383).
IGSF1
IGSF1 (X-linked immunoglobulin superfamily, member 1) gene encodes a transmembrane immunoglobulin superfamily glycoprotein that is highly expressed in the Rathke’s pouch, adult anterior pituitary cells, and the hypothalamus. Loss of function mutations in IGSF1 result in a variable spectrum of anterior pituitary dysfunction, including central hypothyroidism and hypoprolactinemia (384,385). More recently, effects of IGSF1 deficiency on somatroph function were characterized in adult males harboring hemizygous IGSF1 loss-of-function mutations and Igsf1-deficient mice (386). It was shown that IGFS1-deficient patients develop acromegaloid facial features accompanied by elevated IGF-1 concentrations and GH profile. Similar biochemical profiles were also observed in the male Igsf1-deficient mice. The exact mechanism of how IGSF1 regulates or influence GH secretion has not been elucidated.
Kisspeptin
Kisspeptin is a peptide hormone that binds to the G-protein coupled receptor GPR54. Although it was originally characterized as a ‘metastasis suppressor’ gene, its most well-characterized role is in stimulating the secretion of GnRH from GnRH neurons, in turn leading to gonadotrophin production from pituitary gonadotrophs. In addition to this, kisspeptin stimulates GH release from somatotrophs (387,388). These positive effects of kisspeptin are seen when given in vivo to cows or sheep (389), but so far have not been seen when given intravenously in small studies in human volunteers (390), although this may be because the GH stimulatory effects are only observed with central administration.
Catecholamines
In general, alpha-adrenergic pathways stimulate GH secretion, by stimulation of GHRH release and inhibition of somatostatinergic tone, while beta-adrenergic pathways inhibit secretion by increasing somatostatin release (391,392). The alpha2-adrenoceptor agonist clonidine can therefore be used as a provocative test of GH secretion (393,394)although clinical experience suggests that this is an unreliable stimulatory test for GH secretion in practice. L-dopa stimulates GH secretion; however, this action does not appear to be mediated via dopamine receptors as specific blockade of these receptors with pimozide does not alter the GH response to L-dopa (395). Instead, L-dopa’s effects appear to depend on conversion to noradrenaline or adrenaline, as alpha-adrenoceptor blockade with phentolamine disrupts the GH response to L-dopa (396).
Acetylcholine
Muscarinic pathways are known to stimulate GH secretion, probably by modulating somatostatinergic tone (397). Pyridostigmine, an indirect agonist which blocks acetylcholinesterase, increases the 24 hour secretion of GH by selectively increasing GH pulse mass (398). On the other hand, the muscarinic antagonist atropine is able to blunt the GH release associated with slow wave sleep (399) and that associated with GHRH administration (400). Passive immunization with anti-somatostatin antibodies abolishes the pyridostigmine induced rise in GH in rats, but not immunization with anti-GHRH antibodies, supporting the central role of somatostatinergic tone in mediating this response (401).
Dopamine
Continuous infusion of dopamine into normal healthy men leads to an increase in mean GH secretion comparable to that observed with GHRH. When given together, dopamine and GHRH have additive effects on GH secretion, and similarly the dopamine agonist bromocriptine augments the effects of GHRH (402).
Endogenous Opioids
Endorphins and enkephalins are able to stimulate GH secretion in man (403), and blockade with opiate antagonists can attenuate the GH response to exercise (404). Passive immunization against GHRH in rats inhibits GH release in response to an enkephalin analogue, which argues for stimulation of GHRH in response to these compounds (405). In keeping with this, a recent study demonstrated close juxtapositions between the enkephalinergic/ endorphinergic/ dynorphinergic axonal varicosities and GHRH-immunoreactive perikarya in the human hypothalamus (406). Morphologically, the majority of contacts between the GHRH perikarya and endogenous opiates were enkephalinergic while only few dynorphin- and endorphin-GHRH interactions were detected. Enkephalinergic-GHRH interactions and fibers are known to be densely populated in the infundibular nucleus and anterior periventricular area, thereby suggesting that enkephalin regulates not only the activity of GHRH- but also somatostatin-synthesizing neurons (407). The balance between the activation of GHRH and somatostatin neuronal systems may determine if enkephalin stimulates or inhibits or has no effect on pituitary GH secretion. Unfortunately, the study was unable to detect the presence of synapses between the enkephalinergic/ endorphinergic/ dynorphinergic and GHRH neurons because the immunocytochemistry was carried out under light microscope. Electron microscopy was not applied in the study due to the long post-mortem period. Nevertheless, these findings demonstrated the presence of intimate associations between the endogenous opioid and GHRH systems in the human hypothalamus, as well as indicated the significant differences between the regulatory roles of endogenous opioids on growth in humans.
Stimulation of GHRH by endorphins and enkephalins cannot be the only mechanism increasing GH release, however, as the met-enkephalin analogue DAMME is able to increase GH release over and above the levels released during maximal stimulation by a GHRH analogue (408). It is possible that the actions of endogenous opioids occur via an interaction with the GHS-R, as the original GH secretagogues characterized were derived from the enkephalins (336).
Endocannabinoids
As with ACTH/cortisol, the endocannabinoids may also influence the release of GH. Somatotroph cells bear the CB1 receptor (101). The administration of THC for 14 days suppresses GH secretion in response to hypoglycemia in healthy human subjects (104). Oddly enough, THC and anandamide appear to have opposing effects on GH levels in ovariectomized rats: THC increases and anandamide decreases GH secretion in this context (409). However, the treatment of anterior pituitary cells in primary culture with THC does not seem to influence the release of GH and prolactin to GHRH and TRH, suggesting that the effects of THC are mediated via the hypothalamus and not directly on the anterior pituitary (410), perhaps by stimulating somatostatin release (411).
Ghrelin and the Endocannabinoid System
Ghrelin and the endocannabinoid system interact in a bidirectional fashion. The intraperitoneal administration of cannabinoids results in increased plasma ghrelin levels and stomach ghrelin expression in rats (412) and CB1 receptor antagonism with rimonabant reduces ghrelin levels (413), suggesting that the orexigenic effects of cannabinoids may also be connected to an increase in ghrelin secretion from the gastric X/A-like cells. The effects of ghrelin on appetite were also abolished in CB1 knockout or in the presence of the CB1 antagonist rimonabant (414-416). In addition, the effects of cannabinoids are also abolished in the absence of the ghrelin receptor GHS-R1a (417). These findings confirm that both ghrelin and cannabinoid signaling pathways have to intact to mediate the effects of these two systems on appetite. Interestingly, in vivo and in vitro GH release is intact in response to ghrelin in CB1-knockout animals (415). These findings are intriguing because they suggest that the effects of ghrelin on GH release are somehow modulated differently at the receptor-binding stage of the pathway compared to its orexigenic and metabolic effects. Moreover, it has also been proposed that the bidirectional relationship of the ghrelin and endocannabinoid system might be potentially mediated by the interaction (e.g. heterodimerization) between GHS-R1a and CB1 receptors (417). However, further molecular and functional studies are needed to elucidate the exact mechanism of interaction between these two systems.
Free Fatty Acids
The negative feedback regulation of plasma free fatty acids on growth hormone secretion has long been studied (418). Low free fatty acids have been shown to stimulate GH release, although there is a lag period between these two phenomena. Similarly, high plasma levels of free fatty acids have been shown to stimulate splanchnic somatostatin, thereby affecting GH secretion (419). Studies on both hypothalamic and cortical cell cultures have shown marked decrease in somatostatin mRNA levels when both the neuronal cells are treated with free fatty acids, thereby indicating the possible role for free fatty acids in the regulation of the GH secretion centrally (420).
Other Neuropeptides and Factors Affecting GH Secretion
Many neuropeptides, including the ones in the following paragraphs, have been shown to influence GH secretion in various contexts. For the most part, however, their physiological role in man is not well characterized.
Infusion of galanin, a 29 amino acid peptide originally isolated from the small intestine, causes stimulation of GH secretion when infused alone and also enhances GHRH-stimulated GH secretion (421).
Calcitonin, the 32 amino acid peptide secreted from the C cells of the thyroid gland, appears to inhibit the stimulated secretion of GH by GHRH, arginine, and insulin-induced hypoglycemia (422,423).
Neuropeptide Y (NPY) is an orexigenic peptide that has been shown to inhibit GH secretion in rats (424-426), from human somatotroph tumor cells in culture (427), and from rat hypothalamic explants (428). When infused into patients with prolactin-secreting pituitary adenomas, 9 out of 15 patients showed a paradoxical rise in GH levels (429). However, when infused into healthy young men overnight, NPY did not have any significant effect on GH secretion (430).
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a hypothalamic C-terminally amidated 38 residue peptide hormone originally characterized on the basis of its ability to stimulate cAMP accumulation from anterior pituitary cells (431). In rats, PACAP stimulates GH release from pituitary cell lines and also when infused in vivo (432-434). When infused into human volunteers, however, GH levels do not appear to be affected (435).
Klotho, a transmembrane protein that is classically known for its ‘co-receptor’ activity with fibroblast growth hormone receptors, has recently been characterized as a possible secretagogue for GH. Although it is usually attached to membranes, the extracellular region can be shed from the cell surface, and there is some evidence for endocrine activity. Klotho knockout mice exhibit reduced growth in the context of a ‘early aging’ phenotype, and histopathological examination of their somatotrophs demonstrate reduced numbers of secretory granules. Klotho treatment of somatotrophs in vitro has been demonstrated to increase GH secretion, but at present its physiological role is yet to be fully elucidated (436).
GPR101, an orphan GPCR that is constitutively coupled to Gs, has been shown to induce GH secretion through the activation of protein kinase A and protein kinase C in the Gs and Gq/11 pathways (437). Transgenic mice with overexpression of pituitary-specific Gpr101 develops gigantism phenotype and has hypersecretion of GH, in the absence of pituitary hyperplasia or tumorigenesis, thereby indicating that the role of Gpr101 in the pituitary enhances secretion rather than enhancing proliferation (437). In humans, duplication of the GPR101 gene and thus, overexpression of GPR101, leads to a severe form of pituitary gigantism known as X-linked acrogigantism (X-LAG) (438-441). X-LAG is characterized by infant-onset somatotroph tumors or hyperplasia with high levels of GH and in most cases prolactin as well.
Nesfatins, and nesfatin-like-peptides, are hypothalamic and brainstem peptides speculated to be involved in energy homeostasis (442). They have been shown to inhibit GH release in mammalian somatotroph cell lines (443). Both of these peptides bind to the membrane of the GH3 cells, thereby indicating the possibility of a GPCR-mediated action (443). Interestingly, their effects on GH synthesis seem to be concentration-dependent, as low and high concentrations of nesfatins downregulate the expression of GH mRNA, while medium concentrations of nesfatins does not produce this effect (443). Their physiological significance in humans has not yet been established.
FEEDBACK LOOPS OF GH SECRETION
Multiple negative feedback loops exist to autoregulate the GH axis (Figure 7).
- Somatostatin auto-inhibits its own secretion (444).
- GHRH auto-inhibits its own secretion by stimulating somatostatin release (445).
- GH auto-regulates its own secretion in short term by stimulating somatostatin release and inhibiting GHRH-stimulated GH release (446-448). There is also a negative feedback on stomach ghrelin release by GH (449). More recently, it is demonstrated that in long-term feedback situation, the inhibition of GH release is most likely due to feedback inhibition by IGF-1 (450).
- IGF-1, whose production is stimulated by GH, inhibits GH release in a biphasic manner: (1) by stimulating hypothalamic somatostatin release early, and (2) by inhibiting GH release after 24 hours, probably by inhibiting GH mRNA transcription (451,452). Interestingly, IGF-1 infusion suppresses GHRH-induced GH release in males but not in females, suggesting a sexually dimorphic effect (450).
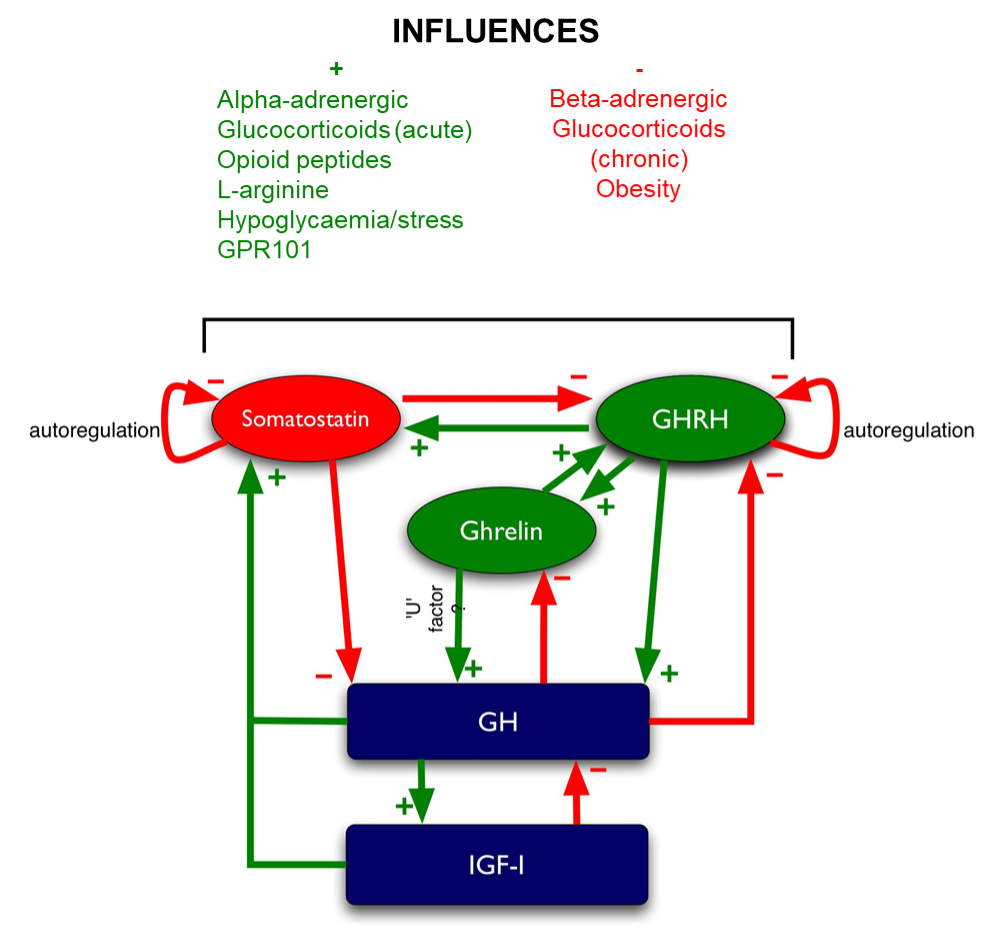
Figure 7. Regulation of GH. Green arrows denote stimulatory influences, red arrows denote inhibitory influences.
PHYSIOLOGY OF GH SECRETION
Pulsatility of GH Secretion
The secretory pattern of GH was first elucidated in rats (453). Circulating GH levels are pulsatile, with high peaks separated by valleys where the GH is undetectable by conventional RIAs or IRMAs (Figure 8). The recent development of sensitive chemiluminescent assays for GH with high frequency sampling and deconvolution analysis has allowed the detailed study of GH secretion. This shows that there are detectable levels of basal GH secretion in the ‘valleys’ (454). On average, there are 10 pulses of GH secretion per day lasting a mean of 96.4 mins with 128 mins between each pulse (455). The diurnal secretory pattern of GH in human is fully developed after puberty, demonstrating a major peak at late night/early morning which is associated with NREM (slow wave)-sleep, and a number of peaks during the light hours of the day, but with quite large individual difference (456).
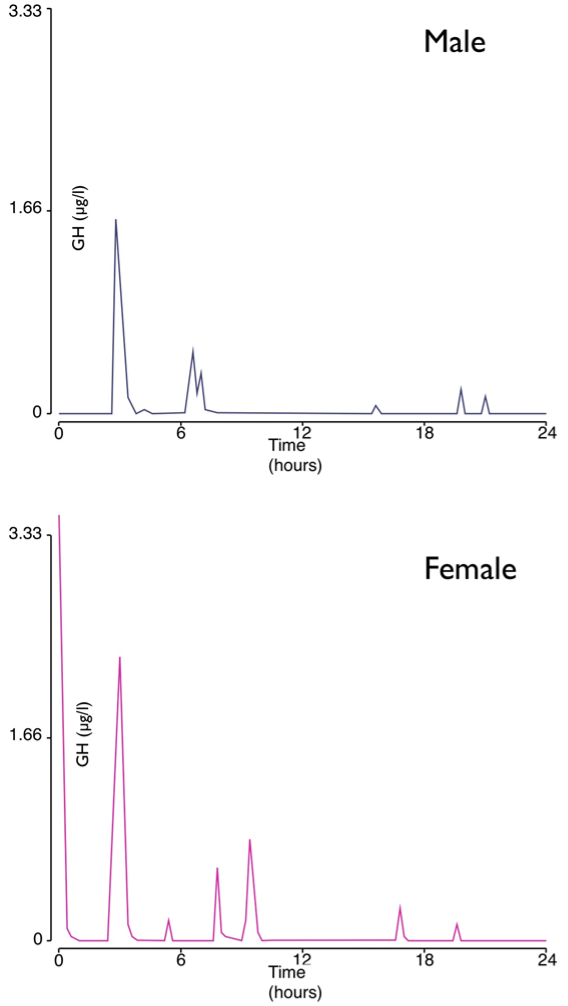
Figure 8. Pulsatility of circulating GH levels in adult men and women.
There is a dynamic interplay of pulsatile GHRH and somatostatin secretion:
- Via crosstalk: GHRH neurons receive inhibitory inputs from somatostatin neurons, whilst somatostatin neurons receive direct stimulatory inputs from GHRH neurons
- Via synergistic actions on somatotrophs: Pre-exposure to somatostatin enhances GHRH-stimulated secretion of GH (457).
Further studies in animals have revealed that somatotropin releasing inhibiting factor regulates the magnitude of the troughs of GH as well as the amplitude of the peaks, whereas GHRH functions as the main regulator of the pulsatile pattern (450,458,459). Interestingly, continuous GHRH administration in human volunteers does not affect the pulsatility of GH secretion (460). Moreover, patients with an inactivating mutation of the GHRH receptor continue to show pulsatile GH secretion, suggesting that somatostatin pulsatility is sufficient to determine GH pulsatility (461). These observations suggest that the mechanisms involved in humans may differ from the animal models.
GH and Sexual Dimorphism
The technical developments in sensitive detection of GH and deconvolution analysis referred to above have elucidated differences in secretion between men and women. Women have higher mean GH levels throughout the day than men due to higher incremental and maximal GH peak amplitudes (Figure 8), but show no significant difference in GH half-life, interpulse times, or pulse frequency (462). The higher basal GH levels may underlie the higher nadir GH levels seen in normal women after GH suppression with oral glucose (463). Recent evidence suggests that there are sexual differences in the expression of somatostatin and somatostatin receptor subtypes in the rat pituitary, which would clearly cause differences in the physiological regulation of GH release (464).
