ABSTRACT
Thyroid disorders are a major cause of non-communicable diseases in developing nations, with the tropical regions presenting unique challenges due to diverse environmental, socio-economic, and cultural factors. Iodine deficiency remains a significant public health concern, leading to conditions such as endemic goiter and cretinism. The prevalence of iodine deficiency disorders has declined due to salt iodization programs, but inconsistent implementation continues to affect many tropical areas. Autoimmune thyroid diseases, including Hashimoto's thyroiditis and Graves' disease are influenced by genetic and environmental factors in the tropics with a minor increase in prevalence following iodization. Thyroiditis, often associated with infections and inflammatory conditions prevalent in tropical regions, add to the complexity. Congenital hypothyroidism, the leading cause of preventable intellectual disability, is challenging to address due to limited newborn screening programs. A multifaceted approach is needed to address these concerns, including improving healthcare infrastructure, increasing public awareness, ensuring consistent iodine supplementation, and enhancing training for healthcare providers. These measures can significantly improve thyroid disorder-related outcomes in tropical nations.
INTRODUCTION
Thyroid disorders are one of the leading cause of non-communicable ailments in the developing nations (1). However, the manifestation and management of thyroid disorders is not uniform across geographic regions. Tropical climate presents a distinct milieu characterized by diverse environmental, socio-economic, and cultural factors that alter the spectrum of thyroid diseases. A confluence of factors ranging from dietary practices to endemic diseases, imparts unique characteristics to the epidemiology, clinical presentation, and management of thyroid disorders. Iodine deficiency disorders (IDDs) remain a significant public health concern in many tropical countries, where suboptimal iodine intake precipitates a spectrum of thyroid abnormalities, including endemic goiter, hypothyroidism, and cretinism. Additionally, the occurrence of autoimmune thyroid diseases (AITDs), such as Hashimoto's thyroiditis and Graves' disease, underscores the interplay between genetic susceptibility, environmental triggers, and immune dysregulation. This chapter highlights the altered presentation of thyroid disorders and concerns specific to the tropics.
ETIOPATHOGENESIS IN TROPICS
Thyroid disorders can be influenced by a variety of environmental factors in tropical countries. These aspects can interact with genetic predisposition and individual health behaviors to impact the functioning of the thyroid gland.
Iodine Deficiency Disorders
Iodine deficiency significantly contributes to the global thyroid disease burden and leads to various metabolic and growth-related diseases (2). Though the prevalence of IDDs has decreased with iodization of salt, the condition still poses a significant health challenge in many tropical nations. In 2018 and 2019, the global age-standardized prevalence rate of iodine deficiency remained relatively stable at around 2218 and 2216 per 100,000 population, respectively. Over the period from 1990 to 2019, there was a notable decrease in this prevalence rate, with an estimated average annual percent change (EAPC) of -0.690. When examining specific countries, Somalia had the highest age-standardized prevalence rate in 2019, followed by the Democratic Republic of the Congo, Djibouti, and the Republic of the Congo. Interestingly, several countries, including the Philippines, Pakistan, and South Sudan, exhibited an increasing trend in iodine deficiency prevalence. In 2019, Central sub-Saharan Africa and South Asia reported the highest prevalence rates.(3).
Despite the environmental abundance of iodine, its low bioavailability in tropics remains a primary factor influencing the prevalence of IDD. The heavy rainfall characteristic of tropical climate accelerates rock weathering, clay formation, and soil leaching. Clayey materials and humic substances, which bind iodine strongly, act as geochemical goitrogens and significantly affect iodine bioavailability (4).
Endocrine Disrupting Chemicals
Tropical countries encounter challenges due to exposure to endocrine disrupting chemicals (EDCs), which can adversely affect thyroid function (5–7). These chemicals, found in pesticides, plastics, and industrial pollutants, stimulate or interfere with hormone signaling pathways, potentially leading to thyroid dysfunction (8). Agricultural practices, industrialization, and inadequate environmental regulations can contribute to higher EDC exposure in tropics, aggravating thyroid disorders (9).
Environmental Pollution
Household cooking with solid fuels significantly contributes to air pollution in Asian countries (10,11). Air pollution from both household and industrial sources is a major concern in the tropics (12,13). A recent study found a positive correlation between thyroid cancer incidence and air pollution, including particulate matter (r=0.23, P < 0.001) and household air pollution (r=0.52, P ≤ 0.001) (14). This suggests that air pollution may play a role in the development of thyroid cancer and calls for more research to understand the connection.
Infections
Tropical countries often face a higher burden of infectious diseases that can affect thyroid function (15). Primary infection of the thyroid is extremely uncommon. Bacteria are the typical causative organisms but fungal, parasitic, and viral infections have also been described (16). Furthermore, rare cases of mycobacterial cold abscesses have been reported, highlighting the diverse range of microorganisms that can affect the thyroid gland (17).
Nutritional Factors
The unique dietary patterns and environmental conditions in the tropics significantly influence thyroid disorders. Iodine deficiency as already discussed is a prevalent problem. Additionally, diets high in goitrogens, such as cassava, millet, and certain cruciferous vegetables, can exacerbate thyroid dysfunction by interfering with iodine uptake (18). Selenium deficiency, another concern in tropical regions, further complicates thyroid hormone production (19,20). Thiocyanate overload has been documented as a goitrogen in Central Africa. When coupled with selenium deficiency, it is a risk factor for endemic myxedematous cretinism (21). Addressing these nutritional deficiencies through diet diversification and supplementation is crucial for mitigating thyroid disorders.
IODINE DEFICIENCY DISORDERS
Goiter and cretinism are two health conditions prevalent in tropical countries resulting from iodine deficiency.
Endemic Goiter
ETIOLOGY
Endemic goiter refers to a visible enlargement of the thyroid gland in regions of environmental iodine deficiency. The condition is defined by the presence of goiter in more than 5% of children aged 6–12 years. It occurs as a maladaptive response to iodine deficiency (22). The role of iodine deficiency in causation is evidenced by its correlation with low iodine levels in food and water in affected regions, reduction in goiter incidence with iodine supplementation, and expected metabolic pattern in individuals that results from iodine deficiency. Other factors such as excess thiocyanates and selenium deficiency may also play a role (2).
PATHOPHYSIOLOGY
If iodine intake is low, the thyroid undergoes significant adaptive changes to maintain adequate hormone production. These adjustments include increased iodide trapping and enhanced intrathyroidal iodine metabolism, primarily driven by elevated levels of TSH. The initial functional response to iodine deficiency involves heightened iodide uptake by the thyroid, mediated by the sodium iodide symporter (NIS), often accompanied by increased TSH levels. Usually, severe iodine deficiency is required to consistently elevate TSH levels. The thyroid's sensitivity to TSH appears to vary with iodine availability, influencing thyroglobulin secretion and iodine clearance rates. Effective adjustment to iodine deficiency can occur without goiter in certain populations, indicating varying degrees of iodine entrapment and hormone synthesis capacity (23).
THYROID HORMONE PROFILE
Iodine deficiency causes thyroid gland enlargement and alters hormone production. The abnormal thyroglobulin in the thyroid releases poorly iodinated compounds such as monoiodotyrosine (MIT) and triiodothyronine (T3), and decreased levels of diiodotyrosine (DIT) and thyroxine (T4). This shift increases the MIT/DIT and T3/T4 ratios, reflecting the severity of iodine depletion. In response to iodine deficiency, the thyroid may secrete T3 and T4 in proportions found within the gland, and preferentially produce T3 or convert more T4 to T3 peripherally. The adaptation is critical as T3 is metabolically more potent and requires less iodine for synthesis, aiding in mitigating iodine deficiency effects (23).
DIAGNOSIS
In childhood, thyroid glands are often diffusely enlarged, while in adults, they tend to be nodular. Common laboratory findings include increased radioiodine uptake (RAIU) by the thyroid, normal or low T4 and free T4 (FT4), normal or elevated T3 and TSH, and reduced urinary iodine excretion. RAIU can usually be suppressed with thyroid hormone treatment. Scans with radioiodine or technetium show a mottled isotope distribution.
PREVENTION AND TREATMENT
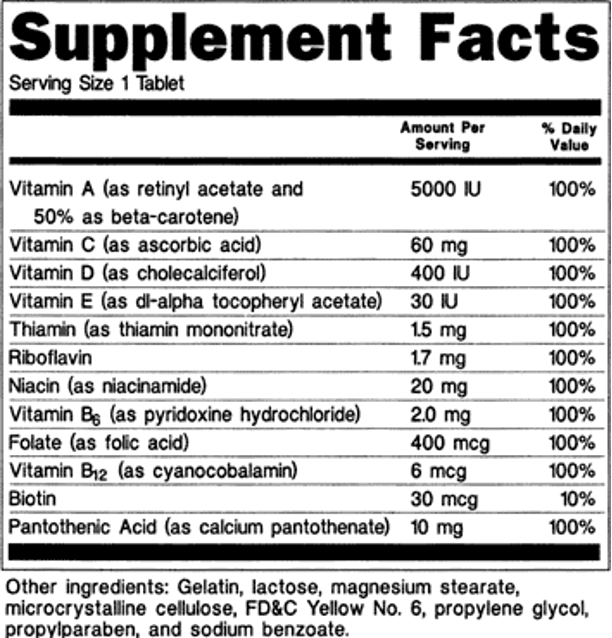
The recommended dietary allowance (RDA) for iodine is 150 mg per day for adults and 250 mg per day during pregnancy. Currently, 20-40 mg of iodine per kg salt can provide the RDA.
Goiters regress in most cases when treated with iodine (24). Generally, treatment with iodine is sufficient and surgery can be avoided. However, individuals with goiters that do not regress, exhibit rebound growth after three months, or present with pressure symptoms may require surgical intervention (25).
There is a small but consistent risk of iodine supplementation causing hyperthyroidism, known as Jod-Basedow disease. Despite the risk, the benefits of iodine treatment outweigh the potential drawbacks. Individuals over the age of 50 years with latent thyroid disease, particularly those with multinodular goiters, are more likely to develop this form of thyrotoxicosis. Typically, iodine-induced thyrotoxicosis resolves naturally, sometimes within 12 months, but it can take up to 3–4 years (26).
