ABSTRACT
Hypopituitarism is described as partial or complete loss of isolated or combined deficits of pituitary hormones. The clinical presentation of this condition varies depending on the age of onset, underlying cause of the disease, severity of deficiency and number of affected hormones. Investigation to confirm the diagnosis is based on basal hormone status and dynamic stimulatory tests. In this review, we will provide a concise background knowledge regarding underlying etiologies, clinical manifestations, hormonal investigations, and related treatments which necessitate close lifelong monitoring.
INTRODUCTION
Hypopituitarism refers to complete or partial failure of secretion of anterior and/or posterior pituitary hormones. It may arise as a result of congenital defects in the development of individual anterior pituitary cell types or hypothalamic function, acquired disease of the pituitary or hypothalamus, or from infundibular lesions which interfere with the hypothalamic control of the pituitary. The multiple aspects of normal pituitary function serve to predict the wide range of clinical manifestations of hypopituitarism which are determined by the severity, extent and duration of the condition. Onset may occur during childhood or adult life and is generally permanent, requiring one or more specific hormone replacements.
ETIOLOGY OF HYPOPITUITARISM
The major causes of hypopituitarism are shown in Table 1.
|
Table 1. Causes of Hypopituitarism |
||
|
Congenital |
Isolated pituitary hormone deficiency |
· Receptor mutation: GHRH, CRH, GnRH, TRH receptor mutations · Transcription factor defect: PITX2, TBX19, DAX1, NR5A1, NR0B1 · Hormone mutation: GH1, bio-inactive GH, FSHb, LHb, TSHb, POMC, POMC processing defect · Prader-Willi syndrome · Bardet-Biedl syndrome · Kallmann syndrome |
|
Multiple pituitary hormone deficiency |
· Transcription factor defect: HESX1, SOX 2/3, LHX3/4, PROP1, POU1F1, IGSF1 mutations · Prohormone convertase enzyme mutation: PC1 |
|
|
Neoplastic |
Pituitary adenoma |
· Functioning and Non-functioning |
|
Peri-pituitary tumors |
· Parasellar lesion: craniopharyngioma, Rathke's cleft cyst · Non-adenomatous neoplasm: meningioma, glioma, germ cell tumor · Metastases, especially breast, renal, bronchus |
|
|
Vascular |
Infarction/ hemorrhage |
· Sheehan's syndrome · Pituitary apoplexy · Aneurysms |
|
Inflammatory / Infiltrative / Immunological |
|
· Hypophysitis: lymphocytic, granulomatous, xanthomatous, necrotizing, IgG4-related, immunotherapy-induced (CTLA-4 inhibitors), other immune-associated · Hemochromatosis · Sarcoidosis · Wegener's granulomatosis · Giant cell granuloma · Langerhans cell histiocytosis |
|
Infectious |
|
· Bacterial: tuberculosis, syphilis, leptospirosis · Fungal: candidiasis, aspergillosis · Viral: Herpes/Varicella infection, SARS-CoV-2 virus |
|
Post-irradiation |
|
· Pituitary · Nasopharyngeal · Cranial |
|
Miscellaneous |
|
· Empty sella · Traumatic brain injury · Medications |
Congenital
Formation of the normal pituitary during embryonic development depends on the juxtaposition of cells of neuroectodermal origin, which form the posterior pituitary, and endodermal cells derived from the primitive stomodeum, which form the anterior pituitary. Congenital forms of hypopituitarism are best considered as being derived either from hypothalamic or pituitary origin. Defects of RPX/HESX1, PROP1, and PIT1 are associated with varying degrees of inherited hypopituitarism in humans (1). Autosomal dominant mutations of the arginine vasopressin-neurophysin II gene give rise to familial antidiuretic hormone (ADH) deficiency (central diabetes insipidus, CDI). Congenital combined pituitary hormone deficiency (CPHD) (or multiple pituitary hormone deficiency (MPHD)) may be associated with hypoplasia of the anterior pituitary and ectopic siting of the posterior pituitary in a superior position. The underlying mechanism for this condition, once thought to be a consequence of birth trauma, remains undetermined but a congenital molecular defect is likely. Furthermore, since some genetic mutations affect early development, they may include extra-pituitary features or midline anomalies as a part of a syndrome. For instance, the transcription factors, including HESX1, SOX2, SOX3, and OTX2, are the most common genes involved in the etiology of septo-optic dysplasia. Hypogonadotropic hypogonadism is a recognized feature of both Prader-Willi and Bardet-Biedl syndromes. The salient features of the currently described transcription factor defects are indicated below.
HESX1 MUTATIONS
HESX1 is a member of the homeobox gene family. It is one of the earliest markers of the pituitary primodium expressed as a prelude to the development of Rathke's pouch. Mutations of the HESX1 gene in humans are associated with septo-optic dysplasia and evolving hypopituitarism. Septo-optic dysplasia is characterized by the classical triad of optic nerve hypoplasia, midline neuroradiological abnormalities such as agenesis of the corpus callosum, and pituitary hypoplasia with consequent panhypopituitarism (2). Expression of the HESX1 gene precedes expression of PROP1 and PIT1, and its consequences are more extensive.
PROP1 DEFECT
The PROP1 gene encodes a transcription factor with a single paired-like DNA-binding domain. Individuals with a single inactivating mutation in PROP1 have deficiencies of luteinizing hormone (LH), follicle stimulating hormone (FSH), GH, prolactin, and TSH (3). Their pituitary glands may be small, normally sized, or large with extrasellar extension. Pituitary degeneration may produce acquired deficiency of adrenocorticotropic hormone (ACTH). Growth restriction and GH responses to GHRH stimulation are more variable in children with PROP1 mutation. This variability does not seem to be dependent on the type of PROP1 mutation because variability exists within sibships. Recessive mutation in PROP1 is the most frequent genetic cause of CPHD and appears to be much more common than POU1F1 mutation (4,5). Clinical suspicion of these mutations should be high in any individual with early onset CPHD, even in those with an intrasellar or suprasellar mass lesion. However, the clinical manifestations of each hormone may vary in onset and severity. GHD is usually present in the first years of life; whereas TSH and gonadotropin deficiency might manifest at birth or later in life. Most patients do not experience adrenal insufficiency during the first years of life although this can evolve later.
PIT1 DEFECT
PIT1, also known as POU1F1, is a pituitary specific transcription factor found in somatotrophs, lactotrophs, and thyrotrophs in the anterior pituitary gland from early fetal development and throughout life. Autosomal recessive defects of PIT1 are associated with combined deficiencies of growth hormone (GH), prolactin, and thyrotropin stimulating hormone (TSH) (6,7). Patients with PIT1 mutations (coded by POU1F1 gene) tend to firstly manifest with GHD and prolactin deficiency in the first year of life and generally do not release detectable amounts of GH after GHRH stimulation. TSH deficiency typically develops later in life (8).
SOX2 MUTATIONS
SOX2 belongs to the SRY-related HMG box (SOX) family of transcription factors which are expressed in various stages of embryonic development and cell differentiation, and play critical roles from the earliest stages of development, in particular expression of anterior neuroectoderm. De novo truncating mutations of SOX2 are found in individuals with bilateral anophthalmia/microphthalmia, small corpus callosum, hippocampal abnormalities, variable mental retardation, anterior pituitary hypoplasia and often hypopituitarism (9-11)
SOX3 MUTATIONS
SOX3 also belongs to the SRY-related HMG box (SOX) family of transcription factors. This dosage sensitive gene is essential for normal hypothalamic-pituitary development (12). Patients manifest with variable hypopituitarism, infundibular hypoplasia, abnormal corpus callosum, hypoplasia of anterior pituitary, absent stalk, ectopic/undescended posterior pituitary, and intellectual disability (13).
OTX2 MUTATION
OTX2 belongs to the orthodenticle homeobox (OTX) and plays an essential role in brain, eyes, and pituitary development. Patients with OTX2 mutations manifest with variable degrees of pituitary dysfunction, intellectual disability, ocular abnormalities including microphthalmia, anophthalmia, and abnormal peripapillary pigmentation (14).
DAX1 MUTATIONS
Mutations in DAX1 (coded by NR0B1), which is another X chromosome gene, cause hypogonadotropic hypogonadism in association with congenital adrenal hypoplasia in males. DAX1 encodes an orphan nuclear hormone receptor with a novel DNA-binding domain, that has a critical role in the development of the hypothalamus, pituitary, adrenal, and gonads (15). DAX1 appears to influence the maintenance of testicular epithelial integrity and spermatogenesis.
KAL1 MUTATION
Isolated gonadotropin deficiency with hyposmia (Kallmann syndrome) may be inherited as an autosomal X-linked disorder or occur sporadically. The X-linked disorder occurs in approximately 1/10,000 to 60,000 live births (16) and a mutation in the KAL1 gene is the basis for this disease. Gonadotropin-releasing hormone (GnRH) neurons develop in the medial olfactory placode and migrate to the hypothalamus during embryonic development; the KAL protein is essential for the initiation and maintenance of this process. Patients with the disorder usually present in late adolescence with delayed pubertal initiation or progression and characteristically exhibit hyposmia. They may develop eunuchoid proportions and usually demonstrate small testes and a short penis; other features which are specific to X-linked Kallmann syndrome include unilateral renal agenesis and synkinesis (mirror movements), whereas midline facial defects, choanal atresia, short metacarpals, malrotation of the gut, and ocular coloboma are more common in autosomal and sporadic forms of the condition.
GH1 MUTATIONS
Mutations in the major GH gene, GH1, can occur as deletions of different size, resulting in severe isolated GH deficiency and may be familial with either a dominant or recessive pattern of inheritance.
TBX19 MUTATION
TBX19, also known as TPIT, is essential for both pro-opiomelanocortin (POMC) transcription and terminal differentiation of POMC-expressing cells. A mutation of TBX19 results in congenital isolated ACTH deficiency and this mutation is discovered in 65% of patients with neonatal-onset congenital isolated ACTH deficiency (17).
Other single gene mutations affecting individual anterior pituitary hormones or pituitary receptors for hypothalamic peptides are indicated in Table 1.
Tumors
Pituitary adenomas are the most common cause of adult-onset hypopituitarism (18,19). Craniopharyngiomas are numerically the next most prevalent and may present as sellar and/or suprasellar masses.
PITUITARY ADENOMA
Pituitary tumors can be either non-functioning or secrete one or more anterior pituitary hormones. They are classified according to size and the presence of extrasellar extension; magnetic resonance imaging (MRI) permits accurate measurement of maximum diameter and tumors are conventionally classified as microadenomas (less than 10mm), and macroadenomas (more than 20mm in diameter) (20). The reported prevalence of pituitary tumors is approximately 10 per million with an average annual incidence of approximately 30 per million depending on tumor type, age, and gender with the highest incidence occurring in pre-menopausal women. Pituitary adenomas may cause typical clinical syndromes resulting from hypersecretion of one or more anterior pituitary hormones. Approximately 25-30% will not present with these symptoms, so they will only be detected when tumor expansion and local compression of the surrounding structures emerges. Post-mortem studies reveal a prevalence of incidental intrasellar adenomas of 10-20%.