Differences in GH secretion patterns between the sexes, with male ‘pulsatile’ secretion versus female ‘continuous’ secretion, can cause different patterns of gene activation in target tissues, e.g. induction of linear growth patterns, gain of body weight, induction of liver enzymes and STAT 5b signaling pathway activity (465).
GH and Aging
GH and IGF-1 levels are known to decline continuously with age and to very low levels in those aged ≥60 years (466). This phenomenon, known as the ‘somatopause’, is also seen in other mammals and has led to the speculation that GH treatment can be a potent anti-aging therapy (467). Conversely, decreased GH/IGF-1 signaling has also been shown to extend longevity in a wide variety of species such as worms, fruit flies, mice, and yeast (468), thus raising the question of whether decreased activity of the GH/IGF-1 axis might be beneficial for human longevity. Somatopause might therefore be nature’s way of sustaining the aging individual (469).
It is also suggested that the anorexia associated with aging is due to the decline in the level of acylated ghrelin in older adults. This is supported by a recent study that showed an age-dependent decline in both circulating acyl-ghrelin and growth hormone levels in older adults (aged 62-74 years, BMI range 20.9-29 kg/m2) compared to young adults (aged 18-28 years, BMI range 20.6-26.2 kg/m2) (470). By estimating the correlations between amplitudes of individual GH secretory events and the average acyl-ghrelin concentration in the 60-minute interval preceding each GH burst, the ghrelin/GH association was more than 3-fold lower in the older group compared with the young adults, thus suggesting that with normal aging, endogenous acyl-ghrelin levels are less tightly linked to GH regulation. In addition, ghrelin mimetics have also been shown to be a potential treatment for the musculoskeletal impairment associated with aging (471).
Sleep
The secretion rate of GH shows a circadian pattern, with peak rates measured during sleep. These are approximately triple the daytime rate (472). GH secretion is especially associated with slow wave sleep (SWS – stages 3 and 4) (473). Deep sleep is also shown to enhance the activity of GH axis and has an inhibitory effect on cortisol levels (474). The decline in GH secretion during aging is paralleled by the decreasing proportion of time spent in SWS, although it is unclear which is cause and which is effect (475). In early data from a clinical trial, GH deficient patients have increased sleep fragmentation and decreased total sleep time, and it is conjectured that such alterations in sleep patterns may be responsible for excessive daytime sleepiness in such patients (476).
Sleep deprivation, in the laboratory or due to travel causing ‘jet lag’, causes two alterations in the GH secretory pattern: the magnitude of secretory spikes is augmented: the return to pre-travel levels takes at least 11 days and is slower to recover after westward travel. The major pulse of GH secretion occurring in early sleep is also shifted to late sleep (477). It is also noted that the GH pulses are more equally distributed throughout 24 hours of sleep deprivation compared to a night-time sleep condition, with large individual pulses occurring during the day (478).
Administration of a GHRH antagonist reduces nocturnal GH pulsatility by 75% (479). Normal subjects remain sensitive to GHRH boluses during the night, however, and the lowering of somatostatinergic tone during the night may be responsible for the increase in GH secretion rate (480). Recent work, however, has also demonstrated that ghrelin levels rise through the night in lean men (481). It is likely, therefore, that a combination of increased GHRH, decreased somatostatin and increased ghrelin levels underlie the circadian variation in GH secretion.
Administration of GHRH augments increased nocturnal GH release and promotes SWS. Somatostatin does not change nocturnal GH release, and does not affect the proportion of SWS, but may increase rapid eye movement (REM) sleep density (482). Ghrelin has been shown to promote slow wave sleep at the expense of REM sleep, accompanied by an increase in GH and prolactin release when administered exogenously (483).
Exercise
Exercise is a powerful stimulus to secretion of GH (484), which occurs by about 15 min from the start of exercise (485). The kinetics may vary between subjects, an effect which is likely to be related to differences in age, sex and body composition (486). Ten minutes of high-intensity exercise is required to stimulate a significant rise in GH (487). Anaerobic exercise causes a larger release of GH than aerobic exercise of the same duration (488).
Acetylcholine, adrenaline, noradrenaline, and endogenous opioids have been implicated in exercise-induced GH release (397). However, ghrelin levels do not rise in acute exercise, indicating that ghrelin may not have a role to play in exercise-induced GH release (489).
Recent evidence also indicates that exercise enhances SWS and thus leads to increase in GH release as well as brain-derived neutrotrophic factors (BDNF) and IGF-1 gene expression and protein levels (490,491). This is thought to improve learning and memory performance, especially in the elderly (490,491). Sleep-deprived individuals seem to have a larger exercise-induced GH response, although the reason behind this is still unclear (492).
Hypoglycemia
Insulin-induced hypoglycemia is another powerful stimulus to GH secretion (Figure 9) (493,494). The peak GH levels achieved during insulin stress testing correlate well with those achieved during slow wave sleep (495). The hypoglycemic response is mediated by alpha2-adrenergic receptors (496) to cause inhibition of somatostatin release (397), although other evidence argues for a role of stimulated GHRH release, as a GHRH receptor antagonist significantly suppressed hypoglycemic GH release (497). Ghrelin is unlikely to be involved in the GH response to insulin-induced hypoglycemia as circulating ghrelin levels are suppressed by the insulin bolus (498).

Figure 9. Normal response of GH to insulin-induced hypoglycemia (≤2.2 mmol/l). Peak GH secreted is ≥6.66 µg/L.
Other Stressors
Other physical stresses such as hypovolemic shock (499) and elective surgery (500) cause increased GH release; alpha-adrenergic dependent mechanisms are thought to underly this, as blockade with phentolamine inhibits the response (500).
Hyperglycemia
In contrast to hypoglycemia, ingestion of an oral glucose load causes an initial suppression of plasma GH levels for 1-3 hours (Figure 10), followed by a rise in GH concentrations at 3-5 hours (501). The initial suppression could be mediated by increased somatostatin release as pyridostigmine, a postulated inhibitor of somatostatin release, blocks this suppression (502). Circulating ghrelin levels also fall following ingestion of glucose (503). The GH response to ghrelin and GHRH infusions is blunted by oral glucose, an effect that is probably mediated by somatostatin (504). The later rise in GH levels is postulated to be due to a decline in somatostatinergic tone plus a reciprocal increase in GHRH, leading to a ‘rebound’ rise (397).
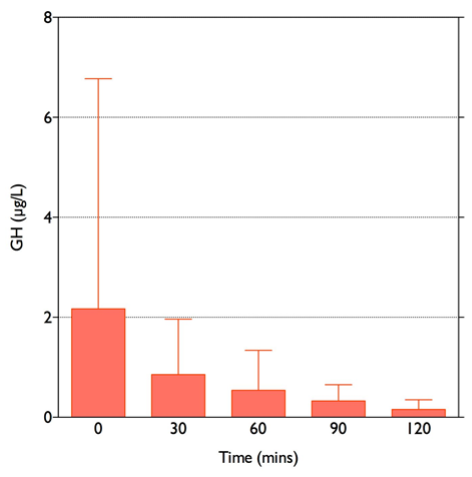
Figure 10. GH response to 75g oral glucose in 8 non-acromegalic, non-diabetic women, given at time 0. Error bars denote SD. Note the high variability of the baseline GH level due to the pulsatile nature of GH secretion. GH levels fall to <0.4 µg/L at 120 minutes.
In type I diabetes mellitus, GH dynamics are disordered, with elevated 24 hour release of GH (505). Deconvolution analysis shows that GH pulse frequencies and maximal amplitudes are increased. The latter is accounted for by higher ‘valley’ levels (506). Better glycemic control appears to normalize these disordered dynamics (507). The pathophysiological mechanism appears to involve reduced somatostatinergic tone (397).
There is conflicting evidence for increased, decreased, or normal GH dynamics in type II diabetics. It is likely that this reflects two factors acting in opposite directions: (1) the confounding factor of obesity in these patients, which leads to hyposecretion of GH; and (2) the hyperglycemia, which leads to hypersecretion (397).
Dietary Restriction and Fasting
Dietary restriction and fasting lead to a significant increase in pituitary secretion of GH (508). A 5-day fast in normal healthy men resulted in a significant increase in the pulse frequency as well as pulse amplitude of GH release. This was coupled with a decrease in expression and secretion of IGF-1, which could explain the lack of feedback inhibitory effect on pituitary GH secretion in the fasting state.
Obesity and Malnutrition
Chronic malnutrition states such as marasmus and kwashiorkor cause a rise in GH levels (509). On the other hand, obesity is known to be associated with lower GH levels, partially due to decreased levels of GH binding protein and partially due to decreased frequency of GH pulses (510). Visceral adiposity, as assessed by CT scanning and dual energy X-ray absorptiometry, seems to be especially important, and correlates negatively with mean 24 hour GH concentrations (511). The mechanism of decreased GH release in obesity has been ascribed to increased somatostatinergic tone, as pyridostigmine is able to reverse this, to some extent, by suppressing somatostatin release (512-514). However, this cannot be the full explanation, as pyridostigmine is not able to fully reverse the hyposomatotropinism of obesity, even when combined with GHRH and the GH secretagogue GHRP-6 (515).
The fasting induced elevation in secretion of GH is blunted in obesity (516,517). Nevertheless, fasting in obese volunteers still induces an appreciable increase in GH secretion, with accompanying increase in lipolysis and insulin resistance. Co-administration of pegvisomant (a GH receptor antagonist) abrogated this phenomenon, suggesting that the elevation in GH during fasting is responsible for the insulin resistance induced by fasting (518).
Although leptin has been shown to be influential on GH secretion in rats (519), this may not be so in humans. Leptin-deficient subjects have been compared with obese non-deficient control subjects in their GH responses when stimulated with GHRH plus GHRP-6. Both these groups have decreased GH peaks compared to non-obese control subjects, as expected. There was no significant difference in mean GH peaks between leptin-deficient and leptin-replete controls, suggesting that leptin does not play a significant role in the GH suppression seen in obese humans, and that the decreased GH secretion of obesity is mediated via other mechanisms (383).
Another candidate for the mechanism linking obesity to GH secretion is ghrelin. Its levels correlate negatively with body fat content (520). A comparative study between 5 lean and 5 obese men employed rapid sampling and pulse analysis of ghrelin levels over 24 hours. Ghrelin levels increased at night in the lean controls but did not in the obese group (481). Weight loss caused circulating ghrelin levels to rise in two studies (521,522). Contradicting this, however, Lindeman and colleagues found that ghrelin levels paradoxically correlated positively with visceral fat area, in contrast with 24-hour GH secretion, which correlated negatively. Moreover, in their study, weight loss increased GH secretion but did not affect ghrelin levels (523). More recently, a study comparing subjects with central obesity only with subjects suffering from the metabolic syndrome showed changes in ghrelin levels not to be associated with central obesity per se but with other components of the metabolic syndrome (524). The response of GH secretion to exogenous ghrelin is significantly blunted in obese patients and this response is restored early on after Roux-en-Y gastric bypass (prior to any major weight loss), suggesting that there is an intrinsic resistance to ghrelin in obesity which is reversed with gastric bypass, and which is not linked to weight loss (525). Therefore, there does not appear to be a simple relationship where obesity-induced reduction in ghrelin levels leads to the reduced secretion of GH.
Amino Acids
GH release is stimulated by a protein meal (526). L-arginine, an essential amino acid, can be used as a provocative test for GH secretion (527). Evidence that L-arginine acts through inhibition of somatostatin release includes the observation that L-arginine can still enhance the GH response to GHRH despite the use of maximal doses of GHRH (528). However, a specific GHRH antagonist blunted the GH response to L-arginine, an observation that supports the notion that L-arginine also acts through stimulation of GHRH secretion (497). Unlike oral glucose, L-arginine does not modify the GH response to ghrelin infusion (504).
REFERENCES
- Marshall J. Control of Pituitary Hormone Secretion – Role of Pulsatility. In: Besser G, Thorner M, eds. Comprehensive Clinical Endocrinology. Edinburgh, UK: Mosby; 2002:19-34.
- Doniach I. Histopathology of the pituitary. Clin Endocrinol Metab. 1985;14(4):765-789.
- McKusick V, Phillips JI, Hamosh A, Tiller G, O'Neill M. Pro-opiomelanocortin. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD, USA: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=176830. Accessed 20 Jan 2007
- Drouin J. 60 YEARS OF POMC: Transcriptional and epigenetic regulation of POMC gene expression. J Mol Endocrinol. 2016;56(4):T99-T112.
- Drouin J, Chamberland M, Charron J, Jeannotte L, Nemer M. Structure of the rat pro-opiomelanocortin (POMC) gene. FEBS Lett. 1985;193(1):54-58.
- Therrien M, Drouin J. Pituitary pro-opiomelanocortin gene expression requires synergistic interactions of several regulatory elements. Mol Cell Biol. 1991;11(7):3492-3503.
- Therrien M, Drouin J. Cell-specific helix-loop-helix factor required for pituitary expression of the pro-opiomelanocortin gene. Mol Cell Biol. 1993;13(4):2342-2353.
- Poulin G, Turgeon B, Drouin J. NeuroD1/beta2 contributes to cell-specific transcription of the proopiomelanocortin gene. Mol Cell Biol. 1997;17(11):6673-6682.
- Lamonerie T, Tremblay JJ, Lanctot C, Therrien M, Gauthier Y, Drouin J. Ptx1, a bicoid-related homeo box transcription factor involved in transcription of the pro-opiomelanocortin gene. Genes Dev. 1996;10(10):1284-1295.
- Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, Drouin J. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell. 2001;104(6):849-859.
- Langlais D, Couture C, Sylvain-Drolet G, Drouin J. A Pituitary-Specific Enhancer of the POMC Gene with Preferential Activity in Corticotrope Cells. Mol Endocrinol. 2011;25(2):348-359.
- Murakami I, Takeuchi S, Kudo T, Sutou S, Takahashi S. Corticotropin-releasing hormone or dexamethasone regulates rat proopiomelanocortin transcription through Tpit/Pitx-responsive element in its promoter. J Endocrinol.2007;193(2):279-290.
- Budry L, Balsalobre A, Gauthier Y, Khetchoumian K, L'Honore A, Vallette S, Brue T, Figarella-Branger D, Meij B, Drouin J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012;26(20):2299-2310.
- Ezzat S, Mader R, Yu S, Ning T, Poussier P, Asa SL. Ikaros integrates endocrine and immune system development. J Clin Invest. 2005;115(4):1021-1029.
- Aguilera G, Harwood JP, Wilson JX, Morell J, Brown JH, Catt KJ. Mechanisms of action of corticotropin-releasing factor and other regulators of corticotropin release in rat pituitary cells. J Biol Chem. 1983;258(13):8039-8045.
- Jin WD, Boutillier AL, Glucksman MJ, Salton SR, Loeffler JP, Roberts JL. Characterization of a corticotropin-releasing hormone-responsive element in the rat proopiomelanocortin gene promoter and molecular cloning of its binding protein. Mol Endocrinol. 1994;8(10):1377-1388.
- Autelitano DJ, Cohen DR. CRF stimulates expression of multiple fos and jun related genes in the AtT-20 corticotroph cell. Mol Cell Endocrinol. 1996;119(1):25-35.
- Loeffler JP, Kley N, Pittius CW, Hollt V. Calcium ion and cyclic adenosine 3',5'-monophosphate regulate proopiomelanocortin messenger ribonucleic acid levels in rat intermediate and anterior pituitary lobes. Endocrinology. 1986;119(6):2840-2847.
- Antoni FA. Interactions between intracellular free Ca2+ and cyclic AMP in neuroendocrine cells. Cell Calcium.2012;51(3-4):260-266.
- Jenks BG. Regulation of proopiomelanocortin gene expression: an overview of the signaling cascades, transcription factors, and responsive elements involved. Ann N Y Acad Sci. 2009;1163:17-30.
- Martens C, Bilodeau S, Maira M, Gauthier Y, Drouin J. Protein-protein interactions and transcriptional antagonism between the subfamily of NGFI-B/Nur77 orphan nuclear receptors and glucocorticoid receptor. Mol Endocrinol.2005;19(4):885-897.
- Boutillier AL, Monnier D, Koch B, Loeffler JP. Pituitary adenyl cyclase-activating peptide: a hypophysiotropic factor that stimulates proopiomelanocortin gene transcription, and proopiomelanocortin-derived peptide secretion in corticotropic cells. Neuroendocrinology. 1994;60(5):493-502.
- Karalis KP, Venihaki M, Zhao J, van Vlerken LE, Chandras C. NF-kappaB participates in the corticotropin-releasing, hormone-induced regulation of the pituitary proopiomelanocortin gene. J Biol Chem.2004;279(12):10837-10840.
- Asaba K, Iwasaki Y, Asai M, Yoshida M, Nigawara T, Kambayashi M, Hashimoto K. High glucose activates pituitary proopiomelanocortin gene expression: possible role of free radical-sensitive transcription factors. Diabetes Metab Res Rev. 2007;23(4):317-323.
- Suda T, Tozawa F, Yamada M, Ushiyama T, Tomori N, Sumitomo T, Nakagami Y, Demura H, Shizume K. Effects of corticotropin-releasing hormone and dexamethasone on proopiomelanocortin messenger RNA level in human corticotroph adenoma cells in vitro. J Clin Invest. 1988;82(1):110-114.
- Wardlaw SL, McCarthy KC, Conwell IM. Glucocorticoid regulation of hypothalamic proopiomelanocortin. Neuroendocrinology. 1998;67(1):51-57.
- Drouin J, Charron J, Gagner JP, Jeannotte L, Nemer M, Plante RK, Wrange O. Pro-opiomelanocortin gene: a model for negative regulation of transcription by glucocorticoids. J Cell Biochem. 1987;35(4):293-304.
- Drouin J, Sun YL, Chamberland M, Gauthier Y, De Lean A, Nemer M, Schmidt TJ. Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. Embo J. 1993;12(1):145-156.
- Autelitano DJ, Lundblad JR, Blum M, Roberts JL. Hormonal regulation of POMC gene expression. Annu Rev Physiol. 1989;51:715-726.
- Pozzoli G, Bilezikjian LM, Perrin MH, Blount AL, Vale WW. Corticotropin-releasing factor (CRF) and glucocorticoids modulate the expression of type 1 CRF receptor messenger ribonucleic acid in rat anterior pituitary cell cultures. Endocrinology. 1996;137(1):65-71.
- Parvin R, Saito-Hakoda A, Shimada H, Shimizu K, Noro E, Iwasaki Y, Fujiwara K, Yokoyama A, Sugawara A. Role of NeuroD1 on the negative regulation of Pomc expression by glucocorticoid. PLoS One. 2017;12(4):e0175435.
- Paez-Pereda M, Kovalovsky D, Hopfner U, Theodoropoulou M, Pagotto U, Uhl E, Losa M, Stalla J, Grubler Y, Missale C, Arzt E, Stalla GK. Retinoic acid prevents experimental Cushing syndrome. The Journal of clinical investigation. 2001;108(8):1123-1131.
- Frederic F, Chianale C, Oliver C, Mariani J. Enhanced endocrine response to novel environment stress and lack of corticosterone circadian rhythm in staggerer (Rora sg/sg) mutant mice. J Neurosci Res. 2006;83(8):1525-1532.