The implementation of iodized salt programs in various countries has led to a significant reduction in the prevalence of goiter. Despite these efforts, many areas remain affected due to inconsistent iodization practices and other socio-economic factors (27–29).
Cretinism
Cretinism, a severe form of IDD, often coexists with endemic goiter in these regions. The condition leads to extreme physical and mental retardation, significantly impacting individuals' quality of life and the overall well-being of communities. Cretinism can be classified into two types: neurological and myxedematous.
NEUROLOGICAL CRETINISM
Neurological cretinism results from severe iodine deficiency during early pregnancy, causing irreversible brain damage in the fetus. Affected individuals display mental retardation, pyramidal signs in a proximal distribution, and extrapyramidal signs, along with a characteristic gait. Other common features included squint, deafness, and primitive reflexes. In iodine-deficient areas, many individuals exhibit intellectual impairments and coordination defects, with a leftward shift in the intelligence curve compared to iodine-sufficient areas (23,30).
MYXEDEMATOUS CRETINISM
Myxedematous cretinism is characterized by overt hypothyroidism from early life, but less severe mental retardation than neurological cretinism. Key features include major growth retardation, immature facial features, mandibular atrophy, puffy and thickened skin, sparse hair, delayed sexual maturation, and typically absent goiter due to thyroid atrophy. Causes include thiocyanate overload from cassava consumption, selenium deficiency leading to thyroid cell destruction, and potential immunological factors. The condition was notably prevalent in Zaire and associated with severe, irreversible hypothyroidism and some neurological signs obscured by the hypothyroid state (30).
The reader is referred to the chapter Iodine Deficiency Disorders in Endotext.com for a detailed review of cretinism and other IDDs (23).
Prevention and Management of Iodine Deficiency Disorders
Public health interventions to address endemic goiter and cretinism in tropical countries have included widespread salt iodization, iodine supplementation programs, and public awareness campaigns. These efforts aim to ensure consistent iodine intake across populations, particularly targeting vulnerable groups such as pregnant women and children. However, challenges remain in ensuring the reach and efficacy of these programs, especially in remote and underserved areas (31).
Iodized salt is the preferred method for iodine fortification due to its widespread use across all socioeconomic groups. It is consumed consistently year-round and produced in large, centralized facilities, allowing for effective and controlled fortification. Potassium iodate, preferred for its stability in humid conditions, and potassium iodide are the two forms used. Direct iodide supplementation, iodized oils, and iodized breads are other means to prevent IDDs (23). Collaborative efforts involving governments, non-governmental organizations, and local communities are crucial for sustaining progress in preventing IDDs.
AUTOIMMUNE THYROID DISORDERS
AITDs arise from an immune system dysregulation, resulting in an immune attack on the thyroid gland. These disorders are T cell-mediated and organ-specific. The prevalence of AITD is around 5%, though the occurrence of antithyroid antibodies might be even higher. AITD primarily manifests in two clinical forms: Graves' disease and Hashimoto's thyroiditis, both marked by lymphocytic infiltration of the thyroid tissue. Clinically, Graves’ disease is characterized by thyrotoxicosis, while Hashimoto’s thyroiditis leads to hypothyroidism. The exact mechanisms initiating the autoimmune response against the thyroid are still not clear. Epidemiological data indicate that an interaction between genetic predisposition and environmental factors is responsible for disrupting immune tolerance and triggering AITD (32). In the tropics, the prevalence, diagnosis, and management of these disorders are influenced by various regional factors.
Hashimoto's Thyroiditis
The classic example of AITD is Hashimoto's thyroiditis, also referred to as chronic lymphocytic thyroiditis. It is associated with chronic inflammation of the thyroid tissue, and causes hypothyroidism in 20-30% of cases (33).
PREVALENCE
In tropical regions, the incidence of Hashimoto's thyroiditis is increasing, which is partially attributed to enhanced iodine intake from iodization programs. While these programs have reduced goiter and cretinism, they have also been linked to an increase in AITD. A study from Sri Lanka found anti-thyroid peroxidase (TPO) antibodies in 10.3%, anti-thyroglobulin (Tg) antibodies in 6.4%, and subclinical hypothyroidism (SCH) int 3%. Median urinary iodine concentration was 138.5 μg/L, indicating iodine sufficiency from universal salt iodization. Despite a high anti-TPO antibody prevalence, SCH remains low. The findings suggest that early post-iodization anti-Tg antibody surge that had gone past 42%, has now settled. Goiter prevalence was 0.6%-1.93% (34).
IODINE CONSUMPTION AND THYROID AUTOIMMUNITY
Several explanations have been proposed to elucidate the link between excessive iodine consumption and thyroid autoimmunity. One hypothesis suggests that iodine may be directly toxic to thyroid tissue and stimulate immune effector cells. The role of free radical damage to thyroid tissue is widely recognized as a contributing factor in the onset or exacerbation of AITD. Experimental studies in genetically predisposed animals have shown that Tg becomes more immunogenic when exposed to excess iodine, potentially triggering autoimmune responses (35).
Previous iodine levels impact thyroid response to increased iodine intake and may be linked to the development of thyroid autoimmunity. The prevalence of thyroid antibodies, however, doesn't always predict thyroid disease. Autoimmune diseases have generally risen in recent decades, making data interpretation challenging. Long-term studies suggest that excessive iodine from uncontrolled iodine prophylaxis can lead to autoimmune thyroiditis. Careful monitoring of iodine prophylaxis is recommended to prevent both iodine deficiency and excess (36).
DIAGNOSIS AND TREATMENT
Individuals with Hashimoto’s thyroiditis often present with a painless goiter, which may occur with or without overt hypothyroidism. For asymptomatic persons, it can be discovered incidentally when a goiter prompts evaluation. Others may experience typical hypothyroidism symptoms, including fatigue, weight gain, cold intolerance, constipation, depression, muscle pain, heavy menstrual bleeding, and dry skin. The diagnosis of Hashimoto’s thyroiditis can be confirmed by a combination of clinical features, thyroid function test results indicating SCH or overt hypothyroidism and elevated Tg and TPO antibodies (37). Anti-TPO antibodies are found in 95% of cases with Hashimoto’s thyroiditis, while anti-Tg antibodies are elevated in 60% to 80%. Treatment involves administration of levothyroxine to correct hypothyroidism (38). The challenge in many tropical regions is the limited access to these diagnostic tests, which may delay identification and treatment.
Graves' Disease
Graves' disease is the most common cause of hyperthyroidism worldwide and involves the immune system producing antibodies (thyroid-stimulating immunoglobulins) that stimulate the thyroid gland to produce excess thyroid hormones (39).
PREVALENCE
Graves' disease accounts for 70-80% of hyperthyroidism cases in iodine-sufficient regions. In contrast, in areas with iodine deficiency, Graves' disease represents about half of all hyperthyroidism cases, with the other half occurring due to nodular thyroid disease (40,41). In Johannesburg in 1981, the incidence of Graves' disease was 5.5 per 100,000/year, significantly lower than global rates. However, over a decade, there was a 60% increase in incidence, possibly due to improved dietary iodine intake among urban migrants (42). Hospital-based studies from Ghana reveal that Graves' disease, contrary to earlier reports, now comprises 54% of all thyroid dysfunction cases, although ascertainment bias might exist. Improved iodine nutrition and better diagnosis have led to increased incidences of both Graves' disease and nodular disease post-iodization in Ghana (43). A systematic review reported higher rates in Asians, and lower incidence in African population compared to Caucasians (44).
PATHOGENESIS IN TROPICS
Genetic predisposition contributes to 79% of Graves’ disease risk, primarily involving genes related to T-cell function, while environmental factors account for remaining 21%. Significant environmental factors include smoking, iodine excess, selenium and vitamin D deficiencies, and viral infections. All these factors hold relevance in the tropical context. Numerous studies have examined whether infectious agents like foamy virus, parvovirus-B19, Epstein-Barr virus (EBV), and hepatitis C virus (HCV) can trigger Graves' disease, with mixed results. EBV reactivation is linked to Graves’ disease recurrence in Japanese patients but not in Caucasians. There is a well-established connection between HCV and thyroid autoimmunity, including hypothyroidism. A link between HCV-related mixed cryoglobulinemia and Graves’ disease has also been demonstrated. Additionally, 2.5 to 20% of individuals with chronic HCV treated with interferon-alpha develop thyroid disorders, including GD (45).
Animal experiments have studied the connection between hygiene hypothesis and Graves’ disease (46). The hygiene hypothesis suggests that a lack of early childhood exposure to infectious agents, symbiotic microorganisms, and parasites can lead to a higher incidence of autoimmune diseases, such as Graves' disease. This hypothesis posits that modern sanitation practices and reduced exposure to microbes may prevent the immune system from developing appropriately, increasing susceptibility to autoimmune conditions. In the context of Graves' disease, the hygiene hypothesis implies that individuals in more sanitized environments might have a higher risk of developing the disease due to an under-stimulated immune system that becomes prone to attacking the body's own thyroid cells. The hygiene hypothesis suggests a potential protective effect on thyroid health in tropical conditions, but clinical evidence is lacking (47).
CLINICAL MANIFESTATIONS
Thyrotoxicosis is the primary presenting feature of Graves' disease, and in this region, it often manifests at a more advanced stage with various complications. Late presentation can be attributed to missed diagnoses and lack of awareness often arising from financial constraints (48). Notably, thyrotoxicosis is a significant cause of cardiac morbidity in tropical countries. In Togo, cardiac complications were reported in 46.6% of patients with thyrotoxicosis (49). Similarly, the heart failure rate was reported to be 42% in Lagos, Nigeria (50).