Clinically non-functioning pituitary adenomas (NFPA) are non- or low grade-secreting tumors which frequently synthesize and secrete free alpha or beta glycoprotein subunits but do not cause clinically recognized symptomatology. Hence, the treatment of choice is surgical debulking, and sometimes followed by external radiation therapy. In contrast to patients with secreting pituitary tumor who present with clinical syndromes of acromegaly, hyperprolactinemia, Cushing's disease, and secondary hyperthyroidism non-functioning tumors present as an incidental finding, hypopituitarism, or due to mass effects of the tumor. The mainstream treatment in these diseases must be surgical therapy, except prolactinoma which is typically responsive to dopamine agonists.
CRANIOPHARYNGIOMAS
Craniopharyngiomas are benign neoplastic lesions which are presumed to originate from the embryological remnants of Rathke's pouch. They may be located in the suprasellar region, within the sella, or both. Due to the infiltration of surrounding structures, they may have extensive adverse consequences. This tumor accounts for 1% of all intracranial tumors in adults and 6-13% of intracranial tumors in children (21). Peak incidence is in the first decade with a subsequent increase in incidence between 50-60 years of age. Adamantinomatous craniopharyngiomas commonly occur in children and typically contain both solid and cystic components containing a lipid rich secretion. By contrast, squamous papillary craniopharyngiomas develop in adults and are rare in younger patients. This type of tumor is predominantly solid tumor with small well-defined cavities containing less lipid rich fluid. Craniopharyngiomas in adults have a generally better prognosis in respect to endocrine, visual, and other neurological deficits than those in children (22).
RATHKE’S CYSTS
Epithelial cysts known as Rathke's cysts are thought to originate from the remnants of Rathke's pouch. Their location can be intrasellar, with or without suprasellar extension and, rarely purely suprasellar. Compared to craniopharyngioma, they are more common in adults (23). Possible symptoms of Rathke's cleft cysts include pituitary hypofunction, hyperprolactinemia brought on by stalk disruption, visual disturbance, and headache. Due to the difficulty in differentiation between craniopharyngiomas and Rathke's cysts from both a clinical and radiologic standpoint, the diagnosis of an intrasellar Rathke’s cyst cannot always be made with absolute certainty. Definitive diagnosis is often only made histologically. Only 6.4% of asymptomatic patients in a recent retrospective cohort showed cystic enlargement over 41 months follow-up and no pituitary deficiency, confirming the safety of conservative management in these patients (24).
PERI-PITUITARY TUMOURS
Peri-sellar meningiomas are well-circumscribed masses, originating from the sphenoid ridge and are associated with varying degrees of hyperostosis. Meningiomas are typically single, but they can occasionally be multiple. However, females are more likely to develop multiple meningiomas.
Primary intracranial germ cell tumors are neoplasms that arise from aberrantly migrated primordial germ cells. According to histology, the tumor types are similar to those occurring in the gonads and are classified into two categories: germinomas, which are comparable to seminomas and dysgerminomas, and non-germinomatous germ cell tumors, which can include teratomas, yolk sac tumors, embryonal carcinomas, and choriocarcinomas. Geographically, their incidence varies; it accounts for 2-3% of all childhood primary CNS tumors in Western countries, whereas the incidence is higher at 4-15% in Japan (25-27). Patients commonly present in the first two decades of life with diabetes insipidus, visual failure, and hypopituitarism. A recent report highlighted the favorable response to a combined chemotherapy-radiotherapy protocol (27).
Metastasis to the pituitary gland may present as an intrasellar mass resulting in hypopituitarism, diabetes insipidus, or pressure effects. In a published series of 500 consecutive autopsy examinations of cancer patients in whom the pituitary fossa and gland were examined, pituitary metastases were found in 3.6% (28). Pituitary metastases have been described in patients with primary malignant tumors of breast, lung, kidney, thyroid, bladder, uterus, pancreas, and colon.
Vascular Causes
The term pituitary apoplexy denotes the clinical consequences of hemorrhage or infarction in a pre-existing adenoma. The expansion of pituitary mass to neighboring cranial nerves, cavernous sinus, optic pathways, or diencephalon results in localizing signs or altered conscious state. It frequently manifests as a sudden onset of severe headache, a visual disturbance, or ophthalmoplegia as a result of cranial nerve III IV or VI palsies (29). The clinical syndrome of pituitary apoplexy usually evolves fully within only hours to two days. The incidence of surgically treated pituitary adenomas ranges from 0.6 to 9.1%, (30) with a broad age range of occurrence from the first to the ninth decade. Although apoplexy usually occurs spontaneously, systolic and/or diastolic hypertension was seen in 26% of patient in a retrospective analysis, making it an important predisposing factor. (31). Other reported predisposing factors include diabetes mellitus, radiation therapy, anticoagulant therapy, bleeding disorders, head trauma, sudden changes in arterial or intracranial pressure, such as during carotid angiography, the use of bromocriptine, and postpartum hemorrhage (Sheehan's syndrome). Sheehan's syndrome, which denotes pituitary necrosis after postpartum hemorrhage and hypovolemia, may result in hypopituitarism, that is either immediate or delayed over several years depending on the extent of tissue destruction. However, this condition is rare with modern obstetric care (32).
Suprasellar or intrasellar aneurysms of the carotid arteries, as well as suprasellar aneurysms of the anterior or posterior communicating arteries can manifest as an expanding mass inside the fossa. Hypopituitarism may result directly from intrasellar aneurysms. Histologically, typical degenerative sclerotic changes of the arterial wall are found along with chronic inflammatory changes. An arteriovenous fistula may occasionally develop in the cavernous sinus as a result of an aneurysm.
Immunological/Inflammatory Disease
Inflammatory lesions of the pituitary gland can clinically and radiologically mimic pituitary tumors, with varying degrees of hypopituitarism and mass effects like headaches and visual field impairment.
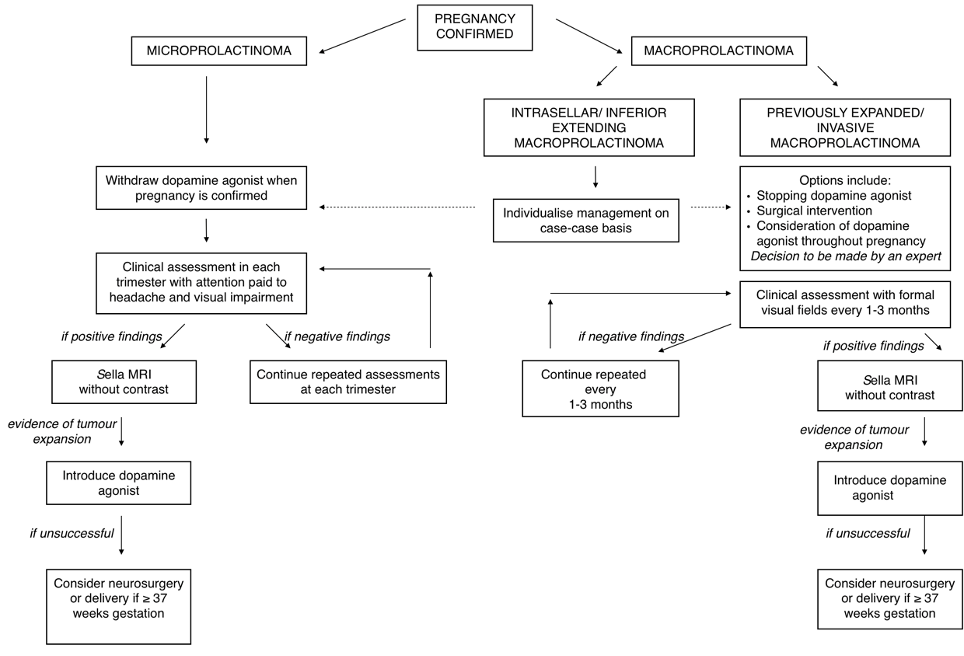
Lymphocytic hypophysitis classically presents as partial hypopituitarism associated with a pituitary mass lesion, particularly in relation to pregnancy (33). It has been speculated that many cases of postpartum pituitary failure, previously attributed to Sheehan's syndrome, may in fact have been caused by lymphocytic hypophysitis (34,35). In this condition, the interstitial tissue of the adenohypophysis is more or less densely infiltrated by lymphocytes and plasma cells. As a result, the anterior pituitary exhibits necrosis and subsequent fibrosis of hormone-producing cells. Since the histological findings are similar to those of other organ-specific autoimmune diseases, the primary etiology is considered to be autoimmune (36,37). Partial hypopituitarism, commonly with isolated ACTH deficiency, or with TSH deficiency, is seen in approximately 80% of patients, with LH and FSH generally remaining unaffected.
Sarcoidosis is a multisystem disorder, which is characterized by the presence of non-caseating granulomas. It most often affects lungs and reticuloendothelial system but can involve any organ. Central nervous system sarcoidosis occurs with an incidence of 5-15% (38,39) and less than 1% of patients have symptoms of a hypothalamic-pituitary disease (40). Neurosarcoidosis affects primarily the leptomeninges of the brain base and posterior fossa; the hypothalamus and/or pituitary subsequently experience local granulomatous infiltration. Coexisting optic nerve infiltration occurs in many patients causing central and peripheral visual defects. In terms of anterior pituitary function, LH, FSH and GH deficiency are frequent, but TSH and ACTH are relatively preserved.
Langerhans cell histiocytosis is a rare disease characterized by aberrant proliferation of a specific dendritic (Langerhans) cell belonging to the monocyte-macrophage system (41). Deposits occur at multiple sites within the body, and frequently involve the hypothalamo-pituitary axis (42). Diabetes insipidus is a well-recognized and common feature of this condition but anterior pituitary dysfunction also occurs frequently.
Ipilimumab, a cytotoxic T-lymphocyte antigen-4 (CTLA-4) inhibitor, a IgG1 monoclonal antibody against CTLA-4 is used for metastatic melanoma and other neoplasms. As a consequence of immune activation, secondary hypophysitis has been reported in 10-15% of ipilimumab treated patients (36). In severe hypophysitis, immune reactions induced extensive necrosis of the adenohypophyseal architecture. Pituitary autoantibodies against thyrotrophs, corticotrophs, and gonadotrophs were identified in patients with ipilimumab induced hypophysitis (37). Patients usually present with headache and fatigue, the diagnosis of hypopituitarism with multiple deficiencies is made 2-3 months or later after initiation of ipilimumab. Central hypothyroidism is the most frequent deficit, followed by central hypocortisolism, and hypogonadism.