- Martin NM, Small CJ, Sajedi A, Liao XH, Weiss RE, Gardiner JV, Ghatei MA, Bloom SR. Abnormalities of the hypothalamo-pituitary-thyroid axis in the pro-opiomelanocortin deficient mouse. Regul Pept. 2004;122(3):169-172.
- Ray DW, Ren SG, Melmed S. Leukemia inhibitory factor (LIF) stimulates proopiomelanocortin (POMC) expression in a corticotroph cell line. Role of STAT pathway. J Clin Invest. 1996;97(8):1852-1859.
- Ray DW, Ren SG, Melmed S. Leukemia inhibitory factor regulates proopiomelanocortin transcription. Ann N Y Acad Sci. 1998;840:162-173.
- Mynard V, Guignat L, Devin-Leclerc J, Bertagna X, Catelli MG. Different mechanisms for leukemia inhibitory factor-dependent activation of two proopiomelanocortin promoter regions. Endocrinology. 2002;143(10):3916-3924.
- Bousquet C, Zatelli MC, Melmed S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J Clin Invest. 2000;106(11):1417-1425.
- Iwasaki Y, Taguchi T, Nishiyama M, Asai M, Yoshida M, Kambayashi M, Takao T, Hashimoto K. Lipopolysaccharide stimulates proopiomelanocortin gene expression in AtT20 corticotroph cells. Endocr J. 2008;55(2):285-290.
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204-220.
- Newell-Price J, King P, Clark AJ. The CpG island promoter of the human proopiomelanocortin gene is methylated in nonexpressing normal tissue and tumors and represses expression. Mol Endocrinol. 2001;15(2):338-348.
- Benjannet S, Rondeau N, Day R, Chretien M, Seidah NG. PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc Natl Acad Sci U S A. 1991;88(9):3564-3568.
- Chretien M, Seidah NG. Chemistry and biosynthesis of pro-opiomelanocortin. ACTH, MSH's, endorphins and their related peptides. Mol Cell Biochem. 1981;34(2):101-127.
- Seidah NG, Chretien M. Complete amino acid sequence of a human pituitary glycopeptide: an important maturation product of pro-opiomelanocortin. Proc Natl Acad Sci U S A. 1981;78(7):4236-4240.
- Seidah NG, Rochemont J, Hamelin J, Benjannet S, Chretien M. The missing fragment of the pro-sequence of human pro-opiomelanocortin: sequence and evidence for C-terminal amidation. Biochem Biophys Res Commun.1981;102(2):710-716.
- Eipper BA, Mains RE. Structure and biosynthesis of pro-adrenocorticotropin/endorphin and related peptides. Endocr Rev. 1980;1(1):1-27.
- Bradbury AF, Smyth DG, Snell CR. Prohormones of beta-melanotropin (beta-melanocyte-stimulating hormone, beta-MSH) and corticotropin (adrenocorticotropic hormone, ACTH): structure and activation. Ciba Found Symp.1976;41:61-75.
- Barnea A, Cho G. Acetylation of adrenocorticotropin and beta-endorphin by hypothalamic and pituitary acetyltransferases. Neuroendocrinology. 1983;37(6):434-439.
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213(4514):1394-1397.
- Shibahara S, Morimoto Y, Furutani Y, Notake M, Takahashi H, Shimizu S, Horikawa S, Numa S. Isolation and sequence analysis of the human corticotropin-releasing factor precursor gene. Embo J. 1983;2(5):775-779.
- Sawchenko PE, Swanson LW. Localization, colocalization, and plasticity of corticotropin-releasing factor immunoreactivity in rat brain. Fed Proc. 1985;44(1 Pt 2):221-227.
- Deussing JM, Chen A. The Corticotropin-Releasing Factor Family: Physiology of the Stress Response. Physiol Rev. 2018;98(4):2225-2286.
- Perrin M, Donaldson C, Chen R, Blount A, Berggren T, Bilezikjian L, Sawchenko P, Vale W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc Natl Acad Sci U S A. 1995;92(7):2969-2973.
- Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci U S A. 1993;90(19):8967-8971.
- Takefuji M, Murohara T. Corticotropin-Releasing Hormone Family and Their Receptors in the Cardiovascular System. Circ J. 2019;83(2):261-266.
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525-557.
- DeBold CR, DeCherney GS, Jackson RV, Sheldon WR, Alexander AN, Island DP, Rivier J, Vale W, Orth DN. Effect of synthetic ovine corticotropin-releasing factor: prolonged duration of action and biphasic response of plasma adrenocorticotropin and cortisol. J Clin Endocrinol Metab. 1983;57(2):294-298.
- Tse A, Lee AK, Tse FW. Ca2+ signaling and exocytosis in pituitary corticotropes. Cell Calcium. 2012;51(3-4):253-259.
- Muglia L, Jacobson L, Dikkes P, Majzoub JA. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373(6513):427-432.
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19(2):162-166.
- Wang X, Su H, Copenhagen LD, Vaishnav S, Pieri F, Shope CD, Brownell WE, De Biasi M, Paylor R, Bradley A. Urocortin-deficient mice display normal stress-induced anxiety behavior and autonomic control but an impaired acoustic startle response. Mol Cell Biol. 2002;22(18):6605-6610.
- Neufeld-Cohen A, Tsoory MM, Evans AK, Getselter D, Gil S, Lowry CA, Vale WW, Chen A. A triple urocortin knockout mouse model reveals an essential role for urocortins in stress recovery. Proc Natl Acad Sci U S A.2010;107(44):19020-19025.
- Chen A, Zorrilla E, Smith S, Rousso D, Levy C, Vaughan J, Donaldson C, Roberts A, Lee KF, Vale W. Urocortin 2-deficient mice exhibit gender-specific alterations in circadian hypothalamus-pituitary-adrenal axis and depressive-like behavior. J Neurosci. 2006;26(20):5500-5510.
- Sugimoto T, Saito M, Mochizuki S, Watanabe Y, Hashimoto S, Kawashima H. Molecular cloning and functional expression of a cDNA encoding the human V1b vasopressin receptor. J Biol Chem. 1994;269(43):27088-27092.
- Raymond V, Leung PC, Veilleux R, Labrie F. Vasopressin rapidly stimulates phosphatidic acid-phosphatidylinositol turnover in rat anterior pituitary cells. FEBS Lett. 1985;182(1):196-200.
- Abou-Samra AB, Catt KJ, Aguilera G. Involvement of protein kinase C in the regulation of adrenocorticotropin release from rat anterior pituitary cells. Endocrinology. 1986;118(1):212-217.
- Plotsky PM. Hypophysiotropic regulation of stress-induced ACTH secretion. Adv Exp Med Biol. 1988;245:65-81.
- Bilezikjian LM, Woodgett JR, Hunter T, Vale WW. Phorbol ester-induced down-regulation of protein kinase C abolishes vasopressin-mediated responses in rat anterior pituitary cells. Mol Endocrinol. 1987;1(8):555-560.
- Levin N, Blum M, Roberts JL. Modulation of basal and corticotropin-releasing factor-stimulated proopiomelanocortin gene expression by vasopressin in rat anterior pituitary. Endocrinology. 1989;125(6):2957-2966.
- Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem. 2006;52(1):112-119.
- Lewandowski KC, Lewinski A, Skowronska-Jozwiak E, Stasiak M, Horzelski W, Brabant G. Copeptin under glucagon stimulation. Endocrine. 2016;52(2):344-351.
- Kacheva S, Kolk K, Morgenthaler NG, Brabant G, Karges W. Gender-specific co-activation of arginine vasopressin and the hypothalamic-pituitary-adrenal axis during stress. Clin Endocrinol (Oxf). 2015;82(4):570-576.
- Evans AN, Liu Y, Macgregor R, Huang V, Aguilera G. Regulation of hypothalamic corticotropin-releasing hormone transcription by elevated glucocorticoids. Mol Endocrinol. 2013;27(11):1796-1807.
- Guardiola-Diaz HM, Kolinske JS, Gates LH, Seasholtz AF. Negative glucorticoid regulation of cyclic adenosine 3', 5'-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol Endocrinol.1996;10(3):317-329.
- Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol. 1999;13(10):1629-1644.
- Malkoski SP, Handanos CM, Dorin RI. Localization of a negative glucocorticoid response element of the human corticotropin releasing hormone gene. Mol Cell Endocrinol. 1997;127(2):189-199.
- Drouin J, Sun YL, Nemer M. Glucocorticoid repression of pro-opiomelanocortin gene transcription. J Steroid Biochem. 1989;34(1-6):63-69.
- Drouin J, Trifiro MA, Plante RK, Nemer M, Eriksson P, Wrange O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol Cell Biol. 1989;9(12):5305-5314.
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505-2511.
- Reul JM, de Kloet ER. Anatomical resolution of two types of corticosterone receptor sites in rat brain with in vitro autoradiography and computerized image analysis. J Steroid Biochem. 1986;24(1):269-272.
- Reul JM, van den Bosch FR, de Kloet ER. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. The Journal of endocrinology.1987;115(3):459-467.
- Edwards CR, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, de Kloet ER, Monder C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet.1988;2(8618):986-989.
- Ratka A, Sutanto W, Bloemers M, de Kloet ER. On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology. 1989;50(2):117-123.
- van Haarst AD, Oitzl MS, de Kloet ER. Facilitation of feedback inhibition through blockade of glucocorticoid receptors in the hippocampus. Neurochem Res. 1997;22(11):1323-1328.
- Brownstein MJ, Russell JT, Gainer H. Synthesis, transport, and release of posterior pituitary hormones. Science.1980;207(4429):373-378.
- Legros JJ, Chiodera P, Demey-Ponsart E. Inhibitory influence of exogenous oxytocin on adrenocorticotropin secretion in normal human subjects. J Clin Endocrinol Metab. 1982;55(6):1035-1039.
- Legros JJ, Chiodera P, Geenen V, von Frenckell R. Confirmation of the inhibitory influence of exogenous oxytocin on cortisol and ACTH in man: evidence of reproducibility. Acta Endocrinol (Copenh). 1987;114(3):345-349.
- Lewis DA, Sherman BM. Oxytocin does not influence adrenocorticotropin secretion in man. J Clin Endocrinol Metab. 1985;60(1):53-56.
- Antoni FA, Holmes MC, Kiss JZ. Pituitary binding of vasopressin is altered by experimental manipulations of the hypothalamo-pituitary-adrenocortical axis in normal as well as homozygous (di/di) Brattleboro rats. Endocrinology.1985;117(4):1293-1299.
- Winter J, Jurek B. The interplay between oxytocin and the CRF system: regulation of the stress response. Cell Tissue Res. 2019;375(1):85-91.
- Gibbs DM, Vale W, Rivier J, Yen SS. Oxytocin potentiates the ACTH-releasing activity of CRF(41) but not vasopressin. Life Sci. 1984;34(23):2245-2249.
- Alexander LD, Evans K, Sander LD. A possible involvement of VIP in feeding-induced secretion of ACTH and corticosterone in the rat. Physiol Behav. 1995;58(2):409-413.
- Alexander LD, Sander LD. Involvement of vasopressin and corticotropin-releasing hormone in VIP- and PHI-induced secretion of ACTH and corticosterone. Neuropeptides. 1995;28(3):167-173.
- Jirikowski GF, Back H, Forssmann WG, Stumpf WE. Coexistence of atrial natriuretic factor (ANF) and oxytocin in neurons of the rat hypothalamus. Neuropeptides. 1986;8(3):243-249.
- King MS, Baertschi AJ. Physiological concentrations of atrial natriuretic factors with intact N-terminal sequences inhibit corticotropin-releasing factor-stimulated adrenocorticotropin secretion from cultured anterior pituitary cells. Endocrinology. 1989;124(1):286-292.
- Kellner M, Wiedemann K, Holsboer F. Atrial natriuretic factor inhibits the CRH-stimulated secretion of ACTH and cortisol in man. Life Sci. 1992;50(24):1835-1842.
- Ur E, Faria M, Tsagarakis S, Anderson JV, Besser GM, Grossman A. Atrial natriuretic peptide in physiological doses does not inhibit the ACTH or cortisol response to corticotrophin-releasing hormone-41 in normal human subjects. J Endocrinol. 1991;131(1):163-167.
- Taylor T, Dluhy RG, Williams GH. beta-endorphin suppresses adrenocorticotropin and cortisol levels in normal human subjects. J Clin Endocrinol Metab. 1983;57(3):592-596.
- Tsagarakis S, Navarra P, Rees LH, Besser M, Grossman A, Navara P. Morphine directly modulates the release of stimulated corticotrophin-releasing factor-41 from rat hypothalamus in vitro [published erratum appears in Endocrinology 1989 Dec;125(6):3095]. Endocrinology. 1989;124(5):2330-2335.
- Jackson RV, Grice JE, Hockings GI, Torpy DJ. Naloxone-induced ACTH release: mechanism of action in humans. Clin Endocrinol (Oxf). 1995;43(4):423-424.
- Pagotto U, Marsicano G, Fezza F, Theodoropoulou M, Grubler Y, Stalla J, Arzberger T, Milone A, Losa M, Di Marzo V, Lutz B, Stalla GK. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor type 1 and synthesize endogenous cannabinoids: first evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J Clin Endocrinol Metab. 2001;86(6):2687-2696.
- Wade MR, Degroot A, Nomikos GG. Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol. 2006;551(1-3):162-167.
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of Cannabinoid Receptor Type 1 for the Basal Modulation of Hypothalamic-Pituitary-Adrenal Axis Function. Endocrinology. 2006.
- Benowitz NL, Jones RT, Lerner CB. Depression of growth hormone and cortisol response to insulin-induced hypoglycemia after prolonged oral delta-9-tetrahydrocannabinol administration in man. J Clin Endocrinol Metab.1976;42(5):938-941.
- al-Damluji S. Adrenergic control of the secretion of anterior pituitary hormones. Baillieres Clin Endocrinol Metab.1993;7(2):355-392.
- Kostoglou-Athanassiou I, Costa A, Navarra P, Nappi G, Forsling ML, Grossman AB. Endotoxin stimulates an endogenous pathway regulating corticotropin-releasing hormone and vasopressin release involving the generation of nitric oxide and carbon monoxide. J Neuroimmunol. 1998;86(1):104-109.
- Pozzoli G, Mancuso C, Mirtella A, Preziosi P, Grossman AB, Navarra P. Carbon monoxide as a novel neuroendocrine modulator: inhibition of stimulated corticotropin-releasing hormone release from acute rat hypothalamic explants. Endocrinology. 1994;135(6):2314-2317.
- Korbonits M, Kaltsas G, Perry LA, Grossman AB, Monson JP, Besser GM, Trainer PJ. Hexarelin as a test of pituitary reserve in patients with pituitary disease. Clin Endocrinol (Oxf). 1999;51(3):369-375.
- Korbonits M, Kaltsas G, Perry LA, Putignano P, Grossman AB, Besser GM, Trainer PJ. The growth hormone secretagogue hexarelin stimulates the hypothalamo-pituitary-adrenal axis via arginine vasopressin. J Clin Endocrinol Metab. 1999;84(7):2489-2495.
- Arvat E, Maccario M, Di Vito L, Broglio F, Benso A, Gottero C, Papotti M, Muccioli G, Dieguez C, Casanueva FF, Deghenghi R, Camanni F, Ghigo E. Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J Clin Endocrinol Metab. 2001;86(3):1169-1174.
- Mozid AM, Tringali G, Forsling ML, Hendricks MS, Ajodha S, Edwards R, Navarra P, Grossman AB, Korbonits M. Ghrelin is released from rat hypothalamic explants and stimulates corticotrophin-releasing hormone and arginine-vasopressin. Horm Metab Res. 2003;35(8):455-459.
- Arvat E, di Vito L, Maccagno B, Broglio F, Boghen MF, Deghenghi R, Camanni F, Ghigo E. Effects of GHRP-2 and hexarelin, two synthetic GH-releasing peptides, on GH, prolactin, ACTH and cortisol levels in man. Comparison with the effects of GHRH, TRH and hCRH. Peptides. 1997;18(6):885-891.
- Kimura T, Shimatsu A, Arimura H, Mori H, Tokitou A, Fukudome M, Nakazaki M, Tei C. Concordant and discordant adrenocorticotropin (ACTH) responses induced by growth hormone-releasing peptide-2 (GHRP-2), corticotropin-releasing hormone (CRH) and insulin-induced hypoglycemia in patients with hypothalamopituitary disorders: evidence for direct ACTH releasing activity of GHRP-2. Endocr J. 2010;57(7):639-644.
- Perras B, Schultes B, Schwaiger R, Metz C, Wesseler W, Born J, Fehm HL. Growth hormone-releasing hormone facilitates hypoglycemia-induced release of cortisol. Regul Pept. 2002;110(1):85-91.
- Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science. 2005;310(5750):996-999.
- Yamamoto D, Ikeshita N, Daito R, Herningtyas EH, Toda K, Takahashi K, Iida K, Takahashi Y, Kaji H, Chihara K, Okimura Y. Neither intravenous nor intracerebroventricular administration of obestatin affects the secretion of GH, PRL, TSH and ACTH in rats. Regul Pept. 2007;138(2-3):141-144.
- McKee KK, Tan CP, Palyha OC, Liu J, Feighner SD, Hreniuk DL, Smith RG, Howard AD, Van der Ploeg LH. Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics. 1997;46(3):426-434.
- Holst B, Egerod KL, Schild E, Vickers SP, Cheetham S, Gerlach LO, Storjohann L, Stidsen CE, Jones R, Beck-Sickinger AG, Schwartz TW. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology.2007;148(1):13-20.
- Luque RM, Cordoba-Chacon J, Ibanez-Costa A, Gesmundo I, Grande C, Gracia-Navarro F, Tena-Sempere M, Ghigo E, Gahete MD, Granata R, Kineman RD, Castano JP. Obestatin plays an opposite role in the regulation of pituitary somatotrope and corticotrope function in female primates and male/female mice. Endocrinology.2014:en20131728.
- Gaillard RC, Favrod-Coune CA, Capponi AM, Muller AF. Corticotropin-releasing activity of the renin-angiotensin system peptides in rat and in man. Neuroendocrinology. 1985;41(6):511-517.
- Rivier C, Vale W. Effect of angiotensin II on ACTH release in vivo: role of corticotropin-releasing factor. Regul Pept.1983;7(3):253-258.
- Murakami K, Ganong WF. Site at which angiotensin II acts to stimulate ACTH secretion in vivo. Neuroendocrinology. 1987;46(3):231-235.
- Sumitomo T, Suda T, Nakano Y, Tozawa F, Yamada M, Demura H. Angiotensin II increases the corticotropin-releasing factor messenger ribonucleic acid level in the rat hypothalamus. Endocrinology. 1991;128(5):2248-2252.
- Armando I, Volpi S, Aguilera G, Saavedra JM. Angiotensin II AT1 receptor blockade prevents the hypothalamic corticotropin-releasing factor response to isolation stress. Brain Res. 2007;1142:92-99.
- Armando I, Carranza A, Nishimura Y, Hoe KL, Barontini M, Terron JA, Falcon-Neri A, Ito T, Juorio AV, Saavedra JM. Peripheral administration of an angiotensin II AT(1) receptor antagonist decreases the hypothalamic-pituitary-adrenal response to isolation Stress. Endocrinology. 2001;142(9):3880-3889.