Ethnicity appears to influence the risk of developing disease complications. For example, Graves' ophthalmopathy is six times more common in Caucasians than in Asians (51). Additionally, the rare but serious complication of hyperthyroidism, thyrotoxic periodic paralysis, is significantly more common in Asian men (52). The genetic basis of this condition has been extensively researched, revealing variations in certain human leukocyte antigen (HLA) haplotypes such as DRw8, A2, Bw22, Aw19, and B17 in affected Asian patients (53).
TREATMENT
Treatment options for graves’ disease include antithyroid medications (e.g., methimazole or propylthiouracil), radioactive iodine therapy (RAI), and surgery. The choice of treatment depends on the severity of the disease, patient preference, and availability of medical resources. In many tropical regions, the lack of access to RAI and surgical facilities limits treatment options, making long-term medication the most feasible approach. In an online global survey conducted in 2023, RAI as the primary treatment for Graves' disease was offered by 13.1% of respondents from Africa and the Middle East and 7.5% of respondents from Latin America, but only in 5% from Asia (54). Rare cases of resistance to anti-thyroid drugs have also been described in tropical regions (55,56). A possible impairment in intrathyroidal drug accumulation could be responsible (57). Interestingly, Asians are more predisposed to insulin autoimmune syndrome after exposure to methimazole because of higher prevalence of HLA allele DRB1*04:06 (58).
SUBACUTE THYROIDITIS
The term “thyroiditis” refers to a group of inflammatory conditions affecting the thyroid gland. The causes can be diverse including autoimmunity, viral, bacterial and fungal infections, iatrogenic, trauma, radiation and idiopathic chronic sclerosing forms such as Riedel thyroiditis (Table 1) (58,59).
|
Table 1. Tropical Infections of the Thyroid Gland |
||
|---|---|---|
|
Type of presentation |
|
Investigations |
|
Acute pyogenic / suppurative thyroiditis |
Bacterial: Streptococcus, Staphylococcus, Enterobacter Fungal: Aspergillus, candida, histoplasma, coccidiodes |
TFT: Normal / mild thyrotoxicosis USG thyroid: Hypoechoic area, abscess FNA followed by staining/culture can identify the causative organism Thyroid scintigraphy: normal function of the unaffected lobe HIV-AIDS to be ruled out in fungal thyroiditis |
|
SAT |
Viral: Adenovirus, echovirus, Epstein Barr virus, coxsackie virus, H1N1 influenza, cytomegalovirus, SARS-CoV-2, dengue. Mycobacterial thyroiditis |
Thyroid scintigraphy: Poor, patchy uptake.# TSH-receptor antibody positivity favors Graves’ disease (may be transiently positive in SAT) High ESR T3 (ng/ml): T4 (mcg/dl) ratio < 20 favors SAT Color Doppler shows decreased vascularity |
|
Thyroid nodule or goiter |
Parasites: Strongyloidiasis, Giardia, Entamoeba, Cryptosporidium, Echinococcus,Trypanosomes |
USG for localization of lesion FNA to detect causative organism Eosinophilia HIV-AIDS to be ruled out in disseminated parasitic infection |
# cut-off value for (99m)Tc-pertechnetate uptake of 1.0% - 1.55% have been proposed to differentiate SAT from Graves’ disease.
TFT- thyroid function test, USG – ultrasonography, FNA – fine needle aspiration, HIV-AIDS – human immunodeficiency virus-acquired immunodeficiency syndrome, SAT – subacute thyroiditis, SARS-CoV-2 - Severe acute respiratory syndrome coronavirus 2, ESR – erythrocyte sedimentation rate.
Etiology and Pathogenesis
Subacute thyroiditis (SAT), also known as de Quervain's thyroiditis or granulomatous thyroiditis, is an inflammatory condition that often follows a viral infection (59). While the clinical presentation and course are generally consistent worldwide, there may be some differences in the pathogenesis and management in tropical countries. SAT has been linked to prior infection with adenovirus, EBV, coxsackie, mumps, measles, H1N1 influenza, dengue, St. Louis encephalitis, hepatitis A , parvovirus B19, cytomegalovirus and severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) (60–64). Cases have been reported after influenza vaccination and following interferon treatment for HCV(65,66). Non-viral infections like malaria, Q fever, and scrub typhus have also been implicated (60,67).
While some of these infections may be more prevalent in tropical regions, there is insufficient evidence to suggest a specific cause that alters the epidemiology.
Infections can induce thyroid autoimmunity by diverse mechanisms like self-antigen modification, mimicry of self-molecules, superantigen mediated polyclonal T-cell activation, immune complex formation, and induction of expression of MHC molecules on thyroid epithelium.
Certain haplotypes like HLA-B*35, HLA-B*18:01 and DRB1* confer susceptibility to SAT in different ethnicities across the world. HLA-B*35 allele has been identified in up to 70% of the cases (68,69), while the alleles HLA-B*18:01 and DRB1*01 have been identified in the remaining cases (70). In an Indian study, HLA-B*35 was reported positive in 55.56% of cases of SAT (71). Further research is needed to understand the variations in HLA haplotypes among different ethnic groups and their relationship with SAT.
Clinical Spectrum
The most common presentation of SAT is temporary thyrotoxicosis followed by transient hypothyroidism and eventual recovery. Permanent hypothyroidism can develop in a minority (72). The inflammatory process disrupts the thyroid follicles causing an enhanced release of the stored T4 and T3 leading to thyrotoxicosis. Continued damage to the follicles may lead to transient or permanent hypothyroidism. In some cases, the disease can recur (73). In a prospective study from India, the prevalence of hypothyroidism after 1 year of SAT was 19.86% (74), while a study from Saudia Arabia reported permanent hypothyroidism in 14.3% (75).
Thyrotoxic features like palpitations, tremulousness, weight loss, heat intolerance and anxiety are present but usually milder in comparison to Graves’ disease (76). Inflammatory features such as neck pain, fever, malaise, fatigue, myalgia and arthralgia often dominate the clinical presentation.
Diagnosis
An upper respiratory tract infection is the usual differential diagnosis. Peripheral levels of thyroid hormones, T4 and T3, are elevated with a ratio of T4 (mcg/dl): T3 (ng/ml) > 20. This is reflective of the proportion of stored thyroid hormones within the gland, though the results may vary with the phase of the disease. Erythrocyte sedimentation rate (ESR) is almost invariably elevated, often to values above 100 mm/hr. Diagnosis is confirmed by a suppressed uptake on a 99m-technectium scintigraphy or radioiodine scan. In many tropical countries, nuclear medicine facilities are not widely available. As a result, diagnosis often relies on clinical and biochemical findings along with a non-elevated TSH-receptor antibody (TRAb). Color flow Doppler ultrasonography shows a hypoechogenic, heterogenous gland with low to normal vascularity (77).
Treatment and Prognosis
The condition is self-limiting and milder cases often require no treatment or nonsteroidal anti-inflammatory agents (NSAIDs) (76). For more severe symptoms, a course of oral prednisolone is recommended at a starting dose of 20-40 mg per day, and tapered over 4-6 weeks (78). Thyrotoxic symptoms respond to β -blockers like propranolol. The reader can refer to the chapter on subacute thyroiditis in endotext.com for a detailed review (59).
SILENT AND POSTPARTUM THYROIDITIS
Silent thyroiditis, similar to SAT, follows a typical triphasic course: initial thyrotoxicosis, followed by hypothyroidism, and finally, a return to normal thyroid function. The condition is presumed to be autoimmune in nature, as levels of anti-TPO and anti-TG antibodies are usually elevated. Histologically, it is characterized by extensive lymphocytic infiltration, sometimes with the formation of lymphoid follicles. Unlike SAT, silent thyroiditis does not present with symptoms of thyroid inflammation such as neck pain or fever. It is more prevalent in iodine-sufficient areas (79).
Postpartum thyroiditis typically occurs within six months of delivery but can rarely present up to 12 months postpartum. It shares many similarities with silent thyroiditis, including an autoimmune etiology (80). Neither silent thyroiditis nor postpartum thyroiditis exhibit any unique characteristics specific to tropical regions.
VIRAL CAUSES OF THYROIDITIS IN TROPICS
Dengue Thyroiditis
SAT can rarely develop as a manifestation of expanded dengue syndrome. This condition should be suspected in patients with dengue fever who develop painful thyroid swelling and thyrotoxic features (61,81). SAT following dengue infection is typically self-limited. In symptomatic cases, an important concern is the high risk of bleeding with the use of aspirin because of the associated thrombocytopenia. Additionally, administration of non-steroidal anti-inflammatory drugs in presence of hypovolemic shock can lead to acute kidney injury. Therefore, oral prednisolone may be preferred for treatment of dengue-associated SAT.
BACTERIAL CAUSES OF THYROIDITIS
Acute Suppurative Thyroiditis
Acute suppurative thyroiditis (AST) or acute pyogenic thyroiditis is a rare but potentially life-threatening form of thyroiditis seen more commonly in tropical countries. They are mostly bacterial in origin, though rarely, fungal and parasitic infestations have also been described (82).
ETIOLOGY
The thyroid gland is generally resistant to infection due to its rich blood supply, thick fibrous capsule, and high levels of iodine and hydrogen peroxide. However, AST can occur in individuals with pre-existing thyroid disorders or in an immunocompromised state. The route of spread is typically hematogenous or lymphatic, but direct spread from the deep fascial spaces of the neck, anterior perforation of the esophagus, or infected thyroglossal cyst can rarely occur (83).
A systematic review of 200 cases of acute suppurative thyroiditis found that 94 cases were from Asia, 24 from Africa, and five from Latin America, suggesting a higher prevalence in tropical regions. Immunocompromised state and pyriform sinus fistulas were the two most common contributing factors. Other causes included disseminated infections and trauma. In 28% of cases, no apparent etiology was identified, although an undiagnosed pyriform sinus fistula remained a possibility (84). It is unclear whether poor hygiene conditions and lack of universal medical facilities in tropical regions contribute to the higher incidence of AST. Rare cases of acute emphysematous thyroiditis in immunocompromised individuals due to infection from Clostridia and Escherichia coli have been reported from tropical countries (85,86).