Infections
Bacterial pituitary sepsis is a rare phenomenon which may arise as a consequence of hematogenous spread or by extension from sinus or meningeal sepsis with subsequent pituitary abscess formation. Small pituitary abscesses have been described at postmortem in patients dying from septicemia. Chronic infection predisposes to necrosis of pituitary tissue with subsequent hypopituitarism. Pituitary tuberculomas may present as space occupying lesions (43)but this appears to be a very rare phenomenon; isolated manifestation of the disease is rare and it is more likely to occur in the context of generalized tuberculous infection with meningitis. Gumma formation, as a manifestation of tertiary syphilis, may occur in the sella region but is extremely rare.
Fungal pituitary infection may occur as a complication of AIDS (44) and pituitary necrosis with hypopituitarism has been described in toxoplasmosis (45).
Radiation Therapy
Hypopituitarism is a common complication of irradiation administered for pituitary adenomas, head and neck tumors, intracranial malignancy, or as adjunctive cranial irradiation for acute lymphoblastic leukemia. Although fractionated radiotherapy with daily doses of <200cGy is well tolerated and safe in terms of neurological sequelae, the majority of patients who have undergone external irradiation will manifest some degree of pituitary failure during long term follow up. In general, this is a relatively late complication and is rarely evident in less than 3 years in patients with completely normal baseline pituitary function. Growth hormone reserve is particularly susceptible and is progressively more likely with time so that periodic assessment of residual pituitary function is mandatory; a five year follow up study after external pituitary irradiation therapy reported, 100% GH deficiency, 91% LH-FSH deficiency, 77% ACTH deficiency and 42% TSH deficiency (46).
Traumatic Brain Injury
Traumatic brain injury (TBI) is common, the prevalence of endocrine dysfunction in these patients ranges from 15-68% (47), with estimated annual incidence 30 patients per 100,000 population per year. Abnormal axes during the acute phase of injury may recover over time, but other pituitary hormone deficits may evolve later even at 6 months after the initial insult. A prospective longitudinal study looking at patients with severe TBI, found that 6% of their patients had proven severe GHD when evaluated after 12 months (48). Multiple pituitary deficiencies or isolated pituitary deficiencies (TSH or ACTH) were very rare.
Empty Sella Syndrome
An enlarged empty sella may be primary (due to a congenital diaphragmatic defect) or secondary to surgery, radiation, or pituitary infarction. The majority of patients with congenital empty sella have normal pituitary function. Hypopituitarism (and/or hyperprolactinemia) may be found in instances due to previous pituitary disease. Occasionally, a cystic pituitary mass may simulate an empty sella on CT or MRI, necessitating a cisternogram for precise delineation.
CLINICAL MANIFESTATION OF HYPOPITUITARISM
The clinical impact of pituitary insufficiency is dependent on the extent and severity of hormone deficiencies (Table 2), the duration of the disease and the age of onset; childhood onset hypopituitarism has consequences for all aspects of somatic development in addition to the pathophysiological effects of specific hormonal deficiencies. In addition, there may be clinical features relating to the mass effect of causative lesion or specific consequences attributable to hypersecretion of prolactin, GH, ACTH or TSH from individual tumor types. Hypopituitarism classically develops in sequential order with the secretion of growth hormone, then gonadotrophins being affected first, subsequently followed by TSH and ACTH. Prolactin deficiency is rarely seen, except in Sheehan's syndrome which is associated with failure of lactation. ADH deficiency is almost never seen as a primary feature of pituitary adenomas but is a usual presenting manifestation of germ cell tumors, pituitary metastases, and granulomatous disorders.
|
Table 2. Summary of Clinical Features of Hypopituitarism |
|
||
|
Hormone deficiency |
Presentation |
Symptoms and signs |
|
|
Adrenocorticotrophic hormone |
Acute |
Fatigue, weakness, dizziness, nausea, vomiting, circulatory failure. As in Addison's disease, except lack of hyperpigmentation, absence of hyperkalemia |
|
|
Chronic |
Tiredness, pallor, anorexia, nausea, weight loss, myalgia, hypoglycemia |
|
|
|
Gonadotropins |
Children |
Delayed puberty |
|
|
Men |
Impaired fertility, impotence, reduced libido, decreased muscle mass and strength, decreased bone mass, decreased erythropoiesis and hair growth, fine wrinkles, testicular hypotrophy |
|
|
|
Women |
Amenorrhea, oligomenorrhea, infertility, loss of libido, dyspareunia, fine wrinkles, breast atrophy, osteoporosis, premature atherosclerosis |
|
|
|
Thyroid-stimulating hormone |
Children |
Growth retardation |
|
|
Adult |
Fatigue, cold intolerance, constipation, weight gain, dry skin, slow relaxing reflexes |
|
|
|
Growth hormone |
Children |
Growth retardation, short stature, increased adiposity |
|
|
Adult |
Reduced exercise capacity, impaired psychological wellbeing, increased cardiovascular risk, increased central obesity, reduced lean body mass |
|
|
|
Prolactin |
Decreased |
Failure of lactation |
|
|
Increased |
Galactorrhea, oligo/amenorrhea, loss of libido |
||
|
Antidiuretic hormone |
|
Polyuria, polydipsia including nocturnal |
|
The clinical features of anterior pituitary hypofunction described in this chapter will concentrate on adult-onset hypopituitarism. Many of the symptoms and signs of a specific pituitary hormone deficiency are similar to those that occur in patients with a primary deficiency of the target gland, but there are exceptions.
Growth Hormone Deficiency
GH deficiency (GHD) in adults is characterized by decreased exercise tolerance, decreased mood and general well-being, decreased quality of life, central adiposity, hyperlipidemia, increased predisposition to atherogenesis, and reduced bone remodeling activity. Fine facial wrinkles may result from a deficiency of growth hormone in addition to hypogonadism. The patient will usually have dry thin skin which contrasts with the thickened skin and increased sweating found in acromegaly. Adults with long-standing GHD are often overweight, have reduced lean body mass, increased fat mass, especially visceral fat, relative insulin resistance, and reduced total bone mass. There is a reduction in cardiac and physical performance. GHD is associated with increases in total cholesterol, LDL-cholesterol and apolipoprotein B (49-51). Studies in Sweden and the UK in patients with hypopituitarism receiving controlled thyroid and steroid hormone replacement but without growth hormone replacement, have demonstrated an approximately two fold increase in cardiovascular mortality compared to the general population. (51) and the increase in standardized mortality ratio is more striking in females (52).
The accumulating evidence suggests that the cardiovascular morbidity cannot be explained solely by suboptimal glucocorticoid, gonadal steroid, or thyroid hormone replacement and unsubstituted growth hormone deficiency is probably an important contributing factor. The decreased bone mineral density found in GHD is associated with increased fracture risk (53).
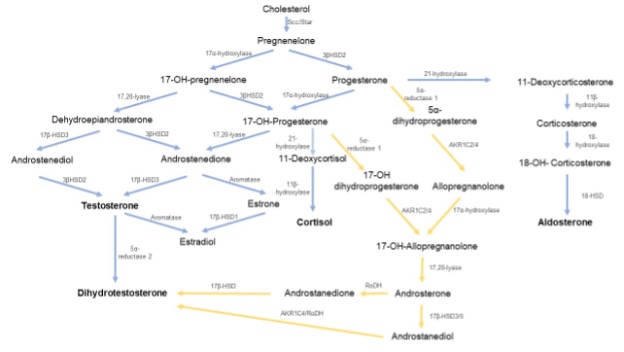
Adrenocorticotrophic Hormone Deficiency
Cortisol and adrenal androgen secretion are ACTH-dependent and are variably decreased in hypopituitarism. Patients with combined ACTH and LH deficiency are completely androgen deficient, a phenomenon that may be particularly important when considering optimum regimens for gonadal steroid replacement in females. Since aldosterone secretion is largely determined by activation of the renin-angiotensin-aldosterone system, it is relatively preserved in the hypopituitary patients. Major symptoms of ACTH deficiency are non-specific, including fatigue, weakness, headache, anorexia, weight loss, nausea, vomiting, abdominal pain, myalgia, and decreased concentration. Hypoglycemia may be present at diagnosis. Hyponatremia is common and is attributable to reduced renal free water clearance as a result of cortisol deficiency with an additional contribution from TSH deficiency if present. Hyperkalemia, which is a frequent finding in primary adrenal failure, does not occur in patients with hypopituitarism due to the relative preservation of aldosterone secretion. Over and above the effects of secondary hypogonadism, reduced adrenal androgen production further exacerbates loss of body hair, particularly in women.
Gonadotrophin Deficiency
Gonadotrophins are responsible for gonadal sex-steroid production, secondary sexual development, maintenance of secondary sexual characteristics, and fertility.
Gonadotrophin deficiency in males’ results in secondary hypogonadism with consequent testosterone deficiency. Clinical features include loss of libido, erectile impotence, oligospermia, reduced erythropoiesis, and decreased lean body mass. Testosterone, via estradiol derived by aromatization, plays an important role in the regulation of bone mineralization, therefore, hypogonadism results in decreased bone mineral density.
Clinical features of secondary hypogonadism in females include oligo/amenorrhea, breast atrophy, diminished secondary sexual hair especially when combined with ACTH deficiency, and a predisposition to osteoporosis.
Thyroid Stimulating Hormone Deficiency
The clinical features of secondary thyroid failure are similar to those of primary thyroid failure with the exception that weight gain is less likely to be a feature if ACTH deficiency is also present. Classical features include cold intolerance, fatigue, and myalgia. Physical examination may demonstrate periorbital edema and delayed reflex relaxation. Additionally, hyponatremia and normochromic normocytic anemia are observed, with the former being exacerbated by cortisol deficit while the latter is aggravated by secondary hypogonadism and GH deficiency.
Prolactin Deficiency
Prolactin deficiency in females result in puerperal alactogenesis, However, isolated prolactin deficiency is uncommon because the great majority of prolactin deficiency states develop as a result of general anterior pituitary problems or as a result of treatment intervention. On the other hand, hyperprolactinemia, which is caused by stalk disruption and results in a lack of dopaminergic regulation from the hypothalamus, is more frequent and can manifest as galactorrhea, abnormal menstruation, loss of libido, and decreased bone density.