- Strowski MZ, Dashkevicz MP, Parmar RM, Wilkinson H, Kohler M, Schaeffer JM, Blake AD. Somatostatin receptor subtypes 2 and 5 inhibit corticotropin-releasing hormone-stimulated adrenocorticotropin secretion from AtT-20 cells. Neuroendocrinology. 2002;75(6):339-346.
- Lamberts SW, Zuyderwijk J, den Holder F, van Koetsveld P, Hofland L. Studies on the conditions determining the inhibitory effect of somatostatin on adrenocorticotropin, prolactin and thyrotropin release by cultured rat pituitary cells. Neuroendocrinology. 1989;50(1):44-50.
- van der Hoek J, Waaijers M, van Koetsveld PM, Sprij-Mooij D, Feelders RA, Schmid HA, Schoeffter P, Hoyer D, Cervia D, Taylor JE, Culler MD, Lamberts SW, Hofland LJ. Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am J Physiol Endocrinol Metab. 2005;289(2):E278-287.
- Silva AP, Schoeffter P, Weckbecker G, Bruns C, Schmid HA. Regulation of CRH-induced secretion of ACTH and corticosterone by SOM230 in rats. Eur J Endocrinol. 2005;153(3):R7-R10.
- Stafford PJ, Kopelman PG, Davidson K, McLoughlin L, White A, Rees LH, Besser GM, Coy DH, Grossman A. The pituitary-adrenal response to CRF-41 is unaltered by intravenous somatostatin in normal subjects. Clin Endocrinol (Oxf). 1989;30(6):661-666.
- Fehm HL, Voigt KH, Lang R, Beinert KE, Raptis S, Pfeiffer EF. Somatostatin: a potent inhibitor of ACTH-hypersecretion in adrenal insufficiency. Klin Wochenschr. 1976;54(4):173-175.
- de Bruin C, Pereira AM, Feelders RA, Romijn JA, Roelfsema F, Sprij-Mooij DM, van Aken MO, van der Lelij AJ, de Herder WW, Lamberts SW, Hofland LJ. Coexpression of dopamine and somatostatin receptor subtypes in corticotroph adenomas. J Clin Endocrinol Metab. 2009;94(4):1118-1124.
- Hofland LJ, van der Hoek J, Feelders R, van Aken MO, van Koetsveld PM, Waaijers M, Sprij-Mooij D, Bruns C, Weckbecker G, de Herder WW, Beckers A, Lamberts SW. The multi-ligand somatostatin analogue SOM230 inhibits ACTH secretion by cultured human corticotroph adenomas via somatostatin receptor type 5. Eur J Endocrinol. 2005;152(4):645-654.
- Feelders RA, de Bruin C, Pereira AM, Romijn JA, Netea-Maier RT, Hermus AR, Zelissen PM, van Heerebeek R, de Jong FH, van der Lely AJ, de Herder WW, Hofland LJ, Lamberts SW. Pasireotide alone or with cabergoline and ketoconazole in Cushing's disease. N Engl J Med. 2010;362(19):1846-1848.
- de Herder WW, Lamberts SW. Is there a role for somatostatin and its analogs in Cushing's syndrome? Metabolism. 1996;45(8 Suppl 1):83-85.
- Redei E, Hilderbrand H, Aird F. Corticotropin release inhibiting factor is encoded within prepro-TRH. Endocrinology. 1995;136(4):1813-1816.
- Redei E, Hilderbrand H, Aird F. Corticotropin release-inhibiting factor is preprothyrotropin-releasing hormone-(178-199). Endocrinology. 1995;136(8):3557-3563.
- Nicholson WE, Orth DN. Preprothyrotropin-releasing hormone-(178-199) does not inhibit corticotropin release. Endocrinology. 1996;137(5):2171-2174.
- Gershengorn MC, Arevalo CO, Geras E, Rebecchi MJ. Thyrotropin-releasing hormone stimulation of adrenocorticotropin production by mouse pituitary tumor cells in culture: possible model for anomalous release of adrenocorticotropin by thyrotropin-releasing hormone in some patients with Cushing's disease and Nelson's syndrome. J Clin Invest. 1980;65(6):1294-1300.
- Bernardini R, Kamilaris TC, Calogero AE, Johnson EO, Gomez MT, Gold PW, Chrousos GP. Interactions between tumor necrosis factor-alpha, hypothalamic corticotropin-releasing hormone, and adrenocorticotropin secretion in the rat. Endocrinology. 1990;126(6):2876-2881.
- Perlstein RS, Mougey EH, Jackson WE, Neta R. Interleukin-1 and interleukin-6 act synergistically to stimulate the release of adrenocorticotropic hormone in vivo. Lymphokine Cytokine Res. 1991;10(1-2):141-146.
- Uehara A, Gottschall PE, Dahl RR, Arimura A. Stimulation of ACTH release by human interleukin-1 beta, but not by interleukin-1 alpha, in conscious, freely-moving rats. Biochem Biophys Res Commun. 1987;146(3):1286-1290.
- Uehara A, Gottschall PE, Dahl RR, Arimura A. Interleukin-1 stimulates ACTH release by an indirect action which requires endogenous corticotropin releasing factor. Endocrinology. 1987;121(4):1580-1582.
- Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev.1996;17(1):64-102.
- Murdoch GH, Potter E, Nicolaisen AK, Evans RM, Rosenfeld MG. Epidermal growth factor rapidly stimulates prolactin gene transcription. Nature. 1982;300(5888):192-194.
- Kontogeorgos G, Stefaneanu L, Kovacs K, Cheng Z. Localization of Epidermal Growth Factor (EGF) and Epidermal Growth Factor Receptor (EGFr) in Human Pituitary Adenomas and Nontumorous Pituitaries: An Immunocytochemical Study. Endocr Pathol. 1996;7(1):63-70.
- Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, Tanaka K, Wieland T, Graf E, Saeger W, Ronchi CL, Allolio B, Buchfelder M, Strom TM, Fassnacht M, Komada M. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet.2015;47(1):31-38.
- Araki T, Liu X, Kameda H, Tone Y, Fukuoka H, Tone M, Melmed S. EGFR Induces E2F1-Mediated Corticotroph Tumorigenesis. J Endocr Soc. 2017;1(2):127-143.
- Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, Melmed S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. The Journal of clinical investigation.2011;121(12):4712-4721.
- Perez-Rivas LG, Theodoropoulou M, Ferrau F, Nusser C, Kawaguchi K, Stratakis CA, Faucz FR, Wildemberg LE, Assie G, Beschorner R, Dimopoulou C, Buchfelder M, Popovic V, Berr CM, Toth M, Ardisasmita AI, Honegger J, Bertherat J, Gadelha MR, Beuschlein F, Stalla G, Komada M, Korbonits M, Reincke M. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing's Disease. J Clin Endocrinol Metab.2015;100(7):E997-1004.
- Perez-Rivas LG, Osswald A, Knosel T, Lucia K, Schaaf C, Hristov M, Fazel J, Kirchner T, Beuschlein F, Reincke M, Theodoropoulou M. Expression and mutational status of USP8 in tumors causing ectopic ACTH secretion syndrome. Endocr Relat Cancer. 2017;24(9):L73-L77.
- Veldhuis JD, Iranmanesh A, Johnson ML, Lizarralde G. Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab. 1990;71(2):452-463.
- Krishnan KR, Ritchie JC, Saunders W, Wilson W, Nemeroff CB, Carroll BJ. Nocturnal and early morning secretion of ACTH and cortisol in humans. Biol Psychiatry. 1990;28(1):47-57.
- Desir D, Van Cauter E, Beyloos M, Bosson D, Golstein J, Copinschi G. Prolonged pulsatile administration of ovine corticotropin-releasing hormone in normal man. J Clin Endocrinol Metab. 1986;63(6):1292-1299.
- Gambacciani M, Liu JH, Swartz WH, Tueros VS, Rasmussen DD, Yen SS. Intrinsic pulsatility of ACTH release from the human pituitary in vitro. Clin Endocrinol (Oxf). 1987;26(5):557-563.
- Walker JJ, Spiga F, Waite E, Zhao Z, Kershaw Y, Terry JR, Lightman SL. The origin of glucocorticoid hormone oscillations. PLoS Biol. 2012;10(6):e1001341.
- Spiga F, Waite EJ, Liu Y, Kershaw YM, Aguilera G, Lightman SL. ACTH-dependent ultradian rhythm of corticosterone secretion. Endocrinology. 2011;152(4):1448-1457.
- Hazell G, Horn G, Lightman SL, Spiga F. Dynamics of ACTH-Mediated Regulation of Gene Transcription in ATC1 and ATC7 Adrenal Zona Fasciculata Cell Lines. Endocrinology. 2019;160(3):587-604.
- Henley DE, Leendertz JA, Russell GM, Wood SA, Taheri S, Woltersdorf WW, Lightman SL. Development of an automated blood sampling system for use in humans. J Med Eng Technol. 2009;33(3):199-208.
- Upton TJ, Zavala E, Methlie P, Kampe O, Tsagarakis S, Oksnes M, Bensing S, Vassiliadi DA, Grytaas MA, Botusan IR, Ueland G, Berinder K, Simunkova K, Balomenaki M, Margaritopoulos D, Henne N, Crossley R, Russell G, Husebye ES, Lightman SL. High-resolution daily profiles of tissue adrenal steroids by portable automated collection. Sci Transl Med. 2023;15(701):eadg8464.
- Kiessling S, Eichele G, Oster H. Adrenal glucocorticoids have a key role in circadian resynchronization in a mouse model of jet lag. J Clin Invest. 2010;120(7):2600-2609.
- Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42(1):201-206.
- Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96(2):271-290.
- Koch CE, Leinweber B, Drengberg BC, Blaum C, Oster H. Interaction between circadian rhythms and stress. Neurobiol Stress. 2017;6:57-67.
- Kornhauser JM, Nelson DE, Mayo KE, Takahashi JS. Photic and circadian regulation of c-fos gene expression in the hamster suprachiasmatic nucleus. Neuron. 1990;5(2):127-134.
- Kornhauser JM, Nelson DE, Mayo KE, Takahashi JS. Regulation of jun-B messenger RNA and AP-1 activity by light and a circadian clock. Science. 1992;255(5051):1581-1584.
- Crosio C, Cermakian N, Allis CD, Sassone-Corsi P. Light induces chromatin modification in cells of the mammalian circadian clock. Nat Neurosci. 2000;3(12):1241-1247.
- Leliavski A, Shostak A, Husse J, Oster H. Impaired glucocorticoid production and response to stress in Arntl-deficient male mice. Endocrinology. 2014;155(1):133-142.
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science.2005;308(5724):1043-1045.
- Yang S, Liu A, Weidenhammer A, Cooksey RC, McClain D, Kim MK, Aguilera G, Abel ED, Chung JH. The role of mPer2 clock gene in glucocorticoid and feeding rhythms. Endocrinology. 2009;150(5):2153-2160.
- Barclay JL, Shostak A, Leliavski A, Tsang AH, Johren O, Muller-Fielitz H, Landgraf D, Naujokat N, van der Horst GT, Oster H. High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am J Physiol Endocrinol Metab. 2013;304(10):E1053-1063.
- Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, Downes M, Evans RM. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480(7378):552-556.
- Watabe T, Tanaka K, Kumagae M, Itoh S, Hasegawa M, Horiuchi T, Miyabe S, Ohno H, Shimizu N. Diurnal rhythm of plasma immunoreactive corticotropin-releasing factor in normal subjects. Life Sci. 1987;40(17):1651-1655.
- Cai A, Wise PM. Age-related changes in the diurnal rhythm of CRH gene expression in the paraventricular nuclei. Am J Physiol. 1996;270(2 Pt 1):E238-243.
- Cunnah D, Jessop DS, Besser GM, Rees LH. Measurement of circulating corticotrophin-releasing factor in man. J Endocrinol. 1987;113(1):123-131.
- Ur E, Capstick C, McLoughlin L, Checkley S, Besser GM, Grossman A. Continuous administration of human corticotropin-releasing hormone in the absence of glucocorticoid feedback in man. Neuroendocrinology.1995;61(2):191-197.
- Yamase K, Takahashi S, Nomura K, Haruta K, Kawashima S. Circadian changes in arginine vasopressin level in the suprachiasmatic nuclei in the rat. Neurosci Lett. 1991;130(2):255-258.
- Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999;96(1):57-68.
- Fahrenkrug J, Hannibal J, Georg B. Diurnal rhythmicity of the canonical clock genes Per1, Per2 and Bmal1 in the rat adrenal gland is unaltered after hypophysectomy. J Neuroendocrinol. 2008;20(3):323-329.
- Ishida A, Mutoh T, Ueyama T, Bando H, Masubuchi S, Nakahara D, Tsujimoto G, Okamura H. Light activates the adrenal gland: timing of gene expression and glucocorticoid release. Cell Metab. 2005;2(5):297-307.
- Oster H, Damerow S, Hut RA, Eichele G. Transcriptional profiling in the adrenal gland reveals circadian regulation of hormone biosynthesis genes and nucleosome assembly genes. J Biol Rhythms. 2006;21(5):350-361.
- Yoder JM, Brandeland M, Engeland WC. Phase-dependent resetting of the adrenal clock by ACTH in vitro. Am J Physiol Regul Integr Comp Physiol. 2014;306(6):R387-393.
- Plotsky PM, Bruhn TO, Vale W. Hypophysiotropic regulation of adrenocorticotropin secretion in response to insulin-induced hypoglycemia. Endocrinology. 1985;117(1):323-329.
- Plotsky PM, Bruhn TO, Vale W. Evidence for multifactor regulation of the adrenocorticotropin secretory response to hemodynamic stimuli. Endocrinology. 1985;116(2):633-639.
- Berkenbosch F, De Goeij DC, Tilders FJ. Hypoglycemia enhances turnover of corticotropin-releasing factor and of vasopressin in the zona externa of the rat median eminence. Endocrinology. 1989;125(1):28-34.
- Jones BJ, Tan T, Bloom SR. Minireview: Glucagon in stress and energy homeostasis. Endocrinology.2012;153(3):1049-1054.
- Meeran K, Hattersley A, Mould G, Bloom SR. Venepuncture causes rapid rise in plasma ACTH. Br J Clin Pract.1993;47(5):246-247.
- Plumpton FS, Besser GM. The adrenocortical response to surgery and insulin-induced hypoglycaemia in corticosteroid-treated and normal subjects. Br J Surg. 1969;56(3):216-219.
- Plumpton FS, Besser GM, Cole PV. Corticosteroid treatment and surgery. 1. An investigation of the indications for steroid cover. Anaesthesia. 1969;24(1):3-11.
- Plumpton FS, Besser GM, Cole PV. Corticosteroid treatment and surgery. 2. The management of steroid cover. Anaesthesia. 1969;24(1):12-18.
- Khoo B, Boshier PR, Freethy A, Tharakan G, Saeed S, Hill N, Williams EL, Moorthy K, Tolley N, Jiao LR, Spalding D, Palazzo F, Meeran K, Tan T. Redefining the stress cortisol response to surgery. Clin Endocrinol (Oxf). 2017.
- Ma XM, Levy A, Lightman SL. Rapid changes in heteronuclear RNA for corticotrophin-releasing hormone and arginine vasopressin in response to acute stress. J Endocrinol. 1997;152(1):81-89.
- Ma XM, Aguilera G. Transcriptional responses of the vasopressin and corticotropin-releasing hormone genes to acute and repeated intraperitoneal hypertonic saline injection in rats. Brain Res Mol Brain Res. 1999;68(1-2):129-140.
- Kim CK, Rivier CL. Nitric oxide and carbon monoxide have a stimulatory role in the hypothalamic-pituitary-adrenal response to physico-emotional stressors in rats. Endocrinology. 2000;141(6):2244-2253.
- Bernstein HG, Keilhoff G, Seidel B, Stanarius A, Huang PL, Fishman MC, Reiser M, Bogerts B, Wolf G. Expression of hypothalamic peptides in mice lacking neuronal nitric oxide synthase: reduced beta-END immunoreactivity in the arcuate nucleus. Neuroendocrinology. 1998;68(6):403-411.
- Yamada K, Emson P, Hokfelt T. Immunohistochemical mapping of nitric oxide synthase in the rat hypothalamus and colocalization with neuropeptides. J Chem Neuroanat. 1996;10(3-4):295-316.
- Keilhoff G, Seidel B, Reiser M, Stanarius A, Huang PL, Bogerts B, Wolf G, Bernstein HG. Lack of neuronal NOS has consequences for the expression of POMC and POMC-derived peptides in the mouse pituitary. Acta Histochem. 2001;103(4):397-412.
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24(4):410-414.
- Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR, Hatton DC, Phillips TJ, Finn DA, Low MJ, Rittenberg MB, Stenzel P, Stenzel-Poore MP. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24(4):403-409.
- Aguilera G, Rabadan-Diehl C. Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: implications for stress adaptation. Regul Pept. 2000;96(1-2):23-29.
- Michalaki M, Margeli T, Tsekouras A, Gogos CH, Vagenakis AG, Kyriazopoulou V. Hypothalamic-pituitary-adrenal axis response to the severity of illness in non-critically ill patients: does relative corticosteroid insufficiency exist? Eur J Endocrinol. 2010;162(2):341-347.
- Tan T, Khoo B, Mills EG, Phylactou M, Patel B, Eng PC, Thurston L, Muzi B, Meeran K, Prevost AT, Comninos AN, Abbara A, Dhillo WS. Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol. 2020.
- Dumbell R, Matveeva O, Oster H. Circadian Clocks, Stress, and Immunity. Front Endocrinol (Lausanne).2016;7:37.
- Itoi K, Mouri T, Takahashi K, Murakami O, Imai Y, Sasaki S, Yoshinaga K, Sasano N. Suppression by glucocorticoid of the immunoreactivity of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of rat hypothalamus. Neurosci Lett. 1987;73(3):231-236.
- Davis LG, Arentzen R, Reid JM, Manning RW, Wolfson B, Lawrence KL, Baldino F, Jr. Glucocorticoid sensitivity of vasopressin mRNA levels in the paraventricular nucleus of the rat. Proc Natl Acad Sci U S A. 1986;83(4):1145-1149.
- Abou-Samra AB, Catt KJ, Aguilera G. Biphasic inhibition of adrenocorticotropin release by corticosterone in cultured anterior pituitary cells. Endocrinology. 1986;119(3):972-977.
- Russell GM, Henley DE, Leendertz J, Douthwaite JA, Wood SA, Stevens A, Woltersdorf WW, Peeters BW, Ruigt GS, White A, Veldhuis JD, Lightman SL. Rapid glucocorticoid receptor-mediated inhibition of hypothalamic-pituitary-adrenal ultradian activity in healthy males. J Neurosci. 2010;30(17):6106-6115.
- Dallman MF, Akana SF, Cascio CS, Darlington DN, Jacobson L, Levin N. Regulation of ACTH secretion: variations on a theme of B. Recent Prog Horm Res. 1987;43:113-173.
- Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5(1):1-24.