DIAGNOSIS AND MANAGEMENT
Individuals with AST present with high grade fever and constitutional symptoms along with severe pain and tenderness over the thyroid gland. However, thyroid function is usually normal and only a subset have laboratory evidence of thyrotoxicosis. Ultrasonography of thyroid reveals an abscess in the gland and needle aspiration may confirm the diagnosis along with identification of responsible organism. 99m-technetium-pertechnetate scan or radioiodine uptake studies show normal function of the unaffected lobe of the thyroid gland, unlike in SAT, where thyroid activity is diffusely suppressed. In children with AST of the left lobe, a barium swallow should be ordered to rule out pyriform sinus fistula connecting the left lobe of thyroid. The left-sided predominance of pyriform sinus fistulas is thought to be due to asymmetrical development during embryogenesis (83).
Antibiotics guided by the culture and sensitivity of the offending organism should be commenced as soon as possible. Needle aspiration to drain the pus from affected lobe may be necessary. Surgical drainage may be required in lesions not responding to antibiotics. Pyriform sinus fistula is treated by endoscopic cauterization or surgical excision to prevent recurrence (87). Prognosis is favorable if managed appropriately without loss of thyroid function. For further reading please refer to the relevant chapter on endotext.com (88).
Tuberculosis
EPIDEMIOLOGY
Tuberculosis of the thyroid is an extremely rare condition, representing less than 1% of all cases of extrapulmonary tuberculosis (89). The mean age of onset is around third to fourth decade. Its incidence is higher in tropical regions where tuberculosis is endemic. Immunocompromised individuals, such as those with HIV/AIDS, are at greater risk. Thyroid tuberculosis can be secondary to miliary spread as part of disseminated tuberculosis, or can manifest as isolated lesion in the gland (90).
CLINICAL FEATURES
Thyroid tuberculosis often presents as a solitary thyroid nodule, with or without a cystic component. It may also present as a thyroid abscess with pain, fever, and other systemic features. The diagnosis can be mistaken as SAT (91). Pyrexia of unknown origin may occasionally lead to a diagnosis of thyroid tuberculosis. In rare cases, the lesion mimics thyroid malignancy, presenting with dysphagia, dysphonia, and recurrent laryngeal nerve palsy (89). A discharging sinus after thyroid surgery has been reported (92). While thyroid function is typically not impaired, extensive involvement can lead to thyrotoxicosis from follicular destruction, potentially progressing to hypothyroidism. Associated cervical lymphadenopathy and rare cases of mediastinal lymph node enlargement have also been described ((93). The pathological varieties include multiple lesions in miliary tuberculosis, goiter with areas of caseation, chronic fibrosing forms, and acute pyogenic or cold abscess (90).
DIAGNOSIS AND MANAGEMENT
Though ultrasonography and computed tomography (CT) scan might aid in localizing the lesion in the gland, the findings are nonspecific and do not help to establish the diagnosis of tuberculosis. Caseous necrosis in fine needle aspiration (FNA) cytology or biopsy implies tuberculosis. In many cases, diagnosis is only evident after surgery when the biopsy specimen reveals epithelioid granulomas and caseous necrosis (89,94). While acid-fast bacilli are diagnostic, they are not typically observed in thyroid specimens. Reverse transcription polymerase chain reaction (RT-PCR), and less commonly culture, can provide microbiologic etiology (91). An X-ray of the chest should be obtained to rule out pulmonary tuberculosis.
Therapy with anti-tuberculous drugs often lead to complete resolution of the infection. The choice of anti-tubercular medications and regimen differs in countries and should be guided by national guidelines. In case of a large abscess, especially with compressive features, surgical intervention may be necessary (89).
FUNGAL CAUSES OF THYROIDITIS
Fungal infections account for 15% of AST cases, with aspergillus being the most common, followed by candida. Other fungal causes include Cryptococcus neoformans, Pseudoallescheria boydii, and Pneumocystis jiroveci. Suppurative thyroiditis due to opportunistic mycoses predominantly occurs in immunocompromised individuals. Endemic mycoses like coccidioidomycosis, paracoccidioidomycosis, and histoplasmosis are usually restricted to certain geographic regions of the tropics.
Thyroid involvement is usually insidious and often accompanies a broader disseminated disease. Symptoms include pain, thyroid swelling, and fever, resembling SAT. Severe involvement can lead to dysphagia and respiratory distress due to esophageal and tracheal obstruction. The condition often starts with thyrotoxicosis and progresses to hypothyroidism, with recovery typically taking weeks to months.
The diagnosis is established by demonstration of fungi on FNA. In many cases the etiology becomes apparent on histopathological examination of a surgically removed specimen. Treatment involves antifungal medications, with or without surgery. The mortality rate for disseminated opportunistic fungal infections is high. For more detailed information, refer to the chapter on “Fungi and Endocrine Dysfunction” in endotext.com (95).
Aspergillus
Thyroid gland is involved in 7-26% of cases disseminated aspergillosis (96). In the past, this condition was typically identified postmortem in immunocompromised individuals. However, advancements in treatment for underlying conditions and improved antifungal therapies have shifted diagnoses to occur more frequently to antemortem stage (97). Thyroid involvement may be silent or can mimic subacute thyroiditis. A characteristic manifestation of aspergillosis is vascular invasion with thrombosis and infarction (98).
The presence of aspergillus hyphae in a thyroid specimen, along with areas of pus, necrotic debris, and hemorrhage, confirms the etiology. Fungal culture of the aspirate might be helpful in equivocal cases. Treatment involves antifungal therapy, sometimes combined with surgical intervention. Voriconazole is recommended as the first-line treatment for invasive aspergillosis, while liposomal amphotericin B and isavuconazole are alternative options (99). The mortality rate remains high, particularly in cases of disseminated infection.
Candida
Candida thyroiditis, is rare and typically results from secondary dissemination in immunocompromised states. Due to coexisting immunosuppression, signs of thyroid inflammation may be minimal. Unlike other fungal thyroiditis, candida infection can be associated with thyroid function abnormalities such as transient thyrotoxicosis followed by hypothyroidism (100,101). Rare cases of thyroid storm leading to heart failure necessitating treatment with plasmapheresis during the thyrotoxic phase has been reported (102).Treatment comprises systemic antifungal therapy, management of thyroid dysfunction, and potentially surgical intervention. The antifungal options include echinocandins, liposomal amphotericin B, fluconazole, and voriconazole. Resistance to azoles in Candida albicans and Candidatropicalis is an emerging threat in Asia and Latin America (103).
Histoplasma
Histoplasmosis is a fungal infection endemic in certain areas of tropics including Central and South America, Africa, and several countries of South East Asia including Thailand, Malaysia, Indonesia, Singapore, and India (104). The infection is associated with exposure to birds or poultry. The condition often presents as SAT, diffuse goiter, or a nodule accompanied by constitutional features like fatigue and weight loss. Diagnosis involves FNA cytology or biopsy to confirm the presence of Histoplasma capsulatum. In biopsy, granulomas may be seen and the fungus appears as oval 2 to 4 micrometers narrow-based budding yeast cells with Gomori methenamine silver or periodic acid. Serology, antigen testing, and molecular techniques like PCR may provide additional information (105). Treatment ranges from oral itraconazole for non-severe cases to liposomal amphotericin B for severe cases. Posaconazole is another therapeutic option (106).
Coccidioides
Coccidioidomycosis, caused by inhaling coccidioides arthroconidia, often presents with flu-like symptoms or pneumonia, with 1-5% of cases disseminating to various organs. Risk factors for extrapulmonary disease include age, African or Filipino ancestry, pregnancy, and immunosuppression (107). The presentation of thyroid involvement can range from asymptomatic to thyroid nodules, or even features of subacute thyroiditis. Imaging studies, FNA, and serologic testing help to ascertain the diagnosis. Treatment typically includes antifungal medication, and in some cases, surgical removal of the affected thyroid tissue. Choice and duration of therapy is not well-defined but generally involves high-dose fluconazole, with itraconazole or posaconazole as alternative options (108).
PARASITIC THYROIDITIS
Parasitic infections of the thyroid gland is extremely rare and characteristically occurs in the setting of disseminated disease in an immunocompromised host. Protozoal infections are caused by Giardia lamblia, Entamoeba histolytica, and Cryptosporidium parvum, in tropical regions (109). Helminthic infections such as Strongyloides stercoralis may cause cystic lesions and resemble thyroglossal cysts (110). Trypanosoma brucei, which causes African Trypanosomiasis (sleeping sickness), has been linked to elevated TSH and low free T4 levels, mimicking primary hypothyroidism (111). Trypanosomes can be identified as spindle-shaped cells in blood or FNA fluid. Filariasis can also affect the thyroid, with microfilariae visible in FNA samples.
THYROID NEOPLASM
Global Trends
The global rise in thyroid cancer incidence has raised concerns. Analysis of data from the 2019 Global Burden of Disease study and the UN’s World Population Prospects 2022, suggests that thyroid cancer incidence increased across all income groups, while mortality modestly decreased except in lower-middle-income groups. The divergent trends in thyroid cancer incidence and mortality suggest potential overdiagnosis in higher-income countries, while highlighting the need to reduce health inequalities and improve access to diagnostic and therapeutic services in tropical lower-middle income countries (112). A study from Central and South America evaluating data between 1997 and 2008, revealed that the incidence of papillary thyroid cancer increased by 9.1-15.0% annually in females, while mortality remained stable. Trends in thyroid cancer among males during this period were stable (113).
Iodine Supplementation and Thyroid Cancer Risk
Iodine supplementation has a complex relationship with thyroid cancer. Iodine deficiency decreases the prevalence of follicular thyroid cancer, while populations with adequate iodine intake have higher rates of papillary thyroid cancers (114). It has been hypothesized, that iodine-induced oxidative stress may lead to genetic alterations, resulting in a higher rate of the BRAF V600E mutation over time in areas with excess iodine intake (115,116). Iodine supplementation has been associated with a decreasing trend in undifferentiated or anaplastic thyroid carcinoma (113,117).