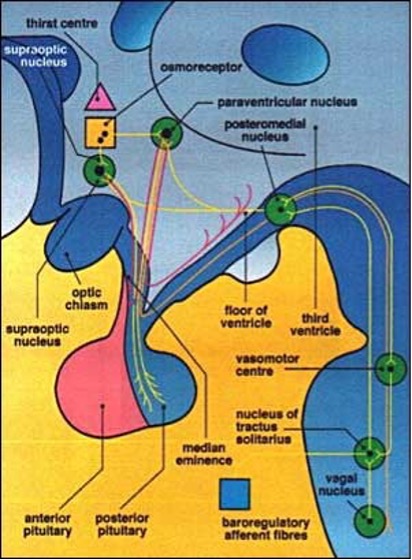
Antidiuretic Hormone Deficiency
Antidiuretic Hormone Deficiency (AHD) previously called central diabetes insipidus (CDI) is characterized by polyuria and polydipsia due to decreased secretion of antidiuretic hormone by the neurosecretory cells terminating in the posterior pituitary. If excessive water excretion exceeds intake, patients will progressively become water-depleted and eventually have a reduction in circulating volume. It occurs more frequently in associated with tumors of the hypothalamus, pituitary metastases, lymphocytic hypophysitis, sarcoidosis, Langerhans cell histiocytosis, craniopharyngiomas, and Rathke's cleft cysts. (54). Additionally, it is commonly observed following neurosurgery and head injury. As cortisol is an essential prerequisite for normal glomerular filtration and free water clearance, it should be highlighted that AHD is masked by co-existing ACTH deficiency and only becomes clinically evident after commencement of glucocorticoid replacement. However, it is extremely unusual for AHD to be a primary manifestation of pituitary adenoma.
INVESTIGATION OF SUSPECTED HYPOPITUITARISM
In hypopituitarism, basal serum hormone measurements are required to confirm the insufficiency; however, dynamic testing is mandatory for the diagnosis of some deficiencies. Pituitary function testing is required for all patients presenting with pituitary disease or in whom an evolving endocrine deficiency is anticipated, e.g., those who have undergone pituitary or cranial radiotherapy.
Baseline Investigations
Basal concentrations of the anterior pituitary hormones and hormones produced by their respective target glands should be measured. Serum samples should be taken unstressed, with no physiological or pharmacological manipulation, between 7 and 9 AM when serum cortisol and testosterone levels are highest. This is important given that the decision to proceed to
testing is based on these levels. The pituitary hormones may remain within the normal range despite low levels of the target hormones indicating that target gland failure is a consequence of inadequate pituitary stimulation. Baseline investigations (Table 3) are sufficient for the diagnosis of secondary hypothyroidism and hypogonadism and will also confirm virtually complete ACTH deficiency (55).
|
Table 3. Baseline investigation of pituitary function |
|
1. Adrenocortical axis: serum cortisol (09.00 AM), ACTH Men - testosterone (09.00 AM), SHBG, LH, FSH; Women - estradiol, LH, FSH, progesterone (Day 21 if menstruating) |
ACTH, adrenocorticotropic hormone, FSH, follicle stimulating hormone; LH, luteinizing hormone; SHBG, sex hormone binding globulin
It should be mentioned that the serum total testosterone measurement should be performed in the morning by a reliable assay and the test should be repeated before verifying the diagnosis of testosterone deficiency. Measuring the level of free or bioavailable testosterone level should be performed in those whose total testosterone levels are close to the lower limit of the normal range or whose SHBG is suspected to have altered (56).
Dynamic Testing
Stimulation tests are used when hypofunction is suspected and are designed to assess the reserve capacity to form and secrete hormone. In contrast, suppression tests are used when endocrine hyperfunction is suspected and are designed to determine whether negative-feedback control is intact, as in the administration of glucocorticoids to inhibit corticotrophin secretion in patients with suspected Cushing’s syndrome or in the administration of glucose in patients with suspected acromegaly. Dynamic pituitary function tests may assess the hypothalamic-pituitary unit (e.g., insulin tolerance test, glucagon, and arginine tests) or directly stimulate the anterior pituitary with pharmacological doses of synthetic hypothalamic peptides and the pituitary hormone response measured (e.g., TRH, GnRH, GHRH tests).
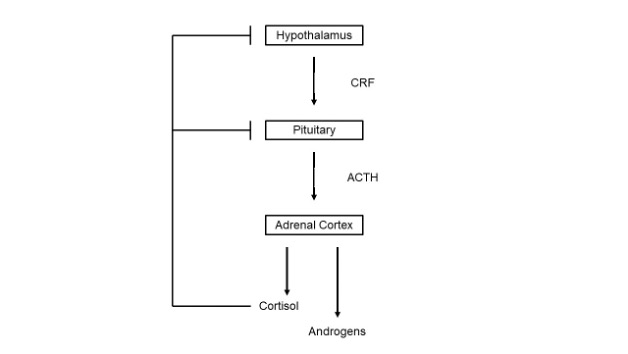
HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
Baseline and dynamic tests are valuable in the diagnosis of ACTH deficiency. With virtually complete ACTH deficiency, the 0900h serum cortisol is less than 100 nmol/L (3.625 µg/dL). In contrast, if the serum cortisol is 400-500nmol/L (14.5 – 18.1µg/dL) or more, ACTH deficiency is unlikely. (57-59). Therefore, dynamic testing of ACTH reserve is required if the basal serum cortisol measured lies between 100 and 400-500nmol/L. The insulin tolerance test (ITT) or glucagon test may be used to assess the adequacy of the hypothalamic-pituitary-adrenal axis. If the patient is taking hydrocortisone this should be discontinued for 18-24 hours; while prednisolone cross reacts in the cortisol assay and therefore should not be administered within 24 hours of investigation of adrenocortical reserve. The rationale of the ITT is to produce physiological stress in a controlled environment by inducing hypoglycemia with intravenous insulin. Hypoglycemia is a powerful stimulus which stimulates GH and ACTH release and a rise in serum cortisol levels in the presence of an intact hypothalamic-pituitary axis. Although the safety of ITT has been questioned, the only absolute contraindications are ischemic heart disease, epilepsy or unexplained loss of consciousness, untreated hypothyroidism or hypoadrenalism, and glycogen storage disease (58). A decrement in plasma glucose to less than 2.2 mmol/L (40 mg/dL) is required for the test to be valid. With normal ACTH reserve the serum cortisol should rise to at least 550 nmol/L (20 µg/dL). Patients who show a normal cortisol response can withstand major surgery without corticosteroid replacement; while patients with subnormal responses but satisfactory basal values (> 250 nmol/L (9 µg/dL) may not require regular replacement therapy but should be fully informed and carry a steroid card. The ITT should only be performed by fully trained staff in designated units. Hypoglycemia may occasionally require reversal with intravenous glucose to avoid the risk of cerebral edema, which affects 50% of adults but limited to 10% of children.
The glucagon stimulation test (GST) may be used for the assessment of ACTH/cortisol and GH reserve simultaneously when the ITT is contraindicated (60). The subcutaneous injection of glucagon causes a transient rise in plasma glucose. During the subsequent fall in plasma glucose, ACTH and GH are released and measured. Serum cortisol cut-off values are similar to those described for the ITT. Glucagon is less reliable than the ITT as a test of ACTH/ cortisol reserve; it is a less powerful stress stimulus and hence false positive results are a recognized problem.
The short synacthen (Cortrosyn in the USA) test (SST) was originally introduced as a test for primary adrenal failure. It involves the intramuscular or intravenous injection of a pharmacological dose (250μg) of synthetic ACTH, with measurement of the serum cortisol response at 30 and 60 minutes later. The test does not distinguish primary from secondary adrenal insufficiency, and it cannot directly assess pituitary ACTH reserve. The 30-minute serum cortisol response is advocated as a surrogate test of ACTH reserve and has been widely used because of its simplicity. However, there is no study showing that a normal SST indicates that the hypothalamic-pituitary-adrenal axis is capable of responding normally to major illness or stress. Demonstrations of good correlations between peak serum cortisol responses on ITT and 30-minute responses on SST have been published and are intuitively predictable. However, false negatives with the SST are well recognized although this may be partially obviated by the use of lower synacthen doses (1μg). Nonetheless, the concern about false negative results and the fact that assessment of GH reserve is also frequently required serve to limit the use of the SST in the investigation of pituitary function.
A recent study comparing the ITT, low dose ACTH, and glucagon stimulation test in the evaluation of the HPA axis and GH-IGF-1 axis in patients with pituitary disorders concluded that all three tests were well correlated in terms of peak cortisol and GH response (61). However, low dose ACTH stimulation gave a higher peak cortisol response. Therefore, the cut-off level for the diagnosis of insufficiency of the HPA axis needs to be individualized for each test. See Chapter Adrenal Insufficiency
GROWTH HORMONE
Normal GH secretion is pulsatile, with four or six pulses per 24 hours, mostly at night in association with REM sleep. A single measurement of serum GH is rarely useful although the fortuitous coincidence of blood sampling at the time of a GH secretory peak may exclude GHD. A stimulatory test is therefore usually required to assess somatotroph reserve. Most, actions of GH are mediated through hepatically or locally-derived insulin-like growth factor-1 (IGF-1). Measurement of baseline serum IGF-1 is a specific but insensitive test of GHD in patients with pituitary disease. Importantly, IGF-1 declines with normal aging. In adult onset GHD, 30% of patients may have serum IGF-1 levels in the lower half of the age-related reference range, with the percentage increasing with age (62). Therefore, a low serum IGF-1, in an adequately nourished patient without liver dysfunction, strongly supports a diagnosis of GHD, but a normal serum IGF-1 cannot exclude it.
A rigid and precise biochemical diagnosis of GHD is required in the context of justification of growth hormone replacement. The ITT is one of the most reliable provocative tests of GH secretion and currently remains the test of choice for this purpose since it has the advantage of assessing ACTH reserve simultaneously. Severe GHD is defined as a peak GH response to insulin induced hypoglycemia of less than 3ng/mL (9mU/L) (63). Obesity may blunt the GH response to dynamic testing so that the diagnosis of GHD should only be made in the context of structural pituitary disease or previous cranial irradiation and/or additional pituitary hormone deficits.
If the ITT is contraindicated, an alternative test for assessment of GH reserve is required. Glucagon, arginine, a combination of arginine and GHRH or growth hormone-releasing peptides may all be used for this purpose. The combination of GHRH plus arginine is the most powerful provocative test of GH secretion but appropriate normative data are required (64-66). Recently, the FDA approved macimorelin, which is a non-peptidyl agonist of GH secretagogue receptor 1a as a diagnostic agent for diagnosing GH deficiency, and macimorelin stimulation test was known to have good sensitivity and specificity in comparison with ITT (67). However, this oral agent is not widely used due to its cost and limited availability.
PITUITARY-THYROID AXIS
In the appropriate clinical context, secondary hypothyroidism can be diagnosed on the basis of a low serum thyroxine (T4, total or free) in the presence of low or low normal TSH (TSH is rarely undetectable in hypopituitarism). However, it should be borne in mind that any systemic illness may produce a reversible reduction in serum T4 (sick euthyroid state).
Dynamic testing using thyrotrophin releasing hormone (TRH) has no diagnostic value for secondary hypothyroidism or predicting a risk of developing TSH deficiency. Although patients with hypothalamic disease may show a delayed response to TRH, this test is seldom used in pituitary reserve assessment. Furthermore, intravenous TRH may precipitate hemorrhagic infarction of pituitary adenomas.