- Deng Q, Riquelme D, Trinh L, Low MJ, Tomic M, Stojilkovic S, Aguilera G. Rapid Glucocorticoid Feedback Inhibition of ACTH Secretion Involves Ligand-Dependent Membrane Association of Glucocorticoid Receptors. Endocrinology. 2015;156(9):3215-3227.
- Taylor AD, Cowell AM, Flower J, Buckingham JC. Lipocortin 1 mediates an early inhibitory action of glucocorticoids on the secretion of ACTH by the rat anterior pituitary gland in vitro. Neuroendocrinology. 1993;58(4):430-439.
- Morris DG, Kola B, Borboli N, Kaltsas GA, Gueorguiev M, McNicol AM, Ferrier R, Jones TH, Baldeweg S, Powell M, Czirjak S, Hanzely Z, Johansson JO, Korbonits M, Grossman AB. Identification of adrenocorticotropin receptor messenger ribonucleic acid in the human pituitary and its loss of expression in pituitary adenomas. J Clin Endocrinol Metab. 2003;88(12):6080-6087.
- Drouin J, Bilodeau S, Vallette S. Of old and new diseases: genetics of pituitary ACTH excess (Cushing) and deficiency. Clin Genet. 2007;72(3):175-182.
- Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, Meij B, Drouin J. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. 2006;20(20):2871-2886.
- Suda T, Tomori N, Yajima F, Ushiyama T, Sumitomo T, Nakagami Y, Demura H, Shizume K. A short negative feedback mechanism regulating corticotropin-releasing hormone release. J Clin Endocrinol Metab. 1987;64(5):909-913.
- Sawchenko PE, Arias C. Evidence for short-loop feedback effects of ACTH on CRF and vasopressin expression in parvocellular neurosecretory neurons. J Neuroendocrinol. 1995;7(9):721-731.
- Roussel-Gervais A, Couture C, Langlais D, Takayasu S, Balsalobre A, Rueda BR, Zukerberg LR, Figarella-Branger D, Brue T, Drouin J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J Clin Endocrinol Metab. 2016;101(2):513-522.
- Quigley ME, Yen SS. A mid-day surge in cortisol levels. J Clin Endocrinol Metab. 1979;49(6):945-947.
- Follenius M, Brandenberger G, Hietter B. Diurnal cortisol peaks and their relationships to meals. J Clin Endocrinol Metab. 1982;55(4):757-761.
- Stimson RH, Mohd-Shukri NA, Bolton JL, Andrew R, Reynolds RM, Walker BR. The postprandial rise in plasma cortisol in men is mediated by macronutrient-specific stimulation of adrenal and extra-adrenal cortisol production. J Clin Endocrinol Metab. 2014;99(1):160-168.
- Al-Damluji S, Iveson T, Thomas JM, Pendlebury DJ, Rees LH, Besser GM. Food-induced cortisol secretion is mediated by central alpha-1 adrenoceptor modulation of pituitary ACTH secretion. Clin Endocrinol (Oxf).1987;26(5):629-636.
- Korbonits M, Trainer PJ, Nelson ML, Howse I, Kopelman PG, Besser GM, Grossman AB, Svec F. Differential stimulation of cortisol and dehydroepiandrosterone levels by food in obese and normal subjects: relation to body fat distribution. Clin Endocrinol (Oxf). 1996;45(6):699-706.
- Wake DJ, Homer NZ, Andrew R, Walker BR. Acute in vivo regulation of 11beta-hydroxysteroid dehydrogenase type 1 activity by insulin and intralipid infusions in humans. J Clin Endocrinol Metab. 2006;91(11):4682-4688.
- Benedict C, Hallschmid M, Scheibner J, Niemeyer D, Schultes B, Merl V, Fehm HL, Born J, Kern W. Gut protein uptake and mechanisms of meal-induced cortisol release. J Clin Endocrinol Metab. 2005;90(3):1692-1696.
- Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology. 1997;138(10):4445-4455.
- Ryan AS, Egan JM, Habener JF, Elahi D. Insulinotropic hormone glucagon-like peptide-1-(7-37) appears not to augment insulin-mediated glucose uptake in young men during euglycemia. J Clin Endocrinol Metab.1998;83(7):2399-2404.
- Gil-Lozano M, Perez-Tilve D, Alvarez-Crespo M, Martis A, Fernandez AM, Catalina PA, Gonzalez-Matias LC, Mallo F. GLP-1(7-36)-amide and Exendin-4 stimulate the HPA axis in rodents and humans. Endocrinology.2010;151(6):2629-2640.
- Lacroix A, Bolte E, Tremblay J, Dupre J, Poitras P, Fournier H, Garon J, Garrel D, Bayard F, Taillefer R, et al. Gastric inhibitory polypeptide-dependent cortisol hypersecretion--a new cause of Cushing's syndrome. N Engl J Med. 1992;327(14):974-980.
- Gogebakan O, Andres J, Biedasek K, Mai K, Kuhnen P, Krude H, Isken F, Rudovich N, Osterhoff MA, Kintscher U, Nauck M, Pfeiffer AF, Spranger J. Glucose-dependent insulinotropic polypeptide reduces fat-specific expression and activity of 11beta-hydroxysteroid dehydrogenase type 1 and inhibits release of free fatty acids. Diabetes.2012;61(2):292-300.
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4(2):141-194.
- Ferrari E, Magri F. Role of neuroendocrine pathways in cognitive decline during aging. Ageing Res Rev.2008;7(3):225-233.
- Aguilera G. HPA axis responsiveness to stress: implications for healthy aging. Exp Gerontol. 2011;46(2-3):90-95.
- Kasckow JW, Lupien SJ, Behan DP, Welge J, Hauger RJ. Circulating human corticotropin-releasing factor-binding protein levels following cortisol infusions. Life Sci. 2001;69(2):133-142.
- Tizabi Y, Aguilera G, Gilad GM. Age-related reduction in pituitary corticotropin-releasing hormone receptors in two rat strains. Neurobiol Aging. 1992;13(2):227-230.
- Lupien S, Lecours AR, Schwartz G, Sharma S, Hauger RL, Meaney MJ, Nair NP. Longitudinal study of basal cortisol levels in healthy elderly subjects: evidence for subgroups. Neurobiol Aging. 1996;17(1):95-105.
- Linkowski P, Van Onderbergen A, Kerkhofs M, Bosson D, Mendlewicz J, Van Cauter E. Twin study of the 24-h cortisol profile: evidence for genetic control of the human circadian clock. Am J Physiol. 1993;264(2 Pt 1):E173-181.
- Franz CE, York TP, Eaves LJ, Mendoza SP, Hauger RL, Hellhammer DH, Jacobson KC, Levine S, Lupien SJ, Lyons MJ, Prom-Wormley E, Xian H, Kremen WS. Genetic and environmental influences on cortisol regulation across days and contexts in middle-aged men. Behav Genet. 2010;40(4):467-479.
- Cai A, Scarbrough K, Hinkle DA, Wise PM. Fetal grafts containing suprachiasmatic nuclei restore the diurnal rhythm of CRH and POMC mRNA in aging rats. Am J Physiol. 1997;273(5 Pt 2):R1764-1770.
- Cooper MS. 11beta-Hydroxysteroid dehydrogenase: a regulator of glucocorticoid response in osteoporosis. J Endocrinol Invest. 2008;31(7 Suppl):16-21.
- Holmes MC, Carter RN, Noble J, Chitnis S, Dutia A, Paterson JM, Mullins JJ, Seckl JR, Yau JL. 11beta-hydroxysteroid dehydrogenase type 1 expression is increased in the aged mouse hippocampus and parietal cortex and causes memory impairments. J Neurosci. 2010;30(20):6916-6920.
- Lesniewska B, Miskowiak B, Nowak M, Malendowicz LK. Sex differences in adrenocortical structure and function. XXVII. The effect of ether stress on ACTH and corticosterone in intact, gonadectomized, and testosterone- or estradiol-replaced rats. Res Exp Med (Berl). 1990;190(2):95-103.
- Lesniewska B, Nowak M, Malendowicz LK. Sex differences in adrenocortical structure and function. XXVIII. ACTH and corticosterone in intact, gonadectomised and gonadal hormone replaced rats. Horm Metab Res.1990;22(7):378-381.
- Jezova D, Kvetnansky R, Vigas M. Sex differences in endocrine response to hyperthermia in sauna. Acta Physiol Scand. 1994;150(3):293-298.
- Jezova D, Jurankova E, Mosnarova A, Kriska M, Skultetyova I. Neuroendocrine response during stress with relation to gender differences. Acta Neurobiol Exp (Wars). 1996;56(3):779-785.
- Vamvakopoulos NC, Chrousos GP. Evidence of direct estrogenic regulation of human corticotropin-releasing hormone gene expression. Potential implications for the sexual dimophism of the stress response and immune/inflammatory reaction. J Clin Invest. 1993;92(4):1896-1902.
- Kirschbaum C, Wust S, Faig HG, Hellhammer DH. Heritability of cortisol responses to human corticotropin-releasing hormone, ergometry, and psychological stress in humans. J Clin Endocrinol Metab. 1992;75(6):1526-1530.
- Matthews KA, Gump BB, Owens JF. Chronic stress influences cardiovascular and neuroendocrine responses during acute stress and recovery, especially in men. Health Psychol. 2001;20(6):403-410.
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477-501.
- Pariante CM. Why are depressed patients inflamed? A reflection on 20 years of research on depression, glucocorticoid resistance and inflammation. Eur Neuropsychopharmacol. 2017;27(6):554-559.
- Beck-Friis J, Ljunggren JG, Thoren M, von Rosen D, Kjellman BF, Wetterberg L. Melatonin, cortisol and ACTH in patients with major depressive disorder and healthy humans with special reference to the outcome of the dexamethasone suppression test. Psychoneuroendocrinology. 1985;10(2):173-186.
- Reus VI, Joseph M, Dallman M. Regulation of ACTH and cortisol in depression. Peptides. 1983;4(5):785-788.
- Dziurkowska E, Wesolowski M. Cortisol as a Biomarker of Mental Disorder Severity. J Clin Med. 2021;10(21).
- Nemeroff CB, Krishnan KR, Reed D, Leder R, Beam C, Dunnick NR. Adrenal gland enlargement in major depression. A computed tomographic study. Arch Gen Psychiatry. 1992;49(5):384-387.
- Rubin RT, Phillips JJ, Sadow TF, McCracken JT. Adrenal gland volume in major depression. Increase during the depressive episode and decrease with successful treatment. Arch Gen Psychiatry. 1995;52(3):213-218.
- Holsboer F, von Bardeleben U, Wiedemann K, Muller OA, Stalla GK. Serial assessment of corticotropin-releasing hormone response after dexamethasone in depression. Implications for pathophysiology of DST nonsuppression. Biol Psychiatry. 1987;22(2):228-234.
- Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48(8):693-699.
- Young EA, Carlson NE, Brown MB. Twenty-four-hour ACTH and cortisol pulsatility in depressed women. Neuropsychopharmacology. 2001;25(2):267-276.
- Amasi-Hartoonian N, Sforzini L, Cattaneo A, Pariante CM. Cause or consequence? Understanding the role of cortisol in the increased inflammation observed in depression. Curr Opin Endocr Metab Res. 2022;24:100356.
- Misiak B, Loniewski I, Marlicz W, Frydecka D, Szulc A, Rudzki L, Samochowiec J. The HPA axis dysregulation in severe mental illness: Can we shift the blame to gut microbiota? Prog Neuropsychopharmacol Biol Psychiatry.2020;102:109951.
- Huang MC, Chuang SC, Tseng MM, Chien YL, Liao SC, Chen HC, Kuo PH. Cortisol awakening response in patients with bipolar disorder during acute episodes and partial remission: A pilot study. Psychiatry Res.2017;258:594-597.
- Walker EF, Brennan PA, Esterberg M, Brasfield J, Pearce B, Compton MT. Longitudinal changes in cortisol secretion and conversion to psychosis in at-risk youth. J Abnorm Psychol. 2010;119(2):401-408.
- Labad J, Stojanovic-Perez A, Montalvo I, Sole M, Cabezas A, Ortega L, Moreno I, Vilella E, Martorell L, Reynolds RM, Gutierrez-Zotes A. Stress biomarkers as predictors of transition to psychosis in at-risk mental states: roles for cortisol, prolactin and albumin. J Psychiatr Res. 2015;60:163-169.
- Shah JL, Malla AK. Much ado about much: stress, dynamic biomarkers and HPA axis dysregulation along the trajectory to psychosis. Schizophr Res. 2015;162(1-3):253-260.
- Chaumette B, Kebir O, Mam-Lam-Fook C, Morvan Y, Bourgin J, Godsil BP, Plaze M, Gaillard R, Jay TM, Krebs MO. Salivary cortisol in early psychosis: New findings and meta-analysis. Psychoneuroendocrinology.2016;63:262-270.
- Nordholm D, Rostrup E, Mondelli V, Randers L, Nielsen MO, Wulff S, Norbak-Emig H, Broberg BV, Krakauer K, Dazzan P, Zunszain PA, Nordentoft M, Glenthoj B. Multiple measures of HPA axis function in ultra high risk and first-episode schizophrenia patients. Psychoneuroendocrinology. 2018;92:72-80.
- Corcoran CM, Smith C, McLaughlin D, Auther A, Malaspina D, Cornblatt B. HPA axis function and symptoms in adolescents at clinical high risk for schizophrenia. Schizophr Res. 2012;135(1-3):170-174.
- Horvath E, Kovacs K. Fine structural cytology of the adenohypophysis in rat and man. J Electron Microsc Tech.1988;8(4):401-432.
- Cohen LE, Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev. 2002;23(4):431-442.
- Kapali J, Kabat BE, Schmidt KL, Stallings CE, Tippy M, Jung DO, Edwards BS, Nantie LB, Raeztman LT, Navratil AM, Ellsworth BS. Foxo1 Is Required for Normal Somatotrope Differentiation. Endocrinology. 2016;157(11):4351-4363.
- Zhu X, Zhang J, Tollkuhn J, Ohsawa R, Bresnick EH, Guillemot F, Kageyama R, Rosenfeld MG. Sustained Notch signaling in progenitors is required for sequential emergence of distinct cell lineages during organogenesis. Genes Dev. 2006;20(19):2739-2753.
- Li CH, Papkoff H. Preparation and properties of growth hormone from human and monkey pituitary glands. Science. 1956;124(3235):1293-1294.
- Li CH, Dixon JS. Human pituitary growth hormone. 32. The primary structure of the hormone: revision. Arch Biochem Biophys. 1971;146(1):233-236.
- Niall HD, Hogan ML, Sauer R, Rosenblum IY, Greenwood FC. Sequences of pituitary and placental lactogenic and growth hormones: evolution from a primordial peptide by gene reduplication. Proc Natl Acad Sci U S A.1971;68(4):866-870.
- Owerbach D, Rutter WJ, Martial JA, Baxter JD, Shows TB. Genes for growth hormone, chorionic somatommammotropin, and growth hormones-like gene on chromosome 17 in humans. Science.1980;209(4453):289-292.
- McKusick V, Phillips JI, Bottani A, Antonarakis S, O'Neill M, Kniffin C, Hartz P, Gross M. GROWTH HORMONE 1; GH1. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD, USA: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=139250. Accessed 20 Jan 2007
- Frankenne F, Rentier-Delrue F, Scippo ML, Martial J, Hennen G. Expression of the growth hormone variant gene in human placenta. J Clin Endocrinol Metab. 1987;64(3):635-637.
- Liebhaber SA, Urbanek M, Ray J, Tuan RS, Cooke NE. Characterization and histologic localization of human growth hormone-variant gene expression in the placenta. J Clin Invest. 1989;83(6):1985-1991.
- McKusick V, Phillips JI. GROWTH HORMONE 2; GH2. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=139240. Accessed 20 Jan 2007
- McKusick V, Phillips JI, Rasooly R. CHORIONIC SOMATOMAMMOTROPIN HORMONE 1; CSH1. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=150200. Accessed 20 Jan 2007
- McKusick V, Rasooly R. CHORIONIC SOMATOMAMMOTROPIN HORMONE 2; CSH2. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=118820. Accessed 20 Jan 2007
- Rasooly R. CHORIONIC SOMATOMAMMOTROPIN HORMONE-LIKE 1; CSHL1. In: McKusick V, ed. Online Mendelian Inheritance in Man. Baltimore, MD: Johns Hopkins University; 2007: http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=603515. Accessed 20 Jan 2007
- Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE, Seeburg PH. The human growth hormone locus: nucleotide sequence, biology, and evolution. Genomics. 1989;4(4):479-497.
- Bodner M, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. The pituitary-specific transcription factor GHF-1 is a homeobox-containing protein. Cell. 1988;55(3):505-518.
- Lemaigre FP, Peers B, Lafontaine DA, Mathy-Hartert M, Rousseau GG, Belayew A, Martial JA. Pituitary-specific factor binding to the human prolactin, growth hormone, and placental lactogen genes. DNA. 1989;8(3):149-159.
- Lemaigre FP, Courtois SJ, Durviaux SM, Egan CJ, LaFontaine DA, Rousseau GG. Analysis of cis- and trans-acting elements in the hormone-sensitive human somatotropin gene promoter. J Steroid Biochem. 1989;34(1-6):79-83.
- Nachtigal MW, Nickel BE, Klassen ME, Zhang WG, Eberhardt NL, Cattini PA. Human chorionic somatomammotropin and growth hormone gene expression in rat pituitary tumour cells is dependent on proximal promoter sequences. Nucleic Acids Res. 1989;17(11):4327-4337.
- Li S, Crenshaw EB, 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature. 1990;347(6293):528-533.
- Fox SR, Jong MT, Casanova J, Ye ZS, Stanley F, Samuels HH. The homeodomain protein, Pit-1/GHF-1, is capable of binding to and activating cell-specific elements of both the growth hormone and prolactin gene promoters. Mol Endocrinol. 1990;4(7):1069-1080.
- Tansey WP, Catanzaro DF. Sp1 and thyroid hormone receptor differentially activate expression of human growth hormone and chorionic somatomammotropin genes. J Biol Chem. 1991;266(15):9805-9813.
- Nickel BE, Nachtigal MW, Bock ME, Cattini PA. Differential binding of rat pituitary-specific nuclear factors to the 5'-flanking region of pituitary and placental members of the human growth hormone gene family. Mol Cell Biochem.1991;106(2):181-187.
- Walker WH, Fitzpatrick SL, Saunders GF. Human placental lactogen transcriptional enhancer. Tissue specificity and binding with specific proteins. J Biol Chem. 1990;265(22):12940-12948.
- Nachtigal MW, Nickel BE, Cattini PA. Pituitary-specific repression of placental members of the human growth hormone gene family. A possible mechanism for locus regulation. J Biol Chem. 1993;268(12):8473-8479.
- Jones BK, Monks BR, Liebhaber SA, Cooke NE. The human growth hormone gene is regulated by a multicomponent locus control region. Mol Cell Biol. 1995;15(12):7010-7021.
- Shewchuk BM, Liebhaber SA, Cooke NE. Specification of unique Pit-1 activity in the hGH locus control region. Proc Natl Acad Sci U S A. 2002;99(18):11784-11789.