Other Factors
Environmental radiation exposure, particularly in iodine-deficient areas, is associated with an increased risk of papillary thyroid cancer. However, specific data analyzing these factors in tropical nations are lacking (118).
Bisphenol A (BPA), a widely used organic compound in manufacturing processes in tropical countries, acts as an endocrine disruptor by binding to thyroid hormone receptors and inhibiting thyroid hormone-regulated gene expression. A study investigating the link between BPA levels, excessive iodine intake, and papillary thyroid cancer found that both urinary iodine concentration and urinary BPA concentration were higher in individuals with papillary thyroid carcinoma compared to controls. BPA and iodine may interact through shared pathways in the development of papillary thyroid carcinoma (119).
Management
Practice patterns for thyroid cancer care differ across countries due to variations in ethnic and racial populations, healthcare systems, economies, and cultures. The expertise and outcomes of thyroid surgery can vary significantly by region. While radioiodine treatment is available in many countries, its accessibility varies. Laboratory services for thyroid function monitoring are generally available, and most countries offer thyroglobulin assays. Recombinant thyrotropin is available in only a few countries, Advanced imaging technologies, such as positron emission tomography (PET), are limited to certain countries (120).
CONGENITAL HYPOTHYROIDISM
Congenital hypothyroidism (CH) is the primary cause of preventable intellectual disability. Since the 1970s, newborn screening for CH has been implemented in many parts of the world. Despite its proven benefits, universal screening for CH is yet to be adopted in many resource-limited tropical countries (121–124). Thyroid dysgenesis is identified as the most common cause of congenital CH in North America and Europe (125). Studies from Asia, however, indicate a higher prevalence of dyshormonogenesis (121,126,127). Cord blood screening with a TSH cut-off of 20 mIU/L has been employed for screening in many tropical nations due to its cost‐effectiveness, immediate action, and lower recall rate(121,128). Levothyroxine at a dosage of 10-15 μg/kg should be started immediately after diagnosis (129).
DRUG-INDUCED THYROID DYSFUNCTION
Iodine Induced Thyroid Dysfunction
A number of medications containing high amounts of iodine are still used in the tropical countries. These include the anti-arrhythmic agent amiodarone (75 mg iodine/tablet), expectorants containing iodinated glycerol (15 mg/tablet), topical antiseptics containing povidone iodine (10 mg/ml), and the anti-amoebic agent iodoquinol (134 mg/tablet). Additionally, traditional and alternative medicines are sometimes rich in iodine. Iodine inhibits T4 and T3 formation and release, predominantly by downregulation of the NIS. A healthy thyroid gland escapes the down-regulation, known as the Wolff Chaikoff effect. However, in the presence of underlying thyroid disorders like Hashimoto’s thyroiditis, non-systemic illnesses like chronic kidney disease, thalassemia major, and exposure to drugs like interferons and lithium, this compensatory mechanism fails, leading to persistent Wolff Chaikoff effect and occurrence of hypothyroidism (130). The resultant increase in TSH leads to further increase in iodide entry into the gland triggering a vicious cycle and leading to hypothyroidism and goiter.
Other Agents
Rifampicin, commonly used in tropical countries for tuberculosis, leprosy, and meningococcal prophylaxis, enhances T4 metabolism, and may increase levothyroxine requirement. Hence, close thyroid function monitoring is prudent in such situations. Ethionamide, another anti-tuberculosis drug, inhibits thyroid hormone synthesis and release (131).
Prolonged use of minocycline has been reported to cause blackening of the thyroid (132). Biotin, frequently used as a hair fall supplement in many tropical countries, occupy the streptavidin-binding sites in biotin-streptavidin two-site sandwich ELISA assay for TSH, leading to falsely low TSH. On the other hand, T4 is measured by competitive immunoassay and therefore, the falsely low signal due to biotin binding to streptavidin-binding sites, is interpreted as falsely elevated T4. Therefore, it is necessary to withhold biotin-containing supplements with doses of up to 10 mg once daily, for 8-h. If biotin intake is more than 10 mg per day, sampling should be delayed for a more extended period (up to 73 h) (133,134).
CHALLENGES TO MANAGEMENT IN THE TROPICS
Managing thyroid diseases in the tropics presents a unique set of challenges that encompass a range of socio-economic, healthcare infrastructure, and environmental factors. (Table 2).
Delayed Diagnosis
One of the primary challenges is the late presentation of patients, which is often due to limited access to healthcare facilities. In many tropical regions, healthcare services are scarce, particularly in rural areas, leading to delays in diagnosis and treatment. This delay can result in the progression of thyroid diseases to more severe stages, making management more complex.
Non-Availability Of Ethnicity Specific Reference Ranges For Thyroid Hormones
Inter-population differences in thyroid function are significant, as thyroid hormone reference intervals are influenced by both genetic factors, such as TSH polymorphisms, and environmental factors. Most tropical countries lack population-specific ranges and rely on Western standards.
A study found that a small fraction of South Asians could have a functionally normal TSH variant due to a novel TSHβ point mutation, to which some monoclonal antibodies fail to bind, resulting in falsely undetectable TSH levels and potential mistreatment as hyperthyroidism (135).
Iodine Deficiency And National Iodization Policies
Several factors, including dietary habits of consuming alternative salts, overheating during cooking, salt packaging, and economic and administrative issues, can hinder the goal of effective iodization. Studies in tropical countries found iodine deficiency in 30 out of 143 countries. Clinical observations in Uganda, Cambodia, Ethiopia, Cameroon, Mali, Burkina Faso, and India still show prevalence of large goiters causing neck compression (136).
Urinary iodine measurement is considered the standard for iodine intake assessment at the population level. The limitations of urinary iodine estimation include specimen integrity issues, sample authenticity concerns, and high pre-analytic variability. To address these limitations, alternative epidemiologic markers of iodine deficiency should be considered, such as percent of newborns with TSH values > 5 mIU/L, serum thyroglobulin measurement, thyroid volume measurement by palpation or ultrasonography. These markers may provide an alternative assessment of iodine status at the population level (137).
Screening Programs For Pregnant Women
Untreated hypothyroidism in pregnancy has short- and long-term consequences. While, universal screening for maternal hypothyroidism during pregnancy isn't generally agreed upon, testing during pregnancy can be beneficial, particularly in tropical regions where thyroid disorders might be underdiagnosed.
Neonatal Screening For Congenital Hypothyroidism
Many tropical countries lack national newborn screening programs for congenital hypothyroidism due to inadequate infrastructure for collecting, transporting, and analyzing dried blood spots, as well as challenges in follow-up care. TSH stability in these samples can be compromised by temperature and humidity, and confirmatory tests require advanced equipment and trained personnel. Increasing the use of point-of-care TSH assays and telecommunication could help overcome these obstacles and improve screening efforts.
Challenges To Diagnosis And Management Of Thyroid Cancer
In the tropics, there is inadequate access to molecular diagnostics and radioactive iodine facilities. Due to unavailability of radioiodine ablative facilities, management of differentiated thyroid cancer relies mostly on surgical management in many places. In cases of indeterminate nodules like BETHESDA 3,4 the use of molecular techniques to detect somatic mutations in 8-gene panel comprising BRAF-V600E, RET/PTC3, RET/PTC1, TERT promoter, HRAS, NRAS, KRAS, and PAX8-PPARG is recommended. However, this is not available in most tropical countries outside research settings, making decision-making difficult and leading to unnecessary surgeries. In cases with medullary thyroid cancers, genetic analysis of RET gene is not easily available and thus prophylactic thyroidectomy is often delayed.
|
Table 2. Challenges and Proposed Solutions to Management of Thyroid Disorders in the Tropics |
||
|---|---|---|
|
Challenges |
Description |
Solutions |
|
Delayed diagnosis |
Often due to limited healthcare access, leading to advanced disease stages at diagnosis. |
Improve healthcare infrastructure and community outreach to promote earlier diagnosis. |
|
Non-availability of ethnicity-specific reference ranges |
Lack of local reference ranges for thyroid hormones, causing potential misdiagnosis. |
Develop local reference ranges based on regional population data. |
|
Iodine deficiency and national iodization policies |
Inadequate iodization and challenges in maintaining iodine intake standards.
|
Implement more robust and monitored iodization programs to ensure optimum iodization avoiding both deficiencies and excess |
|
Storage and stability of thyroid medications |
High temperature and humidity can affect the potency of thyroid medications, making them less effective |
Levothyroxine tablets should be stored in a cool dry place away from light below 25°C. Ensure adherence to global standards of packaging in tropical nations |
|
Infection and thyroid disorders |
Tropical infections can directly or indirectly affect thyroid gland |
Focused research and increased awareness to understand and manage the complex interplay between infection and thyroid functioning |
|
Drug interactions and thyroid disorders |
Many drugs used for treatment for tuberculosis and other tropical disorders can potentially affect thyroid functioning and laboratory results |
Closely monitor for potential drug interactions between thyroid medications and treatments for infectious diseases and other conditions prevalent in tropics |
|
Endocrine disrupting chemicals |
Chemicals in pesticides and herbicides, industrial by-products, and plasticizers can impact thyroid functioning |
Regulatory measures, monitoring policies, sustainable practices, and focused research can help to mitigate the effect on thyroid disorders |
|
Screening programs for pregnant women |
Inadequate screening for maternal hypothyroidism, impacting maternal and fetal health |
Comprehensive screening guided by prevalence and individual and regional risk factors |
|
Neonatal screening for congenital hypothyroidism |
Lack of newborn screening programs for congenital hypothyroidism due to infrastructural limitations |
Universal screening with methodology guided by local resources and healthcare infrastructure |
|
Challenges to diagnosis and management of thyroid cancer |
Limited access to molecular diagnostics and radioactive iodine facilities
|
Increase access to necessary diagnostic tools and technologies, and develop protocols to manage cases with limited resources |
|
Lack of trained medical professional |
Trained healthcare providers are often not available in remote areas |
Leveraging telemedicine to connect patients in remote areas with endocrinologists and specialists. |
CONCLUSION
Thyroid disorders in the tropics present unique challenges due to socio-economic, healthcare infrastructure, and environmental factors. Late diagnosis, financial constraints, and limited healthcare access exacerbate the severity of these conditions. Iodine deficiency remains a significant issue, contributing to the prevalence of goiter and cretinism. Autoimmune thyroid diseases are influenced by environmental triggers and genetic susceptibility. Additionally, congenital hypothyroidism remains underdiagnosed due to inadequate newborn screening programs. Addressing these challenges requires improving healthcare infrastructure, ensuring consistent iodine supplementation, and enhancing public and professional awareness to improve the management and outcomes of thyroid disorders in tropical regions.