PITUITARY-GONADAL AXIS
Symptoms of sex steroid deficiency, menstrual disturbance, low serum estradiol or low serum testosterone levels in the presence of normal or low concentrations of FSH/LH are the mainstay of diagnosis of hypogonadotropic hypogonadism. The gonadotrophin releasing hormone (GnRH) test has virtually no diagnostic value but is used to confirm gonadotroph reserve in the setting of pulsatile GnRH therapy for infertility.
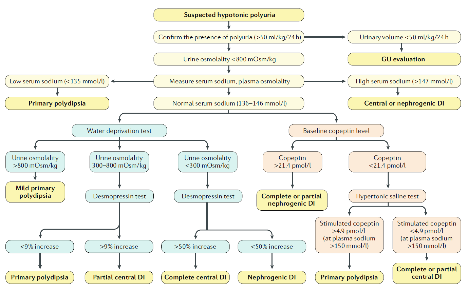
POSTERIOR PITUITARY FUNCTION
Basal investigations include plasma and urine osmolalities obtained simultaneously. In overt AHD, plasma osmolality is usually raised in the presence of inappropriately diluted urine. The diagnosis of partial AHD is confirmed by means of a water deprivation test followed by demonstration of a response to desmopressin. Copeptin is the most recent clinical diagnostic marker for AHD due to its strong correlation with plasma arginine vasopressin (AVP) (68). Copeptin is secreted in equimolar ratio to AVP, mirroring AVP concentrations in the circulation. A copeptin level of 4.9pmol/L stimulated with hypertonic saline infusion differentiates between AHD and primary polydipsia with a high diagnostic accuracy and is superior to the water deprivation test (69). See Chapters on Posterior Pituitary.
Imaging the Pituitary
Magnetic resonance imaging (MRI) is the optimum method of imaging, with computerized tomography (CT) as an acceptable alternative. The only disadvantage to MRI is its insensitivity in defining pathological calcification and lack of signal from corticated bone. CT may be required to demonstrate calcification in craniopharyngiomas and hyperostosis; it is also used by many surgeons to define skeletal anatomy prior to surgery.
MANAGEMENT OF HYPOPITUITARISM
Hypopituitarism, once established, is usually permanent. However, resection of pituitary tumors may, on occasion, result in resumption of normal pituitary function.
Although a seemingly straightforward clinical exercise, hormone replacement therapy cannot simulate normal physiology precisely. With the exception of GH replacement and treatment of infertility, replacement therapy is achieved by administering target hormones. The aim of hormone replacement is to safely eliminate or minimize the symptoms and clinical signs of specific hormone deficiencies. The Endocrine Society Clinical Practice Guidelines committee have issued recommended treatment option of each target hormone deficiency either primary or secondary for the management of hypopituitarism (70).
Hypocortisolism
Hydrocortisone is now the most widely used form of glucocorticoid replacement in patients with primary and secondary adrenal insufficiency. There is no universal agreement regarding the appropriate dose, timing, and monitoring of hydrocortisone replacement. Normal individuals demonstrate undetectable serum cortisol and ACTH when asleep at midnight, with a rise during the early hours of the morning to reach a peak at 0800-0900h, followed by a steady decline throughout the rest of the waking day. There are variable peaks of cortisol secretion due to other factors such as stress, meals, and exercise. Since cortisone acetate requires conversion into hydrocortisone under the influence of 11ß-hydroxysteroid dehydrogenase type 1 (11β-HSD1), and the fact that the enzyme activity is altered in patients with GHD; therefore, cortisone renders intuitively less satisfactory than hydrocortisone for replacement purposes in this condition. Prednisolone has been advocated by some on account of its longer duration of action than hydrocortisone. However, because it cannot be routinely measured, it is impossible to fine tune the replacement dose. Dexamethasone, the alternative synthetic glucocorticoid replacement, is even less satisfactory because of wide interindividual variations in metabolic clearance rates.
Traditional hydrocortisone replacement utilized twice daily dosing but many patients report fatigue or headache in the afternoon on this regimen. There is evidence that many patients 'feel better' on thrice daily regimes (71). Recent cross-sectional studies did not demonstrate any superiority between three times a day versus twice daily replacement in terms of quality of life (72). The average daily requirement is approximately 20mg of hydrocortisone. This should be given as 10mg on waking, 5mg at lunchtime and 5mg in the early evening. Enzyme-inducing drugs, especially, phenytoin, carbamazepine, and rifampicin can increase the metabolism of corticosteroids and should prompt an increment in replacement doses. Doses are therefore fine-tuned according to patient’s well-being and multiple serum cortisol levels (hydrocortisone day curve, HCDC) taken during the day in many centers. (55,73). Some centers have attempted to utilize urine free cortisol measurements to adjust the hydrocortisone dose; however, this is unreliable because saturation of cortisol binding globulin (CBG) following oral hydrocortisone results in supraphysiological urine free cortisol excretion (74). The HCDC should demonstrate adequate levels of cortisol throughout the day, without excess peak (cortisol e.g., >1000 nmol/L (36.25 µg/dL)) or trough (e.g., <100 nmol/L (3.625 µg/dL)) levels before or after doses.
In clinical practice, there is no reliable measure to be certain if patients are receiving optimal glucocorticoid replacement therapy. As a result, patients may be over- or under- treated with resultant morbidity (65). There is a significant increase in 11β-HSD1 activity resulting in abnormalities in corticosteroid metabolism in patients with ACTH deficiency treated with conventional doses of hydrocortisone (66). In ACTH-deficient patients daily hydrocortisone dose exceeding 20mg/day is associated with increase waist to hip ratio. The induction of 11β-HSD1 is associated with central adiposity and has an important role in the development of the metabolically adverse hypopituitary phenotype.
Conventional hydrocortisone cannot mimic the circadian rhythms of cortisol release; in particular the early morning rise in cortisol which slowly declines throughout the day. This has led to the development of a modified release formulation of hydrocortisone (MR-HC, Chronocort®) which can be taken late at night thus allowing a delayed and sustained release (75). A study in healthy men demonstrated that MR-HC 20mg and 10mg, given at 2300 and 0700 hours respectively, could achieve a near normal cortisol circadian rhythm (76). A subsequent phase II study demonstrated that the MR-HC mimics the normal circadian pattern closer to physiological baseline (77); however, the studied preparation is no longer available. An alternative modified formulation (Plenadren®) incorporates an immediate release and delayed release components, which displays diurnal plasma cortisol levels similar to physiological profile. Once daily Plenadren® has been shown to reduce weight, blood pressure and improve glucose metabolism when compared with thrice daily dosing (78).
Another newer oral immediate-release granule formulation of HC (Alkindi®) was approved in EU in 2018. This is an immediate-release hydrocortisone preparation specifically designed for pediatric dosing, which is available in four doses: 0.5mg, 1mg, 2mg and 5mg (79).
Table 5 summarizes the glucocorticoid compounds currently available for adrenal insufficiency.
|
Table 5. Compounds Available for Adrenal Insufficiency |
||
|
Glucocorticoid |
Timing |
Dose |
|
Hydrocortisone |
2-3 times/day; waking, early PM, and at least 6 hours from bedtime |
10-20mg |
|
Prednisolone |
Once in morning |
3-5mg |
|
Modified release hydrocortisone (Chronocort®) |
2times/day; waking and before bed |
10mg in AM and 20mg at bedtime |
|
Modified release hydrocortisone (Plenadren®) |
Once daily in early morning |
Equal to standard HC dose or a 20% increase |
|
Oral immediate release granules (Alkindi®) |
Once daily in early morning |
Equal to appropriate pediatric hydrocortisone dosing |
The effects of continuous subcutaneous hydrocortisone infusion (CSHI) have been compared with conventional oral hydrocortisone in patients with Addison’s disease (80). CSHI produced a more physiological circadian rhythm, normalization of morning ACTH and restoration of nocturnal serum cortisol levels. These infusions are cumbersome and impractical outside of expert centers. Despite a more circadian pattern of cortisol exposure, subcutaneous hydrocortisone infusion was not associated with improved quality of life scores, casting doubt on the potential quality of life effects of circadian cortisol delivery (81).
There is a push to design orally active delayed or sustained release formulations of hydrocortisone to aid the physiological replacement of hydrocortisone and ultimately improve quality of life and side effect profiles in patients requiring lifelong glucocorticoid replacement.
Patients with ACTH deficiency receiving hydrocortisone therapy are unable to respond to surgery, trauma, infections, and severe illnesses by increasing their cortisol concentrations. They therefore require supplemental hydrocortisone therapy with increased oral doses during minor illness, or administration of intramuscular hydrocortisone 100mg four times per day with more severe disease or if oral intake is compromised. When intramuscular injections are contraindicated, a continuous intravenous infusion of hydrocortisone at a rate of 1-3mg per hour provides satisfactory replacement for the severely ill patient. Maintenance mineralocorticoid is not required since aldosterone secretion is usually preserved.
Patients should carry a 'steroid card' and wear a 'medic-alert' bracelet to indicate their requirement for supplemental hydrocortisone in the event of severe illness or trauma. They and their families should understand the importance of life-long compliance, be taught to double the hydrocortisone dose in the event of pyrexial illness, and understand the need for parenteral glucocorticoid replacement if vomiting or diarrhea occurs. An 'emergency' ampoule of hydrocortisone should be provided for domiciliary emergency intramuscular injection and the patient instructed on its use.
Dehydroepiandrosterone (DHEA) is an androgen produced by the adrenal cortex and is also under the regulation of corticotrophin, and is thus deficient in hypopituitarism. In hypopituitarism, DHEA supplementation (25-50mg per day) has shown benefit with respect to well-being and sexual function (82,83). Furthermore, in patients who are replaced with GH, DHEA can augment the IGF-1 response hence leading to a reduction of GH dose in females (84). Currently, there is no licensed preparation of DHEA available, and it is considered to be a food supplement rather than a bioactive drug. However, not all patients respond. Moreover, the androgenic side effects such as greasy skin, acne, and increased body hair may be a limiting factor although generally responsive to dose reduction.
Secondary Hypothyroidism
Synthetic levothyroxine sodium (for example, Synthroid) is the preferred form of replacement. It has a long half-life, allowing a once daily dose. Liothyronine (T3) displays superior gastrointestinal absorption, but its short half-life requires two to three daily doses. Use of T3 is largely restricted to thyroid cancer patients undergoing frequent isotopic imaging or treatment, and occasionally the initiation of thyroid hormone replacement when a gradual increase is desired. Although some experts consider using a T4/T3 combination as an experimental treatment for patients with persistent hypothyroid symptoms on LT4 since patients with normal thyrotropin levels may have lower T3 levels, the evidence is primarily limited to athyreotic patients.(85,86)
Commencing thyroid hormone replacement in patients with severe, untreated ACTH deficiency may result in hypoadrenal crisis. In the situation of combined deficiency, hydrocortisone should always be commenced before thyroxine. The duration of hypopituitarism and presence of co-morbidities, especially ischemic heart disease, should be considered. Young patients with a short history of hypopituitarism and TSH deficiency may commence an initial thyroxine dose of 100µg daily. On the other hand, in patients with a long history of hypopituitarism or elderly patients, a low dose of 25-50µg daily should be commenced, in order to minimize the risk of precipitation of cardiac events.