- Elefant F, Cooke NE, Liebhaber SA. Targeted recruitment of histone acetyltransferase activity to a locus control region. J Biol Chem. 2000;275(18):13827-13834.
- Ho Y, Elefant F, Cooke N, Liebhaber S. A defined locus control region determinant links chromatin domain acetylation with long-range gene activation. Mol Cell. 2002;9(2):291-302.
- Ho Y, Tadevosyan A, Liebhaber SA, Cooke NE. The juxtaposition of a promoter with a locus control region transcriptional domain activates gene expression. EMBO Rep. 2008;9(9):891-898.
- Gil-Puig C, Seoane S, Blanco M, Macia M, Garcia-Caballero T, Segura C, Perez-Fernandez R. Pit-1 is expressed in normal and tumorous human breast and regulates GH secretion and cell proliferation. Eur J Endocrinol.2005;153(2):335-344.
- Frohman LA, Burek L, Stachura MA. Characterization of growth hormone of different molecular weights in rat, dog and human pituitaries. Endocrinology. 1972;91(1):262-269.
- Baumann G, Winter RJ, Shaw M. Circulating molecular variants of growth hormone in childhood. Pediatr Res.1987;22(1):21-22.
- Baumann G, Stolar MW, Amburn K. Molecular forms of circulating growth hormone during spontaneous secretory episodes and in the basal state. J Clin Endocrinol Metab. 1985;60(6):1216-1220.
- Herington AC, Ymer S, Stevenson J. Identification and characterization of specific binding proteins for growth hormone in normal human sera. J Clin Invest. 1986;77(6):1817-1823.
- Leung DW, Spencer SA, Cachianes G, Hammonds RG, Collins C, Henzel WJ, Barnard R, Waters MJ, Wood WI. Growth hormone receptor and serum binding protein: purification, cloning and expression. Nature.1987;330(6148):537-543.
- Zhang Y, Jiang J, Black RA, Baumann G, Frank SJ. Tumor necrosis factor-alpha converting enzyme (TACE) is a growth hormone binding protein (GHBP) sheddase: the metalloprotease TACE/ADAM-17 is critical for (PMA-induced) GH receptor proteolysis and GHBP generation. Endocrinology. 2000;141(12):4342-4348.
- Baumann G, Shaw MA. Plasma transport of the 20,000-dalton variant of human growth hormone (20K): evidence for a 20K-specific binding site. J Clin Endocrinol Metab. 1990;71(5):1339-1343.
- Ross RJ, Esposito N, Shen XY, Von Laue S, Chew SL, Dobson PR, Postel-Vinay MC, Finidori J. A short isoform of the human growth hormone receptor functions as a dominant negative inhibitor of the full-length receptor and generates large amounts of binding protein. Mol Endocrinol. 1997;11(3):265-273.
- Gossard F, Dihl F, Pelletier G, Dubois PM, Morel G. In situ hybridization to rat brain and pituitary gland of growth hormone cDNA. Neurosci Lett. 1987;79(3):251-256.
- Aberg ND, Brywe KG, Isgaard J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. ScientificWorldJournal. 2006;6:53-80.
- Thorner MO, Perryman RL, Cronin MJ, Rogol AD, Draznin M, Johanson A, Vale W, Horvath E, Kovacs K. Somatotroph hyperplasia. Successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J Clin Invest. 1982;70(5):965-977.
- Mayo K, Vale W, Rivier J, Rosenfeld M, Evans R. Expression-cloning and sequence of a cDNA encoding human growth hormone-releasing factor. Nature. 1983;306(5938):86-88.
- Gubler U, Monahan J, Lomedico P, Bhatt R, Collier K, Hoffman B, Bohlen P, Esch F, Ling N, Zeytin F, Brazeau P, Poonian M, Gage L. Cloning and sequence analysis of cDNA for the precursor of human growth hormone-releasing factor, somatocrinin. Proc Natl Acad Sci U S A. 1983;80(14):4311-4314.
- Mayo K. Molecular cloning and expression of a pituitary-specific receptor for growth hormone-releasing hormone. Mol Endocrinol. 1992;6(10):1734-1744.
- Barinaga M, Yamonoto G, Rivier C, Vale W, Evans R, Rosenfeld M. Transcriptional regulation of growth hormone gene expression by growth hormone-releasing factor. Nature. 1983;306(5938):84-85.
- Fukata J, Diamond D, Martin J. Effects of rat growth hormone (rGH)-releasing factor and somatostatin on the release and synthesis of rGH in dispersed pituitary cells. Endocrinology. 1985;117(2):457-467.
- Campbell R, Scanes C. Evolution of the growth hormone-releasing factor (GRF) family of peptides. Growth Regul.1992;2(4):175-191.
- Shen L, Pictet R, Rutter W. Human somatostatin I: sequence of the cDNA. Proc Natl Acad Sci U S A.1982;79(15):4575-4579.
- Siler T, VandenBerg G, Yen S, Brazeau P, Vale W, Guillemin R. Inhibition of growth hormone release in humans by somatostatin. J Clin Endocrinol Metab. 1973;37(4):632-634.
- Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science. 1973;179(68):77-79.
- Tannenbaum GS, Epelbaum J, Bowers CY. Interrelationship between the Novel Peptide Ghrelin and Somatostatin/Growth Hormone-Releasing Hormone in Regulation of Pulsatile Growth Hormone Secretion. Endocrinology. 2003;144(3):967-974.
- Broglio F, Koetsveld Pv P, Benso A, Gottero C, Prodam F, Papotti M, Muccioli G, Gauna C, Hofland L, Deghenghi R, Arvat E, Van Der Lely AJ, Ghigo E. Ghrelin secretion is inhibited by either somatostatin or cortistatin in humans. J Clin Endocrinol Metab. 2002;87(10):4829-4832.
- Norrelund H, Hansen TK, Orskov H, Hosoda H, Kojima M, Kangawa K, Weeke J, Moller N, Christiansen JS, Jorgensen JO. Ghrelin immunoreactivity in human plasma is suppressed by somatostatin. Clin Endocrinol (Oxf).2002;57(4):539-546.
- Shimada M, Date Y, Mondal MS, Toshinai K, Shimbara T, Fukunaga K, Murakami N, Miyazato M, Kangawa K, Yoshimatsu H, Matsuo H, Nakazato M. Somatostatin suppresses ghrelin secretion from the rat stomach. Biochem Biophys Res Commun. 2003;302(3):520-525.
- Vale W, Rivier C, Brazeau P, Guillemin R. Effects of somatostatin on the secretion of thyrotropin and prolactin. Endocrinology. 1974;95(4):968-977.
- Siler TM, Yen SC, Vale W, Guillemin R. Inhibition by somatostatin on the release of TSH induced in man by thyrotropin-releasing factor. J Clin Endocrinol Metab. 1974;38(5):742-745.
- Liddle R. Physiology of somatostatin and its analogues. UpToDate version 14.3. Wellesley, MA, United States of America: UpToDate, Inc.; 2006.
- Patel Y, Greenwood M, Panetta R, Hukovic N, Grigorakis S, Robertson L, Srikant C. Molecular biology of somatostatin receptor subtypes. Metabolism. 1996;45(8 Suppl 1):31-38.
- Peineau S, Guimiot F, Csaba Z, Jacquier S, Fafouri A, Schwendimann L, de Roux N, Schulz S, Gressens P, Auvin S, Dournaud P. Somatostatin receptors type 2 and 5 expression and localization during human pituitary development. Endocrinology. 2014;155(1):33-39.
- Reisine T, Bell GI. Molecular biology of somatostatin receptors. Endocr Rev. 1995;16(4):427-442.
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656-660.
- Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, Rigby M, Sirinathsinghji DJ, Dean DC, Melillo DG, Van der Ploeg LH, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273(5277):974-977.
- Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab. 2002;87(6):2988.
- Chan CB, Cheng CH. Identification and functional characterization of two alternatively spliced growth hormone secretagogue receptor transcripts from the pituitary of black seabream Acanthopagrus schlegeli. Mol Cell Endocrinol. 2004;214(1-2):81-95.
- Hosoda H, Kojima M, Matsuo H, Kangawa K. Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun. 2000;279(3):909-913.
- Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell. 2008;132(3):387-396.
- Lim CT, Kola B, Grossman A, Korbonits M. The expression of ghrelin O-acyltransferase (GOAT) in human tissues. Endocr J. 2011;58(8):707-710.
- Momany FA, Bowers CY, Reynolds GA, Chang D, Hong A, Newlander K. Design, synthesis, and biological activity of peptides which release growth hormone in vitro. Endocrinology. 1981;108(1):31-39.
- Beaumont NJ, Skinner VO, Tan TM, Ramesh BS, Byrne DJ, MacColl GS, Keen JN, Bouloux PM, Mikhailidis DP, Bruckdorfer KR, Vanderpump MP, Srai KS. Ghrelin Can Bind to a Species of High Density Lipoprotein Associated with Paraoxonase. J Biol Chem. 2003;278(11):8877-8880.
- De Vriese C, Gregoire F, Lema-Kisoka R, Waelbroeck M, Robberecht P, Delporte C. Ghrelin degradation by serum and tissue homogenates: identification of the cleavage sites. Endocrinology. 2004;145(11):4997-5005.
- Korbonits M, Bustin SA, Kojima M, Jordan S, Adams EF, Lowe DG, Kangawa K, Grossman AB. The expression of the growth hormone secretagogue receptor ligand ghrelin in normal and abnormal human pituitary and other neuroendocrine tumors. J Clin Endocrinol Metab. 2001;86(2):881-887.
- Caminos JE, Nogueiras R, Blanco M, Seoane LM, Bravo S, Alvarez CV, Garcia-Caballero T, Casanueva FF, Dieguez C. Cellular distribution and regulation of ghrelin messenger ribonucleic acid in the rat pituitary gland. Endocrinology. 2003;144(11):5089-5097.
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656-660.
- Muccioli G, Tschop M, Papotti M, Deghenghi R, Heiman M, Ghigo E. Neuroendocrine and peripheral activities of ghrelin: implications in metabolism and obesity. Eur J Pharmacol. 2002;440(2-3):235-254.
- Wren A, Small C, Abbott C, Dhillo W, Seal L, Cohen M, Batterham R, Taheri S, Stanley S, Ghatei M, Bloom S. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50(11):2540-2547.
- Wren A, Seal L, Cohen M, Brynes A, Frost G, Murphy K, Dhillo W, Ghatei M, Bloom S. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86(12):5992.
- Date Y, Nakazato M, Hashiguchi S, Dezaki K, Mondal M, Hosoda H, Kojima M, Kangawa K, Arima T, Matsuo H, Yada T, Matsukura S. Ghrelin is present in pancreatic alpha-cells of humans and rats and stimulates insulin secretion. Diabetes. 2002;51(1):124-129.
- Takaya K, Ariyasu H, Kanamoto N, Iwakura H, Yoshimoto A, Harada M, Mori K, Komatsu Y, Usui T, Shimatsu A, Ogawa Y, Hosoda K, Akamizu T, Kojima M, Kangawa K, Nakao K. Ghrelin strongly stimulates growth hormone release in humans. J Clin Endocrinol Metab. 2000;85(12):4908-4911.
- Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol.2003;23(22):7973-7981.
- Hassouna R, Zizzari P, Tomasetto C, Veldhuis JD, Fiquet O, Labarthe A, Cognet J, Steyn F, Chen C, Epelbaum J, Tolle V. An early reduction in GH peak amplitude in preproghrelin-deficient male mice has a minor impact on linear growth. Endocrinology. 2014;155(9):3561-3571.
- Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, Wiegand SJ, Sleeman MW. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci U S A. 2004;101(21):8227-8232.
- Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101(13):4679-4684.
- Xie TY, Ngo ST, Veldhuis JD, Jeffery PL, Chopin LK, Tschop M, Waters MJ, Tolle V, Epelbaum J, Chen C, Steyn FJ. Effect of Deletion of Ghrelin-O-Acyltransferase on the Pulsatile Release of Growth Hormone in Mice. J Neuroendocrinol. 2015;27(12):872-886.
- Baldanzi G, Filigheddu N, Cutrupi S, Catapano F, Bonissoni S, Fubini A, Malan D, Baj G, Granata R, Broglio F, Papotti M, Surico N, Bussolino F, Isgaard J, Deghenghi R, Sinigaglia F, Prat M, Muccioli G, Ghigo E, Graziani A. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol. 2002;159(6):1029-1037.
- Cassoni P, Ghe C, Marrocco T, Tarabra E, Allia E, Catapano F, Deghenghi R, Ghigo E, Papotti M, Muccioli G. Expression of ghrelin and biological activity of specific receptors for ghrelin and des-acyl ghrelin in human prostate neoplasms and related cell lines. Eur J Endocrinol. 2004;150(2):173-184.
- Muccioli G, Pons N, Ghe C, Catapano F, Granata R, Ghigo E. Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis in rat adipocytes via a non-type 1a growth hormone secretagogue receptor. Eur J Pharmacol. 2004;498(1-3):27-35.
- Tsubota Y, Owada-Makabe K, Yukawa K, Maeda M. Hypotensive effect of des-acyl ghrelin at nucleus tractus solitarii of rat. Neuroreport. 2005;16(2):163-166.
- Asakawa A, Inui A, Fujimiya M, Sakamaki R, Shinfuku N, Ueta Y, Meguid MM, Kasuga M. Stomach regulates energy balance via acylated ghrelin and desacyl ghrelin. Gut. 2005;54(1):18-24.
- Neary NM, Druce MR, Small CJ, Bloom SR. Acylated ghrelin stimulates food intake in the fed and fasted states but desacylated ghrelin has no effect. Gut. 2006;55(1):135.
- Chen CY, Inui A, Asakawa A, Fujino K, Kato I, Chen CC, Ueno N, Fujimiya M. Des-acyl ghrelin acts by CRF type 2 receptors to disrupt fasted stomach motility in conscious rats. Gastroenterology. 2005;129(1):8-25.
- Popovic V, Damjanovic S, Micic D, Djurovic M, Dieguez C, Casanueva FF. Blocked growth hormone-releasing peptide (GHRP-6)-induced GH secretion and absence of the synergic action of GHRP-6 plus GH-releasing hormone in patients with hypothalamopituitary disconnection: evidence that GHRP-6 main action is exerted at the hypothalamic level. J Clin Endocrinol Metab. 1995;80(3):942-947.
- Seoane LM, Tovar S, Baldelli R, Arvat E, Ghigo E, Casanueva FF, Dieguez C. Ghrelin elicits a marked stimulatory effect on GH secretion in freely-moving rats. Eur J Endocrinol. 2000;143(5):R7-9.
- Bowers CY, Sartor AO, Reynolds GA, Badger TM. On the actions of the growth hormone-releasing hexapeptide, GHRP. Endocrinology. 1991;128(4):2027-2035.
- Garcia JM, Swerdloff R, Wang C, Kyle M, Kipnes M, Biller BM, Cook D, Yuen KC, Bonert V, Dobs A, Molitch ME, Merriam GR. Macimorelin (AEZS-130)-stimulated growth hormone (GH) test: validation of a novel oral stimulation test for the diagnosis of adult GH deficiency. J Clin Endocrinol Metab. 2013;98(6):2422-2429.
- Ge X, Yang H, Bednarek MA, Galon-Tilleman H, Chen P, Chen M, Lichtman JS, Wang Y, Dalmas O, Yin Y, Tian H, Jermutus L, Grimsby J, Rondinone CM, Konkar A, Kaplan DD. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018;27(2):461-469 e466.
- Mani BK, Puzziferri N, He Z, Rodriguez JA, Osborne-Lawrence S, Metzger NP, Chhina N, Gaylinn B, Thorner MO, Thomas EL, Bell JD, Williams KW, Goldstone AP, Zigman JM. LEAP2 changes with body mass and food intake in humans and mice. The Journal of clinical investigation. 2019;129(9):3909-3923.
- Casanueva FF, Burguera B, Muruais C, Dieguez C. Acute administration of corticoids: a new and peculiar stimulus of growth hormone secretion in man. J Clin Endocrinol Metab. 1990;70(1):234-237.
- Burguera B, Muruais C, Penalva A, Dieguez C, Casanueva FF. Dual and selective actions of glucocorticoids upon basal and stimulated growth hormone release in man. Neuroendocrinology. 1990;51(1):51-58.
- Strickland AL, Underwood LE, Voina SJ, French FS, Van Wyk JJ. Growth retardation in Cushing's syndrome. Am J Dis Child. 1972;123(3):207-213.
- Giustina A, Wehrenberg WB. The role of glucocorticoids in the regulation of Growth Hormone secretion: mechanisms and clinical significance. Trends Endocrinol Metab. 1992;3(8):306-311.
- Veldhuis JD, Roemmich JN, Richmond EJ, Bowers CY. Somatotropic and gonadotropic axes linkages in infancy, childhood, and the puberty-adult transition. Endocr Rev. 2006;27(2):101-140.
- Kok P, Paulo RC, Cosma M, Mielke KL, Miles JM, Bowers CY, Veldhuis JD. Estrogen supplementation selectively enhances hypothalamo-pituitary sensitivity to ghrelin in postmenopausal women. J Clin Endocrinol Metab.2008;93(10):4020-4026.
- Leung KC, Johannsson G, Leong GM, Ho KK. Estrogen regulation of growth hormone action. Endocr Rev.2004;25(5):693-721.
- Amit Tanna RM, Harinderjeet Sandhu, Jake Powrie, Stephen Thomas, Anna Brackenridge, Louise Breen & Paul Carroll. Oral and transdermal oestrogen treatments have differing effects on GH sensitivity in hypopituitary women receiving GH replacement. Endocrine Abstract. 2010;21 P286
- Weissberger AJ, Ho KK, Lazarus L. Contrasting effects of oral and transdermal routes of estrogen replacement therapy on 24-hour growth hormone (GH) secretion, insulin-like growth factor I, and GH-binding protein in postmenopausal women. J Clin Endocrinol Metab. 1991;72(2):374-381.
- Isotton AL, Wender MC, Casagrande A, Rollin G, Czepielewski MA. Effects of oral and transdermal estrogen on IGF1, IGFBP3, IGFBP1, serum lipids, and glucose in patients with hypopituitarism during GH treatment: a randomized study. Eur J Endocrinol. 2014;166(2):207-213.
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425-432.
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269(5223):540-543.
- Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661-671.
- Jin L, Burguera BG, Couce ME, Scheithauer BW, Lamsan J, Eberhardt NL, Kulig E, Lloyd RV. Leptin and leptin receptor expression in normal and neoplastic human pituitary: evidence of a regulatory role for leptin on pituitary cell proliferation. J Clin Endocrinol Metab. 1999;84(8):2903-2911.
- Korbonits M, Chitnis MM, Gueorguiev M, Norman D, Rosenfelder N, Suliman M, Jones TH, Noonan K, Fabbri A, Besser GM, Burrin JM, Grossman AB. The release of leptin and its effect on hormone release from human pituitary adenomas. Clin Endocrinol (Oxf). 2001;54(6):781-789.
- Kristiansen MT, Clausen LR, Nielsen S, Blaabjerg O, Ledet T, Rasmussen LM, Jorgensen JO. Expression of leptin receptor isoforms and effects of leptin on the proliferation and hormonal secretion in human pituitary adenomas. Horm Res. 2004;62(3):129-136.