REFERENCES
- Fualal J, Ehrenkranz J. Access, availability, and infrastructure deficiency: The current management of thyroid disease in the developing world. Rev Endocr Metab Disord. 2016 Dec;17(4):583–9.
- Zimmermann MB, Boelaert K. Iodine deficiency and thyroid disorders. Lancet Diabetes Endocrinol. 2015 Apr;3(4):286–95.
- Han X, Ding S, Lu J, Li Y. Global, regional, and national burdens of common micronutrient deficiencies from 1990 to 2019: A secondary trend analysis based on the Global Burden of Disease 2019 study. EClinicalMedicine. 2022 Feb 12;44:101299.
- Dissanayake CB, Chandrajith R. Medical geology in tropical countries with special reference to Sri Lanka. Environ Geochem Health. 2007 Apr;29(2):155–62.
- Starling MCVM, Amorim CC, Leão MMD. Occurrence, control and fate of contaminants of emerging concern in environmental compartments in Brazil. J Hazard Mater. 2019 Jun 15;372:17–36.
- Ngeno E, Ongulu R, Orata F, Matovu H, Shikuku V, Onchiri R, et al. Endocrine disrupting chemicals in wastewater treatment plants in Kenya, East Africa: Concentrations, removal efficiency, mass loading rates and ecological impacts. Environ Res. 2023 Nov 15;237(Pt 2):117076.
- Chakraborty P, Bharat GK, Gaonkar O, Mukhopadhyay M, Chandra S, Steindal EH, et al. Endocrine-disrupting chemicals used as common plastic additives: Levels, profiles, and human dietary exposure from the Indian food basket. Sci Total Environ. 2022 Mar 1;810:152200.
- Zoeller RT. Endocrine disrupting chemicals and thyroid hormone action. Adv Pharmacol San Diego Calif. 2021;92:401–17.
- Gifford R, Siribaddana S, Forbes S, Eddleston M. Endocrine-disrupting chemicals and the diabetes epidemic in countries in the WHO South-East Asia region. Lancet Diabetes Endocrinol. 2015 Dec;3(12):925–7.
- Apte K, Salvi S. Household air pollution and its effects on health. F1000Research. 2016;5:F1000 Faculty Rev-2593.
- Chafe ZA, Brauer M, Klimont Z, Van Dingenen R, Mehta S, Rao S, et al. Household Cooking with Solid Fuels Contributes to Ambient PM2.5 Air Pollution and the Burden of Disease. Environ Health Perspect. 2014 Dec;122(12):1314–20.
- Hasan S, Tamim AR, Patwary MM, Hasan M, Rahman MA, Bardhan M, et al. Heatwaves and Air Pollution: A Deadly Combination for Human Health in South Asia. Prehospital Disaster Med. 2023 Apr;38(2):274–5.
- Makoni M. Air pollution in Africa. Lancet Respir Med. 2020 Jul;8(7):e60–1.
- Maleki Z, Hassanzadeh J, Méndez-Arriaga F, Ghaem H. Environmental factors and incidence of thyroid cancer in the world (1990-2019): an ecological study. Environ Sci Pollut Res Int. 2023 Sep;30(44):100072–7.
- Zumla A, Ustianowski A. Tropical Diseases. Infect Dis Clin North Am. 2012 Jun;26(2):195–205.
- Shah SS, Baum SG. Diagnosis and Management of Infectious Thyroiditis. Curr Infect Dis Rep. 2000 Apr;2(2):147–53.
- DV K, Gunasekaran K, Mishra AK, Iyyadurai R. Disseminated tuberculosis presenting as cold abscess of the thyroid gland—a case report. Oxf Med Case Rep. 2017 Sep 11;2017(9):omx049.
- Babiker A, Alawi A, Al Atawi M, Al Alwan I. The role of micronutrients in thyroid dysfunction. Sudan J Paediatr. 2020;20(1):13–9.
- Phiri FP, Ander EL, Bailey EH, Chilima B, Chilimba ADC, Gondwe J, et al. The risk of selenium deficiency in Malawi is large and varies over multiple spatial scales. Sci Rep. 2019 Apr 25;9(1):6566.
- Ventura M, Melo M, Carrilho F. Selenium and Thyroid Disease: From Pathophysiology to Treatment. Int J Endocrinol. 2017;2017:1297658.
- Vanderpas J. Nutritional epidemiology and thyroid hormone metabolism. Annu Rev Nutr. 2006;26:293–322.
- Prevention and control of iodine deficiency disorders. Lancet Lond Engl. 1986 Aug 23;2(8504):433–4.
- Eastman CJ, Zimmermann MB. The Iodine Deficiency Disorders. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2024 Jun 3]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK285556/
- Führer D, Bockisch A, Schmid KW. Euthyroid Goiter With and Without Nodules—Diagnosis and Treatment. Dtsch Ärztebl Int. 2012 Jul;109(29–30):506–16.
- Watters DAK, Wall J. Thyroid surgery in the tropics. ANZ J Surg. 2007 Nov;77(11):933–40.
- Farebrother J, Zimmermann MB, Andersson M. Excess iodine intake: sources, assessment, and effects on thyroid function. Ann N Y Acad Sci. 2019 Jun;1446(1):44–65.
- Dessie G, Amare D, Dagnew AB, Mulugeta H, Haile Kassa D, Negesse A, et al. Prevalence of goiter among children in Ethiopia and associated factors: a systematic review and meta-analysis. BMC Public Health. 2019 Aug 29;19:1191.
- Speeckaert MM, Speeckaert R, Wierckx K, Delanghe JR, Kaufman JM. Value and pitfalls in iodine fortification and supplementation in the 21st century. Br J Nutr. 2011 Oct;106(7):964–73.
- Vanderpas JB, Moreno-Reyes R. Historical aspects of iodine deficiency control. Minerva Med. 2017 Apr;108(2):124–35.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab. 2010 Feb;24(1):39–50.
- Lisco G, De Tullio A, Triggiani D, Zupo R, Giagulli VA, De Pergola G, et al. Iodine Deficiency and Iodine Prophylaxis: An Overview and Update. Nutrients. 2023 Feb 16;15(4):1004.
- Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev. 2015 Feb;14(2):174–80.
- Ragusa F, Fallahi P, Elia G, Gonnella D, Paparo SR, Giusti C, et al. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract Res Clin Endocrinol Metab. 2019 Dec;33(6):101367.
- Jayatissa R, Okosieme OE, Ranasinghe S, Carter JL, Gunatunga IP, Lazarus JH, et al. Thyroid Autoimmunity and Dysfunction in Sri Lankan Children and Adolescents After 22 Years of Sustained Universal Salt Iodization. Thyroid Off J Am Thyroid Assoc. 2021 Jul;31(7):1105–13.
- Kalarani IB, Veerabathiran R. Impact of iodine intake on the pathogenesis of autoimmune thyroid disease in children and adults. Ann Pediatr Endocrinol Metab. 2022 Dec;27(4):256–64.
- Teti C, Panciroli M, Nazzari E, Pesce G, Mariotti S, Olivieri A, et al. Iodoprophylaxis and thyroid autoimmunity: an update. Immunol Res. 2021 Apr;69(2):129–38.
- Martinez Quintero B, Yazbeck C, Sweeney LB. Thyroiditis: Evaluation and Treatment. Am Fam Physician. 2021 Dec 1;104(6):609–17.
- Caturegli P, De Remigis A, Rose NR. Hashimoto thyroiditis: clinical and diagnostic criteria. Autoimmun Rev. 2014;13(4–5):391–7.
- Antonelli A, Fallahi P, Elia G, Ragusa F, Paparo SR, Ruffilli I, et al. Graves’ disease: Clinical manifestations, immune pathogenesis (cytokines and chemokines) and therapy. Best Pract Res Clin Endocrinol Metab. 2020 Jan;34(1):101388.
- Vanderpump MPJ. The epidemiology of thyroid disease. Br Med Bull. 2011;99:39–51.
- Taylor PN, Albrecht D, Scholz A, Gutierrez-Buey G, Lazarus JH, Dayan CM, et al. Global epidemiology of hyperthyroidism and hypothyroidism. Nat Rev Endocrinol. 2018 May;14(5):301–16.
- Kalk WJ. Thyrotoxicosis in urban black Africans: a rising incidence. East Afr Med J. 1981 Feb;58(2):109–16.
- Sarfo-Kantanka O, Kyei I, Sarfo FS, Ansah EO. Thyroid Disorders in Central Ghana: The Influence of 20 Years of Iodization. J Thyroid Res. 2017;2017:7843972.
- McGrogan A, Seaman HE, Wright JW, De Vries CS. The incidence of autoimmune thyroid disease: a systematic review of the literature. Clin Endocrinol (Oxf). 2008;69(5):687–96.
- Antonelli A, Ferrari SM, Ragusa F, Elia G, Paparo SR, Ruffilli I, et al. Graves’ disease: Epidemiology, genetic and environmental risk factors and viruses. Best Pract Res Clin Endocrinol Metab. 2020 Jan;34(1):101387.
- Nagayama Y, McLachlan SM, Rapoport B, Oishi K. Graves’ hyperthyroidism and the hygiene hypothesis in a mouse model. Endocrinology. 2004 Nov;145(11):5075–9.