Alteration of the hypothalamic-pituitary-thyroid axis following growth hormone replacement is well documented (87-89). GHD will mask central hypothyroidism in a significant proportion of hypopituitary patients both in children and adults. It has been observed that apparently euthyroid hypopituitary patients will require commencement or an increase in thyroxine replacement following initiation of GH replacement. A higher target serum free T4 in the upper half of reference range is appropriate in the GH deficient patient who is not on GH replacement.
Unlike primary hypothyroidism, in which serum TSH is a sensitive marker of under-or over-replacement, there is no biochemical marker to indicate the optimum level of replacement in TSH deficiency. The serum free T4 is the best marker for assessing replacement adequacy in hypopituitarism. By analogy with serum T4 levels in adequately replaced primary hypothyroidism, a conventional recommendation is to maintain serum free T4 in the upper part of the reference range for normal individuals (90). Serum total T4 levels are elevated artefactually by conditions which increase serum thyronine binding globulin, especially estrogen administration.
Gonadotrophin Deficiency
Choice of replacement ranges from oral, transdermal, intramuscular, or subcutaneous administration of gonadal steroids to gonadotropin or gonadotropin-releasing hormone therapy if and when fertility is desired.
WOMEN
Estrogen replacement should be offered to all women with secondary hypogonadism under the age of 50 years in order to avoid immediate symptoms of estrogen deficiency and prevent premature reduction in bone mineral density. The addition of progesterone is mandatory if the uterus is intact in order to avoid unopposed estrogen stimulation of the endometrium with the attendant risk of hyperplasia and neoplasia. The standard regimen for replacement involves the daily administration of estrogen with progesterone co-administrated for 12-14 consecutive days during a 4-week cycle; menses occur cyclically after progesterone withdrawal. Alternatively, a continuous regimen may be employed in which estrogen and progesterone are combined. The latter may be preferred by older patients and there are no adverse effects described apart from unpredictable menstrual bleeding during the initial few months of therapy in a minority of patients. However, since the risk-benefit profile of estrogen therapy is influenced by numerous factors, such as age, treatment-onset since menopause, and existing comorbidities, shared decision making is essential when determining what and route to administer as well as when to stop this treatment.
Estrogen replacement can be administrated via the oral, transdermal, and subcutaneous routes. Oral estrogens undergo extensive hepatic first-pass metabolism necessitating average doses of 1-2mg estradiol per day, or equivalent.
Transdermal preparations are usually applied twice weekly and provide 50-100µg mcg of estradiol per 24 hours in a cyclical combination with a progestogen. Skin irritation may occur but transdermal therapy is the first choice in patients with complex pituitary disease since it avoids the effects of oral estrogen on other hormone binding proteins. Furthermore, in women on concomitant GH replacement, IGF-1 generation is greater when transdermal rather than oral estrogen is used, therefore decreasing the dose of GH required. Subcutaneous implants are inserted every six months, but tachyphylaxis is a frequent problem and limits the value of this regimen.
In addition to relieve vasomotor symptoms and reduce risk of bone loss, hormone therapy may be prescribed on the market to seeking for cardioprotection. According to recent research from the combined Women’s Health Initiative (WHI) studies, women who start hormone therapy closer to the period of natural menopause have a decreased risk of coronary heart disease than women who wait longer after menopause. However, this replacement is not advised for either primary or secondary cardiovascular disease prevention (91).
Concerning the negative effects of estrogen replacement, it has been well established that estrogen, particularly oral estrogen, increases the risk of venous thromboembolism; although transdermal hormone therapy does not seem to enhance this risk (92). In terms of breast cancer, risk varies depending on hormonal formulation and length of treatment. In light of this, selective estrogen receptor modulators (SERMs) have an important role and have obtained licensing for this purpose. Raloxifene has very little effect on the vasomotor symptoms of estrogen deficiency, but is beneficial in prevention and treatment of osteoporosis as well as chemoprevention of breast cancer. Tibolone, an agent with estrogenic, progestogenic, and weak androgenic activity, can provide an alternative treatment for postmenopausal symptoms and also exerts favorable effects on bone.
In some patients with combined LH/FSH and ACTH deficiency, low libido may persist despite conventional estrogen and progesterone replacement. This could be a consequence of complete androgen deficiency and some studies have shown that low dose testosterone replacement, e.g., 50-100mg subcutaneous implants every 6 months, is effective for treating hypoactive sexual desire disorder. The guideline also recommends considering a 6-month therapeutic trial of transdermal testosterone for women who fit this diagnosis (93). Oral combinations of estradiol and testosterone are also available. FSH and LH injections are necessary for fertility; recombinant forms of both are now available and this treatment is usually supervised through fertility clinics.
MEN
Apart from relief of the symptoms of hypogonadism, androgen replacement is also important in maintaining bone integrity, muscle mass, and normal erythropoiesis (94).
The most common method of androgen replacement is as an intramuscular depot injection of testosterone ester (e.g., testosterone enanthate, 250mg intramuscularly three weekly). The ensuing supraphysiological peaks and troughs of serum testosterone may lead to fluctuations in mood, libido, and energy levels but treatment is generally very well tolerated. Depot testosterone injection has become available; testosterone undecanoate (Nebido), can be administrated intramuscularly every three months (95), and maintains physiological testosterone levels without major fluctuation.
Oral testosterone undecanoate is administered two or three times a day. It is extensively metabolized to dihydrotestosterone in the intestine and is absorbed via the lymphatic system. It is generally well tolerated and is most useful in patients with partial hypogonadism or in those who are unable to tolerate depot injections.
Testosterone pellets, implanted subcutaneously at a dose between 400-600mg, will provide normal testosterone levels as well as physiological levels of estradiol and dihydrotestosterone for up to six months (96). Peak serum testosterone levels are seen 2-4 weeks after placement with a gradual decline thereafter. The main disadvantage is the need for a skin incision and the occasional complication of local infection and extrusion of the pellets.
Several other systems for testosterone delivery are available and include patches (97) and gels (98) applied to the skin and buccal bioadhesive tablets (99). Transdermal gel (Testogel, Testim) has become very popular; it must be applied daily to maintain desirable serum testosterone levels. Patches can cause skin irritation in approximately 50% of patients. Buccal testosterone (Striant) is placed on the buccal mucosa above the incisor tooth; testosterone is slowly released over 12 hours. However, gum irritation and inconvenience have been reported.
Induction of spermatogenesis requires injections of FSH and LH; our own practice is to administer FSH 300 units three times weekly and LH 1500 units twice weekly; increases in sperm density are not evident for at least 4 months.
Monitoring Testosterone Replacement
Testosterone levels can be measured in blood as a guide of the adequacy of replacement. With intramuscular depot injections, serum testosterone usually peaks at approximately one week after injection with a nadir prior to the next injection. The nadir serum testosterone concentration should approximate the lower end of the normal reference range and this may require adjustment of the frequency of injections. Random serum testosterone is often low or low normal on oral testosterone undecanoate, but the additional measurement of serum dihydrotestosterone is useful.
Published guidelines (100) addressed the concerns of the risk for either benign or malignant prostate disease. There is no evidence that the incidence of prostate carcinoma in patients on testosterone replacement is greater than background population risk. Referral to a urologist is recommended if the patient has prostatic symptoms or an abnormal digital rectal examination or elevated serum prostate specific antigen (PSA) increasing by more than 1.4ng/mL over 12 months. With the increasing use of long-acting testosterone, it is important to monitor the hematocrit to detect polycythemia. The hematocrit should return to normal before testosterone may be reinstituted at a lower dose.
The available evidence suggests that testosterone replacement should be offered cautiously in patients with a low risk of recurrence for prostatic neoplasm who have been treated with radical prostatectomy and in whom the PSA has normalized (101). Similarly, a retrospective study of patients treated with prostatectomy as primary management with a low PSA at baseline of testosterone therapy and concurrent use of 5α reductase reported that there was no increase in PSA at 15 months (56). However, patients with no definitive surgery treated with brachytherapy or external beam radiation and a raised PSA at baseline should not be offered this treatment.
Growth Hormone Deficiency
The rationale and protocol for growth hormone replacement in adults is discussed in detail in the "Adult Growth Hormone Deficiency" chapter.
Diabetes Insipidus
1-desamino-8-D-arginine-vasopressin (DDAVP) is a synthetic analogue of arginine vasopressin which produces prolonged antidiuresis after intravenous, intranasal or oral administration in patients with AHD. A therapeutic trial of DDAVP, 10-20 mcg intranasally should control polyuria for up to 16 hours. Patients with AHD must have instant improvement in symptoms. In the acute clinical setting, for example where diabetes insipidus follows pituitary surgery, DDAVP is best administrated via the subcutaneous route at a dose of 0.5-1μg. Doses of DDAVP that are too high can lead to hyponatremia if patients continue to drink inappropriately despite antidiuresis. Patient education is required to achieve optimum symptom control particularly at night and to maintain a normal serum osmolality and sodium concentration (102). Slight undertreatment, with normal water homeostasis being maintained by thirst mechanisms, is the preferred approach. Many patients demonstrate adequate control of the condition with a single bedtime dose of intranasal DDAVP but an additional morning dose may be required. Mild degrees of AHD may be treated with oral DDAVP up to 600µg daily in divided doses. If ADH deficiency is accompanied by a reduced thirst threshold, it is most important to monitor body weight and urine output on a fixed dose of DDAVP and adjust fluid intake accordingly.
A recent guideline has been developed for inpatient treatment of diabetes insipidus highlighting that this can develop into a life-threatening situation if inappropriately managed (103). This again highlights that there is an increased risk of mortality and adverse outcomes in patient with hypopituitarism admitted for acute medical conditions especially ones with diabetes insipidus (104).
GUIDELINES
Wierman ME, Arlt W, Basson R, Davis SR, Miller KK, Murad MH, Rosner W, Santoro N. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014 Oct;99(10):3489-510.
Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, Samuels MH. Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016 Nov;101(11):3888-3921.
Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018 May 1;103(5):1715-1744
REFERENCES
- Achermann JC, Jameson JL. Fertility and infertility: genetic contributions from the hypothalamic-pituitary-gonadal axis. Mol Endocrinol 1999;13:812-818
- Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet 1998;19:125-133
- Parks JS, Brown MR, Hurley DL, Phelps CJ, Wajnrajch MP. Heritable disorders of pituitary development. J Clin Endocrinol Metab 1999;84:4362-4370
- Fofanova O, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, Evgrafov OV, Goncharov NP, Bulatov AA, Dedov II, Yamashita S. Compound heterozygous deletion of the PROP-1 gene in children with combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998;83:2601-2604
- Deladoey J, Fluck C, Buyukgebiz A, Kuhlmann BV, Eble A, Hindmarsh PC, Wu W, Mullis PE. "Hot spot" in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 1999;84:1645-1650
- Radovick S, Nations M, Du Y, Berg LA, Weintraub BD, Wondisford FE. A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency. Science 1992;257:1115-1118
- Aarskog D, Eiken HG, Bjerknes R, Myking OL. Pituitary dwarfism in the R271W Pit-1 gene mutation. Eur J Pediatr 1997;156:829-834
- Cohen LE, Wondisford FE, Salvatoni A, Maghnie M, Brucker-Davis F, Weintraub BD, Radovick S. A "hot spot" in the Pit-1 gene responsible for combined pituitary hormone deficiency: clinical and molecular correlates. J Clin Endocrinol Metab 1995;80:679-684
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van H, V, Fitzpatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet 2003;33:461-463
- Kelberman D, de Castro SC, Huang S, Crolla JA, Palmer R, Gregory JW, Taylor D, Cavallo L, Faienza MF, Fischetto R, Achermann JC, Martinez-Barbera JP, Rizzoti K, Lovell-Badge R, Robinson IC, Gerrelli D, Dattani MT. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab 2008;93:1865-1873
- Errichiello E, Gorgone C, Giuliano L, Iadarola B, Cosentino E, Rossato M, Kurtas NE, Delledonne M, Mattina T, Zuffardi O. SOX2: Not always eye malformations. Severe genital but no major ocular anomalies in a female patient with the recurrent c.70del20 variant. Eur J Med Genet 2018;61:335-340
- Stankiewicz P, Thiele H, Schlicker M, Cseke-Friedrich A, Bartel-Friedrich S, Yatsenko SA, Lupski JR, Hansmann I. Duplication of Xq26.2-q27.1, including SOX3, in a mother and daughter with short stature and dyslalia. Am J Med Genet A 2005;138:11-17
- Takagi M, Ishii T, Torii C, Kosaki K, Hasegawa T. A novel mutation in SOX3 polyalanine tract: a case of Kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary2014;17:569-574
- Jones GE, Robertson L, Warman P, Craft EV, Cresswell L, Vasudevan PC. 14q22.3 Microdeletion encompassing OTX2 in a five-generation family with microphthalmia, pituitary abnormalities, and intellectual disability. Ophthalmic Genet 2016;37:352-353
- Burris TP, Guo W, McCabe ER. The gene responsible for adrenal hypoplasia congenita, DAX-1, encodes a nuclear hormone receptor that defines a new class within the superfamily. Recent Prog Horm Res 1996;51:241-259
- Bick D, Franco B, Sherins RJ, Heye B, Pike L, Crawford J, Maddalena A, Incerti B, Pragliola A, Meitinger T, . Brief report: intragenic deletion of the KALIG-1 gene in Kallmann's syndrome. N Engl J Med 1992;326:1752-1755
- Couture C, Saveanu A, Barlier A, Carel JC, Fassnacht M, Fluck CE, Houang M, Maes M, Phan-Hug F, Enjalbert A, Drouin J, Brue T, Vallette S. Phenotypic homogeneity and genotypic variability in a large series of congenital isolated ACTH-deficiency patients with TPIT gene mutations. J Clin Endocrinol Metab 2012;97:E486-495
- Katznelson L, Alexander JM, Klibanski A. Clinical review 45: Clinically nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 1993;76:1089-1094
- Kovacs K, Scheithauer BW, Horvath E, Lloyd RV. The World Health Organization classification of adenohypophysial neoplasms. A proposed five-tier scheme. Cancer 1996;78:502-510
- Adams CB. The management of pituitary tumours and post-operative visual deterioration. Acta Neurochir (Wien ) 1988;94:103-116
- DeVile CJ, Grant DB, Hayward RD, Stanhope R. Growth and endocrine sequelae of craniopharyngioma. Arch Dis Child 1996;75:108-114
- Adamson TE, Wiestler OD, Kleihues P, Yasargil MG. Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J Neurosurg 1990;73:12-17
- Ross DA, Norman D, Wilson CB. Radiologic characteristics and results of surgical management of Rathke's cysts in 43 patients. Neurosurgery 1992;30:173-178
- Sala E, Moore JM, Amorin A, Carosi G, Martinez H, Jr., Harsh GR, Arosio M, Mantovani G, Katznelson L. Natural history of Rathke's cleft cysts: A retrospective analysis of a two centres experience. Clin Endocrinol (Oxf) 2018;89:178-186
- Takeuchi J, Handa H, Nagata I. Suprasellar germinoma. J Neurosurg 1978;49:41-48
- Ho DM, Liu HC. Primary intracranial germ cell tumor. Pathologic study of 51 patients. Cancer 1992;70:1577-1584
- Janmohamed S, Grossman AB, Metcalfe K, Lowe DG, Wood DF, Chew SL, Monson JP, Besser GM, Plowman PN. Suprasellar germ cell tumours: specific problems and the evolution of optimal management with a combined chemoradiotherapy regimen. Clin Endocrinol (Oxf) 2002;57:487-500
- Max MB, Deck MD, Rottenberg DA. Pituitary metastasis: incidence in cancer patients and clinical differentiation from pituitary adenoma. Neurology 1981;31:998-1002
- Bills DC, Meyer FB, Laws ER, Jr., Davis DH, Ebersold MJ, Scheithauer BW, Ilstrup DM, Abboud CF. A retrospective analysis of pituitary apoplexy. Neurosurgery 1993;33:602-608
- Wakai S, Fukushima T, Teramoto A, Sano K. Pituitary apoplexy: its incidence and clinical significance. J Neurosurg 1981;55:187-193
- Randeva HS, Schoebel J, Byrne J, Esiri M, Adams CB, Wass JA. Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol (Oxf) 1999;51:181-188
- Barkan AL. Pituitary atrophy in patients with Sheehan's syndrome. Am J Med Sci 1989;298:38-40
- Jenkins PJ, Chew SL, Lowe DG, Afshart F, Charlesworth M, Besser GM, Wass JA. Lymphocytic hypophysitis: unusual features of a rare disorder. Clin Endocrinol (Oxf) 1995;42:529-534
- Asa SL, Bilbao JM, Kovacs K, Josse RG, Kreines K. Lymphocytic hypophysitis of pregnancy resulting in hypopituitarism: a distinct clinicopathologic entity. Ann Intern Med 1981;95:166-171
- Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab 2001;86:1048-1053
- Thodou E, Asa SL, Kontogeorgos G, Kovacs K, Horvath E, Ezzat S. Clinical case seminar: lymphocytic hypophysitis: clinicopathological findings. J Clin Endocrinol Metab 1995;80:2302-2311
- Annane D, Sebille V, Troche G, Raphael JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA 2000;283:1038-1045
- Oksanen V. Neurosarcoidosis: clinical presentations and course in 50 patients. Acta Neurol Scand 1986;73:283-290
- Chapelon C, Ziza JM, Piette JC, Levy Y, Raguin G, Wechsler B, Bitker MO, Bletry O, Laplane D, Bousser MG, . Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine (Baltimore) 1990;69:261-276
- Stern BJ, Krumholz A, Johns C, Scott P, Nissim J. Sarcoidosis and its neurological manifestations. Arch Neurol 1985;42:909-917
- Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, Gilliland DG. Langerhans'-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med 1994;331:154-160
- Kaltsas GA, Powles TB, Evanson J, Plowman PN, Drinkwater JE, Jenkins PJ, Monson JP, Besser GM, Grossman AB. Hypothalamo-pituitary abnormalities in adult patients with langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab 2000;85:1370-1376
- Basaria S, Ayala AR, Guerin C, Dobs AS. A rare pituitary lesion. J Endocrinol Invest 2000;23:189-192
- Ferreiro J, Vinters HV. Pathology of the pituitary gland in patients with the acquired immune deficiency syndrome (AIDS). Pathology 1988;20:211-215
- Milligan SA, Katz MS, Craven PC, Strandberg DA, Russell IJ, Becker RA. Toxoplasmosis presenting as panhypopituitarism in a patient with the acquired immune deficiency syndrome. Am J Med 1984;77:760-764
- Littley MD, Shalet SM, Beardwell CG, Ahmed SR, Applegate G, Sutton ML. Hypopituitarism following external radiotherapy for pituitary tumours in adults. Q J Med 1989;70:145-160
- Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: a systematic review. JAMA 2007;298:1429-1438
- Personnier C, Crosnier H, Meyer P, Chevignard M, Flechtner I, Boddaert N, Breton S, Mignot C, Dassa Y, Souberbielle JC, Piketty M, Laborde K, Jais JP, Viaud M, Puget S, Sainte-Rose C, Polak M. Prevalence of pituitary dysfunction after severe traumatic brain injury in children and adolescents: a large prospective study. J Clin Endocrinol Metab 2014;99:2052-2060
- de BH, Blok GJ, van d, V. Clinical aspects of growth hormone deficiency in adults. Endocr Rev 1995;16:63-86
- Christ ER, Carroll PV, Russell-Jones DL, Sonksen PH. The consequences of growth hormone deficiency in adulthood, and the effects of growth hormone replacement. Schweiz Med Wochenschr 1997;127:1440-1449
- Carroll PV, Christ ER, Bengtsson BA, Carlsson L, Christiansen JS, Clemmons D, Hintz R, Ho K, Laron Z, Sizonenko P, Sonksen PH, Tanaka T, Thorne M. Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. Growth Hormone Research Society Scientific Committee. J Clin Endocrinol Metab 1998;83:382-395
- Bulow B, Hagmar L, Eskilsson J, Erfurth EM. Hypopituitary females have a high incidence of cardiovascular morbidity and an increased prevalence of cardiovascular risk factors. J Clin Endocrinol Metab 2000;85:574-584
- Rosen T, Wilhelmsen L, Landin-Wilhelmsen K, Lappas G, Bengtsson BA. Increased fracture frequency in adult patients with hypopituitarism and GH deficiency. Eur J Endocrinol 1997;137:240-245
- Maghnie M, Cosi G, Genovese E, Manca-Bitti ML, Cohen A, Zecca S, Tinelli C, Gallucci M, Bernasconi S, Boscherini B, Severi F, Arico M. Central diabetes insipidus in children and young adults. N Engl J Med 2000;343:998-1007
- Trainer PJ, Besser GM. The Bart's Protocols. Churchill Livingstone; 1995.
- Leibowitz RL, Dorff TB, Tucker S, Symanowski J, Vogelzang NJ. Testosterone replacement in prostate cancer survivors with hypogonadal symptoms. BJU Int 2010;105:1397-1401
- SR P, A G, DE P, SR A, J F-S, TA H. A retrospective audit of the combined pituitary function test, using the insulin stress test, TRH and GnRH in a district laboratory. Clin Endocrinol (Oxf). Vol 361992:135-139.