- Saleri R, Giustina A, Tamanini C, Valle D, Burattin A, Wehrenberg WB, Baratta M. Leptin stimulates growth hormone secretion via a direct pituitary effect combined with a decreased somatostatin tone in a median eminence-pituitary perifusion study. Neuroendocrinology. 2004;79(4):221-228.
- Tannenbaum GS, Lapointe M, Gurd W, Finkelstein JA. Mechanisms of impaired growth hormone secretion in genetically obese Zucker rats: roles of growth hormone-releasing factor and somatostatin. Endocrinology.1990;127(6):3087-3095.
- Ozata M, Dieguez C, Casanueva FF. The inhibition of growth hormone secretion presented in obesity is not mediated by the high leptin levels: a study in human leptin deficiency patients. J Clin Endocrinol Metab.2003;88(1):312-316.
- Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, Cambridge E, White JK, le Tissier P, Gharavy SN, Martinez-Barbera JP, Stokvis-Brantsma WH, Vulsma T, Kempers MJ, Persani L, Campi I, Bonomi M, Beck-Peccoz P, Zhu H, Davis TM, Hokken-Koelega AC, Del Blanco DG, Rangasami JJ, Ruivenkamp CA, Laros JF, Kriek M, Kant SG, Bosch CA, Biermasz NR, Appelman-Dijkstra NM, Corssmit EP, Hovens GC, Pereira AM, den Dunnen JT, Wade MG, Breuning MH, Hennekam RC, Chatterjee K, Dattani MT, Wit JM, Bernard DJ. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012;44(12):1375-1381.
- Joustra SD, Roelfsema F, Endert E, Ballieux BE, van Trotsenburg AS, Fliers E, Corssmit EP, Bernard DJ, Oostdijk W, Wit JM, Pereira AM, Biermasz NR. Pituitary Hormone Secretion Profiles in IGSF1 Deficiency Syndrome. Neuroendocrinology. 2016;103(3-4):408-416.
- Joustra SD, Roelfsema F, van Trotsenburg ASP, Schneider HJ, Kosilek RP, Kroon HM, Logan JG, Butterfield NC, Zhou X, Toufaily C, Bak B, Turgeon MO, Brule E, Steyn FJ, Gurnell M, Koulouri O, Le Tissier P, Fontanaud P, Duncan Bassett JH, Williams GR, Oostdijk W, Wit JM, Pereira AM, Biermasz NR, Bernard DJ, Schoenmakers N. IGSF1 Deficiency Results in Human and Murine Somatotrope Neurosecretory Hyperfunction. J Clin Endocrinol Metab. 2020;105(3).
- Gutierrez-Pascual E, Martinez-Fuentes AJ, Pinilla L, Tena-Sempere M, Malagon MM, Castano JP. Direct pituitary effects of kisspeptin: activation of gonadotrophs and somatotrophs and stimulation of luteinising hormone and growth hormone secretion. J Neuroendocrinol. 2007;19(7):521-530.
- Kadokawa H, Suzuki S, Hashizume T. Kisspeptin-10 stimulates the secretion of growth hormone and prolactin directly from cultured bovine anterior pituitary cells. Anim Reprod Sci. 2008;105(3-4):404-408.
- Whitlock BK, Daniel JA, Wilborn RR, Maxwell HS, Steele BP, Sartin JL. Interaction of kisspeptin and the somatotropic axis. Neuroendocrinology. 2010;92(3):178-188.
- Jayasena CN, Comninos AN, Narayanaswamy S, Bhalla S, Abbara A, Ganiyu-Dada Z, Busbridge M, Ghatei MA, Bloom SR, Dhillo WS. Acute and chronic effects of kisspeptin-54 administration on GH, prolactin and TSH secretion in healthy women. Clin Endocrinol (Oxf). 2014;81(6):891-898.
- Massara F, Camanni F. Effect of various adrenergic receptor stimulating and blocking agents on human growth hormone secretion. J Endocrinol. 1972;54(2):195-206.
- Chihara K, Minamitani N, Kaji H, Kodama H, Kita T, Fujita T. Noradrenergic modulation of human pancreatic growth hormone-releasing factor (hpGHRF1-44)-induced growth hormone release in conscious male rabbits: involvement of endogenous somatostatin. Endocrinology. 1984;114(4):1402-1406.
- Milner RD, Burns EC. Investigation of suspected growth hormone deficiency. On behalf of the Health Services Human Growth Hormone Committee. Arch Dis Child. 1982;57(12):944-947.
- Grossman A, Weerasuriya K, Al-Damluji S, Turner P, Besser GM. Alpha 2-adrenoceptor agonists stimulate growth hormone secretion but have no acute effects on plasma cortisol under basal conditions. Horm Res. 1987;25(2):65-71.
- Masala A, Delitala G, Alagna S, Devilla L. Effect of pimozide on levodopa-induced growth hormone release in man. Clin Endocrinol (Oxf). 1977;7(3):253-256.
- Cammani F, Massara F. Phentolamine inhibition of human growth hormone secretion induced by L-DOPA. Horm Metab Res. 1972;4(2):128.
- Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev. 1998;19(6):717-797.
- Friend K, Iranmanesh A, Login IS, Veldhuis JD. Pyridostigmine treatment selectively amplifies the mass of GH secreted per burst without altering GH burst frequency, half-life, basal GH secretion or the orderliness of GH release. Eur J Endocrinol. 1997;137(4):377-386.
- Taylor BJ, Smith PJ, Brook CG. Inhibition of physiological growth hormone secretion by atropine. Clin Endocrinol (Oxf). 1985;22(4):497-501.
- Massara F, Ghigo E, Demislis K, Tangolo D, Mazza E, Locatelli V, Muller EE, Molinatti GM, Camanni F. Cholinergic involvement in the growth hormone releasing hormone-induced growth hormone release: studies in normal and acromegalic subjects. Neuroendocrinology. 1986;43(6):670-675.
- Wehrenberg WB, Wiviott SD, Voltz DM, Giustina A. Pyridostigmine-mediated growth hormone release: evidence for somatostatin involvement. Endocrinology. 1992;130(3):1445-1450.
- Vance ML, Kaiser DL, Frohman LA, Rivier J, Vale WW, Thorner MO. Role of dopamine in the regulation of growth hormone secretion: dopamine and bromocriptine augment growth hormone (GH)-releasing hormone-stimulated GH secretion in normal man. J Clin Endocrinol Metab. 1987;64(6):1136-1141.
- Grossman A. Brain opiates and neuroendocrine function. Clin Endocrinol Metab. 1983;12(3):725-746.
- Moretti C, Fabbri A, Gnessi L, Cappa M, Calzolari A, Fraioli F, Grossman A, Besser GM. Naloxone inhibits exercise-induced release of PRL and GH in athletes. Clin Endocrinol (Oxf). 1983;18(2):135-138.
- Miki N, Ono M, Shizume K. Evidence that opiatergic and alpha-adrenergic mechanisms stimulate rat growth hormone release via growth hormone-releasing factor (GRF). Endocrinology. 1984;114(5):1950-1952.
- Olsen J, Peroski M, Kiczek M, Grignol G, Merchenthaler I, Dudas B. Intimate associations between the endogenous opiate systems and the growth hormone-releasing hormone system in the human hypothalamus. Neuroscience. 2014;258:238-245.
- Dudas B, Merchenthaler I. Topography and associations of leu-enkephalin and luteinizing hormone-releasing hormone neuronal systems in the human diencephalon. J Clin Endocrinol Metab. 2003;88(4):1842-1848.
- Delitala G, Tomasi PA, Palermo M, Ross RJ, Grossman A, Besser GM. Opioids stimulate growth hormone (GH) release in man independently of GH-releasing hormone. J Clin Endocrinol Metab. 1989;69(2):356-358.
- Wenger T, Toth BE, Martin BR. Effects of anandamide (endogen cannabinoid) on anterior pituitary hormone secretion in adult ovariectomized rats. Life Sci. 1995;56(23-24):2057-2063.
- Rettori V, Wenger T, Snyder G, Dalterio S, McCann SM. Hypothalamic action of delta-9-tetrahydrocannabinol to inhibit the release of prolactin and growth hormone in the rat. Neuroendocrinology. 1988;47(6):498-503.
- Rettori V, Aguila MC, Gimeno MF, Franchi AM, McCann SM. In vitro effect of delta 9-tetrahydrocannabinol to stimulate somatostatin release and block that of luteinizing hormone-releasing hormone by suppression of the release of prostaglandin E2. Proc Natl Acad Sci U S A. 1990;87(24):10063-10066.
- Zbucki RL, Sawicki B, Hryniewicz A, Winnicka MM. Cannabinoids enhance gastric X/A-like cells activity. Folia Histochem Cytobiol. 2008;46(2):219-224.
- Senin LL, Al-Massadi O, Folgueira C, Castelao C, Pardo M, Barja-Fernandez S, Roca-Rivada A, Amil M, Crujeiras AB, Garcia-Caballero T, Gabellieri E, Leis R, Dieguez C, Pagotto U, Casanueva FF, Seoane LM. The gastric CB1 receptor modulates ghrelin production through the mTOR pathway to regulate food intake. PLoS One.2013;8(11):e80339.
- Kola B, Farkas I, Christ-Crain M, Wittmann G, Lolli F, Amin F, Harvey-White J, Liposits Z, Kunos G, Grossman AB, Fekete C, Korbonits M. The orexigenic effect of ghrelin is mediated through central activation of the endogenous cannabinoid system. PLoS One. 2008;3(3):e1797.
- Kola B, Wittman G, Bodnar I, Amin F, Lim CT, Olah M, Christ-Crain M, Lolli F, van Thuijl H, Leontiou CA, Fuzesi T, Dalino P, Isidori AM, Harvey-White J, Kunos G, Nagy GM, Grossman AB, Fekete C, Korbonits M. The CB1 receptor mediates the peripheral effects of ghrelin on AMPK activity but not on growth hormone release. FASEB J.2013;27(12):5112-5121.
- Lim CT, Kola B, Korbonits M. The ghrelin/GOAT/GHS-R system and energy metabolism. Rev Endocr Metab Disord. 2011;12(3):173-186.
- Lim CT, Kola B, Feltrin D, Perez-Tilve D, Tschop MH, Grossman AB, Korbonits M. Ghrelin and cannabinoids require the ghrelin receptor to affect cellular energy metabolism. Mol Cell Endocrinol. 2013;365(2):303-308.
- Quabbe HJ, Bratzke HJ, Siegers U, Elban K. Studies on the relationship between plasma free fatty acids and growth hormone secretion in man. The Journal of clinical investigation. 1972;51(9):2388-2398.
- Wasada T, Howard B, McCorkle K, Harris V, Unger RH. High plasma free fatty acid levels contribute to the hypersomatostatinemia of insulin deficiency. Diabetes. 1981;30(4):358-361.
- Senaris RM, Lewis MD, Lago F, Dominguez F, Scanlon MF, Dieguez C. Effects of free fatty acids on somatostatin secretion, content and mRNA levels in cortical and hypothalamic fetal rat neurones in monolayer culture. J Mol Endocrinol. 1993;10(2):207-214.
- Giustina A, Licini M, Schettino M, Doga M, Pizzocolo G, Negro-Vilar A. Physiological role of galanin in the regulation of anterior pituitary function in humans. Am J Physiol. 1994;266(1 Pt 1):E57-61.
- Cantalamessa L, Catania A, Reschini E, Peracchi M. Inhibitory effect of calcitonin on growth hormone and insulin secretion in man. Metabolism. 1978;27(8):987-992.
- Petralito A, Lunetta M, Liuzzo A, Mangiafico RA, Fiore CE. Effects of salmon calcitonin on insulin-induced growth hormone release in man. Horm Metab Res. 1979;11(11):641-643.
- Harfstrand A, Eneroth P, Agnati L, Fuxe K. Further studies on the effects of central administration of neuropeptide Y on neuroendocrine function in the male rat: relationship to hypothalamic catecholamines. Regul Pept.1987;17(3):167-179.
- Rettori V, Milenkovic L, Aguila MC, McCann SM. Physiologically significant effect of neuropeptide Y to suppress growth hormone release by stimulating somatostatin discharge. Endocrinology. 1990;126(5):2296-2301.
- Catzeflis C, Pierroz DD, Rohner-Jeanrenaud F, Rivier JE, Sizonenko PC, Aubert ML. Neuropeptide Y administered chronically into the lateral ventricle profoundly inhibits both the gonadotropic and the somatotropic axis in intact adult female rats. Endocrinology. 1993;132(1):224-234.
- Adams EF, Venetikou MS, Woods CA, Lacoumenta S, Burrin JM. Neuropeptide Y directly inhibits growth hormone secretion by human pituitary somatotropic tumours. Acta Endocrinol (Copenh). 1987;115(1):149-154.
- Korbonits M, Little JA, Forsling ML, Tringali G, Costa A, Navarra P, Trainer PJ, Grossman AB. The effect of growth hormone secretagogues and neuropeptide Y on hypothalamic hormone release from acute rat hypothalamic explants. J Neuroendocrinol. 1999;11(7):521-528.
- Watanobe H, Tamura T. Stimulation by neuropeptide Y of growth hormone secretion in prolactinoma in vivo. Neuropeptides. 1996;30(5):429-432.
- Antonijevic IA, Murck H, Bohlhalter S, Frieboes RM, Holsboer F, Steiger A. Neuropeptide Y promotes sleep and inhibits ACTH and cortisol release in young men. Neuropharmacology. 2000;39(8):1474-1481.
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164(1):567-574.
- Goth MI, Lyons CE, Canny BJ, Thorner MO. Pituitary adenylate cyclase activating polypeptide, growth hormone (GH)-releasing peptide and GH-releasing hormone stimulate GH release through distinct pituitary receptors. Endocrinology. 1992;130(2):939-944.
- Jarry H, Leonhardt S, Schmidt WE, Creutzfeldt W, Wuttke W. Contrasting effects of pituitary adenylate cyclase activating polypeptide (PACAP) on in vivo and in vitro prolactin and growth hormone release in male rats. Life Sci.1992;51(11):823-830.
- Propato-Mussafiri R, Kanse SM, Ghatei MA, Bloom SR. Pituitary adenylate cyclase-activating polypeptide releases 7B2, adrenocorticotrophin, growth hormone and prolactin from the mouse and rat clonal pituitary cell lines AtT-20 and GH3. J Endocrinol. 1992;132(1):107-113.
- Chiodera P, Volpi R, Capretti L, Caffarri G, Magotti MG, Coiro V. Effects of intravenously infused pituitary adenylate cyclase-activating polypeptide on adenohypophyseal Hormone secretion in normal men. Neuroendocrinology.1996;64(3):242-246.
- Shahmoon S, Rubinfeld H, Wolf I, Cohen ZR, Hadani M, Shimon I, Rubinek T. The aging suppressor klotho: a potential regulator of growth hormone secretion. Am J Physiol Endocrinol Metab. 2014;307(3):E326-334.
- Abboud D, Daly AF, Dupuis N, Bahri MA, Inoue A, Chevigne A, Ectors F, Plenevaux A, Pirotte B, Beckers A, Hanson J. GPR101 drives growth hormone hypersecretion and gigantism in mice via constitutive activation of Gs and Gq/11. Nat Commun. 2020;11(1):4752.
- Iacovazzo D, Caswell R, Bunce B, Jose S, Yuan B, Hernandez-Ramirez LC, Kapur S, Caimari F, Evanson J, Ferrau F, Dang MN, Gabrovska P, Larkin SJ, Ansorge O, Rodd C, Vance ML, Ramirez-Renteria C, Mercado M, Goldstone AP, Buchfelder M, Burren CP, Gurlek A, Dutta P, Choong CS, Cheetham T, Trivellin G, Stratakis CA, Lopes MB, Grossman AB, Trouillas J, Lupski JR, Ellard S, Sampson JR, Roncaroli F, Korbonits M. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol Commun. 2016;4(1):56.
- Iacovazzo D, Korbonits M. Gigantism: X-linked acrogigantism and GPR101 mutations. Growth Horm IGF Res.2016;30-31:64-69.
- Beckers A, Lodish MB, Trivellin G, Rostomyan L, Lee M, Faucz FR, Yuan B, Choong CS, Caberg JH, Verrua E, Naves LA, Cheetham TD, Young J, Lysy PA, Petrossians P, Cotterill A, Shah NS, Metzger D, Castermans E, Ambrosio MR, Villa C, Strebkova N, Mazerkina N, Gaillard S, Barra GB, Casulari LA, Neggers SJ, Salvatori R, Jaffrain-Rea ML, Zacharin M, Santamaria BL, Zacharieva S, Lim EM, Mantovani G, Zatelli MC, Collins MT, Bonneville JF, Quezado M, Chittiboina P, Oldfield EH, Bours V, Liu P, W WdH, Pellegata N, Lupski JR, Daly AF, Stratakis CA. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer.2015;22(3):353-367.
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, Castermans E, Villa C, Dimopoulos A, Chittiboina P, Xekouki P, Shah N, Metzger D, Lysy PA, Ferrante E, Strebkova N, Mazerkina N, Zatelli MC, Lodish M, Horvath A, de Alexandre RB, Manning AD, Levy I, Keil MF, Sierra Mde L, Palmeira L, Coppieters W, Georges M, Naves LA, Jamar M, Bours V, Wu TJ, Choong CS, Bertherat J, Chanson P, Kamenicky P, Farrell WE, Barlier A, Quezado M, Bjelobaba I, Stojilkovic SS, Wess J, Costanzi S, Liu P, Lupski JR, Beckers A, Stratakis CA. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371(25):2363-2374.
- Cowley MA, Grove KL. To be or NUCB2, is nesfatin the answer? Cell Metab. 2006;4(6):421-422.
- Velez EJ, Unniappan S. Nesfatin-1 and nesfatin-1-like peptide suppress growth hormone synthesis via the AC/PKA/CREB pathway in mammalian somatotrophs. Sci Rep. 2020;10(1):16686.
- Peterfreund RA, Vale WW. Somatostatin analogs inhibit somatostatin secretion from cultured hypothalamus cells. Neuroendocrinology. 1984;39(5):397-402.
- Aguila MC, McCann SM. The influence of hGRF, CRF, TRH and LHRH on SRIF release from median eminence fragments. Brain Res. 1985;348(1):180-182.
- Sheppard MC, Kronheim S, Pimstone BL. Stimulation by growth hormone of somatostatin release from the rat hypothalamus in vitro. Clin Endocrinol (Oxf). 1978;9(6):583-586.
- Ross RJ, Tsagarakis S, Grossman A, Nhagafoong L, Touzel RJ, Rees LH, Besser GM. GH feedback occurs through modulation of hypothalamic somatostatin under cholinergic control: studies with pyridostigmine and GHRH. Clin Endocrinol (Oxf). 1987;27(6):727-733.
- Aguila MC, McCann SM. Growth hormone increases somatostatin release and messenger ribonucleic acid levels in the rat hypothalamus. Brain Res. 1993;623(1):89-94.