- Raizada N. Helminths and Endocrinology. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2024 Jul 13]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK569325/
- Ogbera AO, Kuku SF. Epidemiology of thyroid diseases in Africa. Indian J Endocrinol Metab. 2011 Jul;15(Suppl2):S82–8.
- Akossou SY, Napporn A, Goeh-Akue E, Hillah A, Sokpoh-Diallo K, Soussou B, et al. [Problems in the management of thyrotoxicosis in Black Africa: the Tongolese experience]. Ann Endocrinol. 2001 Dec;62(6):516–20.
- Ogbera A, Isiba A. The scope of cardiac complications of thyrotoxicosis in Lagos, Nigeria. Endocr Abstr [Internet]. 2007 Mar 1 [cited 2024 Jun 5];13. Available from: https://www.endocrine-abstracts.org/ea/0013/ea0013p301
- Tellez M, Cooper J, Edmonds C. Graves’ ophthalmopathy in relation to cigarette smoking and ethnic origin. Clin Endocrinol (Oxf). 1992 Mar;36(3):291–4.
- Okinaka S, Shizume K, Iino S, Watanabe A, Irie M, Noguchi A, et al. The association of periodic paralysis and hyperthyroidism in Japan. J Clin Endocrinol Metab. 1957 Dec;17(12):1454–9.
- Tamai H, Tanaka K, Komaki G, Matsubayashi S, Hirota Y, Mori K, et al. HLA and thyrotoxic periodic paralysis in Japanese patients. J Clin Endocrinol Metab. 1987 May;64(5):1075–8.
- Villagelin D, Cooper DS, Burch HB. A 2023 International Survey of Clinical Practice Patterns in the Management of Graves Disease: A Decade of Change. J Clin Endocrinol Metab. 2024 Apr 5;dgae222.
- Jin M, Jang A, Kim CA, Young Kim T, Bae Kim W, Kee Shong Y, et al. Long-term follow-up result of antithyroid drug treatment of Graves’ hyperthyroidism in a large cohort. Eur Thyroid J. 2023 Mar 17;12(2):e220226.
- Diack ND, Ndiaye N, Sene M, Ba M, Thiam NF, Samb K, et al. Resistance to Anti-Thyroid Drugs in Graves’ Disease: Clinical-Biological Characteristics and Alternative Therapy in Tropical Area. Open J Endocr Metab Dis. 2020 Nov 29;10(11):147–53.
- Ata F, Khan AA, Tahir S, Al Amer Z. Carbimazole-Resistant Grave’s Thyrotoxicosis is a Diagnostic and Therapeutic Dilemma, Case Report with Literature Review. Int Med Case Rep J. 2023 Nov 28;16:783–90.
- Lin M, Chen Y, Ning J. Insulin Autoimmune Syndrome: A Systematic Review. Int J Endocrinol. 2023 Feb 15;2023:1225676.
- Hennessey J v. Subacute Thyroiditis. In: Endotext [Internet] [Internet]. MDText.com, Inc.; 2018 [cited 2024 Jul 16]. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK279084/
- VOLPÉ R, ROW VV, EZRIN C. Circulating Viral and Thyroid Antibodies in Subacute Thyroiditis1. J Clin Endocrinol Metab. 1967 Sep 1;27(9):1275–84.
- Assir MZK, Jawa A, Ahmed HI. Expanded dengue syndrome: subacute thyroiditis and intracerebral hemorrhage. BMC Infect Dis. 2012 Oct 3;12(1):240.
- Dimos G, Pappas G, Akritidis N. Subacute thyroiditis in the course of novel H1N1 influenza infection. Endocrine. 2010 Jun 1;37(3):440–1.
- A. Al Maawali, S. Al Yaarubi, A. Al Futaisi. An Infant with Cytomegalovirus-induced Subacute Thyroiditis. J Pediatr Endocrinol Metab. 2008 Feb 1;21(2):191–4.
- Engkakul P, Mahachoklertwattana P, Poomthavorn P. de Quervain thyroiditis in a young boy following hand-foot-mouth disease. Eur J Pediatr. 2011 Apr;170(4):527–9.
- Hernán Martinez J, Corder E, Uzcategui M, Garcia M, Sostre S, Garcia A. Subacute thyroiditis and dyserythropoesis after influenza vaccination suggesting immune dysregulation. Boletin Asoc Medica P R. 2011 Apr 1;103(2):48–52.
- Falaschi P, Martocchia A, D’Urso R, Proietti A. Subacute thyroiditis during interferon-alpha therapy for chronic hepatitis C. J Endocrinol Invest. 1997 Jan 1;20(1):24–8.
- Kim S hee, Park TS, Baek HS, Jin HY. Subacute painful thyroiditis accompanied by scrub typhus infection. Endocrine. 2013 Oct;44(2):546–8.
- Nyulassy S, Hnilica P, Buc M, Guman M, Hirschová V, Stefanovic J. Subacute (de Quervain’s) thyroiditis: association with HLA-Bw35 antigen and abnormalities of the complement system, immunoglobulins and other serum proteins. J Clin Endocrinol Metab. 1977 Aug;45(2):270–4.
- Ohsako N, Tamai H, Sudo T, Mukuta T, Tanaka H, Kuma K, et al. Clinical characteristics of subacute thyroiditis classified according to human leukocyte antigen typing. J Clin Endocrinol Metab. 1995 Dec;80(12):3653–6.
- Stasiak M, Tymoniuk B, Michalak R, Stasiak B, Kowalski ML, Lewiński A. Subacute Thyroiditis is Associated with HLA-B*18:01, -DRB1*01 and -C*04:01—The Significance of the New Molecular Background. J Clin Med. 2020 Feb;9(2):534.
- Khan SH, Mahajan A, Laway BA, Rasool R, Rather TA. Technetium-99m thyroid scintigraphy and human leukocyte antigen – B35 in sub-acute thyroiditis. Indian J Nucl Med. 2018 Oct 1;33(4):306.
- DeGroot LJ, Larsen PR, Hennemann G. Acute and subacute thyroiditis. In: DeGroot LJ, Larsen PR, Hennemann G, eds. The Thyroid and Its Diseases. 6th ed. New York: Churchill Livingstone; 1996:705.). In.
- Fatourechi V, Aniszewski JP, Fatourechi GZE, Atkinson EJ, Jacobsen SJ. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003 May;88(5):2100–5.
- Kalra P, Kumar KP. Primary hypothyroidism on follow-up in a cohort of Indian patients with subacute thyroiditis. Thyroid Res Pract. 2021 Jan 1;18(1):1.
- Alfadda AA, Sallam RM, Elawad GE, AlDhukair H, Alyahya MM. Subacute Thyroiditis: Clinical Presentation and Long Term Outcome. Int J Endocrinol. 2014;2014(1):794943.
- Nishihara E, Ohye H, Amino N, Takata K, Arishima T, Kudo T, et al. Clinical characteristics of 852 patients with subacute thyroiditis before treatment. Intern Med Tokyo Jpn. 2008;47(8):725–9.
- Hiromatsu Y, Ishibashi M, Miyake I, Soyejima E, Yamashita K, Koike N, et al. Color Doppler ultrasonography in patients with subacute thyroiditis. Thyroid Off J Am Thyroid Assoc. 1999 Dec;9(12):1189–93.
- Mizukoshi T, Noguchi S, Murakami T, Futata T, Yamashita H. Evaluation of recurrence in 36 subacute thyroiditis patients managed with prednisolone. Intern Med Tokyo Jpn. 2001 Apr;40(4):292–5.
- Samuels MH. Subacute, silent, and postpartum thyroiditis. Med Clin North Am. 2012 Mar;96(2):223–33.
- Inaba H, Akamizu T. Postpartum Thyroiditis. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2024 Jul 16]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK278999/
- Mangaraj S. Subacute thyroiditis complicating dengue fever – Case report and brief review of literature. Trop Doct. 2021 Apr 1;51(2):254–6.
- Paes JE, Burman KD, Cohen J, Franklyn J, McHenry CR, Shoham S, et al. Acute bacterial suppurative thyroiditis: a clinical review and expert opinion. Thyroid Off J Am Thyroid Assoc. 2010 Mar;20(3):247–55.
- Yolmo D, Madana J, Kalaiarasi R, Gopalakrishnan S, Kiruba Shankar M, Krishnapriya S. Retrospective case review of pyriform sinus fistulae of third branchial arch origin commonly presenting as acute suppurative thyroiditis in children. J Laryngol Otol. 2012 Jul;126(7):737–42.
- Lafontaine N, Learoyd D, Farrel S, Wong R. Suppurative thyroiditis: Systematic review and clinical guidance. Clin Endocrinol (Oxf). 2021 Aug;95(2):253–64.
- Gigot JF, Mannell A. Acute emphysematous thyroiditis. Br J Surg. 1983 May;70(5):256–8.
- Idrees S, Godi P, Kumar R, Chand G, Agarwal G, Singh AK, et al. An uncommon encounter in an unusual location – A case report on acute emphysematous suppurative thyroid abscess. Surg Case Rep. 2024 Mar 1;1:100010.
- Pereira KD, Davies JN. Piriform sinus tracts in children. Arch Otolaryngol Head Neck Surg. 2006 Oct;132(10):1119–21.
- Majety P, Hennessey JV. Acute and Subacute, and Riedel’s Thyroiditis. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2024 Jul 17]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK285553/
- Baidya A, Singha A, Bhattacharjee R, Dalal BS. Tuberculosis of the thyroid gland: two case reports. Oxf Med Case Rep. 2015 Apr 16;2015(4):262–4.
- Majid U, Islam N. Thyroid Tuberculosis: A Case Series and a Review of the Literature. J Thyroid Res. 2011 Apr 14;2011:359864.
- Varghese A, Suneha S, Shaha A. Primary tuberculosis of thyroid. Am J Otolaryngol. 2015;36(6):808–9.
- Shekhawat KK, Rathore VS. Discharging Sinus of Neck after Thyroid Surgery: A Rare Case Report. Ann Indian Acad Otorhinolaryngol Head Neck Surg. 2017 Dec;1(2):29.