- Jones SL, Trainer PJ, Perry L, Wass JA, Bessser GM, Grossman A. An audit of the insulin tolerance test in adult subjects in an acute investigation unit over one year. Clin Endocrinol (Oxf) 1994;41:123-128
- Hurel SJ, Thompson CJ, Watson MJ, Harris MM, Baylis PH, Kendall-Taylor P. The short Synacthen and insulin stress tests in the assessment of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol (Oxf) 1996;44:141-146
- Littley MD, Gibson S, White A, Shalet SM. Comparison of the ACTH and cortisol responses to provocative testing with glucagon and insulin hypoglycaemia in normal subjects. Clin Endocrinol (Oxf) 1989;31:527-533
- Simsek Y, Karaca Z, Tanriverdi F, Unluhizarci K, Selcuklu A, Kelestimur F. A comparison of low-dose ACTH, glucagon stimulation and insulin tolerance test in patients with pituitary disorders. Clin Endocrinol (Oxf) 2015;82:45-52
- Hoffman DM, O'Sullivan AJ, Baxter RC, Ho KK. Diagnosis of growth-hormone deficiency in adults. Lancet 1994;343:1064-1068
- Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab2000;85:3990-3993
- Ghigo E, Bellone J, Aimaretti G, Bellone S, Loche S, Cappa M, Bartolotta E, Dammacco F, Camanni F. Reliability of provocative tests to assess growth hormone secretory status. Study in 472 normally growing children. J Clin Endocrinol Metab 1996;81:3323-3327
- Valetto MR, Bellone J, Baffoni C, Savio P, Aimaretti G, Gianotti L, Arvat E, Camanni F, Ghigo E. Reproducibility of the growth hormone response to stimulation with growth hormone-releasing hormone plus arginine during lifespan. Eur J Endocrinol 1996;135:568-572
- Maghnie M, Cavigioli F, Tinelli C, Autelli M, Arico M, Aimaretti G, Ghigo E. GHRH plus arginine in the diagnosis of acquired GH deficiency of childhood-onset. J Clin Endocrinol Metab 2002;87:2740-2744
- Garcia JM, Biller BMK, Korbonits M, Popovic V, Luger A, Strasburger CJ, Chanson P, Medic-Stojanoska M, Schopohl J, Zakrzewska A, Pekic S, Bolanowski M, Swerdloff R, Wang C, Blevins T, Marcelli M, Ammer N, Sachse R, Yuen KCJ. Macimorelin as a Diagnostic Test for Adult GH Deficiency. J Clin Endocrinol Metab 2018;103:3083-3093
- Fenske W, Refardt J, Chifu I, Schnyder I, Winzeler B, Drummond J, Ribeiro-Oliveira A, Jr., Drescher T, Bilz S, Vogt DR, Malzahn U, Kroiss M, Christ E, Henzen C, Fischli S, Tonjes A, Mueller B, Schopohl J, Flitsch J, Brabant G, Fassnacht M, Christ-Crain M. A Copeptin-Based Approach in the Diagnosis of Diabetes Insipidus. N Engl J Med 2018;379:428-439
- Christ-Crain M. Diabetes Insipidus: New Concepts for Diagnosis. Neuroendocrinology 2020;110:859-867
- Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, Samuels MH. Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016;101:3888-3921
- Groves RW, Toms GC, Houghton BJ, Monson JP. Corticosteroid replacement therapy: twice or thrice daily? J R Soc Med 1988;81:514-516
- Bleicken B, Hahner S, Loeffler M, Ventz M, Decker O, Allolio B, Quinkler M. Influence of hydrocortisone dosage scheme on health-related quality of life in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2010;72:297-304
- Peacey SR, Guo CY, Robinson AM, Price A, Giles MA, Eastell R, Weetman AP. Glucocorticoid replacement therapy: are patients over treated and does it matter? Clin Endocrinol (Oxf) 1997;46:255-261
- Swords FM, Drake WM, Carroll PV, Monson JP. Hormonal therapy for hypopituitarism. Encyclopaedia of Endocrine Disease. Vol 22004:662-671.
- Newell-Price J, Whiteman M, Rostami-Hodjegan A, Darzy K, Shalet S, Tucker GT, Ross RJ. Modified-release hydrocortisone for circadian therapy: a proof-of-principle study in dexamethasone-suppressed normal volunteers. Clin Endocrinol (Oxf) 2008;68:130-135
- Debono M, Ghobadi C, Rostami-Hodjegan A, Huatan H, Campbell MJ, Newell-Price J, Darzy K, Merke DP, Arlt W, Ross RJ. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009;94:1548-1554
- Verma S, Vanryzin C, Sinaii N, Kim MS, Nieman LK, Ravindran S, Calis KA, Arlt W, Ross RJ, Merke DP. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2010;72:441-447
- Johannsson G, Nilsson AG, Bergthorsdottir R, Burman P, Dahlqvist P, Ekman B, Engstrom BE, Olsson T, Ragnarsson O, Ryberg M, Wahlberg J, Biller BM, Monson JP, Stewart PM, Lennernas H, Skrtic S. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation. J Clin Endocrinol Metab 2012;97:473-481
- Porter J, Withe M, Ross RJ. Immediate-release granule formulation of hydrocortisone, Alkindi(R), for treatment of paediatric adrenal insufficiency (Infacort development programme). Expert Rev Endocrinol Metab 2018;13:119-124
- Oksnes M, Bjornsdottir S, Isaksson M, Methlie P, Carlsen S, Nilsen RM, Broman JE, Triebner K, Kampe O, Hulting AL, Bensing S, Husebye ES, Lovas K. Continuous subcutaneous hydrocortisone infusion versus oral hydrocortisone replacement for treatment of addison's disease: a randomized clinical trial. J Clin Endocrinol Metab 2014;99:1665-1674
- Ho W, Druce M. Quality of life in patients with adrenal disease: A systematic review. Clin Endocrinol (Oxf) 2018;89:119-128
- Johannsson G, Burman P, Wiren L, Engstrom BE, Nilsson AG, Ottosson M, Jonsson B, Bengtsson BA, Karlsson FA. Low dose dehydroepiandrosterone affects behavior in hypopituitary androgen-deficient women: a placebo-controlled trial. J Clin Endocrinol Metab 2002;87:2046-2052
- Brooke AM, Kalingag LA, Miraki-Moud F, Camacho-Hubner C, Maher KT, Walker DM, Hinson JP, Monson JP. Dehydroepiandrosterone improves psychological well-being in male and female hypopituitary patients on maintenance growth hormone replacement. J Clin Endocrinol Metab 2006;91:3773-3779
- Brooke AM, Kalingag LA, Miraki-Moud F, Camacho-Hubner C, Maher KT, Walker DM, Hinson JP, Monson JP. Dehydroepiandrosterone (DHEA) replacement reduces growth hormone (GH) dose requirement in female hypopituitary patients on GH replacement. Clin Endocrinol (Oxf) 2006;65:673-680
- Evered D, Young ET, Ormston BJ, Menzies R, Smith PA, Hall R. Treatment of hypothyroidism: a reappraisal of thyroxine therapy. Br Med J 1973;3:131-134
- Gullo D, Latina A, Frasca F, Le Moli R, Pellegriti G, Vigneri R. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS One 2011;6:e22552
- Agha A, Walker D, Perry L, Drake WM, Chew SL, Jenkins PJ, Grossman AB, Monson JP. Unmasking of central hypothyroidism following growth hormone replacement in adult hypopituitary patients. Clin Endocrinol (Oxf) 2007;66:72-77
- Martins MR, Doin FC, Komatsu WR, Barros-Neto TL, Moises VA, Abucham J. Growth hormone replacement improves thyroxine biological effects: implications for management of central hypothyroidism. J Clin Endocrinol Metab 2007;92:4144-4153
- Losa M, Scavini M, Gatti E, Rossini A, Madaschi S, Formenti I, Caumo A, Stidley CA, Lanzi R. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid 2008;18:1249-1254
- Gammage M, Franklyn J. Hypothyroidism, thyroxine treatment, and the heart. Heart 1997;77:189-190
- Shufelt CL, Manson JE. Menopausal Hormone Therapy and Cardiovascular Disease: The Role of Formulation, Dose, and Route of Delivery. J Clin Endocrinol Metab 2021;106:1245-1254
- Mehta J, Kling JM, Manson JE. Risks, Benefits, and Treatment Modalities of Menopausal Hormone Therapy: Current Concepts. Front Endocrinol (Lausanne) 2021;12:564781
- Wierman ME, Arlt W, Basson R, Davis SR, Miller KK, Murad MH, Rosner W, Santoro N. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014;99:3489-3510
- Bhasin S, Storer TW, Berman N, Yarasheski KE, Clevenger B, Phillips J, Lee WP, Bunnell TJ, Casaburi R. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab 1997;82:407-413
- Harle L, Basaria S, Dobs AS. Nebido: a long-acting injectable testosterone for the treatment of male hypogonadism. Expert Opin Pharmacother 2005;6:1751-1759
- Handelsman DJ, Conway AJ, Boylan LM. Pharmacokinetics and pharmacodynamics of testosterone pellets in man. J Clin Endocrinol Metab 1990;71:216-222
- Arver S, Dobs AS, Meikle AW, Caramelli KE, Rajaram L, Sanders SW, Mazer NA. Long-term efficacy and safety of a permeation-enhanced testosterone transdermal system in hypogonadal men. Clin Endocrinol (Oxf) 1997;47:727-737
- Swerdloff RS, Wang C, Cunningham G, Dobs A, Iranmanesh A, Matsumoto AM, Snyder PJ, Weber T, Longstreth J, Berman N. Long-term pharmacokinetics of transdermal testosterone gel in hypogonadal men. J Clin Endocrinol Metab 2000;85:4500-4510
- Korbonits M, Slawik M, Cullen D, Ross RJ, Stalla G, Schneider H, Reincke M, Bouloux PM, Grossman AB. A comparison of a novel testosterone bioadhesive buccal system, striant, with a testosterone adhesive patch in hypogonadal males. J Clin Endocrinol Metab 2004;89:2039-2043
- Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM. Testosterone therapy in adult men with androgen deficiency syndromes: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2006;91:1995-2010
- Landau D, Tsakok T, Aylwin S, Hughes S. Should testosterone replacement be offered to hypogonadal men treated previously for prostatic carcinoma? Clin Endocrinol (Oxf) 2012;76:179-181
- Odeh M, Oliven A. Coma and seizures due to severe hyponatremia and water intoxication in an adult with intranasal desmopressin therapy for nocturnal enuresis. J Clin Pharmacol 2001;41:582-584
- Levy M, Prentice M, Wass J. Diabetes insipidus. BMJ 2019;364:l321
- Ebrahimi F, Kutz A, Wagner U, Illigens B, Siepmann T, Schuetz P, Christ-Crain M, Mueller B, Christ ER. Excess Mortality Among Hospitalized Patients With Hypopituitarism-A Population-Based, Matched-Cohort Study. J Clin Endocrinol Metab 2020;105