- Qi X, Reed J, Englander EW, Chandrashekar V, Bartke A, Greeley GH, Jr. Evidence that growth hormone exerts a feedback effect on stomach ghrelin production and secretion. Exp Biol Med (Maywood). 2003;228(9):1028-1032.
- Goldenberg N, Barkan A. Factors regulating growth hormone secretion in humans. Endocrinol Metab Clin North Am. 2007;36(1):37-55.
- Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL. Somatomedin-C mediates growth hormone negative feedback by effects on both the hypothalamus and the pituitary. Science. 1981;212(4500):1279-1281.
- Yamashita S, Melmed S. Insulin-like growth factor I action on rat anterior pituitary cells: suppression of growth hormone secretion and messenger ribonucleic acid levels. Endocrinology. 1986;118(1):176-182.
- Eden S, Bolle P, Modigh K. Monoaminergic control of episodic growth hormone secretion in the rat: effects of reserpine, alpha-methyl-p-tyrosine, p-chlorophenylalanine, and haloperidol. Endocrinology. 1979;105(2):523-529.
- Iranmanesh A, Grisso B, Veldhuis JD. Low basal and persistent pulsatile growth hormone secretion are revealed in normal and hyposomatotropic men studied with a new ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab. 1994;78(3):526-535.
- Toogood AA, Nass RM, Pezzoli SS, O'Neill PA, Thorner MO, Shalet SM. Preservation of growth hormone pulsatility despite pituitary pathology, surgery, and irradiation. J Clin Endocrinol Metab. 1997;82(7):2215-2221.
- Surya S, Symons K, Rothman E, Barkan AL. Complex rhythmicity of growth hormone secretion in humans. Pituitary. 2006;9(2):121-125.
- Tannenbaum GS, Painson JC, Lengyel AM, Brazeau P. Paradoxical enhancement of pituitary growth hormone (GH) responsiveness to GH-releasing factor in the face of high somatostatin tone. Endocrinology.1989;124(3):1380-1388.
- Plotsky PM, Vale W. Patterns of growth hormone-releasing factor and somatostatin secretion into the hypophysial-portal circulation of the rat. Science. 1985;230(4724):461-463.
- Cataldi M, Magnan E, Guillaume V, Dutour A, Conte-Devolx B, Lombardi G, Oliver C. Relationship between hypophyseal portal GHRH and somatostatin and peripheral GH levels in the conscious sheep. J Endocrinol Invest.1994;17(9):717-722.
- Vance ML, Kaiser DL, Martha PM, Jr., Furlanetto R, Rivier J, Vale W, Thorner MO. Lack of in vivo somatotroph desensitization or depletion after 14 days of continuous growth hormone (GH)-releasing hormone administration in normal men and a GH-deficient boy. J Clin Endocrinol Metab. 1989;68(1):22-28.
- Roelfsema F, Biermasz NR, Veldman RG, Veldhuis JD, Frolich M, Stokvis-Brantsma WH, Wit JM. Growth hormone (GH) secretion in patients with an inactivating defect of the GH-releasing hormone (GHRH) receptor is pulsatile: evidence for a role for non-GHRH inputs into the generation of GH pulses. J Clin Endocrinol Metab.2001;86(6):2459-2464.
- van den Berg G, Veldhuis JD, Frolich M, Roelfsema F. An amplitude-specific divergence in the pulsatile mode of growth hormone (GH) secretion underlies the gender difference in mean GH concentrations in men and premenopausal women. J Clin Endocrinol Metab. 1996;81(7):2460-2467.
- Chapman IM, Hartman ML, Straume M, Johnson ML, Veldhuis JD, Thorner MO. Enhanced sensitivity growth hormone (GH) chemiluminescence assay reveals lower postglucose nadir GH concentrations in men than women. J Clin Endocrinol Metab. 1994;78(6):1312-1319.
- Cordoba-Chacon J, Gahete MD, Castano JP, Kineman RD, Luque RM. Somatostatin and its receptors contribute in a tissue-specific manner to the sex-dependent metabolic (fed/fasting) control of growth hormone axis in mice. Am J Physiol Endocrinol Metab. 2011;300(1):E46-54.
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A.1997;94(14):7239-7244.
- Zadik Z, Chalew SA, McCarter RJ, Jr., Meistas M, Kowarski AA. The influence of age on the 24-hour integrated concentration of growth hormone in normal individuals. J Clin Endocrinol Metab. 1985;60(3):513-516.
- Rudman D, Feller AG, Nagraj HS, Gergans GA, Lalitha PY, Goldberg AF, Schlenker RA, Cohn L, Rudman IW, Mattson DE. Effects of human growth hormone in men over 60 years old. N Engl J Med. 1990;323(1):1-6.
- Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science.2010;328(5976):321-326.
- Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. 2013;9(6):366-376.
- Nass R, Farhy LS, Liu J, Pezzoli SS, Johnson ML, Gaylinn BD, Thorner MO. Age-dependent decline in acyl-ghrelin concentrations and reduced association of acyl-ghrelin and growth hormone in healthy older adults. J Clin Endocrinol Metab. 2014;99(2):602-608.
- Nass R. Growth hormone axis and aging. Endocrinol Metab Clin North Am. 2013;42(2):187-199.
- Van Cauter E, Kerkhofs M, Caufriez A, Van Onderbergen A, Thorner MO, Copinschi G. A quantitative estimation of growth hormone secretion in normal man: reproducibility and relation to sleep and time of day. J Clin Endocrinol Metab. 1992;74(6):1441-1450.
- Holl RW, Hartman ML, Veldhuis JD, Taylor WM, Thorner MO. Thirty-second sampling of plasma growth hormone in man: correlation with sleep stages. J Clin Endocrinol Metab. 1991;72(4):854-861.
- Brandenberger G, Gronfier C, Chapotot F, Simon C, Piquard F. Effect of sleep deprivation on overall 24 h growth-hormone secretion. Lancet. 2000;356(9239):1408.
- Van Cauter E, Leproult R, Plat L. Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. Jama. 2000;284(7):861-868.
- Van Cauter E, Latta F, Nedeltcheva A, Spiegel K, Leproult R, Vandenbril C, Weiss R, Mockel J, Legros JJ, Copinschi G. Reciprocal interactions between the GH axis and sleep. Growth Horm IGF Res. 2004;14 Suppl A:S10-17.
- Golstein J, Van Cauter E, Desir D, Noel P, Spire JP, Refetoff S, Copinschi G. Effects of "jet lag" on hormonal patterns. IV. Time shifts increase growth hormone release. J Clin Endocrinol Metab. 1983;56(3):433-440.
- Vgontzas AN, Mastorakos G, Bixler EO, Kales A, Gold PW, Chrousos GP. Sleep deprivation effects on the activity of the hypothalamic-pituitary-adrenal and growth axes: potential clinical implications. Clin Endocrinol (Oxf).1999;51(2):205-215.
- Jaffe CA, Friberg RD, Barkan AL. Suppression of growth hormone (GH) secretion by a selective GH-releasing hormone (GHRH) antagonist. Direct evidence for involvement of endogenous GHRH in the generation of GH pulses. J Clin Invest. 1993;92(2):695-701.
- Jaffe CA, Turgeon DK, Friberg RD, Watkins PB, Barkan AL. Nocturnal augmentation of growth hormone (GH) secretion is preserved during repetitive bolus administration of GH-releasing hormone: potential involvement of endogenous somatostatin--a clinical research center study. J Clin Endocrinol Metab. 1995;80(11):3321-3326.
- Yildiz BO, Suchard MA, Wong M-L, McCann SM, Licinio J. Alterations in the dynamics of circulating ghrelin, adiponectin, and leptin in human obesity. PNAS. 2004;101(28):10434-10439.
- Steiger A, Guldner J, Hemmeter U, Rothe B, Wiedemann K, Holsboer F. Effects of growth hormone-releasing hormone and somatostatin on sleep EEG and nocturnal hormone secretion in male controls. Neuroendocrinology.1992;56(4):566-573.
- Weikel JC, Wichniak A, Ising M, Brunner H, Friess E, Held K, Mathias S, Schmid DA, Uhr M, Steiger A. Ghrelin promotes slow-wave sleep in humans. Am J Physiol Endocrinol Metab. 2003;284(2):E407-415.
- Sutton J, Lazarus L. Growth hormone in exercise: comparison of physiological and pharmacological stimuli. J Appl Physiol. 1976;41(4):523-527.
- Lassarre C, Girard F, Durand J, Raynaud J. Kinetics of human growth hormone during submaximal exercise. J Appl Physiol. 1974;37(6):826-830.
- Raynaud J, Capderou A, Martineaud JP, Bordachar J, Durand J. Intersubject variability in growth hormone time course during different types of work. J Appl Physiol. 1983;55(6):1682-1687.
- Felsing NE, Brasel JA, Cooper DM. Effect of low and high intensity exercise on circulating growth hormone in men. J Clin Endocrinol Metab. 1992;75(1):157-162.
- Vanhelder WP, Goode RC, Radomski MW. Effect of anaerobic and aerobic exercise of equal duration and work expenditure on plasma growth hormone levels. Eur J Appl Physiol Occup Physiol. 1984;52(3):255-257.
- Schmidt A, Maier C, Schaller G, Nowotny P, Bayerle-Eder M, Buranyi B, Luger A, Wolzt M. Acute exercise has no effect on ghrelin plasma concentrations. Horm Metab Res. 2004;36(3):174-177.
- Chennaoui M, Leger D, Gomez-Merino D. Sleep and the GH/IGF-1 axis: Consequences and countermeasures of sleep loss/disorders. Sleep Med Rev. 2020;49:101223.
- Naylor E, Penev PD, Orbeta L, Janssen I, Ortiz R, Colecchia EF, Keng M, Finkel S, Zee PC. Daily social and physical activity increases slow-wave sleep and daytime neuropsychological performance in the elderly. Sleep.2000;23(1):87-95.
- Ritsche K, Nindl BC, Wideman L. Exercise-Induced growth hormone during acute sleep deprivation. Physiol Rep.2014;2(10).
- Roth J, Glick SM, Yalow RS, Berson SA. Hypoglycemia: a potent stimulus to secretion of growth hormone. Science. 1963;140:987-988.
- Greenwood FC, Landon J, Stamp TC. The plasma sugar, free fatty acid, cortisol, and growth hormone response to insulin. I. In control subjects. J Clin Invest. 1966;45(4):429-436.
- Hindmarsh PC, Smith PJ, Taylor BJ, Pringle PJ, Brook CG. Comparison between a physiological and a pharmacological stimulus of growth hormone secretion: response to stage IV sleep and insulin-induced hypoglycaemia. Lancet. 1985;2(8463):1033-1035.
- Tatar P, Vigas M. Role of alpha 1- and alpha 2-adrenergic receptors in the growth hormone and prolactin response to insulin-induced hypoglycemia in man. Neuroendocrinology. 1984;39(3):275-280.
- Jaffe CA, DeMott-Friberg R, Barkan AL. Endogenous growth hormone (GH)-releasing hormone is required for GH responses to pharmacological stimuli. J Clin Invest. 1996;97(4):934-940.
- Broglio F, Prodam F, Gottero C, Destefanis S, Me E, Riganti F, Giordano R, Picu A, Balbo M, Van der Lely AJ, Ghigo E, Arvat E. Ghrelin does not mediate the somatotroph and corticotroph responses to the stimulatory effect of glucagon or insulin-induced hypoglycaemia in humans. Clin Endocrinol (Oxf). 2004;60(6):699-704.
- Carey LC, Cloutier CT, Lowery BD. Growth hormone and adrenal cortical response to shock and trauma in the human. Ann Surg. 1971;174(3):451-460.
- Vigas M, Malatinsky J, Nemeth S, Jurcovicova J. Alpha-adrenergic control of growth hormone release during surgical stress in man. Metabolism. 1977;26(4):399-402.
- Roth A, Cligg SM, Yalow RS, Berson SA. Secretion of growth hormone: physiological and experimental modification. Metabolism. 1963;12:557-559.
- Penalva A, Burguera B, Casabiell X, Tresguerres JA, Dieguez C, Casanueva FF. Activation of cholinergic neurotransmission by pyridostigmine reverses the inhibitory effect of hyperglycemia on growth hormone (GH) releasing hormone-induced GH secretion in man: does acute hyperglycemia act through hypothalamic release of somatostatin? Neuroendocrinology. 1989;49(5):551-554.
- Shiiya T, Nakazato M, Mizuta M, Date Y, Mondal MS, Tanaka M, Nozoe S, Hosoda H, Kangawa K, Matsukura S. Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J Clin Endocrinol Metab. 2002;87(1):240-244.
- Broglio F, Benso A, Gottero C, Prodam F, Grottoli S, Tassone F, Maccario M, Casanueva FF, Dieguez C, Deghenghi R, Ghigo E, Arvat E. Effects of glucose, free fatty acids or arginine load on the GH-releasing activity of ghrelin in humans. Clin Endocrinol (Oxf). 2002;57(2):265-271.
- Hayford JT, Danney MM, Hendrix JA, Thompson RG. Integrated concentration of growth hormone in juvenile-onset diabetes. Diabetes. 1980;29(5):391-398.
- Asplin CM, Faria AC, Carlsen EC, Vaccaro VA, Barr RE, Iranmanesh A, Lee MM, Veldhuis JD, Evans WS. Alterations in the pulsatile mode of growth hormone release in men and women with insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1989;69(2):239-245.
- Salgado LR, Semer M, Nery M, Knoepfelmacher M, Lerario AC, Povoa G, Jana S, Villares SM, Wajchenberg BL, Liberman B, Nicolau W. Effect of glycemic control on growth hormone and IGFBP-1 secretion in patients with type I diabetes mellitus. J Endocrinol Invest. 1996;19(7):433-440.
- Ho KY, Veldhuis JD, Johnson ML, Furlanetto R, Evans WS, Alberti KG, Thorner MO. Fasting enhances growth hormone secretion and amplifies the complex rhythms of growth hormone secretion in man. J Clin Invest.1988;81(4):968-975.
- Soliman AT, Hassan AE, Aref MK, Hintz RL, Rosenfeld RG, Rogol AD. Serum insulin-like growth factors I and II concentrations and growth hormone and insulin responses to arginine infusion in children with protein-energy malnutrition before and after nutritional rehabilitation. Pediatr Res. 1986;20(11):1122-1130.
- Veldhuis JD, Iranmanesh A, Ho KK, Waters MJ, Johnson ML, Lizarralde G. Dual defects in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J Clin Endocrinol Metab.1991;72(1):51-59.
- Vahl N, Jorgensen JO, Skjaerbaek C, Veldhuis JD, Orskov H, Christiansen JS. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am J Physiol. 1997;272(6 Pt 1):E1108-1116.
- Ghigo E, Mazza E, Corrias A, Imperiale E, Goffi S, Arvat E, Bellone J, De Sanctis C, Muller EE, Camanni F. Effect of cholinergic enhancement by pyridostigmine on growth hormone secretion in obese adults and children. Metabolism. 1989;38(7):631-633.
- Loche S, Pintor C, Cappa M, Ghigo E, Puggioni R, Locatelli V, Muller EE. Pyridostigmine counteracts the blunted growth hormone response to growth hormone-releasing hormone of obese children. Acta Endocrinol (Copenh).1989;120(5):624-628.
- Cordido F, Casanueva FF, Dieguez C. Cholinergic receptor activation by pyridostigmine restores growth hormone (GH) responsiveness to GH-releasing hormone administration in obese subjects: evidence for hypothalamic somatostatinergic participation in the blunted GH release of obesity. J Clin Endocrinol Metab. 1989;68(2):290-293.
- Cordido F, Penalva A, Peino R, Casanueva FF, Dieguez C. Effect of combined administration of growth hormone (GH)-releasing hormone, GH-releasing peptide-6, and pyridostigmine in normal and obese subjects. Metabolism.1995;44(6):745-748.
- Maccario M, Aimaretti G, Grottoli S, Gauna C, Tassone F, Corneli G, Rossetto R, Wu Z, Strasburger CJ, Ghigo E. Effects of 36 hour fasting on GH/IGF-I axis and metabolic parameters in patients with simple obesity. Comparison with normal subjects and hypopituitary patients with severe GH deficiency. Int J Obes Relat Metab Disord.2001;25(8):1233-1239.
- Grottoli S, Gauna C, Tassone F, Aimaretti G, Corneli G, Wu Z, Strasburger CJ, Dieguez C, Casanueva FF, Ghigo E, Maccario M. Both fasting-induced leptin reduction and GH increase are blunted in Cushing's syndrome and in simple obesity. Clin Endocrinol (Oxf). 2003;58(2):220-228.
- Pedersen MH, Svart MV, Lebeck J, Bidlingmaier M, Stodkilde-Jorgensen H, Pedersen SB, Moller N, Jessen N, Jorgensen JOL. Substrate Metabolism and Insulin Sensitivity During Fasting in Obese Human Subjects: Impact of GH Blockade. J Clin Endocrinol Metab. 2017;102(4):1340-1349.
- Carro E, Senaris R, Considine RV, Casanueva FF, Dieguez C. Regulation of in vivo growth hormone secretion by leptin. Endocrinology. 1997;138(5):2203-2206.
- Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50(4):707-709.
- Hansen TK, Dall R, Hosoda H, Kojima M, Kangawa K, Christiansen JS, Jorgensen JO. Weight loss increases circulating levels of ghrelin in human obesity. Clin Endocrinol (Oxf). 2002;56(2):203-206.
- Leidy HJ, Gardner JK, Frye BR, Snook ML, Schuchert MK, Richard EL, Williams NI. Circulating Ghrelin Is Sensitive to Changes in Body Weight during a Diet and Exercise Program in Normal-Weight Young Women. J Clin Endocrinol Metab. 2004;89(6):2659-2664.
- Lindeman JH, Pijl H, Van Dielen FM, Lentjes EG, Van Leuven C, Kooistra T. Ghrelin and the hyposomatotropism of obesity. Obes Res. 2002;10(11):1161-1166.
- Yu AP, Ugwu FN, Tam BT, Lee PH, Ma V, Pang S, Chow AS, Cheng KK, Lai CW, Wong CS, Siu PM. Obestatin and growth hormone reveal the interaction of central obesity and other cardiometabolic risk factors of metabolic syndrome. Sci Rep. 2020;10(1):5495.
- Tamboli RA, Antoun J, Sidani RM, Clements A, Harmata EE, Marks-Shulman P, Gaylinn BD, Williams B, Clements RH, Albaugh VL, Abumrad NN. Metabolic responses to exogenous ghrelin in obesity and early after Roux-en-Y gastric bypass in humans. Diabetes Obes Metab. 2017;19(9):1267-1275.
- Sukkar MY, Hunter WM, Passmore R. Changes in plasma levels of insulin and growth-hormone levels after a protein meal. Lancet. 1967;2(7524):1020-1022.
- Parker ML, Hammond JM, Daughaday WH. The arginine provocative test: an aid in the diagnosis of hyposomatotropism. J Clin Endocrinol Metab. 1967;27(8):1129-1136.
- Alba-Roth J, Muller OA, Schopohl J, von Werder K. Arginine stimulates growth hormone secretion by suppressing endogenous somatostatin secretion. J Clin Endocrinol Metab. 1988;67(6):1186-1189.