- Lioté HA, Spaulding C, Bazelly B, Milleron BJ, Akoun GM. Thyroid tuberculosis associated with mediastinal lymphadeniitis. Tubercle. 1987 Sep 1;68(3):229–31.
- Kataria SP, Tanwar P, Singh S, Kumar S. Primary tuberculosis of the thyroid gland: a case report. Asian Pac J Trop Biomed. 2012 Oct;2(10):839–40.
- Bhattacharya S, Kubiha S, Tyagi P. Fungi and Endocrine Dysfunction. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2023 Jul 25]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK572246/
- Alvi MM, Meyer DS, Hardin NJ, deKay JG, Marney AM, Gilbert MP. Aspergillus Thyroiditis: A Complication of Respiratory Tract Infection in an Immunocompromised Patient. Case Rep Endocrinol. 2013;2013:741041.
- Nguyen J, Manera R, Minutti C. Aspergillus thyroiditis: a review of the literature to highlight clinical challenges. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2012 Dec;31(12):3259–64.
- Gowing NF, Hamlin IM. Tissue reactions to Aspergillus in cases of Hodgkin’s disease and leukaemia. J Clin Pathol. 1960 Sep;13(5):396–413.
- Jenks JD, Hoenigl M. Treatment of Aspergillosis. J Fungi. 2018 Aug 19;4(3):98.
- Da’as N, Lossos IS, Yahalom V, Rund D, Wolf DG, Zelig O, et al. Candida abscess of the thyroid in a patient with acute lymphocytic leukemia. Eur J Med Res. 1997 Aug 28;2(8):365–6.
- Gandhi RT, Tollin SR, Seely EW. Diagnosis of Candida thyroiditis by fine needle aspiration. J Infect. 1994 Jan;28(1):77–81.
- Niles D, Boguniewicz J, Shakeel O, Margolin J, Chelius D, Gupta M, et al. Candida tropicalis Thyroiditis Presenting With Thyroid Storm in a Pediatric Patient With Acute Lymphocytic Leukemia. Pediatr Infect Dis J. 2019 Oct;38(10):1051–3.
- Lima R, Ribeiro FC, Colombo AL, de Almeida JN. The emerging threat antifungal-resistant Candida tropicalis in humans, animals, and environment. Front Fungal Biol. 2022;3:957021.
- Baker J, Setianingrum F, Wahyuningsih R, Denning DW. Mapping histoplasmosis in South East Asia – implications for diagnosis in AIDS. Emerg Microbes Infect. 8(1):1139–45.
- Araúz AB, Papineni P. Histoplasmosis. Infect Dis Clin North Am. 2021 Jun;35(2):471–91.
- Barros N, Wheat JL, Hage C. Pulmonary Histoplasmosis: A Clinical Update. J Fungi. 2023 Feb 10;9(2):236.
- Raza N, Heidari A. A Case of Thyroidal Coccidioidal Infection. J Investig Med High Impact Case Rep. 2022 Apr 15;10:23247096221090840.
- Galgiani JN, Ampel NM, Blair JE, Catanzaro A, Geertsma F, Hoover SE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis Off Publ Infect Dis Soc Am. 2016 Sep 15;63(6):e112-146.
- Yue S, Min L. [Strongyloides stercoralis infection with hypothyroidism: one case report]. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi Chin J Schistosomiasis Control. 2017 Apr 27;29(3):393–4.
- Akbulut S, Demircan F, Sogutcu N. Hydatid Cyst Disease of the Thyroid Gland: Report of Two Cases. Int Surg. 2015 Apr;100(4):643–7.
- Reincke M, Allolio B, Petzke F, Heppner C, Mbulamberi D, Vollmer D, et al. Thyroid dysfunction in African trypanosomiasis: a possible role for inflammatory cytokines. Clin Endocrinol (Oxf). 1993;39(4):455–61.
- Wang C, Wu Z, Lei L, Dong X, Cao W, Luo Z, et al. Geographic disparities in trends of thyroid cancer incidence and mortality from 1990 to 2019 and a projection to 2030 across income-classified countries and territories. J Glob Health. 13:04108.
- Sierra MS, Soerjomataram I, Forman D. Thyroid cancer burden in Central and South America. Cancer Epidemiol. 2016 Sep;44 Suppl 1:S150–7.
- Mitro SD, Rozek LS, Vatanasapt P, Suwanrungruang K, Chitapanarux I, Srisukho S, et al. Iodine deficiency and thyroid cancer trends in three regions of Thailand, 1990-2009. Cancer Epidemiol. 2016 Aug;43:92–9.
- Kowalska A, Walczyk A, Kowalik A, Pałyga I, Trybek T, Kopczyński J, et al. Increase in Papillary Thyroid Cancer Incidence Is Accompanied by Changes in the Frequency of the BRAF V600E Mutation: A Single-Institution Study. Thyroid Off J Am Thyroid Assoc. 2016 Apr;26(4):543–51.
- Zhang X, Zhang F, Li Q, Feng C, Teng W. Iodine nutrition and papillary thyroid cancer. Front Nutr. 2022;9:1022650.
- Harach HR, Ceballos GA. Thyroid cancer, thyroiditis and dietary iodine: a review based on the Salta, Argentina model. Endocr Pathol. 2008;19(4):209–20.
- Nettore IC, Colao A, Macchia PE. Nutritional and Environmental Factors in Thyroid Carcinogenesis. Int J Environ Res Public Health. 2018 Aug 13;15(8):1735.
- Zhou Z, Zhang J, Jiang F, Xie Y, Zhang X, Jiang L. Higher urinary bisphenol A concentration and excessive iodine intake are associated with nodular goiter and papillary thyroid carcinoma. Biosci Rep. 2017 Aug 31;37(4):BSR20170678.
- Sundram F, Robinson BG, Kung A, Lim-Abrahan MA, Bay NQ, Chuan LK, et al. Well-Differentiated Epithelial Thyroid Cancer Management in the Asia Pacific Region: A Report and Clinical Practice Guideline. Thyroid®. 2006 May;16(5):461–9.
- Anne RP, Rahiman EA. Congenital hypothyroidism in India: A systematic review and meta-analysis of prevalence, screen positivity rates, and etiology. Lancet Reg Health Southeast Asia. 2022 Oct;5:100040.
- Yarhere IE, Jaja T, Briggs D, Iughetti L. Newborn screening in Nigeria: Associating the screening of congenital hypothyroidism and sickle cell disease can be a winning choice? Acta Bio-Medica Atenei Parm. 2019 May 23;90(2):316–20.
- Alladin BA, Mohamed-Rambaran P, Grey V, Hunter A, Chakraborty P, Henderson M, et al. Cross-sectional prospective feasibility study of newborn screening for sickle cell anaemia and congenital hypothyroidism in Guyana. BMJ Open. 2022 Feb 22;12(2):e046240.
- Pezzuti IL, Lima PP de, Dias VMA. Congenital hypothyroidism: the clinical profile of affected newborns identified by the Newborn Screening Program of the State of Minas Gerais, Brazil. J Pediatr (Rio J). 2009;85(1):72–9.
- Segni M. Congenital Hypothyroidism. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000 [cited 2024 Jul 21]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK279004/
- Nagendra L, Bhavani N, Pavithran PV, Shenoy M, Menon UV, Abraham N, et al. Etiological Profile, Targeted Levothyroxine Dosing and Impact of Partial Newborn Screening in Congenital Hypothyroidism-A Single Centre Experience. Indian J Endocrinol Metab. 2023;27(5):445–9.
- Liu R, Tian JL, Huang XL, Song YZ. Genetic Factors Causing Thyroid Dyshormonogenesis as the Major Etiologies for Primary Congenital Hypothyroidism: Clinical and Genetic Characterization of 33 Patients. J Clin Med. 2022 Dec 9;11(24):7313.
- Alameer S, Althobaiti E, Alshaikh S, Turjoman M, Badriq F, AlSofyani A, et al. Diagnostic comparison between cord blood and filter paper for the screening of congenital hypothyroidism. J Clin Lab Anal. 2021 Dec 3;36(1):e24149.
- Sudhanshu S, Riaz I, Sharma R, Desai MP, Parikh R, Bhatia V. Newborn Screening Guidelines for Congenital Hypothyroidism in India: Recommendations of the Indian Society for Pediatric and Adolescent Endocrinology (ISPAE) - Part II: Imaging, Treatment and Follow-up. Indian J Pediatr. 2018 Jun;85(6):448–53.
- Leung AM, Braverman LE. Iodine-induced thyroid dysfunction. Curr Opin Endocrinol Diabetes Obes. 2012 Oct;19(5):414–9.
- Burch HB. Drug Effects on the Thyroid. N Engl J Med. 2019 Aug 22;381(8):749–61.
- Goto Y, Ohba K, Sasaki S, Nishino N. Minocycline and black thyroid. QJM Int J Med. 2022 Jun 1;115(6):403–4.
- Bowen R, Benavides R, Colón-Franco JM, Katzman BM, Muthukumar A, Sadrzadeh H, et al. Best practices in mitigating the risk of biotin interference with laboratory testing. Clin Biochem. 2019 Dec;74:1–11.
- Ardabilygazir A, Afshariyamchlou S, Mir D, Sachmechi I. Effect of High-dose Biotin on Thyroid Function Tests: Case Report and Literature Review. Cureus. 2018 Jun 20;10(6):e2845.
- Drees JC, Stone JA, Reamer CR, Arboleda VE, Huang K, Hrynkow J, et al. Falsely undetectable TSH in a cohort of South Asian euthyroid patients. J Clin Endocrinol Metab. 2014 Apr;99(4):1171–9.
- BIBAN B, LICHIARDOPOL C. Iodine Deficiency, Still a Global Problem? Curr Health Sci J. 2017;43(2):103–11.
- Wainwright P, Cook P. The assessment of iodine status - populations, individuals and limitations. Ann Clin Biochem. 2019 Jan;56(1):7–14.